
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe atomic-level regulation of single-atom site catalysts for the electrochemical CO2 reduction reaction
Qingyun
Qu
,
Shufang
Ji
,
Yuanjun
Chen
*,
Dingsheng
Wang
 * and
Yadong
Li
* and
Yadong
Li
Department of Chemistry, Tsinghua University, Beijing 100084, China. E-mail: chenyuan15@mails.tsinghua.edu.cn; wangdingsheng@mail.tsinghua.edu.cn
First published on 20th February 2021
Abstract
The electrochemical CO2 reduction reaction (CO2RR) is viewed as a promising way to remove the greenhouse gas CO2 from the atmosphere and convert it into useful industrial products such as methane, methanol, formate, ethanol, and so forth. Single-atom site catalysts (SACs) featuring maximum theoretical atom utilization and a unique electronic structure and coordination environment have emerged as promising candidates for use in the CO2RR. The electronic properties and atomic structures of the central metal sites in SACs will be changed significantly once the types or coordination environments of the central metal sites are altered, which appears to provide new routes for engineering SACs for CO2 electrocatalysis. Therefore, it is of great importance to discuss the structural regulation of SACs at the atomic level and their influence on CO2RR activity and selectivity. Despite substantial efforts being made to fabricate various SACs, the principles of regulating the intrinsic electrocatalytic performances of the single-atom sites still needs to be sufficiently emphasized. In this perspective article, we present the latest progress relating to the synthesis and catalytic performance of SACs for the electrochemical CO2RR. We summarize the atomic-level regulation of SACs for the electrochemical CO2RR from five aspects: the regulation of the central metal atoms, the coordination environments, the interface of single metal complex sites, multi-atom active sites, and other ingenious strategies to improve the performance of SACs. We highlight synthesis strategies and structural design approaches for SACs with unique geometric structures and discuss how the structure affects the catalytic properties.
 Qingyun Qu | Qingyun Qu is currently a senior student at Tsinghua University, majoring in Chemistry. Her current research interests are the rational design and synthesis of single-atom site catalysts and their application in electrocatalysis. |
 Shufang Ji | Shufang Ji received her B.S. (2011), M.S. (2014), and Ph.D. (2018) degrees in chemistry from Qufu Normal University, Soochow University, and Tsinghua University, respectively. She is currently working in Prof. Yadong Li's group as a postdoctoral fellow in the Department of Chemistry at Tsinghua University. Her research interests involve the synthesis and applications of well-defined single-atom site catalysts and supported cluster catalysts. |
 Yuanjun Chen | Yuanjun Chen received his BS degree from the College of Chemical Engineering at Beijing University of Chemical Technology in 2015, and his PhD degree from the Department of Chemistry, Tsinghua University in 2020 under the supervision of Prof. Yadong Li. His research interests focus on the syntheses and applications of functional nanocatalysts and single-atom site catalysts. |
 Dingsheng Wang | Dingsheng Wang received his BS degree from the Department of Chemistry and Physics, University of Science and Technology of China in 2004, and his PhD degree from the Department of Chemistry, Tsinghua University in 2009, under the supervision of Prof. Yadong Li. He did his postdoctoral research at the Department of Physics, Tsinghua University with Prof. Shoushan Fan. He joined the faculty of the Department of Chemistry, Tsinghua University in 2012. |
 Yadong Li | Yadong Li received his B.S. degree from the Department of Chemistry at Anhui Normal University in 1986 and his Ph.D. from the Department of Chemistry at the University of Science and Technology of China in 1998. He joined the faculty of the Department of Chemistry at Tsinghua University in 1999. He was elected as a member of the Chinese Academy of Sciences in 2011. |
1. Introduction
The scalable applications of fossil fuels have generated huge technological developments and resulted in significant improvements in people's living conditions. However, it also causes a high concentration of CO2 in the atmosphere, which contributes to the aggravation of the greenhouse effect, thus shifting ecological balance and accelerating ice sheet melting in Eastern Antarctica.1 There is a general agreement that efforts should be made to control and reduce the CO2 concentration in the atmosphere. Although fossil fuel is a significant part of our energy structure, CO2 emission is inevitable. Capturing, storing and converting CO2 has become a promising way to remove CO2 from the atmosphere.2 Significant efforts have been made in this field, the CO2 reduction reaction (CO2RR) has become one of the hottest topics among them.3 The products of the CO2RR are mainly methane, methanol, formate, ethanol, and so on, which are widely used in industrial production. The valuable products of CO2RR have attracted extensive attention in this field in recent years. There are various approaches used to achieve CO2 reduction, compared to thermocatalysis, biocatalysis and photocatalysis, electrocatalysis has more moderate reaction conditions, lower costs and higher yields.4–10 The electrochemical reduction of CO2 can use electricity generated from renewable energy, such as wind energy and hydro-energy.11 The conversion from electrical energy to the chemical energy of CO2RR products can be viewed as a storage form of renewable energy, which is safe and convenient for transportation and long-term storage.However, there still remains great challenges for the CO2RR, as it has sluggish kinetics, a complex multi-electron mechanism and a hydrogen evolution reaction (HER) competition reaction.12,13 The CO2RR performance of catalysts is related to the rate determining steps, including CO2 adsorption, *COOH and *CO formation on the active sites, which is decided by the adsorption free energy of CO2, the binding energy of the intermediates, the electron transfer rate and so on.14,15 For further reactions, the binding energy of intermediates, especially *CO is also crucial to the formation of C2+ products. Therefore, it is of great importance to manipulate the electronic and geometric structure of the catalysts, which can regulate the rate determining steps by changing the binding and formation energies of the intermediates, thus changing the activity and selectivity of the CO2RR.16,17
Among all the kinds of catalysts used, single-atom site catalysts (SACs) have drawn significant attention owing to their unique structure and properties, which have great potential to bridge the gap between homogenous and heterogeneous catalysts.18–22 SACs feature isolated metal atoms dispersed on substrates as active sites, which result in multiple advantages: (i) they have the maximum theoretical atom utilization.23–25 (ii) Single metal atoms, as active centers, generate low-coordination environments, as well as unique electronic and geometric structures, leading to enhanced catalytic activities.26–30 (iii) The uniformity of the electronic and geometric structure of the active sites in SACs endows them with similar spatial and electronic interactions with substrate molecules, which facilitates an improvement in the catalytic selectivity.31,32 (iv) Furthermore, the structural simplicity and uniformity of the SACs are conducive to the characterization and identification of catalytic active sites, which enables SACs to serve as ideal models to study reaction mechanisms.33–36 This also indicates opportunities to design and regulate the active sites of SACs at the atomic level.37–42 (v) Specifically, in the electrochemical CO2RR, atomically dispersed metal atoms are suitable for single or coupled proton-electron transfer, and enhance the CO2 adsorption and activation, leading to a high selectivity for C1 products.43–46 Meanwhile, isolated metal atoms offer an individual single adsorption site for CO2, thus C2+ products are more difficult to obtain on SACs.47–49 Despite substantial efforts being made to fabricate various SACs, the principles of regulating the intrinsic electrocatalytic performance of the single-atom active sites still needs to be sufficiently investigated.50–52
In this perspective article, we first focus on the controlled synthesis of SACs at the atomic scale for electrochemical CO2 reduction reactions with regards to five aspects: regulation of the central metal atom, the coordination environment, the interface of single metal complex sites, multi-atom active sites and other ingenious strategies to improve the performance of the SACs. Finally, the challenges and potential for precise control over the synthesis and accurate characterization and identification of the active sites of SACs, as well as a study of the reaction mechanisms on electrochemical CO2RR, will be discussed.
2. Design and synthesis of SACs for the CO2RR
The active sites of SACs can be divided into central metal atoms and the surrounding coordination environment, the influences of which on the activity and selectivity will be discussed first. Then, we will focus on the kind of SACs that constitute anchored metal complexes with constructed structures on the substrates, which are different from the traditional SACs that involve the building of coordination structures on substrates by anchoring metal atoms. Moreover, as the active sites of traditional SACs are separated atomically, they are expected to be more favorable to the C1 product. In the fourth section, we will discuss multi-atom active sites which have a huge potential to obtain C2+ products as they have multiple CO2 adsorption sites which can introduce multiple CO2 molecules into a single reaction. Finally, we will review other ingenious strategies to improve the CO2RR performance of the SACs.2.1 Regulation of the central metal
Although SACs based on noble metals have revealed a good activity and selectivity, the shortage of noble metal resources and their expensive price has motivated researchers to investigate non-noble metal SACs.54,55 Cu and transition metals such as Fe, Ni, Co and so on are viewed as promising candidates because of their rich d orbital electrons and the variation in the valence state. Although the electrochemical CO2RR has multiple products, CO, as the simplest one, is often used to evaluate the performance of the central metals. Li et al. investigated the different catalytic performances of metal–Nx (MNx, M = Mn, Fe, Co, Ni, and Cu) moieties on the M–N–C catalysts.56 Catalytic experiments revealed a volcano trend between the atomic number of the tested metal and their electrochemical CO2RR performance, which can be explained by the Gibbs free energy changes of the rate determined steps. Based on different electrochemical potentials, Fe and/or Co are located at the top of the volcano (Fig. 1a). In contrast, there is no obvious trend towards faradaic efficiency for CO. The high selectivity of Fe and Co (FECO > 80%) can be explained by the binding energy of 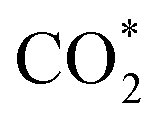 and H*, which are associated with the electron number of d-antibonding. Ju et al. investigated the CO2-to-CO electrochemical reduction catalytic performance of the M–N–C catalysts with different metal centers (M = Mn, Fe, Co, Ni, Cu).40 Fe–N–C showed a maximum FECO of 65% at −0.55 V versus RHE (VRHE), and Ni–N–C exhibited a maximum FECO of 85% at −0.78 VRHE. Combined with the density functional theory (DFT) calculations, Fe–N–C and Ni–N–C were predicted to be promising SACs for CO2-to-CO (Fig. 1b). Zheng et al. developed a universal principle to synthesize a series of SACs–M–N–C (M = Fe, Co, Ni, Cu) using in situ pyrolysis.57 The experimental results revealed Ni > Fe > Cu > Co as the catalytic performance sequence of SAs–M–N–C in electrochemical CO2-to-CO conversion. They attributed this result to the intrinsic property of the pyrrole-type M–N4 structures. Pan et al. synthesized the M–N–C (M = Fe, Co) catalysts with M–N4 sites from the Fe- or Co-doped MOFs precursors.58 Fe–N–C reached a maximum FECO of 93% at −0.58 VRHE, which is much higher than the maximum FECO of Co–N–C, which was 45% at −0.59 VRHE (Fig. 2a and b). This experimental result is consistent with the results of the DFT calculations. In conclusion, regardless of the nuances which may be caused by different substrates, Ni and Fe showed the most promising catalytic activities among the M–N–C catalysts for the CO2-to-CO electrochemical CO2RR, which was verified using multiple experiments.59–61
and H*, which are associated with the electron number of d-antibonding. Ju et al. investigated the CO2-to-CO electrochemical reduction catalytic performance of the M–N–C catalysts with different metal centers (M = Mn, Fe, Co, Ni, Cu).40 Fe–N–C showed a maximum FECO of 65% at −0.55 V versus RHE (VRHE), and Ni–N–C exhibited a maximum FECO of 85% at −0.78 VRHE. Combined with the density functional theory (DFT) calculations, Fe–N–C and Ni–N–C were predicted to be promising SACs for CO2-to-CO (Fig. 1b). Zheng et al. developed a universal principle to synthesize a series of SACs–M–N–C (M = Fe, Co, Ni, Cu) using in situ pyrolysis.57 The experimental results revealed Ni > Fe > Cu > Co as the catalytic performance sequence of SAs–M–N–C in electrochemical CO2-to-CO conversion. They attributed this result to the intrinsic property of the pyrrole-type M–N4 structures. Pan et al. synthesized the M–N–C (M = Fe, Co) catalysts with M–N4 sites from the Fe- or Co-doped MOFs precursors.58 Fe–N–C reached a maximum FECO of 93% at −0.58 VRHE, which is much higher than the maximum FECO of Co–N–C, which was 45% at −0.59 VRHE (Fig. 2a and b). This experimental result is consistent with the results of the DFT calculations. In conclusion, regardless of the nuances which may be caused by different substrates, Ni and Fe showed the most promising catalytic activities among the M–N–C catalysts for the CO2-to-CO electrochemical CO2RR, which was verified using multiple experiments.59–61
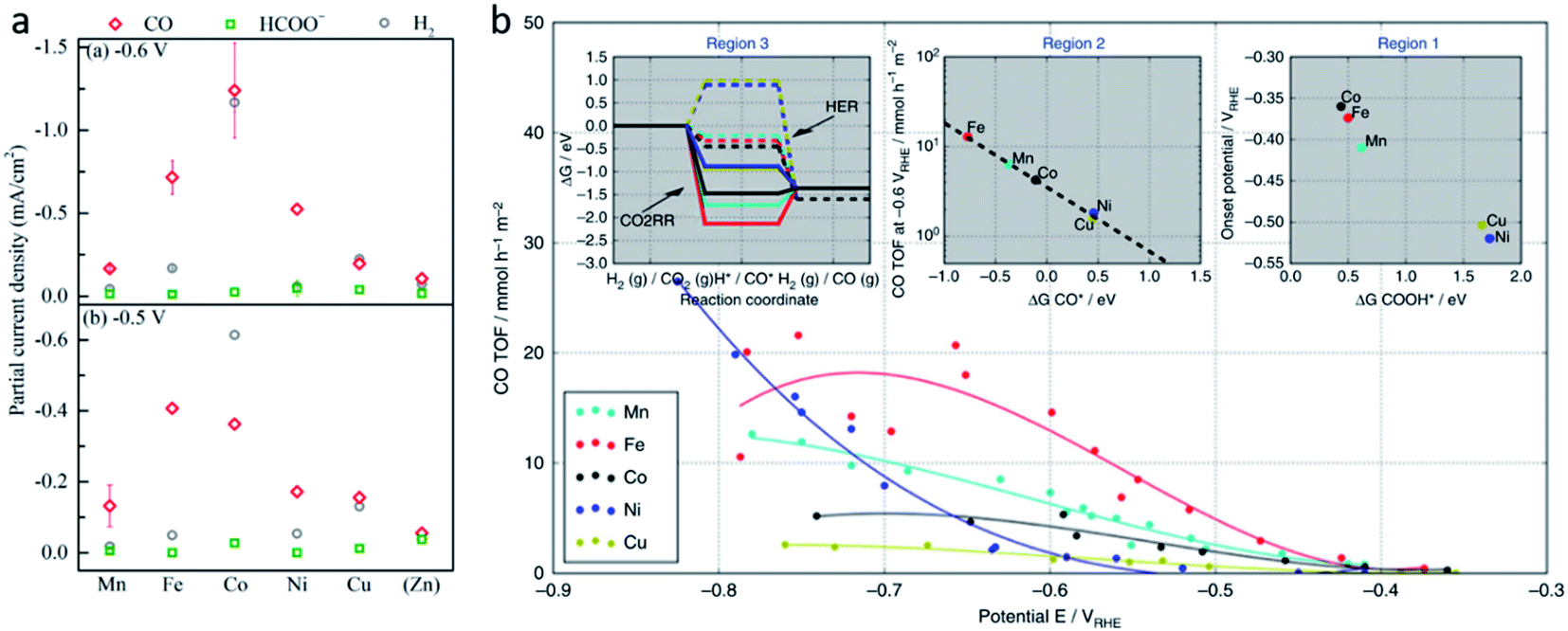 | ||
| Fig. 1 (a) The volcano trend of M–N–C catalysts in terms of electrochemical CO2RR performance. Partial current densities of M–N–C catalysts for CO, HCOO−, and H2 products at −0.6 VRHE and −0.5 VRHE. Reproduced from ref. 56 with permission from the American Chemical Society, copyright 2019. (b) Experimental CO TOFs of M–N–C catalysts. Reproduced from ref. 40 with permission from Springer Nature, copyright 2017. | ||
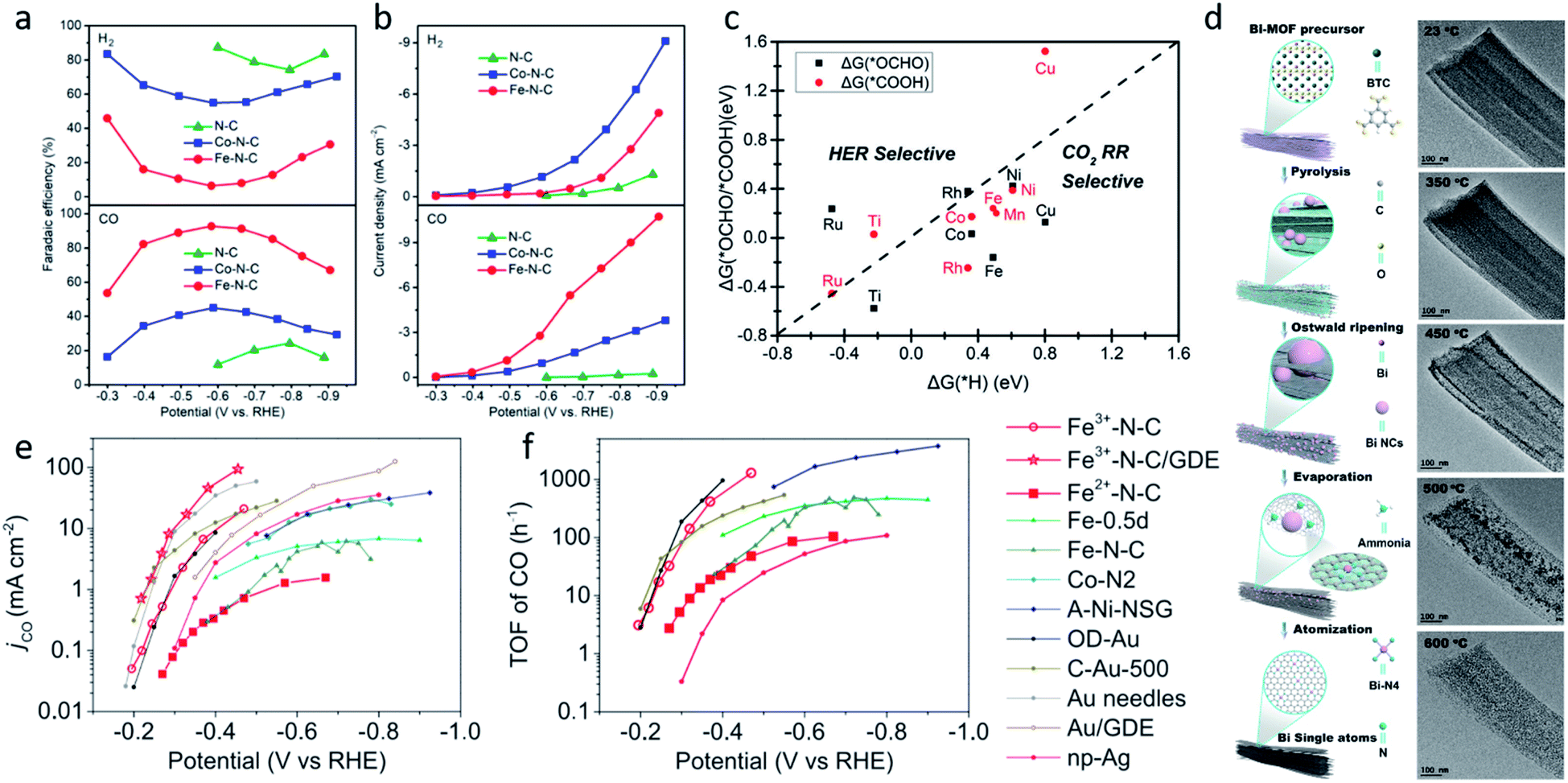 | ||
| Fig. 2 The Faradaic efficiencies for CO and H2 (a) and the current densities (b) of N–C, Co–N–C, and Fe–N–C. Reproduced from ref. 58 with permission from the American Chemical Society, copyright 2018. (c) Gibbs free energy changes for the first protonation step. Reproduced from ref. 63 with permission from The Royal Society of Chemistry, copyright 2018. (d) A schematic illustration of the process of Bi-MOF transformation to single Bi atoms and TEM images of Bi-MOF pyrolyzed at different temperatures. Reproduced from ref. 64 with permission from the American Chemical Society, copyright 2019. (e) CO current density (jCO) and (f) apparent TOFs of CO production of Fe3+–N–C and of Fe2+–N–C in comparison with other reported catalysts. Reproduced from ref. 59 with permission from the American Association for the Advancement of Science, copyright 2019. | ||
Furthermore, different central metals have different binding energies with intermediates such as *CO, *CO2−, and *COOH, which may lead to different products. For example, a high *COOH binding energy and weak *CO binding energy leads to CO, the Sn, Pb species with low *CO2− binding energy lead to formate or formic acid, and metallic Cu with an enhanced *CO and *COOH binding energy leads to hydrocarbons.49,56,62 Zhang et al. synthesized N-doped porous carbon supported Cu single atom catalysts for electrochemical CO2 reduction to acetone, which exhibited a maximum FEacetone of 36.7% at −0.36 VRHE.32 Han et al. designed microporous N-doped carbon supported Zn single atom catalysts for reducing CO2 to CH4, which reached a maximum FECH4 of 85% at −1.8 VSCE.45
However, the design of SACs for complicated products is still hindered by complicated mechanisms and various by-products. With the help of theoretical calculations, some researchers have provided guidance for future experimental efforts. Cui et al. used DFT calculations and a computational hydrogen electrode (CHE) model to investigate C2N–graphene supported SACs with 12 different metal centers (M@C2N).63 They conducted DFT calculations with the models of the metal atom confined in the N6 cavity as the active sites. Among the 12 metals, Ti, Mn, Fe, Co, Ni, Cu, Rh, and Ru preferred the CO2RR to the HER under 0 V (Fig. 2c). Furthermore, different M@C2N were predicted to produce methane (M = Ti, Mn) and methanol (M = Fe, Co, Ni, Ru), as their different carbophilicity and oxophilicity could determine the formation of the important intermediates *CH3O and *CH2OH, leading to the products CH4 and *CH3OH, respectively.
In addition to the transition metals, some SACs with main group elements also reveal an excellent performance in the electrochemical CO2RR. Zhang et al. prepared a Bi-single-atom catalyst supported on N-doped carbon networks (Bi SAs/NC) for CO2 reduction to CO.64 Bi SAs/NC was synthesized by a bismuth-based metal organic framework (MOF) and dicyandiamide (DCD) thermal decomposition. During the decomposition, Bi nanoparticles (NPs) were generated first, the ammonia generated from DCD decomposition promoted the atomization of the Bi NPs (Fig. 2d). Under −0.5 VRHE, Bi SAs/NC exhibits a maximum FECO of 97%, and a current density of 3.9 mA cm−2. DFT calculations illustrated that the Bi–N4 sites act as the active centers, which promotes CO2 activation and *COOH formation.
2.2 Regulation of the coordination environment
The coordination atoms, coordination number and coordination steric structure are the main influencing factors in the coordination environment of the single-atom sites, and these play important roles in regulating the electronic and geometric structures of the single-atom sites. Different coordination atoms directly influence the strength of the EMSI, changing the valence electron distribution of single-atom sites, thus leading to a different catalytic performance for the CO2RR. Nitrogen atoms are the most common coordination atoms owing to the significant development of M–N–C catalysts.69–74 Therefore, plenty of studies have investigated the hybrid coordination environment of the nitrogen and carbon atoms. Hossain et al. explored a series of Ni-single-atom catalysts with different N and C hybrid coordination environments of the Ni sites (Ni–N2C2, Ni–N3C1, Ni–N4) on a graphene support.75 The grand canonical potential kinetics (GCP-K) methodology was developed to predict their catalytic activities for CO2 reduction to CO and H2 production. The prediction revealed the HER performance is correlated with the number of coordinated C atoms, indicating Ni–N4 is least favorable for HER. Ni–N4 exhibited the best performance for CO2 reduction, reaching 40 mA cm−2 with FECO ≈ 100% at −1.05 VRHE (Fig. 3a). Research on Co1–N4−xCx conducted by Geng et al. reported similar results.76 Their study revealed that compared to Co1–N4−xCx, Co1–N4 exhibited a higher binding strength with CO2. This resulted in the easier activation of CO2, and led to a higher current density and TOF of Co1–N4. However, other studies may come to a different conclusion. Zhao et al. applied ab initio molecular dynamics (AIMD) to investigate the catalytic kinetics of Ni-single-atom catalysts supported by N-doped graphene toward CO2-to-CO electrochemical reduction via a “slow-growth” sampling approach.77 Calculations predicted that the higher Fermi level of the Ni–N4 site at the charge-neutral state led to its lower capacity, suggesting its higher electrochemical barriers and lower activity. The activity of Ni–C4 and Ni–C3N1 sites differs by the CO desorption energy, the lower desorption barrier of the Ni–C3N1 sites, resulting in its higher activity. As a result, the coordination environment with one nitrogen and three carbon atoms is believed to have the best activity and selectivity for CO2RR (Fig. 3b). The different results could be attributed to several factors. Firstly, the different calculation models, which focused on different rate determined steps and are inaccurate compared to the real experiment. Secondly, the different catalytic activity evaluation indicators, such as the FE, current density, TOF, and so on. Moreover, the different substrates and their synthesis process may also influence the activity, even though single-atom sites have a similar composition.
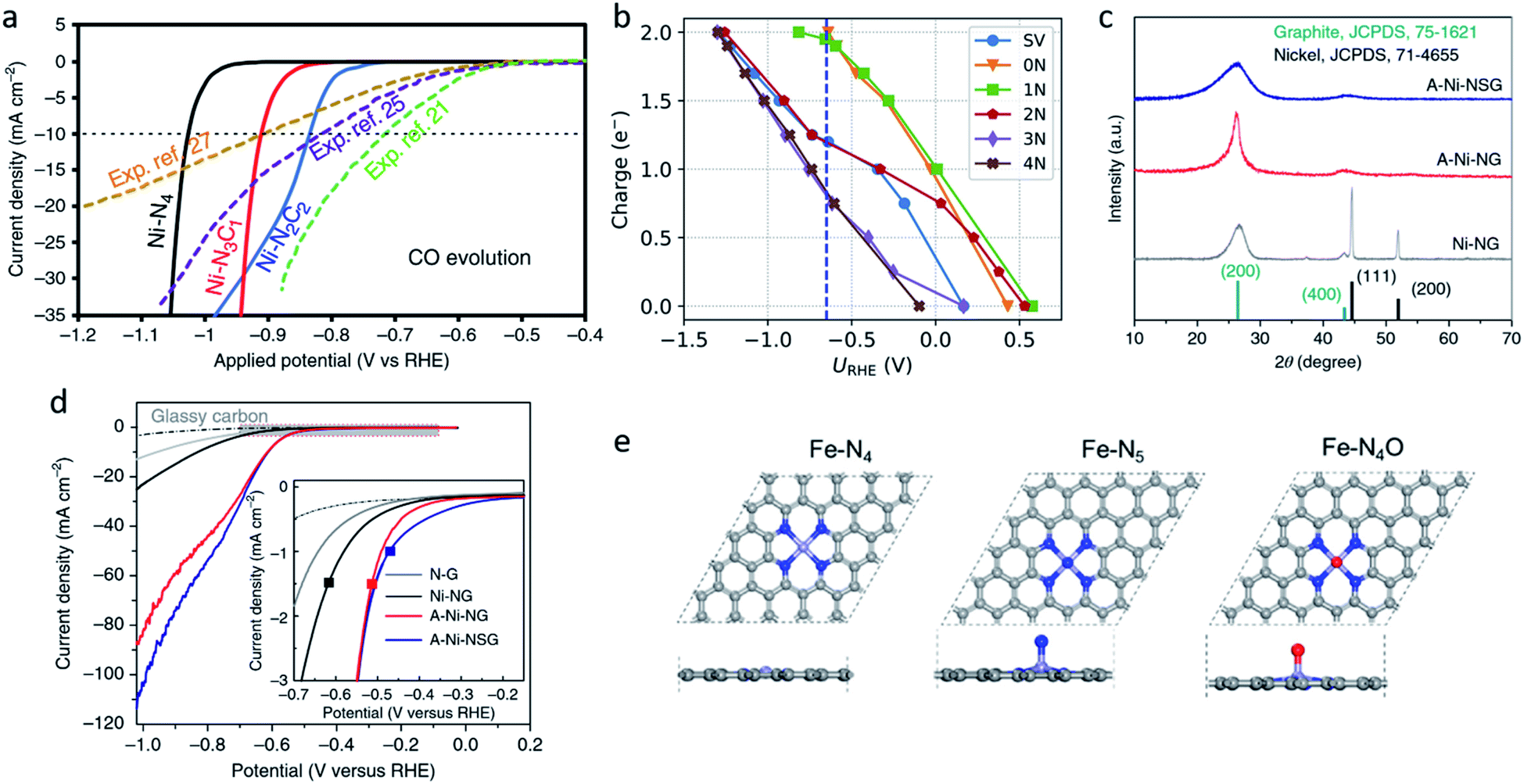 | ||
| Fig. 3 (a) Calculated jCO values in the CO2RR on Ni–N2C2, Ni–N3C1, and Ni–N4. Reproduced from ref. 75 with permission from Springer Nature, copyright 2020. (b) Charge capacities of different sites under different potentials for *COOH. Reproduced from ref. 77 with permission from the American Chemical Society, copyright 2020. (c) XRD patterns and (d) LSV curves for A-Ni-NSG, A-Ni-NG, and Ni-NG. Reproduced from ref. 26 with permission from Springer Nature, copyright 2018. (e) Top views and side views of the Fe–N4, Fe–N5 and Fe–N4O sites. Reproduced from ref. 78 with permission from Elsevier, copyright 2020. | ||
Oxygen and sulphur atoms, which are electron-rich and easy to dope, can also be introduced to the hybrid coordination environment. The hybrid coordination environment not only regulates the electronic structure of single-atom sites, but also creates defects on the substrate, leading to a better catalytic activity and stability. Yang et al. reported a Ni single-atom site catalyst supported on N,S-doped graphene (A-Ni-NSG) and N-doped graphene (A-Ni-NG).26 The weaker and wider X-ray diffractometry (XRD) peaks indicated A-Ni-NSG has a defect-rich structure compared to A-Ni-NG (Fig. 3c). Multiple characterizations revealed the existence of Ni–S bonds in A-Ni-NSG. Compared to A-Ni-NG, A-Ni-NSG exhibited a lower onset overpotential, larger current density and better chemical stability, suggesting the introduction of S atoms into the coordination environment improved the catalytic activity (Fig. 3d). Wang et al. synthesized a nanoporous carbon supported Fe single-atom catalyst with a Fe–N4O structure.78 The extended X-ray absorption fine structure (EXAFS) wavelet transform (WT) results confirmed the existence of Fe–O. In the Fe–N4O structure, four N atoms were coordinated in a planar position to Fe, while the O atom was coordinated out-of-plane (Fig. 3e). This coordination structure was optimized using DFT calculations as the lowest free energy structure. DFT calculations also revealed the CO desorption barrier on Fe–N4O is relatively low, leading to easier CO desorption and alteration of the rate determining step (RDS). The RDS changed from *CO-to-CO to CO2-to-*COOH, indicating that Fe–N4O had a better performance at a high overpotential. The Fe–N4O catalyst reached FECO ≈ 96% at −0.57 VRHE, suggesting the Fe–N4O structure could be a promising direction for designing M–N–C CO2RR catalysts. Also, there are some novel coordination structures, such as the π → d coordination interactions which are not viewed as an atom coordination, but as a bond coordination instead. Shen et al. reported the novel structure of a triphenylene–graphdiyne supported Cu-single-atom catalyst (Cu@TP-GDY) using DFT calculations.79 The DFT calculations unveiled the atomically dispersed Cu species were confined in triangular pores by π → d interactions between diacetylenic linkages and Cu atoms. CO2-to-COOH* was predicted to be the rate determined steps on Cu@TP-GDY. Cu@TP-GDY was expected to show a better catalytic performance towards CO2RR, as its overpotential was found to be lower than most of the Cu-based catalysts.
The change of the coordination number also significantly affects the activity of single-atom sites, which will not only influence the geometric structure, but also reattribute the electronic structures, leading to different catalytic performances. Fan et al. constructed a Ni single atom catalyst (NC-CNTs (Ni)) through the pyrolysis of Ni particles containing commercial multi-walled carbon nanotubes (MWCNTs) coated with a polymeric layer (Fig. 4a).68 EXAFS fitting results indicated the Ni–N coordination number is 2.5 ± 0.2. The authors viewed NiN3 moieties as the active center in the NC-CNTs (Ni). DFT calculations revealed the formation of *COOH has a lower free energy on NiN3 (pyrrolic) than on NiN4, which means the NC-CNTs (Ni) exhibit a better catalytic performance for electrochemical CO2-to-CO conversion (Fig. 4b).
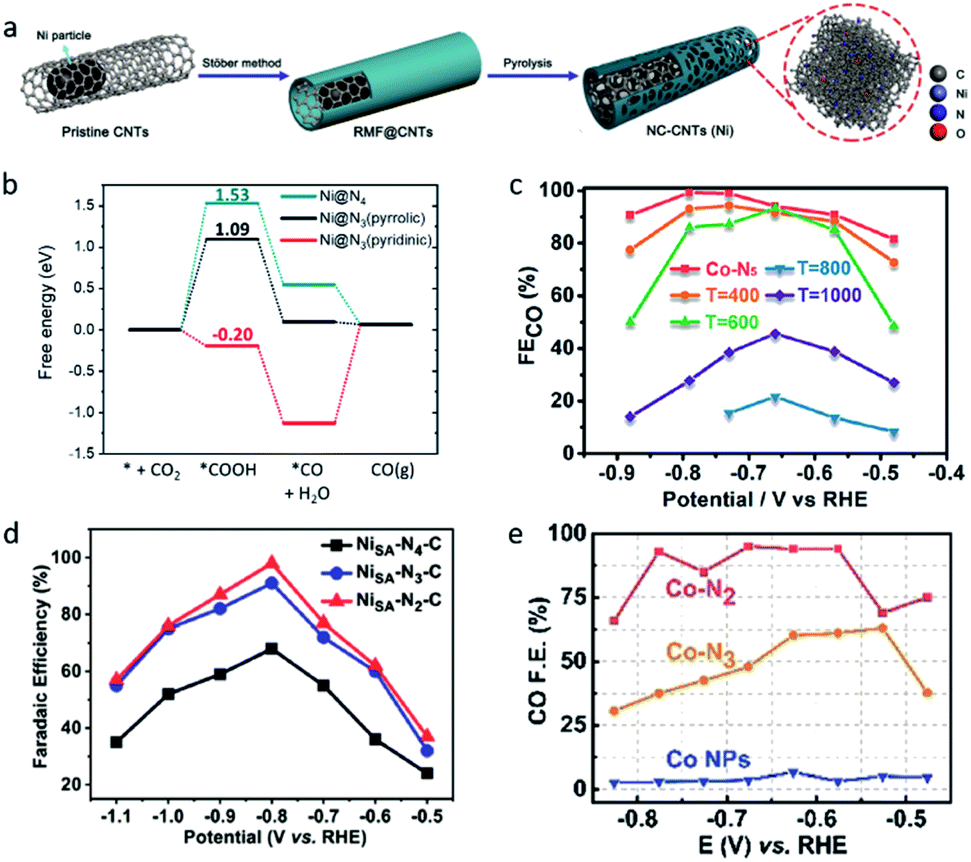 | ||
| Fig. 4 (a) A schematic illustration of the formation of NC-CNTs (Ni). (b) Free energies of the CO2RR to CO on Ni@N4 and Ni@N3. Reproduced from ref. 68 with permission from John Wiley and Sons, copyright 2019. (c) FECO values of Co–N5/HNPCSs-T at different applied potentials. Reproduced from ref. 35 with permission from the American Chemical Society, copyright 2018. (d) FECO values of Ni–Nx–C SACs (x = 2,3,4) at different applied potentials. Reproduced from ref. 38 with permission from John Wiley and Sons, copyright 2019. (e) FECO values of Co–Nx SACs (x = 2,3,4) at different applied potentials. Reproduced from ref. 39 with permission from John Wiley and Sons, copyright 2018. | ||
The most common strategy to create SACs with a lower coordination number is high temperature pyrolysis. Pan et al. synthesized polymer-derived hollow N-doped porous carbon spheres (HNPCSs) supported Co–N5 sites catalysts.35 Cobalt phthalocyanines are uniformly anchored on the HNPCSs, together with the strong electronic interactions which were revealed by Raman spectroscopy. As the pyrolysis temperature was raised to 400 and 600 °C, the FECO of Co–N5/HNPCSs dropped and the coordination number decreased to 4 and 3 (Fig. 4c). The Co–N5/HNPCSs catalyst exhibited a high FECO (>90%) over a wide potential range (−0.57–0.88 VRHE), which can be attributed to the low *COOH formation free energy and moderate binding energy of CO on the Co–N5 sites. In contrast, a negative correlation between the N coordination numbers and catalytic activity was reported. In research published by Gong et al., a series of N-doped carbon supported Ni SACs (Ni–Nx–C, x = 2,3,4), Ni–N2–C achieved the highest FECO of 98% and the highest TOF of 1622 h−1 (Fig. 4d).38 Wang et al. reported Co single-atom catalysts on N-doped porous carbon (Co–Nx SACs, x = 2, 3, 4), the Co–N2 SACs exhibited a high FECO of 94% and a high current density (Fig. 4e).39 The uncertain correlation between the N coordination numbers and the catalytic performance in a different series of SACs may be explained by their different coordination structures, owing to the different synthesis processes. Co–Nx/HNPCSs (x = 3, 4) was obtained by the pyrolysis of Co–N5/HNPCSs, while Ni–Nx–C SACs (x = 2, 3, 4) and Co–Nx SACs (x = 2, 3, 4) were obtained by the pyrolysis of the MOFs precursors under different temperatures.
Although it is difficult to control the coordination number exactly using the pyrolysis method, some well-designed strategies have been developed to produce SACs with a defined coordination number. Rong et al. developed a strategy to synthesize vacancy-defect Ni SACs (Ni–N3–V).37 They first prepared Ni–N3O single-atom catalysts under 500 °C. As the temperature rises, the coordinated oxygen atom on the substrate can be gradually evaporated leaving vacancy-defects, thus the Ni–N3–V SACs were formed (Fig. 5a). The structure of the Ni–N3–V sites was confirmed using Fourier transform (FT)-EXAFS fitting in the R space. At −0.9 VRHE, Ni–N3–V exhibited a high FECO of 90%, a high current density of 65 mA cm−2 and a high TOF of 1.35 × 105 h−1. DFT calculations were performed on Ni–N4, Ni–N3, (in which the Ni atom was coordinated with three nitrogen atoms and exhibited a symmetrical structure) and Ni–N3–V (in which the Ni atom was coordinated with three nitrogen atoms and a vacancy, and exhibited an asymmetrical structure). Ni–N3–V and Ni–N3 both exhibited a lower free energy in the RDS (the formation of *COOH), but Ni–N3 encountered difficulties when releasing the CO, suggesting the vacancy-defect structure in Ni–N3–V greatly promoted the catalysis of CO2 reduction. Zhang et al. reported structured FeN5 SACs on the N-doped graphene support through the pyrolysis of hemin and melamine.28 A large amount of melamine was added, which served as a nitrogen source to promote the formation of Fe–N5 sites. The N atoms in the adjacent layers provided axial ligand sites for FeN4 to convert to FeN5 (Fig. 5b). The FeN5 catalyst showed a greater performance towards FeN4 in CO2-to-CO conversion owing to the *CO weaker binding strength on FeN5, and reached a maximum FECO of 97.0% at −0.46 VRHE.
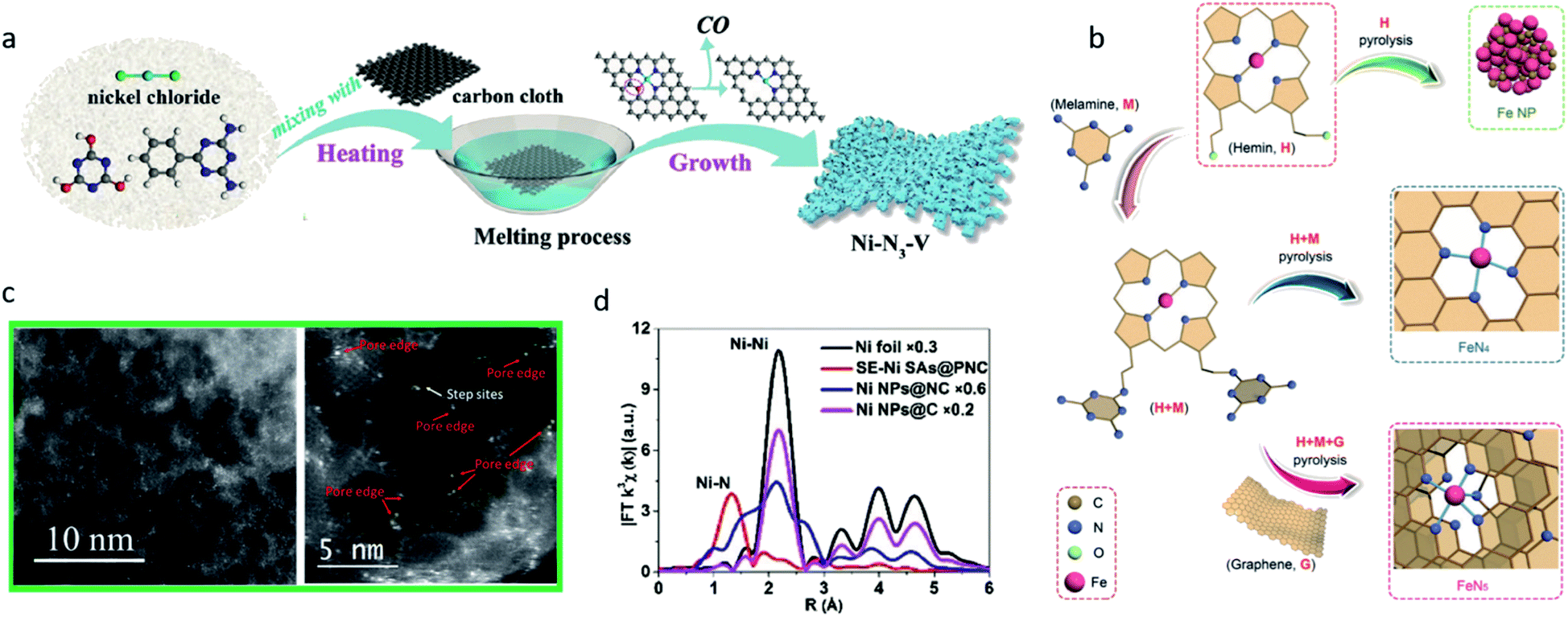 | ||
| Fig. 5 (a) A schematic illustration of the synthesis of Ni–N3–V. Reproduced from ref. 37 with permission from John Wiley and Sons, copyright 2020. (b) A schematic illustration of the synthesis of FeN4 and FeN5 SACs. Reproduced from ref. 28 with permission from John Wiley and Sons, copyright 2019. (c) AC-STEM images of Ni–N-MEGO illustrating that the single atoms of Ni are predominately anchored on the edges of the nanopores. Reproduced from ref. 80 with permission from Elsevier, copyright 2018. (d) FT-EXAFS spectra of Ni foil, SE-Ni SAs@PNC, Ni NPs@NC, and Ni NPs@C. Reproduced from ref. 81 with permission from John Wiley and Sons, copyright 2018. | ||
The effect of the coordination steric structure towards active sites is often overlooked because of its close connection with the coordination atoms and coordination number.78 However, it is of great importance to investigate the appropriative coordination steric structure. This provides a new strategy to regulate the electronic structure, directly modifying the accessibility of the single-atom sites, and eventually improving the selectivity and activity. Cheng et al. developed microwave exfoliated graphene oxide supported Ni SACs (Ni–N-MEGO).80 Aberration-corrected scanning transmission electron microscopy (AC-STEM) combined with X-ray absorption spectroscopy (XAS) indicated the atomically dispersed Ni atoms were anchored on the edges of nanopores (Fig. 5c). The DFT calculations suggested the three N coordinated edge-anchored Ni sites have a better performance than in-plane structures owing to their moderate CO2 activation energy and CO desorption energy.
In addition to all the aforementioned improvements in the catalytic performance, the rational design of the coordination environment can also contribute to the synthesis process, and even improve the durability of SACs. Yang et al. investigated the process of defect-containing N-doped carbon supported Ni NPs converted to Ni single atoms through thermal atomization.81 Aberration corrected high-angle annular dark-field scanning transmission electron microscope (HAADF-STEM) measurements and environmental transmission electron microscopy (TEM) were employed to characterize the process. The average size of the Ni NPs was gradually increased as the temperature rose. When the temperature was above 673 K, the Ni NPs started the atomization process, distributed to Ni single atoms. EXAFS revealed the dominant peak of Ni–N at 1.32 Å appeared after the thermal process, while the Ni–Ni peak at 2.15 Å disappeared, suggesting the Ni–N strong coordination promoted the Ni NPs atomization process (Fig. 5d).
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds breaks, the product, CO, is adsorbed on the M site, while OH is adsorbed on the dangling bonds of the adjacent C atom. This result supports the lower energy of the *COOH dissociation on the M–N2+2–C8 sites, leading to its high catalytic performance. In addition to the C atoms, uncoordinated N atoms, as a rich species on the M–N–C catalysts, may also serve as independent active centers. In the research reported by Song et al., SACs with Co–C2N2 moieties were synthesized by the pyrolysis of ZnO@ZIF to catalyze both the CO2RR and HER.33 At −0.7 VRHE to 1.0 VRHE, it exhibited an almost 100% FECO and an ideal CO/H2 ratio of 1/2. Potassium thiocyanate (KSCN) poisoning experiments and DFT calculations confirmed that the Co–C2N2 moieties served as the active sites for CO2RR, while the additional N functional groups served as the active sites for HER.
O bonds breaks, the product, CO, is adsorbed on the M site, while OH is adsorbed on the dangling bonds of the adjacent C atom. This result supports the lower energy of the *COOH dissociation on the M–N2+2–C8 sites, leading to its high catalytic performance. In addition to the C atoms, uncoordinated N atoms, as a rich species on the M–N–C catalysts, may also serve as independent active centers. In the research reported by Song et al., SACs with Co–C2N2 moieties were synthesized by the pyrolysis of ZnO@ZIF to catalyze both the CO2RR and HER.33 At −0.7 VRHE to 1.0 VRHE, it exhibited an almost 100% FECO and an ideal CO/H2 ratio of 1/2. Potassium thiocyanate (KSCN) poisoning experiments and DFT calculations confirmed that the Co–C2N2 moieties served as the active sites for CO2RR, while the additional N functional groups served as the active sites for HER.
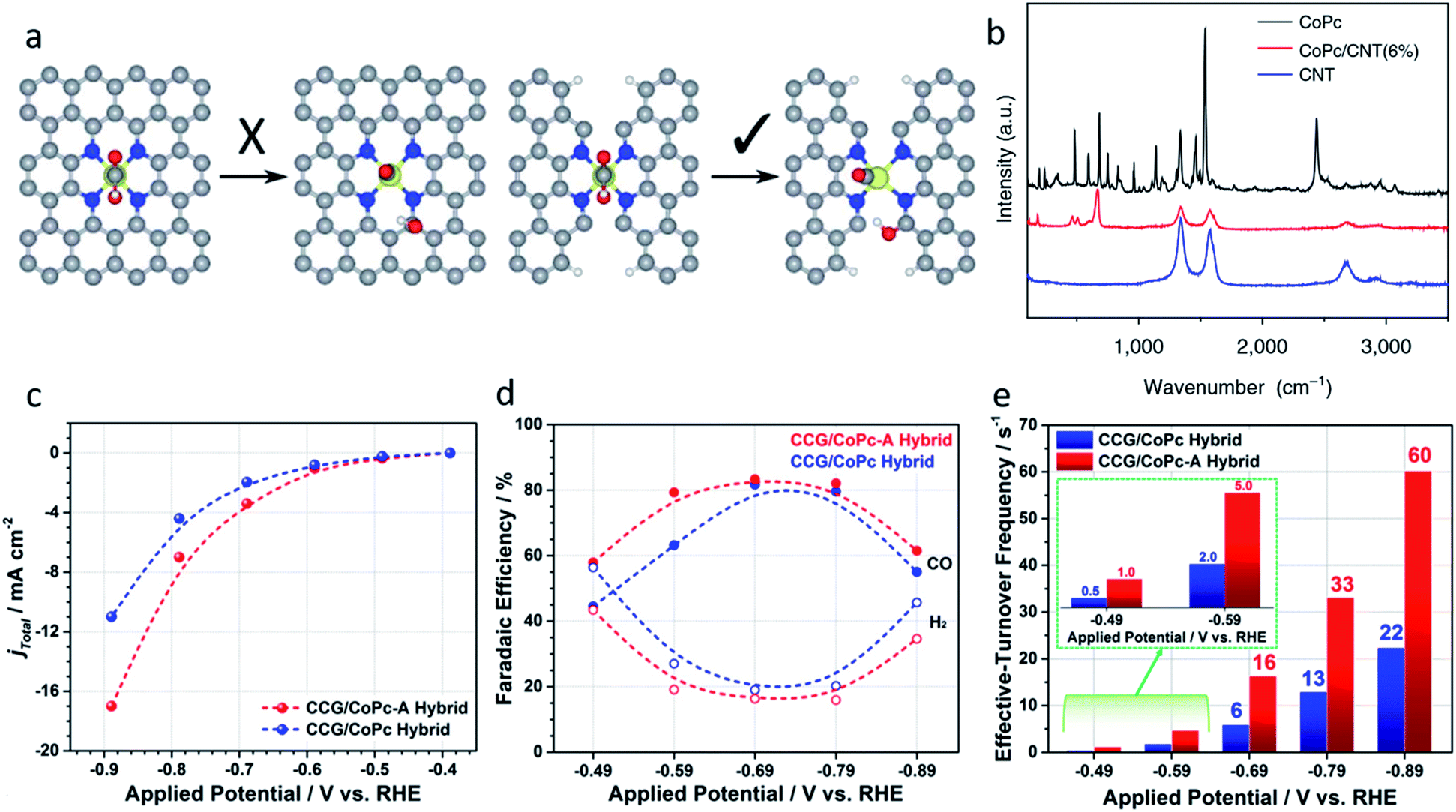 | ||
| Fig. 6 (a) Ni–N4–C10 sites (left) are inactive for the *COOH dissociation reaction, Ni–N2+2–C8 sites are active because of the dangling bonds on the adjacent C atoms. Reproduced from ref. 82 with permission from the Royal Society of Chemistry, copyright 2019. (b) Raman spectra of CoPc, the CoPc/CNT(6%) hybrid, and CNTs. Reproduced from ref. 31 with permission from Springer Nature, copyright 2017. (c) Total current density, (d) faradic efficiencies, and (e) effective turnover frequencies of CCG/CoPc-A and CCG/CoPc hybrids. Reproduced from ref. 85 with permission from the American Chemical Society, copyright 2019. | ||
2.3 Interface design of the single metal complexes
Traditional SACs were synthesized through anchoring metal atoms on the prepared substrate with coordination atoms to construct a coordination structure such as M–N4, which was proved to be analogous to molecular catalysts.83 Recently, a novel strategy was reported to synthesize SACs by anchoring metal complexes with a formed M–N4 structure on a carbon substrate.84 The M–N4 structure in the metal precursor can integrally anchor on the substrate, leading to the formation of SACs with well-defined active sites. Also, as the metal complexes offer M–N4, there is no need for the substrate to provide extra coordination atoms, therefore carbon materials (e.g., graphene, carbon nanotubes, and MOFs) have become the most popular substrates in metal complex derived SACs. Metal phthalocyanine is a promising candidate for this kind of synthesis strategy. Zhang et al. anchored a cobalt phthalocyanine (CoPc) molecule on a carbon nanotubes (CNT) to synthesize a CoPc/CNT catalyst.31 Inductively coupled plasma mass spectrometry (ICP-MS) demonstrated the amount of Co in the prepared CoPc/CNT was 0.63 wt%. Material characterizations also revealed the C and N of CoPc was distributed on the sidewalls of the CNTs that overlapped with the nanotube structures. Some of the vibrational modes of the CoPc are prohibited, indicting the strong electronic interaction between CoPc and CNT (Fig. 6b). CoPc/CNT exhibited a high activity, stability and selectivity in the CO2-to-CO conversion. The phthalocyanine molecule offers several sites for organic modification, leading to improvements in the catalytic performance. The researchers further prepared a CoPc-CN/CNT hybrid catalyst using cobalt-2,3,7,8,12,13,17,18-octacyano-phthalocyanine (CoPc-CN). CoPc-CN/CNT delivered an even better catalytic performance for the CO2-to-CO electrochemical reaction than CoPc/CNT. CoPc-CN/CNT maintained a FECO value of over 95% at −0.53–0.63 VRHE. Choi et al. anchored cobalt(+2) octa-alkoxy phthalocyanine (CoPc-A) on chemically converted graphene (CCG) via π–π stacking.85 Compared to the graphene supported cobalt phthalocyanine catalyst (CCG/CoPc), the octa-alkoxy groups on phthalocyanine helped to suppress its aggregation, leading to its significantly enhanced catalytic activity. CCG/CoPc-A exhibited a higher total current density, a higher FECO, higher eTOFs for CO production and over 30 h of stable CO conversion (Fig. 6c–e). As with the other SACs, the metal loading will largely effect the performance of the catalysts. Yang et al. synthesized a CoTAPc–ZIF-90 hybrid catalyst through anchoring cobalt phthalocyanine on the surface of ZIF-90 using the Schiff base reaction.86 The SEM images confirmed the CoTAPc units were distributed uniformly, and the XANES spectra demonstrated the Co–N4 structure. Catalytic control experiments revealed the catalytic performance of CoTAPc–ZIF-90, which was quantified by the CO partial current density, could be improved by increasing the content of CoTAPc from ZIF-90 (0 mg CoTAPc) to ZIF-90-4 (30 mg CoTAPc) (Fig. 7a). ZIF-90-4, synthesized using 30 mg of CoTAPc and 100 mg of ZIF-90, exhibited a high FECO of 90% at −0.97 VRHE and a large current density of 13 mA cm−2.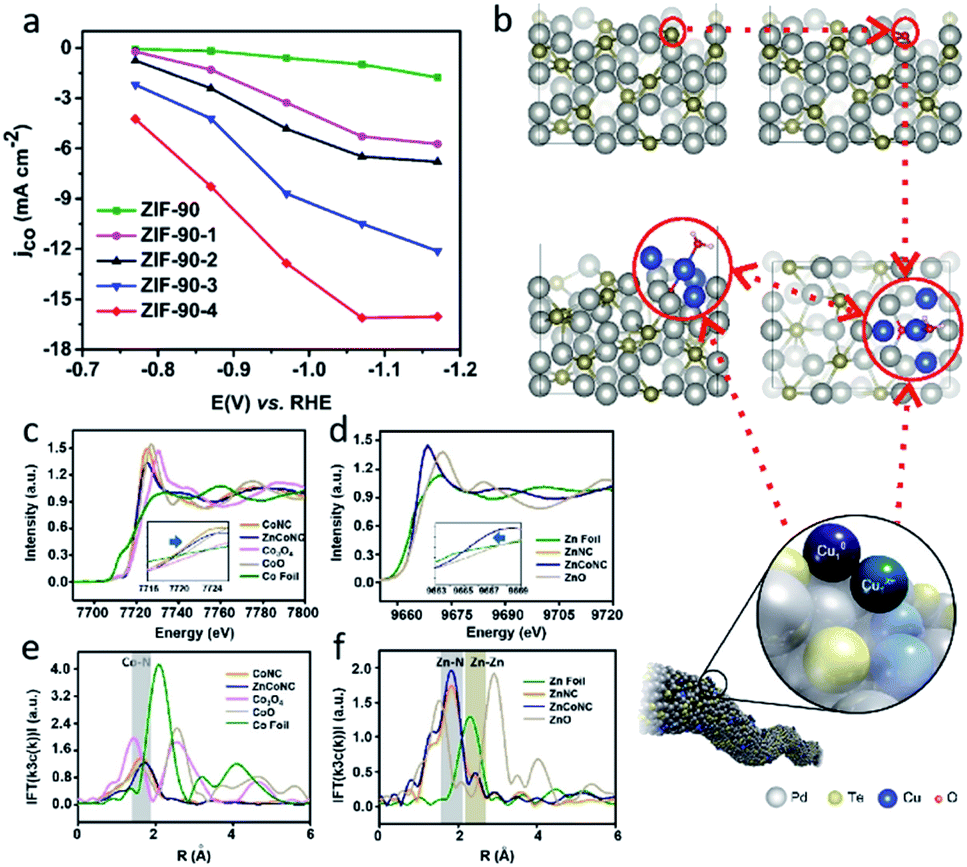 | ||
| Fig. 7 (a) jCO values of ZIF-90, ZIF-90-1, ZIF-90-2, ZIF-90-3, and ZIF-90-4. Reproduced from ref. 86 with permission from the Royal Society of Chemistry, copyright 2020. (b) A schematic illustration of Cu-doped Pd10Te3 nanowires. Reproduced from ref. 91 with permission from Springer Nature, copyright 2019. Co (c) K-edge XANES and (e) Fourier-transform EXAFS spectra of ZnCoNC. Zn (d) K-edge XANES and (f) Fourier-transform EXAFS spectra of ZnCoNC. Reproduced from ref. 27 with permission from John Wiley and Sons, copyright 2020. | ||
Additionally, the catalytic performance of the phthalocyanines derived SACs with different metal centers was investigated. Wang et al. reported a series of SACs (Me-SAC (Pc), Me = Co, Fe, Ni) for CO2RR, synthesized by the pyrolysis of the encapsulating cyano-substituted metal phthalocyanines (MePc-CN) in Zeolitic imidazolate frameworks (ZIFs).87 Me–N/C could enable metal loading up to approximately 2.2 wt% without aggregation. The sequence of the Me-SAC (Pc) current densities under certain potentials in the H-cell is Co-SAC (Pc) > Fe-SAC (Pc) > Ni-SAC (Pc). In the gas-diffusion electrode (GDE) setups, Ni-SAC (Pc) operated steadily from −10 up to −200 mA cm−2 with a FECO of over 96.0%, while Fe-SAC (Pc) only exhibited 50.1% of FECO at −100 mA cm−2. The results suggested the promising potential of Ni-SAC (Pc) for use in practical electrochemical CO2RR devices.
In addition to metal phthalocyanines, some other molecular complexes, such as metal porphyrins, also have a defined M–N4 structure. Kim et al. reported carbon substrate supported Ni molecular complexes with two different porphyrins ligands, tetraphenylporphyrin (N4–TPP) and 21-oxatetraphenylporphyrin (N3O-TPP)88 Spectroscopic and computational studies of Ni–N4-TPP and Ni(–Cl)–N3O-TPP revealed that the destruction of the ligand field symmetry could increase the redox potential from 0 to +1, leading to a better performance in the CO2RR. The additional ligand for the central atoms could also be a modification strategy to regulate the interfacial electronic structure from the axial direction. Wang et al. immobilized tetraphenylporphyrin cobalt (PCo) on graphene by π–π interactions only89 They discovered that diphenyl sulfide (Di-S) could act as an axial ligand to the Co single atom sites. Di-S enhanced the electron transfer between the interface of graphene and Co complexes by parallel stacking geometry, leading to better generation of [PCo]˙− as active sites for CO2RR.
Compared with the traditional synthesis of SACs, using metal complexes such as metal phthalocyanines and metal porphyrins as precursors shows several advantages. First, metal complexes can be anchored by π–π stacking without pyrolysis. Second, the M–N4 structure in the metal complexes can be anchored onto the substrate intact to form SACs with clear active sites. Third, the steric hindrance of the phthalocyanine and porphyrin ring are large enough to alleviate the aggregation of the single atoms. Overall, the interface design of the single metal complexes offers a novel method for the construction of SACs and a bridge to connect molecular homogenous catalysts with heterogonous catalysts directly.
2.4 Design of multi-atom site catalysts
Although SACs exhibit a great performance in the CO2RR, most of them catalyze reactions that only involve single molecules. On traditional SACs, the single metal atoms are dispersed on substrates in isolation with no interaction between each other. When the neighboring single atom is close enough, there is a chances that C–C coupling reactions will be enabled, leading to C2+ products. Guan et al. reported electrochemical CO2 reduction to C2H4 on single-atom Cu catalysts supported on nitrogen-doped carbon.90 When the Cu concentration was below approximately 2.4%mol, the Cu–Nx species were isolated, leading to the C1 products of CH4. When the Cu concentration reached approximately 4.9%mol, the adjacent Cu–Nx species was close enough to enable a synergistic effect between the neighboring Cu single atoms and resulted in the dimerization of the *CO intermediates. If the distance between the single-atom site continues to reduce, the extreme condition of it will result in multi-atoms site catalysts. Several multi-atoms site catalysts have been reported in recent research, in which the active sites were constructed by two or more metal atoms instead of only one metal atom in the SACs. The multiple atoms offer additional sites for CO2 adsorption, involving additional CO2 in a single reaction process, and leading to the production of C2+ products. Jiao et al. reported a Cu atom-pair catalyst (Cu-APC) on Pd10Te3 nanowires for the CO2 electrochemical reduction to CO.91 By combining the EXAFS and DFT calculations, they revealed that in Cu4, which is the smallest stable configuration, Cu1x+ was formed via a single Cu atom binding with an O atom, then a Cu10–Cu1x+ atom pair was constructed as an active site by Cu1x+ together with an adjacent Cu atom (Fig. 7b). Further calculations indicted that the Cu10–Cu1x+ sites catalytic mechanism is a ‘biatomic activating bimolecular’ mechanism. At −0.78 VRHE, Cu-APC with a 0.10% Cu loading exhibited a FECO of 92%, with only a corresponding FEH2 of 3%. The theoretical calculation also confirmed that diatomic sites catalysts promote the formation of C2+ products. Zhao et al. investigated whether the porous C2N layer supported a series of transition metal dimers catalysts (Cu2@C2N) for CO2RR using DFT calculations.92 The results showed a maximum positive free energy change of 0.76 eV in the formation of the COCO* species, indicating that Cu2@C2N is preferred to C2H4 in the CO2RR.In addition, multi-atoms sites can be constructed using different transmission metal atoms. Zhu et al. reported neighboring Zn/Co monomers dispersed on N doped carbon (ZnCoNC) for electrochemical CO2 reduction to CO.27 The indirect electron interaction of the neighboring Zn/Co through the N atoms was confirmed using XANES and EXAFS analysis (Fig. 7c–f). In situ attenuated total reflection-infrared spectroscopy (ATR-IR) and DFT calculations suggested that the electronic effect of the neighboring Zn/Co reduced the *COOH formation energy barrier, leading to easier CO production. ZnCoNC exhibited a FECO of 93.2% at −0.5 VRHE in a 30 h test. Ren et al. synthesized a diatomic Ni–Fe sites catalyst supported by nitrogenated carbon (Ni/Fe–N–C) via an ion-exchange strategy.93 The obvious metal–metal peak for Ni–Fe coordination was observed at 2.06 Å in FT EXAFS spectra. Compared with Ni–N–C and Fe–N–C synthesized using similar methods, Ni/Fe–N–C exhibited a higher faradaic efficiency of CO, a higher current density of CO, and a higher TOF. Moreover, the Ni/Fe–N–C reached a maximum FECO of 98% at −0.7 VRHE, and maintained a FECO of 99% after 30 h of continuous electrolysis, suggesting its outstanding catalytic performance and durability. DFT calculations revealed the diatomic Ni–Fe sites of Ni/Fe–N–C provided an additional active site for the second CO2 activation. Compared with bare Ni/Fe–N–C, CO-adsorbed Ni/Fe–N–C has a weaker binding strength for COOH* and CO*, leading to a lower energy barrier for CO production.
Apart from dual-atom site catalysts with metal–metal bonds, there are some dual-atom site catalysts which are composed of two types of single-atomic sites with two different metal centers. Xie et al. reported hierarchical integrated electrodes supported NiSn atomic pair catalysts (NiSnAPC).94 XAFS and XPS analysis suggested that NiSnAPC contains a separately dispersed Ni–N4 and Sn–N4 configuration and the Ni–Sn bond was not observed. However, the isolated Ni–N4 and Sn–N4 configuration could allow them to modify each other's electronic structure. Compared with Sn-SAC, NiSn-APC exhibited a TOF of 4752 h−1, and revealed a better activity and selectivity for CO2RR compared to formate. Lin et al. synthesized a CoPc and Zn–N–C (CoPc@Zn–N–C) tandem catalyst for the CO2RR to form CH4.95 CoPc@Zn–N–C showed a CH4/CO production rate that was over 100 times greater than that of the CoPc or Zn–N–C catalysts. Electrochemical experiments and DFT calculations revealed the two-step tandem mechanism. CO2 was first reduced to CO on CoPc, then diffused onto Zn–N–C and converted to CH4. CoPc contributed to the reservoir of adsorbed *H on ZnN4, which is critical to the high CH4 production rate.
A change in the number of central atoms in multi-atoms site catalysts may also influence the selectivity of C1 products. He et al. investigated the activity and selectivity of a series of multi-Fe atoms (Fen, n = 1–4) doped graphdiyne catalysts for CO2 electrochemical reduction using ab initio studies.96 Theoretical calculations revealed Fe2 and Fe3 exhibited a better performance in CO2-to-CH4, CO2-to-HCOOH, respectively. Multi-atoms site catalysts may catalyze the synthesis of C2+ products via multiple metal active sites. From the perspective of SACs, introducing novel metal atoms in single-atom sites also offers a novel strategy to regulate the electronic structure of the active sites.
2.5 Strategies to improve SAC performance
All of the aforementioned approaches have focused on improving the intrinsic nature of a single-atom site. Moreover, improving the accessibility of single-atom sites and increasing the density of single-atom sites are also promising strategies. Owing to the different synthesis strategies, there may be part of the single-atom sites that were embedded in substrates in some SACs, leading to significant loss of catalytic activities. Ye et al. compared the electrocatalytic performances of C-AFC©ZIF-8 and C-AFC@ZIF-8.97 The precursor AFC©ZIF-8 was prepared by immersing ZIF-8 in ammonium ferric citrate (AFC) aqueous solution for 24 h, AFC@ZIF-8 was obtained by sealing AFC in ZIF-8. X-ray photoelectron spectroscopy (XPS) confirmed C-AFC©ZIF-8 had more Fe–N active sites on the surface than C-AFC@ZIF-8, leading to its better performance in the CO2RR. Further experiments confirmed the activities of the catalysts towards the CO2RR exhibited a positive correlation with the content of the exposed Fe–N active sites. Therefore, increasing the quantity of single-atom sites on the surface is viewed as an efficient way to improve the catalytic performance of SACs.Two dimensional materials such as nanofibers constructed by one-dimension carbon materials are good choices for substrates because of their high surface areas, which can provide more space to support and anchor metal single-atom sites. Yang et al. reported a strategy for large-scale synthesis of a single Cu atoms catalyst supported on through-hole carbon nanofibers (CuSAs/TCNFs) for electrochemical CO2-to-CO reduction.98 Cu/ZIF-8 nanoparticles were first synthesized using the double-solvent method, and were then attached to polyacrylonitrile (PAN) nanofibers via the electro-spinning method (Fig. 8a). The through-hole structure offered large surface areas and improved the accessibility for Cu atom sites. At −0.9 VRHE, CuSAs/TCNFs exhibited a maximum FEMeOH of 44%, and CO (56%) was obtained in the gas-phase. Later, Yang et al. synthesized cobalt single atoms (CoSA) supported by free-standing carbon nanofibers (HCNFs) via a similar strategy (Fig. 8b).99 CoSA/HCNFs reached an extremely high current density of 211 mA cm−2 with a 92% FE in the flow cell. MOFs with a well-defined spatial structure that can form porous carbon nanoframes through one-step pyrolysis are also viewed as a facile choice. Chen et al. reported SACs with Fe–Nx active sites supported on mesoporous carbon nanoframes synthesized by pyrolysis of Fe-doped MOF precursors (Fig. 8c).100 In the pyrolysis process, spatially isolated Fe2+ in ZIF precursors triggered the Kirkendall effect, which generated the formation of mesoporous carbon nanoframes with hierarchical pore sizes, improved the accessibility of high-density single atom Fe sites and the mass and charge transport abilities, and resulted in an extraordinary electrocatalytic performance for both oxygen and carbon dioxide reduction. The template-assisted method is also a common strategy used to synthesize high surface area substrates with the advantages of reactants and products diffusion. Zhang et al. proposed a template-assisted method to synthesize a Fe single-atom site catalyst confined by carbon foam (Fe–N4/CF) with FePc as a precursor and SiO2 as a template (Fig. 8d).12 The pore-enriched structure boosted the diffusion of the reactants and products. Fe–N4/CF reached a maximum FECO of 94.9% at −0.5 VRHE.
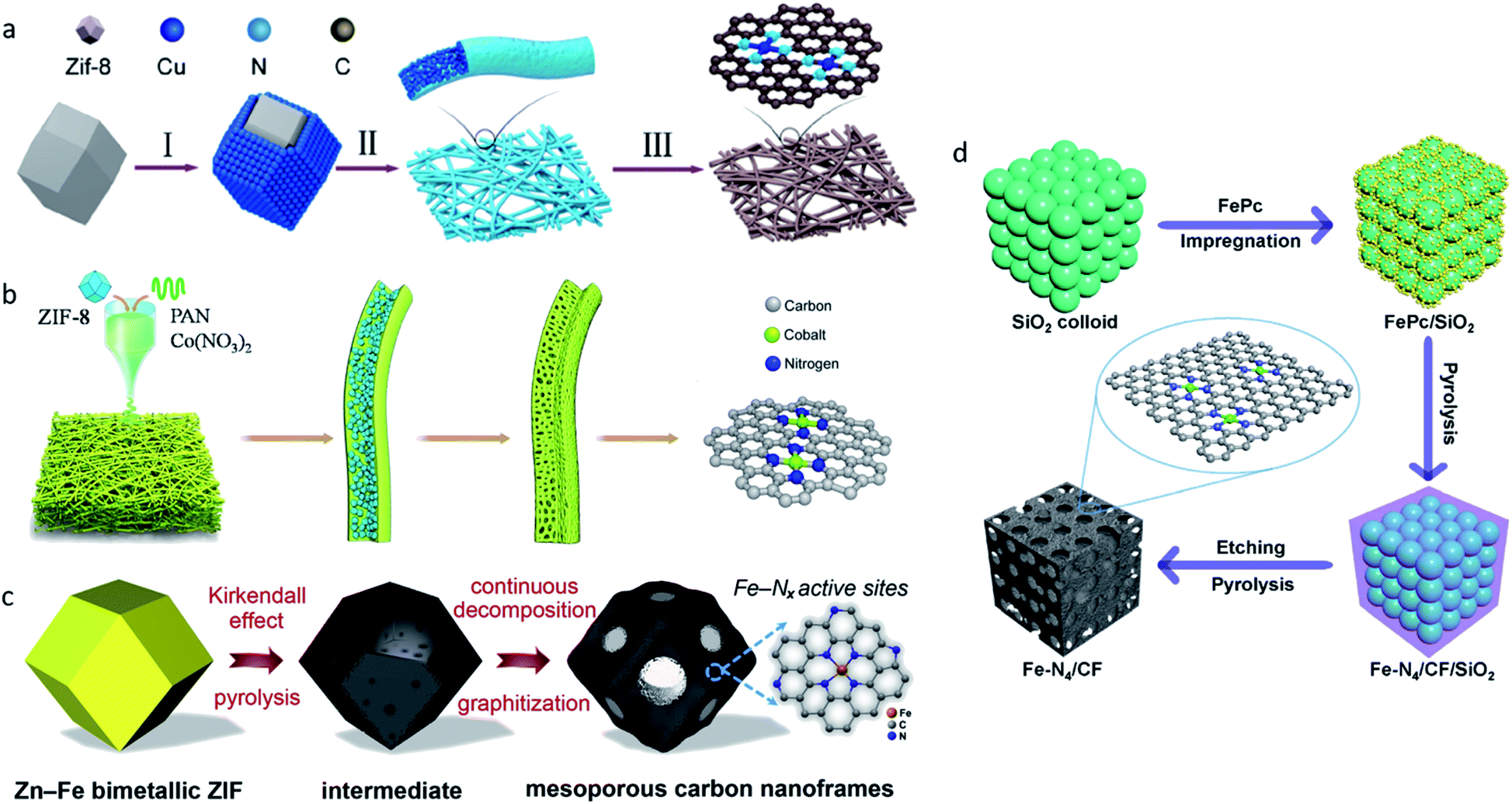 | ||
| Fig. 8 (a) A schematic illustration of the synthesis of CuSAs/THCF. Reproduced from ref. 98 with permission from American Chemical Society, copyright 2019. (b) A schematic illustration of the synthesis of CoSA/HCNFs. Reproduced from ref. 99 with permission from Elsevier, copyright 2020. (c) A schematic illustration of the synthesis of FeSAs/CNF-900. Reproduced from ref. 100 with permission from Elsevier, copyright 2020. (d) A schematic illustration of the synthesis of Fe–N4/CF. Reproduced from ref. 12 with permission from Springer Nature, copyright 2019. | ||
The increasing density of single-atom sites directly revealed an increase in the metal loading in SACs. The main challenge is that the aggregation tendency of the single metal atoms will increase as the distances between isolated single-atom sites become smaller owing to the increased metal contents. Chen et al. reported a Fe and N doped porous carbon nanosphere catalyst (Fe–N–PC) with a high Fe content of 3.9 wt%.101 SiO2 was used as the template and the protecting shell to prevent the aggregation of Fe single atoms and the volatilization of the N-containing species, leading to a high density of Fe single atoms with only a few Fe clusters. Fe–N–PC exhibited a high FECO of 90% at −0.49 VRHE, with a partial CO current density of 11.44 mA cm−2. Zhao et al. later proposed a universal seeding strategy to synthesize a series of single metal atoms (e.g., Ni, Co, Fe, Cu, Ag, Pd) and bimetallic NiCu atoms catalysts supported on 2D materials (e.g., GO, MoS2, BN nanosheets) with metal contents between 2.8–7.9 wt%.102 In particular, a Ni-single-atom site catalyst supported on graphene oxide (SANi-GO) was still able to maintain an extraordinary CO2RR performance after a 50 h cycling test, suggesting the excellent stability.
3. Outlook
The electrochemical CO2RR provides a novel way to convert electricity, which can be generated from renewable energy, to chemical energy in the forms of CO, CH4, and even more complicated hydrocarbon fuels. As a bridge between homogenous catalysts and heterogeneous catalysts, SACs feature single-atom sites similar to molecular catalysts with great uniformity and unique geometric and electronic structures, resulting in their outstanding performances in the CO2RR.In order to design high-performance SACs accurately, the necessity for the atomic-level regulation of SACs must be recognized. In this perspective article, the importance of the regulation of SACs at the atomic level is stressed from several aspects. The design of SACs can be divided into the regulation of SAC structures to improve the catalytic activities and selectivity. Enhancing the intrinsic nature of each single-atom site and increasing the density of the single-atom sites are the two main strategies used to improve SAC catalytic performance. The choice of the metal center and the regulation of the coordination environment are vital for the structural and electronic properties of SACs. Recently, dual- and multiple-atom sites have drawn significant attention owing to their great performance in the CO2RR, especially C2+ products, and they are also important in SACs for achieving highly efficient CO2RR performance. Agglomeration is a huge unavoidable hinderance to increasing the density of single-atom sites. The fine regulation of single-atom sites from both geometric and electronic angles can relieve aggregation through interactions between metal atoms and substrates. Synthetic methods also influence the performance of SACs via determining the accessibility of single-atom sites.
Although the development of SACs holds promise, there are still multiple challenges that must be noted:
(1) Although a large number of SACs have shown good performance for the CO2RR, there is still the large potential for the improvement of the activity and selectivity, especially for C2+ products and liquid hydrocarbon fuels, which are more valuable for industrial use. Although most SACs for the CO2RR catalyze CO2-to-CO conversion, more effort should be made to explore C–C coupling on SACs. Also, the HER, as a common side reaction to the CO2RR, should receive more attention. With appropriate regulation, the HER could work alongside the CO2RR to produce syngas.103
(2) Although single-atom sites have great uniformity, their actual condition still remains ambiguous. An understanding of the reaction mechanism and active sites, which depends on further developments in characterization methods and the assistance of theoretical computation, is vital to the regulation of SACs at the atomic level. Advanced characterization techniques, such as operando characterization, are urgently needed, which can reveal the actual active sites during the catalysis reaction, thus contributing to our understanding of the mechanism, enabling the final regulation of SACs. Theoretical computation is built on known mechanisms and ideal reaction conditions; accordingly, there are differences between the results of theoretical computations and laboratory experiments. The process used to diminish the differences also offers better understanding of the reaction mechanism.
(3) It should be emphasized that there is still a long way to go for SACs to move from laboratory tests into industrial production. The scaling up of synthesis and complicated reaction conditions are the main hinderances to the practical application of SACs. Recently, several mass production methods for SACs have been reported; facile strategies or commercial reactants were used to meet industrial-scale synthesis demand.104 However, there is a large discrepancy between the applications of SACs in half-cells and full-cells, which may be caused by transport loss and ohmic loss. In addition, SACs have to overcome the reaction conditions, which are much more unstable and impure in full-cell and industrial applications, owing to low-cost requirements.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2018YFA0702003), the National Natural Science Foundation of China (21890383, 21671117, 21871159), Science and Technology Key Project of Guangdong Province of China (2020B010188002), Beijing Municipal Science & Technology Commission No. Z191100007219003, the National Postdoctoral Program for Innovative Talents (BX20180160), and the China Postdoctoral Science Foundation (2018M640113).Notes and references
- J. D. Shakun, P. U. Clark, F. He, S. A. Marcott, A. C. Mix, Z. Liu, B. Otto-Bliesner, A. Schmittner and E. Bard, Nature, 2012, 484, 49–54 CrossRef CAS PubMed.
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. Mac Dowell, J. C. Minx, P. Smith and C. K. Williams, Nature, 2019, 575, 87–97 CrossRef CAS PubMed.
- N. Corbin, J. Zeng, K. Williams and K. Manthiram, Nano Res., 2019, 12, 2093–2125 CrossRef CAS.
- F. P. G. de Arquer, D. Cao-Thang, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, N. Dae-Hyun, C. Gabardo, A. Seifitokaldani, X. Wang, Y. C. Li, F. Li, J. Edwards, L. J. Richter, S. J. Thorpe, D. Sinton and E. H. Sargent, Science, 2020, 367, 661–666 CrossRef PubMed.
- S. Lin, C. S. Diercks, Y.-B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- M. Liu, Y. Pang, B. Zhang, P. De Luna, O. Voznyy, J. Xu, X. Zheng, C. T. Dinh, F. Fan, C. Cao, F. P. G. de Arquer, T. S. Safaei, A. Mepham, A. Klinkova, E. Kumacheva, T. Filleter, D. Sinton, S. O. Kelley and E. H. Sargent, Nature, 2016, 537, 382–386 CrossRef CAS PubMed.
- J. Graciani, K. Mudiyanselage, F. Xu, A. E. Baber, J. Evans, S. D. Senanayake, D. J. Stacchiola, P. Liu, J. Hrbek, J. Fernandez Sanz and J. A. Rodriguez, Science, 2014, 345, 546–550 CrossRef CAS PubMed.
- D. Yang, B. Ni and X. Wang, Adv. Energy Mater., 2020, 10, 2001142 CrossRef CAS.
- S. Ji, Y. Qu, T. Wang, Y. Chen, G. Wang, X. Li, J. Dong, Q. Chen, W. Zhang, Z. Zhang, S. Liang, R. Yu, Y. Wang, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2020, 59, 10651–10657 CrossRef CAS PubMed.
- G. Wang, C.-T. He, R. Huang, J. Mao, D. Wang and Y. Li, J. Am. Chem. Soc., 2020, 142, 19339–19345 CrossRef CAS PubMed.
- M. Li, H. Wang, W. Luo, P. C. Sherrell, J. Chen and J. Yang, Adv. Mater., 2020, 32, e2001848 CrossRef PubMed.
- Z. Zhang, C. Ma, Y. Tu, R. Si, J. Wei, S. Zhang, Z. Wang, J.-F. Li, Y. Wang and D. Deng, Nano Res., 2019, 12, 2313–2317 CrossRef CAS.
- Q. Wang, Y. Lei, D. Wang and Y. Li, Energy Environ. Sci., 2019, 12, 1730–1750 RSC.
- Y. Ouyang, L. Shi, X. Bai, Q. Li and J. Wang, Chem. Sci., 2020, 11, 1807–1813 RSC.
- F. Li, A. Thevenon, A. Rosas-Hernandez, Z. Wang, Y. Li, C. M. Gabardo, A. Ozden, D. Cao Thang, J. Li, Y. Wang, J. P. Edwards, Y. Xu, C. McCallum, L. Tao, Z.-Q. Liang, M. Luo, X. Wang, H. Li, C. P. O'Brien, C.-S. Tan, D.-H. Nam, R. Quintero-Bermudez, T.-T. Zhuang, Y. C. Li, Z. Han, R. D. Britt, D. Sinton, T. Agapie, J. C. Peters and E. H. Sargent, Nature, 2020, 577, 509–513 CrossRef CAS PubMed.
- R. Lin, X. Ma, W.-C. Cheong, C. Zhang, W. Zhu, J. Pei, K. Zhang, B. Wang, S. Liang, Y. Liu, Z. Zhuang, R. Yu, H. Xiao, J. Li, D. Wang, Q. Peng, C. Chen and Y. Li, Nano Res., 2019, 12, 2866–2871 CrossRef CAS.
- X. Li, H. Rong, J. Zhang, D. Wang and Y. Li, Nano Res., 2020, 13, 1842–1855 CrossRef CAS.
- N. Fu, X. Liang, Z. Li, W. Chen, Y. Wang, L. Zheng, Q. Zhang, C. Chen, D. Wang, Q. Peng, L. Gu and Y. Li, Nano Res., 2020, 13, 947–951 CrossRef CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- Z. Zhuang, Q. Kang, D. Wang and Y. Li, Nano Res., 2020, 13, 1856–1866 CrossRef CAS.
- N. Zhang, C. Ye, H. Yan, L. Li, H. He, D. Wang and Y. Li, Nano Res., 2020, 13, 3165–3182 CrossRef.
- S. Ji, Y. Chen, X. Wang, Z. Zhang, D. Wang and Y. Li, Chem. Rev., 2020, 120, 11900–11955 CrossRef CAS PubMed.
- H. Rong, S. Ji, J. Zhang, D. Wang and Y. Li, Nat. Commun., 2020, 11, 5884 CrossRef CAS PubMed.
- Z. Li, Y. Chen, S. Ji, Y. Tang, W. Chen, A. Li, J. Zhao, Y. Xiong, Y. Wu, Y. Gong, T. Yao, W. Liu, L. Zheng, J. Dong, Y. Wang, Z. Zhuang, W. Xing, C.-T. He, C. Peng, W.-C. Cheong, Q. Li, M. Zhang, Z. Chen, N. Fu, X. Gao, W. Zhu, J. Wan, J. Zhang, L. Gu, S. Wei, P. Hu, J. Luo, J. Li, C. Chen, Q. Peng, X. Duan, Y. Huang, X.-M. Chen, D. Wang and Y. Li, Nat. Chem., 2020, 12, 764–772 CrossRef CAS PubMed.
- Z. Li, S. Ji, Y. Liu, X. Cao, S. Tian, Y. Chen, Z. Niu and Y. Li, Chem. Rev., 2020, 120, 623–682 CrossRef CAS PubMed.
- H. B. Yang, S.-F. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H.-Y. Wang, W. Cai, R. Chen, J. Gao, X. Yang, W. Chen, Y. Huang, H. M. Chen, C. M. Li, T. Zhang and B. Liu, Nat. Energy, 2018, 3, 140–147 CrossRef CAS.
- W. Zhu, L. Zhang, S. Liu, A. Li, X. Yuan, C. Hu, G. Zhang, W. Deng, K. Zang, J. Luo, Y. Zhu, M. Gu, Z.-J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2020, 59, 12664–12668 CrossRef CAS PubMed.
- H. Zhang, J. Li, S. Xi, Y. Du, X. Hai, J. Wang, H. Xu, G. Wu, J. Zhang, J. Lu and J. Wang, Angew. Chem., Int. Ed., 2019, 58, 14871–14876 CrossRef CAS PubMed.
- S. Tian, M. Hu, Q. Xu, W. Gong, W. Chen, J. Yang, Y. Zhu, C. Chen, J. He, Q. Liu, H. Zhao, D. Wang and Y. Li, Sci. China Mater., 2021, 64, 642–650 CrossRef CAS.
- J. Mao, C.-T. He, J. Pei, Y. Liu, J. Li, W. Chen, D. He, D. Wang and Y. Li, Nano Lett., 2020, 20, 3442–3448 CrossRef CAS PubMed.
- X. Zhang, Z. Wu, X. Zhang, L. Li, Y. Li, H. Xu, X. Li, X. Yu, Z. Zhang, Y. Liang and H. Wang, Nat. Commun., 2017, 8, 14675 CrossRef PubMed.
- K. Zhao, X. Nie, H. Wang, S. Chen, X. Quan, H. Yu, W. Choi, G. Zhang, B. Kim and J. G. Chen, Nat. Commun., 2020, 11, 2455 CrossRef CAS PubMed.
- X. Song, H. Zhang, Y. Yang, B. Zhang, M. Zuo, X. Cao, J. Sun, C. Lin, X. Li and Z. Jiang, Adv. Sci., 2018, 5, 1800177 CrossRef PubMed.
- K. Jiang, S. Siahrostami, T. Zheng, Y. Hu, S. Hwang, E. Stavitski, Y. Peng, J. Dynes, M. Gangisetty, D. Su, K. Attenkofer and H. Wang, Energy Environ. Sci., 2018, 11, 893–903 RSC.
- Y. Pan, R. Lin, Y. Chen, S. Liu, W. Zhu, X. Cao, W. Chen, K. Wu, W.-C. Cheong, Y. Wang, L. Zheng, J. Luo, Y. Lin, Y. Liu, C. Liu, J. Li, Q. Lu, X. Chen, D. Wang, Q. Peng, C. Chen and Y. Li, J. Am. Chem. Soc., 2018, 140, 4218–4221 CrossRef CAS PubMed.
- J. Zhang, C. Zheng, M. Zhang, Y. Qiu, Q. Xu, W.-C. Cheong, W. Chen, L. Zheng, L. Gu, Z. Hu, D. Wang and Y. Li, Nano Res., 2020, 13, 3082–3087 CrossRef.
- X. Rong, H.-J. Wang, X.-L. Lu, R. Si and T.-B. Lu, Angew. Chem., Int. Ed., 2020, 59, 1961–1965 CrossRef CAS PubMed.
- Y.-N. Gong, L. Jiao, Y. Qian, C.-Y. Pan, L. Zheng, X. Cai, B. Liu, S.-H. Yu and H.-L. Jiang, Angew. Chem., Int. Ed., 2020, 59, 2705–2709 CrossRef CAS PubMed.
- X. Wang, Z. Chen, X. Zhao, T. Yao, W. Chen, R. You, C. Zhao, G. Wu, J. Wang, W. Huang, J. Yang, X. Hong, S. Wei, Y. Wu and Y. Li, Angew. Chem., Int. Ed., 2018, 57, 1944–1948 CrossRef CAS PubMed.
- W. Ju, A. Bagger, G. P. Hao, A. S. Varela, I. Sinev, V. Bon, B. Roldan Cuenya, S. Kaskel, J. Rossmeisl and P. Strasser, Nat. Commun., 2017, 8, 944 CrossRef PubMed.
- H. Shang, W. Sun, R. Sui, J. Pei, L. Zheng, J. Dong, Z. Jiang, D. Zhou, Z. Zhuang, W. Chen, J. Zhang, D. Wang and Y. Li, Nano Lett., 2020, 20, 5443–5450 CrossRef CAS PubMed.
- T. Sun, Y. Li, T. Cui, L. Xu, Y.-G. Wang, W. Chen, P. Zhang, T. Zheng, X. Fu, S. Zhang, Z. Zhang, D. Wang and Y. Li, Nano Lett., 2020, 20, 6206–6214 CrossRef CAS PubMed.
- S. Back, J. Lim, N.-Y. Kim, Y.-H. Kim and Y. Jung, Chem. Sci., 2017, 8, 1090–1096 RSC.
- C. Zhang, S. Yang, J. Wu, M. Liu, S. Yazdi, M. Ren, J. Sha, J. Zhong, K. Nie, A. S. Jalilov, Z. Li, H. Li, B. I. Yakobson, Q. Wu, E. Ringe, H. Xu, P. M. Ajayan and J. M. Tour, Adv. Energy Mater., 2018, 8, 1703487 CrossRef.
- L. Han, S. Song, M. Liu, S. Yao, Z. Liang, H. Cheng, Z. Ren, W. Liu, R. Lin, G. Qi, X. Liu, Q. Wu, J. Luo and H. L. Xin, J. Am. Chem. Soc., 2020, 142, 12563–12567 CrossRef CAS PubMed.
- H. Shang, T. Wang, J. Pei, Z. Jiang, D. Zhou, Y. Wang, H. Li, J. Dong, Z. Zhuang, W. Chen, D. Wang, J. Zhang and Y. Li, Angew. Chem., Int. Ed., 2020, 59, 22465–22469 CrossRef CAS PubMed.
- D. Tan, J. Zhang, L. Yao, X. Tan, X. Cheng, Q. Wan, B. Han, L. Zheng and J. Zhang, Nano Res., 2020, 13, 768–774 CrossRef CAS.
- D. Gao, R. M. Aran-Ais, H. S. Jeon and B. Roldan Cuenya, Nat. Catal., 2019, 2, 198–210 CrossRef CAS.
- T. Zheng, K. Jiang and H. Wang, Adv. Mater., 2018, 30, e1802066 CrossRef PubMed.
- T. Sun, L. Xu, D. Wang and Y. Li, Nano Res., 2019, 12, 2067–2080 CrossRef CAS.
- J. Yang, W. Li, D. Wang and Y. Li, Small Struct, 2021, 2, 2000051 CrossRef.
- Y. Chen, R. Gao, S. Ji, H. Li, K. Tang, P. Jiang, H. Hu, Z. Zhang, H. Hao, Q. Qu, X. Liang, W. Chen, J. Dong, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 3212–3221 CrossRef CAS PubMed.
- L. Gong, D. Zhang, C.-Y. Lin, Y. Zhu, Y. Shen, J. Zhang, X. Han, L. Zhang and Z. Xia, Adv. Energy Mater., 2019, 9, 1902625 CrossRef CAS.
- Q. He, J. H. Lee, D. Liu, Y. Liu, Z. Lin, Z. Xie, S. Hwang, S. Kattel, L. Song and J. G. Chen, Adv. Funct. Mater., 2020, 30, 2000407 CrossRef CAS.
- T. Wang, Q. Zhao, Y. Fu, C. Lei, B. Yang, Z. Li, L. Lei, G. Wu and Y. Hou, Small Methods, 2019, 3, 1900210 CrossRef CAS.
- J. Li, P. Prslja, T. Shinagawa, A. J. Martin Fernandez, F. Krumeich, K. Artyushkova, P. Atanassov, A. Zitolo, Y. Zhou, R. Garcia-Muelas, N. Lopez, J. Perez-Ramirez and F. Jaouen, ACS Catal., 2019, 9, 10426–10439 CrossRef CAS.
- W. Zheng, F. Chen, Q. Zeng, Z. Li, B. Yang, L. Lei, Q. Zhang, F. He, X. Wu and Y. Hou, Nano-Micro Lett., 2020, 12, 108 CrossRef CAS PubMed.
- F. Pan, H. Zhang, K. Liu, D. Cullen, K. More, M. Wang, Z. Feng, G. Wang, G. Wu and Y. Li, ACS Catal., 2018, 8, 3116–3122 CrossRef CAS.
- J. Gu, C.-S. Hsu, L. Bai, H. M. Chen and X. Hu, Science, 2019, 364, 1091–1094 CrossRef CAS PubMed.
- Y. Hou, Y.-L. Liang, P.-C. Shi, Y.-B. Huang and R. Cao, Appl. Catal., B, 2020, 271, 118929 CrossRef CAS.
- H.-Y. Jeong, M. Balamurugan, V. S. K. Choutipalli, E.-s. Jeong, V. Subramanian, U. Sim and K. T. Nam, J. Mater. Chem. A, 2019, 7, 10651–10661 RSC.
- W. Zhang, Y. Hu, L. Ma, G. Zhu, Y. Wang, X. Xue, R. Chen, S. Yang and Z. Jin, Adv. Sci., 2018, 5, 1700275 CrossRef PubMed.
- X. Cui, W. An, X. Liu, H. Wang, Y. Men and J. Wang, Nanoscale, 2018, 10, 15262–15272 RSC.
- E. Zhang, T. Wang, K. Yu, J. Liu, W. Chen, A. Li, H. Rong, R. Lin, S. Ji, X. Zhene, Y. Wang, L. Zheng, C. Chen, D. Wang, J. Zhang and Y. Li, J. Am. Chem. Soc., 2019, 141, 16569–16573 CrossRef CAS PubMed.
- J. Yang, W. Li, D. Wang and Y. Li, Adv. Mater., 2020, 32, 2003300 CrossRef CAS PubMed.
- C. F. Wen, F. Mao, Y. Liu, X. Y. Zhang, H. Q. Fu, L. R. Zheng, P. F. Liu and H. G. Yang, ACS Catal., 2020, 10, 1086–1093 CrossRef CAS.
- S. He, D. Ji, J. Zhang, P. Novello, X. Li, Q. Zhang, X. Zhang and J. Liu, J. Phys. Chem. B, 2020, 124, 511–518 CrossRef CAS PubMed.
- Q. Fan, P. Hou, C. Choi, T.-S. Wu, S. Hong, F. Li, Y.-L. Soo, P. Kang, Y. Jung and Z. Sun, Adv. Energy Mater., 2020, 10, 1903068 CrossRef CAS.
- X. Qin, S. Zhu, F. Xiao, L. Zhang and M. Shao, ACS Energy Lett., 2019, 4, 1778–1783 CrossRef CAS.
- F. Yang, P. Song, X. Liu, B. Mei, W. Xing, Z. Jiang, L. Gu and W. Xu, Angew. Chem., Int. Ed., 2018, 57, 12303–12307 CrossRef CAS PubMed.
- K. Mou, Z. Chen, X. Zhang, M. Jiao, X. Zhang, X. Ge, W. Zhang and L. Liu, Small, 2019, 15, e1903668 CrossRef PubMed.
- Y. Chen, Y. Yao, Y. Xia, K. Mao, G. Tang, Q. Wu, L. Yang, X. Wang, X. Sun and Z. Hu, Nano Res., 2020, 13, 2777–2783 CrossRef CAS.
- M. Kuang, A. Guan, Z. Gu, P. Han, L. Qian and G. Zheng, Nano Res., 2019, 12, 2324–2329 CrossRef CAS.
- H. Zhong, F. Meng, Q. Zhang, K. Liu and X. Zhang, Nano Res., 2019, 12, 2318–2323 CrossRef CAS.
- M. D. Hossain, Y. Huang, T. H. Yu, W. A. Goddard Iii and Z. Luo, Nat. Commun., 2020, 11, 2256 CrossRef CAS PubMed.
- Z. Geng, Y. Cao, W. Chen, X. Kong, Y. Liu, T. Yao and Y. Lin, Appl. Catal., B, 2019, 240, 234–240 CrossRef CAS.
- X. Zhao and Y. Liu, J. Am. Chem. Soc., 2020, 142, 5773–5777 CrossRef CAS PubMed.
- X. Wang, Y. Pan, H. Ning, H. Wang, D. Guo, W. Wang, Z. Yang, Q. Zhao, B. Zhang, L. Zheng, J. Zhang and M. Wu, Appl. Catal., B, 2020, 266, 118630 CrossRef.
- H. Shen and Q. Sun, J. Phys. Chem. C, 2019, 123, 29776–29782 CrossRef CAS.
- Y. Cheng, S. Zhao, H. Li, S. He, J.-P. Veder, B. Johannessen, J. Xiao, S. Lu, J. Pan, M. F. Chisholm, S.-Z. Yang, C. Liu, J. G. Chen and S. P. Jiang, Appl. Catal., B, 2019, 243, 294–303 CrossRef CAS.
- J. Yang, Z. Qiu, C. Zhao, W. Wei, W. Chen, Z. Li, Y. Qu, J. Dong, J. Luo, Z. Li and Y. Wu, Angew. Chem., Int. Ed., 2018, 57, 14095–14100 CrossRef CAS PubMed.
- F. Pan, H. Zhang, Z. Liu, D. Cullen, K. Liu, K. More, G. Wu, G. Wang and Y. Li, J. Mater. Chem. A, 2019, 7, 26231–26237 RSC.
- D. M. Koshy, S. Chen, D. U. Lee, M. B. Stevens, A. M. Abdellah, S. M. Dull, G. Chen, D. Nordlund, A. Gallo, C. Hahn, D. C. Higgins, Z. Bao and T. F. Jaramillo, Angew. Chem., Int. Ed., 2020, 59, 4043–4050 CrossRef CAS PubMed.
- Z. Jiang, Y. Wang, X. Zhang, H. Zheng, X. Wang and Y. Liang, Nano Res., 2019, 12, 2330–2334 CrossRef CAS.
- J. Choi, P. Wagner, S. Gambhir, R. Jalili, D. R. MacFarlane, G. G. Wallace and D. L. Officer, ACS Energy Lett., 2019, 4, 666–672 CrossRef CAS.
- Z. Yang, X. Zhang, C. Long, S. Yan, Y. Shi, J. Han, J. Zhang, P. An, L. Chang and Z. Tang, CrystEngComm, 2020, 22, 1619–1624 RSC.
- Y. Wang, Z. Jiang, X. Zhang, Z. Niu, Q. Zhou, X. Wang, H. Li, Z. Lin, H. Zheng and Y. Liang, ACS Appl. Mater. Interfaces, 2020, 12, 33795–33802 CrossRef CAS PubMed.
- H. Kim, D. Shin, W. Yang, D. H. Won, H.-S. Oh, M. W. Chung, D. Jeong, S. H. Kim, K. H. Chae, J. Y. Ryu, J. Lee, S. J. Cho, J. Seo, H. Kim and C. H. Choi, J. Am. Chem. Soc., 2021, 143, 925–933 CrossRef CAS PubMed.
- L. Lin, T. Liu, J. Xiao, H. Li, P. Wei, D. Gao, B. Nan, R. Si, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2020, 59, 22408–22413 CrossRef CAS PubMed.
- A. Guan, Z. Chen, Y. Quan, C. Peng, Z. Wang, T.-K. Sham, C. Yang, Y. Ji, L. Qian, X. Xu and G. Zheng, ACS Energy Lett., 2020, 5, 1044–1053 CrossRef CAS.
- J. Jiao, R. Lin, S. Liu, W.-C. Cheong, C. Zhang, Z. Chen, Y. Pan, J. Tang, K. Wu, S.-F. Hung, H. M. Chen, L. Zheng, Q. Lu, X. Yang, B. Xu, H. Xiao, J. Li, D. Wang, Q. Peng, C. Chen and Y. Li, Nat. Chem., 2019, 11, 222–228 CrossRef CAS.
- J. Zhao, J. Zhao, F. Li and Z. Chen, J. Phys. Chem. C, 2018, 122, 19712–19721 CrossRef CAS.
- W. Ren, X. Tan, W. Yang, C. Jia, S. Xu, K. Wang, S. C. Smith and C. Zhao, Angew. Chem., Int. Ed., 2019, 58, 6972–6976 CrossRef CAS PubMed.
- W. Xie, H. Li, G. Cui, J. Li, Y. Song, S. Li, X. Zhang, J. Y. Lee, M. Shao and M. Wei, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202014655.
- J. Wang, X. Huang, S. Xi, H. Xu and X. Wang, Angew. Chem., Int. Ed., 2020, 59, 19162–19167 CrossRef CAS PubMed.
- T. He, L. Zhang, G. Kour and A. Du, J. CO2 Util., 2020, 37, 272–277 CrossRef CAS.
- Y. F. Ye, F. Cai, H. B. Li, H. H. Wu, G. X. Wang, Y. S. Li, S. Miao, S. H. Xie, R. Si, J. Wang and X. H. Bao, Nano Energy, 2017, 38, 281–289 CrossRef CAS.
- H. Yang, Y. Wu, G. Li, Q. Lin, Q. Hu, Q. Zhang, J. Liu and C. He, J. Am. Chem. Soc., 2019, 141, 12717–12723 CrossRef CAS PubMed.
- H. Yang, Q. Lin, Y. Wu, G. Li, Q. Hu, X. Chai, X. Ren, Q. Zhang, J. Liu and C. He, Nano Energy, 2020, 70, 104454 CrossRef CAS.
- X. Chen, D.-D. Ma, B. Chen, K. Zhang, R. Zou, X.-T. Wu and Q.-L. Zhu, Appl. Catal., B, 2020, 267, 118720 CrossRef CAS.
- Y. Chen, L. Zou, H. Liu, C. Chen, Q. Wang, M. Gu, B. Yang, Z. Zou, J. Fang and H. Yang, J. Phys. Chem. C, 2019, 123, 16651–16659 CrossRef CAS.
- S. Zhao, G. Chen, G. Zhou, L.-C. Yin, J.-P. Veder, B. Johannessen, M. Saunders, S.-Z. Yang, R. De Marco, C. Liu and S. P. Jiang, Adv. Funct. Mater., 2020, 30, 1906157 CrossRef CAS.
- M. Zhang, Z. Hu, L. Gu, Q. Zhang, L. Zhang, Q. Song, W. Zhou and S. Hu, Nano Res., 2020, 13, 3206–3211 CrossRef.
- L. Zhao, Y. Zhang, L.-B. Huang, X.-Z. Liu, Q.-H. Zhang, C. He, Z.-Y. Wu, L.-J. Zhang, J. Wu, W. Yang, L. Gu, J.-S. Hu and L.-J. Wan, Nat. Commun., 2019, 10, 1278 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2021 |