
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEmergent synthetic methods for the modular advancement of sp3-rich fragments
Max J.
Caplin
 and
Daniel J.
Foley
*
and
Daniel J.
Foley
*
School of Physical and Chemical Sciences, University of Canterbury, Christchurch, New Zealand. E-mail: daniel.foley@canterbury.ac.nz
First published on 2nd March 2021
Abstract
Fragment-based drug discovery is an important and increasingly reliable technology for the delivery of clinical candidates. Notably, however, sp3-rich fragments are a largely untapped resource in molecular discovery, in part due to the lack of general and suitably robust chemical methods available to aid their development into higher affinity lead and drug compounds. This Perspective describes the challenges associated with developing sp3-rich fragments, and succinctly highlights recent advances in C(sp3)–H functionalisations of high potential value towards advancing fragment hits by ‘growing’ functionalised rings and chains from unconventional, carbon-centred vectors.
 Max J. Caplin | Max Caplin achieved his Master's in Chemistry at Univ. Manchester (2019), which included a Master's research project supervised by Prof. N. J. Turner at the Manchester Institute of Biotechnology. He began his PhD studies at the University of Canterbury in 2019 under the supervision of Dr Daniel Foley, and his research is focused on the development of new methods to enable sp3-rich fragment elaboration. |
 Daniel J. Foley | Dan Foley earned his Master's in Chemistry at Univ. Manchester (2011), including industrial experience at Syngenta. He carried out his PhD (2015) with Profs S. Marsden and A. Nelson at Univ. Leeds, and was subsequently awarded an EPRSC Doctoral Prize Fellowship (2015–2017), which he carried out at Leeds and the Diamond Light Source. Further postdoctoral studies (2017–2018) were completed with with Prof. H. Waldmann at the Max Planck Institute of Molecular Physiology, Dortmund, where he held a Marie Skłodowska–Curie European Fellowship. Dan recently joined the faculty at the University of Canterbury, New Zealand, where his research is focused on the development of new synthetic methods of value to molecular discovery and medicinal chemistry. |
Introduction
Fragment-based drug discovery (FBDD) is an established approach for the development of small molecule tools and drugs.1–3 More than forty compounds derived from FBDD approaches have entered clinical trials, and four drugs have been approved.4 In contrast to traditional high-throughput screening, which relies upon the availability of vast numbers (∼106 to 109) of lead or drug sized compounds, FBDD enables the efficient5,6 exploration of chemical space7–11 by screening smaller collections (∼103) of smaller compounds (typically <20 heavy atoms).Fragments are usually screened against pre-defined molecular targets using biophysical techniques, and generally exhibit high hit rates.1 Fragment hits tend to be weak binders (typically μmol to mmol affinity), yet ligand-to-target interactions can often be maintained from hit-to-lead.12 For instance, the initial hit that was developed into vemurafenib had ∼200 μM affinity.13 The essential role of synthetic chemistry in FBDD is ascertained by the range of strategies required for hit advancement,14,15 commonly including ‘growing’ hits, but also linking and merging fragments.16
The molecular properties of modern fragment libraries are often carefully controlled, e.g. by adhering to Astex's ‘rule of three’,17 or even tighter constraints.15 However, with respect to their molecular shapes, commercially available libraries have historically been dominated by flat (het)aryl compounds,1,7,18,19 and sp3-rich fragments have tended to be underrepresented in screening collections.1,18
It has been suggested that the introduction of shape-diverse fragments into screening collections may enable modulation of intractable molecular targets,1,15,18,20 and allow the construction of unique chemotypes that may constitute valuable intellectual property. Drug candidates featuring fractions of sp3-hybridised carbons in the range 0.4–0.5 have been associated with increased likelihood of clinical progression.21 The proposed introduction of sp3 enriched fragments into screening libraries has raised concerns regarding their potential hit rates,7,22,23 however, reports detailing appreciable hit rates for sp3-enriched fragments are beginning to emerge.24,25 Furthermore, the identification of distinctively shaped hits may provide increased certainty about the validity of their binding.18
In recent years sp3-rich fragment libraries have been designed to include ‘functional handles’ to allow elaboration from specific, often heteroatom-centred (e.g. amine) vectors using robust, well-established transformations (e.g. amide bond formations; reductive aminations).26–29 However, in the absence of functionalisable synthetic handles, concerns may arise about the direct synthetic developability of sp3-rich fragments due to a lack of predictable synthetic methods to aid their advancement.30 Indeed, pre-emptive development and exemplification of synthetic chemistries for direct fragment elaboration has recently been framed as a pressing challenge for the synthetic chemistry community.15,30 While sp3-rich fragments pose challenges with regards to their synthetic (and biological) advancement, they also offer exciting opportunities for the development of new synthetic methods.14,15,24,30–33 This review highlights recent enabling synthetic methods that are likely to be useful towards the advancement of sp3-rich fragment hits into higher affinity lead and drug compounds.
Defining ‘valuable’ methods for fragment advancement
In a typical fragment screening campaign, a hit with suitable binding affinity against a given biomolecular target is selected for development. Structural analogues of the fragment hit are prepared (or sourced) and screened against the target, with a view towards rationalising the exploitable chemical space around the hit for structural advancement of the ligand. In particular, enhancements to the hit's binding affinity and ligand-to-target binding interaction networks are sought after, ideally leading to improvements in ligand efficiency.1For typical commercial fragments, structural analogues of hit compounds may be readily prepared by ‘growing’ the molecule from heteroatom-centred vectors, allowing variation of the peripheral functionalities and ring-systems (Fig. 1, orange arrows). Well-established synthetic methods such as amide bond formations, alkylations etc. are typically used.34 However, elaboration of fragments from heteroatom vectors may disturb crucial fragment-to-target binding interactions, hampering development.15
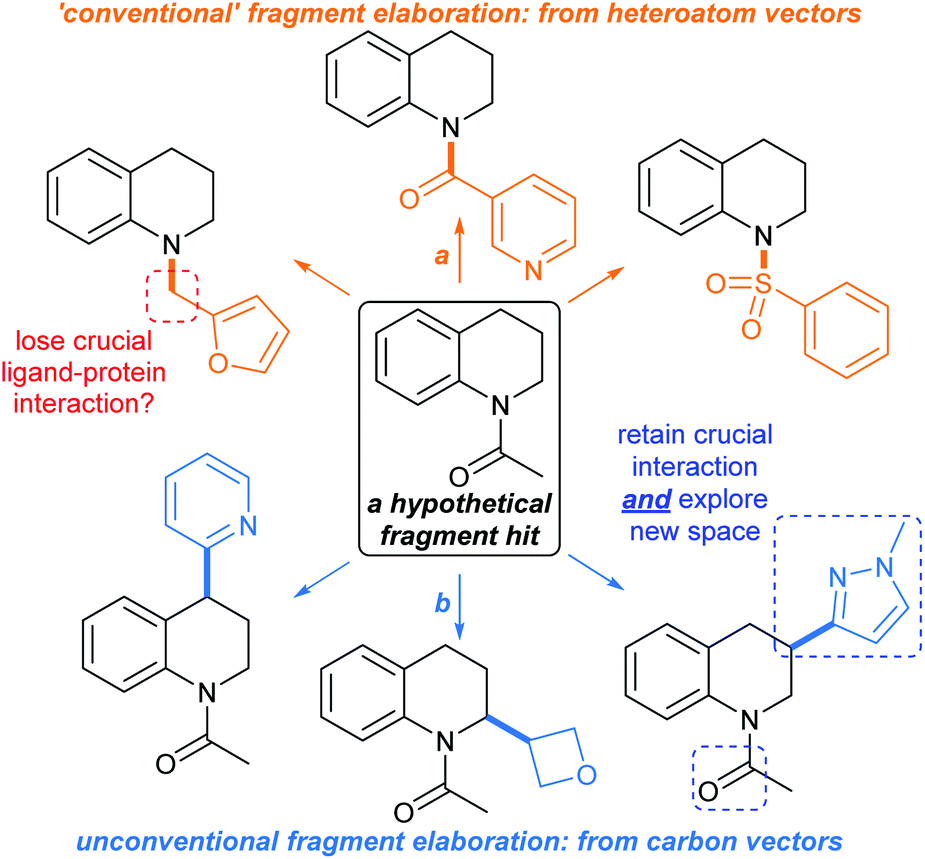 | ||
| Fig. 1 Methods for fragment elaboration. (a) Conventional elaboration exploiting heteroatom-centred vectors; (b) unconventional elaboration by C(sp3)–H functionalisations (focus of this Perspective). | ||
Due to their weak affinities, and general lack of cellular activity, fragment hits are situated towards the lower end of the synthesis-value trajectory in drug discovery,14 and therefore synthetic chemists typically want to avoid esoteric and/or tedious synthetic routes to deliver follow-up compounds, as these may only have limited value towards progressing a hit compound.
One approach to fragment elaboration has been to pre-emptively develop reactions that enable the exploration of chemical space around specific scaffolds.35 In addition, we propose that general synthetic toolkits should be developed for the advancement of sp3-rich fragment hits, and indeed herein show that many relevant methods are already in existence. Ideally these toolkits should tolerate functionalities that are likely to maintain ligand-to-target binding interactions,15 without the need for protecting groups, with the aim of either (1) generating new binding interactions between the fragment and the target or (2) reinforcing existing binding interactions (Fig. 1, blue arrows). Such synthetic toolkits should ensure that fragments can be reliably advanced in predictable, modular, and additive ways.
Suitable synthetic toolkits to achieve these aims differ slightly from late-stage optimisation toolkits where, in general, specific ‘point mutations’ to a lead compound are often sought for, such as the introduction of small functional groups (e.g. alcohols, amines), and only occasionally entire ring systems.36
In order to grow fragments into higher affinity ligands, new ring systems generally need to be added to drive improvements in affinity and develop high quality polar ligand-to-target binding interactions. Furthermore, the augmentation of fragment hits with functionalised aliphatic chains may offer opportunities to link hits together and generate lead compounds.1
This review serves to provide a snapshot of the current state-of-the-art for elaborating sp3-rich heterocycles by forming new C–C bonds via C(sp3)–H functionalisations, specifically with regard to methods that are likely to have value towards introducing new heteroatom-containing ring systems and linkers into sp3-rich fragments. Due to the emphasis of FBDD on ligand structure, this Perspective is focused on overall synthetic transformations, rather than in-depth discussion of the mechanistic aspects of the reactions highlighted. The methods summarised herein may be considered an ‘early-stage optimisation toolkit’ for fragment development.
Nascent methods for fragment development
Heteroatoms either imbedded in, or exocyclic to, a fragment framework tend to enable crucial intermolecular binding interactions between a fragment and its biomolecular target. Synthetic methodologies that can exploit fragment functionalities whilst maintaining pre-existing fragment-to-target binding interactions are therefore likely to have high utility in hit advancement.15 The following sections focus on new C–C bond forming methodologies that exploit C(sp3)–H functionalisations to enable the incorporation of functionalised ring-systems and chains to sp3-rich frameworks, with a view to increasing the potential for hits to interact with their intended molecular targets and increase their affinity (Fig. 2). Attention is given to recent methodologies that can introduce new vectors into sp3-rich frameworks using innate selectivity (i.e. reactions exploiting the intrinsic reactivity of C–H bonds), and guided selectivity (i.e. harnessing pre-existing functionalities adorning a framework to either proximally or distally connect new C–C bonds).36,37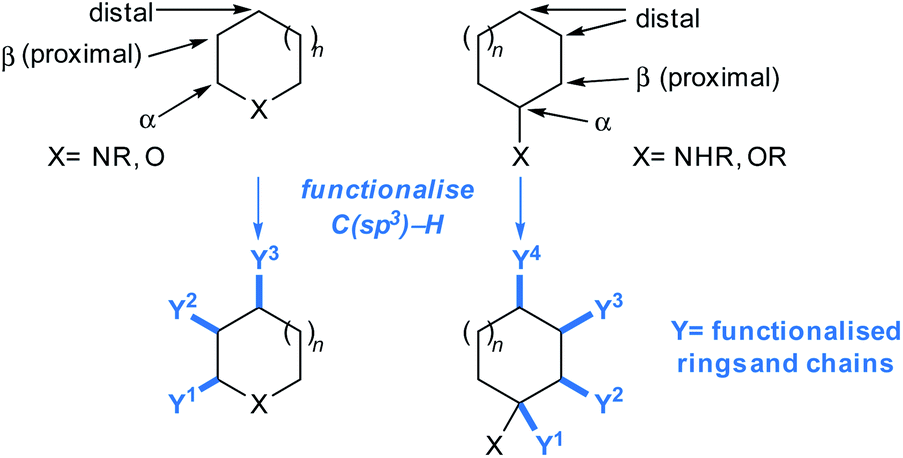 | ||
| Fig. 2 Functionalisation types highlighted in this Perspective. | ||
In choosing the methodologies that are discussed herein, key considerations were (1) the potential ability of the methodology to rapidly grow fragment hits and increase their potential for interactions with a biomolecule in a previously unprecedented manner; (2) robustness with respect to functional group-tolerance (where possible we have included examples demonstrating compatibility with unprotected amines and nitrogenous hetaryls); and (3) the atom efficiency of the transformation, where possible avoiding discussion of reactions where a large excess of a bespoke fragment hit would be required. With regards to their downstream synthetic development, chromatographically inseparable mixtures of diastereomers (and/or regioisomers) have the potential to frustrate the fragment-to-lead optimisation process. Therefore, where similar overall transformations are described, we offer guidance towards selecting methods that generally provide higher regio- and diastereocontrol. We wish to emphasise that the methodologies that we have chosen to discuss were not necessarily developed with the underlying intention of advancing fragments, and may have some limitations in this regard, however, we hope that the general framework provided herein can provide guidance to those developing new methodologies with this purpose in mind.
C(sp3)–H functionalisations with (het)aryls
By introducing rigid rings with specifically placed heteroatoms, incorporation of (het)aryl ring systems to Fsp3-rich fragments offers the potential for opportunities to strengthen the network of intermolecular interactions between a fragment hit and biological target. For instance, this may be achieved by introducing π–π stacking interactions with aromatic amino acid residues in a protein backbone; and, where heteroatoms feature in or on the aromatic ring, through introducing new hydrogen-bonding interactions. Inversely, functionalisation of aromatic fragments with sp3-rich systems might be used as a means to enhance their aqueous solubility.38Arylation strategies exploiting innate selectivity
 | ||
Scheme 2 Arylations α- to alicyclic heteroatoms, mediated by a decatungstate photocatalyst.44 Unless otherwise stated, rr (regiomeric ratio) and dr are >95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5. :5. | ||
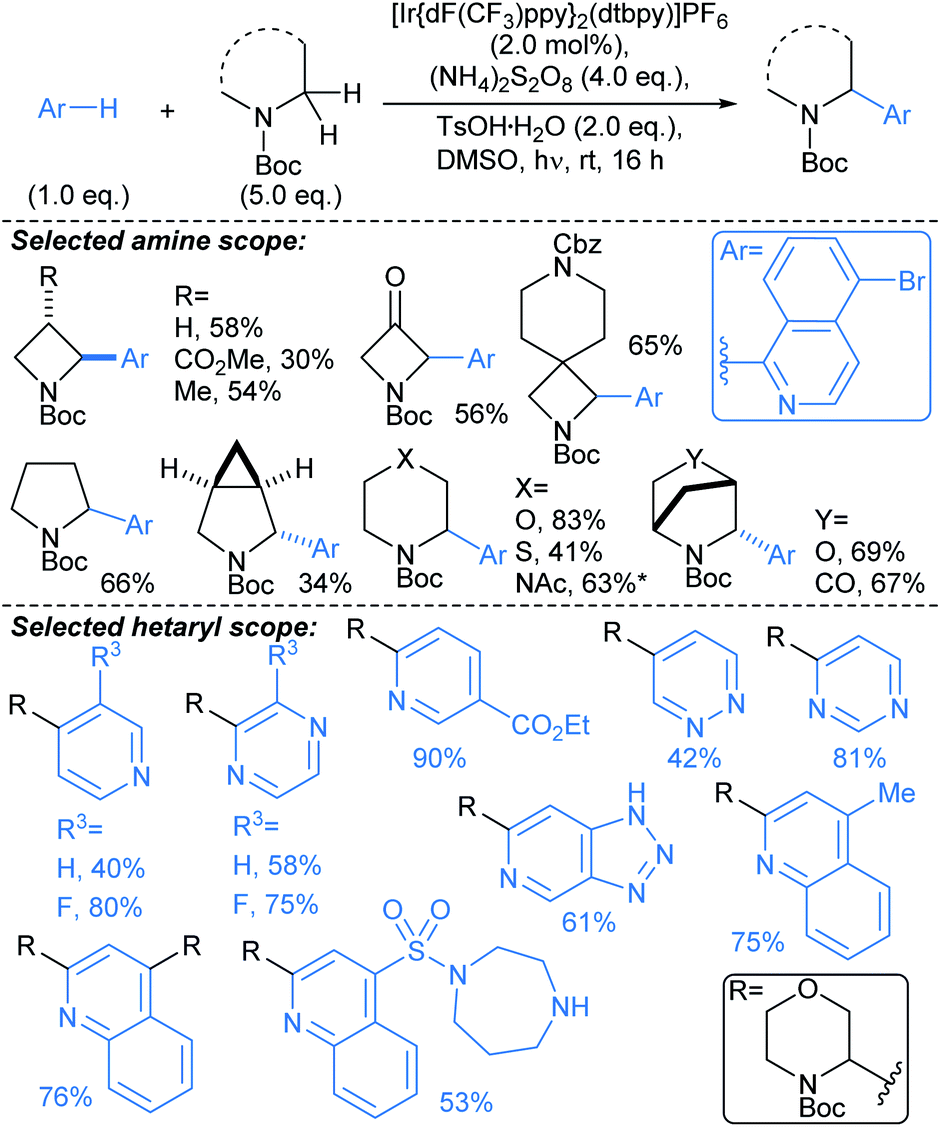
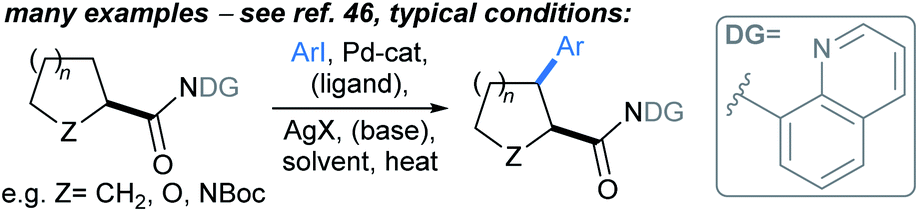
:11–97:3 er. A range of aryl iodides were tolerated in the protocol, including substrates bearing boronate ester (Bpin), cyano, amine, (protected) alcohol, and halide functional groups. Without further optimisation, the method was immediately applicable to 4–7-membered saturated azacycles, including a piperazine (→2), and in general high enantioselectivities were achieved (up to 98:2 er). Remarkably, functionalisation of tetrahydroisoquinoline gave complete regioselectivity for arylation at the non-benzylic methylene (→3). The procedure was also carried out using a non-chiral ligand (PCy3) to give comparable yields of racemic tetrahydroquinolines. A marked advantage of this approach over previous46 guided C(sp3)–H activation approaches is that the directing group can be removed following the arylation by treatment NaOMe. Related protocols were developed by Yu47 and Gong.48
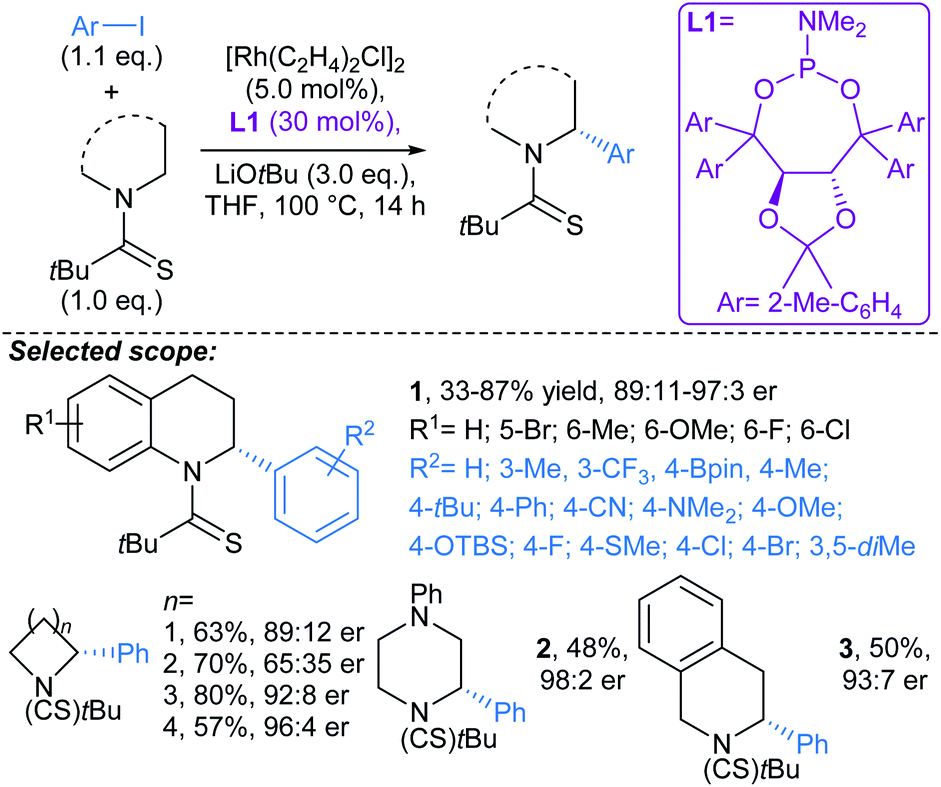 | ||
| Scheme 3 Rh-catalysed arylation of nitrogenous heterocycles.45 | ||
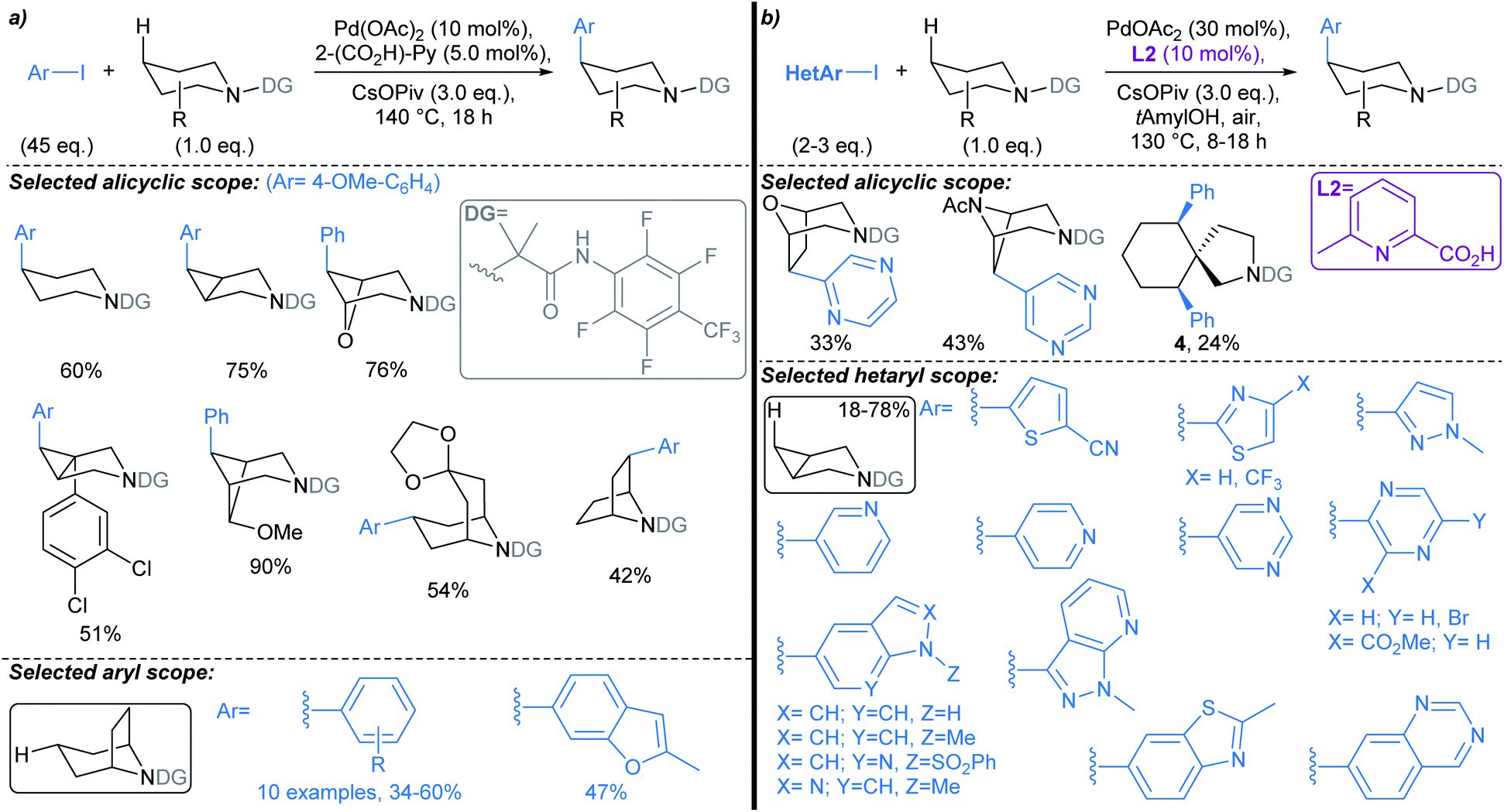 | ||
| Scheme 4 Pd-catalysed directed C(sp3)–H arylations of alicyclic amines.50,52 (a) Original procedure developed by Sanford; (b) next generation protocol by Li, Dechanstreiter, and Dandapandi, incorporating ligand L2, and demonstrating substantial scope with respect to the aryl iodide coupling partner. | ||
Arylations distal to exocyclic functional groups
 | ||
| Scheme 5 Pd-catalysed directed C(sp3)–H α-arylations distal to alicyclic carboxylic acids.46 | ||
Yu first described Pd-catalysed γ-arylation of secondary exocyclic amines using aryl iodides and 2-hydroxynicotinaldehyde as a transient directing group (TDG) in 2016.55 Development of this reaction to enable compatibility with hetaryl iodides was achieved through the cooperative effect of a TDG in combination with a 2-pyridone ligand (e.g.L3, L4), which acted as an internal base to accelerate Pd insertion into C(sp3)–H bonds, and mitigated unproductive side reactions.56 Both γ- and δ-arylations were possible using the latter methodology (Scheme 6a and b, respectively), with the regioselectivity controlled through careful choice of TDG and ligand. TDGs that coordinate Pd(II) and form a six-membered chelate were selective for γ-C–H bond activation (e.g.TDG1), whereas TDGs that coordinate with Pd(II) via a five-membered chelate activated δ-C–H bonds (e.g.TDG2). γ-Arylation of cyclohexylamine 5 was demonstrated using hetaryl iodides, including functionalised pyridines, pyrimidines, quinolines, and quinoxalines (Scheme 6a). In general, complete diastereoselectivity for the γ-cis arylated products was achieved in monocyclic aliphatic ring systems, with the exception of cyclopentylamine, which gave the γ-arylated product in 50:50 dr. Similar scope coupling hetaryl bromides was achieved using an alternative TDG. The δ-arylation protocol proceeded in with high diastereoselectivity (up to >91:9 dr), showing preference for activation of methyl C–H bonds (Scheme 6b), but δ-methylene arylation was possible in the absence of δ-methyl groups (→6).
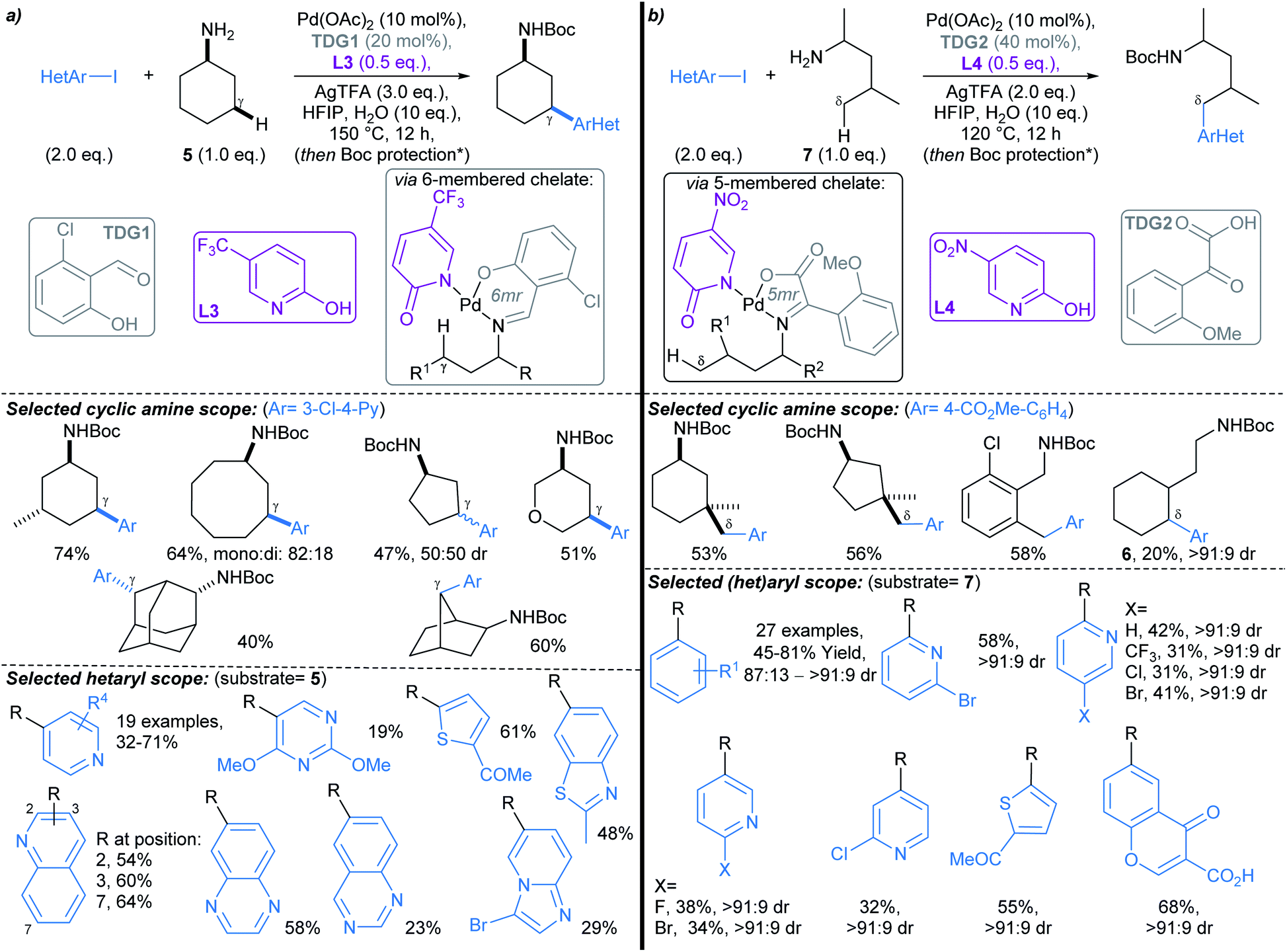 :5 dr), but poorer stereocontrol was observed for 6- and 7-membered ring alcohols (60:40 to 67:33 dr). Due to the wide prevalence of alcohols in pharmaceuticals, this transformation is likely to have high value for fragment functionalisation. It is worth noting that while the directing group is not transient, it could be readily installed in a single operation, and was removed by Pd(OH)2/C catalysed hydrogenolysis.
:5 dr), but poorer stereocontrol was observed for 6- and 7-membered ring alcohols (60:40 to 67:33 dr). Due to the wide prevalence of alcohols in pharmaceuticals, this transformation is likely to have high value for fragment functionalisation. It is worth noting that while the directing group is not transient, it could be readily installed in a single operation, and was removed by Pd(OH)2/C catalysed hydrogenolysis.
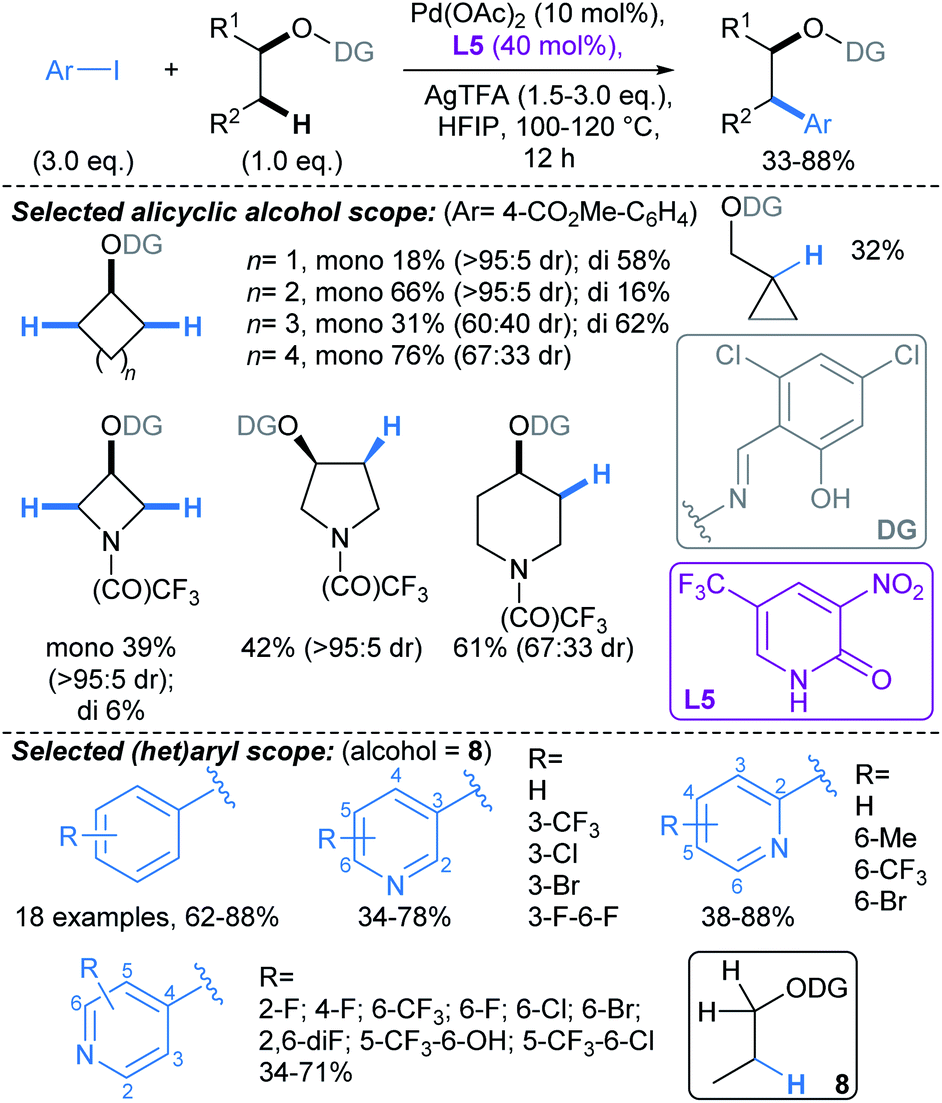 | ||
| Scheme 7 Pd-catalysed directed β-C(sp3)–H arylations of alcohols using an O–N tethered directing group.57 | ||
Yu developed a conceptually related Pd-catalysed approach, mediated by an O–N tethered iminopyruvate-derived directing group, that enabled C(sp3)–H arylation γ- to aliphatic alcohols (Scheme 8). This proceeds via the formation of a six-membered palladacycle.58 Combination of DG1 with 2-pyridone ligand L6, enabled arylation of substrates without active C–H bonds β- to the alcohol (i.e. substrates with no β-methyl or -methylene groups, Scheme 8a). For substrates with active C–H bonds β- to the alcohol, use of DG2, without a 2-pyridone ligand, allowed selective activation of γ-methylene C–H bonds (Scheme 8b). Interestingly, in the latter protocol, γ-C–H arylation was less favourable in smaller rings due to their rigid geometric strain: β:γ selectivity for small ring alcohols, 9 and 10, were >95:5 and 67:33, respectively, versus β:γ selectivity of 20:80 and <5:95 for the medium (11) and large (12) ring substrates. The directing group could be installed in a two-step, one-pot procedure, and was removed by Pd/C catalysed hydrogenolysis.
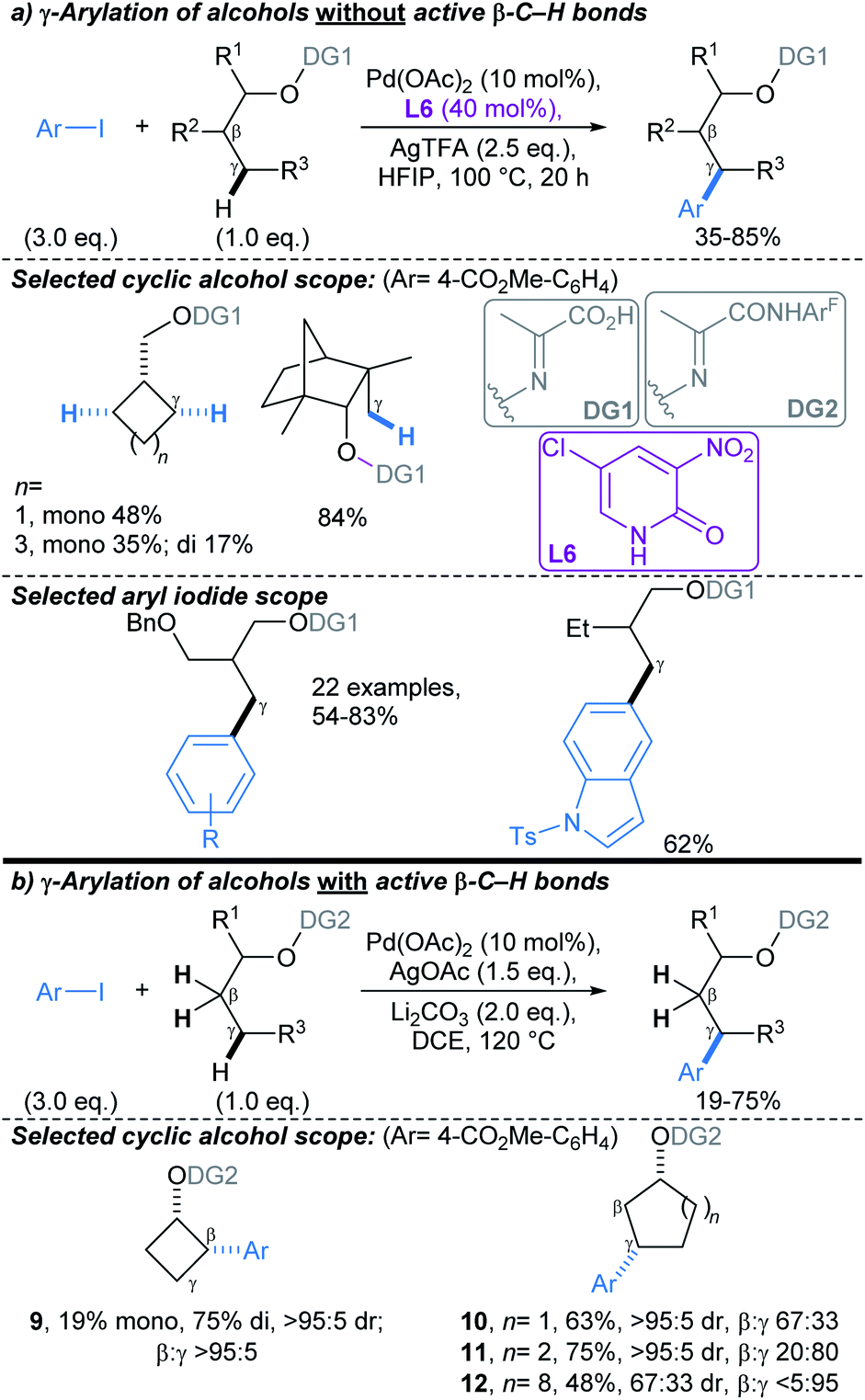 | ||
| Scheme 8 Pd-catalysed directed γ-C(sp3)–H arylations of alcohols using an O-tethered directing group.58 (a) γ-Arylation of alcohols without active β-C–H bonds; (b) γ-arylation of alcohols with active β-C–H bonds. ArF = 2,3,5,6-tetrafluoro-4-(trifluoromethyl) phenyl. | ||
C(sp3)–H functionalisations with aliphatic ring-systems and chains
Incorporation of aliphatic rings and chains to Fsp3-rich fragments offers the potential for opportunities to strengthen the network of intermolecular interactions between a fragment hit and biological target, for instance, by enabling new hydrogen-bonding interactions between specifically placed heteroatoms on or in the heteroaliphatic ring. In addition, incorporation of appropriately oriented functionalised aliphatic chains may enable fragment hits binding to adjacent protein sub-pockets to be linked1 together to accelerate ligand development.Alkylation strategies exploiting innate selectivity
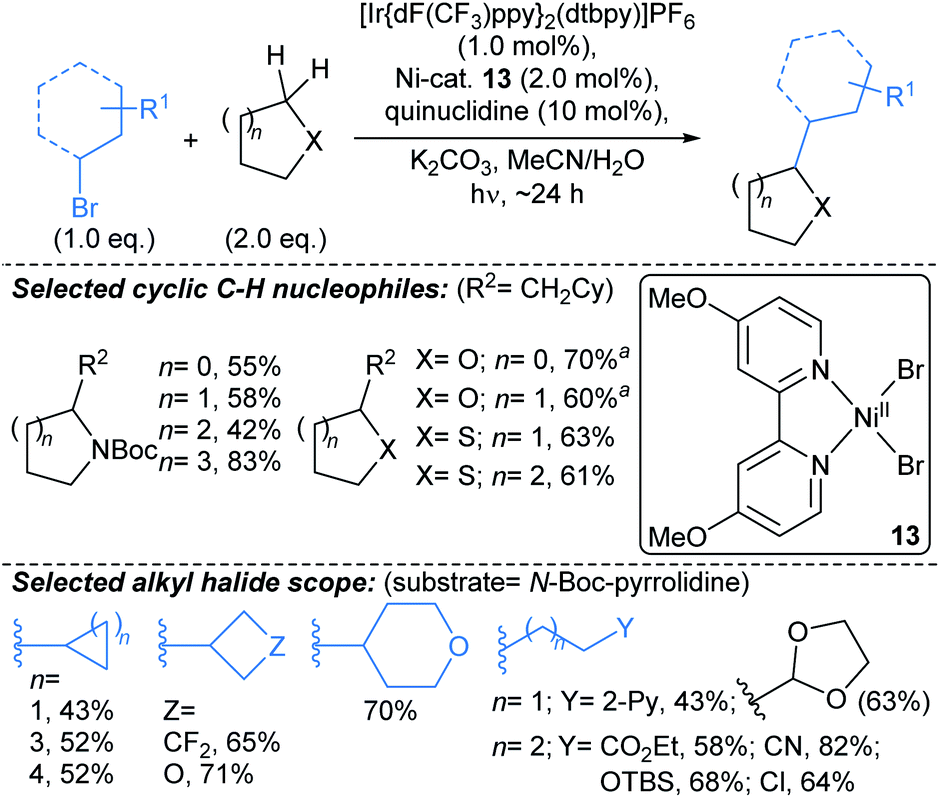 | ||
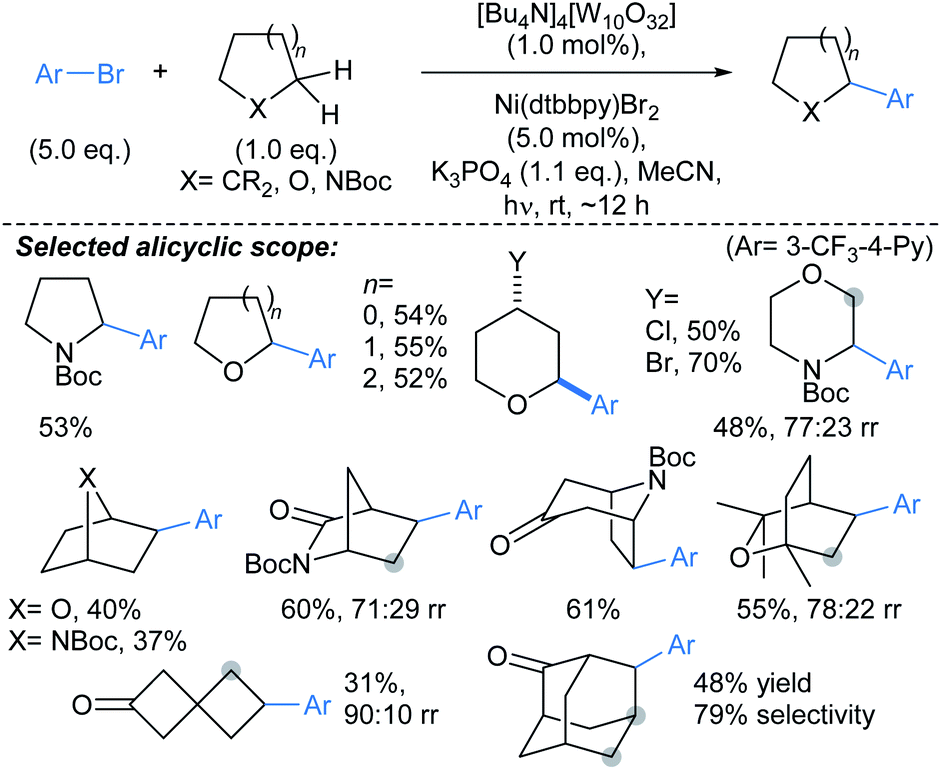
| Scheme 9 Alkylations α- to alicyclic heteroatoms using a Ir/quinuclidine/Ni catalyst system.60†a50 eq. of cyclic ether used in the reaction. | ||
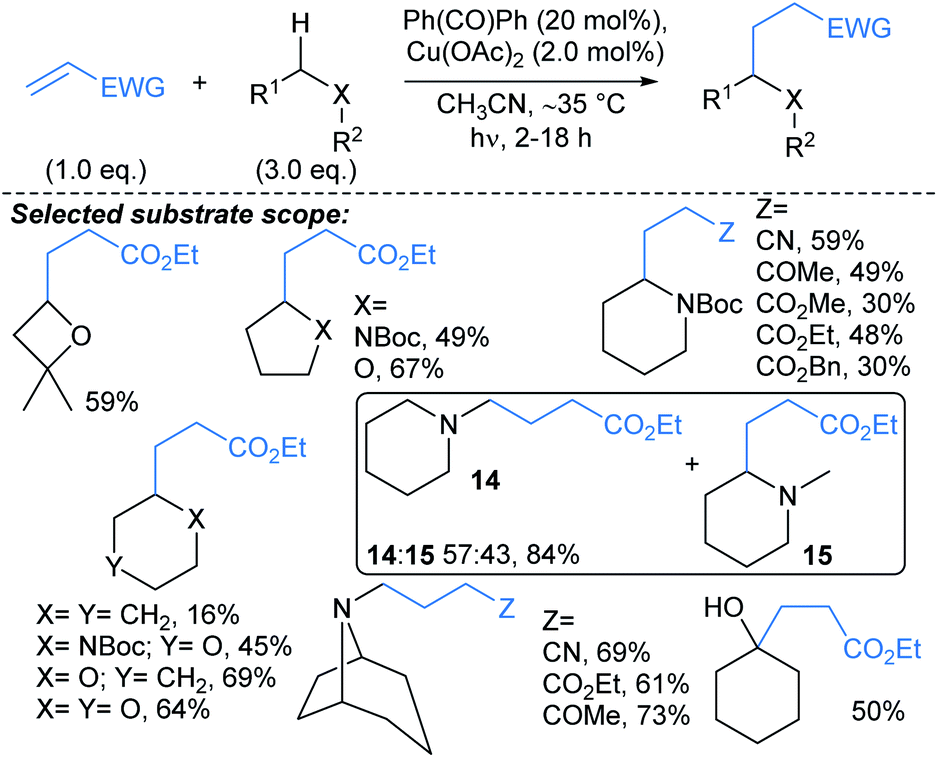
Toullec and Vincent used an approach combining the photoreactivity of benzophenone with Cu catalysis to couple alkyl radicals and electron deficient alkenes under UV-A irradiation (Scheme 10).61 A range of electron deficient alkenes were found to be productive coupling partners in the reaction, including alkyl acrylates, acrylonitriles and vinyl ketones. Interestingly in the absence of Cu(OAc)2, the alkenes polymerised (>95%), suggesting that Cu plays a role in limiting alkene polymerisation. The reaction was regioselective for functionalisation α- to (N,O) heteroatoms, including next to unprotected tertiary amines, Boc-protected amines, ethers, and unprotected primary and secondary alcohols. Further selectivity studies showed that for N-Boc morpholine, functionalisation α- to Boc-protected amine was favoured over the ether. In addition, N-methylpiperidine showed a slight preference (57:43) for methyl (→14) versus α-methylene alkylation (→15). Noteworthy protocols that deliver similar overall transformations were developed by Barriault,62 Knowles and Alexanian,63 and Nicewicz.64,65
Zhang, Yang, and Walsh described cross-dehydrogenative coupling of saturated heterocycles with benzophenone imines (Scheme 11).66 2-Azaallyl anions are highly reducing, and by reducing aryl iodide 16 to the corresponding aryl radical they can generate a sterically hindered sacrificial hydrogen atom abstractor. This aryl radical subsequently enables H-abstraction from a suitable donor such as an ether, unprotected amine (or toluene derivative, not shown), followed by combination of the resulting radical with the 2-azaallyl radical. Although the donor was generally used as solvent, five equivalents of more expensive substrates were effectively coupled using benzene as solvent (e.g. →17). Interestingly, for N-methyl cyclic amines, coupling was only observed adjacent to the amino group on the ring. No or poor (up to 74:26 dr) diastereoselectivity is generally observed in the products, so with regard to use of this method for fragment-to-lead optimisation, next generation protocols would benefit from inducing stereocontrol in the procedure. Related protocols have been developed using photoredox catalysis,66 but the methodology highlighted offers the advantage of avoiding transition metal catalysts. The benzophenone imine products can be readily hydrolysed to provide the free primary amines.
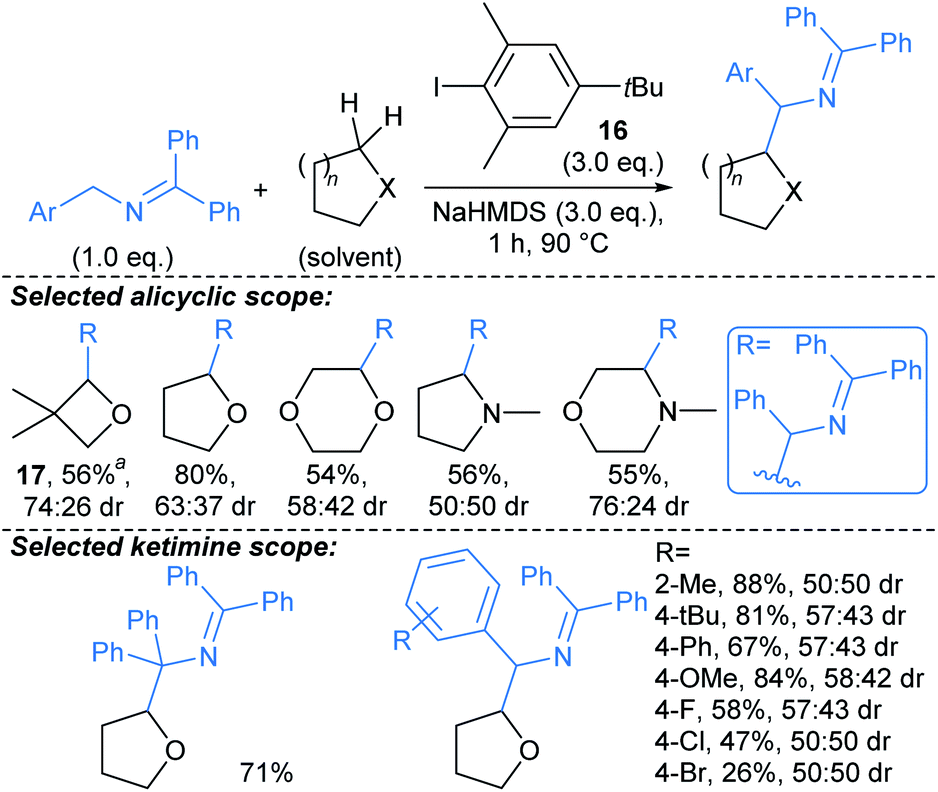 | ||
| Scheme 11 Cross-dehydrogenative couplings between saturated heterocycles and benzophenone imines.66†a5.0 eq. heteroaliphatic donor used. | ||
A related protocol developed by Wang enabled coupling of α-amino radicals with benzaldehydes and aryl ketones to give 1,2-amino alcohols, under irradiation from white light LEDs (Scheme 12).67 This was postulated to proceed by a Brønsted acid-activated proton-coupled electron transfer pathway. The optimised conditions exhibited tolerance of a range of functionalities on benzaldehyde coupling partners, including nitrile and ester functionalities, along with nitrogenous heteroaromatic benzaldehydes (4-pyridyl; 5-pyrimidinyl). The substrate scope with respect to ketones included substituted acetophenones and benzophenones. Aldimines were also suitable coupling partners, furnishing vicinal diamines (not shown). The demonstrated scope of this methodology was limited to coupling anilines with aldehydes and ketones, therefore for the development of next generation protocols, compatibility with amines bearing removable protecting groups would offer a marked advantage. As for the procedure above (Scheme 11), no or poor (up to 60:40 dr) diastereoselectivity is observed for this transformation, which, without further development might limit its utility in fragment-to-lead optimisation.
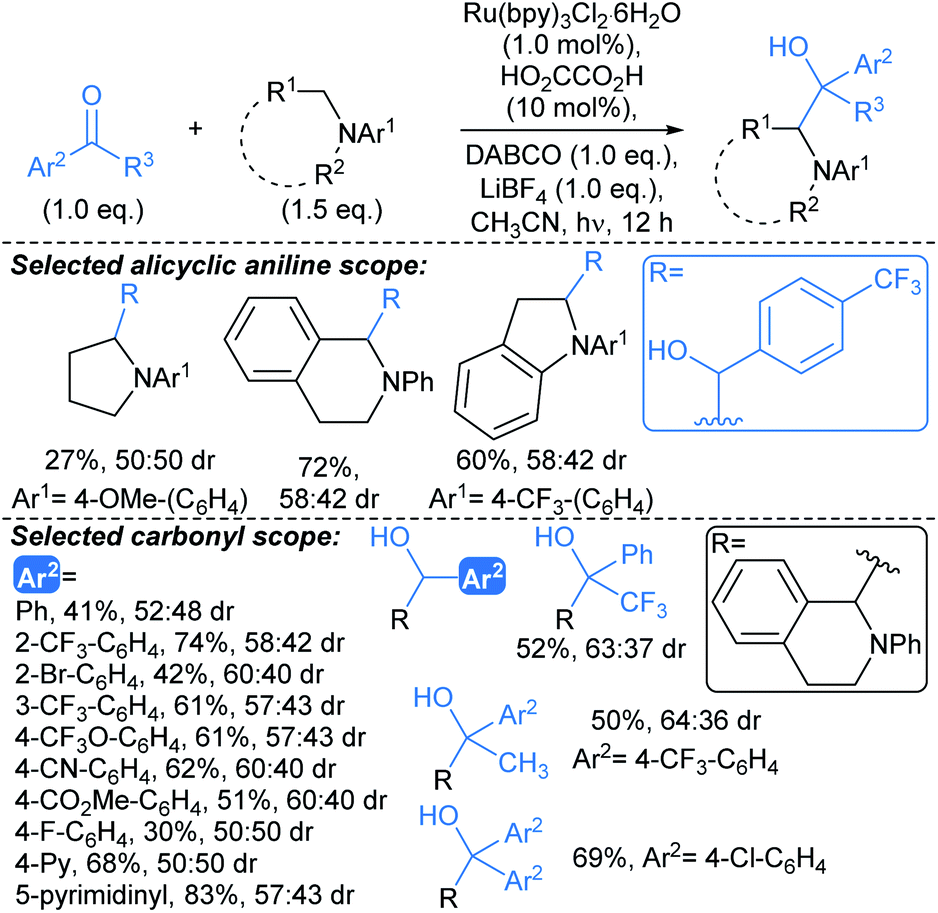
Alkylation strategies exploiting guided selectivity
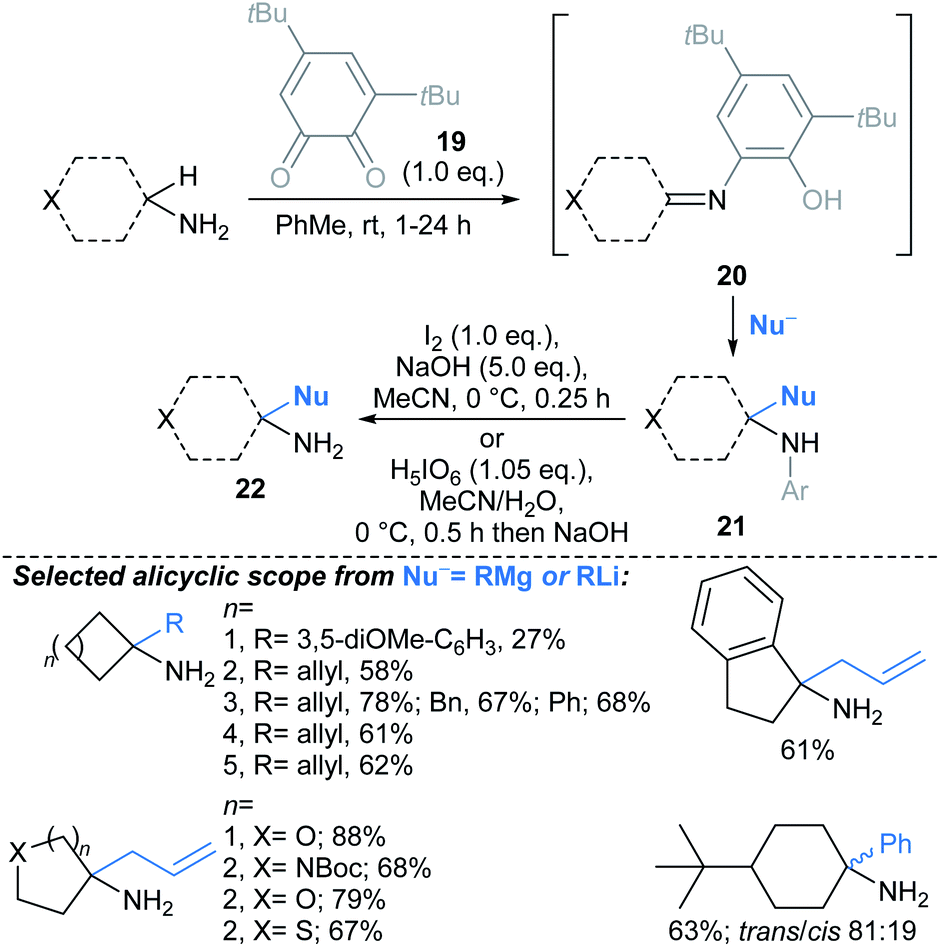 | ||
| Scheme 14 Quinone-mediated oxidation of primary amines followed by addition of nucleophiles, furnishing primary α-tertiary amines.70† Conditions for Grignard addition: RMgX or RLi (6.0 eq.), TMEDA (1.0 eq.), 0–25 °C, 1–24 h. | ||
Creswell developed a catalytic approach for C(sp3)–H alkylation of unprotected primary amines (Scheme 15).71 Under blue LED irradiation, alicyclic primary amines were alkylated at the α-position with alkyl acrylates, using a process combining an organophotocatalyst with Bu4N+N3− as a HAT catalyst. Treatment of the alkylated products 23 with Et3N delivered spirocyclic γ-lactams 24, in a one-pot telescoped procedure. The approach was successfully applied to molecules containing polar functionalities such as unprotected alcohols, (thio)ethers, sulfonamides, sulfones, and nitrogenous hetaryl rings, along with protected amines and ketones. The reaction was compatible with functionalised acrylates and styrenes. Notably, the alkylation also worked using a continuous flow photoreactor (reaction times 10–20 min, versus 20 h for the batch set up).
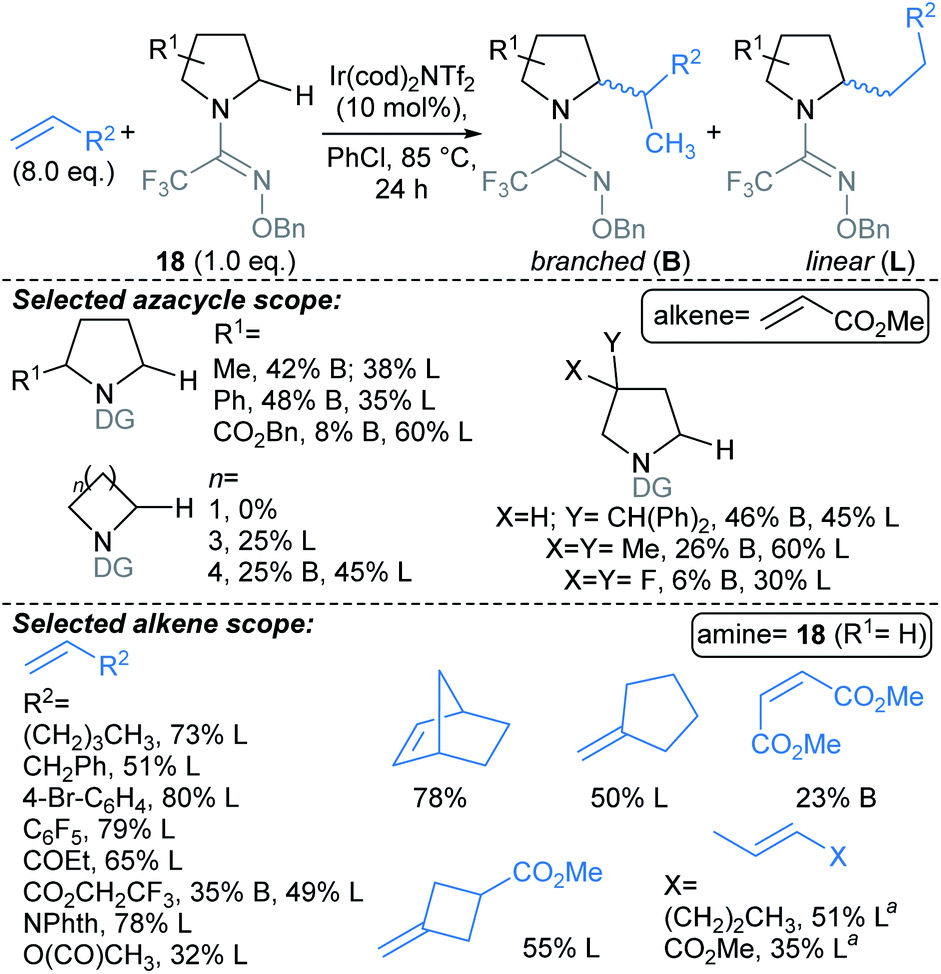
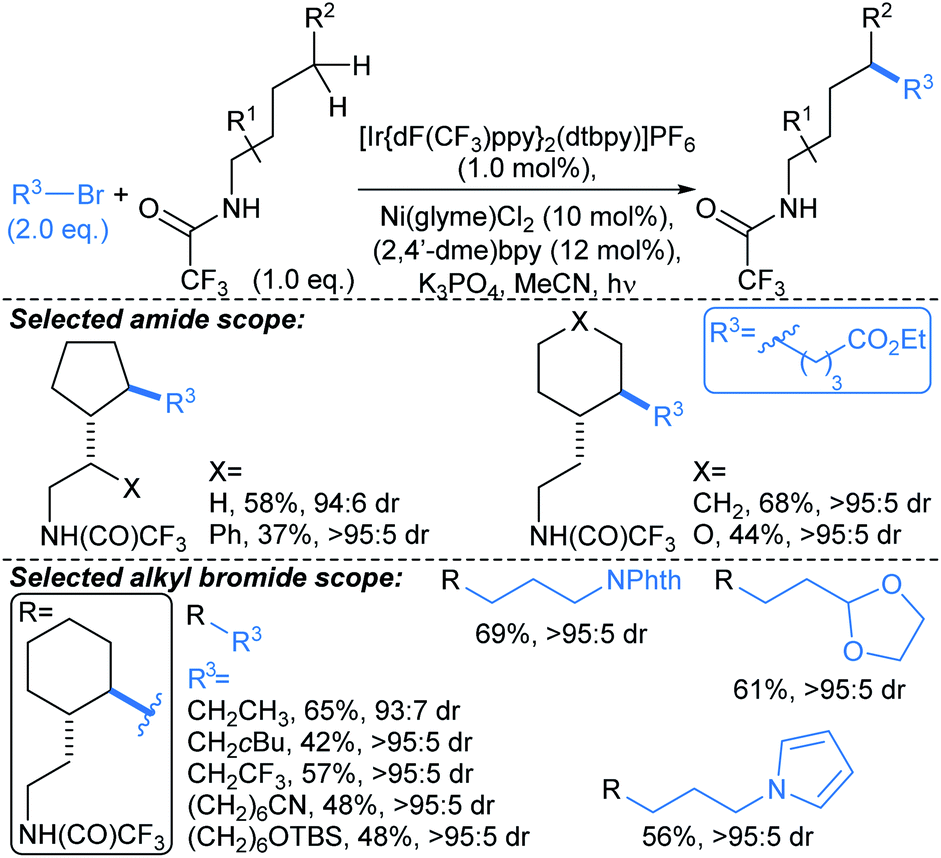
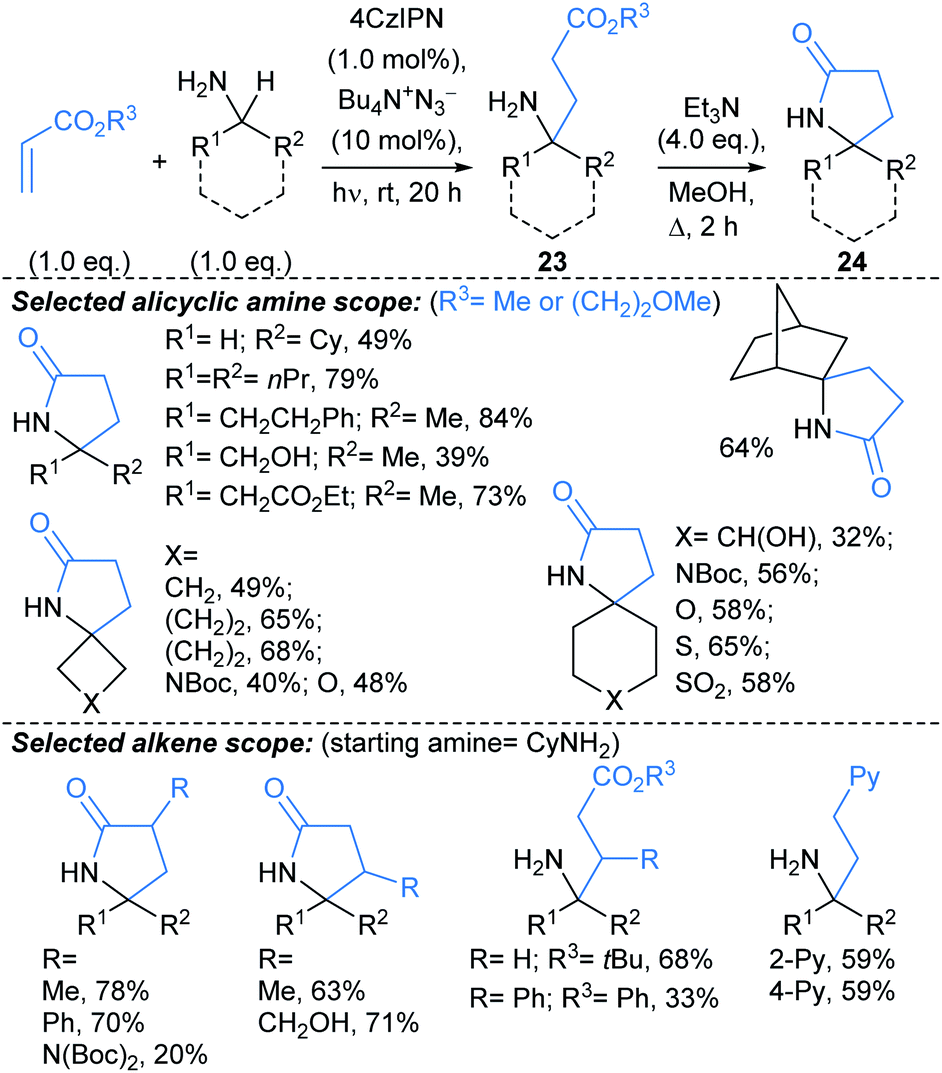 :5 dr. This methodology might be harnessed to introduce fragment linkers to an existing fragment before deprotection and derivatisation at the amide nitrogen. Related approaches by Rovis,73 and Knowles,74 use Michael acceptors to trap the δ-methylenyl alkyl radicals.
:5 dr. This methodology might be harnessed to introduce fragment linkers to an existing fragment before deprotection and derivatisation at the amide nitrogen. Related approaches by Rovis,73 and Knowles,74 use Michael acceptors to trap the δ-methylenyl alkyl radicals.
Future outlook
In this Perspective we outlined the need for methods to elaborate sp3-rich fragments, and have identified existing methodologies that are likely to be useful towards achieving this challenging goal. As an illustrative “thought experiment”, we have considered how the methods discussed might be applied to develop known sp3-rich fragment hits,4,75–78 without an unreasonable level of prior manipulation to the hit's structure (Fig. 3). Since the fragments shown were already successfully developed79–82 into lead compounds, we are not suggesting that it would necessarily be biologically relevant to develop these hits in the manners shown. However, we believe that Fig. 3 showcases potential instances where the methods discussed could prove to be useful as synthetic tools for fragment-to-lead development. A limitation of this exercise is that it is likely that there are sp3-rich fragment hits that were not previously developed into leads due to the confines of the available methodologies for fragment elaboration at the time of hit discovery,30 but such hits might now be developable using methodologies highlighted in this manuscript.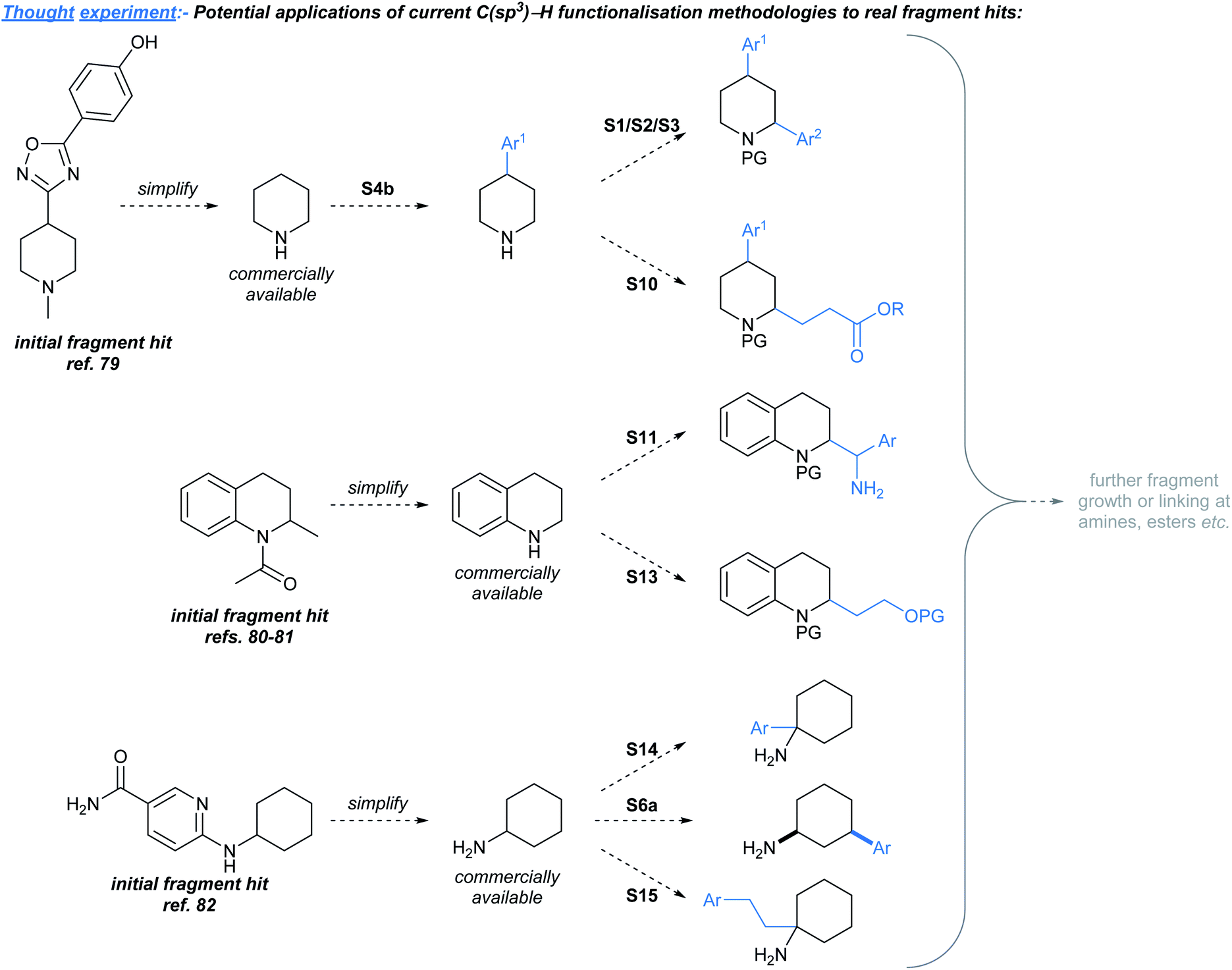 | ||
| Fig. 3 A “thought experiment” showing how known sp3-rich fragment hits might be structurally advanced using methodologies highlighted in this Perspective. “S#” refers to the specific scheme number in this manuscript for the transformation shown. For brevity, protecting group manipulation strategies are not shown. PG = protecting group. | ||
Almost all of the methods discussed in this review were not specifically developed for the purpose of advancing fragments, and in some instances, the presently available methods have deficiencies with respect to their regio- and diastereoselectivity, atom efficiency, and compatibility with functional groups typically found in fragment hits (e.g. unprotected amines). Moving forwards, practitioners of fragment-based drug discovery will significantly benefit from the development of robust methods that resolve such challenges. Nonetheless the state-of-the-art hints at a bright future for both mission-driven14,15,30 and ‘blue skies’ methodology development for sp3-rich fragment-to-lead optimisation.
High yielding synthetic transformations that harness the bespoke fragment component as the limiting reagent, and that allow functionalisations of heteroalicyclic systems without the requirement for installation and removal of protecting groups and/or fixed directing groups, will revolutionise the efficiency of sp3-rich fragment advancement. In some instances, ligand development may allow improvements to the regio- and diastereoselectivity of the methods discussed in this review. Reaction conditions that tolerate exposure to air and water will have high utility in array and automated reactions, enabling systematic growth and diversification at specific fragment vectors to explore biologically relevant chemical space, establish structure–activity relationships, and drive hit advancement. Biocatalysis is also likely to play an important future role in fragment elaboration, as recently highlighted by Cosgrove and Turner.33
Conclusions
We have summarised exciting recent synthetic developments that are likely to have high value towards elaborating sp3-rich fragments. There remains huge scope for the establishment of efficient new classes of reactions that can enable fragment growth from specific vectors, incorporating structural motifs that can further their potential for interaction with their molecular targets. Future advances in this field will drive marked improvements in the development of fragment hits into higher affinity compounds, and ultimately new drugs.Author contributions
D. J. F. conceived the idea for the Perspective. D. J. F. and M. J. C. conducted the literature investigation and wrote the draft manuscript. D. J. F. wrote the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the following organisations for funding: the Royal Society of Chemistry (RSC Research Fund R19-5035), the Biomolecular Interaction Centre, and the University of Canterbury. We are grateful to the Univ. Canterbury College of Science for a Doctoral Scholarship (to M. J. C.). We thank Jerram Wood for proofreading this manuscript.Notes and references
- D. A. Erlanson, S. W. Fesik, R. E. Hubbard, W. Jahnke and H. Jhoti, Twenty Years on: The Impact of Fragments on Drug Discovery, Nat. Rev. Drug Discovery, 2016, 15(9), 605–619, DOI:10.1038/nrd.2016.109
.
- B. C. Doak, R. S. Norton and M. J. Scanlon, The Ways and Means of Fragment-Based Drug Design, Pharmacol. Ther., 2016, 167, 28–37, DOI:10.1016/j.pharmthera.2016.07.003
- M. Baker, Fragment-Based Lead Discovery Grows Up, Nat. Rev. Drug Discovery, 2012, 121, 2012 Search PubMed
- W. Jahnke, D. A. Erlanson, I. J. P. de Esch, C. N. Johnson, P. N. Mortenson, Y. Ochi and T. Urushima, Fragment-to-Lead Medicinal Chemistry Publications in 2019, J. Med. Chem., 2020, 57, 47, DOI:10.1021/acs.jmedchem.0c01608
- I. D. Kuntz, K. Chen, K. A. Sharp and P. A. Kollman, The Maximal Affinity of Ligands, Proc. Natl. Acad. Sci. U. S. A., 1999, 96(18), 9997–10002, DOI:10.1073/pnas.96.18.9997
- A. L. Hopkins, C. R. Groom and A. Alex, Ligand Efficiency: A Useful Metric for Lead Selection, Drug Discovery Today, 2004, 9(10), 430–431, DOI:10.1016/S1359-6446(04)03069-7
- R. J. Hall, P. N. Mortenson and C. W. Murray, Efficient Exploration of Chemical Space by Fragment-Based Screening, Prog. Biophys. Mol. Biol., 2014, 116(2–3), 82–91, DOI:10.1016/J.PBIOMOLBIO.2014.09.007
- L. Ruddigkeit, R. van Deursen, L. C. Blum and J.-L. Reymond, Enumeration of 166 Billion Organic Small Molecules in the Chemical Universe Database GDB-17, J. Chem. Inf. Model., 2012, 52(11), 2864–2875, DOI:10.1021/ci300415d
- A. H. Lipkus, Q. Yuan, K. A. Lucas, S. A. Funk, W. F. Bartelt, R. J. Schenck and A. J. Trippe, Structural Diversity of Organic Chemistry. A Scaffold Analysis of the CAS Registry, J. Org. Chem., 2008, 73(12), 4443–4451, DOI:10.1021/jo8001276
- T. D. Downes, S. P. Jones, H. F. Klein, M. C. Wheldon, M. Atobe, P. S. Bond, J. D. Firth, N. S. Chan, L. Waddelove and R. E. Hubbard,
et al., Design and Synthesis of 56 Shape-Diverse 3D Fragments, Chem.–Eur. J., 2020, 26(41), 8969–8975, DOI:10.1002/chem.202001123
- S. L. Kidd, T. J. Osberger, N. Mateu, H. F. Sore and D. R. Spring, Recent Applications of Diversity-Oriented Synthesis Toward Novel, 3-Dimensional Fragment Collections, Front. Chem., 2018, 6, 460, DOI:10.3389/fchem.2018.00460
- C. W. Murray and D. C. Rees, The Rise of Fragment-Based Drug Discovery, Nat. Chem., 2009, 1(3), 187–192, DOI:10.1038/nchem.217
- G. Bollag, J. Tsai, J. Zhang, C. Zhang, P. Ibrahim, K. Nolop and P. Hirth, Vemurafenib: The First Drug Approved for BRAF-Mutant Cancer, Nat. Rev. Drug Discovery, 2012, 11(11), 873–886, DOI:10.1038/nrd3847
- D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Organic Synthesis Provides Opportunities to Transform Drug Discovery, Nat. Chem., 2018, 10(4), 383–394, DOI:10.1038/s41557-018-0021-z
- C. W. Murray and D. C. Rees, Opportunity Knocks: Organic Chemistry for Fragment-Based Drug Discovery (FBDD), Angew. Chem., Int. Ed., 2016, 55(2), 488–492, DOI:10.1002/anie.201506783
- D. E. Scott, A. G. Coyne, S. A. Hudson and C. Abell, Fragment-Based Approaches in Drug Discovery and Chemical Biology, Biochemistry, 2012, 51(25), 4990–5003, DOI:10.1021/bi3005126
- M. Congreve, R. Carr, C. Murray and H. Jhoti, A ‘Rule of Three’ for Fragment-Based Lead Discovery?, Drug Discovery Today, 2003, 8(19), 876–877, DOI:10.1016/S1359-6446(03)02831-9
- A. D. Morley, A. Pugliese, K. Birchall, J. Bower, P. Brennan, N. Brown, T. Chapman, M. Drysdale, I. H. Gilbert and S. Hoelder,
et al., Fragment-Based Hit Identification: Thinking in 3D, Drug Discovery Today, 2013, 18(23–24), 1221–1227, DOI:10.1016/j.drudis.2013.07.011
- S. Barelier and I. Krimm, Ligand Specificity, Privileged Substructures and Protein Druggability from Fragment-Based Screening, Curr. Opin. Chem. Biol., 2011, 15(4), 469–474, DOI:10.1016/J.CBPA.2011.02.020
- A. W. Hung, A. Ramek, Y. Wang, T. Kaya, J. A. Wilson, P. A. Clemons and D. W. Young, Route to Three-Dimensional Fragments Using Diversity-Oriented Synthesis, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(17), 6799–6804, DOI:10.1073/pnas.1015271108
- F. Lovering, J. Bikker and C. Humblet, Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success, J. Med. Chem., 2009, 52(21), 6752–6756, DOI:10.1021/jm901241e
- M. M. Hann, A. R. Leach and G. Harper, Molecular Complexity and Its Impact on the Probability of Finding Leads for Drug Discovery, J. Chem. Inf. Model., 2001, 41(3), 856–864, DOI:10.1021/ci000403i
- F. Lovering, Escape from Flatland 2: Complexity and Promiscuity, Medchemcomm, 2013, 4(3), 515–519, 10.1039/C2MD20347B
- P. C. Ray, M. Kiczun, M. Huggett, A. Lim, F. Prati, I. H. Gilbert and P. G. Wyatt, Fragment Library Design, Synthesis and Expansion: Nurturing a Synthesis and Training Platform, Drug Discovery Today, 2017, 22(1), 43–56, DOI:10.1016/J.DRUDIS.2016.10.005
- D. J. Foley, R. G. Doveston, I. Churcher, A. Nelson and S. P. Marsden, A Systematic Approach to Diverse, Lead-like Scaffolds from α,α-Disubstituted Amino Acids, Chem. Commun., 2015, 51(56), 11174–11177, 10.1039/C5CC03002A
- D. G. Twigg, N. Kondo, S. L. Mitchell, W. R. J. D. Galloway, H. F. Sore, A. Madin and D. R. Spring, Partially Saturated Bicyclic Heteroaromatics as an Sp3 -Enriched Fragment Collection, Angew. Chem., Int. Ed., 2016, 55(40), 12479–12483, DOI:10.1002/anie.201606496
- R. Zhang, P. J. McIntyre, P. M. Collins, D. J. Foley, C. Arter, F. von Delft, R. Bayliss, S. Warriner and A. Nelson, Construction of a Shape-Diverse Fragment Set: Design, Synthesis and Screen against Aurora-A Kinase, Chem.–Eur. J., 2019, 25(27), 6831–6839, DOI:10.1002/chem.201900815
- D. J. Foley, P. G. E. Craven, P. M. Collins, R. G. Doveston, A. Aimon, R. Talon, I. Churcher, F. von Delft, S. P. Marsden and A. Nelson, Synthesis and Demonstration of the Biological Relevance of Sp3-Rich Scaffolds Distantly Related to Natural Product Frameworks, Chem.–Eur. J., 2017, 23(60), 15227–15232, DOI:10.1002/chem.201704169
- A. R. Hanby, N. S. Troelsen, T. J. Osberger, S. L. Kidd, K. T. Mortensen and D. R. Spring, Fsp3-Rich and Diverse Fragments Inspired by Natural Products as a Collection to Enhance Fragment-Based Drug Discovery, Chem. Commun., 2020, 56(15), 2280–2283, 10.1039/C9CC09796A
- J. D. St. Denis, R. J. Hall, C. W. Murray, T. D. Heightman and D. C. Rees, Fragment-Based Drug Discovery: Opportunities for Organic Synthesis, RSC Med. Chem., 2021 10.1039/D0MD00375A
- G. M. Keserű, D. A. Erlanson, G. G. Ferenczy, M. M. Hann, C. W. Murray and S. D. Pickett, Design Principles for Fragment Libraries: Maximizing the Value of Learnings from Pharma Fragment-Based Drug Discovery (FBDD) Programs for Use in Academia, J. Med. Chem., 2016, 59(18), 8189–8206, DOI:10.1021/acs.jmedchem.6b00197
- Q. Michaudel, Y. Ishihara and P. S. Baran, Academia-Industry Symbiosis in Organic Chemistry, Acc. Chem. Res., 2015, 48(3), 712–721, DOI:10.1021/ar500424a
- J. I. Ramsden, S. C. Cosgrove and N. J. Turner, Is It Time for Biocatalysis in Fragment-Based Drug Discovery?, Chem. Sci., 2020, 11(41), 11104–11112, 10.1039/D0SC04103C
- O. B. Cox, T. Krojer, P. Collins, O. Monteiro, R. Talon, A. Bradley, O. Fedorov, J. Amin, B. D. Marsden and J. Spencer,
et al., A Poised Fragment Library Enables Rapid Synthetic Expansion Yielding the First Reported Inhibitors of PHIP(2), an Atypical Bromodomain, Chem. Sci., 2016, 7(3), 2322–2330, 10.1039/C5SC03115J
- N. Palmer, T. M. Peakman, D. Norton and D. C. Rees, Design and Synthesis of Dihydroisoquinolones for Fragment-Based Drug Discovery (FBDD), Org. Biomol. Chem., 2016, 14(5), 1599–1610, 10.1039/C5OB02461G
- T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, The Medicinal Chemist's Toolbox for Late Stage Functionalization of Drug-like Molecules, Chem. Soc. Rev., 2016, 45(3), 546–576, 10.1039/C5CS00628G
- T. Brückl, R. D. Baxter, Y. Ishihara and P. S. Baran, Innate and Guided C-H Functionalization Logic, Acc. Chem. Res., 2012, 45(6), 826–839, DOI:10.1021/ar200194b
- T. J. Ritchie and S. J. F. Macdonald, The Impact of Aromatic Ring Count on Compound Developability - Are Too Many Aromatic Rings a Liability in Drug Design?, Drug Discovery Today, 2009, 14(21–22), 1011–1020, DOI:10.1016/j.drudis.2009.07.014
- J. Jin and D. W. C. MacMillan, Direct α-Arylation of Ethers through the Combination of Photoredox-Mediated C-H Functionalization and the Minisci Reaction, Angew. Chem., Int. Ed., 2015, 54(5), 1565–1569, DOI:10.1002/anie.201410432
- R. A. Garza-Sanchez, A. Tlahuext-Aca, G. Tavakoli and F. Glorius, Visible Light-Mediated Direct Decarboxylative C-H Functionalization of Heteroarenes, ACS Catal., 2017, 7(6), 4057–4061, DOI:10.1021/acscatal.7b01133
- R. S. J. Proctor, H. J. Davis and R. J. Phipps, Catalytic Enantioselective Minisci-Type Addition to Heteroarenes, Science, 2018, 360(6387), 419–422, DOI:10.1126/science.aar6376
- R. Grainger, T. D. Heightman, S. V. Ley, F. Lima and C. N. Johnson, Enabling Synthesis in Fragment-Based Drug Discovery by Reactivity Mapping: Photoredox-Mediated Cross-Dehydrogenative Heteroarylation of Cyclic Amines, Chem. Sci., 2019, 10(8), 2264–2271, 10.1039/C8SC04789H
- M. H. Shaw, V. W. Shurtleff, J. A. Terrett, J. D. Cuthbertson and D. W. C. MacMillan, Native Functionality in Triple Catalytic Cross-Coupling: Sp3 C-H Bonds as Latent Nucleophiles, Science, 2016, 352(6291), 1304–1308, DOI:10.1126/science.aaf6635
- I. B. Perry, T. F. Brewer, P. J. Sarver, D. M. Schultz, D. A. DiRocco and D. W. C. MacMillan, Direct Arylation of Strong Aliphatic C–H Bonds, Nature, 2018, 560(7716), 70–75, DOI:10.1038/s41586-018-0366-x
- S. Greßies, F. J. R. Klauck, J. H. Kim, C. G. Daniliuc and F. Glorius, Ligand-Enabled Enantioselective Csp3 -H Activation of Tetrahydroquinolines and Saturated Aza-Heterocycles by Rh I, Angew. Chem., Int. Ed., 2018, 57(31), 9950–9954, DOI:10.1002/anie.201805680
- D. Antermite and J. A. Bull, Transition Metal-Catalyzed Directed C(Sp3)–H Functionalization of Saturated Heterocycles, Synthesis, 2019, 51(17), 3171–3204, DOI:10.1055/s-0037-1611822
- P. Jain, P. Verma, G. Xia and J.-Q. Yu, Enantioselective Amine α-Functionalization via Palladium-Catalysed C–H Arylation of Thioamides, Nat. Chem., 2017, 9(2), 140–144, DOI:10.1038/nchem.2619
- L. Gong, H.-J. Jiang, X.-M. Zhong, J. Yu, Y. Zhang, X. Zhang and Y.-D. Wu, Assembling a Hybrid Pd-Catalyst from Chiral Anionic Co(III)-Complex and Ligand for Asymmetric C(Sp3)-H Functionalization, Angew. Chem., Int. Ed., 2018 DOI:10.1002/anie.201812426
- J. J. Topczewski, P. J. Cabrera, N. I. Saper and M. S. Sanford, Palladium-Catalysed Transannular C–H Functionalization of Alicyclic Amines, Nature, 2016, 531(7593), 220–224, DOI:10.1038/nature16957
- P. J. Cabrera, M. Lee and M. S. Sanford, Second-Generation Palladium Catalyst System for Transannular C–H Functionalization of Azabicycloalkanes, J. Am. Chem. Soc., 2018, 140(16), 5599–5606, DOI:10.1021/jacs.8b02142
- M. Lee, A. Adams, P. Cox and M. Sanford, Access to 3D Alicyclic Amine-Containing Fragments through Transannular C–H Arylation, Synlett, 2019, 30(4), 417–422, DOI:10.1055/s-0037-1610861
- Z. Li, M. Dechantsreiter and S. Dandapani, Systematic Investigation of the Scope of Transannular C–H Heteroarylation of Cyclic Secondary Amines for Synthetic Application in Medicinal Chemistry, J. Org. Chem., 2020, 85(10), 6747–6760, DOI:10.1021/acs.joc.0c00870
- C. He, W. G. Whitehurst and M. J. Gaunt, Palladium-Catalyzed C(Sp3)–H Bond Functionalization of Aliphatic Amines, Chem, 2019, 5(5), 1031–1058, DOI:10.1016/j.chempr.2018.12.017
- T. Bhattacharya, S. Pimparkar and D. Maiti, Combining Transition Metals and Transient Directing Groups for C-H Functionalizations, RSC Adv., 2018, 8(35), 19456–19464, 10.1039/C8RA03230K
- Y. Wu, Y. Q. Chen, T. Liu, M. D. Eastgate and J. Q. Yu, Pd-Catalyzed γ-C(Sp3)-H Arylation of Free Amines Using a Transient Directing Group, J. Am. Chem. Soc., 2016, 138(44), 14554–14557, DOI:10.1021/jacs.6b09653
- Y. Q. Chen, Z. Wang, Y. Wu, S. R. Wisniewski, J. X. Qiao, W. R. Ewing, M. D. Eastgate and J. Q. Yu, Overcoming the Limitations of γ- And δ-C-H Arylation of Amines through Ligand Development, J. Am. Chem. Soc., 2018, 140(51), 17884–17894, DOI:10.1021/jacs.8b07109
- G. Xia, Z. Zhuang, L. Y. Liu, S. L. Schreiber, B. Melillo and J. Q. Yu, Ligand-Enabled β-Methylene C(Sp3)–H Arylation of Masked Aliphatic Alcohols, Angew. Chem., Int. Ed., 2020, 59(20), 7783–7787, DOI:10.1002/anie.202000632
- G. Xia, J. Weng, L. Liu, P. Verma, Z. Li and J. Q. Yu, Reversing Conventional Site-Selectivity in C(Sp 3)–H Bond Activation, Nat. Chem., 2019, 11(6), 571–577, DOI:10.1038/s41557-019-0245-6
- J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, The Merger of Transition Metal and Photocatalysis, Nat. Rev. Chem., 2017, 1(7), 0052, DOI:10.1038/s41570-017-0052
- C. Le, Y. Liang, R. W. Evans, X. Li and D. W. C. MacMillan, Selective Sp 3 C-H Alkylation via Polarity-Match-Based Cross-Coupling, Nature, 2017, 547(7661), 79–83, DOI:10.1038/nature22813
- B. Abadie, D. Jardel, G. Pozzi, P. Toullec and J. M. Vincent, Dual Benzophenone/Copper-Photocatalyzed Giese-Type Alkylation of C(Sp3)–H Bonds, Chem.–Eur. J., 2019, 25(70), 16120–16127, DOI:10.1002/chem.201904111
- S. Rohe, A. O. Morris, T. McCallum and L. Barriault, Hydrogen Atom Transfer Reactions via Photoredox Catalyzed Chlorine Atom Generation, Angew. Chem., Int. Ed., 2018, 57(48), 15664–15669, DOI:10.1002/anie.201810187
- C. M. Morton, Q. Zhu, H. Ripberger, L. Troian-Gautier, Z. S. D. Toa, R. R. Knowles and E. J. C.-H. Alexanian, Alkylation via Multisite-Proton-Coupled Electron Transfer of an Aliphatic C-H Bond, J. Am. Chem. Soc., 2019, 141(33), 13253–13260, DOI:10.1021/jacs.9b06834
- J. B. McManus, N. P. R. Onuska and D. A. Nicewicz, Generation and Alkylation of α-Carbamyl Radicals via Organic Photoredox Catalysis, J. Am. Chem. Soc., 2018, 140(29), 9056–9060, DOI:10.1021/jacs.8b04890
- J. B. McManus, N. P. R. Onuska, M. S. Jeffreys, N. C. Goodwin and D. A. Nicewicz, Site-Selective C-H Alkylation of Piperazine Substrates via Organic Photoredox Catalysis, Org. Lett., 2020, 22(2), 679–683, DOI:10.1021/acs.orglett.9b04456
- Z. Liu, M. Li, G. Deng, W. Wei, P. Feng, Q. Zi, T. Li, H. Zhang, X. Yang and P. J. Walsh, Transition-Metal-Free C(Sp3)-H/C(Sp3)-H Dehydrogenative Coupling of Saturated Heterocycles with: N-Benzyl Imines, Chem. Sci., 2020, 11(29), 7619–7625, 10.1039/D0SC00031K
- Q. Xia, H. Tian, J. Dong, Y. Qu, L. Li, H. Song, Y. Liu and Q. Wang, N-Arylamines Coupled with Aldehydes, Ketones, and Imines by Means of Photocatalytic Proton-Coupled Electron Transfer, Chem.–Eur. J., 2018, 24(37), 9269–9273, DOI:10.1002/chem.201801886
- P. Verma, J. M. Richter, N. Chekshin, J. X. Qiao and J. Q. Yu, Iridium(I)-Catalyzed α-C(Sp3)-H Alkylation of Saturated Azacycles, J. Am. Chem. Soc., 2020, 142(11), 5117–5125, DOI:10.1021/jacs.9b12320
- A. T. Tran and J.-Q. Yu, Practical Alkoxythiocarbonyl Auxiliaries for Iridium(I)-Catalyzed C–H Alkylation of Azacycles, Angew. Chem., Int. Ed., 2017, 56(35), 10530–10534, DOI:10.1002/anie.201704755
- D. Vasu, A. L. Fuentes de Arriba, J. A. Leitch, A. De Gombert and D. J. Dixon, Primary α-Tertiary Amine Synthesis via α-C-H Functionalization, Chem. Sci., 2019, 10(11), 3401–3407, 10.1039/C8SC05164J
- A. S. H. Ryder, W. B. Cunningham, G. Ballantyne, T. Mules, A. G. Kinsella, J. Turner-Dore, C. M. Alder, L. J. Edwards, B. S. J. McKay and M. N. Grayson,
et al., Photocatalytic α-Tertiary Amine Synthesis via C–H Alkylation of Unmasked Primary Amines, Angew. Chem., Int. Ed., 2020, 59(35), 14986–14991, DOI:10.1002/anie.202005294
- S. M. Thullen, S. M. Treacy and T. Rovis, Regioselective Alkylative Cross-Coupling of Remote Unactivated C(Sp3)-H Bonds, J. Am. Chem. Soc., 2019, 141(36), 14062–14067, DOI:10.1021/jacs.9b07014
- J. C. K. Chu and T. Rovis, Amide-Directed Photoredox-Catalysed C–C Bond Formation at Unactivated Sp3 C–H Bonds, Nature, 2016, 539(7628), 272–275, DOI:10.1038/nature19810
- G. J. Choi, Q. Zhu, D. C. Miller, C. J. Gu and R. R. Knowles, Catalytic Alkylation of Remote C-H Bonds Enabled by Proton-Coupled Electron Transfer, Nature, 2016, 539(7628), 268–271, DOI:10.1038/nature19811
- C. N. Johnson, D. A. Erlanson, C. W. Murray and D. C. Rees, Fragment-to-Lead Medicinal Chemistry Publications in 2015, J. Med. Chem., 2017, 89–99, DOI:10.1021/acs.jmedchem.6b01123
- C. N. Johnson, D. A. Erlanson, W. Jahnke, P. N. Mortenson and D. C. Rees, Fragment-to-Lead Medicinal Chemistry Publications in 2016, J. Med. Chem., 2018, 1774–1784, DOI:10.1021/acs.jmedchem.7b01298
- P. N. Mortenson, D. A. Erlanson, I. J. P. De Esch, W. Jahnke and C. N. Johnson, Fragment-to-Lead Medicinal Chemistry Publications in 2017, J. Med. Chem., 2018, 62(8), 3857–3872, DOI:10.1021/acs.jmedchem.8b01472
- D. A. Erlanson, I. J. P. De Esch, W. Jahnke, C. N. Johnson and P. N. Mortenson, Fragment-to-Lead Medicinal Chemistry Publications in 2018, J. Med. Chem., 2020, 63(9), 4430–4444, DOI:10.1021/acs.jmedchem.9b01581
- P. Di Lello, R. Pastor, J. M. Murray, R. A. Blake, F. Cohen, T. D. Crawford, J. Drobnick, J. Drummond, L. Kategaya and T. Kleinheinz,
et al., Discovery of Small-Molecule Inhibitors of Ubiquitin Specific Protease 7 (USP7) Using Integrated NMR and in Silico Techniques, J. Med. Chem., 2017, 60(24), 10056–10070, DOI:10.1021/acs.jmedchem.7b01293
- D. S. Millan, K. J. Kayser-Bricker, M. W. Martin, A. C. Talbot, S. E. R. Schiller, T. Herbertz, G. L. Williams, G. P. Luke, S. Hubbs and M. A. Alvarez Morales,
et al., Design and Optimization of Benzopiperazines as Potent Inhibitors of BET Bromodomains, ACS Med. Chem. Lett., 2017, 8(8), 847–852, DOI:10.1021/acsmedchemlett.7b00191
- R. P. Law, S. J. Atkinson, P. Bamborough, C. W. Chung, E. H. Demont, L. J. Gordon, M. Lindon, R. K. Prinjha, A. J. B. Watson and D. J. Hirst, Discovery of Tetrahydroquinoxalines as Bromodomain and Extra-Terminal Domain (BET) Inhibitors with Selectivity for the Second Bromodomain, J. Med. Chem., 2018, 61(10), 4317–4334, DOI:10.1021/acs.jmedchem.7b01666
- R. Caldwell, L. Liu-Bujalski, H. Qiu, I. Mochalkin, R. Jones, C. Neagu, A. Goutopoulos, R. Grenningloh, T. Johnson and B. Sherer,
et al., Discovery of a Novel Series of Pyridine and Pyrimidine Carboxamides as Potent and Selective Covalent Inhibitors of Btk, Bioorg. Med. Chem. Lett., 2018, 28(21), 3419–3424, DOI:10.1016/j.bmcl.2018.09.033
Footnote |
| † Acyclic aliphatic scope not (fully) shown. |
| This journal is © The Royal Society of Chemistry 2021 |