
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric total synthesis of (+)-xestoquinone and (+)-adociaquinones A and B†
Xiao-Long
Lu‡
a,
Yuanyou
Qiu‡
a,
Baochao
Yang
a,
Haibing
He
 b and
Shuanhu
Gao
*ab
b and
Shuanhu
Gao
*ab
aShanghai Key Laboratory of Green Chemistry and Chemical Processes, School of Chemistry and Molecular Engineering, East China Normal University, 3663N Zhongshan Road, Shanghai 200062, China. E-mail: hbhe@chem.ecnu.edu.cn; shgao@chem.ecnu.edu.cn
bShanghai Engineering Research Center of Molecular Therapeutics and New Drug Development, East China Normal University, 3663N Zhongshan Road, Shanghai 200062, China
First published on 19th February 2021
Abstract
The asymmetric total synthesis of (+)-xestoquinone and (+)-adociaquinones A and B was achieved in 6–7 steps using an easily accessible meso-cyclohexadienone derivative. The [6,6]-bicyclic decalin B–C ring and the all-carbon quaternary stereocenter at C-6 were prepared via a desymmetric intramolecular Michael reaction with up to 97% ee. The naphthalene diol D–E ring was constructed through a sequence of Ti(Oi-Pr)4-promoted photoenolization/Diels–Alder, dehydration, and aromatization reactions. This asymmetric strategy provides a scalable route to prepare target molecules and their derivatives for further biological studies.
Various halenaquinone-type natural products with promising biological activity have been isolated from marine sponges of the genus Xestospongia1 from the Pacific Ocean. (+)-Halenaquinone (1),2,3 (+)-xestoquinone (2), and (+)-adociaquinones A (3) and B (4)4,5 bearing a naphtha[1,8-bc]furan core (Fig. 1) are the most typical representatives of this family. Naturally occurring (−)-xestosaprol N (5) and O (6)6,7 have the same structure as 3 and 4 except for a furan ring, while a naphtha[1,8-bc]furan core can also be found in fungus-isolated furanosteroids (−)-viridin (7) and (+)-nodulisporiviridin E (8)8,9 (Fig. 1). Halenaquinone (1) was first isolated from the tropical marine sponge Xestospongia exigua2 and it shows antibiotic activity against Staphylococcus aureus and Bacillus subtilis. Xestoquinone (2) and adociaquinones A (3) and B (4) were firstly isolated, respectively, from the Okinawan marine sponge Xestospongia sp.4a and the Truk Lagoon sponge Adocia sp.,4b and they show cardiotonic,4a,c cytotoxic,4b,i antifungal,4i antimalarial,4j and antitumor4l activities. These compounds inhibit the activity of pp60v-src protein tyrosine kinase,4d topoisomerases I4e and II,4f myosin Ca2+ ATPase,4c,g and phosphatases Cdc25B, MKP-1, and MKP-3.4h,k
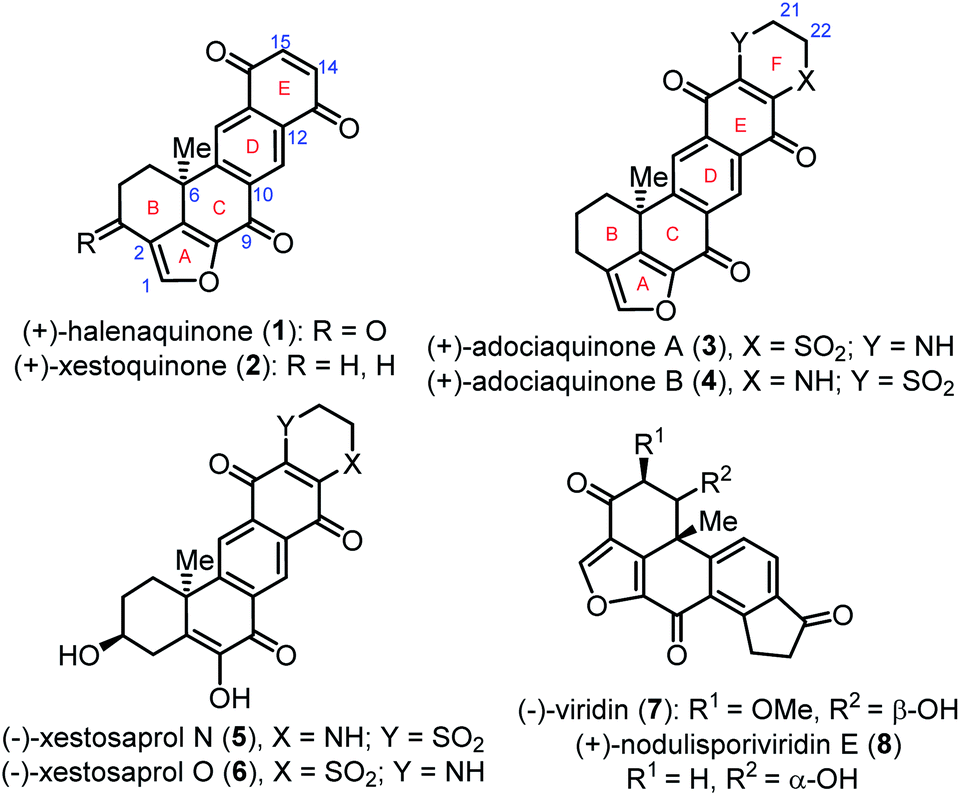 | ||
| Fig. 1 Structure of halenaquinone-type natural products and viridin-type furanosteroids. | ||
Owing to their diverse bioactivities, the synthesis of this family of natural compounds has been extensively studied, with published pathways making use of Diels–Alder,3a,d,e,5a–c,e,g furan ring transfer,5b Heck,3b,c,5f,7,9b,d palladium-catalyzed polyene cyclization,5d Pd-catalyzed oxidative cyclization,3f and hydrogen atom transfer (HAT) radical cyclization9c reactions. In this study, we report the asymmetric total synthesis of (+)-xestoquinone (2), (−)-xestoquinone (2′), and (+)-adociaquinones A (3) and B (4) (Fig. 1).
The construction of the fused tetracyclic B–C–D–E skeleton and the all carbon quaternary stereocenter at C-6 is a major challenge towards the total synthesis of xestoquinone (2) and adociaquinones A (3) and B (4). Based on our retrosynthetic analysis (Scheme 1), the all-carbon quaternary carbon center at C-6 of cis-decalin 12 could first be prepared stereoselectively from the achiral aldehyde 13via an organocatalytic desymmetric intramolecular Michael reaction.10,11 The tetracyclic framework 10 could then be formed via a Ti(Oi-Pr)4-promoted photoenolization/Diels–Alder (PEDA) reaction12–16 of 11 and enone 12. Acid-mediated cyclization of 10 followed by oxidation state adjustment could be subsequently applied to form the furan ring A of xestoquinone (2). Finally, based on the biosynthetic pathway of (+)-xestoquinone (2)4b,5c and our previous studies,7 the heterocyclic ring F of adociaquinones A (3) and B (4) could be prepared from 2via a late-stage cyclization with hypotaurine (9).
 | ||
| Scheme 1 Retrosynthetic analysis of (+)-xestoquinone and (+)-adociaquinones A and B. | ||
The catalytic enantioselective desymmetrization of meso compounds has been used as a powerful strategy to generate enantioenriched molecules bearing all-carbon quaternary stereocenters.10,11 For instance, two types of asymmetric intramolecular Michael reactions were developed using a cysteine-derived chiral amine as an organocatalyst by Hayashi and co-workers,11a,b while a desymmetrizing secondary amine-catalyzed asymmetric intramolecular Michael addition was later reported by Gaunt and co-workers to produce enantioenriched decalin structures.11c Prompted by these pioneering studies and following the suggested retrosynthetic pathway (Scheme 1), we first screened conditions for organocatalytic desymmetric intramolecular Michael addition of meso-cyclohexadienone 13 (Table 1) in order to form the desired quaternary stereocenter at C-6. Compound 13 was easily prepared on a gram scale via a four-step process (see details in the ESI†).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. (equiv.) | Additive (equiv.) | Solvent | Time | Yield/d.r. at C2b | e.e.c |
a All reactions were performed using 13 (5.8 mg, 0.03 mmol, 1.0 equiv., and 0.1 M) and a catalyst at room temperature in analytical-grade solvents, unless otherwise noted.
b The yields and diastereoisomeric ratios (d.r.) were determined from the crude 1H NMR spectrum of 14 using CH2Br2 as an internal standard, unless otherwise noted.
c The enantiomeric excess (e.e.) values were determined by chiral high-performance liquid chromatography (Chiralpak IG-H).
d Compound 13: 9.6 mg, 0.05 mmol, and 0.1 M.
e Isolated combined yield of 14a + 14b.
f Compound 13: 192 mg, 1.0 mmol, and 0.1 M.
g The d.r. values decreased to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 after purification by silica gel column chromatography.
h Compound 13: 1.31 g, 6.82 mmol, and 0.1 M.
i Isolated combined yield of 12a + 12b.
j The d.r. values were determined from the crude 1H NMR spectrum of 12 obtained from the one-pot process. :1 after purification by silica gel column chromatography.
h Compound 13: 1.31 g, 6.82 mmol, and 0.1 M.
i Isolated combined yield of 12a + 12b.
j The d.r. values were determined from the crude 1H NMR spectrum of 12 obtained from the one-pot process.
|
||||||
| 1 | (R)-cat.I (0.5) | — | Toluene | 10.0 h | 52%/10.3:1 |
14a: 96%; 14b: 75% |
| 2 | (R)-cat.I (1.0) | — | Toluene | 4.0 h | 60%/10.0:1 |
14a: 93%; 14b: 75% |
| 3 | (R)-cat.I (1.0) | — | MeOH | 4.0 h | 47%/5.5:1 |
14a: 86%; 14b: −3% |
| 4 | (R)-cat.I (1.0) | — | DCM | 10.0 h | 28%/24.0:1 |
14a: 91%; 14b: 7% |
| 5 | (R)-cat.I (1.0) | — | Et2O | 10.0 h | 22%/22.0:1 |
14a: 91%; 14b: 65% |
| 6 | (R)-cat.I (1.0) | — | MeCN | 10.0 h | 12%/2.6:1 |
14a: 90%; 14b: 62% |
| 7 | (R)-cat.I (1.0) | — | Toluene/MeOH (2:1) |
4.0 h | 47%/10.0:1 |
14a: 87%; 14b: −38% |
| 8d | (R)-cat.I (1.0) | AcOH (5.0) | Toluene | 4.0 h | 60%e/2.1:1 |
14a: 96%; 14b: 95% |
| 9d | (R)-cat.I (0.5) | AcOH (2.0) | Toluene | 6.0 h | 75%e/4.0:1 |
14a: 97%; 14b: 91% |
| 10d | (R)-cat.I (0.5) | AcOH (0.2) | Toluene | 6.0 h | 73%e/4.3:1 |
14a: 96%; 14b: 92% |
| 11f | (R)-cat.I (0.5) | AcOH (0.2) | Toluene | 6.0 h | 75%e/8.0:1g |
14a: 95%; 14b: 93% |
| 12h | (R)-cat.I (0.2) | AcOH (0.2) | Toluene | 9.0 h | 80%i/6.0:1j |
14a: 97%; 14b: 91% |

We initially investigated the desymmetric intramolecular Michael addition of 13 using (S)-Hayashi–Jørgensen catalysts,17 and found that the absolute configuration of the obtained cis-decalin was opposite to the required stereochemistry of the natural products (see Table S1 in the ESI†). In order to achieve the desired absolute configuration of the angular methyl group at C-6, (R)-cat.I was used for further screening. In the presence of this catalyst, the intramolecular Michael addition afforded 14a (96% e.e.) and 14b (75% e.e.) in a ratio of 10.3:1 and 52% combined yield (entry 1, Table 1). We assumed that the enantioselectivity of the reaction was controlled by the more sterically hindered aromatic group of (R)-cat.I, which protected the upper enamine face and allowed an endo-like attack by the si-face of cyclohexadienone, as shown in the transition state TS-A (Table 1). In order to increase the yield of this reaction and improve the enantioselectivity of 14b, we further screened solvents and additives. Increasing the catalyst loading from 0.5 to 1.0 equivalents and screening various reaction solvents did not improve the enantiomeric excess of 14b (entries 2–7, Table 1). Therefore, based on previous studies,11d,e we added 5.0 equivalents of acetic acid (AcOH) to a solution of compound 13 and (R)-cat.I in toluene, which improved the enantiomeric excess of 14b to 95% with a 60% combined yield (entry 8, Table 1). And, the stability of (R)-cat.I has also been verified in the presence of AcOH (see Table S2 in the ESI†). Further adjustment of the (R)-cat.I and AcOH amount and ratio (entries 9–12, Table 1) indicated that 0.2 equivalents each of (R)-cat.I and AcOH were the best conditions to achieve high enantioselectivity for both 14a and 14b, and it also increased the reaction yield (entry 12, Table 1). The enantioselectivity was not affected when the optimized reaction was performed on a gram scale: 14a (97% e.e.) and 14b (91% e.e.) were obtained in 80% isolated yield (entry 12, Table 1). We also found that the gram-scale experiments needed a longer reaction time which led a slight decrease of the diastereoselectivity. The purification of the cyclized products by silica gel flash column chromatography indicated that the major product 14a was epimerized and slowly converted to the minor product 14b (entry 11, Table 1). Both 14a and 14b are useful in the syntheses because the stereogenic center at C-2 will be converted to sp2 hybridized carbon in the following transformations. Therefore, the aldehyde group of analogues 14a and 14b was directly protected with 1,3-propanediol to give the respective enones 12a and 12b for use in the subsequent PEDA reaction.
Afterward, we selected the major cyclized cis-decalins 12a and 12a′ (obtained by using (S)-cat.I in desymmetric intramolecular Michael addition, see Table S1 in the ESI†) as the dienophiles to prepare the tetracyclic naphthalene framework 10 through a sequence of Ti(Oi-Pr)4-promoted PEDA, dehydration, and aromatization reactions (Scheme 2). When using 3,6-dimethoxy-2-methylbenzaldehyde (11) as the precursor of diene, no reaction occurred between 12a/12a′ and 11 under UV irradiation at 366 nm in the absence of Ti(Oi-Pr)4 (Scheme 2A). In contrast, the 1,2-dihydronaphthalene compounds 16a and 16a′ were successfully synthesized when 3.0 equivalents of Ti(Oi-Pr)4 were used. Based on our previous studies,13a,e the desired hydroanthracenol 15a was probably generated through the chelated intermediate TS-B and the cycloaddition occurred through an endo direction (Scheme 2B).18 The newly formed β-hydroxyl ketone groups in 15a and 15a′ could then be dehydrated with excess Ti(Oi-Pr)4 to form enones 16a and 16a′. These results confirmed the pivotal role of Ti(Oi-Pr)4 in this PEDA reaction: it stabilized the photoenolized hydroxy-o-quinodimethanes and controlled the diastereoselectivity of the reaction.
 | ||
| Scheme 2 PEDA reaction of 11 and enone 12. | ||
Subsequent aromatization of compounds 16a and 16a′ with 2,3-dichloro-5,6-dicyanobenzoquinone (DDQ) at 80 °C afforded compounds 10a and 10a′ bearing a fused tetracyclic B–C–D–E skeleton. The stereochemistry and absolute configuration of 10a were confirmed by X-ray diffraction analysis of single crystals (Scheme 3). The synthesis of (+)-xestoquinone (2) and (+)-adociaquinones A (3) and B (4) was completed by forming the furan A ring. Compound 10 was oxidized using bubbling oxygen gas in the presence of t-BuOK to give the unstable diosphenol 17a, which was used without purification in the next step. The subsequent acid-promoted deprotection of the acetal group led to the formation of an aldehyde group, which reacted in situ with enol to furnish the pentacyclic compound 18 bearing the furan A ring. The stereochemistry and absolute configuration of 18 were confirmed by X-ray diffraction analysis of single crystals (Scheme 3). Further oxidation of 18 with ceric ammonium nitrate afforded (+)-xestoquinone (2) in 82% yield. Following the same reaction process, (−)-xestoquinone (2′) was also synthesized from 10a′ in order to determine in the future whether xestoquinone enantiomers differ in biological activity. Further heating of a solution of (+)-xestoquinone (2) with hypotaurine (9) at 50 °C afforded a mixture of (+)-adociaquinones A (3) (21% yield) and B (4) (63% yield). We also tried to optimize the selectivity of this condensation by tuning the reaction temperature and pH of reaction mixtures (see Table S3 in the ESI†). The 1H and 13C NMR spectra, high-resolution mass spectrum, and optical rotation of synthetic (+)-xestoquinone (2), (+)-adociaquinones A (3) and B (4) were consistent with those data reported by Nakamura,4a,g Laurent,4j Schmitz,4b Harada5a,c and Keay.5d
 | ||
| Scheme 3 Total synthesis of (+)-xestoquinone and (+)-adociaquinones A and B. | ||
Conclusions
In summary, we developed a concise approach for the asymmetric total synthesis of (+)-xestoquinone (2) in 6 steps and of (+)-adociaquinones A (3) and B (4) in 7 steps from a known compound 13. Organocatalytic desymmetric intramolecular Michael addition was used to construct the cis-decalin skeleton bearing the all-carbon quaternary carbon center at the C-6 position. The B–C–D–E tetracyclic framework was then prepared through a Ti(Oi-Pr)4-mediated PEDA reaction, and further modifications led to the desired naphtha[1,8-bc]furan core. The application of this strategy to the synthesis of structurally related natural products is currently under investigation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (21971068 and 21772044), the “National Young Top-Notch Talent Support Program”, the Program of Shanghai Academic/Technology Research Leader (18XD1401500), the Program of Shanghai Science and Technology Committee (18JC1411303), the “Shuguang Program” supported by the Shanghai Education Development Foundation (grant numbers 19SG21), the Program for Changjiang Scholars and Innovative Research Team in University and “the Fundamental Research Funds for the Central Universities” for generous financial support.Notes and references
- For reviews of the natural products isolated from Xestospongia gens marine sponges and their bioactivities, see: (a) X. Zhou, T. Xu, X.-W. Yang, R. Huang, B. Yang, L. Tang and Y. Liu, Chem. Biodiversity, 2010, 7, 2201–2227 CrossRef CAS; (b) F. L. Liang, H. Liu, Y. Li, W. Ma, Y. Guo and W. He, Yaoxue Xuebao, 2014, 49, 1218–1237 Search PubMed.
- For the first isolation and bioactivities of halenaquinone, see: D. M. Roll, P. J. Scheuer, G. K. Matsumoto and J. Clardy, J. Am. Chem. Soc., 1983, 105, 6177–6178 CrossRef CAS.
- For the synthesis of halenaquinone, see: (a) N. Harada, T. Sugioka, Y. Ando, H. Uda and T. Kuriki, J. Am. Chem. Soc., 1988, 110, 8483–8487 CrossRef CAS; (b) A. Kojima, T. Takemoto, M. Sodeoka and M. Shibasaki, J. Org. Chem., 1996, 61, 4876–4877 CrossRef CAS; (c) A. Kojima, T. Takemoto, M. Sodeoka and M. Shibasaki, Synthesis, 1998, 581–589 CrossRef CAS; (d) H. S. Sutherland, F. E. S. Souza and R. G. A. Rodrigo, J. Org. Chem., 2001, 66, 3639–3641 CrossRef CAS; (e) M. A. Kienzler, S. Suseno and D. Trauner, J. Am. Chem. Soc., 2008, 130, 8604–8605 CrossRef CAS; (f) S. Goswami, K. Harada, M. F. El-Mansy, R. Lingampally and R. G. Carter, Angew. Chem., Int. Ed., 2018, 57, 9117–9121 CrossRef CAS.
- For the isolation and bioactivities of xestoquinone and adociaquinones A and B see: (a) H. Nakamura, J. i. Kobayashi, M. Kobayashi, Y. Ohizumi and Y. Hirata, Chem. Lett., 1985, 713–716 CrossRef CAS; (b) F. J. Schmitz and S. J. Bloor, J. Org. Chem., 1988, 53, 3922–3925 CrossRef CAS; (c) M. Kobayashi, A. Muroyama, H. Nakamura, J. Kobayashi and Y. Ohizumi, J. Pharmacol. Exp. Ther., 1991, 257, 90–94 CAS; (d) K. A. Alvi, J. Rodriguez, M. C. Diaz, R. Moretti, R. S. Wilhelm, R. H. Lee, D. L. Slate and P. Crews, J. Org. Chem., 1993, 58, 4871–4880 CrossRef CAS; (e) M. A. Bae, T. Tsuji, K. Kondo, T. Hirase, M. Ishibashi, H. Shigemori and J. Kobayashi, Biosci., Biotechnol., Biochem., 1993, 57, 330–331 CrossRef CAS; (f) G. P. Concepcion, T. A. Foderaro, G. S. Eldredge, E. Lobkovsky, J. Clardy, L. R. Barrows and C. M. Ireland, J. Med. Chem., 1995, 38, 4503–4507 CrossRef CAS; (g) M. Nakamura, T. Kakuda, Y. Oba, M. Ojika and H. Nakamura, Bioorg. Med. Chem., 2003, 11, 3077–3082 CrossRef CAS; (h) S. Cao, C. Foster, M. Brisson, J. S. Lazo and D. G. Kingston, Bioorg. Med. Chem., 2005, 13, 999–1003 CrossRef CAS; (i) M. Nakamura, T. Kakuda, J. Qi, M. Hirata, T. Shintani, Y. Yoshioka, T. Okamoto, Y. Oba, H. Nakamura and M. Ojika, Biosci., Biotechnol., Biochem., 2005, 69, 1749–1752 CrossRef CAS; (j) D. Laurent, V. Jullian, A. Parenty, M. Knibiehler, D. Dorin, S. Schmitt, O. Lozach, N. Lebouvier, M. Frostin, F. Alby, S. Maurel, C. Doerig, L. Meijer and M. Sauvain, Bioorg. Med. Chem., 2006, 14, 4477–4482 CrossRef CAS; (k) S. Cao, B. T. Murphy, C. Foster, J. S. Lazo and D. G. I. Kingston, Bioorg. Med. Chem., 2009, 17, 2276–2281 CrossRef CAS; (l) L. Du, F. Mahdi, S. Datta, M. B. Jekabsons, Y. D. Zhou and D. G. Nagle, J. Nat. Prod., 2012, 75, 1553–1559 CrossRef CAS.
- For the synthesis of xestoquinone and adociaquinones A and B, see: (a) N. Harada, T. Sugioka, H. Uda and T. Kuriki, J. Org. Chem., 1990, 55, 3158–3163 CrossRef CAS; (b) K. Kanematsu, S. Soejima and G. Wang, Tetrahedron Lett., 1991, 32, 4761–4764 CrossRef CAS; (c) N. Harada, T. Sugioka, T. Soutome, N. Hiyoshi, H. Uda and T. Kuriki, Tetrahedron: Asymmetry, 1995, 6, 375–376 CrossRef CAS; (d) S. P. Maddaford, N. G. Andersen, W. A. Cristofoli and B. A. Keay, J. Am. Chem. Soc., 1996, 118, 10766–10773 CrossRef CAS; (e) R. Carlini, K. Higgs, C. Older, S. Randhawa and R. Rodrigo, J. Org. Chem., 1997, 62, 2330–2331 CrossRef CAS; (f) F. Miyazaki, K. Uotsu and M. Shibasaki, Tetrahedron, 1998, 54, 13073–13078 CrossRef CAS; (g) H. S. Sutherland, K. C. Higgs, N. J. Taylor and R. Rodrigo, Tetrahedron, 2001, 57, 309–317 CrossRef CAS.
- For the isolation and bioactivities of xestosaprol N and O, see: (a) H.-S. Lee, Y.-J. Lee, C.-K. Kim, S.-K. Park, J. Soon Kang, J.-S. Lee and H. Jae Shin, Heterocycles, 2012, 85, 895–901 CrossRef; (b) R. M. Centko, A. Steinø, F. I. Rosell, B. O. Patrick, N. de Voogd, A. G. Mauk and R. J. Andersen, Org. Lett., 2014, 16, 6480–6483 CrossRef CAS.
- For the synthesis of xestosaprol N and O, see: Y. Shi, Y. Ji, K. Xin and S. Gao, Org. Lett., 2018, 20, 732–735 CrossRef CAS.
- For the isolation and bioactivities of viridin and nodulisporiviridin E, see: (a) P. W. Brian and J. G. McGowan, Nature, 1945, 156, 144–145 CrossRef CAS; (b) J. S. Moffatt, J. Chem. Soc. C, 1966, 734–743 RSC; (c) J. F. Grove, P. McCloskey and J. S. Moffatt, J. Chem. Soc. C, 1966, 743–747 RSC; (d) Q. Zhao, G.-D. Chen, X.-L. Feng, Y. Yu, R.-R. He, X.-X. Li, Y. Huang, W.-X. Zhou, L.-D. Guo, Y.-Z. Zheng, X.-S. Yao and H. Gao, J. Nat. Prod., 2015, 78, 1221–1230 CrossRef CAS.
- For the synthesis of viridin and nodulisporiviridin E, see: (a) E. A. Anderson, E. J. Alexanian and E. J. Sorensen, Angew. Chem., Int. Ed., 2004, 43, 1998–2001 CrossRef CAS; (b) M. Del Bel, A. R. Abela, J. D. Ng and C. A. Guerrero, J. Am. Chem. Soc., 2017, 139, 6819–6822 CrossRef CAS; (c) Y. Ji, Z. Xin, H. He and S. Gao, J. Am. Chem. Soc., 2019, 141, 16208–16212 CrossRef CAS; (d) Y. Ji, Z. Xin, Y. Shi, H. He and S. Gao, Org. Chem. Front., 2020, 7, 109–112 RSC.
- For the reviews of desymmetrization, see: (a) G. Maertens, M.-A. Ménard and S. Canesi, Synthesis, 2014, 46, 1573–1582 CrossRef; (b) X. P. Zeng, Z. Y. Cao, Y. H. Wang, F. Zhou and J. Zhou, Chem. Rev., 2016, 116, 7330–7396 CrossRef CAS.
- For the selected studies of organocatalytic desymmetric Michael addition, see: (a) Y. Hayashi, H. Gotoh, T. Hayashi and M. Shoji, Angew. Chem., Int. Ed., 2005, 44, 4212–4215 CrossRef CAS; (b) Y. Hayashi, H. Gotoh, T. Tamura, H. Yamaguchi, R. Masui and M. Shoji, J. Am. Chem. Soc., 2005, 127, 16028–16029 CrossRef CAS; (c) N. T. Vo, R. D. M. Pace, F. O'Har and M. J. Gaunt, J. Am. Chem. Soc., 2008, 130, 404–405 CrossRef CAS; (d) K. Patora-Komisarska, M. Benohoud, H. Ishikawa, D. Seebach and Y. Hayashi, Helv. Chim. Acta, 2011, 94, 719–745 CrossRef CAS; (e) H. Chen, X.-H. Li, J. Gong, H. Song, X.-Y. Liu and Y. Qin, Tetrahedron, 2016, 72, 347–353 CrossRef CAS.
- For the first study of the PEDA reaction, see: N. C. Yang and C. Rivas, J. Am. Chem. Soc., 1961, 83, 2213 CrossRef CAS.
- For the studies of the PEDA reaction in the Gao group, see: (a) B. Yang, K. Lin, Y. Shi and S. Gao, Nat. Commun., 2017, 8, 622 CrossRef; (b) B. Yang and S. Gao, Chem. Soc. Rev., 2018, 47, 7926–7953 RSC; (c) D. Xue, M. Xu, C. Zheng, B. Yang, M. Hou, H. He and S. Gao, Chin. J. Chem., 2019, 37, 135–139 CrossRef CAS; (d) D. Jiang, K. Xin, B. Yang, Y. Chen, Q. Zhang, H. He and S. Gao, CCS Chem., 2020, 2, 800–812 CrossRef; (e) X.-L. Lu, B. Yang, H. He and S. Gao, Org. Chem. Front., 2021 10.1039/d0qo01346c.
- For the studies of the PEDA reaction in the Melchiorre group, see: (a) L. Dell'Amico, A. Vega-Peñaloza, S. Cuadros and P. Melchiorre, Angew. Chem., Int. Ed., 2016, 55, 3313–3317 CrossRef; (b) L. Dell'Amico, V. M. Fernández-Alvarez, F. Maseras and P. Melchiorre, Angew. Chem., Int. Ed., 2017, 56, 3304–3308 CrossRef; (c) S. Cuadros, L. Dell'Amico and P. Melchiorre, Angew. Chem., Int. Ed., 2017, 56, 11875–11879 CrossRef CAS; (d) H. B. Hepburn, G. Magagnano and P. Melchiorre, Synthesis, 2017, 49, 76–86 CAS; (e) S. Cuadros and P. Melchiorre, Eur. J. Org. Chem., 2018, 2018, 2884–2891 CrossRef CAS.
- For the studies of the PEDA reaction in the Nicolaou group, see: (a) K. C. Nicolaou and D. Gray, Angew. Chem., Int. Ed., 2001, 40, 761–763 CrossRef CAS; (b) K. C. Nicolaou, D. Gray and J. Tae, Angew. Chem., Int. Ed., 2001, 40, 3675–3678 CrossRef CAS; (c) K. C. Nicolaou, D. Gray and J. Tae, Angew. Chem., Int. Ed., 2001, 40, 3679–3683 CrossRef CAS; (d) K. C. Nicolaou and D. L. F. Gray, J. Am. Chem. Soc., 2004, 126, 607–612 CrossRef CAS; (e) K. C. Nicolaou, D. L. F. Gray and J. Tae, J. Am. Chem. Soc., 2004, 126, 613–627 CrossRef CAS.
- For the selected studies of the PEDA reaction in other groups, see: (a) T. Durst, E. C. Kozma and J. L. Charlton, J. Org. Chem., 1985, 50, 4829–4833 CrossRef CAS; (b) K. Hashimoto, M. Horikawa and H. Shirahama, Tetrahedron Lett., 1990, 31, 7047–7050 CrossRef CAS; (c) A. G. Griesbeck and S. Stadtmüller, Chem. Ber., 1993, 126, 2149–2150 CrossRef CAS; (d) T. J. Connolly and T. Durst, Tetrahedron, 1997, 53, 15969–15982 CrossRef CAS; (e) M. Sobczak and P. J. Wagner, Tetrahedron Lett., 1998, 39, 2523–2526 CrossRef CAS; (f) B. Grosch, C. N. Orlebar, E. Herdtweck, W. Massa and T. Bach, Angew. Chem., Int. Ed., 2003, 42, 3693–3696 CrossRef CAS; (g) O. A. Mukhina, N. N. Bhuvan Kumar, T. M. Arisco, R. A. Valiulin, G. A. Metzel and A. G. Kutateladze, Angew. Chem., Int. Ed., 2011, 50, 9423–9428 CrossRef CAS; (h) N. N. B. Kumar, O. A. Mukhina and A. G. Kutateladze, J. Am. Chem. Soc., 2013, 135, 9608–9611 CrossRef CAS; (i) O. A. Mukhina, D. M. Kuznetsov, T. M. Cowger and A. G. Kutateladze, Angew. Chem., Int. Ed., 2015, 54, 11516–11520 CrossRef CAS; (j) O. A. Mukhina and A. G. Kutateladze, J. Am. Chem. Soc., 2016, 138, 2110–2113 CrossRef CAS; (k) D. M. Kuznetsov, O. A. Mukhina and A. G. Kutateladze, Angew. Chem., Int. Ed., 2016, 55, 6988–6991 CrossRef CAS; (l) A. Mavroskoufis, K. Rajes, P. Golz, A. Agrawal, V. Ruß, J. P. Götze and M. N. Hopkinson, Angew. Chem., Int. Ed., 2020, 59, 3190–3194 CrossRef CAS.
- For the reviews of Hayashi–Jørgensen catalysts, see: (a) K. L. Jensen, G. Dickmeiss, H. Jiang, Ł. Albrecht and K. A. Jørgensen, Acc. Chem. Res., 2012, 45, 248–264 CrossRef CAS; (b) G. J. Reyes-Rodríguez, N. M. Rezayee, A. Vidal-Albalat and K. A. Jørgensen, Chem. Rev., 2019, 119, 4221–4260 CrossRef.
- Based on our previous studies (ref. 13a and e), we concluded that Ti(Oi-Pr)4 plays a key role in the PEDA reaction, which chelated with the photo-generated Z-dienol and ortho methoxy group of 11, forming a relatively stable complex. This complex may exist as a dimeric titanium complex, given the observed relationships between the Ti dosage and reaction yield (ref. 13a). The ortho methoxy may serve as a key neighbouring group that helps to stabilize the short-lived photoenolized hydroxy-o-quinodimethane diene, which then interacts with dienophile 12a to give a chelated intermediate TS-B. Then the activated enone reacts with the diene component from the endo direction. After dissociation, cycloaddition product 15a was generated stereospecifically.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2050867–2050869. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc07089k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |