
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorescent polymer cubosomes and hexosomes with aggregation-induced emission†
Hui
Chen
 a,
Yujiao
Fan
a,
Nian
Zhang
ab,
Sylvain
Trépout
c,
Bergam
Ptissam
c,
Annie
Brûlet
d,
Ben Zhong
Tang
e and
Min-Hui
Li
*a
a,
Yujiao
Fan
a,
Nian
Zhang
ab,
Sylvain
Trépout
c,
Bergam
Ptissam
c,
Annie
Brûlet
d,
Ben Zhong
Tang
e and
Min-Hui
Li
*a
aChimie ParisTech, PSL Université Paris, CNRS, Institut de Recherche de Chimie Paris, UMR8247, 11 rue Pierre et Marie Curie, 75005 Paris, France. E-mail: min-hui.li@chimieparistech.psl.eu
bBeijing Advanced Innovation Center for Soft Matter Science and Engineering, Beijing University of Chemical Technology, 15 North Third Ring Road, Chaoyang District, 100029 Beijing, China
cInstitut Curie, Université Paris-Saclay, Inserm US43, CNRS UMS2016, Centre Universitaire, Bât. 101B-110-111-112, Rue Henri Becquerel, CS 90030, 91401 Orsay Cedex, France
dLaboratoire Léon Brillouin, Université Paris-Saclay, UMR12 CEA-CNRS, CEA Saclay, 91191 Gif sur Yvette Cedex, France
eDepartment of Chemistry, Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong, China
First published on 3rd March 2021
Abstract
Fluorescent polymer cubosomes and hexosomes with aggregation-induced emission (AIE) were prepared from amphiphilic block copolymers PEG-b-PTPEMA where the hydrophobic block PTPEMA was a polymethacrylate with tetraphenylethene (TPE) as the AIE side group. Four highly asymmetric block copolymers with hydrophilic block weight ratio fPEG ≤ 20% were synthesized. Cubosomes and hexosomes with strong fluorescence emission were obtained by nanoprecipitation of polymers with fPEG < 9% in dioxane/water and THF/water systems. Their ordered internal structures were studied by electron microscopy (cryo-EM, SEM and TEM) and the X-ray scattering technique (SAXS). To elucidate the formation mechanisms of these inverted colloids, other parameters influencing the morphologies, like the water content during self-assembly and the organic solvent composition, were also investigated. This study not only inspires people to design novel building blocks for the preparation of functional cubosomes and hexosomes, but also presents the first AIE fluorescent polymer cubosome and hexosome with potential applications in bio-related fields.
Introduction
Amphiphilic block copolymers (ABCPs) can self-assemble in a selective solvent (water being the mostly used) into various forms of colloids like spherical micelles, cylindrical micelles, vesicles, cubosomes and hexosomes, depending on the overall shape of ABCPs in solution.1–5 This shape, by analogy with low-molecular-weight surfactants, can be described with the packing parameter p in the first approximation, p = v/al, where v is the volume of the core segment, l its length, and a the equilibrium area at the core/corona interface.6 Most efforts have long since been dedicated to exploring polymer micelles and polymer vesicles (polymersomes) with p < 1, which belong to the normal (oil-in-water) self-assembled nanostructures. Very recently, polymer cubosomes and hexosomes with p > 1, which belong to inverted (water-in-oil) self-assembled nanostructures, have attracted more and more attention.7 Compared to spherical/cylindrical micelles and vesicles, these colloids with inverted morphologies present numerous advantages due to their uniform, tunable and highly ordered porous structure, such as higher loading volume for both hydrophilic and hydrophobic substances, larger specific surface areas, and much better colloidal, chemical and mechanical stability.8,9 Moreover, because of the high molecular weight of polymers, polymer cubosomes and hexosomes present lattice parameters of at least one order of magnitude higher than lipid cubosomes and hexosomes, and their pore size can be adjusted from 101 to 102 nm by regulating the molecular weight of polymers. Therefore, polymer cubosomes and hexosomes are interesting candidates as functional materials for gated uptake, release, separation, catalysts, electrodes, photonics, etc.7,10,11Since the first report on polymer hexosomes, which were named at that time hexagonally packed hollow hoops (HHH),12 polymer cubosomes and hexosomes have now been rationalized as part of the morphological diagram of ABCPs in a selective solvent.3,7 Nevertheless, this field is still in its infancy, with many remaining challenges and exciting opportunities. The polymer cubosomes and hexosomes reported in the literature include the following categories according to the architectures of ABCPs: (1) linear diblock copolymers;13–22 (2) side-chain diblock copolymers23–33 with side-chains exhibiting special features (crystalline,23–28 liquid crystalline,29 nucleobase-pair interaction,30 halogen bonding interaction,31etc.); (3) diblock copolymers with dendrimer blocks.34–40 The commonly used hydrophilic blocks are poly(acrylic acid) (PAA) and poly(ethylene glycol) (including linear PEG, dendrimers with oligomer OEG branches, or polymethacrylate or polynorbornene with OEG side-chains). In the case of PAA as the hydrophilic block, the negatively charged pores and channels of cubosomes made from PAA-b-polystyrene (PS) were used to encapsulate positively charged proteins with high loading capacity, and the pore size could be adjusted by pH variation for sustained release.16 The most studied hydrophobic blocks include either amorphous polymers like PS16–21 and poly(n-butyl methacrylate) (PBMA),15 or crystalline polymers like poly(n-octadecyl methacrylate) (PODMA)24–27 and poly(n-docosyl methacrylate) (PDSMA).28 The melting point of the crystalline block was employed to perform the thermally controlled release of ibuprofen previously encapsulated in cubosomes.28 Driven by the increasing research interest in these polymer colloids with inverted nanostructures, the development of new ABCPs for the controllable preparation of cubosomes and hexosomes with innovative potential applications is highly desirable.
In this work, we report fluorescent polymer cubosomes and hexosomes with aggregation-induced emission (AIE) made from a new ABCP, PEG-b-PTPEMA, in which the hydrophobic block PTPEMA is a side chain polymethacrylate with pendants of tetraphenylethane (TPE) (Fig. 1). The TPE is a typical AIE luminogen, which is faintly emissive in solution but emits strong fluorescence in the aggregated state. It offers an extraordinary remedy to tackle the aggregation-caused quenching (ACQ) issue of conventional luminogens. Owing to their unique characteristics, a variety of AIE fluorescent materials, including self-assembled nanomaterials, have been developed for extensive applications.41–47 Here, the combination of AIE properties with polymer cubosomes and hexosomes creates original fluorescent polymer materials for potential applications in theranostics and bioimaging. As far as we are aware, this is the first report on AIE fluorescent polymer cubosomes and hexosomes.
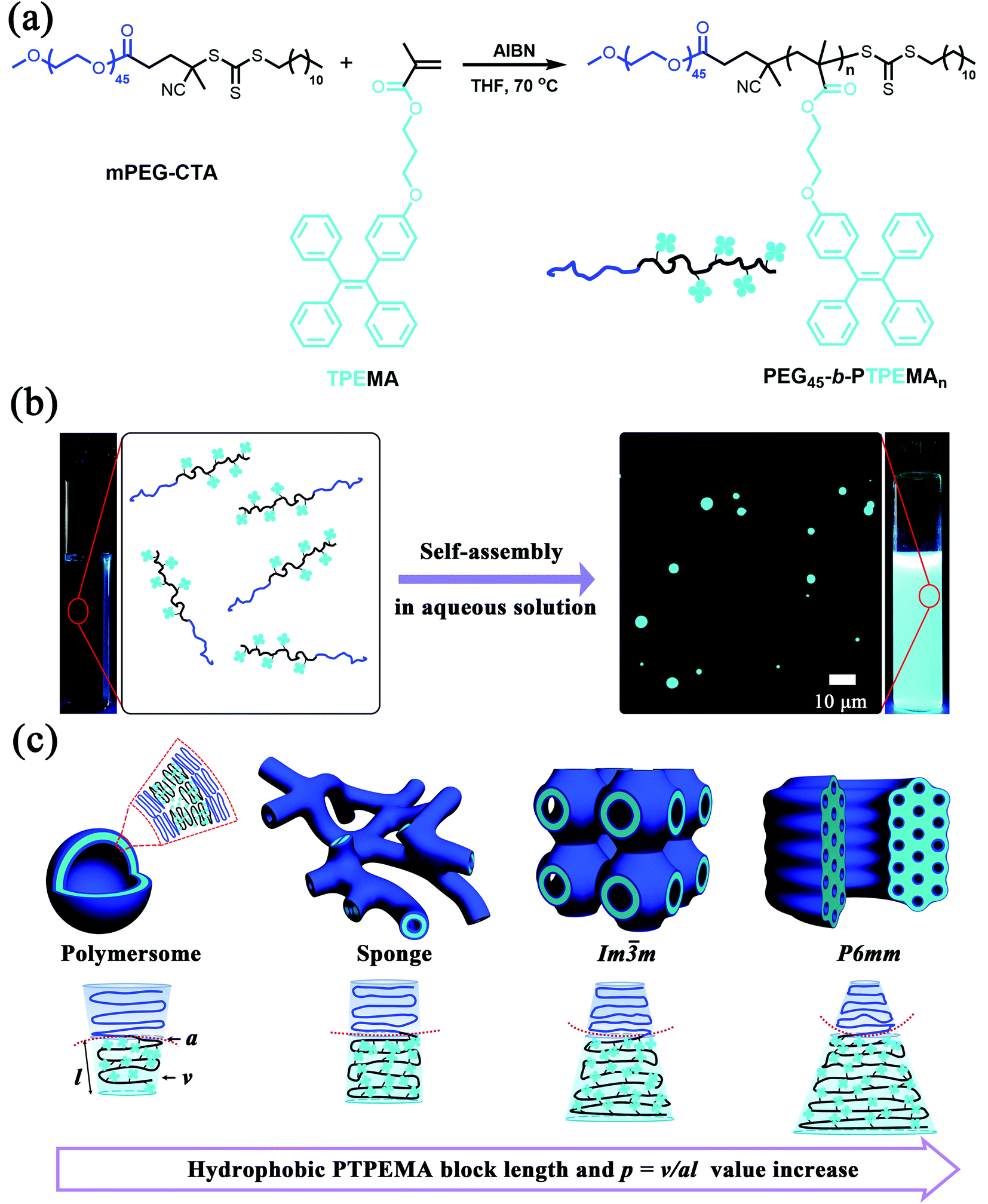 | ||
| Fig. 1 (a) Synthesis of PEG45-b-PTPEMAn by RAFT polymerization. (b) Schematic representation of the cyan fluorescence light-up of PEG45-b-PTPEMAn self-assemblies. Left: polymer in dioxane solution; right: self-assemblies dispersed in water. Photographs were taken under UV light (λ = 365 nm), and the micrograph of particles with cyan emission was taken by epifluorescence microscopy (excitation: λex = 365 nm). (c) Schematic illustration of the morphological transformation of PEG45-b-PTPEMAn assemblies with the length increase of the hydrophilic block PTPEMA which corresponds to the increase of packing parameter p. | ||
Results and discussion
Syntheses of amphiphilic block copolymers PEG-b-PTPEMA
One of the prerequisites for the formation of bicontinuous double cubic phases (primitive Im![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, diamond Pnm, and gyroid Iad) or a hexagonal phase (P6mm) is constructing highly asymmetric ABCPs with a small hydrophilic block weight ratio (e.g., <25% or lower depending on the detailed chemical structure). Here, amphiphilic block copolymers PEG-b-PTPEMA with a hydrophobic block much larger than its corresponding hydrophilic block were synthesized by reversible addition–fragmentation chain transfer (RAFT) polymerization of monomer TPEMA in the presence of the macro-chain transfer agent mPEG45-CTA (Mn = 2400 Da) and the initiator 2,2′-azobis(isobutyronitrile) (AIBN) (Fig. 1a). TPEMA and mPEG45-CTA were synthesized as described previously.48 By adjusting the feeding ratios of mPEG-CTA/TPEMA, four amphiphilic block copolymers PEG45-b-PTPEMAn (n = 16, 29, 42, 122) with narrow molecular weight distribution (Đ = Mw/Mn = 1.1–1.2) were prepared, which had PEG block weight ratios fPEG (wt%) = 20, 12.3, 8.9, and 3.3%, respectively. The details of synthesis and characterization by NMR, SEC and DSC are described in the ESI (Fig. S1–S3, and Table S1†). The hydrophobic block PTPEMA exhibited a high glassy temperature Tg = 93 °C as measured in PEG45-b-PTPEMA122 (Fig. S3†).
m, diamond Pnm, and gyroid Iad) or a hexagonal phase (P6mm) is constructing highly asymmetric ABCPs with a small hydrophilic block weight ratio (e.g., <25% or lower depending on the detailed chemical structure). Here, amphiphilic block copolymers PEG-b-PTPEMA with a hydrophobic block much larger than its corresponding hydrophilic block were synthesized by reversible addition–fragmentation chain transfer (RAFT) polymerization of monomer TPEMA in the presence of the macro-chain transfer agent mPEG45-CTA (Mn = 2400 Da) and the initiator 2,2′-azobis(isobutyronitrile) (AIBN) (Fig. 1a). TPEMA and mPEG45-CTA were synthesized as described previously.48 By adjusting the feeding ratios of mPEG-CTA/TPEMA, four amphiphilic block copolymers PEG45-b-PTPEMAn (n = 16, 29, 42, 122) with narrow molecular weight distribution (Đ = Mw/Mn = 1.1–1.2) were prepared, which had PEG block weight ratios fPEG (wt%) = 20, 12.3, 8.9, and 3.3%, respectively. The details of synthesis and characterization by NMR, SEC and DSC are described in the ESI (Fig. S1–S3, and Table S1†). The hydrophobic block PTPEMA exhibited a high glassy temperature Tg = 93 °C as measured in PEG45-b-PTPEMA122 (Fig. S3†).
Hexosomes, primitive cubosomes and polymersomes formed in the dioxane/water system
The self-assembly of PEG45-b-PTPEMAn was performed by the nanoprecipitation method using firstly dioxane as the cosolvent (see ESI† for experimental details). Typically, a solution of copolymer at a concentration of 0.25 wt% was prepared in dioxane, and then water was added progressively into the solution until the volume content of water (fwater) reached 83%. The final self-assemblies were obtained by dialyzing the mixture against a large volume of water (refreshing the water 3 times) to remove all organic solvent.Taking PEG45-b-PTPEMA42 (fPEG = 8.9%) as an example, we started to study the intermediate morphologies of self-assemblies formed at different water contents during the nanoprecipitation. At fwater = 15, 20, 60, and 83%, small aliquots (10 μL) were withdrawn from the self-assembly mixture, respectively. Each aliquot was immediately injected into a large excess of water (∼1 mL) to quench the self-assembly. The obtained colloids were then examined by scanning electron microscopy (SEM), transmission electron microscopy (TEM) and cryogenic-electron microscopy (cryo-EM). The cryo-EM images clearly illustrate spherical polymersomes and irregular giant polymersomes at fwater = 15% (Fig. 2A). Increasing fwater from 15% to 20%, large compound vesicles (LCVs) are observed as shown in Fig. 2B, presumably due to the coalescence of polymersomes. Upon further increase of fwater from 20% to 60%, these LCVs undergo structural refinement, forming sponge-like particles (Fig. 2C). Subsequently, the sponge structures tend to rearrange into spherical cubosomes with rather regular pores on the surface as shown in Fig. 2D for fwater = 83% (the cubic phase structures will be discussed in detail below). All these observations reveal the pathway of morphological evolution from polymersomes, to sponge particles and to cubosomes, which indicates the formation mechanism of cubosomes (hexosomes) and guides us for their preparation.
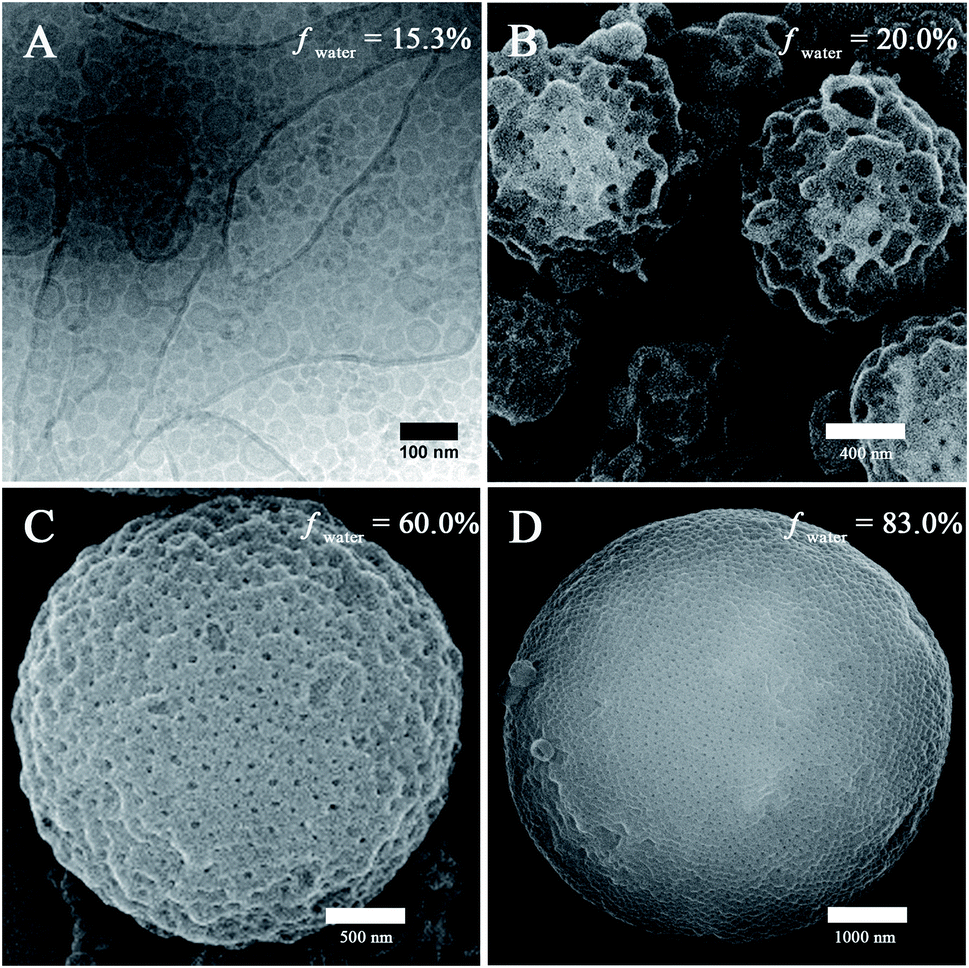 | ||
| Fig. 2 The intermediate morphologies of particles obtained at different water contents (fwater) in the self-assembly process of PEG45-b-PTPEMA42 by nanoprecipitation in dioxane/water. (A) Cryo-EM image and (B–D) SEM images. | ||
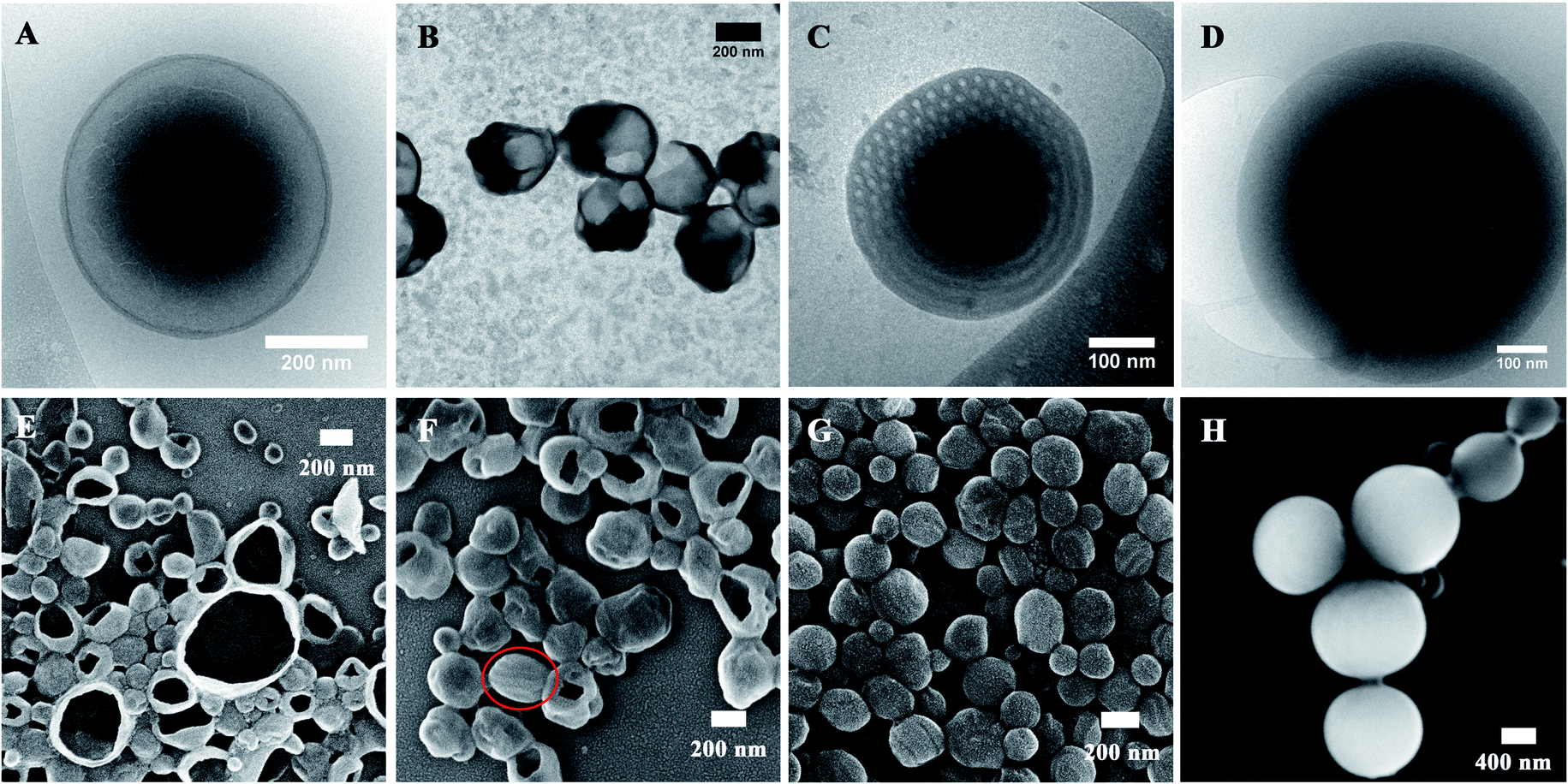
The final self-assemblies of PEG45-b-PTPEMAn (n = 16, 29, 42 and 122) were then prepared using the above-discussed nanoprecipitation until added water reaching fwater = 83% and then dialyzed against a large quantity of water. Their morphologies were studied by cryo-EM, TEM and SEM as shown in Fig. 3. PEG45-b-PTPEMAn with n = 16, 29, 42 and 122 formed polymersomes, sponge structures, cubosomes, and hexosomes, respectively. The spherical vesicles of PEG45-b-PTPEMA16 are clearly revealed by the cryo-EM image (Fig. 3A) and confirmed by the SEM image showing broken empty spheres (Fig. 3E). The membrane thickness (hydrophobic part) of polymersomes is 8.5 ± 0.5 nm, as measured by statistical analysis of 30 different polymersomes from the collected cryo-EM images. On the other hand, the length of the stretched hydrophobic part PTPEMA16 was about 6.5 nm (calculated using Chem3D software). A perfect tail-to-tail bilayer should have the membrane thickness of 13.0 nm which is higher than the measured one. This indicates that the conformation of the hydrophobic chain PTPEMA16 should be coil-like in the membrane and the chain entanglement would also be possible between two leaflets. The average hydrodynamic diameter of polymersomes is 160 nm, as measured by DLS (Fig. S4A†). When the length of the hydrophobic block PTPEMAn increased to n = 29, sponge-like nanoparticles with disordered porous structures, together with polymersomes, appeared as shown by TEM and SEM images (Fig. 3B and F). On further increasing the hydrophobic block length to n = 42 and 122, the PEG45-b-PTPEMA42 and PEG45-b-PTPEMA122 self-assembled into cubosomes (Fig. 3C and G) and hexosomes (Fig. 3D and H), respectively, with significantly larger hydrodynamic diameters up to 5 μm (DLS curves in Fig. S4C and D†) than those of polymersomes. The inset in Fig. 3G (SEM) shows that the cubosome has tetragonal pores on its surface. Fig. 3D (cryo-EM) shows the typical curved cylinders of hexosomes, while Fig. 3H (SEM) displays a non-spherical morphology with packed cylinders. The tendency of the formation of polymersomes, cubosomes and hexosomes with the gradual decrease of hydrophilic ratio fPEG (from 20% to 3.3%) and the increase of the hydrophobic block length is in agreement with the classical phase diagram according to packing parameter p changing from p ≤ 1 for normal vesicular structures to p > 1 for the colloids with internally inverse cubic phase and hexagonal phase (Fig. 1c).3
 | ||
| Fig. 3 Self-assemblies formed by PEG45-b-PTPEMAn using dioxane as the cosolvent with an initial concentration of 0.25 wt%. (A and E) Polymersomes of PEG45-b-PTPEMA16. (B and F) Mixed morphologies of sponge structures and polymersomes of PEG45-b-PTPEMA29. (C and G) Primitive cubosomes of PEG45-b-PTPEMA42 (the inset in G shows the amplified images of a part of the surface. Scale bar: 100 nm). (D and H) Hexosomes of PEG45-b-PTPEMA122. (A and D) Cryo-EM, (B and C) TEM, and (E–H) SEM images. The black circle in (H) shows where an orientational change of the cylinders takes place. | ||
More SEM and cryo-EM images of cubosomes and hexosomes are given in Fig. 4 together with small-angle X-ray scattering (SAXS) results obtained from dried powder samples. The SAXS profile of PEG45-b-PTPEMA42 cubosomes (Fig. 4A and S5A†) displays a characteristic ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:32 for the square of scattering vector (q2), which corresponds to (110), (200), and (440) reflections of primitive cubic structure Imm. Their lattice parameter thus deduced was aP = 46 nm. Note that the SAXS signal of powder samples is the sum of two contributions: (1) a huge scattering from interfaces between the cubosome grains and the air, which generally decreases as q−4 (Porod law). (2) Bragg peaks arising from the ordered structure inside the cubosome grains (Bragg law). The q4*I(q) versus q representation allows then the Bragg peaks to be highlighted as shown in Fig. S5A.†Fig. 4B and C show the SEM images of cubosomes of PEG45-b-PTPEMA42 broken by liquid nitrogen freezing during the sample preparation, where the ordered internal domains along the [100] and [110] directions are exposed (insets in Fig. 4B and C). TEM was also applied to examine the internal structure of cubosomes (Fig. 3C and S6B†). In the thin places of the cubosome, the organization along the [100] direction of the primitive cubic phase is visible (Fig. S6B†). The average unit cell size of the primitive cubic structure was measured as aP = 45 nm from the TEM image, which is in good agreement with that evaluated by SAXS.
:4:32 for the square of scattering vector (q2), which corresponds to (110), (200), and (440) reflections of primitive cubic structure Imm. Their lattice parameter thus deduced was aP = 46 nm. Note that the SAXS signal of powder samples is the sum of two contributions: (1) a huge scattering from interfaces between the cubosome grains and the air, which generally decreases as q−4 (Porod law). (2) Bragg peaks arising from the ordered structure inside the cubosome grains (Bragg law). The q4*I(q) versus q representation allows then the Bragg peaks to be highlighted as shown in Fig. S5A.†Fig. 4B and C show the SEM images of cubosomes of PEG45-b-PTPEMA42 broken by liquid nitrogen freezing during the sample preparation, where the ordered internal domains along the [100] and [110] directions are exposed (insets in Fig. 4B and C). TEM was also applied to examine the internal structure of cubosomes (Fig. 3C and S6B†). In the thin places of the cubosome, the organization along the [100] direction of the primitive cubic phase is visible (Fig. S6B†). The average unit cell size of the primitive cubic structure was measured as aP = 45 nm from the TEM image, which is in good agreement with that evaluated by SAXS.
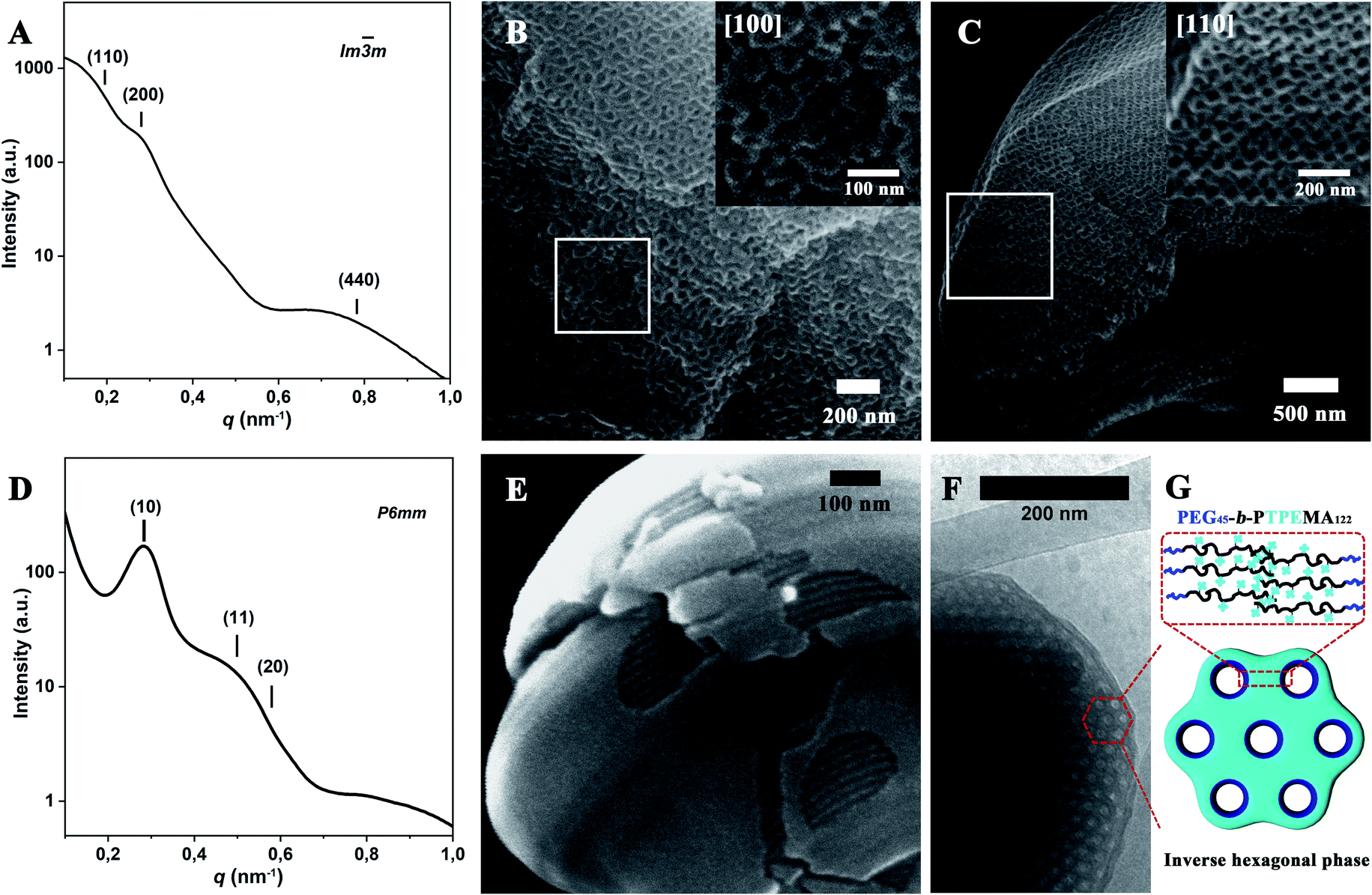 | ||
| Fig. 4 Structural characterization of PEG45-b-PTPEMAn cubosomes and hexosomes formed with dioxane as co-solvent with an initial concentration of 0.25 wt%. SAXS profile (A) and SEM (B and C) images of Imm cubosomes of PEG45-b-PTPEMA42. SAXS profile (D), SEM (E) and cryo-EM (F) images of hexosomes of PEG45-b-PTPEMA122. Peak positions of SAXS profiles were more clearly determined using a q4*I(q) versus q representation (see Fig. S5†). (G) is the schematic of the cross-section of the inverse hexagonal phase. The insets in (B) and (C) are the SEM images of Imm cubosomes in the [100] and [110] directions, respectively. | ||
The SAXS profile of PEG45-b-PTPEMA122 hexosomes (Fig. 4D for I(q) ∼ q and Fig. S5B† for q4*I(q) ∼ q) gives q2 ratios of 1:3:4 corresponding to (10), (11), and (20) reflections of hexagonal structure P6mm. Their lattice parameter thus deduced was aH = 25 nm. Since the 1st order Bragg peak (10) is well defined (Fig. 4D), the correlation length of the P6mm hexagonal structure perpendicular to the long axes of cylinders can be calculated from the full width at half maximum (FWHM) of the peak (10), which is 120 nm for PEG45-b-PTPEMA122 hexosomes (see ESI 6.† SAXS data analysis for calculation details). Fig. 4E and F show the SEM and cryo-EM images of hexosomes of PEG45-b-PTPEMA122. The SEM image in Fig. 4E displays one hexosome with a broken surface, which clearly reveals the tightly packed cylindrical channels inside the colloid. The hexagonal packing of these cylinders is visible in cryo-EM images (Fig. 4F and S7A†) in the regions of cross-sections. The average unit cell parameter of the P6mm structure was measured as aH = 24 nm from the cryo-EM image, which is also in good agreement with that evaluated by SAXS.
It is interesting to note that the shape of hexosomes is not spherical: faceted contours are shown by the cryo-EM image (Fig. 3D and S7A†); twists and turns of cylinders with topological defects in the form of disclinations are revealed at the colloidal surface by SEM images in Fig. 3H, S7C and S16A† (see the section “Effects of co-solvent” below for S16A†). As a matter of fact, the cylindrical hexagonal structure is a kind of two-dimensional (2D) ordering. Packing the parallelly ordered cylinders into a colloid with spherical 3D topology results inevitably in topological defects.49,50 In other words, the introduction of disclinations makes the spherical topology possible by winding cylinders with twists and turns. Other morphologies, such as a spindle-like one composed of hexagonally packed cylinders that rotate along a central axis of symmetry to form circular hoops, were the most reported.12,21,29 Recently, vesicular polymer hexosomes exhibiting topological defects have also been reported, where inverted cylinders formed the shell of hollow colloids with noncircular hoops.51 The morphology of hexosomes should be intricately connected to the self-assembly pathway51 and to the bending energy of the cylinders that depends on the chemical structures of the block copolymers as well as their interaction with the solvent in a complicated way.12,50
Further, the effect of the initial concentration on the self-assembly behaviour was examined by taking PEG45-b-PTPEMA122 in a dioxane/water system as an example with higher initial concentrations c0 = 0.4 wt% and 2 wt% in dioxane, respectively. When c0 = 0.4 wt%, Pnm and P6mm phase structures coexist in most of the colloid particles as revealed by SEM images in Fig. S8A and B.†21 A few of the particles with Imm structure showing tetragonal pores on the surface were also observed (Fig. S8C and D†). On further increasing the concentration to c0 = 2 wt%, polymersomes and particles with sponge structure were obtained (Fig. S9†). In conclusion, the low initial concentration c0 = 0.25 wt% is the most favorable one for the preparation of cubosomes and hexosomes of copolymers PEG45-b-PTPEMAn.
Hexosomes and polymersomes formed in a THF/water system
Instead of dioxane, tetrahydrofuran (THF) was also used as a co-solvent to prepare particles of PEG45-b-PTPEMAn with an initial concentration of 0.25 wt%. Fig. 5 shows the cryo-EM, TEM and SEM images of self-assemblies of PEG45-b-PTPEMAn (n = 16, 29, 42, 122) formed in THF/water systems. As in the case of the dioxane/water system, the PEG45-b-PTPEMA16 also forms spherical vesicles, as displayed by the cryo-EM image (Fig. 5A) and confirmed by the SEM image showing broken open capsules (Fig. 5E). The membrane thickness of polymersomes is e = 11.0 ± 0.5 nm, as measured by cryo-EM, and the average hydrodynamic diameter of polymersomes is Dh = 250 nm, as measured by DLS (Fig. S10A†). Interestingly, the thickness e = 11.0 nm here is still lower but remarkably close to the membrane thickness of 13.0 nm for a perfect tail-to-tail bilayer. These characteristic values (e and Dh) of polymersomes PEG45-b-PTPEMA16 obtained in THF/water are larger than those obtained in dioxane/water where e = 8.5 ± 0.5 nm and Dh = 160 nm. That means the PTPEMA chains are more extended when self-assembling in THF/water. Indeed, a thicker bilayer will bend less easily, and the vesicles obtained have a lower curvature and larger diameter as in the case of polymersomes in THF/water. The reason is that THF is a better solvent for PTPEMA than dioxane and the chain of PTPEMA is more extended in THF than in dioxane (see the next section “Effects of co-solvent on the morphology of self-assembly” for detailed discussion). Increasing the hydrophobic block length to n = 29, polymersomes are still the dominant species based on the SEM and TEM images (Fig. 5B and F). However, in THF/water systems, a sponge-like structure and inverse cubic phase do not occur anymore. The hexosomes directly appear with the co-existence of polymersomes (see Fig. 5B, F and S11†). On further increasing the hydrophobic block length to n = 42, the hexosomes become the major structures (Fig. 5C, G and S12†), with an average hydrodynamic diameter of 220 nm and narrow dispersity of PDI = 0.2 as measured by DLS (Fig. S10†). Cryo-EM images (Fig. 5C and S12C, D†) reveal their internal structures with a hexagonally packed cross-section. The average unit cell parameter of the P6mm structure was measured as aH = 26 nm based on cryo-EM images. This hexagonal structure of PEG45-b-PTPEMA42 when prepared in THF/water was confirmed by the analysis of the SAXS profile, showing peaks with q2 ratios of 1:3:7, assigned to the (10), (11), and (21) reflections of the hexagonal structure with aH = 29 nm as the lattice parameter (Fig. S13†). From the FWHM of the Bragg peak, the correlation length of the P6mm hexagonal structure is estimated as 110 nm for PEG45-b-PTPEMA42 hexosomes formed in the THF/water system. Finally, when the hydrophobic block length increases to n = 122, solid particles with dense structures are obtained according to their cryo-EM and SEM images (Fig. 5D and H).
 | ||
| Fig. 5 Self-assemblies formed by PEG45-b-PTPEMAn using THF as the cosolvent with an initial concentration of 0.25 wt%. (A and E) Polymersomes of PEG45-b-PTPEMA16. (B and F) Polymersomes and hexosomes of PEG45-b-PTPEMA29 (hexosomes indicated by the red circle). (C and G) Hexosomes of PEG45-b-PTPEMA42. (D and H) Solid spheres of PEG45-b-PTPEMA122. (A, C, and D) Cryo-EM, (B) TEM, and (E–H) SEM images. | ||
Effects of co-solvent on the morphology of self-assembly
The above-discussed results suggest that the nature of co-solvent has significant influence on the structure of the colloidal particles. This is expected because the co-solvent with different solubility parameters directly affects the dimensions of both hydrophilic and hydrophobic domains during the self-assembly process.15 The solubility parameters (δ) of THF, dioxane, and the repeat unit of PEG are δTHF = 18.6, δdioxane = 20.5, and δEG = 20.2 MPa1/2, respectively.52 For the PTPEMA block, δTPEMA = 18.9 MPa1/2 is calculated using a group contribution method.52 Typically, the closer are the δ values of the polymer and the solvent, the better is the solubility of the polymer in the solvent. Therefore, PEG is more soluble in dioxane than in THF, while PTPEMA is more soluble in THF than in dioxane. The initial shapes of PEG45-b-PTPEMA42, for example, are different in dioxane/water and THF/water systems, as shown in Fig. 6a. Thus, the inverse curvature toward the PEG interface is higher in the THF case than in the dioxane case. Indeed, PEG45-b-PTPEMA42 self-assembled into cubosomes with a lower negative interface curvature in dioxane/water, but into hexosomes with a higher negative curvature in the THF/water system. Moreover, for a shorter PTPEMA block, PEG45-b-PTPEMA29 formed a sponge structure together with polymersomes in the dioxane/water system, while it formed hexosomes with a highly negative curvature together with polymersomes in the THF/water system. As the solvent in the PEG part is progressively replaced by water (a good solvent for PEG but not miscible at all with PTPEMA), the swollen shape of PTPEMA in THF or in dioxane is the key parameter during the self-assembly process. Table S2† lists the thickness of the hydrophobic part of the bilayer membrane in different morphologies. In general, PEG45-b-PTPEMAn assemblies formed in the THF/water system have a thicker membrane than those constructed in the dioxane/water system, implying that the PTPEMA chain is more extended in THF/water than in the dioxane/water system during the self-assembly. The above results suggest that a cosolvent with higher affinity for the hydrophobic block tends to facilitate the formation of nanostructures with a higher negative curvature. This was further proved by the self-assembly behaviour of PEG45-b-PTPEMA42 in the dioxane/THF/water system. Mixtures of organic solvents dioxane/THF with a THF content of 25%, 40% and 50% were used to prepare the PEG45-b-PTPEMA42 solution with initial concentration c0 = 0.25 wt%. As shown in Table S3,† with the increase of the THF content, the solubility parameter of the binary cosolvent approaches that of the hydrophobic block PTPEMA. At the THF content of 25% (δ = 20.1 MPa1/2), the PEG45-b-PTPEMA42 preferably self-assembled into the spherical Imm cubosomes (Fig. S14†), showing tetragonal pores on its surface (Fig. S14A†). Its ordered internal domains along the [100] direction were also observed (inset in Fig. S14A†). When the THF fraction increased to 40% (δ = 19.7 MPa1/2), non-spherical particles were formed, displaying both regular pores and tightly packed channels on its surface as shown in Fig. S15A.† The tetragonal pores correspond to the Imm structure, while the cylindrical channels belong to the P6mm structure. Interestingly, the particles with mixed structures of Pnm and P6mm phases were also observed as shown in Fig. S15B,† where characteristic polyhedral surfaces of the Pnm structure co-exist with the tightly packed channels of the P6mm structure. The TEM images further confirmed the internal structure of the aggregate, showing cubic and P6mm phases (Fig. S15C†). These intermediate structural mixtures evidence the morphology evolution from cubosomes to hexosomes. On further increasing the proportions of THF to 50% (δ = 19.5 MPa1/2), only hexosomes were observed according to the collected SEM and TEM images (Fig. S16A and B†).
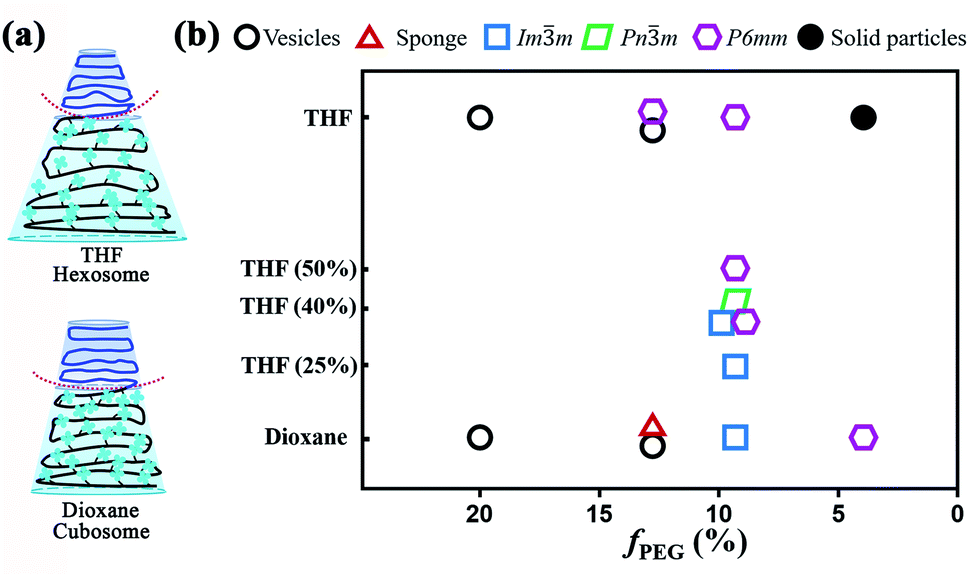 | ||
| Fig. 6 (a) Schematic representation of the molecular shapes of PEG45-b-PTPEMA42 in dioxane/water and THF/water systems, which result in cubosomes and hexosomes, respectively. (b) Morphological phase diagram of the self-assemblies of PEG45-b-PTPEMAn with different fPEG fractions and using different organic co-solvents. | ||
In conclusion, a phase transformation of the self-assembled structure of PEG45-b-PTPEMA42 from cubosomes Imm and Pnm to hexosomes P6mm was observed with the increase of the affinity between the cosolvent and hydrophobic block by adjusting the content of THF in the solvent mixture of dioxane/THF. Together with the morphological transformation upon the increase of the hydrophobic length (the decrease of the fPEG fraction) of PEG45-b-PTPEMAn in pure dioxane and THF as discussed in the previous sections, a phase diagram is drawn in Fig. 6b.
AIE fluorescence of PEG45-b-PTPEMAn self-assemblies
The fluorescence evolution during the self-assembly process by nanoprecipitation was first studied taking PEG45-b-PTPEMA42 in dioxane/water as an example. As shown in Fig. 7A, a slight increase of fluorescence intensity is observed after the addition of water to fwater = 15%, indicating that PEG45-b-PTPEMA42 begins to form self-assemblies.45,53 On further increasing the water fraction, the fluorescence intensity keeps a slow increase rate until ∼50% of water content. At this stage, the mobility of the hydrophobic chains and TPE moieties in the aggregates is still high because of the plasticizing effect of the organic solvent, leading to the slow growth rate of fluorescence. On further increasing the water content, an exponential augmentation of fluorescence intensity is observed, probably due to the removal of the organic solvent from the hydrophobic domains driven by the high osmotic pressure between the outside and the inside of these domains. This solvent removal effectively reduces the mobility of the PTPEMA chain and blocks the intramolecular rotation of TPE units, resulting in a rapid growth of the fluorescence intensity. These assemblies with strong fluorescence also correspond to the organization of the nanostructure into a sponge phase (∼60% of water content) and cubic phase (∼80% of water content) as discussed above (Fig. 2). These observations show clearly the AIE properties of the self-assemblies (the insets in Fig. 7A).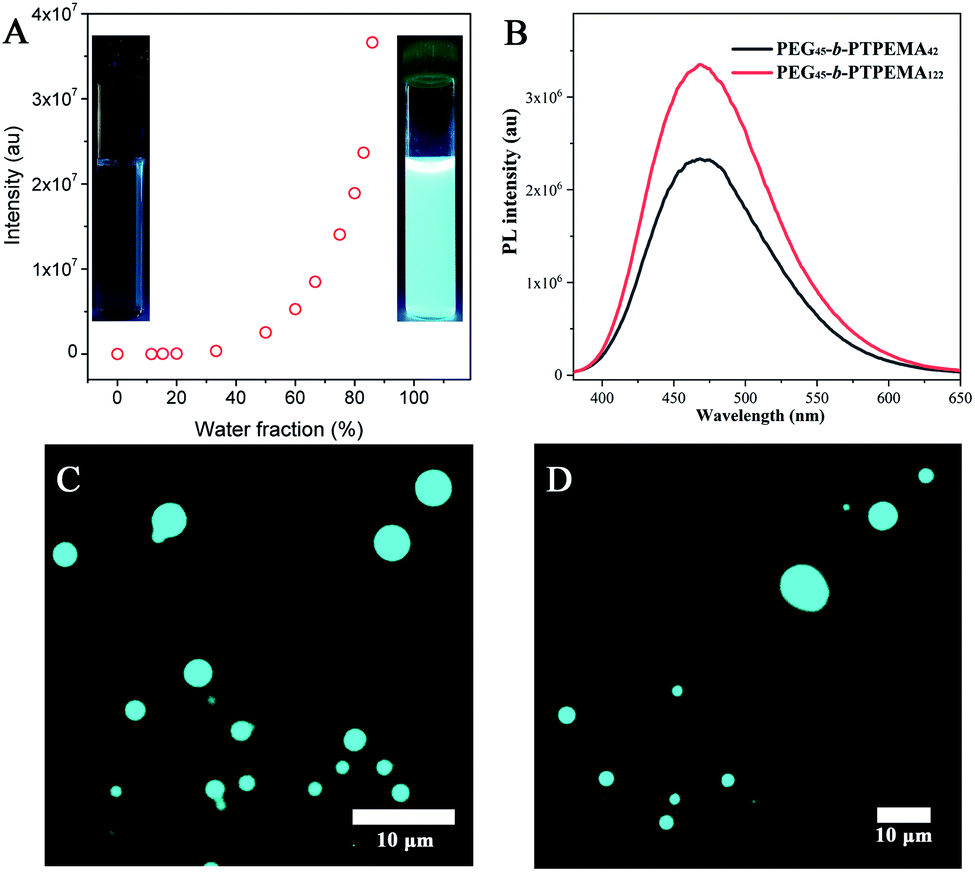 | ||
| Fig. 7 (A) The evolution of fluorescence intensity as a function of water content during the nanoprecipitation of PEG45-b-PTPEMA42 in the dioxane/water system. The insets are the photos taken under UV light of 365 nm for PEG45-b-PTPEMA42 in dioxane solution (left) and its cubosomes in water after dialysis (right). (B) Photoluminescence spectra of the PEG45-b-PTPEMA42 cubosomes and PEG45-b-PTPEMA122 hexosomes (concentration: 0.4 mg mL−1; excitation wavelength: λ = 370 nm). (C and D) The images of PEG45-b-PTPEMA42 cubosomes (C) and PEG45-b-PTPEMA122 hexosomes (D) taken with an epifluorescence microscope (illumination wavelength: λ = 365 nm). The self-assembly was performed in the dioxane/water system. | ||
The final colloidal dispersions of PEG45-b-PTPEMA42 and of PEG45-b-PTPEMA122 in water were then characterized using a fluorospectrometer and fluorescence microscope. PEG45-b-PTPEMA42 cubosomes and PEG45-b-PTPEMA122 hexosomes exhibit high fluorescence (Fig. 7B) with maximal emission at wavelengths of λem = 468 nm and 469 nm, respectively, under the excitation at λex = 370 nm, which corresponds to the maximal wavelength in the excitation spectrum (Fig. S18†). As observed by epifluorescence microscopy, the colloids (cubosomes and hexosomes) emit bright cyan fluorescence under the illumination of the UV lamp (λex = 365 nm) as shown in Fig. 7C and D. The quantum yields of PEG45-b-PTPEMA42 cubosomes and PEG45-b-PTPEMA122 hexosomes were measured as 17% and 23%, respectively, using the integrating sphere method.
Biocompatibility of PEG45-b-PTPEMAn self-assemblies
The biocompatibility of PEG45-b-PTPEMA42 cubosomes and PEG45-b-PTPEMA122 hexosomes was tested on both the A375M melanoma cell line and A549 lung cell line. The cells lines were incubated for four days with PEG45-b-PTPEMAn self-assemblies at different concentrations (for details see the method section in the ESI†). Proliferation assays were performed using an IncuCyte™ live-cell Imaging apparatus. Fig. S19† shows that the percentage of cell death is inferior to 1% under all tested conditions. Overall, the same growth speed was observed for both cell lines. No morphological difference was noticed between control and treated cells (Fig. S20†). All PEG45-b-PTPEMAn cubosomes and hexosomes studied are therefore non-cytotoxic, indicating their excellent biocompatibility.Conclusion
A series of amphiphilic block copolymers PEG45-b-PTPEMAn with the bulky AIE moiety TPE as the side group and with high asymmetry in block lengths (fPEG = 20, 12.3, 8.9, 3.3%) have been synthesized. These ABCPs could form polymersomes, sponge structures, and colloids (cubosomes and hexosomes) with inverse ordered structures including a primitive cubic phase (Imm) and hexagonal phase (P6mm) by increasing the length of the hydrophobic block (decreasing the hydrophilic ratio fPEG). In addition to fPEG, other parameters influencing the self-assembly like the initial polymer concentration, the water content added before the final dialysis, and the co-solvent were studied in detail. The demonstration of the morphological evolution pathway from polymersomes, large complex vesicles, and sponge structures to primitive cubosomes upon the addition of water allows us to understand the formation mechanism of cubosomes. The study on the effect of organic co-solvent guides us to control the final internal structure of colloids by appropriate choice of solvent. In brief, this research provides a platform for a better comprehension of the formation of inverted morphologies and enriches the examples of ABCPs to form cubosomes and hexosomes.
The polymer cubosomes and hexosomes of PEG45-b-PTPEMAn are extremely stable colloids with Tg around 90 °C for the hydrophobic part, which can be kept stable for months under ambient conditions without a morphological change. Moreover, in contrast to polymersomes, they can be freeze-dried and re-dispersed in water without a structural change, as proved by SEM and cryo-EM observation. All polymer cubosomes and hexosomes exhibit bright cyan fluorescence and also show excellent biocompatibility. Here, TPE that emits blue light when excited under UV light was used for the proof of concept of AIE inverted colloids. Other AIEgens excitable at longer wavelengths or AIE photosensitizers can be introduced into the hydrophobic part of cubosomes and hexosomes, which may have potential applications in bioimaging and nanomedicine. This work is in progress.
Author contributions
The manuscript was written through contributions of all authors. H. C. and M.-H. L. designed the research; H.·C., Y.·F., N. Z. and M. H. L. performed the research; H. C. and S. T. contributed to the cryo-EM imaging and analysis; B. P. and S. T. contributed to the cytotoxicity experiments; A. B. contributed to the SAXS experiments; H. C., B. Z. T. and M. H. L. analysed the data; H. C. and M. H. L. wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the French National Research Agency (ANR-16-CE29-0028). Yujiao Fan and Nian Zhang gratefully acknowledge the China Scholarship Council for funding her PhD scholarship. The Multimodal Imaging Center of Institut Curie is acknowledged for providing access to cryo-EM facility in Orsay. We thank Chao Deng (Zhejiang University) for quantum yield measurement, Fan Sun (Chimie ParisTech) for TEM measurement, and Florent Poyer, Méghane Sittewelle, Cathy Pichol-Thievend, and Laetitia Besse (Institut Curie, Orsay) for providing the cell lines and the assistance during the cell culture experiments and the IncuCyte measurement.Notes and references
- M. Antonietti and S. Förster, Adv. Mater., 2003, 15, 1323–1333 CrossRef CAS
.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS PubMed
- A. H. Gröschel and A. Walther, Angew. Chem., Int. Ed., 2017, 56, 10992–10994 CrossRef PubMed
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC
- C. K. Wong, X. Qiang, A. H. E. Müller and A. H. Gröschel, Prog. Polym. Sci., 2020, 102, 101211 CrossRef CAS
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans., 1976, 72, 1525–1568 RSC
- S. D. Allen, S. Bobbala, N. B. Karabin and E. A. Scott, Nanoscale Horiz., 2019, 4, 258–272 RSC
- A. Yaghmur, M. Rappolt and S. W. Larsen, J. Drug Delivery Sci. Technol., 2013, 23, 325–332 CrossRef CAS
- L. de Campo, A. Yaghmur, L. Sagalowicz, M. E. Leser, H. Watzke and O. Glatter, Langmuir, 2004, 20, 5254–5261 CrossRef CAS PubMed
- C. Li, Q. Li, Y. V. Kaneti, D. Hou, Y. Yamauchi and Y. Mai, Chem. Soc. Rev., 2020, 49, 4681–4736 RSC
- H. M. G. Barriga, M. N. Holme and M. M. Stevens, Angew. Chem., Int. Ed., 2019, 58, 2958–2978 CrossRef CAS PubMed
- L. Zhang, C. Bartels, Y. Yu, H. Shen and A. Eisenberg, Phys. Rev. Lett., 1997, 79, 5034–5037 CrossRef CAS
- C. K. Haluska, W. T. Góźdź, H.-G. Döbereiner, S. Förster and G. Gompper, Phys. Rev. Lett., 2002, 89, 238302 CrossRef PubMed
- Y. Chen, J. Du, M. Xiong, H. Guo, H. Jinnai and T. Kaneko, Macromolecules, 2007, 40, 4389–4392 CrossRef CAS
- B. E. McKenzie, J. l. F. de Visser, H. Friedrich, M. J. Wirix, P. H. Bomans, G. de With, S. J. Holder and N. A. Sommerdijk, Macromolecules, 2013, 46, 9845–9848 CrossRef CAS
- H. Yu, X. Qiu, S. P. Nunes and K.-V. Peinemann, Nat. Commun., 2014, 5, 1–10 Search PubMed
- W. J. Zhang, C. Y. Hong and C. Y. Pan, Macromol. Rapid Commun., 2015, 36, 1428–1436 CrossRef CAS PubMed
- Z. Ding, C. Gao, S. Wang, H. Liu and W. Zhang, Polym. Chem., 2015, 6, 8003–8011 RSC
- K. H. Ku, J. M. Shin, D. Klinger, S. G. Jang, R. C. Hayward, C. J. Hawker and B. J. Kim, ACS Nano, 2016, 10, 5243–5251 CrossRef CAS PubMed
- H. He, K. Rahimi, M. Zhong, A. Mourran, D. R. Luebke, H. B. Nulwala, M. Möller and K. Matyjaszewski, Nat. Commun., 2017, 8, 1–8 CrossRef PubMed
- Z. Lin, S. Liu, W. Mao, H. Tian, N. Wang, N. Zhang, F. Tian, L. Han, X. Feng and Y. Mai, Angew. Chem., Int. Ed., 2017, 56, 7135–7140 CrossRef CAS PubMed
- S. D. Allen, S. Bobbala, N. B. Karabin, M. Modak and E. A. Scott, ACS Appl. Mater. Interfaces, 2018, 10, 33857–33866 CrossRef CAS PubMed
- D. Presa-Soto, G. A. Carriedo, R. de la Campa and A. Presa Soto, Angew. Chem., Int. Ed., 2016, 55, 10102–10107 CrossRef CAS PubMed
- B. E. McKenzie, F. Nudelman, P. H. Bomans, S. J. Holder and N. A. Sommerdijk, J. Am. Chem. Soc., 2010, 132, 10256–10259 CrossRef CAS PubMed
- S. J. Holder, G. Woodward, B. McKenzie and N. A. Sommerdijk, RSC Adv., 2014, 4, 26354–26358 RSC
- B. E. McKenzie, J. F. de Visser, G. Portale, D. Hermida-Merino, H. Friedrich, P. H. H. Bomans, W. Bras, O. R. Monaghan, S. J. Holder and N. A. J. M. Sommerdijk, Soft Matter, 2016, 12, 4113–4122 RSC
- B. E. McKenzie, H. Friedrich, M. J. Wirix, J. F. de Visser, O. R. Monaghan, P. H. Bomans, F. Nudelman, S. J. Holder and N. A. Sommerdijk, Angew. Chem., Int. Ed., 2015, 54, 2457–2461 CrossRef CAS PubMed
- O. Monaghan, P. Bomans, N. A. Sommerdijk and S. J. Holder, Polym. Chem., 2017, 8, 5303–5316 RSC
- X. Lyu, A. Xiao, W. Zhang, P. Hou, K. Gu, Z. Tang, H. Pan, F. Wu, Z. Shen and X. H. Fan, Angew. Chem., Int. Ed., 2018, 57, 10132–10136 CrossRef CAS PubMed
- Y. Kang, A. Pitto-Barry, M. S. Rolph, Z. Hua, I. Hands-Portman, N. Kirby and R. K. O'Reilly, Polym. Chem., 2016, 7, 2836–2846 RSC
- A. Vanderkooy, P. Pfefferkorn and M. S. Taylor, Macromolecules, 2017, 50, 3807–3817 CrossRef CAS
- A. L. Parry, P. H. Bomans, S. J. Holder, N. A. Sommerdijk and S. C. Biagini, Angew. Chem., Int. Ed., 2008, 47, 8859–8862 CrossRef CAS PubMed
- S. A. Barnhill, N. C. Bell, J. P. Patterson, D. P. Olds and N. C. Gianneschi, Macromolecules, 2015, 48, 1152–1161 CrossRef CAS
- Y. La, C. Park, T. J. Shin, S. H. Joo, S. Kang and K. T. Kim, Nat. Chem., 2014, 6, 534–541 CrossRef CAS PubMed
- C. Park, Y. La, T. H. An, H. Y. Jeong, S. Kang, S. H. Joo, H. Ahn, T. J. Shin and K. T. Kim, Nat. Commun., 2015, 6, 1–9 Search PubMed
- T. H. An, Y. La, A. Cho, M. G. Jeong, T. J. Shin, C. Park and K. T. Kim, ACS Nano, 2015, 9, 3084–3096 CrossRef CAS PubMed
- A. Cho, Y. La, S. Jeoung, H. R. Moon, J.-H. Ryu, T. J. Shin and K. T. Kim, Macromolecules, 2017, 50, 3234–3243 CrossRef CAS
- A. Cho, Y. La, T. J. Shin, C. Park and K. T. Kim, Macromolecules, 2016, 49, 4510–4519 CrossRef CAS
- M. G. Jeong and K. T. Kim, Macromolecules, 2017, 50, 223–234 CrossRef CAS
- Y. La, J. Song, M. G. Jeong, A. Cho, S.-M. Jin, E. Lee and K. T. Kim, Nat. Commun., 2018, 9, 5327 CrossRef CAS PubMed
- Kenry, B. Z. Tang and B. Liu, Chem, 2020, 6, 1195–1198 CAS
- J. Mei, N. L. Leung, R. T. Kwok, J. W. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed
- J. Li, J. Wang, H. Li, N. Song, D. Wang and B. Z. Tang, Chem. Soc. Rev., 2020, 49, 1144–1172 RSC
- K. Li and B. Liu, Chem. Soc. Rev., 2014, 43, 6570–6597 RSC
- N. Zhang, H. Chen, Y. Fan, L. Zhou, S. Trepout, J. Guo and M. H. Li, ACS Nano, 2018, 12, 4025–4035 CrossRef CAS PubMed
- X. Tao, H. Chen, S. Trepout, J. Cen, J. Ling and M. H. Li, Chem. Commun., 2019, 55, 13530–13533 RSC
- L. Yan, Y. Zhang, B. Xu and W. Tian, Nanoscale, 2016, 8, 2471–2487 RSC
- D. Zhang, Y. Fan, H. Chen, S. Trepout and M. H. Li, Angew. Chem., Int. Ed., 2019, 58, 10260–10265 CrossRef CAS PubMed
- L. Jia, A. Cao, D. Lévy, B. Xu, P.-A. Albouy, X. Xing, M. J. Bowick and M.-H. Li, Soft Matter, 2009, 5, 3446–3451 RSC
- X. Xing, H. Shin, M. J. Bowick, Z. Yao, L. Jia and M.-H. Li, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 5202–5206 CrossRef CAS PubMed
- C. K. Wong, M. Heidelmann, M. Dulle, X. Qiang, S. Forster, M. H. Stenzel and A. H. Groschel, J. Am. Chem. Soc., 2020, 142, 10989–10995 CrossRef CAS PubMed
-
J. Brandrup, E. H. Immergut, E. A. Grulke, A. Abe and D. R. Bloch, Polymer handbook, Wiley New York, 1989 Search PubMed
- T.-L. Nghiem, S. Riebe, I. Maisuls, C. A. Strassert, J. Voskuhl and A. H. Gröschel, Polymer, 2020, 208, 122942 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR, 13C NMR and SEC spectra of PEG-b-PTPEMA block copolymers. Supplemental data of DLS. Supplemental cryo-EM images of the assemblies. Supplemental data of photoluminescence spectra. Details of cell viability assays. See DOI: 10.1039/d1sc00270h |
| This journal is © The Royal Society of Chemistry 2021 |