
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Self-assembled metallasupramolecular cages towards light harvesting systems for oxidative cyclization†
Atul
Kumar
,
Rupak
Saha
and
Partha Sarathi
Mukherjee
 *
*
Department of Inorganic and Physical Chemistry, Indian Institute of Science, Bangalore 560012, India. E-mail: psm@iisc.ac.in
First published on 1st March 2021
Abstract
Designing artificial light harvesting systems with the ability to utilize the output energy for fruitful application in aqueous medium is an intriguing topic for the development of clean and sustainable energy. We report here facile synthesis of three prismatic molecular cages as imminent supramolecular optoelectronic materials via two-component coordination-driven self-assembly of a new tetra-imidazole donor (L) in combination with 180°/120° di-platinum(II) acceptors. Self-assembly of 180° trans-Pt(II) acceptors A1 and A2 with L leads to the formation of cages Pt4L2(1a) and Pt8L2(2a) respectively, while 120°-Pt(II) acceptor A3 with L gives the Pt8L2(3a) metallacage. PF6− analogues (1b, 2b and 3b) of the metallacages possess a high molar extinction coefficient and large Stokes shift. 1b–3b are weakly emissive in dilute solution but showed aggregation induced emission (AIE) in a water/MeCN mixture as well as in the solid state. AIE active 2b and 3b in aqueous (90% water/MeCN mixture) medium act as donors for fabricating artificial light harvesting systems via Förster resonance energy transfer (FRET) with organic dye rhodamine-B (RhB) with high energy efficiency and good antenna effect. The metallacages 2b and 3b represent an interesting platform to fabricate new generation supramolecular aqueous light harvesting systems with high antenna effect. Finally, the harvested energy of the LHSs (2b + RhB) and (3b + RhB) was utilized successfully for efficient visible light induced photo-oxidative cross coupling cyclization of N,N-dimethylaniline (4) with a series of N-alkyl/aryl maleimides (5) in aqueous acetonitrile with dramatic enhancement in yields compared to the reactions with RhB or cages alone.
Introduction
In nature, the photosynthesis process of harvesting solar light and its transformation to chemical energy plays a crucial role in development of life. The process of harvesting light in photosynthesis critically relies on excitation energy migration from the light absorbing pigment ‘chlorophyll’ which is embedded in light harvesting complexes known as antenna proteins to the reaction center ‘carotenoid’ where light energy is utilized for the eventual conversion into chemical energy.1 Fluorescence resonance energy transfer (FRET) is a radiation less transition through the dipole–dipole interaction which emerges as one of the most efficient processes of electronic excitation energy transfer within and between molecules for the development of efficient artificial light harvesting systems (LHSs) that mimic the natural photosynthetic process.2 In recent years, much attention has been focused by synthetic and material chemists to construct artificial LHSs via the FRET process which include conjugated polymers, porphyrin arrays, porous materials, dendrimers, and host–guest assemblies.3 Among them supramolecular systems based on non-covalent interactions draw considerable attention in the synthesis of efficient FRET systems as they avoid a multistep synthetic process which is obligatory in the synthesis of scaffold covalent compounds.4 Although some successful approaches were adopted for an efficient FRET process in supramolecular architectures based on covalent organic frameworks (COFs)5 and metal organic frameworks (MOFs),6 such systems are easy to synthesize but their poor solubility and stability in common solvents restrict their processability for further application. Supramolecular coordination complexes (SCCs) formed by coordination driven self-assembly provide an alternative hope in this regard due to their facile one-pot synthesis, good stability, and high solubility in common solvents.7 Over the past decade supramolecular materials based on SCCs ranging from 2-D macrocycles to 3-D metallacages have been synthesized which found their widespread use in host–guest chemistry, sensing, catalysis, stabilizing reactive intermediates, etc.8–10 The FRET process often requires to be performed at very low concentration in order to reduce molecular aggregation. SCCs based on the metal–ligand interaction which is even effective at micromolar to nanomolar concentration are preferred over other host–guest or H-bonded supramolecular complexes in which interactions become ineffective upon dilution due to their low association constant in common organic solvents.11 For the synthesis of synthetic LHSs, multiple donor molecules are required for a single acceptor, but use of multiple donors in single molecular assembly can cause fluorescence quenching by aggregation. Therefore, development of aggregation induced emissive fluorophores as donors offers an alternative in constructing efficient LHSs.Tetraphenylethene (TPE) is weakly emissive in solution but highly emissive in the aggregate state12 due to the restriction of phenyl ring rotation.13,14 Recently Stang15 and other researchers16 have reported a series of emissive metallacycles and metallacages as AIE active supramolecular complexes by incorporation of TPE functional units within SSCs via coordination driven self-assembly of TPE based donor/(s) and metal-acceptor/(s), which demonstrated their profound use as light emitting and chemical/bio-sensor materials.17 AIE active supramolecular chromophores possess distinct features including bright emission with significant quantum yield and extraordinary stability in aqueous media, which make them ideal applicants as light emitting donors for energy transfer with water soluble organic dyes.2c,4,18 In this regard designing artificial LHSs possessing high energy efficiencies and antenna effect in green aqueous medium with the ability to photo-catalyse chemical transformation under mild conditions by output energy remains a challenging task.
Herein, we report facile synthesis of three prismatic cages via co-ordination driven self-assembly of a newly designed donor L with 180°/120° trans-Pt(II) acceptors. Trans-[Pt(PEt3)2(ONO2)2] (A1) with L leads to the formation of a cage (1a) having a molecular composition of A14L2 while 180° based trans-Pt(II) acceptor A2 and 120° acceptor A3 with L form Pt8 cages 2a (A24L2) and 3a (A34L2), respectively (Scheme 1). PF6− analogues (1b, 2b and 3b) of the metallacages are highly emissive in the aggregate state in aqueous acetonitrile medium due to the formation of spherical nano-aggregates of the cages having AIE active TPE units. AIE active cages 2b and 3b in the aggregate state represent a new platform for fabricating artificial light harvesting systems in the presence of a suitable acceptor dye RhB. As cages 2b/3b possess strong absorption in the UV region and emit strongly at lower wavelengths of the visible region, they serve as antenna sources to efficiently transfer the energy from the UV/Visible light to RhB by the FRET process and hence stimulate its photocatalytic activity. Furthermore, the LHSs ((2b + RhB) and (3b + RhB)) showed dramatic enhancement in visible light mediated photocatalytic activity for the cross coupling cyclization of N,N-dimethylaniline (4) with a series of N-alkyl/aryl maleimides (5) in aqueous acetonitrile medium (90% water/MeCN mixture) as compared to RhB/cages alone. This study provides an efficient and facile approach towards development of aqueous artificial light harvesting materials and use of the harvested energy for visible-light mediated photocatalysis in aqueous acetonitrile medium.
 | ||
| Scheme 1 Schematic representation of the formation of Pt(II) metallacages using a flexible tetra-imidazole donor. | ||
Results and discussion
Synthesis and characterization of L and metallacages
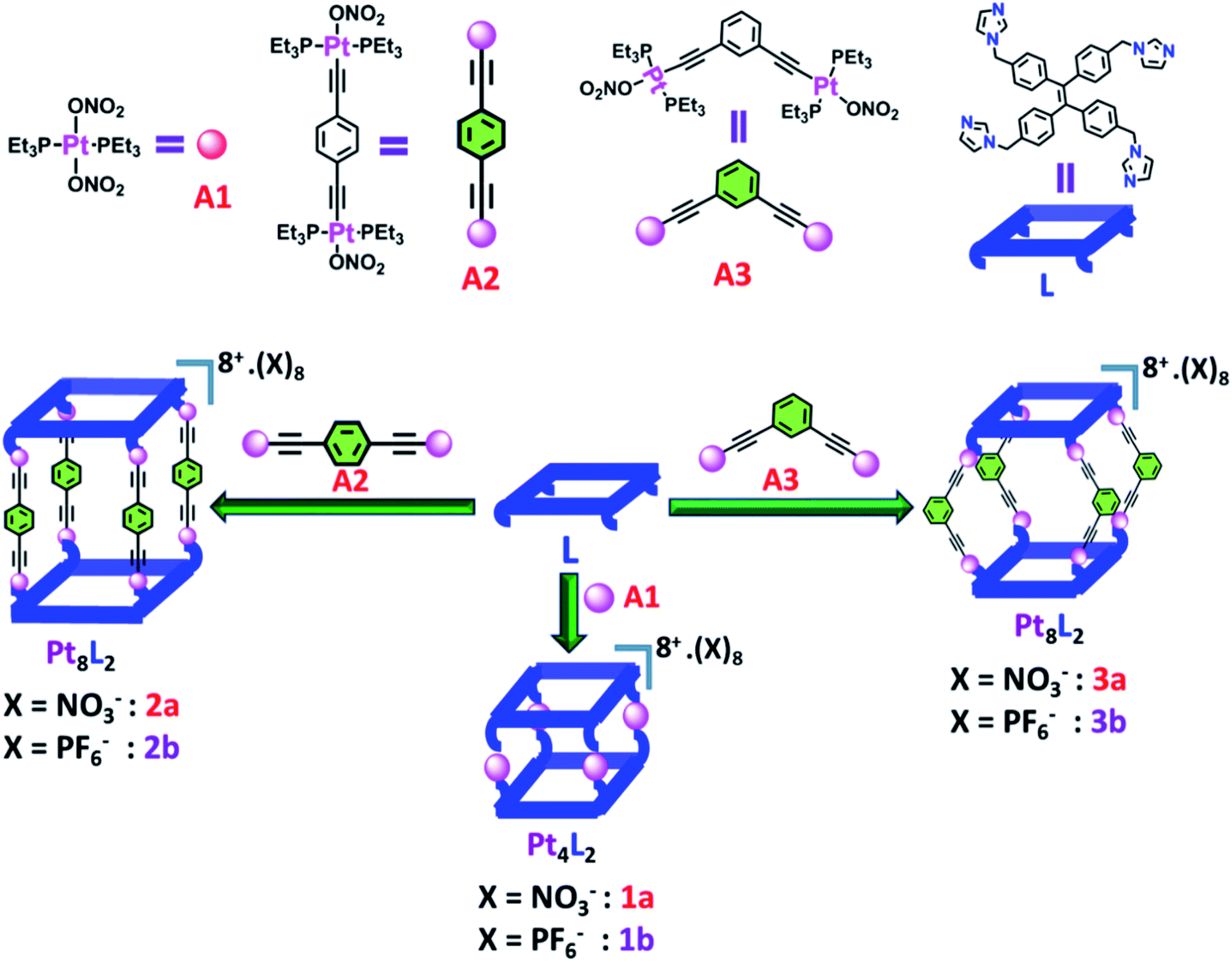
Desired ligand L was synthesized by allylic coupling of imidazole with 1,1,2,2-tetrakis(4-(bromomethyl)phenyl)ethene C1 in the presence of a base in 67% yield (Scheme S1†). Ligand L was fully characterized by NMR and ESI-MS analyses. Acceptors A1, A2 and A3 were prepared according to the reported procedures.19Self-assembled Pt4L2 molecular cage 1a and Pt8L2 based cages 2a and 3a were prepared by co-ordination driven two-component self-assembly of flexible TPE based tetratopic donor L and 180° (for 1a and 2a)/120° (for 3a) based Pt-acceptors. 1a was prepared by self-assembly of trans-[Pt(PEt3)2(ONO2)2] (A1) with L in 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio in methanol (MeOH) with subsequent stirring at 50 °C for 24 h. The resulting clear solution was triturated with diethyl ether to obtain pure self-assembled molecular cage 1a. Similarly, Pt8L2 based 2a and 3a were obtained by self-assembly of donor L with 180° Pt-acceptor A2 or 120° Pt-acceptor A3 respectively, in 1:2 molar ratio in MeOH. 1a–3a were characterized by 1H and 31P NMR. The 31P{1H} NMR spectra (Fig. S6, S10 and S14†) of all the metallacages showed a sharp singlet with associated 195Pt satellites (δ = 1.12, 15.78 and 15.65 for 1a, 2a and 3a respectively), which suggests the formation of a symmetric single product. The upfield shifts of ∼15.94 ppm, 4.43 ppm (Fig. 1) and 4.30 ppm were observed in the 31P{1H} NMR of the metallacages 1a, 2a and 3a respectively, with respect to their corresponding Pt-building blocks (A1, A2 and A3). Such upfield shifts are due to the donation of electron density by the donor atom to the acceptor upon metal–ligand coordination. The 1H NMR spectrum of 2a reveals an upfield shift with broadness of the imidazole proton (Ha) as compared to the free ligand due to metal coordination with the formation of a larger assembly. Other aromatic protons of the TPE unit (Hd and He), imidazole (Hb and Hc) and Pt-acceptor A2 (Hi) merge and give a broad peak at around 7.45–7.56 ppm which is slightly upfield shifted compared to the free ligand. The characteristic CH2 peak of 2a is upfield shifted by Δδ ∼ 0.56 ppm as compared to free L (Fig. 2). Similar observation was also noticed on the formation of other two metallacages 1a and 3a. The nitrate analogues of the cages 1a, 2a and 3a were converted to corresponding PF6− analogues 1b, 2b and 3b respectively, by treating with excess KPF6 in methanol. Formation of 1b, 2b and 3b was further investigated by 1H and 31P NMR, DOSY NMR and ESI-MS. Diffusion-ordered NMR spectroscopy (DOSY) supported the formation of a single product by the appearance of a single band for complexes 1b, 2b and 3b at logD = −9.38 (Fig. S17†), −9.78 (Fig. S18†) and −9.81 (Fig. S19†), respectively.
:1 molar ratio in methanol (MeOH) with subsequent stirring at 50 °C for 24 h. The resulting clear solution was triturated with diethyl ether to obtain pure self-assembled molecular cage 1a. Similarly, Pt8L2 based 2a and 3a were obtained by self-assembly of donor L with 180° Pt-acceptor A2 or 120° Pt-acceptor A3 respectively, in 1:2 molar ratio in MeOH. 1a–3a were characterized by 1H and 31P NMR. The 31P{1H} NMR spectra (Fig. S6, S10 and S14†) of all the metallacages showed a sharp singlet with associated 195Pt satellites (δ = 1.12, 15.78 and 15.65 for 1a, 2a and 3a respectively), which suggests the formation of a symmetric single product. The upfield shifts of ∼15.94 ppm, 4.43 ppm (Fig. 1) and 4.30 ppm were observed in the 31P{1H} NMR of the metallacages 1a, 2a and 3a respectively, with respect to their corresponding Pt-building blocks (A1, A2 and A3). Such upfield shifts are due to the donation of electron density by the donor atom to the acceptor upon metal–ligand coordination. The 1H NMR spectrum of 2a reveals an upfield shift with broadness of the imidazole proton (Ha) as compared to the free ligand due to metal coordination with the formation of a larger assembly. Other aromatic protons of the TPE unit (Hd and He), imidazole (Hb and Hc) and Pt-acceptor A2 (Hi) merge and give a broad peak at around 7.45–7.56 ppm which is slightly upfield shifted compared to the free ligand. The characteristic CH2 peak of 2a is upfield shifted by Δδ ∼ 0.56 ppm as compared to free L (Fig. 2). Similar observation was also noticed on the formation of other two metallacages 1a and 3a. The nitrate analogues of the cages 1a, 2a and 3a were converted to corresponding PF6− analogues 1b, 2b and 3b respectively, by treating with excess KPF6 in methanol. Formation of 1b, 2b and 3b was further investigated by 1H and 31P NMR, DOSY NMR and ESI-MS. Diffusion-ordered NMR spectroscopy (DOSY) supported the formation of a single product by the appearance of a single band for complexes 1b, 2b and 3b at logD = −9.38 (Fig. S17†), −9.78 (Fig. S18†) and −9.81 (Fig. S19†), respectively.
 | ||
| Fig. 1 31P{1H} (162 MHz) NMR comparison of (a) acceptor A2 and (b) cage 2a in CD3OD. | ||
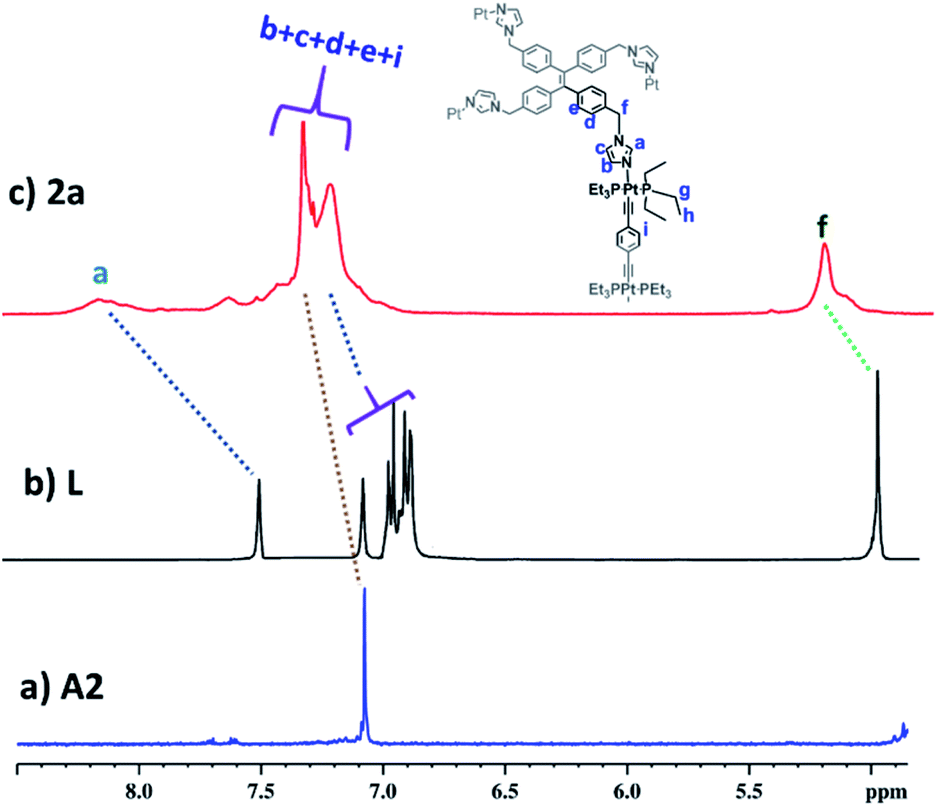 | ||
| Fig. 2 Partial 1H (400 MHz) NMR comparison of (a) acceptor A2, (b) free ligand L and (c) cage 2a in CD3OD. | ||
Stoichiometric compositions of 1b, 2b and 3b (PF6− analogues) were confirmed by ESI-MS analysis. The mass spectrum for 2b showed peaks at m/z = 3061.67, 1992.54, 1458.31 and 1137.49 with isotopic patterns corresponding to [2b-2PF6]2+, [2b-3PF6]3+, [2b-4PF6]4+ and [2b-5PF6]5+ charge fragments, respectively (Fig. 3 and S27†), which suggests the formation of 2b by [4 + 2] self-assembly of A2 and L. Similarly, the ESI-MS results of 1b (Fig. S24†) and 3b (Fig. S29†) were also consistent with the formation of [4 + 2] self-assembled cages of the respective acceptors with donor L. In all the cases calculated isotopic distribution patterns matched well with the theoretical patterns (Fig. S23–S29†). DOSY NMR and ESI-MS spectrometry clearly established the formation of molecular assemblies as [4 + 2] quadrangular 3D cages and ruled out the formation of any other possible structure by [2 + 1] self-assembly of the acceptor and donor.
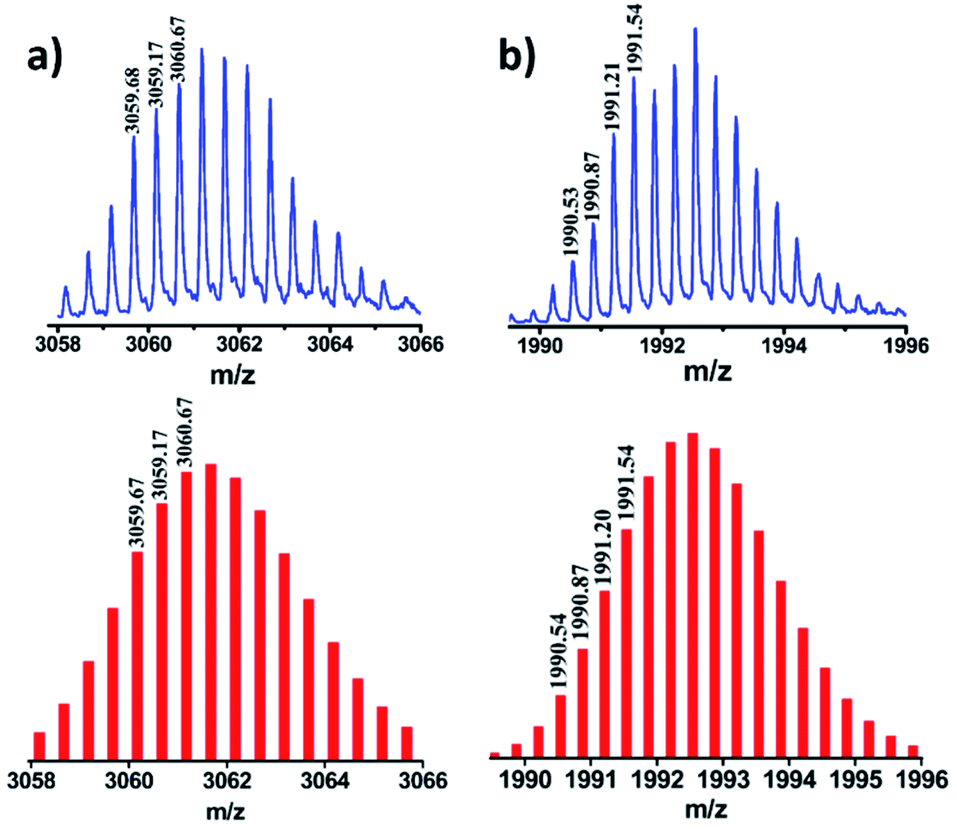 | ||
| Fig. 3 Experimental (top) and theoretical (bottom) isotopic distribution patterns of the peaks corresponding to (a) [2b-2PF6−]2+ and (b) [2b-3PF6−]3+. | ||
Computational method
To obtain further information on the ground-state structures, L, 1, 2 and 3 were optimized using DFT at the B3LYP/6-31g(d) level for P, C, N, H and B3LYP/LanL2DZ for heavy metal Pt, as implemented in the Gaussian 09 program package.20 DFT structures of all the assemblies resemble quadrangular molecular cages which possess different lengths (l), breadths (b) and heights (h). In metallacage 1, two propeller units of TPE are held by four trans-Pt(II) metal acceptors and separated from each other by 9.1 Å(C160 − C159), while in 2 and 3, two units of TPE units are held by 1,4-diethenylbenzene (for 2) and 1,3-diethenylbenzene (for 3) and separated from each other by 20.4 Å (C161 − C158) and 17.8 Å (C158 − C161), respectively. Formation of molecular structures with two different angles either 180° (A1 and A2) or 120° (A3) trans-Pt(II) acceptor becomes feasible due to the flexibility of co- ordinating imidazole because of the CH2 group. The angle between phenyl and imidazole in the case of uncoordinated ligand L is 113.7° (∠C40 − C46 − N69) which increases upon the formation of metallacages as 115.6° (∠C138 − C135 − N171) for 1, 116.6° (∠C68 − C52 − N165) for 2 and 117.4° (∠C58 − C55 − N168) for 3. The overall dimensions (l × b × h) of the cages are ∼(11.8 × 11.6 × 9.1 Å3) for 1, (14.2 × 11.6 × 20.4 Å3) for 2, and (17.2 × 14.4 × 17.8 Å3) for 3 (Fig. 4).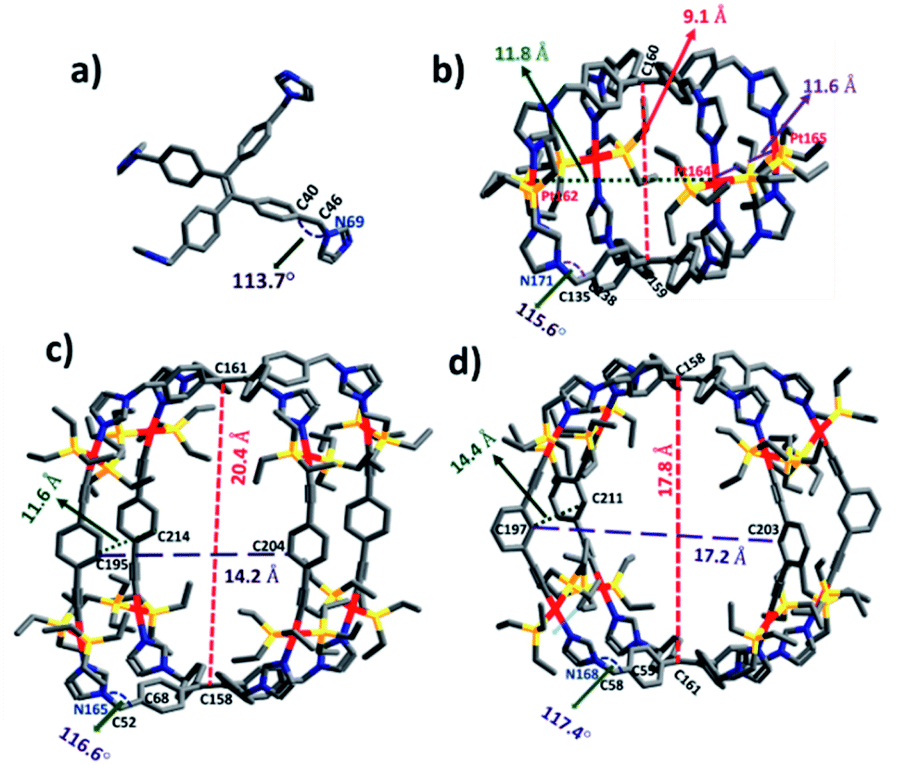 | ||
| Fig. 4 Optimized structures of (a) L, (b) 1, (c) 2 and (d) 3. Color codes: carbon (grey), nitrogen (blue), phosphorus (yellow), and platinum (red). | ||
Photophysical properties
UV-Vis and fluorescence spectroscopic studies of all individual compounds L, 1b, 2b and 3b were monitored in acetonitrile (MeCN) to probe the absorption and emission patterns (Table S1†). Ligand L shows a single absorption band at 280 nm ascribed to the π–π* transition of the TPE moiety. All the metal complexes show dual absorption bands, where the high energy transition corresponds to the π–π* transition similar to L while the low energy transition is ascribed to metal to ligand charge transfer (MLCT) and the n–π* transition (Fig. S34†). It is worth noting that bigger metallacages 2b and 3b show considerable bathochromic shifts of lower energy bands as compared to smaller system 1b. Incorporation of 1,4-diethenylbenzene (for 2b) and 1,3-diethenylbenzene (for 3b) with additional four Pt(II) centers provides more electronic communication throughout its structure which causes red shifts of about 20 nm in 2b (λmax = 345 nm) and 15 nm in 3b (λmax = 340 nm) compared to 1b (λmax = 325 nm). In solid state L showed a broad absorption maximum at 345 nm which is red shifted by about 65 nm from its solution state. All the metal complexes showed red shifts of around 20 nm in the solid state from their corresponding solution state absorption. This appreciable change in red shifts is common in the solid state (Fig. S35†). L showed very low emission with a maximum at 420 nm when excited at 290 nm. Similar large Stokes shifts were also observed for all metallacages (Fig. S34†). 2b and 3b showed lower emission intensity with high bathochromic shifts in emission compared to 1b as they provide better electronic communication within their structures as compared to 1b.AIE behaviour of the metallacages
Since ligand L contains an AIE active TPE unit, to check the AIE activity of the metallacages (PF6− analogues 1b, 2b and 3b) a water/MeCN mixture was selected for supramolecular aggregation. Ligand L is weakly emissive in dilute solution in MeCN due to non-radiative decay and shows emission at 420 nm. However, immobilizing weakly emissive L in the cage structure due to metal-coordination leads to a six-fold enhancement of fluorescence (Fig. 5a) in 1b. Metal complexes 2b and 3b show only ∼two-fold increase in emission intensity compared to free L in MeCN. Due to the greater separation of two TPE units in 2b and 3b as compared to 1b provides more free rotation of the phenyl ring of the TPE units even after co-ordination in the dilute state which leads to small emission enhancement. Upon increasing the water fraction in MeCN, aggregation induced emission enhancement (AIEE) was noticed due to the greater restriction of phenyl ring rotation in the aggregate state. In a 90% water/MeCN mixture maximum emission enhancement was observed for all the cages; 1b showed 5-fold while 2b and 3b showed 25-fold and 16-fold emission intensity enhancement respectively, as compared to their corresponding dilute solutions.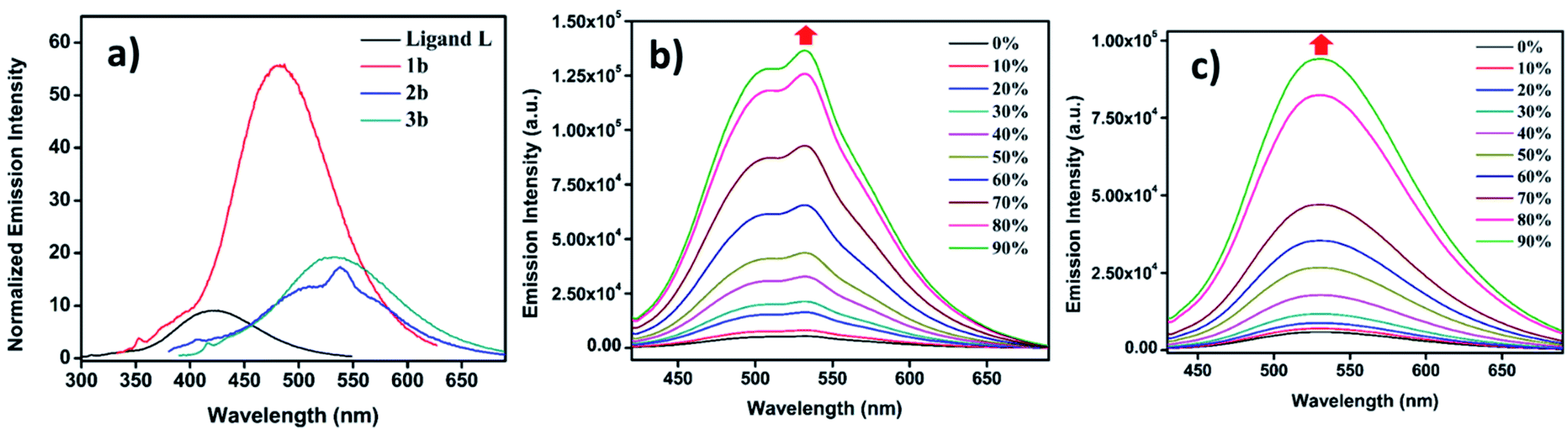 | ||
| Fig. 5 (a) Fluorescence emission of different compounds (c = 10−5 M) recorded in MeCN. Fluorescence emission with increasing water fraction in MeCN for: (b) 2b (λex = 345 nm, c = 10−5 M) and (c) 3b (λex = 340 nm, c = 10−5 M). | ||
2b and 3b showed greater AIE effects due to more rigidification of TPE units in the aggregate state as compared to 1b. AIE properties of the metal complexes were well supported by fluorescence quantum yield (ΦF) measurements (Table S3†). A dilute solution of L in MeCN showed ΦF = 3.46%, which slightly increased upon metal coordination to 4.63% for 2b and 6.27% for 3b, while 1b showed a considerable increase to 12.45% due to more rigidification of TPE units within the formed supramolecular framework (Fig. S48†). In the aggregate state fluorescence quantum yields were further increased to 21.22% for 1b, 25.67% for 2b and 22.34% for 3b (Fig. S49†). Ligand L and all metallacages are highly emissive in the solid state with further red shift in emission as compared to solution where red shifts of about 28 nm for L, 30 nm for 1a, 10 nm for 2b and 12 nm for 3b were observed (Fig. S35†). All metal complexes showed further increase in ΦF to 24.07%, 29.34% and 26.91% for 1b, 2b and 3b respectively in the solid state (Fig. S50†).
Morphological characterization in the aggregate state
The change in emission behavior with change in water fraction for all metallacages is due to the formation of aggregates. Therefore, morphologies of the supramolecular aggregates were investigated by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). For SEM analysis 150 μL of 10−5 M solution of individual metal complexes 1b, 2b and 3b in a 90% water/MeCN mixture was deposited on silicon wafer followed by drying in a vacuum. SEM analysis of 1b (Fig. S53†), 2b (Fig. 6c), and 3b (Fig. 6d) showed the formation of spherical nanoparticles. For TEM analysis, 5 μL of 10−5 M solution in a 90% water/MeCN mixture of respective metal complexes was dropcast on copper grids and dried to afford the final sample. TEM analysis also supports the formation of agglomerated spherical nano-aggregates. The average size of nano-spheres varies from 200 nm to 400 nm for 1b (Fig. S52†), 350 to 500 nm for 2b (Fig. 6a) and 300 to 550 nm for 3b (Fig. 6b). To further support our observation, dynamic light scattering (DLS) analysis was performed on aggregates of all the metallacages in a 90% water/MeCN mixture where we obtained a mean hydrodynamic diameter of around 305 nm for 1b (Fig. S54a†), 462 nm for 2b (Fig. S54b†) and 412 nm for 3b (Fig. S54c†). | ||
| Fig. 6 TEM images of aggregates in the 90% H2O/MeCN fraction for (a) 2b and (b) 3b. SEM images of aggregates in the same medium for (c) 2b and (d) 3b. | ||
Solvatochromism
Photophysical properties of all the assemblies were investigated in different solvents from non-polar toluene to polar DMSO. The UV-Vis spectra of all the metallacages displayed a slight change in absorption profiles (Fig. S39–S41†). Considerable shifts in fluorescence emission profiles were observed for all metallacages (summarized in Table S2†). In non-polar solvents like toluene and chloroform 1b showed emission maxima at 460 and 472 nm respectively, which are red shifted in polar solvents like MeCN (λem = 480) and DMF (λem = 479 nm). All metallacages show high emission intensity in non-polar solvents like toluene and chloroform as compared to polar solvents like MeCN or DMF, since non-polar solvents lead to aggregate formation of metallacages due to their poor solvation (Fig. S39–S41†). As all metallacages have AIE active TPE units, their emission got enhanced upon aggregate formation in non-polar solvents.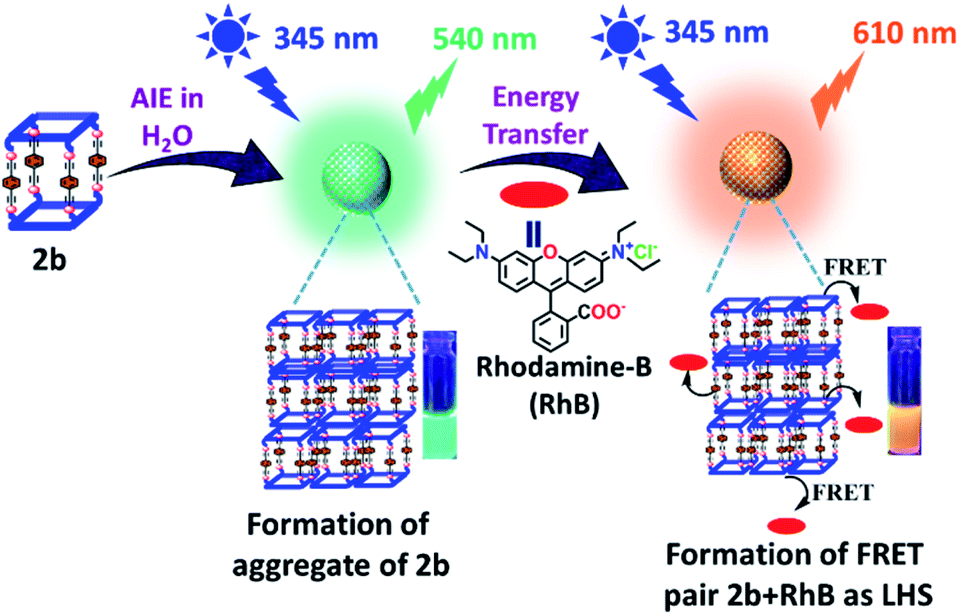 | ||
| Fig. 7 Schematic illustration of AIE and light harvesting function of metallacage 2b. | ||
Artificial light harvesting by Förster resonance energy transfer
Metallacage 1b showed blue emission while 2b and 3b showed light-green emission in the aggregate state. The maximum emission intensity was observed in a 90% water/MeCN fraction for all the metallacages. Therefore, the 90% water/MeCN solvent medium was selected for investigation of the energy transfer process as in this medium the cage is stable and doesn't shows any self-quenching over time due to precipitation. Water soluble organic fluorescent dyes are potential candidates for sensors in biological medium or used in energy transfer for artificial light harvesting. For efficient energy transfer by FRET there should be considerable overlap of donor emission and acceptor absorption as the primary requirement.Here, we have chosen rhodamine-B (RhB) as an acceptor molecule for artificial LHS by the FRET mechanism (Fig. 7). RhB absorbs light from 450 nm to 560 nm with the absorbance maximum at 547 nm and emits light at 580 nm in a 90% water/MeCN mixture (Fig. S42†). Cages 2b (λem = 540 nm)/3b (λem = 530 nm) show a considerable overlap of their broad emission with the absorption of RhB, while 1b (λem = 490 nm) doesn't show such considerable overlap (Fig. 8a and S43†). Therefore, 2b and 3b become possible donors for energy transfer in artificial LHSs for acceptor RhB. The stability of cage 2b in the presence of RhB was first investigated by 1H and 31P NMR titration. Due to poor 1H and 31P peak intensity in lower concentration, we checked the stability of cages in the 10−3 M concentration range in MeCN-d3. Both 1H and 31P NMR suggest that chemical shifts of 2b remain unaffected and only an increase in the 1H signal of RhB was observed upon its gradual addition (up to six equivalents with respect to 2b) to a cage solution of 2b (Fig. S20 and S21†).
The energy transfer process was studied by fluorescence titration of RhB with the solution of 2b/3b. When 20 μL of 2 × 10−5 M (90% water/MeCN mixture) solution of RhB was gradually added to 2 mL of 10−5 M (90% water/MeCN mixture) cage solution of 2b, a sharp decrease in emission intensity at 540 nm corresponds to 2b was observed while fluorescence emission intensity at 610 nm which was further red shifted by 30 nm corresponding to the emission of RhB increased upon excitation at 345 nm (Fig. 8b and c). This decrease in emission intensity of 2b and increase in emission intensity of RhB suggest energy transfer from donor 2b to acceptor RhB. This process of energy transfer got saturated upon 200 μL of 2 × 10−5 M addition of RhB to 2 mL of 10−5 M 2b, suggesting the maximum energy transfer for 5:1 ratio for (2b + RhB). However, under similar conditions a 2 × 10−6 M solution of RhB in a 90% water–MeCN mixture was found to be non-emissive when excited at 345 nm, which rules out the emission of dye by direct excitation (Fig. S46†). The CIE chromaticity diagram also confirms the change in fluorescence colour from light green (2b) [CIE coordinate (0.37:0.59)] to yellow (2b:RhB: 10:1) [CIE coordinate (0.42:0.46)] to orange-red (2b:RhB: 5:1) [CIE coordinate (0.51:0.38)] (Fig. 8d). Similar observation of energy transfer was observed in the case of 3b with decrease in emission intensity at 530 nm and increase in emission intensity at 615 nm with further red shift by about 35 nm in the emission of RhB (Fig. S44†).
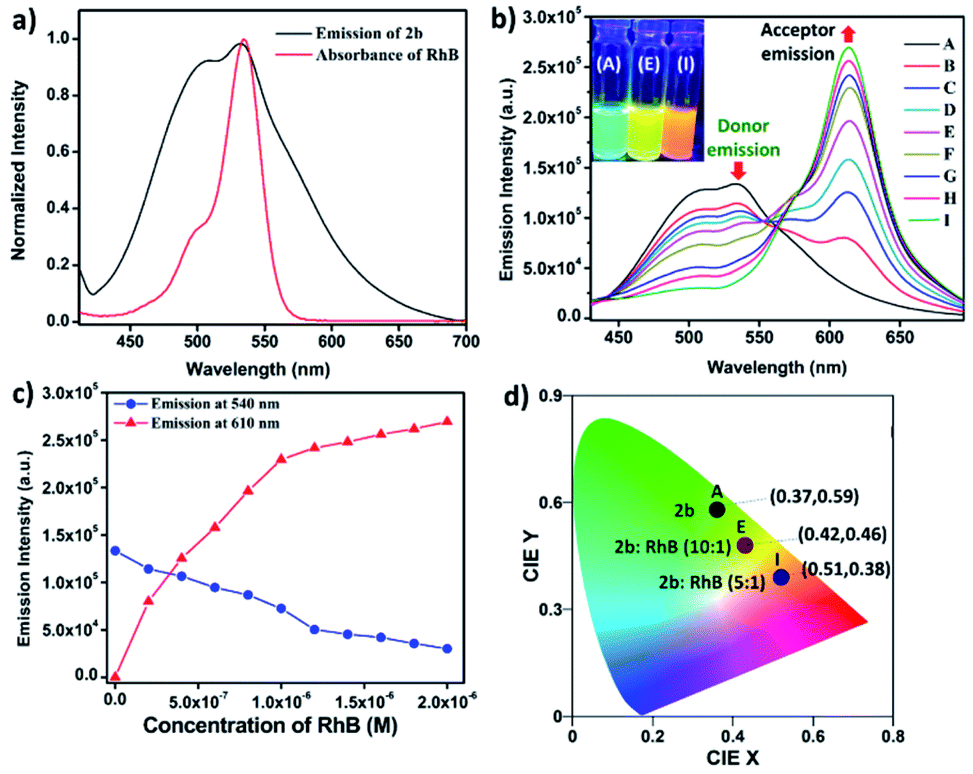 | ||
| Fig. 8 (a) Normalized plot for the emission of 2b and the absorption of RhB. (b) Fluorescence spectra of metallacage 2b with gradual addition of RhB (λex = 345 nm) in a 90% H2O/MeCN mixture. Inset of Fig. 8(b): photograph of 2b (left), (2b + RhB, 10:1 equiv.) (middle), (2b + RhB, 5:1 equiv.) (right) under UV irradiation at 365 nm. (c) Change in fluorescence emission intensity for 2b upon gradual addition of RhB at 540 nm and 610 nm. (d) The 1931 CIE chromaticity coordinate changes of 2b (10−5 M in a 90% water/MeCN mixture) upon titration with RhB (0 to 0.2 equiv.). | ||
The energy transfer process is also evident by the increase in fluorescence quantum yield (ΦF). When energy transfer is maximum {[cage 2b or 3b] = 10−5 M and [RhB] = 2 × 10−6 M} ΦF increases to 32.76% for 2b and 30.54% for 3b, which were substantially high as compared to ΦF of 2b or 3b alone in both aggregate (90% water/MeCN) and solid states (Fig. S51†).
UV-Visible study was performed by gradual addition of a 2 × 10−5 M (90% water/MeCN) solution of RhB to a 10−5 M (90% water/MeCN) cage solution of 2b. Absorption (λmax = 345 nm) corresponding to 2b remains constant while absorption intensity corresponding to RhB at λmax = 547 nm increases with gradual addition of RhB, which suggests the absence of any ground sate interactions between 2b and RhB molecules (Fig. S45†). The process of FRET from cage 2b or 3b to RhB was well supported by measurement of fluorescence lifetime of the donor and donor in the presence of the acceptor which is summarized in Table S4.† The fluorescence life-time was measured by the time-correlated single-photon counting technique (TCSPC) which reveals that the fluorescence life time of cage 2b (τ1 = 0.97 ns, τ2 = 3.90 ns, τav = 1.83 ns) or 3b (τ1 = 0.80 ns, τ2 = 3.98 ns, τav = 2.09 ns) was found to be shorter in the presence of dye RhB ({τ1 = 0.74 ns, τ2 = 2.98 ns, τav = 1.20 ns}) for (2b + RhB) or {τ1 = 0.70 ns, τ2 = 3.21 ns, τav = 1.47 ns} for (3b + RhB) (Fig. 9 and S47c†). The energy transfer efficiencies (ΦET) of fluorescent cages 2b and 3b were calculated (eqn S(2)†) as 77% and 58% at a donor/acceptor ratio of 5:1 in a 90% H2O/MeCN mixture, respectively (Table S5†). In controlled experiments donor–acceptor LHSs (2b + RhB and 3b + RhB) were excited at acceptor's (RhB) absorption maximum at 547 nm (Fig. S55 and S56†) where both LHSs (2b + RhB) [eqn S(3), Table S6†] and (3b + RhB) [eqn S(4), Table S7†] show good antenna effect of 21 and 16, respectively.
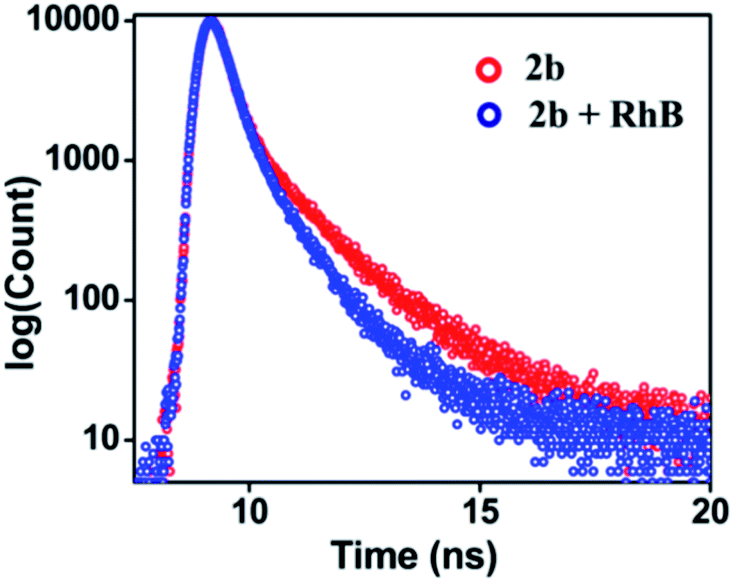 | ||
| Fig. 9 Fluorescence decay profile for 2b (red, λex = 340 nm) and (2b + RhB) (blue, λex = 340 nm) {[2b] = 10−5 M, [RhB] = 2 × 10−6 M} in 90% water/MeCN. | ||
Photocatalytic activity and regulation
To explore the use of a light-harvesting system (2a + RhB) as an efficient energy source, we investigated its photocatalytic activity for the photo-oxidative cross coupling cyclization reaction between N,N-dimethylaniline (4) and N-phenyl maleimide (5a) in aqueous acetonitrile medium (90% water/MeCN mixture) under visible light to afford 1,2,3,4-tetrahydroquinoline, which is an important product for pharmaceutical industries.21 The LH complex (2a + RhB) under optimized conditions showed enhanced photo-oxidative catalytic efficiency with a shorter reaction time in aqueous (90% water/ACN) solution under visible light in comparison to the organic catalyst such as naphthochromenones,22 Eosin Y,23 metal catalyst,24 COF system,25 chlorophyll,26 and catalyst free system (under UV light/fluorescent lamp)27 for the tetrahydroquinoline preparation.After optimization of the reaction conditions (Table 1, Fig. S58–S67†), we observed that the LH complex (2a + RhB) {5 mol% 2a+ 1 mol% RhB} readily catalysed the formation of 6a by the photo-oxidative cyclization reaction of N,N-dimethylaniline 4 and N-phenyl maleimide 5a in aqueous acetonitrile medium (90% water/MeCN) in an excellent yield of 97% under visible light irradiation for 12 h (Fig. 10). In contrast, RhB (1 mol%) alone under similar conditions resulted 6a in just 5% yield (Fig. S58†). Increasing the amount of RhB to 5 mol% marginally increased the product conversion to 13% (Fig. S59†). However, no trace of product (6a) formation was observed when the reaction was performed with 5 mol% of cage 2a alone (Fig. S60†). RhB dye has very low absorption at lower wavelengths of visible and UV regions, while cage 2b possesses strong absorption in the UV region and emits strongly at lower wavelengths of the visible region (λem = 540 nm). Hence, 2b serves as an antenna source to efficiently transfer energy from UV/Visible light to RhB to perform photo-oxidative catalytic reaction. Thus, cage 2b represents an artificial light harvesting system which mimics the natural photosynthetic process. The above photocatalytic reaction was triggered under very mild conditions by using solar light as an energy source (here we used a white light LED as a solar light simulator). However, very low catalytic activity was observed when the reaction was performed in the dark (Table 1) suggesting the importance of visible light in photo sensitization in the catalytic cycle. As shown in Fig. 11, the reaction proceeds by photosensitizer RhB by the single electron transfer (SET) mechanism21–27 whose efficiency is substantially improved by energy transfer (by the FRET mechanism) from cage 2b (Fig. 11). In the above catalytic cycle, molecular oxygen plays a pivotal role in oxidation of the intermediate 7d. Generation of hydrogen peroxide as a side product during catalysis was detected using the KI/starch indicator (Table S8†). Although the LH system (3b + RhB) has lower energy transfer efficiency and antenna effect as compared to (2b + RhB), the product conversion with (3b + RhB) as a photocatalyst under similar conditions gave an excellent yield of 92% (Fig. S68†).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cage 2b (X mol%) | RhB (Y mol%) | Time (in h) | Light source | % yields of 6ab |
| a Reaction conditions: N,N-dimethylaniline 4 (0.04 mmol), N-phenyl maleimide 5 (0.04 mmol), water (4.5 mL) + MeCN (0.5 mL), stirring. b Crude yields determined from 1H NMR based on the starting material with internal standard 1,3,5-trimethoxy benzene. Philips 40 W LED bulb (4000 Lm, 6500 K) has been used as a visible light source. | |||||
| 1 | 5 | 1 | 4 | Visible | 37 |
| 2 | 5 | 1 | 8 | Visible | 59 |
| 3 | 5 | 1 | 12 | Visible | 97 |
| 4 | 5 | 0.5 | 12 | Visible | 55 |
| 5 | 5 | 1.5 | 12 | Visible | 98 |
| 6 | 5 | 1 | 12 | Dark | 13 |
| 7 | None | 1 | 12 | Visible | 05 |
| 8 | None | 5 | 12 | Visible | 16 |
| 9 | 5 | None | 12 | Visible | Trace |
| 10 | Ligand L (5 mol%) | 1 | 12 | Visible | 8 |
| 11 | Cage 3b (5 mol%) | 1 | 12 | Visible | 92 |

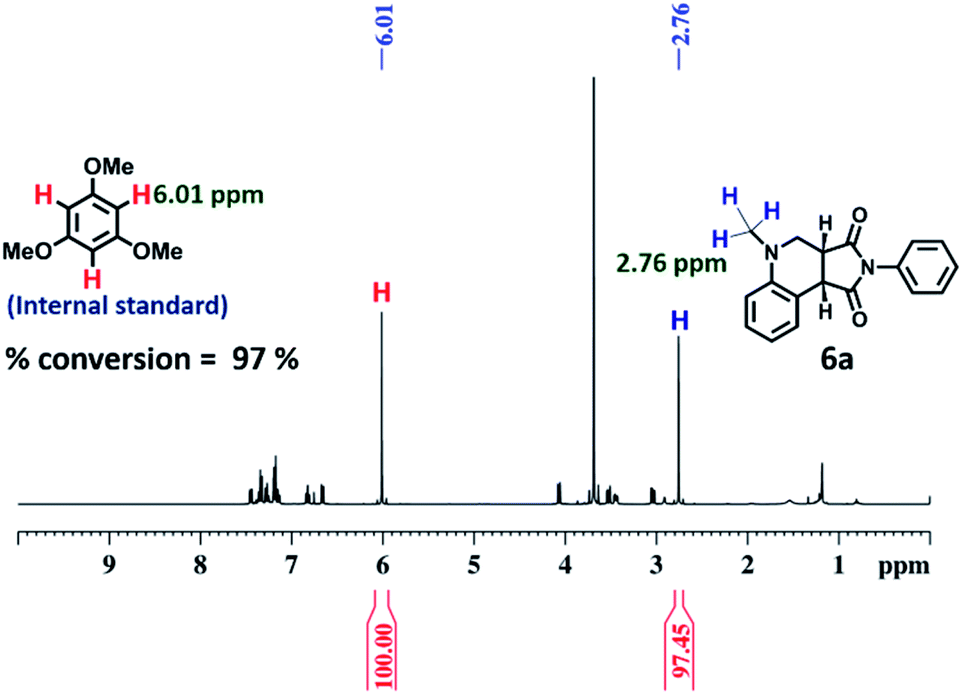 | ||
| Fig. 10 1H NMR (400 MHz, CDCl3) of the reaction of (4 + 5a) using {5 mol% cage 2b + 1 mol% RhB} as the photocatalyst for 12 h under visible light. Percentage conversion of the product 6a was determined from 1H NMR integration based on the starting material with internal standard 1,3,5-trimethoxy benzene. | ||
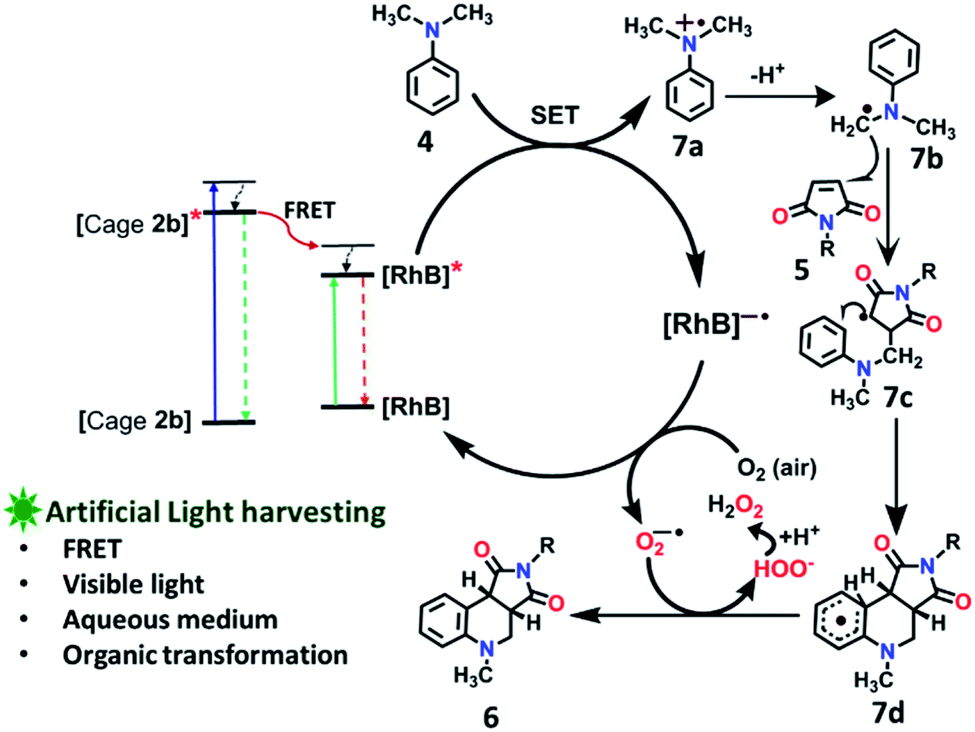 | ||
| Fig. 11 Proposed reaction pathways of photo-oxidative cyclization of N,N-dimethylaniline 4 and N-alkyl/aryl maleimides 5. | ||
Further, the scope of (2a + RhB) as a photocatalyst was examined using a series of N-alkyl/aryl maleimides (5). Substrates with alkyl groups i.e., ethyl (5b) and cyclohexyl (5c), electron withdrawing aryl groups, i.e., 4-Br-Ph (5e), and electron donating aryl groups 4-CH3-Ph (5d) are consistent with this method and the desired products were obtained in excellent yields of 97–99% (Table 2, Fig. S69–S72†). This artificial light harvesting complex (2a + RhB) in a 70% water/MeCN mixture is even successful with bulkier N-pyrenemalemide (5f), which yielded the desired product 6f in a good yield of 74% (Fig. S73†) easily. Recyclability of the cage 2b was well examined in the model reaction of 4 and 5a. After extraction of the reaction mixture with CDCl3, the catalysis reaction was further performed with the recovered cage 2b which catalyses the reaction of 4 and 5a as fast as the first cycle and doesn't lose its catalytic activity up to four cycles (Fig. S57†).
|
|
|||
|---|---|---|---|
| Entry | R | Product (6) | % yieldsb |
| a N,N-Dimethylaniline 4 (0.04 mmol), N-alkyl/aryl maleimides 5 (0.04 mmol), catalyst A (5 mol%), RhB (1 mol%) water (4.5 mL), MeCN (0.5 mL), stirring at rt in open air. b Crude yields determined from 1H NMR based on the starting material with internal standard 1,3,5-trimethoxy benzene. c Reaction was performed in a 70% water/MeCN mixture (1.5 mL MeCN + 3.5 mL water). | |||
| 1 | Ph (5a) | 6a | 97 |
| 2 | Ethyl (5b) | 6b | 98 |
| 3 | Cyclohexyl (5c) | 6c | 98 |
| 4 | 4-CH3-Ph (5d) | 6d | 98 |
| 5 | 4-Br-Ph (5e) | 6e | 99 |
| 6 | 1-Pyrene (5f)c | 6f | 74 |

Conclusions
In summary, we report here facile synthesis of three tetragonal prismatic metallacages via two-component self-assembly of a newly designed flexible tetraimidazole donor (L) with 180°/120° trans-Pt acceptors (A1–A3). Due to the presence of the AIE active TPE backbone, the assemblies are highly emissive in both aggregate and solid states, which is supported by increased quantum yield in aggregate and solid states compared to their dilute solution state. Formation of spherical supramolecular nano-aggregates in 90% H2O/MeCN was confirmed by SEM, TEM and DLS studies. Moreover, AIE active cages 2b and 3b have been utilized for fabricating artificial light harvesting materials in aqueous acetonitrile medium (90% water/MeCN mixture) where they act as donors for energy transfer by the FRET mechanism to organic dye rhodamine-B (RhB). The process of energy transfer is well supported by the increase in quantum yield, decrease in lifetime, colour change from light green to red-orange of the cages in the presence of dye RhB. The maximum energy transfer efficiencies obtained for (2b + RhB) and (3b + RhB) at a donor/acceptor ratio of 5:1 in a 90% H2O/MeCN mixture are as high as 77% and 58%, respectively. Both the LHSs ((2b + RhB) and (3b + RhB)) showed good antenna effect of 21 and 16, respectively. Most importantly, light harvesting materials (2b + RhB)/(3b + RhB) have been successfully utilized as visible-light photocatalysts for cross coupling cyclization of N,N-dimethylaniline and N-alkyl/aryl maleimides in aqueous acetonitrile with much enhanced yields compared to similar reactions with dye or cages alone. Overall, this article demonstrates a facile synthetic strategy to obtain AIE active supramolecular architectures for fabricating artificial light harvesting systems in aqueous acetonitrile medium where the output energy is successfully utilized for catalytic chemical transformation for practical application.
Author contributions
A. K. performed synthesis/characterization of the cages and light harvesting systems. R. S. was involved in data interpretation and catalysis. P. S. M. designed the studies, involved in manuscript preparation. All authors discussed the results and were involved in manuscript writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. S. M. is grateful to SERB for financial support (Grant No. CRG/2018/000315). A. K. sincerely thanks Aryaman Pattanaik for his kind help in ligand synthesis and Soumalya Bhattacharyya for helpful discussion on light harvesting.References
- G. McDermott, S. Prince, A. Freer, A. Hawthornthwaite-Lawless, M. Papiz, R. Cogdell and N. Isaacs, Nature, 1995, 374, 517–521 CrossRef CAS.
- (a) T. Förster, Ann. Phys., 1948, 437, 55–75 CrossRef; (b) G. L. Closs and J. R. Miller, Science, 1988, 240, 440 CrossRef CAS PubMed; (c) Y. Qin, L.-J. Chen, Y. Zhang, Y.-X. Hu, W.-L. Jiang, G.-Q. Yin, H. Tan, X. Li, L. Xu and H.-B. Yang, Chem. Commun., 2019, 55, 11119–11122 RSC; (d) L. Catti, N. Kishida, T. Kai, M. Akita and M. Yoshizawa, Nat. Commun., 2019, 10, 1948 CrossRef PubMed.
- (a) L.-B. Meng, D. Li, S. Xiong, X.-Y. Hu, L. Wang and G. Li, Chem. Commun., 2015, 51, 4643–4646 RSC; (b) Y. Li, Y. Dong, L. Cheng, C. Qin, H. Nian, H. Zhang, Y. Yu and L. Cao, J. Am. Chem. Soc., 2019, 141, 8412–8415 CrossRef CAS PubMed; (c) M.-S. Choi, T. Aida, T. Yamazaki and I. Yamazaki, Chem.–Eur. J., 2002, 8, 2667–2678 CrossRef CAS; (d) H. Imahori, J. Phys. Chem. B, 2004, 108, 6130–6143 CrossRef CAS PubMed; (e) A. Adronov, S. L. Gilat, J. M. J. Fréchet, K. Ohta, F. V. R. Neuwahl and G. R. Fleming, J. Am. Chem. Soc., 2000, 122, 1175–1185 CrossRef CAS; (f) M. Hao, G. Sun, M. Zuo, Z. Xu, Y. Chen, X.-Y. Hu and L. Wang, Angew. Chem., Int. Ed., 2020, 59, 10095–10100 CrossRef CAS.
- A. J. P. Teunissen, C. Pérez-Medina, A. Meijerink and W. J. M. Mulder, Chem. Soc. Rev., 2018, 47, 7027–7044 RSC.
- (a) S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695–704 CrossRef CAS; (b) H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef; (c) H.-C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed; (d) S. Liu, S. Jiang, J. Xu, Z. Huang, F. Li, X. Fan, Q. Luo, W. Tian, J. Liu and B. Xu, Macromol. Rapid Commun., 2019, 40, 1800892 CrossRef PubMed.
- (a) T. R. Cook, Y.-R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734–777 CrossRef CAS PubMed; (b) T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001–7045 CrossRef CAS PubMed; (c) C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Chem. Rev., 2015, 115, 3012–3035 CrossRef CAS PubMed; (d) S. D. P. Fielden, D. A. Leigh and S. L. Woltering, Angew. Chem., Int. Ed., 2017, 56, 11166–11194 CrossRef CAS PubMed; (e) S. Chakraborty and G. R. Newkome, Chem. Soc. Rev., 2018, 47, 3991–4016 RSC; (f) W. Wang, Y.-X. Wang and H.-B. Yang, Chem. Soc. Rev., 2016, 45, 2656–2693 RSC; (g) X. Jing, C. He, L. Zhao and C. Duan, Acc. Chem. Res., 2019, 52, 100–109 CrossRef CAS PubMed; (h) D. Zhang, T. K. Ronson and J. R. Nitschke, Acc. Chem. Res., 2018, 51, 2423–2436 CrossRef CAS PubMed; (i) H. Sepehrpour, W. Fu, Y. Sun and P. J. Stang, J. Am. Chem. Soc., 2019, 141, 14005–14020 CrossRef CAS PubMed.
- (a) C. G. Oliveri, P. A. Ulmann, M. J. Wiester and C. A. Mirkin, Acc. Chem. Res., 2008, 41, 1618–1629 CrossRef CAS PubMed; (b) M. J. Prakash and M. S. Lah, Chem. Commun., 2009, 3326–3341 RSC; (c) W. Zheng, W. Wang, S.-T. Jiang, G. Yang, Z. Li, X.-Q. Wang, G.-Q. Yin, Y. Zhang, H. Tan, X. Li, H. Ding, G. Chen and H.-B. Yang, J. Am. Chem. Soc., 2019, 141, 583–591 CrossRef CAS PubMed; (d) C. Gütz, R. Hovorka, G. Schnakenburg and A. Lützen, Chem.–Eur. J., 2013, 19, 10890–10894 CrossRef PubMed; (e) L. R. Holloway, P. M. Bogie, Y. Lyon, C. Ngai, T. F. Miller, R. R. Julian and R. J. Hooley, J. Am. Chem. Soc., 2018, 140, 8078–8081 CrossRef CAS PubMed; (f) J. Anhäuser, R. Puttreddy, Y. Lorenz, A. Schneider, M. Engeser, K. Rissanen and A. Lützen, Org. Chem. Front., 2019, 6, 1226–1235 RSC; (g) M. Käseborn, J. J. Holstein, G. H. Clever and A. Lützen, Angew. Chem., Int. Ed., 2018, 57, 12171–12175 CrossRef; (h) C. Schulte to Brinke and F. E. Hahn, Dalton Trans., 2015, 44, 14315–14322 RSC; (i) L.-L. Ma, Y.-Y. An, L.-Y. Sun, Y.-Y. Wang, F. E. Hahn and Y.-F. Han, Angew. Chem., Int. Ed., 2019, 58, 3986–3991 CrossRef CAS PubMed.
- (a) J. R. Nitschke, Acc. Chem. Res., 2007, 40, 103–112 CrossRef CAS PubMed; (b) S. Liu, Y.-F. Han and G.-X. Jin, Chem. Soc. Rev., 2007, 36, 1543–1560 RSC; (c) G. H. Clever and M. Shionoya, Coord. Chem. Rev., 2010, 254, 2391–2402 CrossRef CAS; (d) R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS; (e) M. Han, D. M. Engelhard and G. H. Clever, Chem. Soc. Rev., 2014, 43, 1848–1860 RSC; (f) Y.-F. Han and G.-X. Jin, Acc. Chem. Res., 2014, 47, 3571–3579 CrossRef CAS PubMed; (g) M. Yoshizawa and J. K. Klosterman, Chem. Soc. Rev., 2014, 43, 1885–1898 RSC; (h) S. De, K. Mahata and M. Schmittel, Chem. Soc. Rev., 2010, 39, 1555–1575 RSC; (i) M. Schmittel and S. Qinghai, Chem. Commun., 2012, 48, 2707–2709 RSC; (j) M. Schmittel, S. Pramanik and S. De, Chem. Commun., 2012, 48, 11730–11732 RSC; (k) M. L. Saha and M. Schmittel, Inorg. Chem., 2016, 55, 12366–12375 CrossRef CAS PubMed; (l) A. Goswami, S. Saha, P. K. Biswas and M. Schmittel, Chem. Rev., 2020, 120, 125–199 CrossRef CAS PubMed; (m) N. Sinha, L. Stegemann, T. T. Y. Tan, N. L. Doltsinis, C. A. Strassert and F. E. Hahn, Angew. Chem., Int. Ed., 2017, 56, 2785–2789 CrossRef CAS PubMed; (n) N. Sinha, F. Roelfes, A. Hepp and F. E. Hahn, Chem.–Eur. J., 2017, 23, 5939–5942 CrossRef CAS PubMed; (o) L.-Y. Sun, N. Sinha, T. Yan, Y.-S. Wang, T. T. Y. Tan, L. Yu, Y.-F. Han and F. E. Hahn, Angew. Chem., Int. Ed., 2018, 57, 5161–5165 CrossRef CAS PubMed; (p) L.-Y. Sun, T. Feng, R. Das, F. E. Hahn and Y.-F. Han, Chem.–Eur. J., 2019, 25, 9764–9770 CrossRef CAS PubMed.
- (a) C. J. Kuehl, Y. K. Kryschenko, U. Radhakrishnan, S. R. Seidel, S. D. Huang and P. J. Stang, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4932 CrossRef CAS PubMed; (b) A. M. Brown, M. V. Ovchinnikov, C. L. Stern and C. A. Mirkin, J. Am. Chem. Soc., 2004, 126, 14316–14317 CrossRef CAS PubMed; (c) K. Nakabayashi, M. Kawano, M. Yoshizawa, S.-i. Ohkoshi and M. Fujita, J. Am. Chem. Soc., 2004, 126, 16694–16695 CrossRef CAS PubMed; (d) J. Heo and C. A. Mirkin, Angew. Chem., Int. Ed., 2006, 45, 941–944 CrossRef CAS PubMed; (e) K. Harano, S. Hiraoka and M. Shionoya, J. Am. Chem. Soc., 2007, 129, 5300–5301 CrossRef CAS PubMed; (f) A. K. Bar, R. Chakrabarty, G. Mostafa and P. S. Mukherjee, Angew. Chem., Int. Ed., 2008, 47, 8455–8459 CrossRef CAS PubMed; (g) W. Meng, J. K. Clegg, J. D. Thoburn and J. R. Nitschke, J. Am. Chem. Soc., 2011, 133, 13652–13660 CrossRef CAS PubMed; (h) M. Wang, C. Wang, X.-Q. Hao, X. Li, T. J. Vaughn, Y.-Y. Zhang, Y. Yu, Z.-Y. Li, M.-P. Song, H.-B. Yang and X. Li, J. Am. Chem. Soc., 2014, 136, 10499–10507 CrossRef CAS PubMed; (i) A. Kumar and P. S. Mukherjee, Chem.–Eur. J., 2020, 26, 4842–4849 CrossRef CAS PubMed.
- (a) M. Chen, J. Wang, S.-C. Wang, Z. Jiang, D. Liu, Q. Liu, H. Zhao, J. Yan, Y.-T. Chan and P. Wang, J. Am. Chem. Soc., 2018, 140, 12168–12174 CrossRef CAS PubMed; (b) S.-Y. Wang, J.-Y. Huang, Y.-P. Liang, Y.-J. He, Y.-S. Chen, Y.-Y. Zhan, S. Hiraoka, Y.-H. Liu, S.-M. Peng and Y.-T. Chan, Chem.–Eur. J., 2018, 24, 9274–9284 CrossRef CAS PubMed; (c) M. Yamashina, Y. Tanaka, R. Lavendomme, T. K. Ronson, M. Pittelkow and J. R. Nitschke, Nature, 2019, 574, 511–515 CrossRef CAS PubMed; (d) M. Yoshizawa and L. Catti, Acc. Chem. Res., 2019, 52, 2392–2404 CrossRef CAS PubMed; (e) T. Tsutsui, S. Kusaba, M. Yamashina, M. Akita and M. Yoshizawa, Chem.–Eur. J., 2019, 25, 4320–4324 CrossRef CAS PubMed; (f) J. Huang, D. Liu, S.-C. Wang, M. Chen, H. Zhao, K. Li, Y.-T. Chan and P. Wang, Inorg. Chem., 2019, 58, 5051–5057 CrossRef CAS PubMed; (g) J.-H. Fu, S.-Y. Wang, Y.-S. Chen, S. Prusty and Y.-T. Chan, J. Am. Chem. Soc., 2019, 141, 16217–16221 CrossRef CAS PubMed; (h) T. Wu, Y.-S. Chen, M. Chen, Q. Liu, X. Xue, Y. Shen, J. Wang, H. Huang, Y.-T. Chan and P. Wang, Inorg. Chem., 2017, 56, 4065–4071 CrossRef CAS PubMed; (i) A. Baba, T. Kojima and S. Hiraoka, Chem.–Eur. J., 2018, 24, 754 CrossRef CAS; (j) S. Kai, V. Martí-Centelles, Y. Sakuma, T. Mashiko, T. Kojima, U. Nagashima, M. Tachikawa, P. J. Lusby and S. Hiraoka, Chem.–Eur. J., 2018, 24, 663–671 CrossRef CAS PubMed; (k) T. Kai, M. Kishimoto, M. Akita and M. Yoshizawa, Chem. Commun., 2018, 54, 956–959 RSC; (l) S. Prusty, K. Yazaki, M. Yoshizawa and D. K. Chand, Chem.–Eur. J., 2017, 23, 12456–12461 CrossRef CAS PubMed; (m) A. Ahmedova, D. Momekova, M. Yamashina, P. Shestakova, G. Momekov, M. Akita and M. Yoshizawa, Chem.–Asian J., 2016, 11, 474–477 CrossRef CAS PubMed; (n) Z. Chen, Y.-T. Chan, D. Miyajima, T. Kajitani, A. Kosaka, T. Fukushima, J. M. Lobez and T. Aida, Nat. Commun., 2016, 7, 13640 CrossRef CAS PubMed; (o) G.-T. Xu, L.-L. Wu, X.-Y. Chang, T. W. H. Ang, W.-Y. Wong, J.-S. Huang and C.-M. Che, Angew. Chem., Int. Ed., 2019, 58, 16297–16306 CrossRef CAS PubMed; (p) Z. Li, Y. Sei, M. Akita and M. Yoshizawa, Chem.–Asian J., 2014, 9, 1016–1019 CrossRef CAS PubMed; (q) S. Hiraoka, T. Nakamura, M. Shiro and M. Shionoya, J. Am. Chem. Soc., 2010, 132, 13223–13225 CrossRef CAS PubMed; (r) S. Hiraoka, Y. Yamauchi, R. Arakane and M. Shionoya, J. Am. Chem. Soc., 2009, 131, 11646–11647 CrossRef CAS PubMed; (s) S. Hiraoka, Y. Kubota and M. Fujita, Chem. Commun., 2000, 1509–1510 RSC; (t) S. Hiraoka and M. Fujita, J. Am. Chem. Soc., 1999, 121, 10239–10240 CrossRef CAS.
- (a) N. Jiang, Z. Yuan, T. Li, Y. Zhu, Y.-S. Chen, L. Lin, J. Zhang, Y.-T. Chan and J. Wang, J. Org. Chem., 2018, 83, 4824–4830 CrossRef CAS PubMed; (b) C. Wang, X.-Q. Hao, M. Wang, C. Guo, B. Xu, E. N. Tan, Y.-Y. Zhang, Y. Yu, Z.-Y. Li, H.-B. Yang, M.-P. Song and X. Li, Chem. Sci., 2014, 5, 1221–1226 RSC; (c) L.-J. Chen, G.-Z. Zhao, B. Jiang, B. Sun, M. Wang, L. Xu, J. He, Z. Abliz, H. Tan, X. Li and H.-B. Yang, J. Am. Chem. Soc., 2014, 136, 5993–6001 CrossRef CAS PubMed; (d) M. Wang, C. Wang, X.-Q. Hao, J. Liu, X. Li, C. Xu, A. Lopez, L. Sun, M.-P. Song, H.-B. Yang and X. Li, J. Am. Chem. Soc., 2014, 136, 6664–6671 CrossRef CAS; (e) B. Sun, M. Wang, Z. Lou, M. Huang, C. Xu, X. Li, L.-J. Chen, Y. Yu, G. L. Davis, B. Xu, H.-B. Yang and X. Li, J. Am. Chem. Soc., 2015, 137, 1556–1564 CrossRef CAS PubMed; (f) G.-F. Huo, X. Shi, Q. Tu, Y.-X. Hu, G.-Y. Wu, G.-Q. Yin, X. Li, L. Xu, H.-M. Ding and H.-B. Yang, J. Am. Chem. Soc., 2019, 141, 16014–16023 CrossRef CAS PubMed; (g) J.-H. Tang, R. Ni, Y.-Q. He, R. T. Vanderlinden, Y. Li, B. Shi, Z.-Y. Li, H. Wang, X. Li, Y. Sun, Y.-W. Zhong and P. J. Stang, Inorg. Chem., 2019, 58, 13376–13381 CrossRef CAS; (h) Y.-Q. He, W. Fudickar, J.-H. Tang, H. Wang, X. Li, J. Han, Z. Wang, M. Liu, Y.-W. Zhong, T. Linker and P. J. Stang, J. Am. Chem. Soc., 2020, 142, 2601–2608 CrossRef CAS PubMed.
- (a) J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC; (b) J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS; (c) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS; (d) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- (a) Z. Zhao, J. W. Y. Lam and B. Z. Tang, J. Mater. Chem., 2012, 22, 23726–23740 RSC; (b) Y. Dong, J. W. Y. Lam, A. Qin, J. Liu, Z. Li, B. Z. Tang, J. Sun and H. S. Kwok, Appl. Phys. Lett., 2007, 91, 011111 CrossRef; (c) A. Qin, C. K. W. Jim, Y. Tang, J. W. Y. Lam, J. Liu, F. Mahtab, P. Gao and B. Z. Tang, J. Phys. Chem. B, 2008, 112, 9281–9288 CrossRef CAS; (d) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC; (e) V. S. Vyas and R. Rathore, Chem. Commun., 2010, 46, 1065–1067 RSC; (f) C. Zhang, Y. Li, X. Xue, P. Chu, C. Liu, K. Yang, Y. Jiang, W.-Q. Chen, G. Zou and X.-J. Liang, Chem. Commun., 2015, 51, 4168–4171 RSC; (g) V. M. Suresh, A. De and T. K. Maji, Chem. Commun., 2015, 51, 14678–14681 RSC; (h) G. Yu, G. Tang and F. Huang, J. Mater. Chem. C, 2014, 2, 6609–6617 RSC; (i) J. Shi, N. Chang, C. Li, J. Mei, C. Deng, X. Luo, Z. Liu, Z. Bo, Y. Q. Dong and B. Z. Tang, Chem. Commun., 2012, 48, 10675–10677 RSC.
- (a) J. Zhao, D. Yang, Y. Zhao, X.-J. Yang, Y.-Y. Wang and B. Wu, Angew. Chem., Int. Ed., 2014, 53, 6632–6636 CrossRef CAS PubMed; (b) N. B. Shustova, T.-C. Ong, A. F. Cozzolino, V. K. Michaelis, R. G. Griffin and M. Dincă, J. Am. Chem. Soc., 2012, 134, 15061–15070 CrossRef CAS; (c) Z. Wei, Z.-Y. Gu, R. K. Arvapally, Y.-P. Chen, R. N. McDougald, J. F. Ivy, A. A. Yakovenko, D. Feng, M. A. Omary and H.-C. Zhou, J. Am. Chem. Soc., 2014, 136, 8269–8276 CrossRef CAS PubMed; (d) M. Zhang, G. Feng, Z. Song, Y.-P. Zhou, H.-Y. Chao, D. Yuan, T. T. Y. Tan, Z. Guo, Z. Hu, B. Z. Tang, B. Liu and D. Zhao, J. Am. Chem. Soc., 2014, 136, 7241–7244 CrossRef CAS PubMed; (e) Q. Zhang, J. Su, D. Feng, Z. Wei, X. Zou and H.-C. Zhou, J. Am. Chem. Soc., 2015, 137, 10064–10067 CrossRef CAS PubMed.
- (a) X. Yan, T. R. Cook, P. Wang, F. Huang and P. J. Stang, Nat. Chem., 2015, 7, 342–348 CrossRef CAS PubMed; (b) X. Yan, M. Wang, T. R. Cook, M. Zhang, M. L. Saha, Z. Zhou, X. Li, F. Huang and P. J. Stang, J. Am. Chem. Soc., 2016, 138, 4580–4588 CrossRef CAS PubMed; (c) Y. Tian, X. Yan, M. L. Saha, Z. Niu and P. J. Stang, J. Am. Chem. Soc., 2016, 138, 12033–12036 CrossRef CAS PubMed; (d) M. Zhang, S. Li, X. Yan, Z. Zhou, M. L. Saha, Y.-C. Wang and P. J. Stang, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 11100 CrossRef CAS PubMed; (e) G. Yu, M. Zhang, M. L. Saha, Z. Mao, J. Chen, Y. Yao, Z. Zhou, Y. Liu, C. Gao, F. Huang, X. Chen and P. J. Stang, J. Am. Chem. Soc., 2017, 139, 15940–15949 CrossRef CAS; (f) X. Yan, H. Wang, C. E. Hauke, T. R. Cook, M. Wang, M. L. Saha, Z. Zhou, M. Zhang, X. Li, F. Huang and P. J. Stang, J. Am. Chem. Soc., 2015, 137, 15276–15286 CrossRef CAS; (g) W. Zheng, G. Yang, S.-T. Jiang, N. Shao, G.-Q. Yin, L. Xu, X. Li, G. Chen and H.-B. Yang, Mater. Chem. Front., 2017, 1, 1823–1828 RSC.
- (a) P. Das, A. Kumar, P. Howlader and P. S. Mukherjee, Chem.–Eur. J., 2017, 23, 12565–12574 CrossRef CAS PubMed; (b) G.-Q. Yin, H. Wang, X.-Q. Wang, B. Song, L.-J. Chen, L. Wang, X.-Q. Hao, H.-B. Yang and X. Li, Nat. Commun., 2018, 9, 567 CrossRef PubMed; (c) N. Liu, T. Lin, M. Wu, H.-K. Luo, S.-L. Huang and T. S. A. Hor, J. Am. Chem. Soc., 2019, 141, 9448–9452 CrossRef CAS PubMed; (d) Y. Li, Y.-Y. An, J.-Z. Fan, X.-X. Liu, X. Li, F. E. Hahn, Y.-Y. Wang and Y.-F. Han, Angew. Chem., Int. Ed., 2020, 59, 10073–10080 CrossRef CAS PubMed; (e) P. C. Purba, S. Bhattacharyya, M. Maity, S. Mukhopadhyay, P. Howlader and P. S. Mukherjee, Chem. Commun., 2019, 55, 8309–8312 RSC; (f) W.-J. Fan, B. Sun, J. Ma, X. Li, H. Tan and L. Xu, Chem.–Eur. J., 2015, 21, 12947–12959 CrossRef CAS PubMed; (g) L.-J. Chen, Y.-Y. Ren, N.-W. Wu, B. Sun, J.-Q. Ma, L. Zhang, H. Tan, M. Liu, X. Li and H.-B. Yang, J. Am. Chem. Soc., 2015, 137, 11725–11735 CrossRef CAS PubMed.
- M. Zhang, S. Yin, J. Zhang, Z. Zhou, M. L. Saha, C. Lu and P. J. Stang, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 3044 CrossRef CAS PubMed.
- (a) Z. Zhang, Z. Zhao, Y. Hou, H. Wang, X. Li, G. He and M. Zhang, Angew. Chem., Int. Ed., 2019, 58, 8862–8866 CrossRef CAS PubMed; (b) K. Acharyya, S. Bhattacharyya, H. Sepehrpour, S. Chakraborty, S. Lu, B. Shi, X. Li, P. S. Mukherjee and P. J. Stang, J. Am. Chem. Soc., 2019, 141, 14565–14569 CrossRef CAS PubMed; (c) Z. Wang, X. He, T. Yong, Y. Miao, C. Zhang and B. Zhong Tang, J. Am. Chem. Soc., 2020, 142, 512–519 CrossRef CAS PubMed; (d) Z. Zhang, Z. Zhao, L. Wu, S. Lu, S. Ling, G. Li, L. Xu, L. Ma, Y. Hou, X. Wang, X. Li, G. He, K. Wang, B. Zou and M. Zhang, J. Am. Chem. Soc., 2020, 142, 2592–2600 CrossRef CAS; (e) L.-J. Wang, X. Li, S. Bai, Y.-Y. Wang and Y.-F. Han, J. Am. Chem. Soc., 2020, 142, 2524–2531 CrossRef CAS PubMed; (f) C.-B. Huang, L. Xu, J.-L. Zhu, Y.-X. Wang, B. Sun, X. Li and H.-B. Yang, J. Am. Chem. Soc., 2017, 139, 9459–9462 CrossRef CAS PubMed.
- (a) S. Shanmugaraju and P. S. Mukherjee, Chem.–Eur. J., 2015, 21, 6656–6666 CrossRef CAS; (b) Z. Zhou, D.-G. Chen, M. L. Saha, H. Wang, X. Li, P.-T. Chou and P. J. Stang, J. Am. Chem. Soc., 2019, 141, 5535–5543 CrossRef CAS PubMed; (c) X. Chang, Z. Zhou, C. Shang, G. Wang, Z. Wang, Y. Qi, Z.-Y. Li, H. Wang, L. Cao, X. Li, Y. Fang and P. J. Stang, J. Am. Chem. Soc., 2019, 141, 1757–1765 CrossRef CAS PubMed; (d) Z. Yue, H. Wang, Y. Li, Y. Qin, L. Xu, D. J. Bowers, M. Gangoda, X. Li, H.-B. Yang and Y.-R. Zheng, Chem. Commun., 2018, 54, 731–734 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Wallingford CT, 2009.
- J. Tang, G. Grampp, Y. Liu, B.-X. Wang, F.-F. Tao, L.-J. Wang, X.-Z. Liang, H.-Q. Xiao and Y.-M. Shen, J. Org. Chem., 2015, 80, 2724–2732 CrossRef CAS PubMed.
- J. Mateos, F. Rigodanza, A. V. PeÇaloza, A. Sartorel, M. Natali, T. Bortolato, G. Pelosi, X. Companyl, M. Bonchio and L. D. Amico, Angew. Chem., Int. Ed., 2020, 59, 1302–1312 CrossRef CAS PubMed.
- Z. Liang, S. Xu, W. Tian and R. Zhang, Beilstein J. Org. Chem., 2015, 11, 425–430 CrossRef CAS PubMed.
- (a) Y. Abderrazak, J. Organomet. Chem., 2020, 920, 121335 CrossRef CAS; (b) X. –L. Yang, J. –D. Guo, T. Lei, B. Chen, C.-H. Tung and L.-Z. Wu, Org. Lett., 2018, 20, 2916–2920 CrossRef CAS PubMed; (c) T. P. Nicholls, G. E. Constable, J. C. Robertson, M. G. Gardiner and A. C. Bissember, ACS Catal., 2016, 6, 451–457 CrossRef CAS; (d) M. Nishino, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2011, 76, 6447–6451 CrossRef CAS PubMed; (e) X. Ju, D. Li, W. Li, W. Yu and F. Bian, Adv. Synth. Catal., 2012, 354, 3561–3567 CrossRef CAS.
- (a) R. Li, J. Byun, W. Huang, C. Ayed, L. Wang and K. A. I. Zhang, ACS Catal., 2018, 8, 4735–4750 CrossRef CAS; (b) Z. J. Wang, S. Ghasimi, K. Landfester and K. A. I. Zhang, Adv. Synth. Catal., 2016, 358, 2576–2582 CrossRef CAS.
- J.-T. Guo, D.-C. Yang, Z. Guan and Y.-H. He, J. Org. Chem., 2017, 82, 1888–1894 CrossRef CAS PubMed.
- C.-W. Hsu and H. Sunden, Org. Lett., 2018, 20, 2051–2054 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc00097g |
| This journal is © The Royal Society of Chemistry 2021 |