
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultifunctional peptide-assembled micelles for simultaneously reducing amyloid-β and reactive oxygen species†
Li
Lei
ac,
Zhifeng
Zou
a,
Jin
Liu
a,
Zhiai
Xu
 *a,
Ying
Fu
d,
Yang
Tian
a and
Wen
Zhang
*b
*a,
Ying
Fu
d,
Yang
Tian
a and
Wen
Zhang
*b
aSchool of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200241, China. E-mail: zaxu@chem.ecnu.edu.cn
bShanghai Engineering Research Center of Molecular Therapeutics and New Drug Development, East China Normal University, Shanghai 200062, China. E-mail: wzhang@chem.ecnu.edu.cn
cDepartment of Chemistry, School of Basic Medical Science, Guizhou Medical University, Guiyang 550025, China
dDepartment of Chemical Engineering and Biotechnology, University of Cambridge, Philippa Fawcett Drive, Cambridge, CB3 0AS, UK
First published on 13th April 2021
Abstract
The excessive production and deposition of amyloid-β (Aβ) is one of the most important etiologies of Alzheimer's disease (AD). The interaction between Aβ and metal ions produces aberrant reactive oxygen species (ROS), which induce oxidative stress and accelerate the progression of AD. To reduce Aβ plaques and ROS to maintain their homeostasis is an emerging and ingenious strategy for effective treatment of AD. Herein, we report the rational design of multifunctional micelles (MPGLT) based on a polymer-grafted peptide to simultaneously clear Aβ and ROS for AD therapy. The MPGLT integrating three functional peptides as a ROS scavenger (tk-GSH), β-sheet breaker (LP) and an autophagy activator (TK) respectively, could capture and degrade Aβ. Meanwhile, the tk-GSH on the surface of MPGLT effectively scavenges the intracellular ROS. Consequently, MPGLT reduced the cytotoxicity of Aβ and ROS. In vivo animal studies using an AD mouse model further showed that MPGLT could transport across the blood–brain barrier for decreasing the Aβ plaque and eliminating ROS in vivo. This peptide micelle-based synergistic strategy may provide novel insight for AD therapy.
Introduction
Alzheimer's disease (AD) is the most pervasive neurodegenerative disease, which is difficult to prevent, cure or even slow down, and affects human life and health worldwide.1–4 The clinical treatment of AD has been restricted by insufficient understanding of the mechanisms underlying AD onset and progression.5,6 Extracellular senile amyloid-β (Aβ) plaques, oxidative stress related to reactive oxygen species (ROS) and intracellular neurofibrillary tangles containing hyperphosphorylated tau are considered as the characteristic pathological hallmarks of AD.7,8 Over the past few decades, the reported therapies targeting single biomarkers have been proved ineffective in clinical trials due to the synergistic relationship of pathogenetic molecules.9 The crosstalk between Aβ and various molecular signaling pathways, especially the interaction of Aβ with excessive metal ions, induces mitochondrial dysfunction that might be a possible cause of AD.10,11 In addition, oxidative stress may promote Aβ aggregation due to ROS-induced upregulation of the amyloid-β protein precursor (APP).12 Therefore, the abnormal Aβ and ROS accumulation is closely linked to the pathogenesis and progression of AD, and simultaneously reducing Aβ and ROS could be of great value for AD therapy.The recent efforts for AD treatment have been mainly devoted to inhibiting the self-assembly of Aβ into fibrils and reducing the deposition of Aβ, so as to alleviate Aβ-induced neurotoxicity. Antibodies, peptides, metal nanoparticles and various small molecules were used for preventing or reducing Aβ fibrillation.13–17 However, some passive inhibition methods could only control the ratio of the Aβ monomer to aggregates without changing total Aβ accumulation. Autophagy is an intracellular pathway for clearing the metabolites by lysosome-mediated protein degradation, which maintains the homeostasis of the intracellular environment.18 For example, strong evidence has demonstrated the potential of peptide-based nanomaterials for capturing and cleaning Aβ based on autophagy.15 Therefore, a rational design for Aβ clearance based on the autophagy process is highly promising for active treatment of AD.
Along with Aβ deposition, excessive ROS generation in the brain is another challenge for AD therapy. Antioxidant enzymes such as superoxide dismutase, peroxidase, catalase and so on, could eliminate ROS and reduce their cytotoxicity.19 However, the natural enzymes are unstable. The antioxidant enzymes are located in different subcellular organelles and regulated by different signalling pathways. It is difficult to control the reduction activity of several enzymes at the same time.20,21 In contrast, nanomaterial-based artificial enzymes such as nanoceria, mesoporous silica and carbon clusters have the ability to scavenge ROS and have been used for ROS clearance and maintaining the redox balance in the cell and preventing it from oxidative damage in vivo.22–24 In comparison with natural enzymes, the nanomaterial-based artificial enzymes display better stability and are much more cost-efficient. Among them, the organic nanomaterials composed of biocompatible components such as peptides are promising for AD therapy.
Herein, we rationally designed self-assembled peptide micelles (MPGLT) targeting both Aβ and ROS for highly effective AD treatment. MPGLT was constructed by integrating three functional units for degrading extracellular Aβ, activating cellular autophagy and scavenging intracellular ROS, respectively. As shown in Scheme 1, the MPGLT is composed of a polysialic acid (PSA) core decorated with tk-GSH, LPFFD (LD) and TGFQGSHWIHFTANFVNTK (TK). PSA is a natural and biodegradable carbohydrate polymer bearing N-acetylneuraminic acid units for chemical modification. More importantly, it is crucial for central nervous system development by promoting precursor cell migration, axon path finding, and synaptic remodeling.25 PSA could also interact with Aβ via hydrogen bonds and electrostatic interaction.26 Glutathione (GSH), an important antioxidant oligopeptide in the cell, was used to create ROS scavenger tk-GSH via a thioketal (tk) bond. LD is a well-known inhibitor for Aβ aggregation and a fibril disruptor. TK was derived from autophagy protein Beclin 1, which could activate autophagy for degrading Aβ. Thus, the resultant MPGLT could not only capture and dissociate extracellular Aβ aggregates, but also inhibit the formation of toxic Aβ fibrils. After that, MPGLT gains access into cells and upregulates cell autophagy. Meanwhile, the tk-GSH on the surface of MPGLT can consume intracellular ROS with the tk bond and the GSH moiety. Thus, MPGLT can simultaneously reduce Aβ fibrils and eliminate intracellular ROS for AD prevention and treatment. The in vitro experiments confirmed that MPGLT efficiently inhibited Aβ aggregation and reduced ROS. Furthermore, MPGLT encapsulating a near-infrared fluorescence probe IR780 could transport across the blood–brain barrier (BBB) and synchronously reduce Aβ and ROS in the brain of AD transgenic mice. Our study suggests the promising potential of peptide micelles for AD treatment.
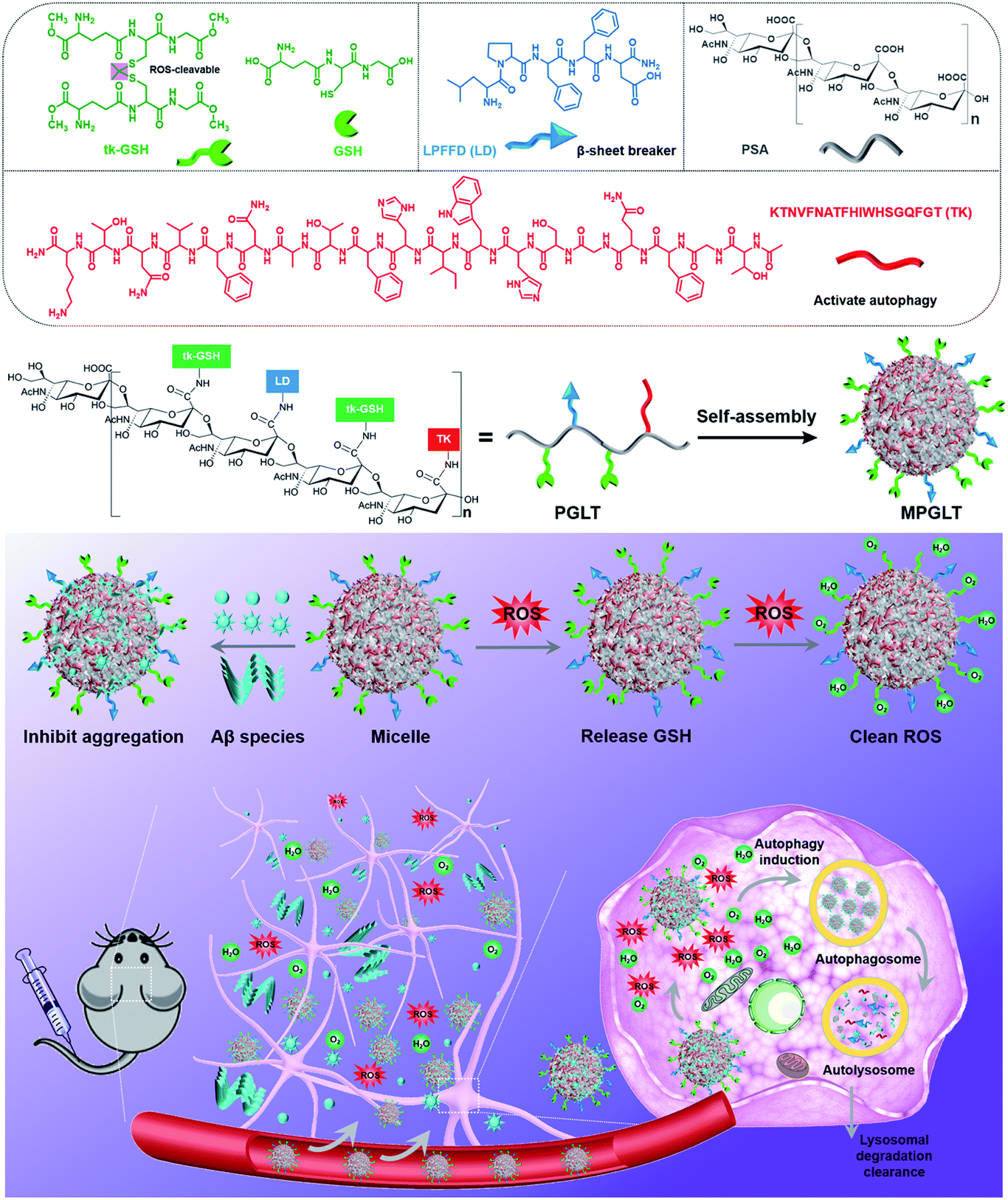 | ||
| Scheme 1 (Top panel) Chemical structures of the functional units and schematic illustration of the peptide-assembled MPGLT micelles. (Bottom panel) Mechanistic illustration of the peptide micelle-based AD treatment. The peptide micelles specifically bind the extracellular amyloid-β (Aβ) fibrils and transport them into cells for autophagy-mediated degradation. Meanwhile, the peptide micelles consume the reactive oxygen species (ROS) with the tk-GSH unit, thus simultaneously reduce Aβ and ROS. | ||
Results and discussion
Design and synthesis of multifunctional peptide micelles
Carbohydrate polymer PSA is an outstanding scaffold material due to its neuroprotective activity, biocompatibility and the α-2,8-linked N-acetyl neuraminic acid units for further modification.27 In this study, an LD or TK peptide was coupled with the carboxylic group of PSA to synthesize the polymer–peptide conjugates of PSA-LD (PLT1) or PSA-TK (PLT5), respectively. The chemical structures of LD, TK, PSA, PLT1, and PLT5 conjugates were characterized by proton nuclear magnetic resonance (1H NMR) measurements. The characteristic peaks assigned to LD, TK and PSA appeared at 7.5–7.0, 8.6–6.7 and 4.2–3.4 ppm in the 1H NMR spectra of PLT1 and PLT5, respectively,28–30 indicating the successful synthesis of PSA–peptide conjugates (Fig. 1A and S1†). Then, a set of peptide–polymer conjugates were synthesized by adjusting the feed molar ratios between LD and TK (i.e., 0.75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.25, 0.5:0.5, and 0.25:0.75). The resultant peptide–polymer conjugates were termed PLT2, PLT3, and PLT4 respectively, and their chemical structures were validated with 1H NMR spectra (Fig. S2 and S3†).
:0.25, 0.5:0.5, and 0.25:0.75). The resultant peptide–polymer conjugates were termed PLT2, PLT3, and PLT4 respectively, and their chemical structures were validated with 1H NMR spectra (Fig. S2 and S3†).
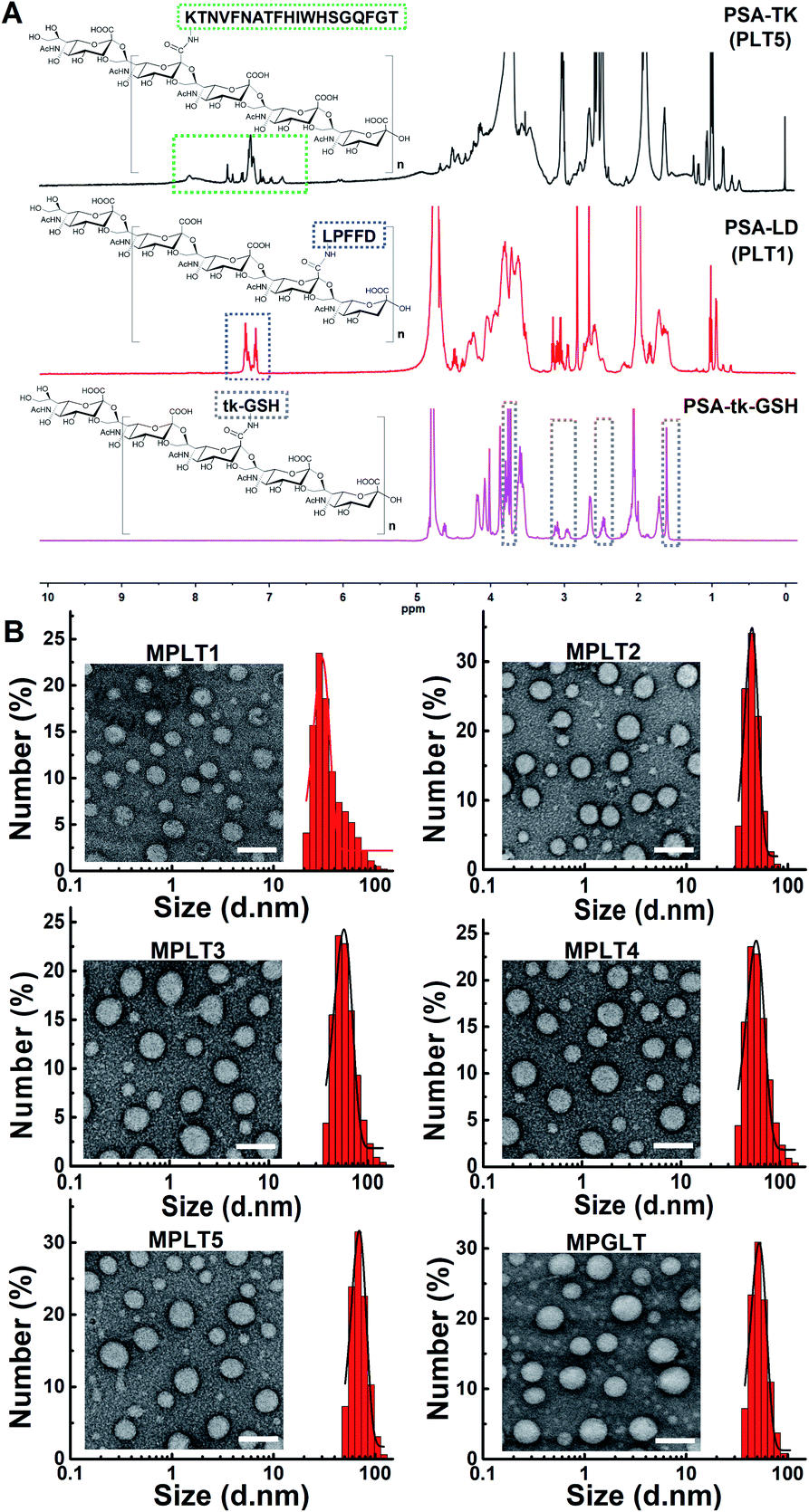 | ||
| Fig. 1 (A) The structure and 1H NMR spectra of PSA-TK (PLT5), PSA-LD (PLT1), and PSA-tk-GSH. (B) TEM images and hydrodynamic size distribution of MPLT1, MPLT2, MPLT3, MPLT4, MPLT5, and MPGLT, scale bar = 100 nm. | ||
A ROS-sensitive tk-GSH peptide with the thioketal group was also synthesized (Fig. S4†), and it was confirmed by 1H NMR spectrum and high-resolution mass spectra (HR-MS) measurements (Fig. S5 and S6†). Then, tk-GSH was covalently conjugated with the carboxyl groups on PSA to obtain PSA-tk-GSH. The 1H NMR spectrum of PSA-tk-GSH displayed the characteristic peaks of tk-GSH at 3.7 ppm, 3.3–2.8 ppm, 2.6–2.4 ppm, and 1.6 ppm, and the characteristic peaks of PSA at 4.4–4.2 ppm, 2.7–2.6 ppm, and 2.0 ppm from PSA,30 verifying the successful synthesis of PSA-tk-GSH (Fig. 1A). The LD and TK peptides with a feed mole ratio of 1:1 were subsequently grafted onto the PSA-tk-GSH to synthesize peptide–polymer PGLT (Fig. S7†). The chemical structure of PGLT was also verified using 1H NMR spectra with appearance of the characteristic peaks of LD, TK and tk-GSH at 8.5–6.5 ppm, and 1.6 ppm, respectively (Fig. S8†).
With the peptide–PSA conjugates in hand, we next investigated their self-assembly profile by transmission electron microscopy (TEM) and dynamic light scattering (DLS) measurements. The micellar nanoparticles prepared from PLT1–5 and PGLT (namely MPLT1–5 and MPGLT, respectively) all displayed spherical morphology with a uniform particle size distribution (Fig. 1B). DLS examination further revealed that the PLT1–5 and PGLT micelles were of particle size around 30 nm for MPLT1, 40 nm for MPLT2, 50 nm for MPLT3, 60 nm for MPLT4, and 70 nm for MPLT5, respectively. The size of multifunctional peptide-assembled micelles increased with the increase of the TK peptide ratio of the peptide–polymer, and the size of MPGLT was similar to that of MPLT3.
Inhibition of Aβ1–42 aggregation by multifunctional peptide micelles
Aβ aggregates can induce various lesions in the brain, and Aβ deposition has been defined as one of the central hypotheses of AD pathogenesis.31 To investigate whether the peptide micelles can inhibit the formation of Aβ1–42 aggregation, the Aβ1–42 monomer was incubated with the peptide micelles under the predetermined conditions. The TEM images demonstrated that Aβ1–42 formed fibril structures upon 48 h incubation in PBS at 37 °C (Fig. S9A†). The thioflavin T (ThT) colorimetric assay displayed strong green fluorescence emission from ThT (Fig. S9B†), verifying that Aβ1–42 formed fibrous aggregates. In contrast, negligible Aβ fibers were observed upon incubation with the peptide micelles containing 100%–50% LD peptide (e.g., MPLT1, MPLT2, and MPLT3) (Fig. 2A). Meanwhile, the morphologies of the MPLT1–3 micelles were altered upon incubation, which may be attributed to the adsorption and co-assembly of the Aβ1–42 monomer into the micelles. However, the Aβ1–42 solution incubated with MPLT4 (with 25% LD peptide) or MPLT5 (without LD peptide) displayed micellar and fibril structures. These results showed that the micelles containing high proportion of the LD peptide had a greater inhibitory effect on the fibrillation of Aβ1–42. MPGLT showed similar influence on Aβ1–42 aggregation to that of MPLT3.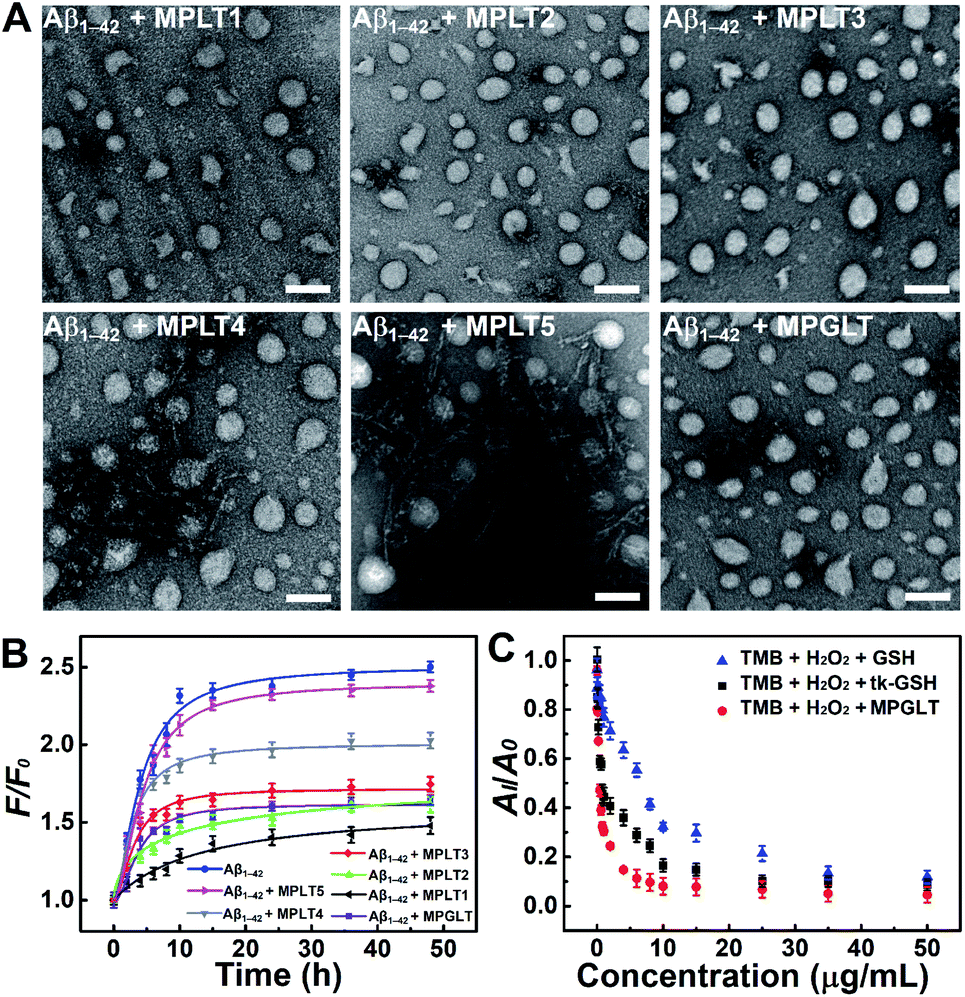 | ||
| Fig. 2 (A) Representative TEM images of Aβ1–42 upon incubation with or without MPLT1, MPLT2, MPLT3, MPLT4, MPLT5, and MPGLT micelles for 24 h, scale bar = 100 nm. (B) ThT assay of Aβ1–42 upon incubation with MPLT1–5 and MPGLT micelles for different time durations, F and F0 represent the fluorescence intensity of ThT with or without Aβ1–42, Aβ1–42 + MPLT1–5, and Aβ1–42 + MPGLT, respectively. (C) The 652 nm light absorption of TMB + H2O2 + GSH, TMB + H2O2 + tk-GSH, and TMB + H2O2 + MPGLT in the presence of hemin. AI and A0 are the 652 nm absorption of TMB + H2O2 with or without GSH, tk-GSH, and MPGLT. Data are presented as mean ± s.d. (n = 3). | ||
Furthermore, a train of curves were obtained by fitting the fluorescence of ThT after incubating with Aβ1–42 in the absence or presence of micelles (Fig. 2B). The fluorescence intensity increased gradually with the extension of the Aβ1–42 incubation time, and the sigmoidal curve of the Aβ1–42 group suggested a typical fibrillation formation process. Importantly, the fluorescence intensity of the Aβ1–42 + MPLT3 group was much lower than that of the Aβ1–42 and Aβ1–42 + MPLT5 groups. ThT fluorescence intensity from MPLT5 to MPLT1 decreased with the increase of the LD peptide content, which validated that LD suppressed Aβ aggregation by recognizing and binding Aβ1–42. The TEM images and ThT assay showed that Aβ1–42 incubated with MPGLT (Fig. 2A and B) was similar and superior to the Aβ1–42/MPLT3 group, further validating that the LD peptide could inhibit Aβ1–42 aggregation, and even degrade Aβ fibrils.
ROS scavenging activity of MPGLT
To evaluate the ROS scavenging properties of the peptide micelles, MPGLT was incubated with H2O2 and the reaction was monitored by 3,3′,5,5′-tetramethyl benzidine (TMB) chromogenic assay. The absorbance of TMB at 652 nm under the catalysis of hemin is a classic method for detecting H2O2.32 An absorption peak appeared at 600–700 nm when TMB was incubated with 1.2 M H2O2. In contrast, the absorption of the peak decreased with the addition of 20 μg mL−1 of tk-GSH (Fig. S10A†). Upon addition of GSH, tk-GSH and MPGLT at an identical concentration gradient, the absorption of TMB at 652 nm declined gradually following the decrease rates of MPGLT > tk-GSH > GSH, indicating that MPGLT displayed the strongest ROS clearance ability (Fig. 2C and S10†). The satisfactory ROS scavenging properties of MPGLT micelles could be explained by the chemical structure of tk-GSH and the topological architecture of the micelle nanoparticles. The tk-GSH groups on the surface of the micellar nanoparticles are more accessible for H2O2 than free tk-GSH, and tk-GSH released two molecule of GSH upon cleavage of the thioketal bond.Biocompatibility and cellular uptake of the peptide-assembled micelles
To study the biological effects of the peptide micelles, we first exploited their biocompatibility by cytotoxicity assay. SHSY-5Y neuroblastoma cells were incubated with MPLT1–5 and MPGLT at a concentration gradient for 24 h, and the cell survival rate was detected by CCK-8 assay.33Fig. 3A shows that over 85.0% of the cells survived at a micelle concentration up to 80 μg mL−1, confirming good biocompatibility of the micelles.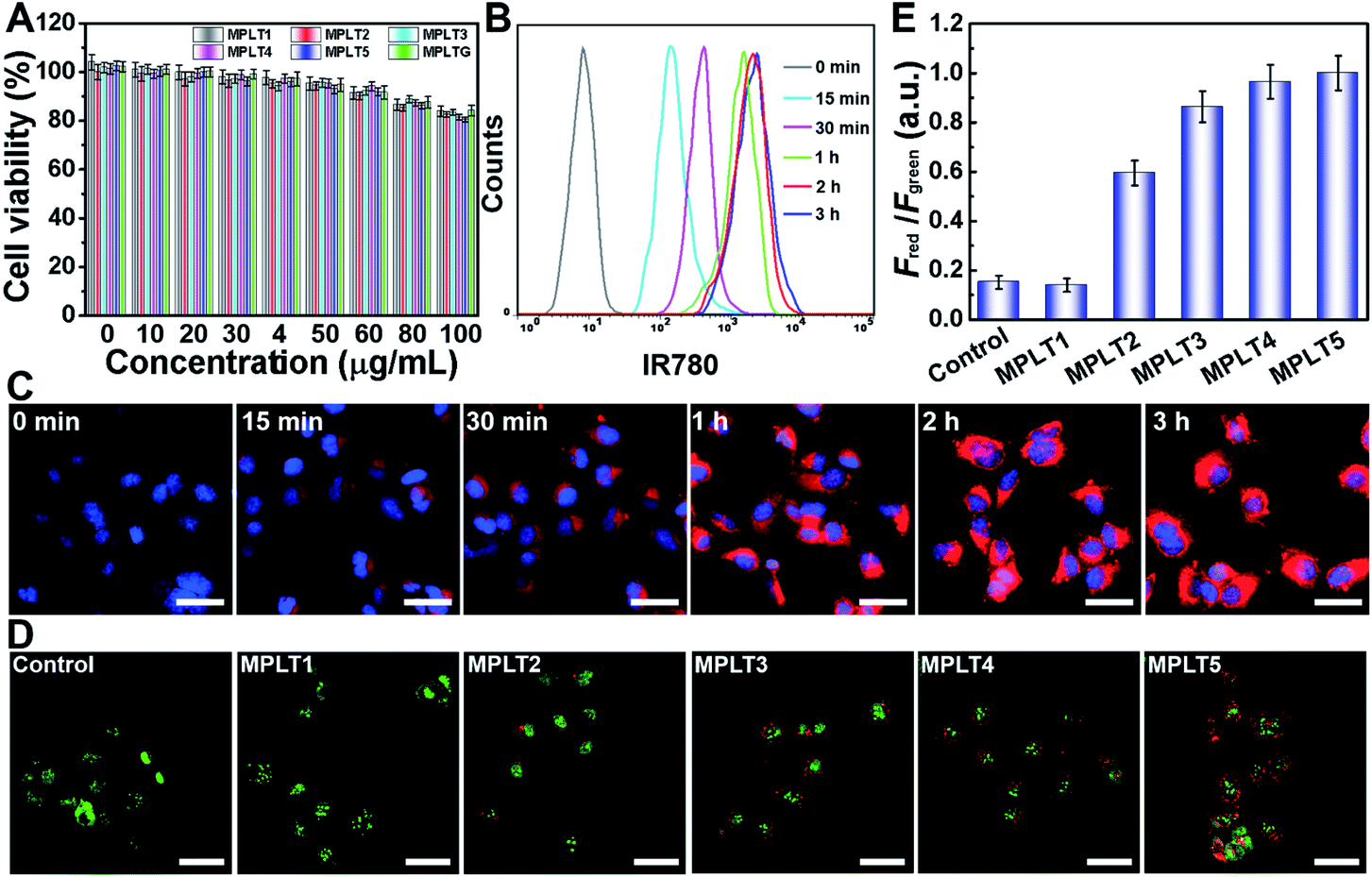 | ||
| Fig. 3 (A) Cell viability estimated by CCK-8 assay; SHSY-5Y cells were incubated with different concentrations of MPLT1–5 and MPGLT for 24 h. Data are presented as mean ± s.d. (n = 3). (B) Flow cytometry and (C) fluorescence images of MPGLT-IR780 internalized into SHSY-5Y cells with different incubation times, scale bar = 50 μm. (D) Acridine orange (AO) staining of SHSY-5Y cells after incubating with MPLT1–5, scale bar = 50 μm. (E) Quantified results of Fred to Fgreen in graph (D): Fred and Fgreen stand for the red (AO marks acidic vesicular organelles)36 and green (AO marks cytoplasm and DNA)36 fluorescence intensity. Data are presented as mean ± s.d. (n = 3). | ||
In order to visualize the cellular uptake and distribution of MPGLT, the MPGLT micelles were loaded with 0.59 (wt)% of near infrared dye IR780 to obtain MPGLT-IR780 according to the absorption spectra (Fig. S11†). The SHSY-5Y cells were incubated with 20 μg mL−1 of the MPGLT-IR780 micelle for predetermined time durations. The cells were then examined by flow cytometry (FCM) and confocal laser scanning microscopy (CLSM) measurements. Fig. 3B and S12† show that the micelle nanoparticles were highly and efficiently internalized into the cells upon 3 h incubation. CLSM examination further displayed increased intracellular accumulation of the nanoparticles in vitro (Fig. 3C).
Up-regulation of cellular autophagy by peptide micelles
To investigate the autophagy regulation performance of the peptide micelles in SHSY-5Y cells, acridine orange (AO) staining and cell imaging were performed. AO is a dye that shows red fluorescence in acidic organelles (e.g., lysosome and autophagosome) while it shows green fluorescence in cytoplasm and nuclei, and is widely used as an autophagy indicator by examining the intensity ratio of red to green fluorescence.34–36 The SHSY-5Y cells were incubated with MPLT1–5 for 4 h, and then stained with the AO dye for 10 min, followed by CLSM observation. As shown in Fig. 3D, abundant red spots emerged in the MPLT5-treated cells, and red fluorescence in MPLT2–5 increased with the increasing proportion of the TK peptide from 25% to 100%. In contrast, negligible red spots appeared in PBS or MPLT1 (without the TK peptide)-treated control group cells. Furthermore, quantification of the red to green fluorescence intensity ratio revealed that MPLT2–5 treatments remarkably increased the fraction of the acidic autophagosome than those of the PBS treated and MPLT1 treated cells, verifying that the peptide micelles activated the autophagy in the SHSY-5Y cells in vitro (Fig. 3E).Anti-Aβ and reducing ROS toxicity in vitro by peptide micelles
Aβ-induced neuronal cell apoptosis is a critical event in AD pathology.37 To determine the anti-Aβ activity of MPLT1–5 and MPGLT, we tested the viability of SHSY-5Y cells upon incubation with 20 μM Aβ1–42 with or without the addition of 20 μg mL−1 of MPLT1–5 and MPGLT. Fig. 4A shows that Aβ1–42 alone reduced the cell viability to 54.8%, confirming the remarkable toxicity of the Aβ monomer and aggregates, which was consistent with previous reports.38,39 However, the toxicity of Aβ1–42 was rapidly reduced in the presence of micelles. The cells treated with MPLT2–4 and MPGLT containing both LD and TK peptides showed obviously higher cell viabilities than the cells of the MPLT1 and MPLT5 groups. It is worth noting that MPLT3 and MPGLT showed higher cell viability of 95.8% and 97.6% respectively, in comparison with that of the control group (100%). The cell viability assay showed that the integration of TK and LD peptides produced an accumulative protective effect.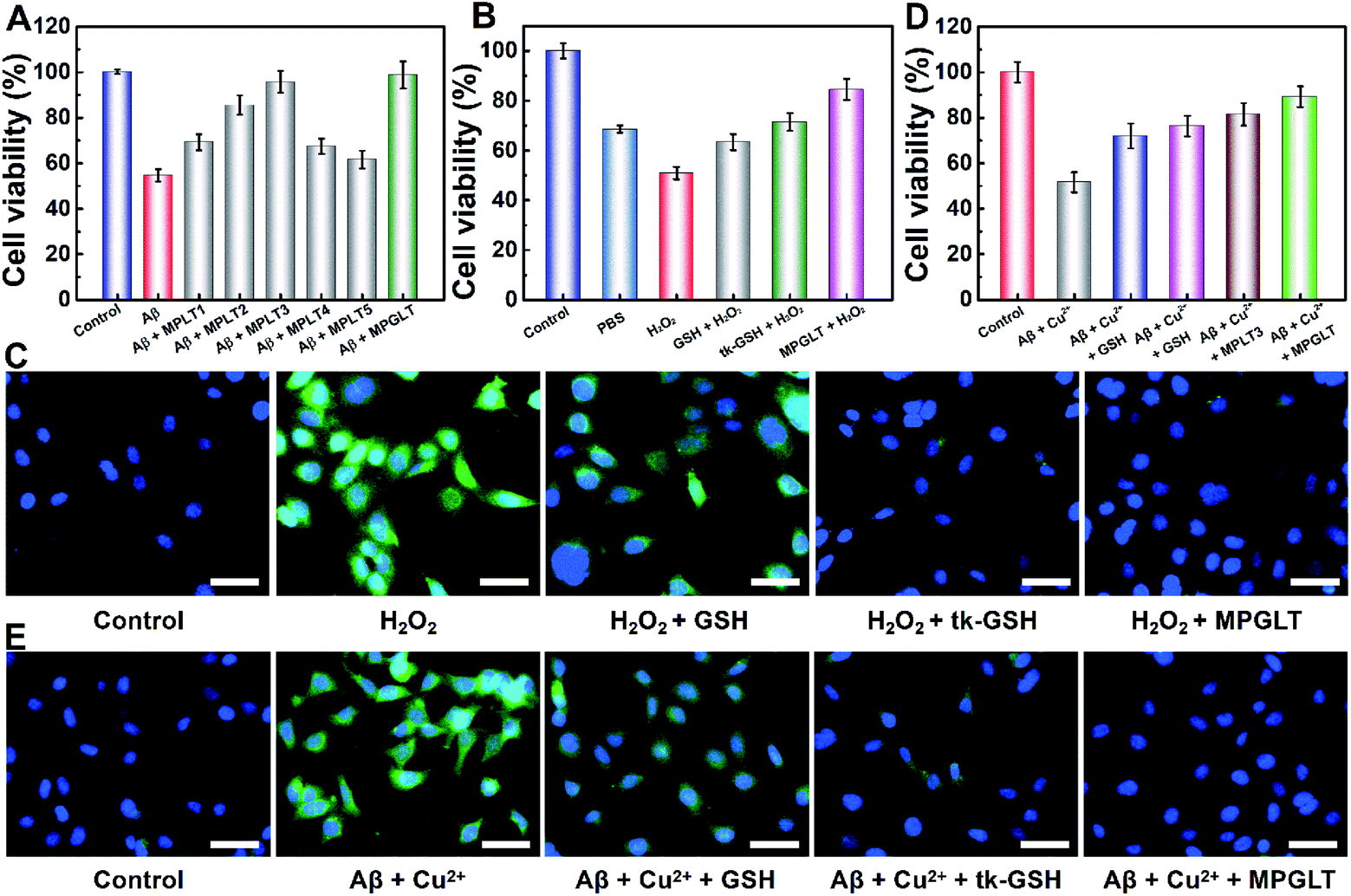 | ||
| Fig. 4 (A) The cytotoxicity of 20 μM Aβ1–42 to SHSY-5Y cells with or without 20 μg mL−1 of MPLT1–5 and MPGLT. (B) The cytotoxicity of 100 μM H2O2 to SHSY-5Y cells with or without GSH, tk-GSH, MPLT3, and MPGLT. (C) CLSM images of ROS generation in SHSY-5Y cells upon incubation with GSH, tk-GSH, or MPGLT. (D) The cytotoxicity of Aβ1–42 (20 μM) + Cu2+ (20 μM) to SHSY-5Y cells in the absence or presence of GSH, tk-GSH, MPLT3, and MPGLT. All cell viabilities were determined by CCK-8 assay. (E) CLSM images of the effect of GSH, tk-GSH and MPGLT on Aβ1–42 (20 μM) + Cu2+ (20 μM) induced production of ROS (scale bar = 50 μm). Data are presented as mean ± s.d. (n = 3). | ||
We next sought to investigate the antioxidant capacity of MPGLT by cytotoxicity assay.40 The intracellular ROS was generated by incubating the SHSY-5Y cells with H2O2. CCK-8 assay demonstrated that changing the H2O2 concentrations could cause different cytotoxicities in vitro (Fig. S13†). The cell survival rate dropped to about 40% when the H2O2 incubation time was extended to 4 h (Fig. S13D†). The cells treated with 100 μM H2O2 for 4 h displayed about 50% of survival rate and was used as the control in the following studies. Fig. 4B shows that GSH, tk-GSH and MPGLT all distinctly affected the cell viability. MPGLT efficiently reduced the toxicity of ROS to cells, and promoted cell viability to more than 85.1%. This indicated that the MPGLT can rescue the cell from injury due to oxidative stress.
2,7-Dichlorofluorescein diacetate (DCFH-DA) can be oxidized by ROS into green fluorescent DCF, which was used to evaluate intracellular levels of ROS by fluorescence imaging.41 CLSM examination displayed a dazzling green fluorescence from DCF in the cells treated with exogenous H2O2, which illustrated high intracellular levels of ROS (Fig. 4C). MPGLT incubation for 3 h resulted in decreased green fluorescence compared with that of GSH and tk-GSH. Quantitative analysis indicated that approximately 83.4% of ROS could be swept by MPGLT (Fig. S14A†). This is significantly higher than the value of 26.5% for the GSH group. The average fluorescence intensity (Fig. S14A†) and FCM performance (Fig. S14B and C†) further demonstrated the highest ROS removal rate of MPGLT. The above data further demonstrated increased protective effects of MPGLT against H2O2-mediated cytotoxicity in SH-SY5Y cells in vitro.
Inhibition of Aβ- and Cu2+-mediated cytotoxicity by peptide micelles
Copper ions were found to play key roles in AD pathogenesis, which not only induce Aβ aggregation, but also create ROS through the formation of an Aβ/Cu complex.42,43 We thus investigated the effects of MPGLT on relieving the cytotoxicity of Aβ aggregates in the presence of Cu2+ in SH-SY5Y cells. A Cu2+ concentration of 20 μM without an apparent toxic effect was selected for the cytotoxicity assay (Fig. S15†).44,45 The subsequent assays were performed under the same conditions, and the data were normalized using the results from the controls (cells without the treatment of Aβ + Cu2+). Fig. 4D shows that the cell viability decreased to 51.6% by incubating with Aβ + Cu2+, and the addition of GSH and tk-GSH slightly reduced the Aβ-induced cytotoxicity. Upon incubation with the MPLT3 micelles, the cell viability increased up to 81.4%. The cell viability was further restored to ∼89.2% in the presence of MPGLT, indicating that MPGLT significantly protected the SH-SY5Y cells from Aβ + Cu2+ induced cytotoxicity.The inhibition of Aβ and Cu2+-mediated cytotoxicity by the peptide micelles could be explained by the elimination of intracellular ROS generation by Aβ + Cu2+ as imaged by CLSM and quantified by FCM examination (Fig. S16†), respectively. Aβ + Cu2+ treatment remarkably increased the intracellular ROS compared to the control group (Fig. 4E). GSH treatment slightly reduced the ROS level, and tk-GSH showed stronger ROS scavenging ability than GSH since the thioketal bond in tk-GSH and 2 times of GSH released by tk-GSH can consume ROS simultaneously. In contrast, MPGLT further reduced the intracellular ROS level.
Reducing Aβ and ROS in vivo by peptide micelles
Given the satisfactory ability of the peptide micelles to neutralize the intracellular ROS and reduce the cytotoxicity of the Aβ fibrils, we next investigated their potential for reducing Aβ and ROS in the brain of AD mice in vivo. Cyclosporin A (CSA), which interacts with P-glycoprotein (P-gp) in the blood–brain barrier (BBB), has been applied to promote the permeability of BBB.46,47 Here, 10 μM CSA was injected into the Balb/c mice intravenously (i.v.), subsequently, a dose of 220 μg mL−1 MPGLT-IR780 was further injected into the mice by i.v. administration. Fluorescence imaging in vivo showed that MPGLT-IR780 had a higher accumulation in the brain compared to the PBS or MPGLT-IR780 only group (Fig. 5A). Ex vivo fluorescence imaging of the major organs further revealed higher brain accumulation of CSA/MPGLT-IR780 compared to MPGLT-IR780, verifying that CSA could facilitate active brain targeting of MPGLT-IR780 (Fig. 5B). Moreover, the relative fluorescence intensity in Fig. 5C and the red fluorescence in Fig. S17† from the CSA/MPGLT-IR780 group show that the MPGLT could cross the BBB with the assistance of CSA.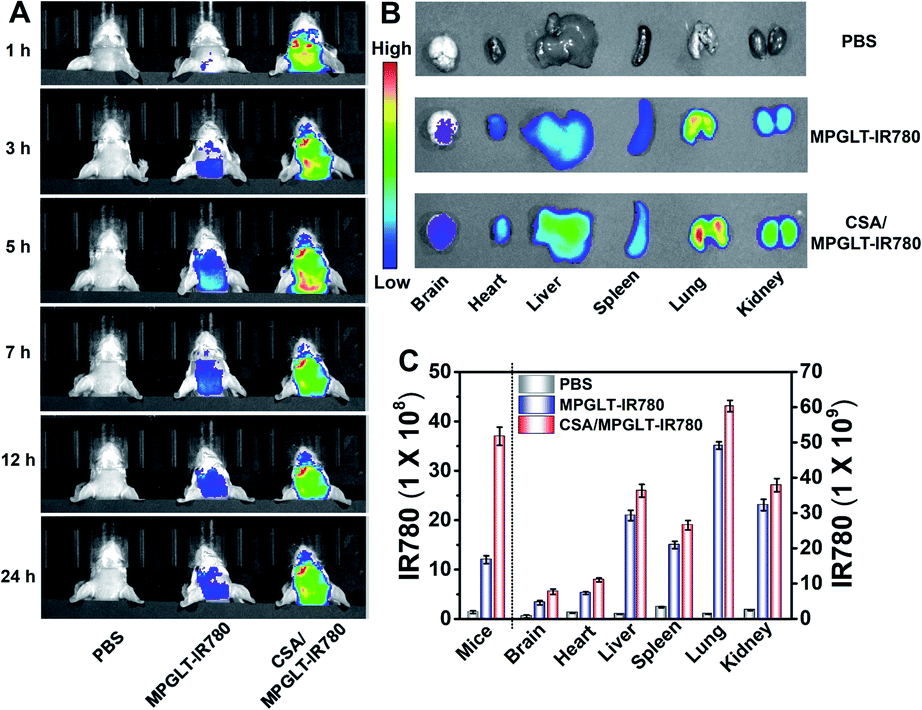 | ||
| Fig. 5 (A) Time course fluorescence imaging in vivo of the mice treated with PBS or 220 μg mL−1 of MPGLT-IR780 with or without the use of cyclosporine A (10 μM). (B) Fluorescence imaging of the major organs and brain ex vivo at 24 h post i.v. injection of MPGLT-IR780 with or without 10 μM CSA pre-treatment. (C) Relative fluorescence intensity of the whole mice, brain and major organs in vivo and ex vivo. Data are presented as mean ± s.d., n = 3. | ||
To validate if the MPGLT could reduce ROS and amyloid plaques in the brains of APPswe/PS1dE9 transgenic (AD) mice, the AD mice were intravenously injected with MPGLT every other day for two weeks. The wild type (WT) and AD mice were then injected with ROS probe DCFH-DA. IVIS fluorescence imaging was employed to investigate the effect of ROS clearance of MPGLT in the brain; bright DCF fluorescence was observed in AD mice compared with the WT control, which indicated excess ROS in the brain of AD mice (Fig. 6A and B). Nevertheless, there was insignificant DCF fluorescence in the brains of AD mice treated with MPGLT, showing that MPGLT had the ability to clean ROS, which was in agreement with the quantitative analysis of fluorescence in the brain. The brain tissue slice imaging showed obvious difference of DCF fluorescence between the WT, AD and MPGLT treated groups, which further demonstrated that ROS could be scavenged by MPGLT (Fig. 6C and S18A†).
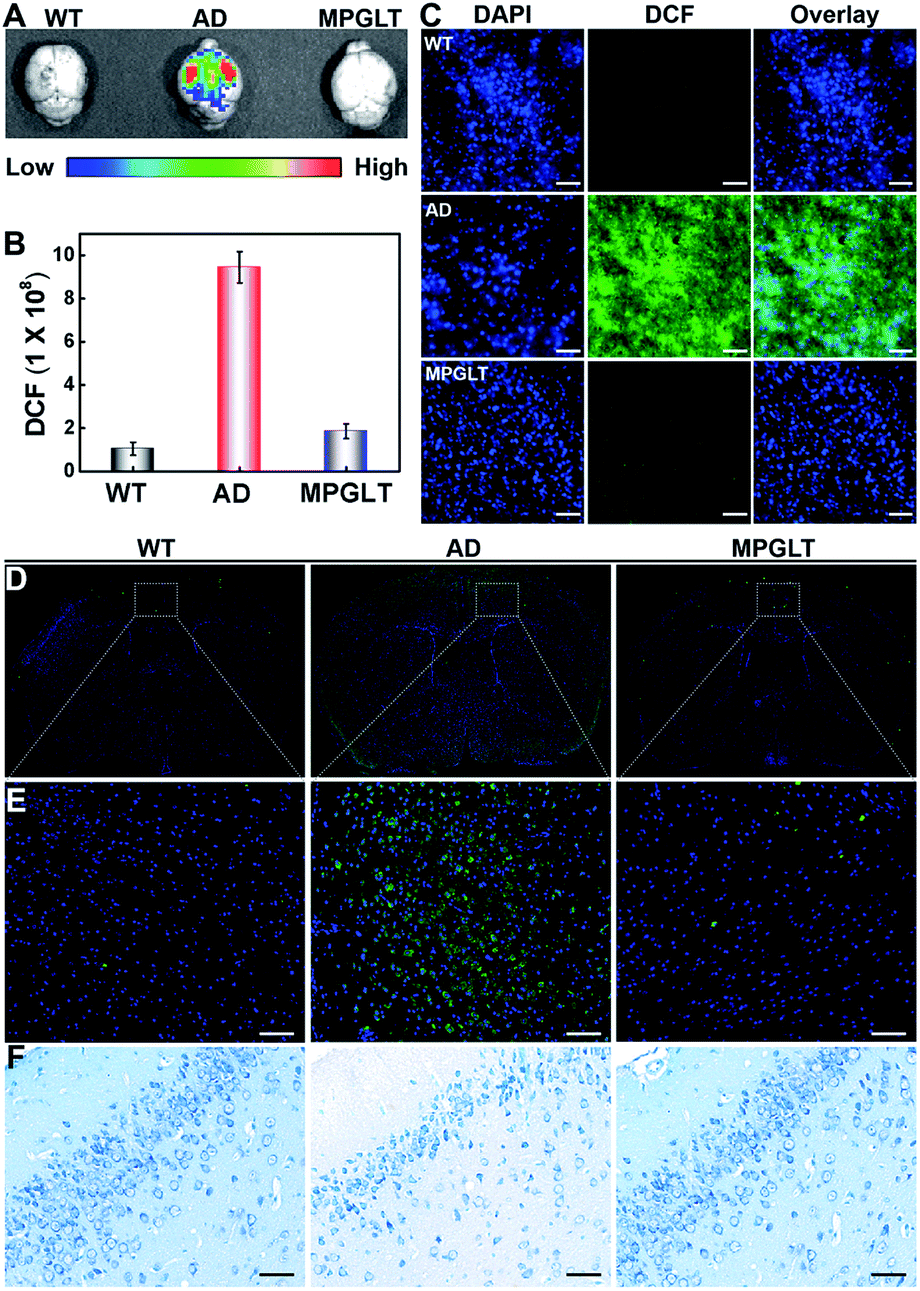 | ||
| Fig. 6 (A) Fluorescence images of ROS distribution in the brain of WT mice, AD mice, and MPGLT-treated AD mice, respectively. (B) Semi-quantitative evaluation of fluorescence intensity of the three groups in graph (A). Data are presented as mean ± s.d., n = 3. (C) Fluorescence analysis of ROS distribution in the brain sections of AD mice treated with PBS (AD group) or MPGLT (MPGLT group), and WT mice treated with PBS as a control, scale bar = 200 μm. (D) and (E) The immunofluorescence analysis of Aβ1–42 aggregation in the brains of WT mice, AD mice, and MPGLT-treated AD mice, scale bar = 100 μm. (F) The Nissl staining of nerve cells in the brains of WT mice, AD mice, and MPGLT-treated AD mice, scale bar = 100 μm. The Nissl bodies were stained blue. | ||
Immunofluorescence assay was further employed to detect Aβ deposition in the brain. Aβ antibody was used as the primary antibody and thioflavine S (ThS)-conjugated goat antirabbit IgG polyclonal antibody was used as the secondary antibody.48,49Fig. 6D, E and S18B† display negligible green fluorescence of ThS in the brain of WT mice. In contrast, 5-fold brighter fluorescence of ThS was observed in the brain of AD mice than the WT mice, indicating a large amount of Aβ deposition. Noticeably, the fluorescence intensity of the MPGLT-treated AD mice remarkably decreased by 74.1% compared to that of the AD mice, verifying that MPGLT treatment dramatically decreased the Aβ plaque in the brains of AD mice.
Nissl staining was further conducted to verify if the side effect of MPGLT can be observed in the brain. Nissl staining targets the Nissl body, which is regarded as a sign of the functional stage of neurons.50 The results of Nissl staining in Fig. 6F show that neuronal hypocellularity in the WT mice brains had large numbers of Nissl bodies (blue), indicating strong metabolic function of the neurons. However, Nissl bodies were reduced and even disappeared in the brains of AD mice, indicating that the neurons were damaged. When the AD mice were treated with MPGLT, Nissl bodies reappeared and increased, and reached the normal level, suggesting that MPGLT treatment obviously attenuated neuron loss in AD mice. Meanwhile, the systemic toxicity of MPGLT was evaluated by hematoxylin and eosin (H&E) assays (Fig. S19†). The good performance of MPGLT in AD mice could be attributed to the contribution of PSA for nerve repair and functional peptides for reducing Aβ and ROS. There was no histological damage observed in the major organs (heart, liver, spleen, lungs, and kidneys) of the MPGLT-treated mice, verifying good biosafety of MPGLT. All of the above results suggested that MPGLT could be potentially used as a neuroprotective nanomaterial with excellent therapeutic effects on AD.
Conclusion
In summary, we successfully constructed multifunctional peptide micelles (MPGLT) by integrating functional peptides of tk-GSH, LD and TK into a PSA-based nanoplatform. The micelles could prevent Aβ aggregation and degrade Aβ aggregates by activating the intracellular autophagy process. Meanwhile, tk-GSH on the MPGLT released GSH to deplete intracellular ROS. Moreover, in vitro and in vivo studies verified excellent performance of the peptide-assembled micelles for eliminating Aβ aggregates and ROS in AD mice without obvious side effects. Overall, this work might provide novel insight for AD therapy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
All animal studies were performed in accordance with the guidelines approved by the Institutional Animal Care and Use Committee of East China Normal University. Financial support from the National Natural Science Foundation of China (No. 21775046, 22074043 and 21635003), Science and Technology Commission of Shanghai Municipality (No. 20142202800) and the fundamental research funds from the central universities is greatfully appreciated.Notes and references
- K. Blennow, M. J. De Leon and H. Zetterberg, Lancet, 2006, 368, 387–403 CrossRef CAS.
- R. B. Maccioni, J. P. Munoz and L. Barbeito, Arch. Med. Res., 2001, 32, 367–381 CrossRef CAS PubMed.
- M. P. Mattson, Nature, 2004, 430, 631–639 CrossRef CAS.
- S. S. Hindo, A. M. Mancino, J. J. Braymer, Y. H. Liu, S. Vivekanandan, A. Ramamoorthy and M. H. Lim, J. Am. Chem. Soc., 2009, 131, 16663–16665 CrossRef CAS PubMed.
- C. Haass and D. J. Selkoe, Nat. Rev. Mol. Cell Biol., 2007, 8, 101–112 CrossRef CAS PubMed.
- J. L. Cummings, T. Morstorf and K. Zhong, Alzheimer's Res. Ther., 2014, 6, 37 CrossRef PubMed.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2006, 75, 333–366 CrossRef CAS.
- B. Dubois, H. H. Feldman, C. Jacova, H. Hampel, J. L. Molinuevo, K. Blennow, S. T. DeKosky, S. Gauthier, D. Selkoe, R. Bateman, S. Cappa, S. Crutch, S. Engelborghs, G. B. Frisoni, N. C. Fox, D. Galasko, M. O. Habert, G. A. Jicha, A. Nordberg and F. Pasquier, Lancet Neurol., 2014, 13, 614–629 CrossRef.
- S. Salloway, R. Sperling, N. C. Fox, K. Blennow, W. Klunk, M. Raskind, M. Sabbagh, L. S. Honig, A. P. Porsteinsson, S. Ferris, M. Reichert, N. Ketter, B. Nejadnik, V. Guenzler, M. Miloslavsky, D. Wang, Y. Lu, J. Lull, I. C. Tudor and E. Liu, N. Engl. J. Med., 2014, 370, 322–333 CrossRef CAS PubMed.
- S. Tu, S.-i. Okamoto, S. A. Lipton and H. Xu, Mol. Neurodegener., 2014, 9, 48–59 CrossRef.
- M. G. Bartley, K. Marquardt, D. Kirchhof, H. M. Wilkins, D. Patterson and D. A. Linseman, J. Alzheimer's Dis., 2012, 28, 855–868 CAS.
- Y. H. Edrey, S. Oddo, C. Cornelius, A. Caccamo, V. Calabrese and R. Buffenstein, J. Neurosci. Res., 2014, 92, 195–205 CrossRef CAS.
- J. McLaurin, R. Cecal, M. E. Kierstead, X. Tian, A. L. Phinney, M. Manea, J. E. French, M. H. L. Lambermon, A. A. Darabie, M. E. Brown, C. Janus, M. A. Chishti, P. Horne, D. Westaway, P. E. Fraser, H. T. J. Mount, M. Przybylski and P. St George-Hyslopet, Nat. Med., 2002, 8, 1263–1269 CrossRef CAS.
- B. Hu, F. Dai, Z. Fan, G. Ma, Q. Tang and X. Zhang, Adv. Mater., 2015, 27, 5499–5505 CrossRef CAS PubMed.
- Q. Luo, Y. X. Lin, P. P. Yang, Y. Qi, G. B. Wang, Z. Y. Qiao, B. N. Li, K. Zhang, J. P. Zhang, L. Wang and H. Wang, Nat. Commun., 2018, 9, 1802 CrossRef.
- Y. Lai, Y. Zhu, Z. Xu, X. Hu, M. Saeed, H. Yu, X. Chen, J. Liu and W. Zhang, Adv. Funct. Mater., 2020, 30, 1908473 CrossRef CAS.
- D. E. Ehrnhoefer, J. Bieschke, A. Boeddrich, M. Herbst, L. Masino, R. Lurz, S. Engemann, A. Pastore and E. E. Wanker, Nat. Struct. Mol. Biol., 2008, 15, 558–566 CrossRef CAS PubMed.
- N. Mizushima, Genes Dev., 2007, 21, 2861–2873 CrossRef CAS PubMed.
- J. Geng, M. Li, L. Wu, J. S. Ren and X. G. Qu, J. Med. Chem., 2012, 55, 9146–9155 CrossRef CAS PubMed.
- G. Grasso, M. L. Giuffrid and E. Rizzarelli, Metallomics, 2012, 4, 937–949 CrossRef CAS.
- J. Wu, X. Wang, Q. Wang, Z. Lou, S. Li, Y. Zhu, L. Qin and H. Wei, Chem. Soc. Rev., 2013, 42, 6060–6093 RSC.
- E. L. Samuel, D. C. Marcano, V. Berka, B. R. Bitner, G. Wu, A. Potter, R. H. Fabian, R. G. Pautler, T. A. Kent, A. L. Tsai and J. M. Tour, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 2343–2348 CrossRef CAS.
- M. Lian, Z. Xue, X. Qiao, C. Liu, S. Zhang, X. Li, C. Huang, Q. Song, W. Yang, X. Chen and T. Wang, Chem, 2019, 5, 2378–2387 CAS.
- Y. Li, X. He, J. J. Yin, Y. Ma, P. Zhang, J. Li, Y. Ding, J. Zhang, Y. Zhao and Z. Chai, Angew. Chem., Int. Ed., 2015, 54, 1832–1835 CrossRef CAS PubMed.
- X. J. Wang, C. H. Peng, S. Zhang, X. L. Xu, G. F. Shu, J. Qi, Y. F. Zhu, D. M. Xu, X. Q. Kang, K. J. Lu, F. Y. Jin, R. S. Yu, X. Y. Ying, J. You, Y. Z. Du and J. S. Ji, Nano Lett., 2019, 19, 829–838 CrossRef PubMed.
- A. Kakio, S. Nishimoto, K. Yanagisawa, Y. Kozutsumi and K. Matsuzaki, Biochemistry, 2002, 41, 7385–7390 CrossRef CAS PubMed.
- A. B. Rebecca, L. S. Angela and Z. Nan, Biomacromolecules, 2011, 12, 314–320 CrossRef.
- Y. Song, E. G. Moore, Y. Guo and J. S. Moore, J. Am. Chem. Soc., 2017, 139, 4298–4301 CrossRef CAS PubMed.
- Y. Wang, Y. X. Lin, Z. Y. Qiao, H. W. An, S. L. Qiao, L. Wang, R. P. Rajapaksha and H. Wang, Adv. Mater., 2015, 27, 2627–2634 CrossRef CAS PubMed.
- M. Liu, X. Luo, Q. Qiu, L. Kang, T. Li, J. Ding, Y. Xiong, Z. Zhao, J. Zan, C. Chang, X. Liu, Y. Song and Y. Deng, Mol. Pharmaceutics, 2018, 15, 5534–5545 CrossRef CAS PubMed.
- D. J. Selkoe, Science, 2019, 365, 540–541 CrossRef CAS PubMed.
- W. Zhang, S. Hu, J. J. Yin, W. He, W. Lu, M. Ma, N. Gu and Y. Zhang, J. Am. Chem. Soc., 2016, 138, 5860–5865 CrossRef CAS PubMed.
- G. Jiao, X. He, X. Li, J. Qiu, H. Xu, N. Zhang and S. Liu, RSC Adv., 2015, 5, 53240–53244 RSC.
- Y. Wang, Y. X. Lin, Z. Y. Qiao, H. W. An, S. L. Qiao, L. Wang, R. P. Rajapaksha and H. Wang, Adv. Mater., 2015, 27, 2627–2634 CrossRef CAS.
- D. J. Klionsky, Autophagy, 2012, 8, 445–544 CrossRef CAS PubMed.
- F. Zoccarato, L. Cavallini and A. Alexandre, J. Neurochem., 1999, 72, 625–633 CrossRef CAS PubMed.
- K. Hsiao, P. Chapman, S. Nilsen, C. Eckman, Y. Harigaya, S. Younkin, F. Yang and G. Cole, Science, 1996, 274, 99–103 CrossRef CAS PubMed.
- N. Gao, H. Sun, K. Dong, J. Ren, T. Duan, C. Xu and X. Qu, Nat. Commun., 2014, 5, 3422 CrossRef PubMed.
- F. Huang, J. Wang, A. Qu, L. Shen, J. Liu, J. Liu, Z. Zhang, Y. An and L. Shi, Angew. Chem., Int. Ed., 2014, 53, 8985–8990 CrossRef CAS PubMed.
- M. J. Akhtar, M. Ahamed, H. A. Alhadlaq and A. Alshamsan, Biochim. Biophys. Acta, Gen. Subj., 2017, 1861, 802–813 CrossRef CAS.
- J. Yao, Y. Cheng, M. Zhou, S. Zhao, S. Lin, X. Wang, J. Wu, S. Li and H. Wei, Chem. Sci., 2018, 9, 2927–2933 RSC.
- T. J. Huat, J. Camats-Perna, E. A. Newcombe, N. Valmas, M. Kitazawa and R. Medeiros, J. Mol. Biol., 2019, 431, 1843–1868 CrossRef CAS PubMed.
- P. Faller, C. Hureau and P. G. La, Acc. Chem. Res., 2014, 47, 2252–2259 CrossRef CAS PubMed.
- G. Caruso, D. A. Distefano, P. Parlascino, C. G. Fresta, G. Lazzarino, S. M. Lunte and V. G. Nicoletti, Mol. Cell. Biochem., 2017, 425, 85–93 CrossRef CAS PubMed.
- J. Zhang, D. Duan, J. Xu and J. Fang, ACS Appl. Mater. Interfaces, 2018, 10, 33010–33021 CrossRef CAS PubMed.
- P. Doze, A. Van Waarde, P. H. Elsinga, N. H. Hendrikse and W. Vaalburg, Synapse, 2000, 36, 66–74 CrossRef CAS.
- N. H. Hendrikse, A. H. Schinkel, E. G. E. De Vries, E. Fluks, W. T. A. Van der Graaf, A. T. M. Willemsen, W. Vaalburg and E. J. F. Franssen, Br. J. Pharmacol., 1998, 124, 1413–1418 CrossRef CAS PubMed.
- R. Guntern, C. Bouras, P. R. Hof and G. Vallet, Cell. Mol. Life Sci., 1992, 48, 8–10 CrossRef CAS PubMed.
- N. Inestrosa, C. Tapia-Rojas, T. Griffith, F. Carvajal, M. Benito, A. Rivera-Dictter, A. Alvarez, F. Serrano, J. Hancke, P. Burgos, J. Parodi and L. Varela-Nallar, Transl. Psychiatry, 2011, 1, e20 CrossRef CAS PubMed.
- A. Kádár, G. Wittmann, Z. Liposits and C. Fekete, J. Neurosci. Methods, 2009, 184, 115–118 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc00153a |
| This journal is © The Royal Society of Chemistry 2021 |