
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Plasmonic photothermal catalysis for solar-to-fuel conversion: current status and prospects
Shunqin
Luo
ab,
Xiaohui
Ren
ab,
Huiwen
Lin
ac,
Hui
Song
*a and
Jinhua
Ye
 *abd
*abd
aInternational Center for Materials Nanoarchitectonics (WPI-MANA), National Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan. E-mail: song.hui@nims.go.jp; Jinhua.YE@nims.go.jp
bGraduate School of Chemical Sciences and Engineering, Hokkaido University, Sapporo 060-0814, Japan
cJiangsu Key Laboratory of Materials and Technology for Energy Conversion, Nanjing University of Aeronautics and Astronautics, Nanjing 210016, P. R. China
dTJU-NIMS International Collaboration Laboratory, School of Material Science and Engineering, Tianjin University, Tianjin 300072, P. R. China
First published on 12th March 2021
Abstract
Solar-to-fuel conversion through photocatalytic processes is regarded as promising technology with the potential to reduce reliance on dwindling reserves of fossil fuels and to support the sustainable development of our society. However, conventional semiconductor-based photocatalytic systems suffer from unsatisfactory reaction efficiencies due to limited light harvesting abilities. Recent pioneering work from several groups, including ours, has demonstrated that visible and infrared light can be utilized by plasmonic catalysts not only to induce local heating but also to generate energetic hot carriers for initiating surface catalytic reactions and/or modulating the reaction pathways, resulting in synergistically promoted solar-to-fuel conversion efficiencies. In this perspective, we focus primarily on plasmon-mediated catalysis for thermodynamically uphill reactions converting CO2 and/or H2O into value-added products. We first introduce two types of mechanism and their applications by which reactions on plasmonic nanostructures can be initiated: either by photo-induced hot carriers (plasmonic photocatalysis) or by light-excited phonons (photothermal catalysis). Then, we emphasize examples where the hot carriers and phonon modes act in concert to contribute to the reaction (plasmonic photothermal catalysis), with special attention given to the design concepts and reaction mechanisms of the catalysts. We discuss challenges and future opportunities relating to plasmonic photothermal processes, aiming to promote an understanding of underlying mechanisms and provide guidelines for the rational design and construction of plasmonic catalysts for highly efficient solar-to-fuel conversion.
1. Introduction
Solar-energy is the most abundant renewable energy resource, and is one of the most promising candidates in the future renewable energy supply framework.1,2 Using solar-energy with photocatalysts to initiate catalytic reactions, the so-called photocatalytic process, is an emerging trend and regarded as a promising strategy to curtail increasing global energy demand while alleviating environmental issues.3 Adopting solar-energy solely as the energy input, CO2, H2O, and other building blocks can be transformed by photocatalysts into value-added chemicals, demonstrating the concept of solar-to-fuel conversion by efficiently storing the solar-energy in chemical bonds.4,5 Due to the wide band gap and limited light–matter interaction, conventional semiconductor-based photocatalysis faces two major challenges: limited photo harvesting and low apparent quantum efficiency (AQE). In our opinion, extending the range of optical response to improve solar light harvesting is highly desirable, and is considered to be an effective approach for enormously enhancing the net reaction efficiency. Previous work has suggested that plasmonic photocatalysis performs with attractive light absorption and charge carrier utilization features.6,7 In plasmonic photocatalysis, the free electrons in a plasmonic metal nanoparticle can be oscillated collectively under light irradiation through the excitation of localized surface plasmon resonance (LSPR).8 In addition to the electron–hole pairs generated from semiconductor photocatalysts, LSPR can serve as an additional energy input for enhanced photocatalytic performance by converting light energy into a hot carrier. Since the resonant wavelength and SPR intensity are determined by the size, shape, and composition of the plasmonic nanostructures, it is possible to fabricate plasmonic nanometals with full solar-spectrum response.9,10Along with the plasmonic photocatalysts which combine plasmonic metals with semiconductors, utilizing plasmonic metals solely as catalysts has emerged as a cutting-edge research area of solar-to-fuel conversion technology.11 The unique capability of plasmonic metals that combines light-harvesting, hot carrier excitation, photo-to-thermal conversion, and surface catalytic reaction in one material endows them with a bright future for catalyzing various chemical reactions. With the assistance of both hot carriers and photothermal heat, several conventional catalytic processes which were originally driven by thermal-energy can be initiated by solar-energy via plasmonic photothermal technology without additional thermal energy input, even demonstrating superior activity compared with traditional thermocatalysis.12–14 Furthermore, the participation of a hot carrier in the surface reaction can preferentially activate desired chemical reactions through specifically targeting electronic excitations, offering great potential to discover additional and more selective reaction pathways that cannot be accessed by the conventional thermocatalytic process.15 These remarkable achievements strongly indicate that plasmonic photothermal technology that takes advantage of both the photon and the thermal features of sunlight holds great promise for addressing increasing global energy demands through solar-to-fuel conversion.
Previous reviews describing the reaction mechanism of plasmon-mediated photocatalysis and photothermal catalysis are already available, and those published reviews mainly included plasmonic or photothermal catalytic reactions for C1 molecules or conversion of other small molecules.12,16–18 Considering the advantages of plasmonic photothermal catalysis over other photocatalytic processes, an up-to-date perspective on current advances is necessary. In this perspective, recent advances in energetically uphill photosynthetic reactions (primarily including the transformation of chemical feedstocks CO2 and/or H2O into valuable fuels) driven by plasmonic photocatalysis, photothermal catalysis, and plasmonic photothermal catalysis are summarized. First, the concepts and reaction mechanisms of plasmonic photocatalysis, purely photothermal catalysis and plasmonic photothermal catalysis, including hot carrier transfer and photothermal conversion, are briefly introduced. Then, we comprehensively discuss the material design strategies and reaction mechanisms based on different plasmon-mediated endothermic reaction concepts: (i) hot carrier-mediated reaction at ambient temperature where the photo-induced hot carriers directly participate in the reaction without the contribution of photothermal heat (Section 3.1); and (ii) photothermal effect dominated catalytic reaction where most of hot carriers dissipate their energy into heat instead of activating the adsorbates (Section 3.2). Moreover, by elaborately constructing plasmonic photothermal catalysts, dual-functional roles (hot carrier excitation and photothermal heat) can synergistically contribute to the chemical reaction, and we particularly emphasize the important role of plasmonic photothermal catalysis in solar-to-fuel conversion (Section 3.3). The design of catalysts and approaches for improving energy conversion efficiency in this field are discussed. Finally, we conclude with the challenges and opportunities in future research on the current topic. This perspective aims to deliver conceptually novel and insightful views toward highly efficient solar-energy-driven catalytic chemical reactions.
2. Fundamentals of plasmonic photothermal catalysis
Surface plasmon resonance (SPR) is a common optical property of several metallic nanoparticles, which can be visualized as the coherent oscillation of conduction electrons established when the frequency of the photons matches the natural frequency of the surface electrons oscillating against the restoring force of positive nuclei.15 After the intra-band (between the Fermi level and the sp conduction band) or inter-band (between the d band and sp conduction band) excitation of plasmonic nanometals, energetic hot carriers are produced from the dephasing of the free electron oscillation through a non-radiative energy dissipation pathway (also known as the Landau damping process).10 Those hot carriers with sufficient energy can inject into the electron-accepting states of nearby surface adsorbates or semiconductors, inducing a surface chemical transformation (at femtosecond to picosecond level), or dissipate their energy into heat through the energy exchange between hot carriers and phonon modes (at picosecond to nanosecond level) to promote the mass transfer and reaction rate.8,11 Depending on the type of main driving force, the SPR-mediated catalytic process over plasmonic nanostructures can be divided into plasmonic photocatalysis and photothermal catalysis, where the former is dominated by charge carrier activation and the latter is governed by photothermal heat. It is noteworthy that through tailoring the optical properties (such as the energy of LSPR) of the plasmonic metal to match the electronic structure of the adsorbates, hot carrier activation and photothermal heat can be constructively coupled by the plasmonic photothermal catalysts under the irradiation of high-intensity light, ultimately initiating the surface reaction efficiently and selectively. Such a kind of reaction process is defined here as plasmonic photothermal catalysis that combines the advantages of plasmonic photocatalysis and photothermal catalysis. The reaction mechanism of plasmonic photothermal catalysis, including hot carrier transfer and photothermal conversion, is introduced briefly in the following section.2.1 Reaction mechanism of plasmonic photocatalysis
Purely plasmonic photocatalytic reactions are generally conducted at ambient temperature or low reaction temperature (less than 100 °C), where photo-induced hot carriers play a dominant role in the reaction, and the influence of photothermal heat is insignificant during the plasmonic photocatalysis. In the plasmonic photocatalytic process, photo-induced hot electrons can be produced over a plasmonic nanoparticle and transfer to an adjacent semiconductor or interact with the molecules adsorbed on the surface of nanoparticle.19–21 For the plasmonic metal/semiconductor system (Fig. 1a), abundant hot electrons with high potential energy are generated from plasmonic metals via SPR excitation, followed by the efficient transfer of hot electrons to the conduction band (CB) of the interfaced semiconductors.8,22 The Schottky barrier (ΦSB) formed at the semiconductor–metal interface prevents the flowback of the hot electrons, extending the lifetime of the hot electrons and accumulated electrons in the CB of the semiconductor, which is beneficial to various surface chemical reactions, such as H2O splitting and CO2 reduction.23–25 On the other hand, for the plasmonic metal/adsorbate system, photo-induced hot carriers are in a distribution of low-energy charge carriers in high concentration. Hot carriers with a suitable energy level can be injected into the adsorbates through accessible orbitals, which is called an indirect hot carrier transfer mechanism (Fig. 1b).26 In addition, the hot carriers can be directly transferred from metals to adsorbates through a hybridized surface electronic state formed at the interface for activating the reactants, without the formation of the excited electron distribution (Fig. 1c).27–29 With the assistance of hot carriers, both the bond activation and the intermediate conversion can be facilitated through transient electronic excitations after photoexcitation of plasmonic nanostructures. In detail, hot carriers help the adsorbate molecules to evolve on the surface of plasmonic metal after energy transfer from plasmon to adsorbates along the excited potential energy surface, providing additional vibration energy to the adsorbates, and thus facilitating the chemical bond activation.29 The LSPR can be established when the light wavelengths are longer than the size of the plasmonic nanoparticles, and the generation process of photo-induced hot carriers has been described by theoretical calculations.30 By using a free electron mode, Nordlander and co-authors simulated the energy distribution of hot electrons and holes in an Ag nanoparticle, and the results indicated that a higher production rate and lower energy of hot carriers could be achieved over a larger nanoparticle.31 In general, with the increasing size of plasmonic nanoparticles, a red-shift of the LSPR absorption peak could be observed.32 The resonant wavelength and the LSPR intensity are dependent on both the intrinsic nature of plasmonic metals and the geometry of plasmonic nanostructures.9,10 Plasmonic nanostructures with sharp features, such as the corners of nanocubes and tips of nanocones, demonstrate significantly higher local electromagnetic field enhancements compared with spherical nanoparticles of similar size.33 Due to the lightning rod effect and reduced radiative damping, sharp features of plasmonic nanostructures can concentrate the incident light energy and increase the production of photo-induced hot carriers, which play important roles for activating surface chemical transformations.34 In additions, through changing the size and shape of plasmonic nanostructures, a shift in the electric field density could be induced, changing the oscillation frequency of the electrons, thereby inducing different cross-sections of the LSPR properties. Therefore, modulating the composition and geometry of the plasmonic nanostructures could ultimately result in an enhanced catalytic property.35 Among all the metallic elements, coinage elements (Cu, Ag, and Au) in group IB demonstrate the best SPR effect. The energy thresholds for the inter-band excitation of Cu and Au nanometals are 2.1 and 2.2 eV, respectively, indicating the visible light absorption capacity of Cu and Au.36–38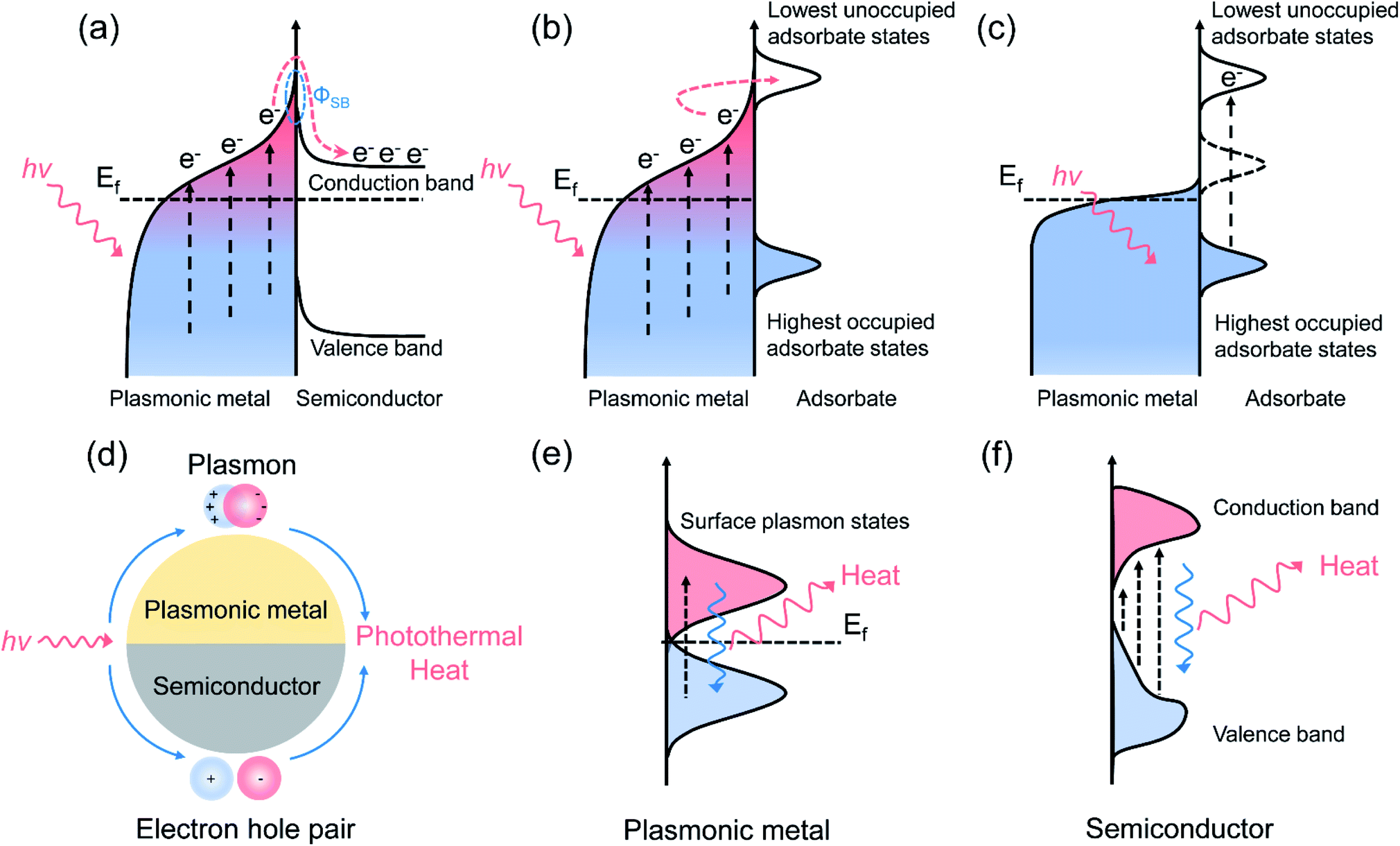 | ||
| Fig. 1 Schematic illustrations of the hot carrier transfer pathway and photothermal conversion. (a) A hot electron transfer process at a plasmonic metal/semiconductor interface. (b) The indirect hot carrier transfer mechanism. (c) The direct hot carrier transfer mechanism. (d) The photo-induced thermal effect of a plasmonic metal and semiconductor via plasmon- and exciton-based processes. (e) The photothermal conversion process over a plasmonic metal. (f) The photothermal conversion process over a semiconductor with LSPR characteristics. | ||
2.2 Reaction mechanism of pure photothermal catalysis
Solar-energy can also be converted into heat through photothermal conversion over plasmonic materials, providing sufficient thermal energy to stimulate the catalytic reactions (Fig. 1d).39 For plasmonic metallic nanostructures, upon light irradiation, the oscillation of conduction electrons dephases and produces hot carriers. An athermal charge-carrier distribution then is formed.40,41 Afterwards, hot carriers cool down through transferring energy to lattice phonons. That is, those energetic charge carriers which do not participate in the charge migration process of the surface reaction would finally dissipate their energy to the phonon modes, inducing a considerable temperature increase on the surface of the plasmonic nanometals (Fig. 1e).15,42 During the photothermal conversion process, the heat can be dissipated through particle–media interfaces after electron–phonon interaction. Therefore, the photothermal conversion efficiency is dependent on the medium and the particle size of the plasmonic nanostructures. Generally, reducing the particle size of plasmonic nanometals can lead to an enhancement in the photothermal conversion efficiency.43 For semiconductor nanomaterials with LSPR characteristics, the photo-induced electrons fall back to the lower energy states through releasing energy into photons or phonons, where the latter can interact with the lattice to produce heat (Fig. 1f).44 Assuming that most of the carriers' energy is converted into heat through energy exchange between electrons and phonons, the reaction obeys the same mechanism as conventional thermocatalytic reactions.45 Unlike plasmonic photocatalysis, pure photothermal catalytic processes generally occur in gas-phase reactions, and light with high intensity (orders of magnitude higher than solar flux) is required to produce high temperatures to initiate the chemical reactions. In pure photothermal catalysis, solar-energy is merely converted into thermal energy, and the activation of surface adsorbates by hot carrier is negligible.2.3 Reaction mechanism of plasmonic photothermal catalysis
In plasmonic photothermal catalysis, through the rational design of plasmonic catalysts, hot carrier-driven reactant activation and photothermal heating co-exist and can be combined to synergistically drive the surface chemical reactions.13,14,46 As described above, initially formed hot carriers after light excitation could redistribute their energy through different plasmon decay pathways.11 Through electron–electron collisions, light-excited charge carriers over plasmonic metals could dissipate their energy, resulting in the secondary excitation of charge carriers; and this could lead to a Fermi–Dirac distribution of hot carriers near the Fermi level of plasmonic metals.15 High-energy charge carriers from such a distribution can participate in the surface chemical reactions through exchanging energy between plasmonic nanostructures and surface adsorbed reactants.15 Alternatively, adsorbate electronic states induced by strong interaction between plasmonic metals and adsorbates can serve as an additional channel for directly dissipating the LSPR energy.29 The prerequisites for realizing such hot electron activation processes have several aspects. First, accessible adsorbate orbitals must exist that could match with the electronic structure of plasmonic metals, or the presence of interfacial electronic states through the strong interaction between adsorbate and plasmonic metal that would enable an ultra-fast and momentum-reserved direct electronic excitation.11 In addition, an intense electric field at the surface of plasmonic metals after light irradiation, which can reroute the charge excitation process, is also considered a precondition for such an energy exchange process.11 Consequently, hot carriers can facilitate the surface reaction through the following reaction roles: (1) activating specific chemical bonds of surface reactants to modify the reaction pathway, which can enhance the selectivity and suppress unintended side reactions;47 (2) accelerating the desorption of certain surface-adsorbed species, which can further promote the catalytic activity and prolong the stability by resisting the coking of metallic nanoparticles.48 The residual hot carriers that do not participate in the surface catalytic reaction could subsequently cool down by transferring their energy to phonon modes, resulting in heating of the plasmonic metal surface.11 Photo-induced heat can also serve as an effective driving force for stimulating the surface reaction through the following functions: (1) exciting the phonon modes of the plasmonic nanoparticle, which could couple with a reaction coordinate, evolving the surface-adsorbed reactants to products;12 (2) promoting the adsorption–desorption process of the surface catalytic process;39 (3) shifting the chemical equilibria toward a higher yield of end products. Compared with plasmonic catalysis and photothermal catalysis, plasmonic photothermal catalysis is much more attractive since both the light and thermal features of solar light are concurrently utilized, resulting in highly efficient solar-to-energy conversion. Under such circumstances, the dual-functional roles of the plasmonic nanostructures (hot carrier-driven reactant activation and photothermal conversion) should be systematically considered and carefully differentiated.There are several distinct signatures that can experimentally differentiate the contribution of hot carriers from photothermal heat: (1) a linear dependence of the reaction rate on light intensity indicates that the surface reaction is induced by hot carriers, and this can be attributed to the fact that the generation rate of a hot carrier increases linearly with the photon flux;15 (2) a higher kinetic isotope effect compared with the reaction driven by thermal energy at same reaction temperature;49 (3) modified selectivity for the chemical reaction induced by photo energy compared with the same reaction induced by thermal energy;48 (4) similar tendencies of apparent quantum efficiency and the optical absorption spectrum of plasmonic metal.50 The contribution of photo-induced hot carriers in the situation where the hot carriers and photothermal heat work synergistically can be evaluated through precisely designed contrast experiments.14 More specifically, through comparing the catalytic activity under purely solar-driven conditions (without additional thermal energy input) and under thermocatalytic condition (without irradiation of light) with external heating temperature equivalent to those achieved under illumination, the contribution of hot carriers can be differentiated quantitively.14 It should be noted that the surface temperature of the catalyst needs to be detected accurately in such a process.51
2.4 Solar-to-energy conversion efficiency
In order to evaluate the efficiency of plasmonic photothermal catalytic systems, two kinds of efficiency, i.e., solar-to-fuel efficiency (STF) and apparent quantum efficiency (AQE), are introduced. For a thermodynamically uphill reaction, STF is an ideal method to calculate the conversion and storage efficiency of the solar light, whereas the AQE is suitable for both thermodynamically uphill and downhill reactions since it involves only the number of reacted electrons.STF can be calculated using eqn (1).
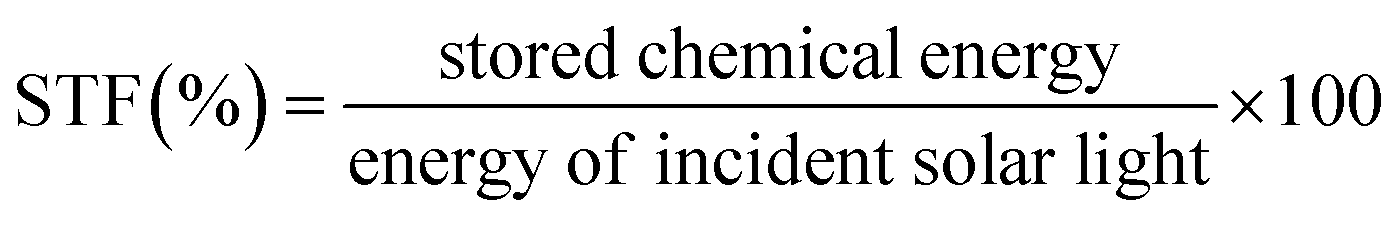 | (1) |
AQE can be calculated by using eqn (2).
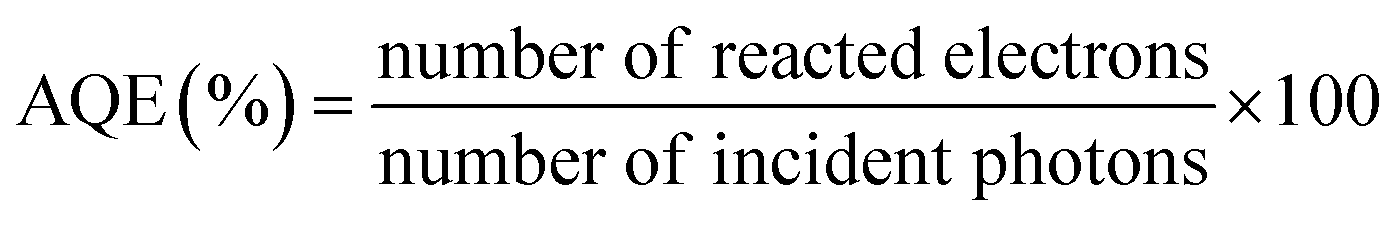 | (2) |
3. Solar-driven uphill reactions on plasmonic catalysts
3.1 Plasmonic photocatalysis
Metal nanoparticles with strong light scattering ability can generate hot electrons under ultraviolet, visible, and near-infrared light irradiation through the decay of surface plasmons. By applying hot electrons as the driving force to initiate the activation of adsorbates or intermediates, the performance of photocatalytic reactions can be greatly enhanced through two different mechanisms, as previously proposed: (1) enhanced light scattering with the presence of metals can improve the light trapping and absorption of semiconductors. Increasing the electron density in the metal can lead to near-field electric enhancement with increased interfacial charge transfer capacity; (2) optically excited hot electrons with sufficient energy can be injected into the conduction band of the semiconductor by overcoming the Schottky barrier and can take part in the reaction.52 With the assistance of hot carriers, various thermodynamically unfavorable reactions can be initiated effectively even at ambient temperature. In this section, we focus mainly on chemical reactions driven by photo-induced hot carriers, primarily plasmonic overall H2O splitting and CO2 reduction with H2O without the participation of photo-induced thermal energy.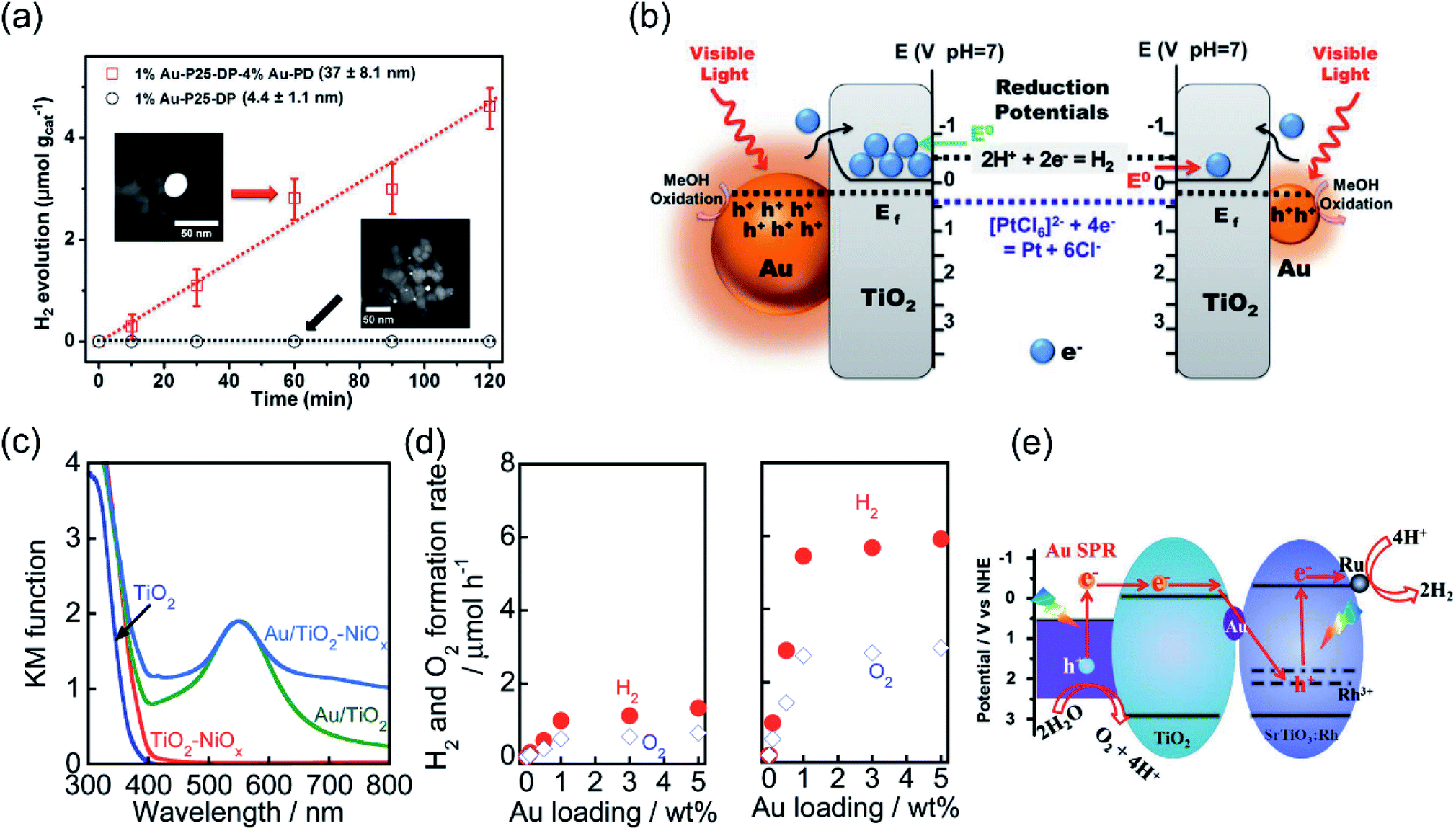 | ||
| Fig. 2 Plasmonic photocatalysis for overall water splitting. (a) The H2 evolution performance over Au–TiO2 with small-size and large-size Au under λ > 435 nm irradiation. (b) The proposed mechanism for the plasmon-mediated electron transfer process. (a and b) Reproduced from ref. 21 with permission from the American Chemical Society, Copyright 2014. (c) Light absorption spectra of TiO2, TiO2–NiOx, Au/TiO2, and Au/TiO2–NiOx photocatalysts. (d) A comparison of Au–TiO2 and Ni-modified Au–TiO2 with different Au loading amounts for overall water splitting. (c and d) Reproduced from ref. 56 with permission from the Royal Society of Chemistry, Copyright 2017. (e) The proposed Z-scheme electron transfer model for pure water splitting. (e) Reproduced from ref. 57 with permission from Elsevier, Copyright 2017. | ||
Further constructing a Z-scheme system based on Au–TiO2 has been proposed by Wang et al.57 Stable H2 (5.6 μmol h−1 g−1) and O2 (2.7 μmol h−1 g−1) evolution was achieved on Au–TiO2/Rh–SrTiO3 under visible light irradiation. Benefiting from electron accumulation, the plasmon-induced hot electrons generated in Au can transfer through the Z-scheme electron pathway and recombine with the holes in SrTiO3, as illustrated in Fig. 2e, which may lead to a decrease in surface potential and efficient charge separation for achieving overall water splitting. By controlling the length and width (aspect ratio) of Au nanorods, the SPR absorption range can be extended from the visible (600 nm) to the longer wavelength NIR (1200 nm) region. Elbanna et al. prepared Au nanorods with different aspect ratios (2.3, 3.2, 4.8 and 7.5) through a seed-growth method.58 After loading Au nanorods on TiO2 mesocrystals, the longitudinal SPR absorption peak red shifted from 660 to 975 nm, resulting in higher H2 production rate (924 μmol h−1 g−1) and prolonged electron lifetime under Vis-NIR light irradiation.
Recent studies have shown that the LSPR excitation of Au–CdS hybrids also assisted electron transfer between the interface of Au and CdS. Naya et al. prepared selective deposited hexagonal CdS (shell) above Au nanoparticles (core) to form an Au–CdS heteroepitaxial junctional structure (Fig. 3a and b) with different mean sizes (dAu) and shell thicknesses (lCdS).59 As shown in Fig. 3c, the Au LSPR showed strong damping around 620 nm in contrast to the original Au signal with the peak located at λ = 520 nm. The H2 production activity was controlled by both the lifetime (τ) of SPR-electrons as well as the size of the CdS nanoparticle (Fig. 3d). Au–CdS (dAu = 12.1 nm, lCdS = 2.1 nm) gave a continuous H2 generation rate of (79.2 ± 2.1 μmol h−1 g−1) with an external quantum yield of 0.24% under excitation by red-light (λ = 640 nm). Benefitting from SPR, hot electrons generated from Au under red-light irradiation can transfer to CdS, endowing a stable stoichiometric water splitting of H2 and O2 (with production ratio 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) for more than 200 h (Fig. 3e and f). The above study further verified that the geometry and the dimension of the plasmonic nanostructures determine the overall efficiency toward water splitting.
:1) for more than 200 h (Fig. 3e and f). The above study further verified that the geometry and the dimension of the plasmonic nanostructures determine the overall efficiency toward water splitting.
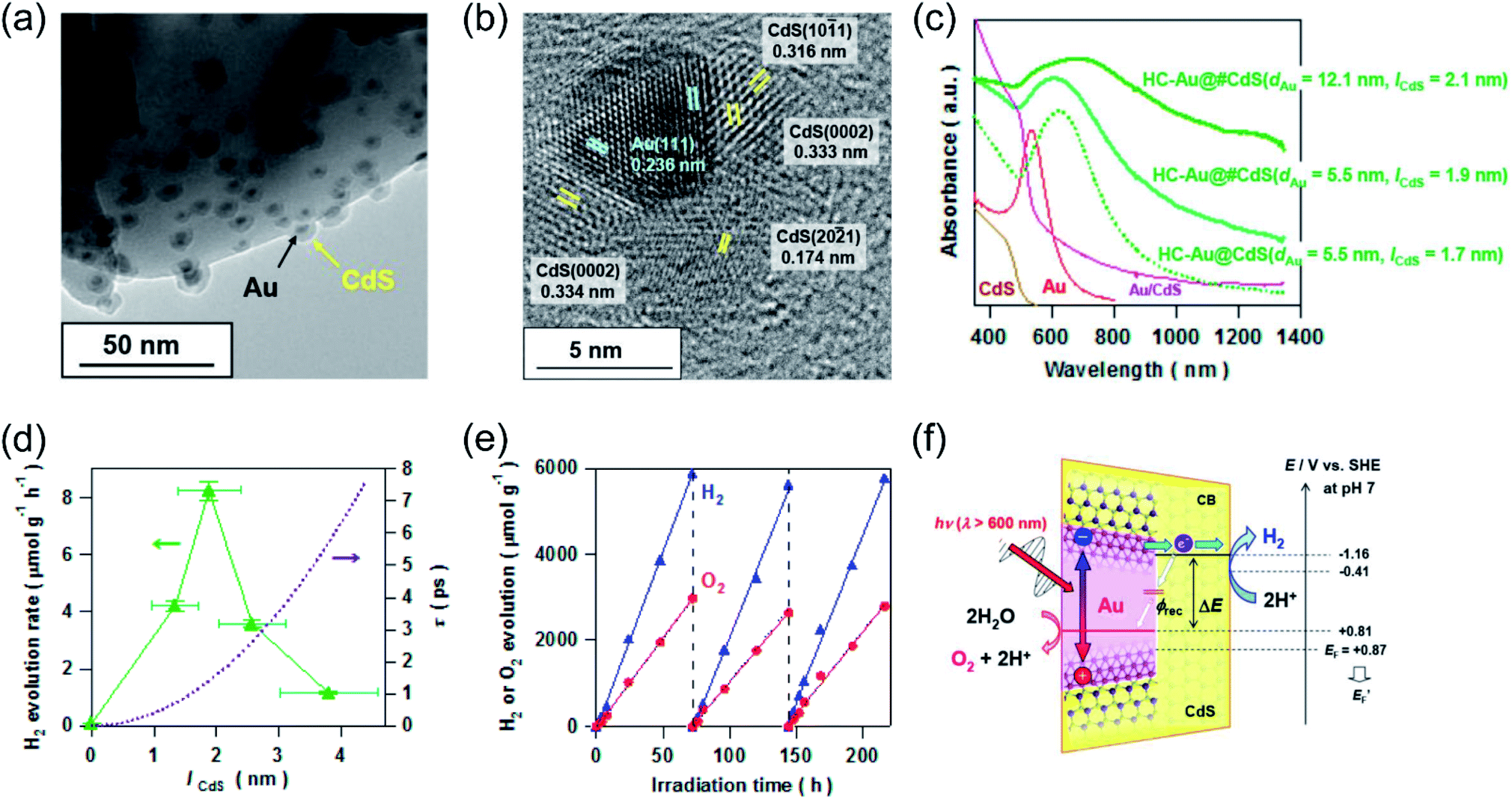 | ||
| Fig. 3 Plasmonic photocatalysis for overall water splitting. (a) and (b) TEM images of a ZnO supported Au@CdS heteroepitaxial junctional structure. (c) UV-vis-NIR absorption spectra of Au@CdS. (d) H2 evolution rate and electron transit time values in relation to CdS thickness. (e) Stability test over the optimal Au@CdS sample under red-light illumination. (f) The proposed LSPR-induced hot electron transfer mechanism. (a–f) Reproduced from ref. 59 with permission from the American Chemical Society, Copyright 2018. | ||
In addition to Au, Ag-modified TiO2 exhibited an absorption peak in the visible region, and the bandgap was reduced from 3.0 to 2.5 eV due to the SPR effect. H2 generation from pure water was dramatically enhanced over Ag-modified TiO2 (470 μmol h−1 g−1) in comparison to bare TiO2 (206 μmol h−1 g−1) under UV light. The co-existence of metallic Ag and Ti–Ag–O species can trigger the SPR effect and form a new electron pathway for efficient electron–hole pair separation.60
Whilst the physical properties of LSPR effects are well understood, the reaction mechanisms of every process in the overall reaction over the real surfaces of catalysts are still controversial. In order to explain interfacial charge dynamics in photocatalytic reactions over plasmonic metal/semiconductor nanocomposites, a combination of in situ near ambient pressure XPS measurements and time-resolved optoelectronic characterization were conducted by O'Shea and co-authors.67 In their work, the reaction mechanism of photoreduction of CO2 using H2O as electron donor over plasmonic Ag/TiO2 nanostructures was systematically investigated. Under UV-light irradiation, an approximately 15-fold enhancement in the CH4 generation rate over Ag/TiO2 compared with bare TiO2 was found (Fig. 4a). In contrast, under visible light irradiation, the main product was shifted to CH3OH over Ag/TiO2 (Fig. 4b). Based on the operando spectroscopic measurements and theoretical calculations, the authors indicated that the surface sub-band gap states formed between the plasmonic Ag nanoparticles and TiO2 through interfacial dielectric coupling was an important factor to promote the separation of photo-induced charge carriers (Fig. 4c). The production of CH4 over Ag/TiO2 under UV light irradiation can be attributed to the electron-scavenging ability of Ag nanoparticles, resulting in an increased lifetime of photo-induced charge carriers compared to the original TiO2 (Fig. 4d and e). Under UV light irradiation, photo-induced electrons from TiO2 could be scavenged by Ag nanoparticles, while the holes could be accumulated at the valence band of TiO2, resulting in the spatial separation of photo-induced electron–hole pairs and an increased lifetime. The accumulation of photo-induced electrons on the surface of the Ag nanoparticle can facilitate the production of electron-demanding products (CH4) over Ag/TiO2. Furthermore, the longer lifetime of photo-induced electron–hole pairs enabled water to be the electron source for CO2 reduction (Fig. 4d). In situ near ambient pressure XPS measurements further verified the formation of C–H intermediates, indicating that the reaction pathway followed the carbene mechanism. Under visible light irradiation, photo-induced electronic transitions from intra-band gap surface states were induced, generating charge carriers in the near-surface region. Compared with the charge-carriers induced by UV light irradiation, those visible light-induced carriers showed shorter-lived transient absorption decay (Fig. 4f), shifting the reaction selectivity to lower electron-demanding products (CH3OH). This work provides an unequivocal explanation of the interfacial charge dynamics and a basic understanding of each process in the CO2 photoreduction.
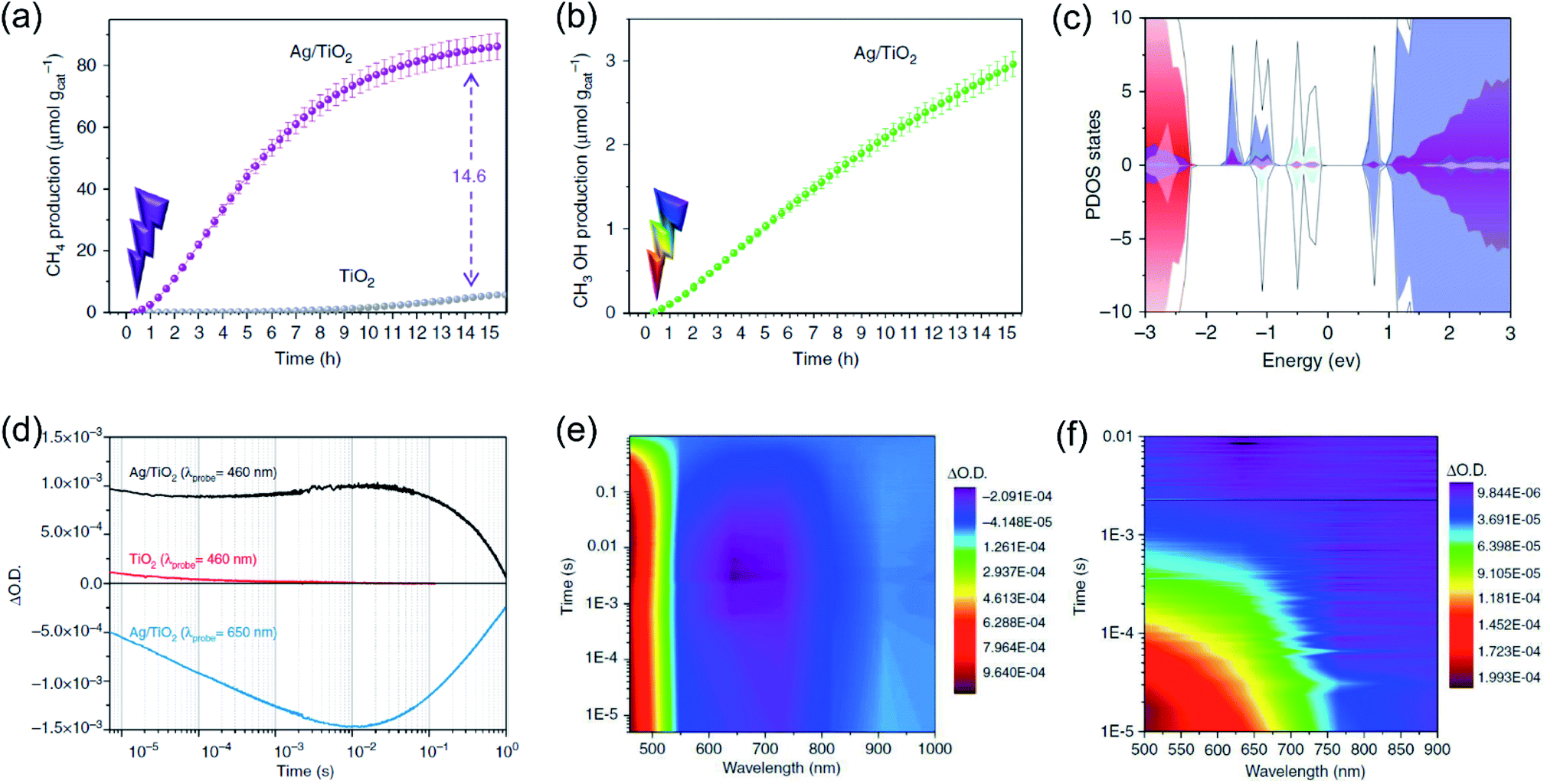 | ||
| Fig. 4 Plasmonic photocatalysis for CO2 reduction. (a) The cumulative production of CH4 under 365 nm irradiation. (b) The cumulative production of CH3OH under 450 nm irradiation. (c) Total DOS (black) and atom-projected DOS (PDOS) for Ag 5s (light blue), Ag 5d (grey), and Ti 3d (purple). (d) Temporal profiles of the transient absorption decay of Ag/TiO2 and TiO2 films after excitation at 355 nm. (e) A two-dimensional plot of the transient absorption spectrum of Ag/TiO2 film after UV excitation. (f) A two-dimensional plot of the transient absorption spectrum of Ag/TiO2 film excited with visible light. (a–f) Reproduced from ref. 67 with permission from Springer Nature, Copyright 2018. | ||
Very recently, Sharma and co-authors reported that a strong endothermic reaction CO2 + C → 2CO which requires a high activation energy (3.1 eV) can be realized at ambient reaction temperature, and the LSPR energy of Al in the deep-UV range was used as the sole driving force to initiate the reaction without additional thermal energy input.68 The localized surface plasmon (LSP) mode of the Al nanoparticle was excited by an electron beam in an environmental scanning transmission electron microscope. The presence of LSP modes was characterized by using the electron energy loss spectroscopy (EELS) spectrum, and the spatial distribution was stimulated by using the discontinuous Galerkin time-domain (DGTD) method. The reaction product was detected by in situ gas chromatography-mass spectrometry (GC-MS). The reaction rate was estimated by measuring carbon gasification near the Al nanoparticle. Under electron beam excitation, an electric field can be induced on the side of the Al nanoparticle. After electron beam illumination, a new receded edge of the graphite flake was formed, which may prove the consumption of carbon during the reaction. From the GC-MS measurements, measurable CO signals were detected both after electron beam illumination at room temperature and after heating at a reaction temperature of 900 °C, confirming that the reaction did happen in both cases. The authors proposed that the plasmon of Al with high energy played a dominant role in driving the reaction at ambient temperature. In this work, a direct excitation and observation of electron-excited LSPR of a plasmonic Al nanoparticle were realized by using an environmental transmission electron microscope. In such a process, both the spatial distribution of LSP modes and the dynamic behavior of the plasmonic nanoparticles are characterized simultaneously. This work demonstrates a new path towards driving other industrially tough reactions at ambient temperature by using a plasmon.
Currently, it is of great interest to use noble metals as plasmonic materials not only for improving light absorption capacity but also for inducing hot carriers to promote catalytic performance over plasmonic photocatalysis. By modulating the species, size, and geometry of plasmonic nanostructures, enhanced photocatalytic activity can be observed. It should be noted that isotope measurements are necessary to confirm that the products indeed come from the reactants, especially when the amounts of the products are quite low.
3.2 Photothermal catalysis
Another strategy to convert solar-energy into chemical energy is via photo-thermochemistry, which utilizes the full spectrum of sunlight to increase the surface temperatures of catalysts and then initiate thermocatalytic reactions.69 Under these circumstances, photo-induced hot carriers play an insignificant role in activating the surface adsorbed reactants, but dissipate their energy into heat through energy exchange between electrons and phonons. Group VIII nanometals are considered promising candidates for photothermal catalysis because of their outstanding photothermal conversion capacity and catalytic property, although their SPR intensities are relatively weak.70 Due to the high photothermal conversion efficiency of plasmonic photothermal catalysts, the surface temperatures of catalysts can be elevated to 300–600 °C under irradiation by focused solar light, providing sufficient energy to stimulate thermodynamically unfavorable reactions, such as CO2 reforming with methane and reverse water gas shift.71–73Photothermal catalytic CO2 reduction over group VIII elements (such as Ru, Rh, Ni, Co) was systematically investigated previously by Ye and co-authors, and the reaction rates for photothermal CO2 reduction (mol h−1 g−1) were found to be several orders of magnitude larger than for conventional photocatalysis.88 The main products of CO2 reduction were CH4 and CO. A maximum CO2 reaction rate (18.16 mol h−1 g−1) was detected over metallic Ru, while Fe demonstrated the best selectivity towards photothermal catalytic RWGS. The authors indicated that the outstanding catalytic performance for VIII elements can be attributed to their excellent photothermal conversion capacity and outstanding catalytic activity. Pd@Nb2O5 was also found to exhibit good catalytic activity towards RWGS.89 After light irradiation, photothermal heat induced over Pd nanocrystals served as the driving force for stimulating RWGS, and a CO production rate of 1.8 mmol g−1 h−1 was observed.
Ozin and co-authors recently reported the fabrication of a nanoneedle-array-based cobalt plasmonic superstructure (Co-PS@SiO2) for photothermal catalytic CO2 reduction.90 In Co-PS@SiO2, each nanoneedle was composed of Co nanocrystals and encapsulated by silica (Fig. 5a), which enabled nearly 100% light absorption efficiency (Fig. 5b). Under intense light irradiation, Co nanocrystals were heated to high temperature through the photothermal effect. Compared with Co@SiO2, L-Co@SiO2, and Co/FTO, the as-prepared Co-PS@SiO2 obtained the highest surface temperature of 383 °C under 20 Suns illumination, indicating the outstanding photon harvesting and photothermal conversion capacity of Co-PS@SiO2. A photothermal CO2 hydrogenation reaction was conducted as the model reaction to evaluate the catalytic activity. The main products for photothermal CO2 hydrogenation over Co-based catalysts were CO and CH4, and the conversion rate of CO2 over Co-PS@SiO2 was measured to be 0.6 mol g−1 h−1 (normalized by the mass of Co) (Fig. 5c). Based on surface analysis and mechanistic studies, the authors indicated that a higher local temperature induced by intense light was essential for the observed higher activity of Co-PS@SiO2. This work provides an innovative strategy to promote photothermal conversion through constructing plasmonic nanostructures with antireflection effects, enabling a complete harvesting of the solar-energy towards highly efficient CO2 reduction.
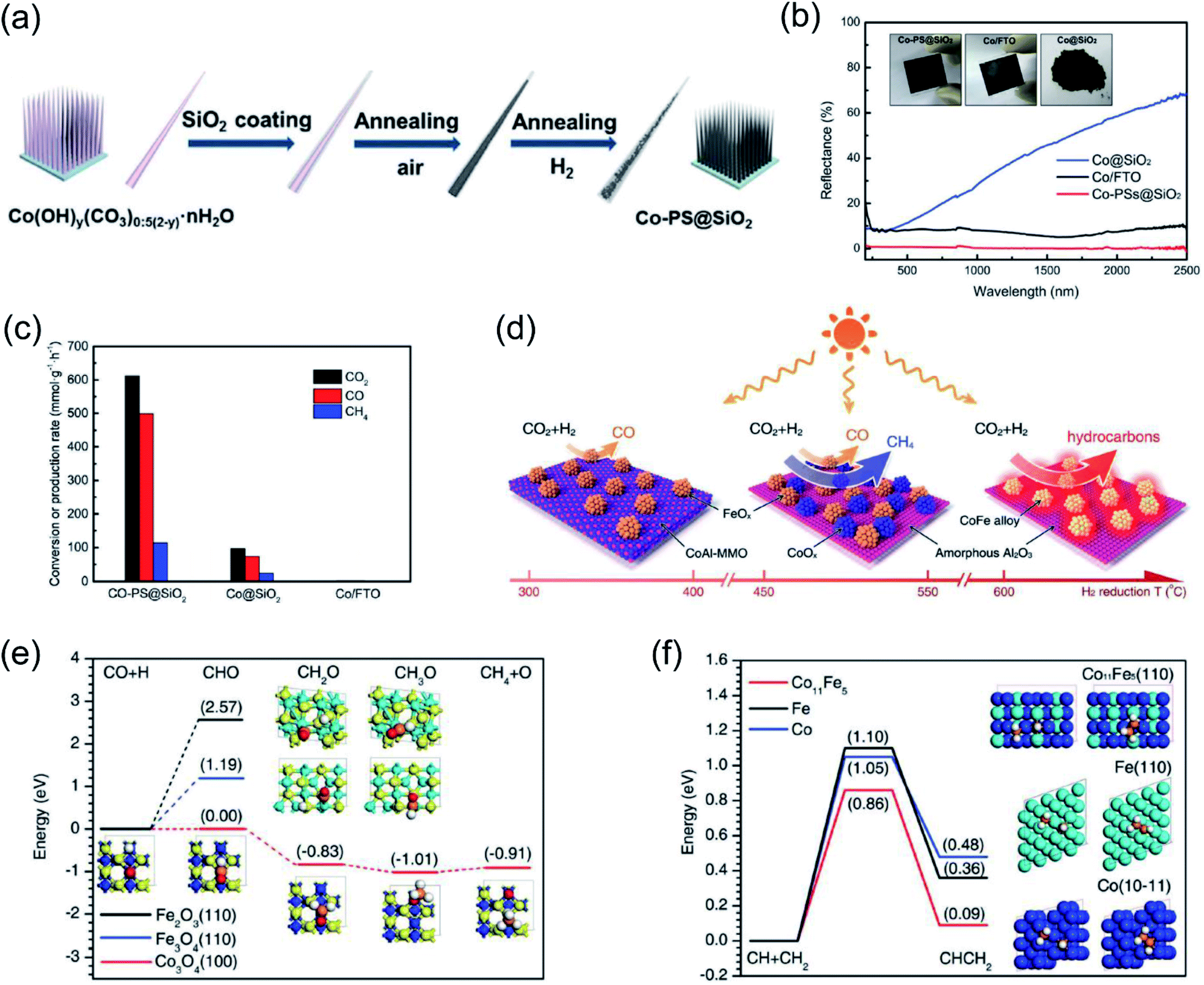 | ||
| Fig. 5 Photothermal catalytic CO2 reduction. (a) A schematic illustration of the fabrication process of Co-PS@SiO2. (b) The optical absorption properties of different cobalt catalysts. (c) The catalytic behavior of different samples normalized by the mass of Co. (a–c) Reproduced from ref. 90 with permission from John Wiley and Sons, Copyright 2020. (d) Different catalysts formed via H2 reduction at different temperatures and their selectivity toward solar-driven CO2 reduction. (e) The reaction paths for CO hydrogenation over different catalysts. (f) The probable C–C coupling path over different catalysts. (d–f) Reproduced from ref. 92 with permission from John Wiley and Sons, Copyright 2018. | ||
It was also reported that the photon–matter interaction can enhance the photothermal conversion efficiency over plasmonic nanostructures, thus promoting the photothermal catalytic RWGS reaction. Ouyang and co-authors employed atomic-scale dispersed Co–N species anchored Co@C hybrid structure (Co@CoN&C) nanomaterials for CO2 photothermal reduction, and a CO2 conversion rate of 132 mmol g−1 h−1 with suppressed undesirable methanation was achieved in a batch-type reaction system under the irradiation of a 300 W Xe lamp.91 However, under the same conditions, Co nanoparticles demonstrated a reaction rate of only 27 mmol g−1 h−1. The temperature of the catalysts during the reaction was measured with an infrared thermometer, and a surface temperature of approximately 518 °C was detected over Co@CoN&C, higher than over Co nanoparticles (330 °C) under the same conditions. These results strongly indicated that Co@CoN&C exhibited higher photothermal conversion efficiency, which favored photothermal CO2 reduction. Contrast experiments suggested that the high selectivity could be attributed to the advantageous selectivity of the CoN&C shell, and the efficient photothermal conversion over the hybrid interfacial structure further promoted the catalytic activity. Based on chemisorption data and surface analysis results, the authors proposed that the enhanced CO2 adsorption capacity and the alleviated hydrogenation of CO2 into HCOO* synergistically facilitated the high conversion rate of CO2. This study further verifies that the thermodynamic and kinetic factors of a photochemical reaction can be modulated by the rational design of microstructures.
Through changing the reduction temperature in an H2 atmosphere, a series of CoFe-based catalysts can be derived from CoFeAl layered-double-hydroxide (LDH) materials, and they were applied in photothermal catalytic CO2 reduction. As presented by Zhang and co-authors, the chemical composition and morphology of CoFe catalysts can be modulated by changing the reduction temperature, which can ultimately control the reaction selectivity.92 With an increase in reduction temperature, the reaction products from photothermal catalytic CO2 reduction over CoFe catalysts shifted from CO to CH4, and eventually to C2+ products (Fig. 5d). A comparison of CO2 conversion rates over CoFe nanoparticles under photothermal heating and direct thermal heating without light irradiation at the same reaction temperature indicated that photothermal heat played a dominant role in the reaction. The authors proposed that the CoFe alloy nanoparticles are responsible for the high selectivity and photothermal effects. According to density functional theory (DFT) calculation results, the authors indicated that during the CO2 hydrogenation process, Fe-based catalysts favored the production of CO, whereas Co-based materials favored the production of CH4 (Fig. 5e). Furthermore, a C–C coupling reaction was promoted by CoFe bimetallic alloy, resulting in the production of C2+ products (Fig. 5f).
Recently, using a photothermal route for driving CO2 reduction with CH4 has attracted tremendous research interest. Group VIII metals (such as Ni, Co, and Pt) have been regarded as ideal catalysts for photothermal catalytic DRM.73,97,98 For example, Li and co-authors designed silica-cluster-modified Ni nanocrystals (SCM-Ni/SiO2) for highly efficient photothermal catalytic DRM.97 Under irradiation by focused light of high intensity, the surface temperature of SCM-Ni/SiO2 could reach approximately 650 °C from photothermal conversion. Photo-induced heating served as the driving force to initiate photothermal catalytic DRM without additional thermal energy input. Under full-spectrum light irradiation, high production rates of H2 and CO (17.1 and 19.9 mmol min−1 g−1) with a solar-to-fuel conversion efficiency up to 12.5% can be achieved. Density functional theory (DFT) calculation was conducted via the constrained occupancy approach to calculate the activation energy (Ea) for each elementary step both in the excited state and the ground state during solar-driven DRM over an Ni/SiO2 slab, and the results revealed that the Ea for CH and CHO dissociation in the excited state were decreased (from 0.67 and 1.11 eV to 0.58 and 0.99 eV, respectively). In addition, the Ea for C and CH oxidation in the excited state also showed a slight decrease compared with the Ea in the ground state, suggesting the existence of a photoactivation effect which was responsible for the acceleration of DRM. In addition, SCM-Ni/SiO2 demonstrated excellent durability. After 700 h of reaction, no carbon deposition or deactivation could be observed over SCM-Ni/SiO2. Based on mechanistic investigations, the authors concluded that the highly efficient catalytic activity of SCM-Ni/SiO2 towards solar-light-driven DRM can be attributed to surface plasmon-induced photothermal conversion and an IR heating effect. The high surface temperature on the catalytically active Ni nanocrystals was responsible for the high activity.
In summary, CO2 conversion can be driven efficiently through photothermal technology without additional energy input. Various plasmonic nanostructured materials, such as Co-PS@SiO2, Co@CoN&C, or CoFe alloys, can be adopted as photothermal catalysts for CO2 conversion. By using photothermal technology, CO2 can be reduced by H2, H2O or CH4, and be converted into energy-containing fuel gas. Mechanistic investigations indicated that the full solar-spectrum, especially infrared light, can be absorbed to heat the catalysts to a high temperature through photothermal conversion, which promoted the activation of reactants and initiated thermodynamically unfavorable reactions such as RWGS and photosynthetic processes.
3.3 Plasmonic photothermal catalysis
After the oscillated electrons have been excited under light irradiation over plasmonic metals, the high energy electrons can relax to the ground state by a non-radiative decay route, following the Frank–Condon principle. The photo-induced hot carriers can either interact with molecules adsorbed on the surface of plasmonic metals by the selective activation of the chemical bonds of the adsorbates, or they can dissipate their energy into the environment to increase the local temperature. Recently, several studies have demonstrated that by the rational design of plasmonic photothermal catalysts, the hot carriers and photothermal heating can contribute to the reaction simultaneously, resulting not only in enhanced activity but also in modulated selectivity.42,99,100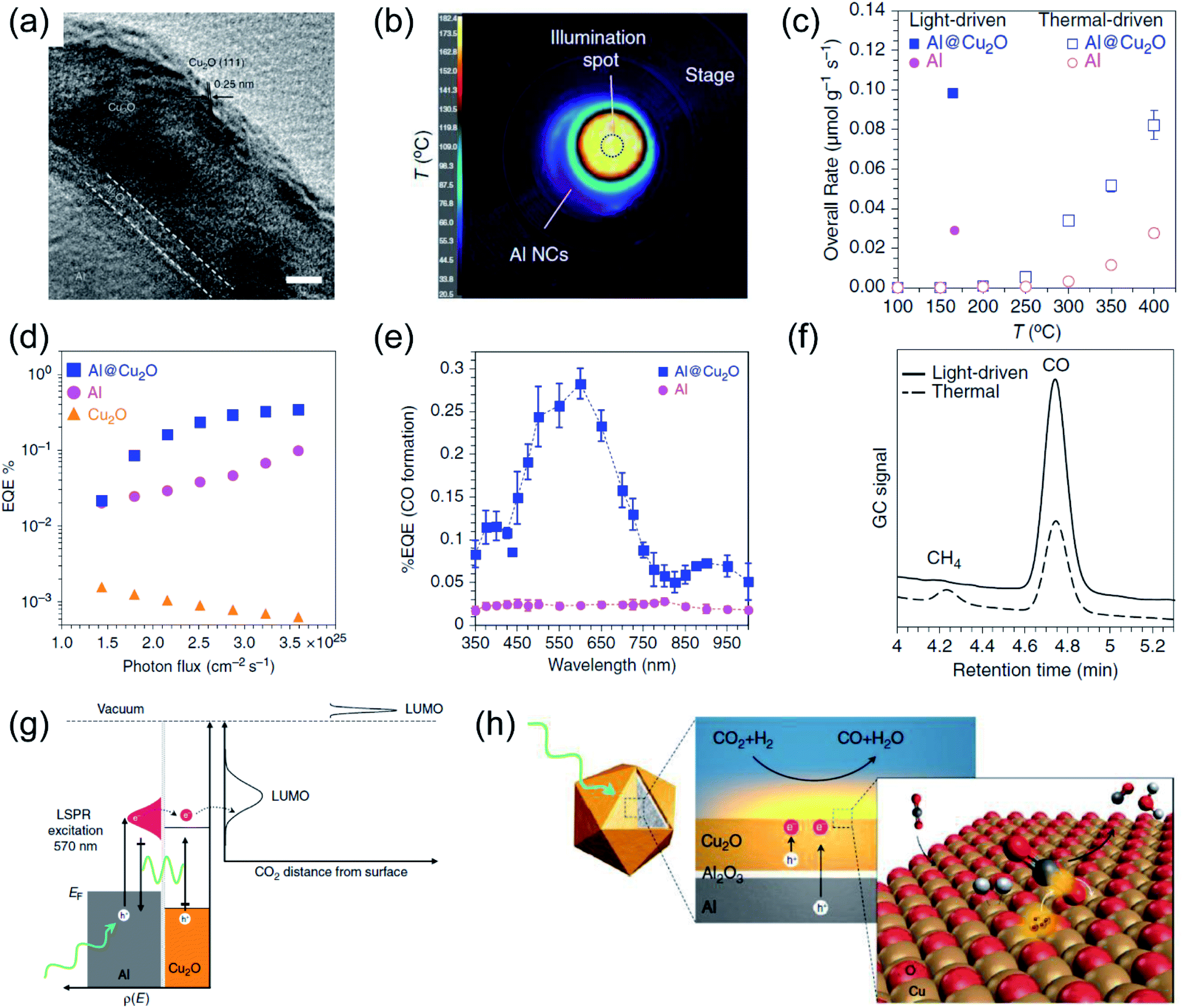 | ||
| Fig. 6 The plasmonic photothermal catalytic RWGS reaction over Al/Al2O3/Cu2O antenna-reactor nanoparticles. (a) A HRTEM image of Al/Al2O3/Cu2O. (b) The spatial temperature distribution on the surface of the catalyst under light irradiation. (c) A comparison of the overall rates of product formation under purely light-driven and thermal-driven conditions. (d) Apparent external quantum efficiency (EQE) plotted against photon flux. (e) Apparent external quantum efficiency (EQE) plotted against illumination wavelength. (f) Gas chromatograms of the chamber output during solar- and thermal-driven processes on Al@Cu2O. (g) The proposed mechanism of using Al@Cu2O for plasmon-mediated charge transfer and adsorbate activation processes. (h) A schematic illustration of hot electron-driven RWGS over Al@Cu2O plasmonic photothermal catalysts. (a–h) Reproduced from ref. 50 with permission from Springer Nature, Copyright 2017. | ||
Our group previously reported that photo-induced hot electrons over plasmonic Fe nanometals coated with a carbon layer (Fe@C) can effectively assist photothermal RWGS.102 Photoirradiation could induce significant heating through efficient photothermal conversion, since a high surface temperature (481 °C) on Fe@C was detected after light irradiation (Fig. 7a). After light irradiation for 2 h, a CO amount of 2196.17 μmol was obtained over the Fe@C catalyst. The authors proposed that apart from photo-induced heat, the hot carriers generated from Fe nanoparticles also participated in the surface reaction. A series of contrast experiments were conducted to reveal the difference between plasmonic photothermal catalysis (without additional thermal energy input) and conventional thermocatalysis (without light irradiation). Interestingly, the production rates for CO under the photo-induced conditions were obviously higher than those under thermal conditions at the same surface temperatures, suggesting the existence of another reaction mechanism other than photo-induced heat. Afterward, UV light was introduced into the reactor during the conventional thermocatalytic process, and a comparable activity to the photoinduced thermal condition was observed, indicating that UV light played a major role in enhancing the RWGS reaction over Fe@C catalysts (Fig. 7b). This result was further confirmed by contrast experiments under irradiation by different wavelengths (Fig. 7c). To investigate the underlying reaction mechanism, several types of theoretical calculation were conducted. The electrostatic calculation results showed that the work function of carbon atoms bonded to Fe9 was decreased by 0.38 eV after loading on graphene (Fig. 7d), which can be attributed to the charge transfer from Fe to graphene. As revealed by density of states (DOS) calculation, the electronic structure near the Fermi level was modified to a metallic state, resulting in a much easier electron transfer from Fe nanoparticles to reactants during the catalytic reaction (Fig. 7e). The adsorption energy of CO on different material surfaces was further investigated, and the results indicated that adsorption of CO on Fe can be modulated effectively after graphene coating, resulting in much easier desorption of CO (Fig. 7f). Electromagnetic-field simulation results revealed that the plasmon–photon coupling was amplified on the surface of the Fe nanoparticles. After coating a few layers of carbon over Fe, the electric field enhancement of Fe was further increased, facilitating the production of hot electrons for activating CO2 electronically (Fig. 7g). In this study, UV-light was captured by Fe nanoparticles to excite the LSPR effect, and the light-induced thermal effect can further promote the reaction. Through rational design of plasmonic photothermal catalysts, hot carrier activation, photothermal conversion and promoted desorption of CO were all integrated effectively in one catalyst, resulting in a highly efficient RWGS reaction.
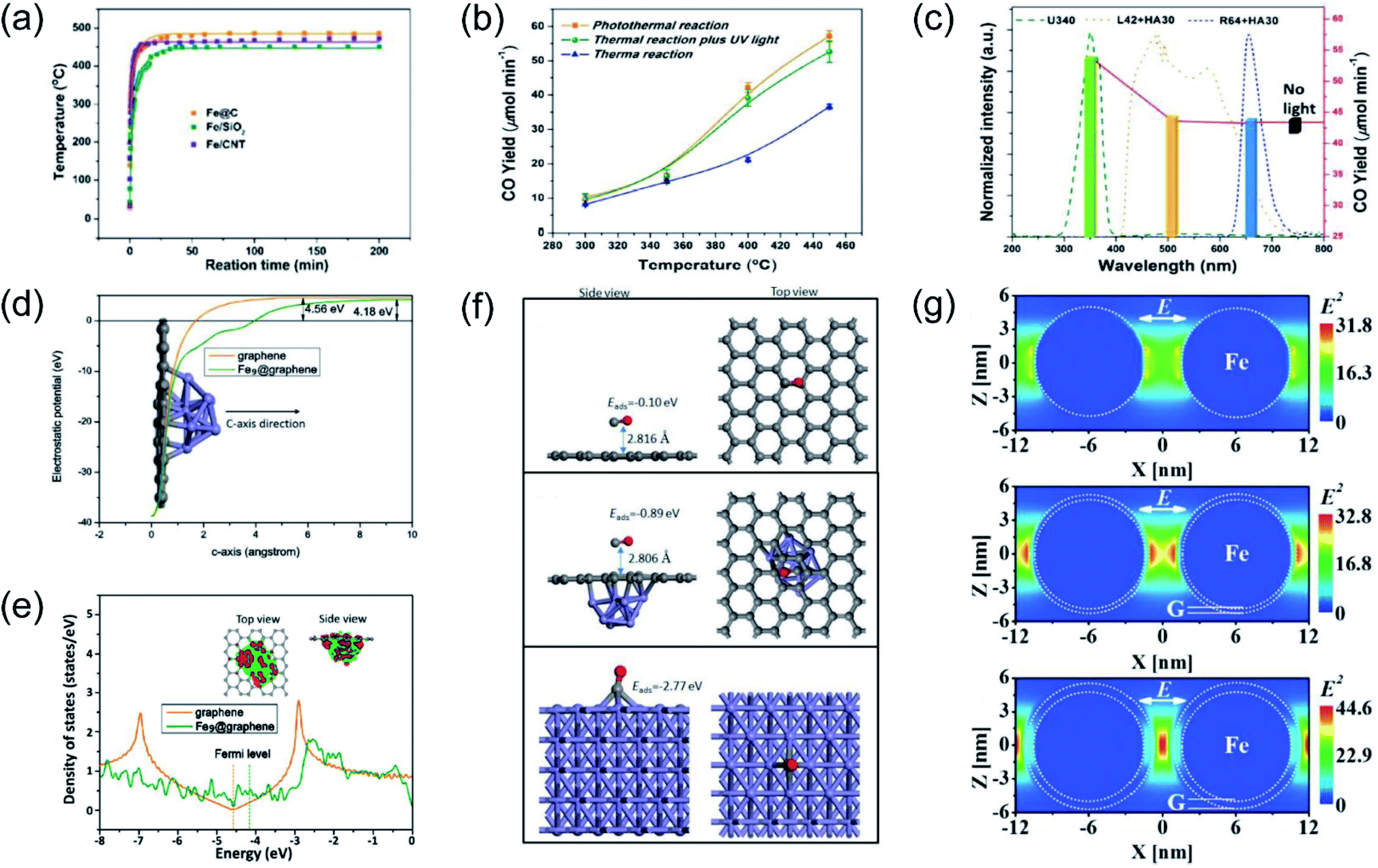 | ||
| Fig. 7 Plasmonic photothermal catalytic RWGS. (a) In situ monitoring of the surface temperature under light irradiation. (b) Comparison of CO generation rates for photo-induced thermal and conventional thermal reactions over an Fe@C catalyst. (c) CO production rates over Fe@C without or with light irradiation in different wavelength ranges at 450 °C. (d) The electrostatic potentials of graphene and Fe9@graphene. (e) Density of states (DOS) calculations involving graphene and Fe9@graphene. (f) CO adsorption configurations on graphene, Fe9@graphene, and Fe (110) surfaces. (g) The spatial distribution of the SPR-induced enhancement of the electric field over an Fe nanoparticle, Fe coated with a single layer of carbon, and Fe coated with multiple layers of carbon. (a–g) Reproduced from ref. 102 with permission from John Wiley and Sons, Copyright 2016. | ||
Plasmonic Au was also utilized as a plasmonic photothermal catalyst for driving RWGS with high selectivity without addition thermal energy input. Jia and co-authors fabricated Au/CeO2 nanocomposites, and found that the reaction rate under photothermal conditions was much higher than under purely thermal conditions.103 Under visible light irradiation, a maximum surface temperature of 400 °C was detected, and the CO2 conversion reached 40%, which was approximately ten times higher than that in the thermal process. Contrast experiments under photothermal and purely thermal conditions revealed a decisive function of Au for high CO2 conversion in the photothermal process. Based on reaction kinetic investigations and the operando DRIFT spectra results, the authors concluded that the photo-induced hot electrons of Au can induce the dissociation of adsorbed H2, which favored the subsequent hydrogenation of CO2 to CO.
Very recently, Halas and co-authors designed a plasmonic single atomic site antenna-reactor consisting of a Cu nanoparticle “antenna” with single Ru active “reactor” sites dispersed on the Cu surface (Cu–Ru) for an efficient plasmonic photothermal DRM reaction.48 The Cu–Ru was synthesized through the co-precipitation method, and the catalytic activity towards photothermal DRM was measured in a flow-type reactor under atmospheric pressure. A supercontinuum laser was applied as the energy supply to drive the photothermal DRM reaction. The surface temperatures of the catalysts under illumination were recorded by a thermal camera. Under irradiation, a surface temperature of 1000 K was detected over Cu19.8Ru0.2. Cu19.8Ru0.2 demonstrated the optimal performance towards photothermal DRM among all the samples, and a turnover frequency of 34 mol H2 (mol Ru)−1 s−1 was achieved under irradiation by white light (19.2 W cm−2) without an external energy input (Fig. 8a). Through a comparison of activity and selectivity over Cu19.8Ru0.2 under photo-driven and thermal-driven conditions at the same surface temperature induced by illumination and external heating, a hot carrier-mediated DRM process was proposed (Fig. 8b). The participation of hot carriers in the surface reaction was further confirmed by investigating the reaction selectivity under different light intensities, and an increased selectivity with light intensity was observed, which was consistent with the hot carrier-mediated reaction mechanism. In a such process, a solar-to-fuel energy conversion efficiency of up to 15% was achieved under irradiation by white light with a strong light intensity of 16 W cm−2. Based on mechanistic investigations and theoretical calculations in the both ground- and excited-states, the authors indicated that the advanced plasmonic photothermal catalytic activity of Cu19.8Ru0.2 can be attributed to two aspects. Firstly, atomically dispersed Ru active sites on the surface of Cu facilitated CH4 dehydrogenation with a suppressed side reaction (such as RWGS) and coke formation. Secondly, C–H bond activation and H2 desorption on the Ru active sites were promoted by the photo-induced hot carriers, resulting in a substantial reduction of the energy barrier for CH4 activation and eventually facilitating the charge transfer onto the detaching H (Fig. 8c). Under intense white light irradiation, the plasmonic Cu could deliver the hot carriers to the surface Ru atoms, powering the surface reaction over Ru active sites. This study highlighted that light-energy might be a more effective form of energy input for driving DRM. By utilizing plasmonic photothermal catalysis, a combination of photo-induced hot carriers and photothermal heating can be achieved for driving tough but industrially valuable reactions.
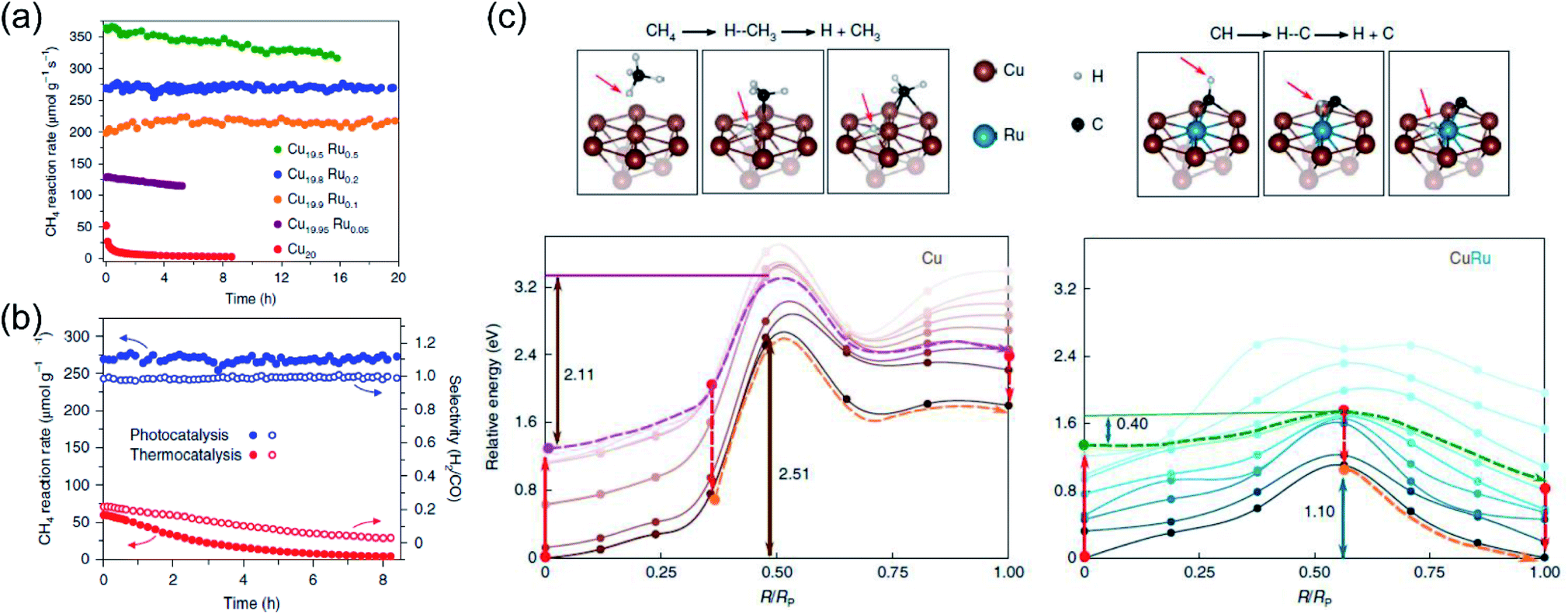 | ||
| Fig. 8 The plasmonic photothermal catalytic DRM reaction over Cu–Ru surface alloy antenna-reactor plasmonic photocatalysts. (a) Reaction activity and long-term stability data for Cu–Ru catalysts with different Ru ratios for plasmonic photothermal catalytic DRM. (b) A comparison of the activity, stability, and selectivity of Cu19.8Ru0.2 under 19.2 W cm−2 white-light illumination and for thermocatalysis at a reaction temperature of 1000 K. (c) Calculated ground- and excited-state energy curves for CH4 and CH dehydrogenation over Cu and Cu–Ru, respectively. (a–c) Reproduced from ref. 48 with permission from Springer Nature, Copyright 2020. | ||
4. Conclusions and outlook
The above studies conducted so far have demonstrated that plasmonic energy can serve as a promising and effective stimulus for converting solar-energy into chemical energy. The full solar-spectrum, including ultraviolet, visible, and even infrared light, can be effectively harnessed by plasmonic catalysts for driving various kinds of reactions, such as H2O splitting, CO2 reduction, and artificial photosynthesis. Depending on the different decay pathways of plasmonic energy, the main driving force for initiating the surface reaction can be disparate, classifying the reaction process into plasmonic photocatalysis, photothermal catalysis and plasmonic photothermal catalysis. During plasmonic photocatalysis where the hot carriers play dominant roles, the solar-to-fuel conversion efficiencies are unsatisfactory. Compared with plasmonic photocatalysis, photothermal catalysis demonstrates better efficiency for solar-to-fuel conversion due to the participation of photothermal heat in the surface reactions, but the activation of adsorbates by hot carriers is negligible. Furthermore, with the assistance of both the hot carrier-induced adsorbate activation and photothermal heating, plasmonic photothermal catalysis enables the initiation of thermodynamically unfavorable reactions efficiently and selectively compared with both conventional photocatalytic and thermocatalytic processes, resulting in a highly boosted STF conversion efficiency. The utilization of plasmonic photothermal technology presents several advantages. First, the most abundant and renewable solar-energy is adopted in the chemical reaction as the driving force, which can effectively reduce the consumption of non-renewable fossil fuels. Second, the reaction pathways can be modified by injecting hot carriers into specific chemical bonds, thus increasing the reaction selectivity. Third, compared with the thermocatalytic process, the milder reaction conditions of the photothermal catalytic process can lead to significant advantages, such as prolonged stability, enhanced safety, and a wider choice of catalysts. Although there have been significant developments in plasmonic catalysis in the past few years, both the STF and AQE efficiency are still rather moderate, due to the limited light-capturing ability and disordered charge transfer process. In order to further promote the efficiency, it is highly desirable to develop plasmonic nanostructures with excellent light response from UV to infrared light, numerous active catalytic sites, and prolonged lifetimes of hot carriers by the modification of plasmonic nanostructures, such as modulating the size and shape of plasmonic metals, and the construction of complex (potentially multi-metallic) plasmonic nanostructures.First, the charge transfer behavior of plasmonic nanostructures is an essential factor for determining the overall catalytic activity and selectivity. Such a phenomenon is attributed to the specific injection of hot carriers from photocatalysts to certain chemical bonds of the adsorbates, resulting in a new mechanistic pathway through the selective initiation of individual elementary steps of chemical reactions. Current investigations suggest that the energy of hot carriers could be modulated by tailoring the optical properties of plasmonic nanostructures (such as SPR band position and intensity) in order to match the specific electronic states and/or vibrational modes of adsorbates, ultimately controlling the reaction selectivity precisely. Toward this end, significant advances in the design and fabrication of plasmonic nanostructures with optimal geometry and composition will be highly desirable to realize a controllable charge transfer process. For instance, combining the plasmonic metal with another chemically reactive metal is considered to be promising for constructing highly efficient plasmonic photothermal catalysts. The optical property of the catalysts is determined by plasmonic metals, and the interaction between adsorbates and reactive metals enables the tuning of the electronic structure of the adsorbates. By matching these optical and electronic properties, the charge transfer behavior over plasmonic metals can be regulated, inducing a preferential charge carrier injection to particular orbital of adsorbates. In addition, great efforts should also be devoted to the development of predictive theories and a basic understanding of the molecular behavior on the surface of plasmonic metals, which will be helpful for us in designing plasmonic photothermal catalysts with optimal nanostructures and compositions.
Second, apart from hot carrier excitation and adsorbate activation, photothermal conversion is also a critical parameter which influences the performance of plasmonic photothermal catalysts. The optical characteristics (such as position of SPR absorption peak and light absorption capacity) of plasmonic nanostructures are the fundamentals to determine the photon energy that can be utilized for participating in the reaction. As for the aspect of photothermal conversion, transforming solar-energy across the entire solar-spectrum, especially low-energy photons in the visible and infrared regions, into heat should be more favorable. One potential avenue for achieving this is to synthesize complex (potentially multi-metallic) plasmonic nanostructures to create more oscillation modes and hotspots, resulting in a full solar-spectrum response. In addition, modification of the nanostructures to improve antireflection may result in enhanced sunlight-harvesting ability, improving the photothermal conversion.
Third, plasmonic photothermal catalysis is a newly developed technology in the broad area of catalysis. Basic understandings of the reaction mechanism, such as hot carrier generation and reactant activation on the plasmonic nanostructures, remain at the preliminary stage owing to the complicated reaction pathways and limited surface characterization techniques. Advanced mechanistic investigations, such as excited-state theoretical calculation and in situ transient surface analysis are expected to provide some convincing evidence for unravelling the interaction process between plasmonic nanostructures and reactants. In addition, in the plasmonic photothermal catalysis system, photothermal heat and hot carrier transfer are likely to act in concert, and the contribution of each factor remains to be clarified qualitatively and quantitively. In order to investigate the underlying thermodynamics and kinetics of plasmonic photothermal catalytic reactions, more technologies are required for not only accurately measuring the surface temperature of catalysts, but also for identifying the transient species generated during the reaction.
Finally, in addition to plasmonic photothermal catalysis that utilizes the full solar spectrum efficiently, it is important to explore other strategies that could combine both light and thermal features of solar energy for highly efficient solar-to-energy conversion. The recently developed solar thermal electrochemical process (STEP) provides an alternative and effective strategy for solar-to-chemical conversion, especially in CO2 reduction.104 In STEP, solar light is concentrated and separated into ultraviolet, visible, and infrared light, in which the ultraviolet-visible portion is utilized to drive a photovoltaic (PV) device to generate electricity for electrolysis, and the infrared portion is utilized to heat an electrolysis cell.105,106 As a result, the required electrical potential can be lowered enormously in the electrolysis process when the reaction temperature rises drastically under infrared light irradiation.107 Several electrolysis reactions which were originally initiated at room temperature, such as CO2 reduction to produce CO or CH4, can be promoted at high temperature via the solar thermal electrochemical process.108–110 STEP provides an alternative strategy for solar-energy conversion, with full utilization efficiency of solar light. At present, several important factors, such as the electrolysis temperature and electrolyte composition, should be extensively investigated to achieve higher product selectivity and conversion efficiency. In addition, it is highly desirable to develop more stable electrode materials to maintain long-term operation under the harsh reaction conditions of STEP.
Author contributions
H. S. and J. Y. conceived the overall idea of the perspective. S. L., X. R., H. L. wrote the initial manuscript. S. L., H. S. and J. Y. revised and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by JSPS KAKENHI (JP18H02065), the World Premier International Research Center Initiative (WPI Initiative) on Materials Nanoarchitectonics (MANA), MEXT (Japan), Photo-excitonic Project in Hokkaido University, National Natural Science Foundation of China (Grant No. 21633004), and State Scholarship Fund by China Scholarship Council (CSC) (201806400044).References
- Y. A. Wu, I. McNulty, C. Liu, K. C. Lau, Q. Liu, A. P. Paulikas, C.-J. Sun, Z. Cai, J. R. Guest, Y. Ren, V. Stamenkovic, L. A. Curtiss, Y. Liu and T. Rajh, Nat. Energy, 2019, 4, 957–968 CrossRef CAS.
- S. Chen, T. Takata and K. Domen, Nat. Rev. Mater., 2017, 2, 17050 CrossRef CAS.
- D. Voiry, H. S. Shin, K. P. Loh and M. Chhowalla, Nat. Rev. Chem., 2018, 2, 0105 CrossRef CAS.
- Y. Wang, X. Shang, J. Shen, Z. Zhang, D. Wang, J. Lin, J. C. S. Wu, X. Fu, X. Wang and C. Li, Nat. Commun., 2020, 11, 3043 CrossRef CAS PubMed.
- X.-B. Li, C.-H. Tung and L.-Z. Wu, Nat. Rev. Chem., 2018, 2, 160–173 CrossRef CAS.
- S. Linic, P. Christopher, H. Xin and A. Marimuthu, Acc. Chem. Res., 2013, 46, 1890–1899 CrossRef CAS PubMed.
- D. F. Swearer, H. Zhao, L. Zhou, C. Zhang, H. Robatjazi, J. M. P. Martirez, C. M. Krauter, S. Yazdi, M. J. McClain, E. Ringe, E. A. Carter, P. Nordlander and N. J. Halas, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 8916–8920 CrossRef CAS PubMed.
- Y. Zhang, S. He, W. Guo, Y. Hu, J. Huang, J. R. Mulcahy and W. D. Wei, Chem. Rev., 2018, 118, 2927–2954 CrossRef CAS PubMed.
- M. A. El-Sayed, Acc. Chem. Res., 2001, 34, 257–264 CrossRef CAS PubMed.
- C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025–1102 CrossRef CAS PubMed.
- U. Aslam, V. G. Rao, S. Chavez and S. Linic, Nat. Catal., 2018, 1, 656–665 CrossRef.
- Z.-j. Wang, H. Song, H. Liu and J. Ye, Angew. Chem., Int. Ed., 2020, 59, 8016–8035 CrossRef CAS PubMed.
- S. Rej, L. Mascaretti, E. Y. Santiago, O. Tomanec, Š. Kment, Z. Wang, R. Zbořil, P. Fornasiero, A. O. Govorov and A. Naldoni, ACS Catal., 2020, 10, 5261–5271 CrossRef CAS.
- L. Zhou, D. F. Swearer, C. Zhang, H. Robatjazi, H. Zhao, L. Henderson, L. Dong, P. Christopher, E. A. Carter, P. Nordlander and N. J. Halas, Science, 2018, 362, 69–72 CrossRef CAS PubMed.
- S. Linic, U. Aslam, C. Boerigter and M. Morabito, Nat. Mater., 2015, 14, 567–576 CrossRef CAS PubMed.
- Y. Zhao, W. Gao, S. Li, G. R. Williams, A. H. Mahadi and D. Ma, Joule, 2019, 3, 920–937 CrossRef CAS.
- A. Gellé, T. Jin, L. de la Garza, G. D. Price, L. V. Besteiro and A. Moores, Chem. Rev., 2020, 120, 986–1041 CrossRef PubMed.
- G. Chen, G. I. N. Waterhouse, R. Shi, J. Zhao, Z. Li, L.-Z. Wu, C.-H. Tung and T. Zhang, Angew. Chem., Int. Ed., 2019, 58, 17528–17551 CrossRef CAS PubMed.
- S. F. Hung, F. X. Xiao, Y. Y. Hsu, N. T. Suen, H. B. Yang, H. M. Chen and B. Liu, Adv. Energy Mater., 2016, 6, 1501339 CrossRef.
- J. S. DuChene, B. C. Sweeny, A. C. Johnston-Peck, D. Su, E. A. Stach and W. D. Wei, Angew. Chem., Int. Ed., 2014, 53, 7887–7891 CrossRef CAS PubMed.
- K. Qian, B. C. Sweeny, A. C. Johnston-Peck, W. Niu, J. O. Graham, J. S. DuChene, J. Qiu, Y.-C. Wang, M. H. Engelhard, D. Su, E. A. Stach and W. D. Wei, J. Am. Chem. Soc., 2014, 136, 9842–9845 CrossRef CAS PubMed.
- J. Sá, G. Tagliabue, P. Friedli, J. Szlachetko, M. H. Rittmann-Frank, F. G. Santomauro, C. J. Milne and H. Sigg, Energy Environ. Sci., 2013, 6, 3584–3588 RSC.
- A. Furube, L. Du, K. Hara, R. Katoh and M. Tachiya, J. Am. Chem. Soc., 2007, 129, 14852–14853 CrossRef CAS PubMed.
- W. Hou, W. H. Hung, P. Pavaskar, A. Goeppert, M. Aykol and S. B. Cronin, ACS Catal., 2011, 1, 929–936 CrossRef CAS.
- Z. Zhang, Z. Wang, S.-W. Cao and C. Xue, J. Phys. Chem. C, 2013, 117, 25939–25947 CrossRef CAS.
- H. Song, X. Meng, Z.-j. Wang, H. Liu and J. Ye, Joule, 2019, 3, 1606–1636 CrossRef CAS.
- M. J. Kale, T. Avanesian and P. Christopher, ACS Catal., 2014, 4, 116–128 CrossRef CAS.
- P. Christopher and M. Moskovits, Annu. Rev. Phys. Chem., 2017, 68, 379–398 CrossRef CAS PubMed.
- M. J. Kale and P. Christopher, Science, 2015, 349, 587–588 CrossRef CAS PubMed.
- A. O. Govorov, H. Zhang and Y. K. Gun’ko, J. Phys. Chem. C, 2013, 117, 16616–16631 CrossRef CAS.
- A. Manjavacas, J. G. Liu, V. Kulkarni and P. Nordlander, ACS Nano, 2014, 8, 7630–7638 CrossRef CAS PubMed.
- S. Link and M. A. El-Sayed, J. Phys. Chem. B, 1999, 103, 4212–4217 CrossRef CAS.
- B. D. Clark, C. R. Jacobson, M. Lou, D. Renard, G. Wu, L. Bursi, A. S. Ali, D. F. Swearer, A.-L. Tsai, P. Nordlander and N. J. Halas, ACS Nano, 2019, 13, 9682–9691 CrossRef CAS PubMed.
- L. Yuan, M. Lou, B. D. Clark, M. Lou, L. Zhou, S. Tian, C. R. Jacobson, P. Nordlander and N. J. Halas, ACS Nano, 2020, 14, 12054–12063 CrossRef CAS PubMed.
- C. Wang and D. Astruc, Chem. Soc. Rev., 2014, 43, 7188–7216 RSC.
- H. Wang, F. Tam, N. K. Grady and N. J. Halas, J. Phys. Chem. B, 2005, 109, 18218–18222 CrossRef CAS PubMed.
- A. Pinchuk, U. Kreibig and A. Hilger, Surf. Sci., 2004, 557, 269–280 CrossRef CAS.
- R. H. Doremus, J. Chem. Phys., 1964, 40, 2389–2396 CrossRef.
- M. Ghoussoub, M. Xia, P. N. Duchesne, D. Segal and G. Ozin, Energy Environ. Sci., 2019, 12, 1122–1142 RSC.
- Z. Xie, Y. Duo, Z. Lin, T. Fan, C. Xing, L. Yu, R. Wang, M. Qiu, Y. Zhang, Y. Zhao, X. Yan and H. Zhang, Adv. Sci., 2020, 7, 1902236 CrossRef CAS PubMed.
- G. Tas and H. J. Maris, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 15046–15054 CrossRef CAS PubMed.
- M. L. Brongersma, N. J. Halas and P. Nordlander, Nat. Nanotechnol., 2015, 10, 25–34 CrossRef CAS PubMed.
- K. Jiang, D. A. Smith and A. Pinchuk, J. Phys. Chem. C, 2013, 117, 27073–27080 CrossRef CAS.
- C. M. Hessel, V. P. Pattani, M. Rasch, M. G. Panthani, B. Koo, J. W. Tunnell and B. A. Korgel, Nano Lett., 2011, 11, 2560–2566 CrossRef CAS PubMed.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS PubMed.
- S. Luo, H. Song, D. Philo, M. Oshikiri, T. Kako and J. Ye, Appl. Catal., B, 2020, 272, 118965 CrossRef CAS.
- E. Cortés, Adv. Opt. Mater., 2017, 5, 1700191 CrossRef.
- L. Zhou, J. M. P. Martirez, J. Finzel, C. Zhang, D. F. Swearer, S. Tian, H. Robatjazi, M. Lou, L. Dong, L. Henderson, P. Christopher, E. A. Carter, P. Nordlander and N. J. Halas, Nat. Energy, 2020, 5, 61–70 CrossRef CAS.
- M. Bonn, S. Funk, C. Hess, D. N. Denzler, C. Stampfl, M. Scheffler, M. Wolf and G. Ertl, Science, 1999, 285, 1042–1045 CrossRef CAS PubMed.
- H. Robatjazi, H. Zhao, D. F. Swearer, N. J. Hogan, L. Zhou, A. Alabastri, M. J. McClain, P. Nordlander and N. J. Halas, Nat. Commun., 2017, 8, 27 CrossRef PubMed.
- G. Baffou, I. Bordacchini, A. Baldi and R. Quidant, Light: Sci. Appl., 2020, 9, 108 CrossRef PubMed.
- L. Yan, F. Wang and S. Meng, ACS Nano, 2016, 10, 5452–5458 CrossRef CAS PubMed.
- Y. Li, S. Wang, P. Wang, Y. He, X. Wang, K. Chang, H. Lin, X. Ding, H. Chen, H. Zhang, Y. Izumi, T. Kako and J. Ye, Nano Energy, 2020, 76, 105077 CrossRef CAS.
- X. Ren, D. Philo, Y. Li, L. Shi, K. Chang and J. Ye, Coord. Chem. Rev., 2020, 424, 213516 CrossRef CAS.
- J.-J. Chen, J. C. S. Wu, P. C. Wu and D. P. Tsai, J. Phys. Chem. C, 2011, 115, 210–216 CrossRef CAS.
- A. Tanaka, K. Teramura, S. Hosokawa, H. Kominami and T. Tanaka, Chem. Sci., 2017, 8, 2574–2580 RSC.
- S. Wang, Y. Gao, Y. Qi, A. Li, F. Fan and C. Li, J. Catal., 2017, 354, 250–257 CrossRef CAS.
- O. Elbanna, S. Kim, M. Fujitsuka and T. Majima, Nano Energy, 2017, 35, 1–8 CrossRef CAS.
- S.-i. Naya, T. Kume, R. Akashi, M. Fujishima and H. Tada, J. Am. Chem. Soc., 2018, 140, 1251–1254 CrossRef CAS PubMed.
- D. Gogoi, A. Namdeo, A. K. Golder and N. R. Peela, Int. J. Hydrogen Energy, 2020, 45, 2729–2744 CrossRef CAS.
- B. Mei, A. Pougin and J. Strunk, J. Catal., 2013, 306, 184–189 CrossRef CAS.
- E. Liu, Y. Hu, H. Li, C. Tang, X. Hu, J. Fan, Y. Chen and J. Bian, Ceram. Int., 2015, 41, 1049–1057 CrossRef CAS.
- M. Tahir, B. Tahir and N. A. S. Amin, Appl. Surf. Sci., 2015, 356, 1289–1299 CrossRef CAS.
- J. Jiao, Y. Wei, Z. Zhao, W. Zhong, J. Liu, J. Li, A. Duan and G. Jiang, Catal. Today, 2015, 258, 319–326 CrossRef CAS.
- M. Tahir, B. Tahir and N. A. S. Amin, Appl. Catal., B, 2017, 204, 548–560 CrossRef CAS.
- S. Yu and P. K. Jain, Nat. Commun., 2019, 10, 2022 CrossRef PubMed.
- L. Collado, A. Reynal, F. Fresno, M. Barawi, C. Escudero, V. Perez-Dieste, J. M. Coronado, D. P. Serrano, J. R. Durrant and V. A. de la Peña O'Shea, Nat. Commun., 2018, 9, 4986 CrossRef PubMed.
- C. Wang, W.-C. D. Yang, D. Raciti, A. Bruma, R. Marx, A. Agrawal and R. Sharma, Nat. Mater., 2021, 20, 346–352 CrossRef CAS.
- L. Zhu, M. Gao, C. K. N. Peh and G. W. Ho, Mater. Horiz., 2018, 5, 323–343 RSC.
- X. Meng, L. Liu, S. Ouyang, H. Xu, D. Wang, N. Zhao and J. Ye, Adv. Mater., 2016, 28, 6781–6803 CrossRef CAS.
- G. Liu, X. Meng, H. Zhang, G. Zhao, H. Pang, T. Wang, P. Li, T. Kako and J. Ye, Angew. Chem., Int. Ed., 2017, 56, 5570–5574 CrossRef CAS PubMed.
- H. Huang, M. Mao, Q. Zhang, Y. Li, J. Bai, Y. Yang, M. Zeng and X. Zhao, Adv. Energy Mater., 2018, 8, 1702472 CrossRef.
- S. Wu, Y. Li, Q. Zhang, Z. Jiang, Y. Yang, J. Wu and X. Zhao, Energy Environ. Sci., 2019, 12, 2581–2590 RSC.
- C. Kim, S. Hyeon, J. Lee, W. D. Kim, D. C. Lee, J. Kim and H. Lee, Nat. Commun., 2018, 9, 3027 CrossRef PubMed.
- P. G. O'Brien, A. Sandhel, T. E. Wood, A. A. Jelle, L. B. Hoch, D. D. Perovic, C. A. Mims and G. A. Ozin, Adv. Sci., 2014, 1, 1400001 CrossRef PubMed.
- L. Wang, M. Ghoussoub, H. Wang, Y. Shao, W. Sun, A. A. Tountas, T. E. Wood, H. Li, J. Y. Y. Loh, Y. Dong, M. Xia, Y. Li, S. Wang, J. Jia, C. Qiu, C. Qian, N. P. Kherani, L. He, X. Zhang and G. A. Ozin, Joule, 2018, 2, 1369–1381 CrossRef CAS.
- Z.-j. Wang, H. Song, H. Pang, Y. Ning, T. D. Dao, Z. Wang, H. Chen, Y. Weng, Q. Fu, T. Nagao, Y. Fang and J. Ye, Appl. Catal., B, 2019, 250, 10–16 CrossRef CAS.
- Y. Qi, L. Song, S. Ouyang, X. Liang, S. Ning, Q. Zhang and J. Ye, Adv. Mater., 2020, 32, 1903915 CrossRef CAS PubMed.
- L. Wang, Y. Dong, T. Yan, Z. Hu, A. A. Jelle, D. M. Meira, P. N. Duchesne, J. Y. Y. Loh, C. Qiu, E. E. Storey, Y. Xu, W. Sun, M. Ghoussoub, N. P. Kherani, A. S. Helmy and G. A. Ozin, Nat. Commun., 2020, 11, 2432 CrossRef CAS PubMed.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- M. D. Porosoff, B. Yan and J. G. Chen, Energy Environ. Sci., 2016, 9, 62–73 RSC.
- A. Goguet, F. C. Meunier, D. Tibiletti, J. P. Breen and R. Burch, J. Phys. Chem. B, 2004, 108, 20240–20246 CrossRef CAS.
- V. Lebarbier, R. Dagle, A. Datye and Y. Wang, Appl. Catal., A, 2010, 379, 3–6 CrossRef CAS.
- A. M. Bahmanpour, F. Héroguel, M. Kılıç, C. J. Baranowski, L. Artiglia, U. Röthlisberger, J. S. Luterbacher and O. Kröcher, ACS Catal., 2019, 9, 6243–6251 CrossRef CAS.
- L. Wang, H. Liu, Y. Chen and S. Yang, Int. J. Hydrogen Energy, 2017, 42, 3682–3689 CrossRef CAS.
- M. J. L. Ginés, A. J. Marchi and C. R. Apesteguía, Appl. Catal., A, 1997, 154, 155–171 CrossRef.
- W. Wang, Y. Zhang, Z. Wang, J.-m. Yan, Q. Ge and C.-j. Liu, Catal. Today, 2016, 259, 402–408 CrossRef CAS.
- X. Meng, T. Wang, L. Liu, S. Ouyang, P. Li, H. Hu, T. Kako, H. Iwai, A. Tanaka and J. Ye, Angew. Chem., Int. Ed., 2014, 53, 11478–11482 CrossRef CAS PubMed.
- J. Jia, P. G. O'Brien, L. He, Q. Qiao, T. Fei, L. M. Reyes, T. E. Burrow, Y. Dong, K. Liao, M. Varela, S. J. Pennycook, M. Hmadeh, A. S. Helmy, N. P. Kherani, D. D. Perovic and G. A. Ozin, Adv. Sci., 2016, 3, 1600189 CrossRef PubMed.
- K. Feng, S. Wang, D. Zhang, L. Wang, Y. Yu, K. Feng, Z. Li, Z. Zhu, C. Li, M. Cai, Z. Wu, N. Kong, B. Yan, J. Zhong, X. Zhang, G. A. Ozin and L. He, Adv. Mater., 2020, 32, 2000014 CrossRef CAS PubMed.
- S. Ning, H. Xu, Y. Qi, L. Song, Q. Zhang, S. Ouyang and J. Ye, ACS Catal., 2020, 10, 4726–4736 CrossRef CAS.
- G. Chen, R. Gao, Y. Zhao, Z. Li, G. I. N. Waterhouse, R. Shi, J. Zhao, M. Zhang, L. Shang, G. Sheng, X. Zhang, X. Wen, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Mater., 2018, 30, 1704663 CrossRef PubMed.
- U. Ulmer, T. Dingle, P. N. Duchesne, R. H. Morris, A. Tavasoli, T. Wood and G. A. Ozin, Nat. Commun., 2019, 10, 3169 CrossRef PubMed.
- D. Liu, Y. Xu, M. Sun, Y. Huang, Y. Yu and B. Zhang, J. Mater. Chem. A, 2020, 8, 1077–1083 RSC.
- C. Wang, Y. Zhao, H. Xu, Y. Li, Y. Wei, J. Liu and Z. Zhao, Appl. Catal., B, 2020, 263, 118314 CrossRef CAS.
- J. Li, Y. Ye, L. Ye, F. Su, Z. Ma, J. Huang, H. Xie, D. E. Doronkin, A. Zimina, J.-D. Grunwaldt and Y. Zhou, J. Mater. Chem. A, 2019, 7, 2821–2830 RSC.
- H. Huang, M. Mao, Q. Zhang, Y. Li, J. Bai, Y. Yang, M. Zeng and X. Zhao, Adv. Energy Mater., 2018, 8, 1702472 CrossRef.
- M. Mao, Q. Zhang, Y. Yang, Y. Li, H. Huang, Z. Jiang, Q. Hu and X. Zhao, Green Chem., 2018, 20, 2857–2869 RSC.
- X. Zhang, X. Li, M. E. Reish, D. Zhang, N. Q. Su, Y. Gutiérrez, F. Moreno, W. Yang, H. O. Everitt and J. Liu, Nano Lett., 2018, 18, 1714–1723 CrossRef CAS PubMed.
- X. Li, X. Zhang, H. O. Everitt and J. Liu, Nano Lett., 2019, 19, 1706–1711 CrossRef CAS PubMed.
- M. Gao, P. K. N. Connor and G. W. Ho, Energy Environ. Sci., 2016, 9, 3151–3160 RSC.
- H. Zhang, T. Wang, J. Wang, H. Liu, T. D. Dao, M. Li, G. Liu, X. Meng, K. Chang, L. Shi, T. Nagao and J. Ye, Adv. Mater., 2016, 28, 3703–3710 CrossRef CAS PubMed.
- B. Lu, F. Quan, Z. Sun, F. Jia and L. Zhang, Catal. Commun., 2019, 129, 105724 CrossRef CAS.
- J. Ren, A. Yu, P. Peng, M. Lefler, F.-F. Li and S. Licht, Acc. Chem. Res., 2019, 52, 3177–3187 CrossRef CAS PubMed.
- B. Wang, D. Gu, J. Dong, D. Yuan and L. Zhu, Energy Convers. Manage., 2017, 152, 99–109 CrossRef CAS.
- S. Licht, Adv. Mater., 2011, 23, 5592–5612 CrossRef CAS PubMed.
- S. Licht, L. Halperin, M. Kalina, M. Zidman and N. Halperin, Chem. Commun., 2003, 3006–3007 RSC.
- F.-F. Li, S. Liu, B. Cui, J. Lau, J. Stuart, B. Wang and S. Licht, Adv. Energy Mater., 2015, 5, 1401791 CrossRef.
- F.-F. Li, J. Lau and S. Licht, Adv. Sci., 2015, 2, 1500260 CrossRef PubMed.
- D. Ji, Z. Li, W. Li, D. Yuan, Y. Wang, Y. Yu and H. Wu, Int. J. Hydrogen Energy, 2019, 44, 5082–5089 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |