
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn unexpected P-cluster like intermediate en route to the nitrogenase FeMo-co†‡
Leon P.
Jenner
 a,
Mickael V.
Cherrier
a,
Patricia
Amara
a,
Luis M.
Rubio
*b and
Yvain
Nicolet
*a
a,
Mickael V.
Cherrier
a,
Patricia
Amara
a,
Luis M.
Rubio
*b and
Yvain
Nicolet
*a
aUniv. Grenoble Alpes, CEA, CNRS, IBS, Metalloproteins Unit, F-38000 Grenoble, France. E-mail: yvain.nicolet@ibs.fr
bCentro de Biotecnología y Genómica de Plantas, Universidad Politécnica de Madrid, Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria, Pozuelo de Alarcón, 28223 Madrid, Spain. E-mail: lm.rubio@upm.es
First published on 4th March 2021
Abstract
The nitrogenase MoFe protein contains two different FeS centers, the P-cluster and the iron–molybdenum cofactor (FeMo-co). The former is a [Fe8S7] center responsible for conveying electrons to the latter, a [MoFe7S9C-(R)-homocitrate] species, where N2 reduction takes place. NifB is arguably the key enzyme in FeMo-co assembly as it catalyzes the fusion of two [Fe4S4] clusters and the insertion of carbide and sulfide ions to build NifB-co, a [Fe8S9C] precursor to FeMo-co. Recently, two crystal structures of NifB proteins were reported, one containing two out of three [Fe4S4] clusters coordinated by the protein which is likely to correspond to an early stage of the reaction mechanism. The other one was fully complemented with the three [Fe4S4] clusters (RS, K1 and K2), but was obtained at lower resolution and a satisfactory model was not obtained. Here we report improved processing of this crystallographic data. At odds with what was previously reported, this structure contains a unique [Fe8S8] cluster, likely to be a NifB-co precursor resulting from the fusion of K1- and K2-clusters. Strikingly, this new [Fe8S8] cluster has both a structure and coordination sphere geometry reminiscent of the fully reduced P-cluster (PN-state) with an additional μ2-bridging sulfide ion pointing toward the RS cluster. Comparison of available NifB structures further unveils the plasticity of this protein and suggests how ligand reorganization would accommodate cluster loading and fusion in the time-course of NifB-co synthesis.
Introduction
In stark contrast to the energy-intensive Haber–Bosch process,1 nitrogen-fixing bacteria and archaea can transform atmospheric N2 to NH3 at ambient temperature using nitrogenases.2 These enzymes carry unique and complex metalloclusters that enable their function.3 The most prominent of these is FeMo-co, the catalytic [MoFe7S9C-(R)-homocitrate] cluster of molybdenum nitrogenase,4–6 which is supplied with electrons by a specialized [Fe8S7] redox site termed the P-cluster.7,8 Each of these complex clusters originates from the combination of two simple [Fe4S4] clusters.9,10 P-cluster maturation occurs in situ on the catalytic MoFe protein component of nitrogenase, typically requiring the dinitrogenase reductase (Fe protein) component, but also achievable in vitro using a strong reducing agent (∼−500 mV vs. standard hydrogen electrode).11,12 FeMo-co formation is more complicated and initially requires the dedicated radical S-adenosyl-L-methionine (SAM) enzyme NifB.13The exact mechanism of NifB has yet to be elucidated, but since the purification of functional enzyme was achieved less than two decades ago13 much has been discovered about its structure and activity.3 NifB can bind three [Fe4S4] clusters, two of which fuse and incorporate additional C and S atoms to produce NifB-co, the [Fe8S9C] cluster that serves as biosynthetic precursor to FeMo-co.13,14 NifB-co is then transferred to the scaffold protein NifEN where Mo and R-homocitrate are inserted through a complex and ill-characterized process. This mature FeMo-co is then transferred to apo-MoFe protein (containing P-clusters but devoid of FeMo-co) to yield catalytically competent enzyme.13,15 Of the three NifB [Fe4S4] clusters, one binds to a typical Cx3Cx2C radical SAM specific motif (the RS-cluster) corresponding to C38, C42 and C45 in Methanobacterium thermoautotrophicum (MthNifB, all residue numbers refer to this organism unless stated otherwise).16 Radical SAM enzymes belong to a superfamily of over 120![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 members catalyzing a broad range of reactions with various substrates.17 Almost all of them use a [Fe4S4]+ cluster to cleave SAM into methionine and a highly reactive 5′-deoxyadenosyl radical (5′-dA˙). In most cases, the 5′-dA˙ abstracts a hydrogen atom from a substrate to trigger a radical-based reaction.
000 members catalyzing a broad range of reactions with various substrates.17 Almost all of them use a [Fe4S4]+ cluster to cleave SAM into methionine and a highly reactive 5′-deoxyadenosyl radical (5′-dA˙). In most cases, the 5′-dA˙ abstracts a hydrogen atom from a substrate to trigger a radical-based reaction.
The remaining two [Fe4S4] clusters of NifB are substrates for the formation of NifB-co. These are believed to be in close proximity at some stage of the reaction and are jointly termed the K-cluster or individually referred to as K1 and K2. Interatomic distances determined by pulsed EPR and X-ray spectroscopy experiments were used to propose C18, C52 and C115 in MthNifB as K1 ligands with occasional ligation from an unknown histidine.10 The recent crystal structure of Methanotrix thermoacetophila (MtNifB) solved by X-ray diffraction18 identified H31, C18 and C115 as K1 ligands, but unexpectedly featured C52 as fourth ligand of the RS cluster. Importantly, C52 was part of a short flexible loop, which also provides E53 as a fourth ligand of the K1-cluster.18 The residues C260 and C263 were proposed as K2-cluster ligands because their replacement with alanine led to [Fe4S4]+ cluster EPR signal disappearance along with impaired NifB-co production.16 Spectroscopically-determined interatomic distances are consistent with this ligand assignment and suggested C264 as an additional K2-cluster ligand in Methanosarcina acetivorans NifB, although this residue is not conserved in MthNifB or MtNifB.10 Equivalent residues to C260 and C263 are present in the sequence of MtNifB, however the published structure does not model these residues as the lack of K2-cluster resulted in disordered C-terminus stretch.18
Radioisotope labeling indicated that the carbide ion of NifB-co originates from one SAM molecule, which is converted to S-adenosyl-L-homocysteine (SAH).19 Combined site-directed mutagenesis and EPR spectroscopy experiments supported that such SAM binding requires that all the [Fe4S4] clusters are reduced beforehand and suggested that the K2-cluster receives the methyl group of SAM.10 Acid treatment of this species liberated methanethiol, consistent with the proposal that one of the K2-cluster sulfides may attack the methyl group of SAM as part of a simple SN2 mechanism not involving radical chemistry.10 A second molecule of SAM then binds and is cleaved into 5′-dA˙, which abstracts a hydrogen atom from the transferred methyl group to form a methylene radical species. This species presumably deprotonates twice and remains inserted into the forming cluster as a carbide ion.19 Both SAM reactions require the cysteine ligands proposed for the RS- and K2-clusters (see above).10 The nascent NifB-co then requires a ninth sulfur atom at the belt position. It was possible to use sulfite and reductant to generate this S atom in vitro.20 However, an enzyme with sulfur transferase activity probably performs this role in vivo.
The first published 1.95 Å resolution MtNifB crystal structure only contained the RS- and K1-clusters and identified a flexible loop bound to the unique iron site of the RS-cluster and to the K1-cluster via C52 and E53, respectively.18 We proposed that this structure corresponds to an early stage of the reaction with only the K1-cluster substrate loaded and the flexible loop acting as a stopper to prevent unwanted SAM binding and cleavage before the K2-cluster is present.18 Replacing the flexible loop ligands C52 and E53 abolished NifB-co production but retained SAM cleavage favoring 5′-dA˙ formation over methyl transfer.18
More recently, a 3.0 Å-resolution crystal structure of NifB from M. thermoautotrophicum was published by Kang and co-workers with a full complement of three [Fe4S4] clusters (RS, K1 and K2) in the absence of SAM.21 However, the authors fail to convincingly model key structural information, especially relating to the [Fe4S4] clusters, which were modeled as perfect cubes in the corresponding electron densities. The resulting geometry and ligand coordination are thus not representative of a true structure and no clear mechanistic conclusions can be drawn from this model (Fig. S1‡). A careful look at the corresponding electron density map clearly indicates that refinement was not carried out to convergence (Fig. S2 and S3‡). Indeed, in addition to a failure in refining the [Fe4S4] clusters with correct geometry and ligand bound restraints, other significant parts of the structure were not correctly modeled. For instance, many omitted side chains have clear corresponding electron density (Fig. S2‡) and the N-terminus stretches for both molecules in the asymmetric unit have electron density present for residues 7–15 that were absent in the previously published model (Fig. S2A‡). The same problem applies to the C-terminus stretch of the chain A molecule (Fig. S2D‡). All these observations led us to reinvestigate the published X-ray data. Here we report an improved processing of the structure, which sheds new light on the first steps of the NifB catalytic cycle and draws interesting parallels to the formation of the P-cluster in nitrogenase.
Results
Our refined model (PDB 7BI7) contains residues 7-285 for chain A and 7-277 for chain B and has Rwork and Rfree factors = 0.202 and 0.239, respectively (in comparison to 0.262 and 0.314 for PDB 7JMB) (see ESI and Table S1‡). The overall structure has an RMSD of 0.38 Å when compared to PDB 7JMB. Many side chains that were missing in the original structure were added when present in the electron density map (Fig. S2, S4 and S5‡). As was previously shown, NifB structure can be best described as a ¾ β-barrel that corresponds to the core RS-domain surrounded by short N- and C-terminus stretches that define K1- and K2-cluster binding domains, respectively (see Fig. 1).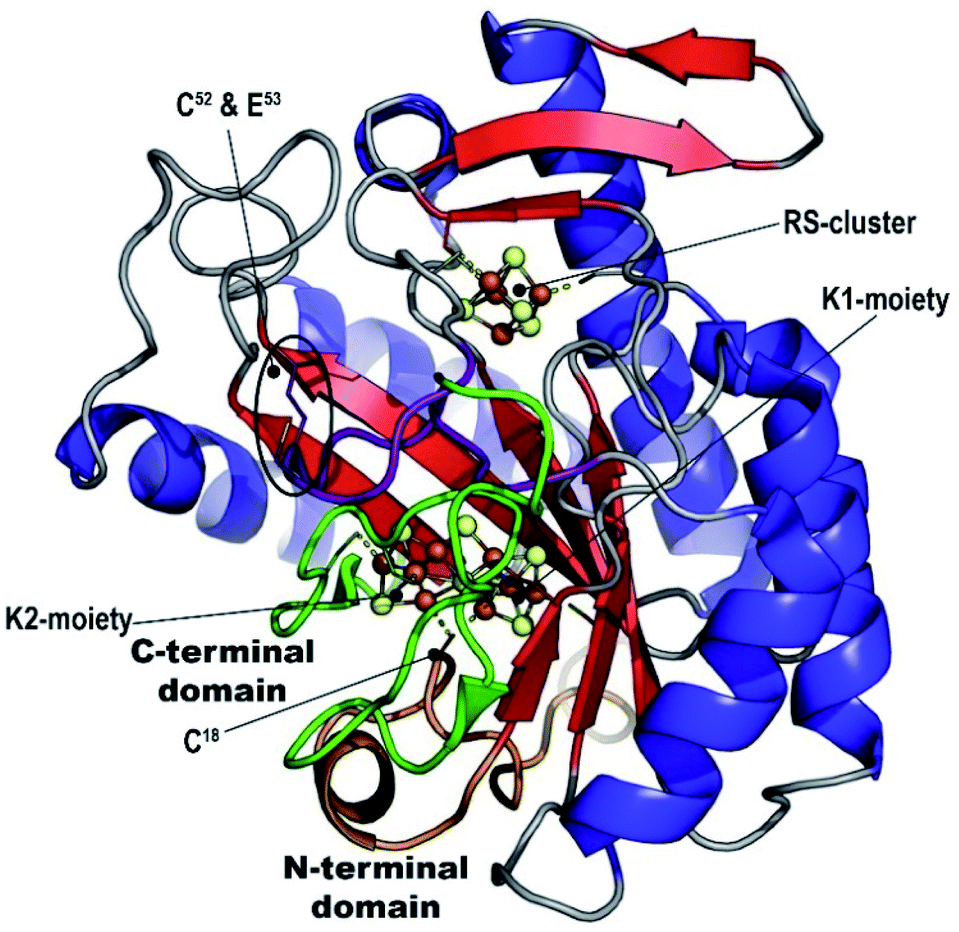 | ||
| Fig. 1 MthNifB crystal structure. The radical SAM core structure β-barrel is depicted in red (strands) and blue (helices). The N- and C-terminal domains are depicted in orange and green, respectively. The FeS-cluster iron and sulfur atoms are represented in brown and yellow spheres, respectively. The bridging cysteine residue C18 that belongs to the N-terminal domain is indicated. The flexible loop is depicted in purple. C52 and E53 are also indicated. | ||
Manual placement of the [Fe4S4] K1- and K2-clusters at reasonable distances to the different ligands was not possible. Similarly, once correct geometry restraints for both the [Fe4S4] clusters and ligands to iron bonds were set (2.3 Å for Cys–S–Fe and 2.4 Å for His–N–Fe bonds), our attempts to refine the K1 and K2 clusters using BUSTER software22 always led to displacements of the two moieties bringing one of their respective sulfide ions closer to the other (Fig. S6‡). This displacement, in addition to the observation of an unexplained strong bump in the electron density map, suggested K1- and K2-clusters might be fused, sharing one of their sulfide ions, and bridged together by a monoatomic ligand occupying the bump. The oblong-shaped electron density surrounded by the strictly conserved residues C18, H31, C115, C260 and C263 has a geometry reminiscent of the coordination sphere of the reduced P-cluster from the nitrogenase MoFe protein (Fig. 2).
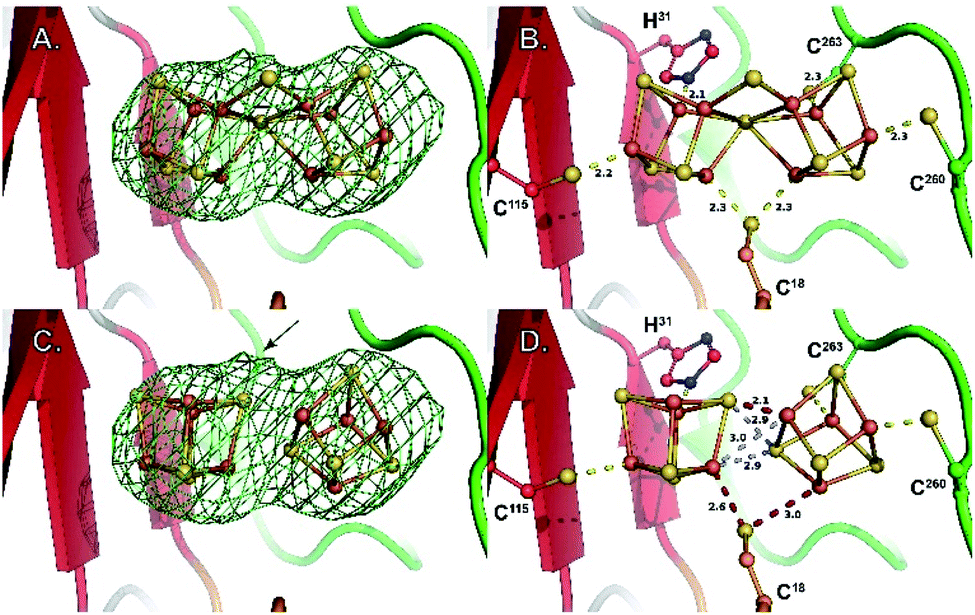 | ||
| Fig. 2 Close-up of the K1 and K2 cluster-binding site. (A) Fo − Fc difference Fourier electron density map (omit map) around the K-cluster contoured at 3σ. (B) Coordination geometry of the K-cluster in the newly refined structure. (C) Model of two independent [Fe4S4] clusters in the same Fo − Fc difference Fourier electron density map (omit map) as in (A), illustrating a lower fit when compared to (A). The arrow indicates the position of the bump observed in the electron density map (see also Fig. S2C‡) (D) coordination geometry obtained when refining two independent [Fe4S4] clusters. Both the ligand coordination and the [Fe4S4] cluster contacts are chemically meaningless. Stereoviews of each panel are available as Fig. S7–S10,‡ respectively. | ||
Replacing the K1 and K2 clusters with a P-cluster model in its reduced PN state (residue CLF from PDB 3U7Q) displays a nearly perfect fit to the electron density. Unexpectedly, when superimposing this P-cluster like center with that from the MoFe protein, a perfect match is also observed between the positions of the ligand residues C18, H31, C115, C260 and C263 (NifB) and α-C88, α-C154, α-C62, β-C70 and β-C153 (MoFe protein; see Fig. 2 and 3). Strikingly, the Sγ of residue β-C95 in MoFe protein is located at the same position as the previously unmodeled bump in the electron density. This bump was therefore modeled as a sulfide ion bridging two iron atoms resulting in a novel [Fe8S8] cluster with P-cluster-like structure (see Fig. 2 and 3). We propose that this model corresponds to the true K-cluster, which, rather than previously proposed discrete [Fe4S4] modules, would be an already fused cluster intermediate during the conversion of K1 and K2 into NifB-co (the L-cluster). Unfortunately, current data resolution does not allow more accurate description of its geometry and would require higher-resolution diffraction data to determine its exact structure. However, it is notable that the external bridging sulfide ion points toward the unique Fe site of the RS-cluster and is kept in place by strictly conserved residues N194 and K192 (see Fig. S6‡). Both residues are located in the crevice defined by the partial β-barrel of the RS-domain. This suggests that this sulfide, already occupying a position analogous to the belt sulfides in FeMo-co, might be the actual acceptor site for methyl transfer from SAM. In addition, at this stage of reaction mechanism, residue C18 from the N-terminus domain previously identified as ligand of the K1-cluster at an earlier stage,18 occupies a bridging position between the two moieties of the K-cluster, analogous to a FeMo-co belt sulfide.
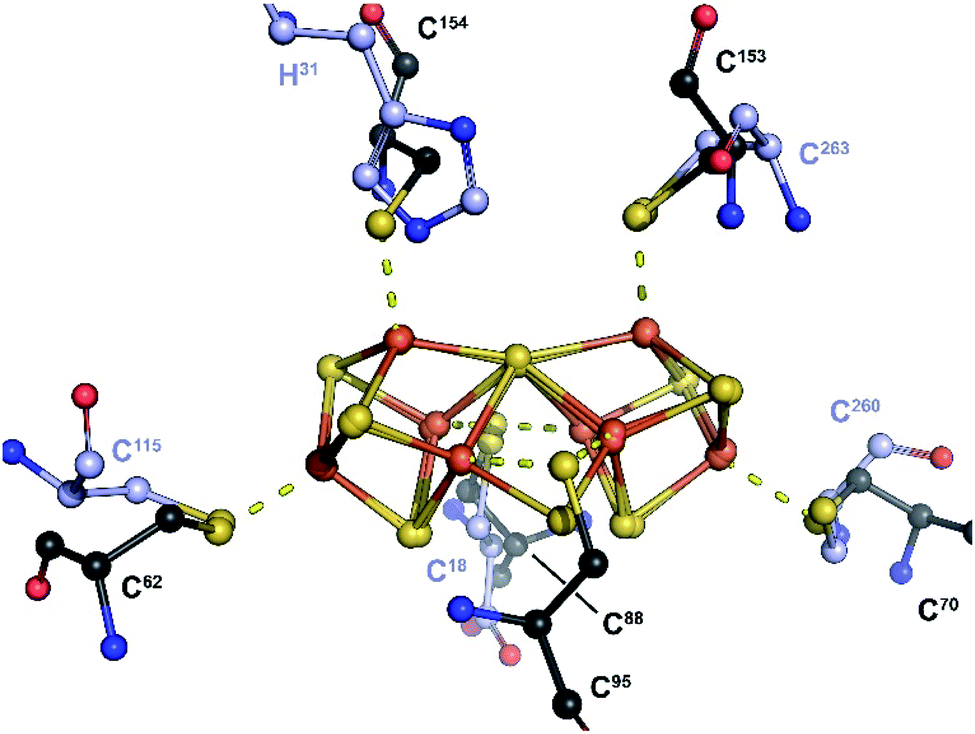 | ||
| Fig. 3 Overlay of the P-cluster from the MoFe protein (PN-state; PDB 3U7Q) and the K-cluster from NifB. Only the P- and K-cluster atoms were used for superposition. The carbon atoms of amino acid residues serving as ligands of the P- and K-clusters are depicted in black and purple, respectively. In the MoFe-protein, residues C62, C88 and C154 belong to chain C (NifD or α chain), while residues C70, C95 and C153 belong to chain D (NifK or β chain). A stereoview is available as Fig. S11.‡ | ||
The distance between the belt sulfide and the RS-cluster unique iron is 10.5 Å (see Fig. 1). As previously shown, despite the fact that no SAM-bound NifB structure is available, all the SAM-binding motifs are present at the RS-cluster site. This suggests that SAM would bind to NifB in the same orientation observed in all other radical SAM enzymes.23 Yet, in that orientation, the SAM methyl would be positioned on the opposite side of the K-cluster. If this were the case, methyl transfer would logically require either an alternative SAM binding mode or a shuttle residue as yet unidentified. We used molecular docking to screen for plausible alternative SAM binding modes (see ESI‡ for details). In the high score poses, the adenine moiety always binds to the classical SAM binding site (Fig. S12‡). However, differences are observed at the ribose moiety, with poses placing the methyl group at an average of 3.7 Å from the belt-sulfide ion, compatible with direct transfer. Unlike most radical SAM enzymes, NifB does not contain a conserved E/D residue that interacts with the 2′ and 3′ hydroxyl groups of the ribose moiety. Instead, NifB has a conserved threonine residue at that position (T141) that always interacts with the ribose 3′ hydroxyl group as S126 does in MoaA (PDB 1TV8).24 This peculiar feature may result in looser interactions that would allow increased flexibility of SAM binding leading to alternative binding modes to allow both methyl transfer and SAM cleavage from the same site. Further experimental evidence is required to settle this question.
Discussion
The K-cluster structure presented here unveils a key step in the mechanism of NifB and is consistent with the requirement of K1- and K2-clusters reduction prior to methyl transfer.25 We propose that, similar to what has been reported for P-cluster synthesis,11 K-cluster formation could be induced by full reduction of two [Fe4S4]2+ centers in proximity. An all-ferrous K-cluster hypothesis is attractive because it would render the external bridging sulfide more nucleophilic, consistent with a subsequent SN2 type attack on the methyl group of SAM. In addition, it might facilitate subsequent carbide insertion as the internal μ6-bound sulfide could be displaced by carbide to another belt position. However, it must be noted that nonenzymatic P-cluster synthesis requires the strong reductant Ti(III) citrate,11 while MthNifB was fully reconstituted with FeS clusters and treated with sodium dithionite as reductant before crystallization.21 In addition, previous EPR analyses of dithionite-reduced NifB reported a three-[Fe4S4]+ state of the enzyme,10,16 although further K1 and K2 reduction to the [Fe4S4]0 state in some NifB molecules cannot be discarded as only the [Fe4S4]+ state is detectable by EPR. The time-scale difference between EPR and X-ray crystallography experiments (seconds versus days) might also allow for complete reduction and cluster fusion to occur before crystallization. Thus, advanced spectroscopic NifB characterization is required to establish the K-cluster reduction state.Structural comparison between the K-cluster bound MthNifB and the K1-cluster bound MtNifB shows that their N-terminal and RS-domains are highly similar with RMSD = 0.96 Å for 234 aligned Cα atoms and about 50% amino acid sequence identities. Residues corresponding to C18, H31 and C115 and the K1-cluster or K1-cluster derived fragment of the K-cluster, sit at the same position in both structures, suggesting only slight motions of that region would occur upon cluster fusion. Conversely, the unique flexible loop that contains strictly conserved residues C52 and E53 (in purple in Fig. 1) is now pushed back out of the active site crevice, freeing the RS-cluster to bind SAM and allowing K-cluster formation, as was previously proposed. Substitution of either of these two residues led to NifB protein variants with greatly reduced levels of NifB-co production that were still able to unproductively cleave SAM.18 We previously proposed that this specific loop would play a role in controlling SAM binding and cleavage depending on the presence of the K2-cluster, a hypothesis also supported by this new structure.
Kang and colleagues proposed that residue H24 was a ligand of the K2-cluster.21 However, this residue is not conserved in all NifB sequences and can sometimes be shifted by one position in the primary amino acid sequence. Examination of NifB amino acid sequence alignments indicates that there is no insertion or deletion in that region between the HPC and Cx2Cx2C motifs (residues 16–18 and 38–45, respectively) and both Mth and MtNifB structures display the same structure for the N-terminal domain. Thus, as evidenced by the MtNifB structure, the shift places H24 too far from the K2-cluster binding site to reasonably be considered a ligand of the K2-cluster.18 Conversely, H24 is often replaced by asparagine. Both residues can establish hydrogen bonds with one of the K-cluster sulfide ions, as suggested in the corrected MthNifB structure presented in this work. Such N–H–S hydrogen bonds are commonly observed in the structures of FeS-cluster containing proteins, so it is more likely that H24 is semi-conserved for this role in stabilizing and tuning redox potential, rather than being a direct ligand. Thus, the K2-cluster has only two identified cysteinyl ligands and no obvious candidates for possible third and fourth ligands.
The MthNifB structure indicates that the C-terminal domain, starting at residue 255, spans over the partial β-barrel. It consists of a short β-strand interacting with the edge of the RS-domain, followed by a loop that contains cysteine residues C260 and C263 and a β-hairpin, which interacts with the first strand of the RS-domain (in green in Fig. 1). Both the apo-MthNifB and K1-bound MtNifB structures highlight that the presence of [Fe4S4]-clusters is key to the folding of their domains.18,21 As we previously proposed, the structural arrangement of the N- and C-terminal domains supports a mechanism where K1 is loaded first sequentially rather than K1 and K2 being loaded simultaneously (see Fig. 4).18 Indeed, considering the partial β-barrel as the structural core of NifB, the N-terminal domain is located at its bottom, placing the K1-cluster deep inside the crevice (see Fig. 1). The short C-terminal domain acts like a strap, closing the side of the β-barrel to stabilize the K2-cluster in a less-buried environment (in green in Fig. 1). These observations suggest that upon [Fe4S4] cluster loading by an iron–sulfur cluster scaffold or chaperone (like NifU in Azotobacter vinelandii),3 the K1-cluster would bind first, stabilizing the N-terminal domain in its observed position in both Mt and MthNifB structures.18,21 Then, the short loop that contains C260 and C263 would accommodate the K2-cluster. It is not clear whether residues E53 and C18 would play a role in this step. Upon reduction, the two clusters would get close enough for fusion to occur with only minor structural changes to NifB itself, such as a slight displacement of the loop with C260 and C263. K-cluster formation would then trigger the release of the flexible loop and allow SAM binding. Since the analogous P-cluster exhibits different structures depending on its redox state, it cannot be excluded that similar intermediate forms may exist during or after K-cluster fusion.26 Further structural characterizations of well-defined redox states at higher resolution would therefore be required to better understand how these clusters fuse.
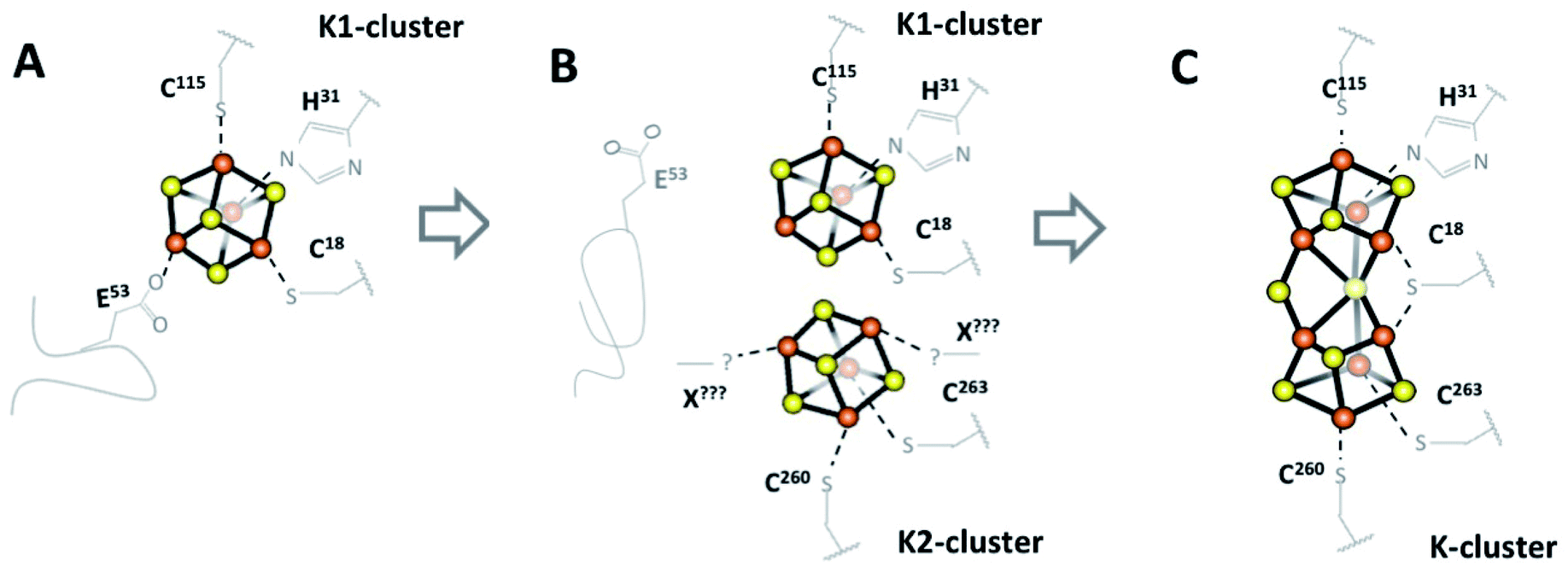 | ||
| Fig. 4 Schematic view of the early steps for NifB-co formation by NifB. (A) NifB initially binds two [Fe4S4] clusters, an RS-cluster (not shown) and a K1-cluster with four ligands including labile E53 on a flexible loop.18 (B) E53 is displaced allowing a second [Fe4S4] cluster, K2, to bind to previously disordered C-terminal residues C260, C263 and likely one or two unknown additional ligands. (C) The ordered coordination environment then forces K1 and K2 into proximity where they fuse into a single [Fe8S8] K-cluster (the structure in this work). NB: This mechanism is not charge balanced, pending further spectroscopic evidence to identify the oxidation states of the clusters. | ||
The K-cluster coordination observed in the MthNifB structure suggests that upon rearrangement from K to NifB-co, H31 and C263 would be displaced by the new belt sulfide in agreement with the previous observation of a loss of a nitrogen ligand.27 C260 and C115 in this case would remain bound to the two terminal iron ions and C18 would still occupy a bridging position where the ninth sulfide ion would bind at a later stage to produce NifB-co. Strikingly, C18 belongs to the N-terminal domain and is thus located at the bottom of the partial β-barrel (see Fig. 1). As was suggested by the X-ray structure of the apo MthNifB (PDB 7JMA),21 when unbound to clusters, the N-terminal domain is disordered. Thus, this domain may move to give access to the active site. In a recent in vitro study, a sulfite ion serves as source of the missing belt sulfide.20 We propose that in this case sulfite would replace C18 and be reduced stepwise by 6 electrons. However, in vivo, a more typical sulfur provider such as NifS or a sulfur transferase could interact with NifB at the same site. Interestingly, a rhodanese-like gene (rhdN) possibly encoding an enzyme with sulfur transferase activity is found in the same transcriptional unit as nifB in Azotobacter vinelandii.28 This would provide a sulfide ion as a cysteine-persulfide species that could be reduced by 2 electrons in a mechanism similar to [Fe2S2] cluster assembly in NifU or IscU.29
Conclusions
Reinvestigating the NifB crystallographic data published by Kang and co-workers, we structurally characterized the K-cluster as a plausible [Fe8S8] intermediate in the time course of FeMo-co biosynthesis. The novel structure of this cluster, as well as knowledge of its protein coordination and analysis of the overall protein structure provides highly valuable information about the mechanism of NifB-co synthesis by supporting a stepwise sequence of FeS cluster fusion followed by methyl transfer to a belt sulfide. It also gives insight into how the protein matrix would assist the structural changes necessary for NifB-co production. Unexpectedly this intermediate is highly similar to the P-cluster, highlighting a common mechanism in the biosynthesis of both the P-cluster and FeMo-co with possible evolutionary relationships between these two metal centers present in the nitrogenase MoFe protein.Author contributions
L. P. J. and M. V.·C. refined the structure. P. A. did the molecular docking calculations. L. M. R. and Y. N. wrote the paper with the help of all the co-authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the French National Research Agency in the framework of the Investissements d’Avenir program (ANR-15-IDEX-02), through the funding of the “Origin of Life” project of the Univ. Grenoble-Alpes to Y. N., M. V. C. and L. P. J. P. A. appreciates the help from the staff of the computing facility provided by the Commissariat à l’Energie Atomique et aux Energies Alternatives (CEA/DRF/GIPSI), Saclay and CCRT, Bruyères-le-Châtel. L. M. R. acknowledges support from FEDER/Ministerio de Ciencia, Innovación y Universidades – Agencia Estatal de Investigación/Proyecto 2017-88475-R.Notes and references
- R. W. Howarth, HABs Eutrophication, 2008, vol. 8, pp. 14–20 Search PubMed.
- B. M. Hoffman, D. Lukoyanov, Z.-Y. Yang, D. R. Dean and L. C. Seefeldt, Chem. Rev., 2014, 114, 4041–4062 CrossRef CAS PubMed.
- S. Buren, E. Jimenez-Vicente, C. Echavarri-Erasun and L. M. Rubio, Chem. Rev., 2020, 120, 4921–4968 CrossRef CAS PubMed.
- V. K. Shah and W. J. Brill, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 3249–3253 CrossRef CAS PubMed.
- O. Einsle, F. A. Tezcan, S. L. A. Andrade, B. Schmid, M. Yoshida, J. B. Howard and D. C. Rees, Science, 2002, 297, 1696–1700 CrossRef CAS PubMed.
- T. Spatzal, M. Aksoyoglu, L. Zhang, S. L. A. Andrade, E. Schleicher, S. Weber, D. C. Rees and O. Einsle, Science, 2011, 334, 940 CrossRef CAS PubMed.
- J. W. Peters, M. H. Stowell, S. M. Soltis, M. G. Finnegan, M. K. Johnson and D. C. Rees, Biochemistry, 1997, 36, 1181–1187 CrossRef CAS PubMed.
- W. N. Lanzilotta and L. C. Seefeldt, Biochemistry, 1997, 36, 12976–12983 CrossRef CAS PubMed.
- M. C. Corbett, Y. Hu, F. Naderi, M. W. Ribbe, B. Hedman and K. O. Hodgson, J. Biol. Chem., 2004, 279, 28276–28282 CrossRef CAS PubMed.
- L. A. Rettberg, J. Wilcoxen, C. C. Lee, M. T. Stiebritz, K. Tanifuji, R. D. Britt and Y. Hu, Nat. Commun., 2018, 9, 2824 CrossRef PubMed.
- K. Rupnik, C. C. Lee, J. A. Wiig, Y. Hu, M. W. Ribbe and B. J. Hales, Biochemistry, 2014, 53, 1108–1116 CrossRef CAS PubMed.
- E. Jimenez-Vicente, Z.-Y. Yang, J. S. Martin Del Campo, V. L. Cash, L. C. Seefeldt and D. R. Dean, J. Biol. Chem., 2019, 294, 6204–6213 CrossRef CAS PubMed.
- L. Curatti, P. W. Ludden and L. M. Rubio, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5297–5301 CrossRef CAS PubMed.
- V. K. Shah, J. R. Allen, N. J. Spangler and P. W. Ludden, J. Biol. Chem., 1994, 269, 1154–1158 CrossRef CAS.
- L. Curatti, J. A. Hernandez, R. Y. Igarashi, B. Soboh, D. Zhao and L. M. Rubio, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17626–17631 CrossRef CAS PubMed.
- J. Wilcoxen, S. Arragain, A. A. Scandurra, E. Jimenez-Vicente, C. Echavarri-Erasun, S. Pollmann, R. D. Britt and L. M. Rubio, J. Am. Chem. Soc., 2016, 138, 7468–7471 CrossRef CAS PubMed.
- H. J. Sofia, G. Chen, B. G. Hetzler, J. F. Reyes-Spindola and N. E. Miller, Nucleic Acids Res., 2001, 29, 1097–1106 CrossRef CAS PubMed.
- A. S. Fajardo, P. Legrand, L. A. Payá-Tormo, L. Martin, M. T. Pellicer Martínez, C. Echavarri-Erasun, X. Vernède, L. M. Rubio and Y. Nicolet, J. Am. Chem. Soc., 2020, 142, 11006–11012 CrossRef CAS PubMed.
- J. A. Wiig, Y. Hu, C. Chung Lee and M. W. Ribbe, Science, 2012, 337, 1672–1675 CrossRef CAS PubMed.
- K. Tanifuji, C. C. Lee, N. S. Sickerman, K. Tatsumi, Y. Ohki, Y. Hu and M. W. Ribbe, Nat. Chem., 2018, 10, 568–572 CrossRef CAS PubMed.
- W. Kang, L. Rettberg, M. Stiebritz, A. Jasniewski, K. Tanifuji, C. C. Lee, M. W. Ribbe and Y. Hu, Angew. Chem., Int. Ed. Engl., 2021, 60, 2364–2370 CrossRef CAS PubMed.
- G. Bricogne, E. Blanc, M. Brandl, C. Flensburg, P. Keller, W. Paciorek, P. Roversi, A. Sharff, O. S. Smart, C. Vonrhein and T. O. Womack, BUSTER version 2.10.3, Global Phasing Ltd, Cambridge, United Kingdom, 2017 Search PubMed.
- Y. Nicolet, Nat. Catal., 2020, 3, 337–350 CrossRef CAS.
- P. Hänzelmann and H. Schindelin, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12870–12875 CrossRef PubMed.
- J. A. Wiig, Y. Hu and M. W. Ribbe, Nat. Commun., 2015, 6, 8034 CrossRef CAS PubMed.
- S. M. Keable, O. A. Zadvornyy, L. E. Johnson, B. Ginovska, A. J. Rasmussen, K. Danyal, B. J. Eilers, G. A. Prussia, A. X. LeVan, S. Raugei, L. C. Seefeldt and J. W. Peters, J. Biol. Chem., 2018, 293, 9629–9635 CrossRef CAS PubMed.
- A. J. Jasniewski, J. Wilcoxen, K. Tanifuji, B. Hedman, K. O. Hodgson, R. D. Britt, Y. Hu and M. W. Ribbe, Angew. Chem., Int. Ed. Engl., 2019, 58, 14703–14707 CrossRef CAS PubMed.
- J. A. Hernandez, S. J. George and L. M. Rubio, Biochemistry, 2009, 48, 9711–9721 CrossRef CAS PubMed.
- P. Yuvaniyama, J. N. Agar, V. L. Cash, M. K. Johnson and D. R. Dean, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 599–604 CrossRef CAS PubMed.
Footnotes |
| † Coordinates have been deposited to the Protein Data Bank with ID 7BI7. |
| ‡ Electronic supplementary information (ESI) available: The experimental material and methods, X-ray refinement statistics, supplementary figures and tables. See DOI: 10.1039/d1sc00289a |
| This journal is © The Royal Society of Chemistry 2021 |