
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of double core chromophore-functionalized nanothreads by compressing azobenzene in a diamond anvil cell†
Sebastiano
Romi
a,
Samuele
Fanetti
 *ab,
Frederico
Alabarse
c,
Antonio M.
Mio
d and
Roberto
Bini
*abe
*ab,
Frederico
Alabarse
c,
Antonio M.
Mio
d and
Roberto
Bini
*abe
aLENS, European Laboratory for Non-linear Spectroscopy, Via N. Carrara 1, I-50019, Sesto Fiorentino, Firenze, Italy. E-mail: fanetti@lens.unifi.it; roberto.bini@unifi.it; Fax: +390554572489; Tel: +390554572436
bICCOM-CNR, Istituto di Chimica dei Composti OrganoMetallici, Via Madonna del Piano 10, I-50019, Sesto Fiorentino, Firenze, Italy
cELETTRA, Elettra Sincrotrone Trieste S.C.p.A, in AREA Science Park 34149 Basovizza, Trieste, Italy
dIMM-CNR, Istituto per la Microelettronica e Microsistemi, VIII Strada 5 - Zona Industriale 95121, Catania, Italy
eDipartimento di Chimica “Ugo Schiff”, Università di Firenze, Via della Lastruccia 3, I-50019, Sesto Fiorentino, Italy
First published on 13th April 2021
Abstract
Carbon nanothreads are likely the most attracting new materials produced under high pressure conditions. Their synthesis is achieved by compressing crystals of different small aromatic molecules, while also exploiting the applied anisotropic stress to favor nontopochemical paths. The threads are nanometric hollow structures of saturated carbon atoms, reminiscent of the starting aromatic molecule, gathered in micron sized bundles. The examples collected so far suggest that their formation can be a general phenomenon, thus enabling the design of functionalities and properties by suitably choosing the starting monomer on the basis of its chemical properties and crystal arrangement. The presence of heteroatoms or unsaturation within the thread is appealing for improving the processability and tuning the electronic properties. Suitable simple chromophores can fulfill these requirements and their controlled insertion along the thread would represent a considerable step forward in tailoring the optical and electronic properties of these mechanically extraordinary materials. Here, we report the synthesis and extensive characterization of double core nanothreads linked by azo groups. This is achieved by compressing azobenzene in a diamond anvil cell, the archetype of a wide class of dyes, and represents a fundamental step in the realization of nanothreads with tailored photochemical and photophysical properties.
Introduction
Carbon nanomaterials have been receiving increasing attention from the scientific community because of the rapidly growing number of their technological applications which are not limited to the impressive number of present and potential uses of graphene.1 The peculiar mechanical and optical properties of nanodiamonds,2 the coexistence of complex sp2–sp3 nanostructures in diaphite,3 the thermoelectric characteristics of nanotubes4 and their potentiality as solid state supercapacitors5 are worth mentioning. In addition, their use as a biomaterial is not any more sporadic, representing their solid prospects in biomedical applications.6 These materials can be classified according to their dimensionality (0D, 1D or 2D) and to the hybridization of the carbon atoms (sp2 or sp3). Among these six different classes, the last to be discovered is that consisting of threads (1D) made from fully saturated carbon atoms. Three different polymorphs of these diamond-like threads have been independently predicted7–9 before the first synthesis, performed by compressing crystalline benzene at room temperature up to 23 GPa in a diamond anvil cell (DAC).10 The interest in the synthesized material is essentially related to its potentially extraordinary mechanical properties which combine flexibility, resilience and tensile strength and the way in which they can be tuned by controlling the quality and length of the threads.11,12 These remarkable properties have recently allowed the use of nanothread bundles for realizing energy storage devices13 or as nanofillers to realize high thermal conductivity composites.14The nanothread formation from benzene crystals has been fully rationalized identifying the crucial non-topochemical reaction step as due to the uniaxial stress which can be realized in the DAC avoiding the use of any hydrostatic compression medium.15,16 When the direction of the applied stress coincides or has an important component along the crystal direction where the rings interact in a parallel configuration, the formation of C–C interring bonds along the stack becomes possible. This reaction path is sharply evidenced by the alignment of the resulting nanothreads along the applied stress direction.10,16 The threads arrange parallel in a pseudohexagonal close-packed 2D lattice (a = 6.5 Å) characterized by a correlation length of several tens of nm, but definitely lower, ≤2.5 nm, along the threads.17,18 A consequence of the mechanism described above leading to the nanothread formation is that the reaction cannot be quantitative, competing with the topochemical one which leads to the formation of amorphous hydrogenated carbon (a-C:H).19,20
After the discovery of nanothread formation from benzene, computational and experimental studies revealed the possibility of synthesising nanothreads from a large number of aromatics and heteroaromatics. Ab initio DFT calculations and classical MD simulations suggested the formation of fully saturated carbon nanothreads from polycyclic aromatic hydrocarbons21 and from substituted benzenes (–CH3, –NH2, –OH, and –F).22 The presence of such groups is extremely attracting in view of exploiting them for functionalization or for tuning the electronic properties without reducing the mechanical properties which are not substantially altered upon functionalization with respect to benzene derived nanothreads. The presence of unsaturation can also play an important role in tuning the electronic properties by changing the gap from the UV to the near infrared, but obviously decreasing both strength and stiffness with respect to fully saturated nanothreads.23 Experimentally, nanothreads have been synthesized from substituted aromatics and heteroaromatics like pyridine,24,25 aniline26 and thiophene,27 and recently also from arene-perfluoroarene co-crystals28,29 and highly strained fully saturated molecules like cubane.30 In spite of the general similarity regarding the mechanism of the nanothread formation, consistent differences in the synthesis conditions, in the thread packing and in the reaction yield can characterize the different systems and are ascribable to the crystal arrangement of the starting monomer. Temperature is an important parameter in these reactions because through its change we can modulate the amplitude of the molecular motions in the crystal. With increasing the temperature above 400 K, the reaction threshold in pyridine has been lowered to 14 GPa improving the quality and yield (≥30%) of the synthesized nanothreads and also modifying the packing arrangement.25 The importance of temperature in the nanothread formation is even more pronounced in the case of aniline26 and cubane,30 where temperatures in excess of 500 K are required for inducing the reaction in spite of pressure conditions around or exceeding 30 GPa. In the cubane case, an excess of energy is required to release the internal strain of the molecule,30 whereas in aniline the reaction threshold experimentally observed, 550 K and 33 GPa,26 nicely fits with a topochemical mechanism where the reactivity is driven by a complex cooperation between lattice motions19,31 and H-bonding32,33 that causes a strongly anisotropic crystal response to compression and favors inter stack bonding while preserving the NH2 groups. With further increasing the temperature and reducing the pressure, the increased amplitude of both thermal oscillation and reaction cavity35 favors the topochemical path, which leads to the formation of amorphous 2D or 3D carbon nitrides both in the case of aniline34 as well as pyridine.25
The presence of heteroatoms or substituents within the threads can be exploited for tuning the electronic properties or to act as potential linkers and active sites for doping. Aromatic or heteroaromatic molecules containing chemically stable or well protected functional groups represent appealing systems for the realization of diamond-like nanothreads where the optical, thermal, electrical and conductivity properties can be suitably adjusted by choosing appropriate substituents. From this point of view, the azobenzene family is extremely attracting in view of the efficient tuning of their absorption, emission and photochemical properties by changing the substituents in the phenyl rings.36 This is the reason of their widespread use as dyes even though the most important characteristic is the efficient photo-isomerization, which makes them attracting building block structures for realizing molecular devices and functional materials,37 molecular photoswitches38–40 and even graphene composites.41 Azobenzene, the simplest member of this family, is characterized, in its trans form, by an excellent chemical stability of the crystal. The structural data, available only at ambient pressure, indicate a monoclinic P21/a structure with two molecules in the asymmetric unit cell (see ESI1†).42 Both molecules lie at inversion centers with one of them showing dynamic orientational disorder.43 Following the behavior observed on cooling, the disorder is expected to reduce with increasing the pressure as indeed supported by Raman experiments under hydrostatic conditions.44 The shortest distance between the centers of mass of equivalent molecules, an arrangement suggestive of a potentially selective Diels–Alder [4 + 2] polymerization, is realized along the b axis and it is 5.756 Å at ambient pressure. The formation of double nanothreads, in which the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N chromophore is preserved and protected, let us envisage the possibility to synthesize diamond-like bundles of nanothreads, where the azo groups, eventually present in low concentration by suitably choosing the starting mixed crystal and the ring substituents, can act as efficient light absorbers giving rise to a nanomaterial that combines the mechanical properties of carbon nanothreads with the tunable optical properties of azocompounds to realize functional ultrahard coatings or ultrafast heat sinks.
N chromophore is preserved and protected, let us envisage the possibility to synthesize diamond-like bundles of nanothreads, where the azo groups, eventually present in low concentration by suitably choosing the starting mixed crystal and the ring substituents, can act as efficient light absorbers giving rise to a nanomaterial that combines the mechanical properties of carbon nanothreads with the tunable optical properties of azocompounds to realize functional ultrahard coatings or ultrafast heat sinks.
In this manuscript we report the formation of double nanothreads by compressing trans-azobenzene crystals. Different pressure and temperature reaction paths have been explored to identify the best conditions for producing high quality bidimensional crystalline packing of double nanothreads linked by unaltered azo groups.
Results and discussion
Only Raman spectroscopy data are available at high pressure for the azobenzene crystal.44,45 Through compression and decompression experiments a phase transition was suggested around 10 GPa on the basis of intensity reduction of the Raman modes and the simultaneous appearance of new bands between 500 and 800 cm−1.45 Further compression above 18 GPa led to an irreversible loss of the Raman spectrum that was associated with the sample amorphization or an irreversible breakdown of the molecular structure. The employment of Raman spectroscopy to study possible chemical changes in the sample presents important drawbacks. The first, common to all the molecular systems, is the usual Raman intensity reduction due to strain caused by the applied anisotropic stress, which prevents a reliable identification of the occurrence of a chemical reaction rather than an amorphization of the system. In addition, Raman spectroscopy is a local probe, and typically the investigated area is of the order of a few squared microns, so in order to obtain reliable information, an appropriate mesh of the sample is required. Finally, the employment of laser light that can be absorbed through two-photon processes35 can give rise to photo-isomerization,46,47 thus representing a potential source of chemical instability of the sample. All these disadvantages are overcome by the employment of Fourier Transform Infrared Spectroscopy (FTIR), which also provides quantitative information about the reaction evolution.IR absorption spectra have been recorded at ambient temperature as a function of pressure up to 31.5 GPa, and at constant pressure (10 and 15 GPa) as a function of temperature. In all these cases, we detected a chemical transformation identifying the reaction threshold conditions and monitoring the kinetics of the transformation. In Fig. 1, we report some representative spectra collected in a room temperature compression and an isobaric heating experiment up the reaction threshold conditions.
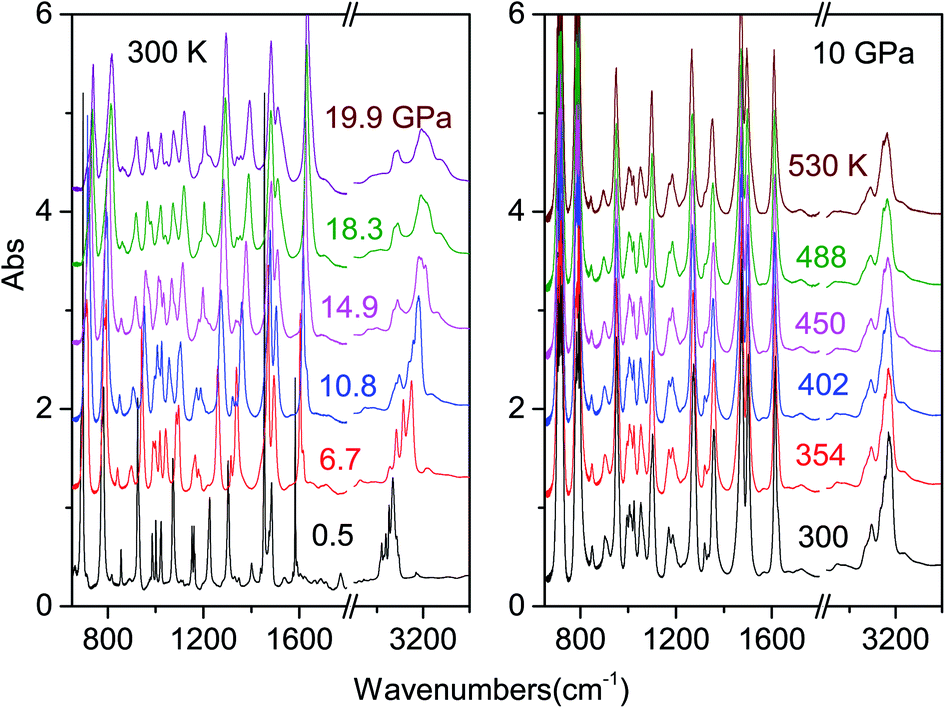 | ||
| Fig. 1 Representative spectra showing the evolution of the IR absorption spectrum of the azobenzene crystal on increasing pressure at ambient temperature (left) and isobarically heating the sample at 10 GPa (right). | ||
As it can be seen from the spectra collected during room temperature compression, the system is chemically stable at least up to 20 GPa. Careful analysis of the pressure evolution of the spectral components and of the relative parameters (peak frequency, FWHM and band area) shows some changes between 10 and 11 GPa (ESI2†), which are potential hints of a structural change as suggested by Raman spectra.45 However, the fact that most of the peaks are saturated and overlapped does not allow for a clear detection of a phase change requiring, in order to solve this issue, a direct structural characterization by X-ray diffraction.
The determination of the reaction's onset is not trivial because of the broadness of the product bands at high pressure, which makes its identification by spectral changes really challenging. To overcome this problem, we increased the pressure in small steps (0.1–0.2 GPa) subtracting from the last registered spectrum the previous one. In this way the reaction occurrence is readily evident especially from the reduction of the monomer bands, which appear as sharp negative peaks in the difference spectrum. An example is reported in Fig. 2 together with the crystal instability boundary accordingly identified. The higher temperature necessary to induce the reactivity at lower pressure is characteristic of the vast majority of molecular systems and is related to the larger amplitude of the thermal motion required, on decreasing the pressure, to balance the increase of intermolecular distances.19,31,34
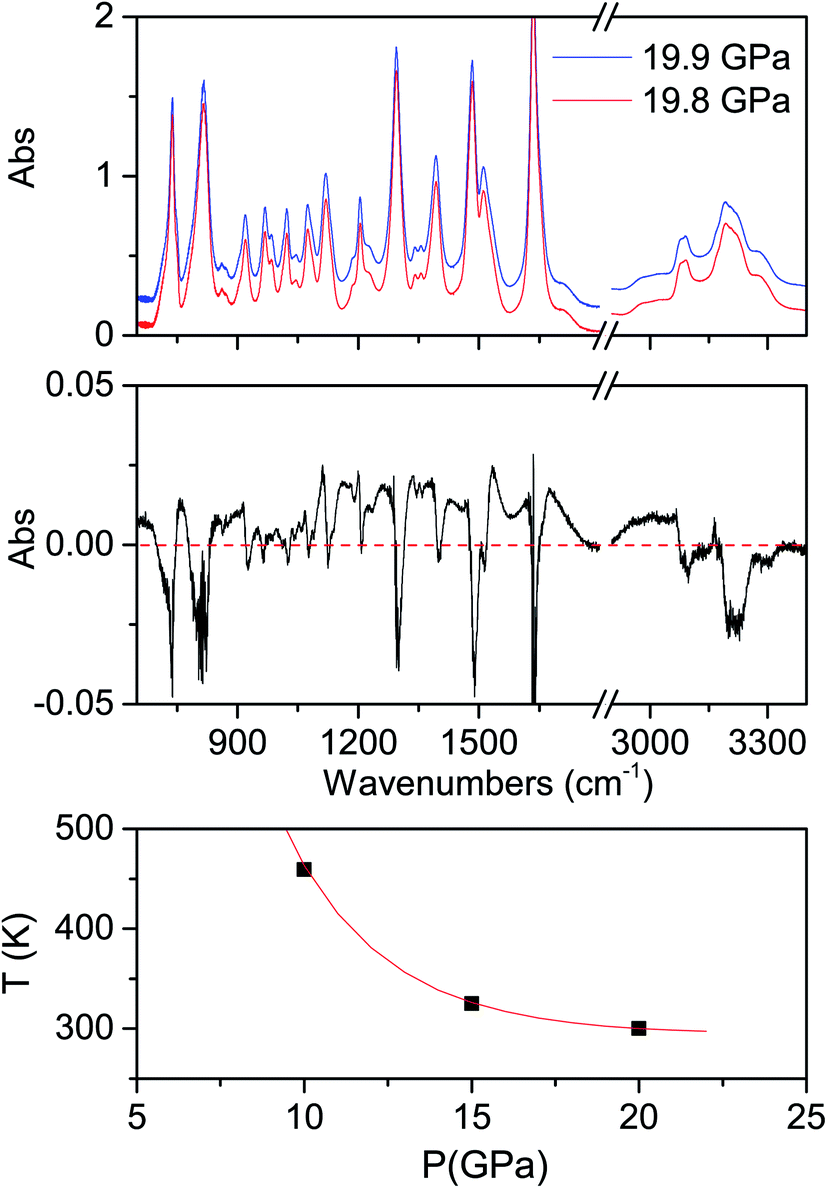 | ||
| Fig. 2 Upper panel: spectra registered during the room temperature compression at the onset of the crystal transformation; the two spectra are separated by a time interval of 20 minutes and the pressure difference is just due to the compression hysteresis. For the sake of clarity the two spectra are vertically shifted. Middle panel: difference spectrum obtained by the subtraction of the two spectra reported above, specifically, subtracting from the absorption IR spectrum recorded at 19.9 GPa that recorded immediately before at 19.8 GPa. The sharp negative peaks are related to the monomer, whose absorption decreases between the two spectra, whereas the broad positive features extending from about 1000 to 1600 cm−1 is due to the product. Lower panel: P–T point where the onset of the chemical transformation of the azobenzene crystal has been determined according to the analysis of the difference spectra; the full line is a guide to the eye. | ||
Once the reaction onset was identified, we measured the kinetics of the process by acquiring IR absorption spectra as a function of time under constant pressure and temperature conditions. In Fig. 3, we report the kinetics measured at ambient temperature and two different pressures, 25.0 and 31.5 GPa. Although the reaction onset was identified at 300 K around 20 GPa, the transformation rate was found to be extremely slow at this pressure; therefore the kinetic studies were performed at higher pressures, conditions realized by quickly (2–4 minutes) increasing the pressure once the threshold was reached. The kinetics were analyzed by using the Avrami model developed to describe the crystal growth from the melt48–50 and then extended to the study of diffusion controlled solid–state reactions.51 The fraction of the azobenzene monomer R reacted as a function of time is described by the following relationship:
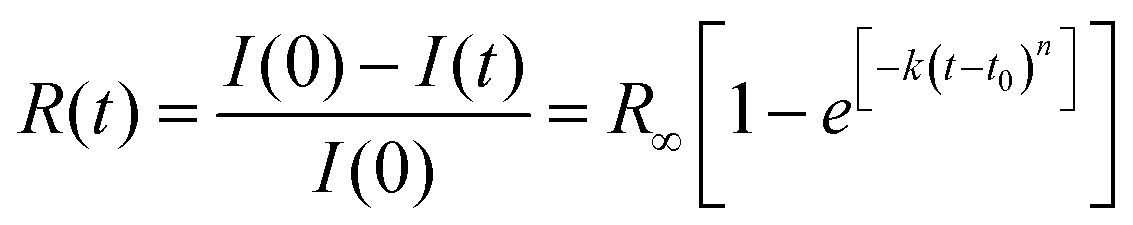 | (1) |
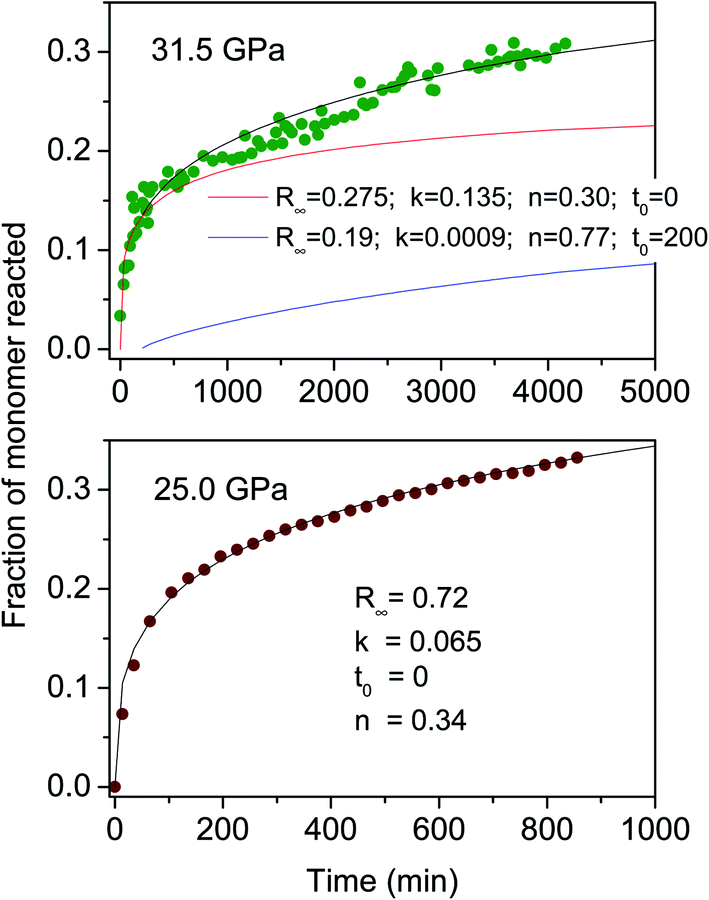 | ||
| Fig. 3 Kinetic evolution of the reactions performed at ambient temperature and 25.0 (lower) and 31.5 GPa (upper). The fraction of the monomer reacted, determined by the absorption of selected azobenzene bands (see the text), is plotted as a function of time. Eqn (1) is employed to reproduce these data; the fitting parameters are reported in both cases in the inset. The data collected at 31.5 GPa present a clear discontinuity after 270–300 minutes and have been reproduced (black line) by using the sum of two kinetic processes (red and blue lines). | ||
The main result of the kinetics' analysis regards the n values, all smaller than 1, thereby indicating a one-dimensional growth of the product as is expected for the formation of nanothreads. The need for using two different kinetic laws for the higher pressure reaction can be easily explained as due to the activation of competing polymerization paths along different directions because of the decreased intermolecular contacts at higher pressure. An analogous behaviour has been observed in the pressure induced polymerization of the ethylene crystal, where the anisotropic reduction of the lattice parameters with pressure activates a competing reaction path leading to gauche defected polymers.52 This additional reaction path, characterized by a rate constant one order of magnitude smaller, is activated when the faster step of the reaction has already progressed. The main (faster) kinetic contribution well agrees with the one measured at 25.0 GPa, exhibiting the same n value and a two times higher rate constant as expected according to the higher pressure value.
Further insight into the reaction mechanism is provided by the analysis of the molecularity of the process. The two sets of kinetic data just presented provide, as described in the ESI4,† a molecularity value consistent with a process, whose rate limiting step involves two units (molecules or oligomers) and therefore with a linear accretion of the product.
Analysis of the recovered products
After the completion of the kinetic studies, the pressure was released while acquiring IR absorption spectra to detect possible changes in the sample. During this step we did not observe any change in the sample, which can be related to an evolution of the chemical reaction. The recovered materials contain a variable amount of unreacted azobenzene, which is almost completely eliminated by opening the cell, to release the residual pressure, and then keeping the sample under vacuum for a few hours. The recovered materials from the different experiments are characterized by a coloration ranging from tangerine to dark red for the samples synthesized at room temperature, and dark grey for those produced under high temperature conditions (ESI5†). They all present an excellent chemical stability upon exposure to the atmosphere, as proved by the recorded IR spectra after several weeks. Some of the spectra measured on the recovered materials produced under different P–T conditions are reported in Fig. 4 together with a spectrum of azobenzene recorded at 0.1 GPa at the beginning of one of the experiments to highlight the amount of monomer left in the recovered product. The C–H stretching region is substantially modified by the reaction because of the hybridization change from sp2 to sp3 of most of the carbon atoms as revealed by the formation of strong absorption bands just below 3000 cm−1. All the products present two pairs of bands, the first at 695 and 750 cm−1, and the second one at 1493 and 1598 cm−1, reminiscent of the monomer spectral structure. The lowest doublet falls, although slightly red shifted, in the region of the azobenzene phenyl out-of-plane C–H bending modes, whereas the other doublet corresponds to the monomer region characteristic of the ring and of NN stretching modes,53 thus suggesting an ordered product and the maintenance of a ring-like structure and the azo group. A further hint of the NN bond preservation is the missed observation of protonated nitrogen (N–H stretching modes and C–N–H bending mode).
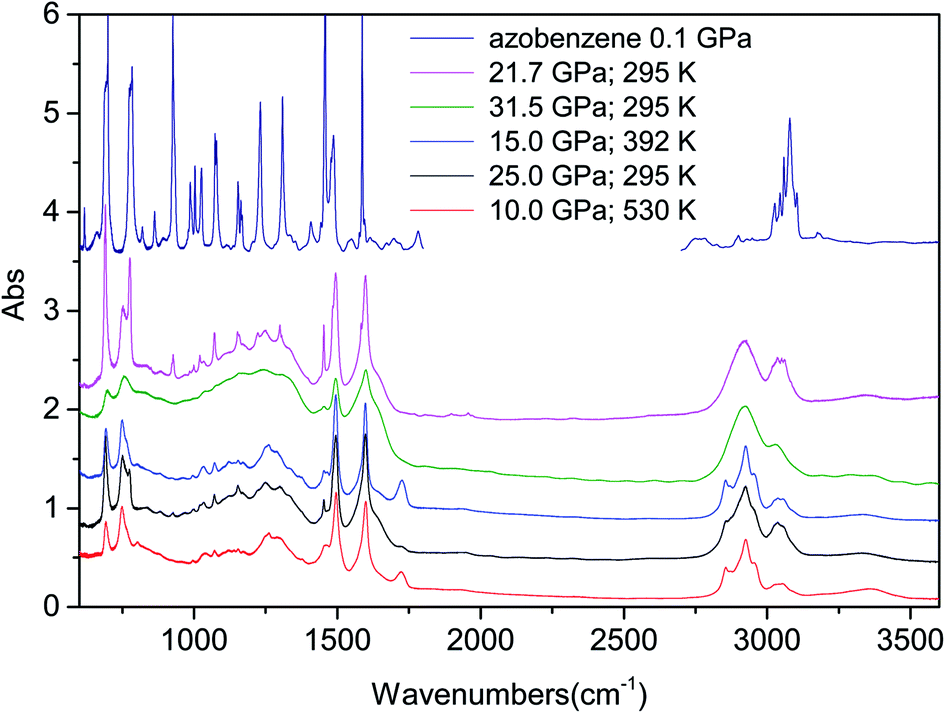 | ||
| Fig. 4 Comparison of the IR absorption spectra recorded on the materials recovered by the different reactions (P–T conditions of the synthesis reported in the legend) after having removed them from the DAC, and that of the pristine material immediately after loading. In this latter case, the region between 1800 and 2700 cm−1 has been removed because of the presence of the strong diamond absorption. The residual monomer peaks present in the product are easily identified as sharp lines, being particularly visible in the trace relative to the product from the reaction performed at ambient temperature and 21.7 GPa (magenta trace). | ||
Clear evidence of the nanothread formation is the characteristic, generally hexagonal, spotty XRD pattern which attests to the 2D ordered lattice in which the nanothreads are arranged.16,24,25 In fact, since the threads develop along the applied stress direction, the same direction of the probing X-ray beam, we have from this technique direct evidence of the 2D arrangement of the threads. In Fig. 5, we have reported the 2D images recorded with synchrotron light of the samples recovered from some of the reactions performed together with the integrated diffraction patterns as a function of the d spacing of the samples synthesized at ambient temperature. Diffraction patterns have been acquired in air with the samples contained in the gasket without the diamonds. The 2D images of the samples produced at ambient temperature exhibit the characteristic pattern of the pseudohexagonal lattice although showing some differences between them, but especially with respect to pyridine24,25 and benzene,10,16 where a single diffraction peak was detected. Here the strongest and sharper (FWHM ∼0.4 Å) peak is observed at 5.6 Å, as in pyridine and benzene, but another peak is observed at a d value (11.2–11.5 Å depending on the sample) slightly larger than the double, and having a FWHM of about 0.9–1.0 Å. In the case of the sample produced at the lower pressure (21.7 GPa), we have a clear doubling of the hexagonal pattern likely due to the presence of slightly differently packed 2D crystallites. It is worth remembering that the X-ray beam employed for this analysis, 80 μm in diameter, probes almost the entire sample, thus suggesting a remarkable order all over the sample. The doubling in the 2D image corresponds to a visible splitting of the main peak (blue trace in Fig. 5) with two maxima at 5.34 and 5.54 Å. This splitting is not observed in the products from the reactions performed at 25.0 and 31.5 GPa, where only a peak at 5.60 Å is observed. The peak present at larger d values is broader, exhibits a considerable dispersion (0.3–0.4 Å) and, in contrast to the lower d one, derives from a spotty powder-like diffraction ring, thus suggesting some kind of disorder in the product along this direction. The fourth 2D image reported in Fig. 5(d) is relative to the recovered sample from the experiment performed at 530 K and 10 GPa. In this case, as also for the other high temperature experiment (15 GPa and 350 K), no diffraction peaks can be identified therefore suggesting the formation of an amorphous material.
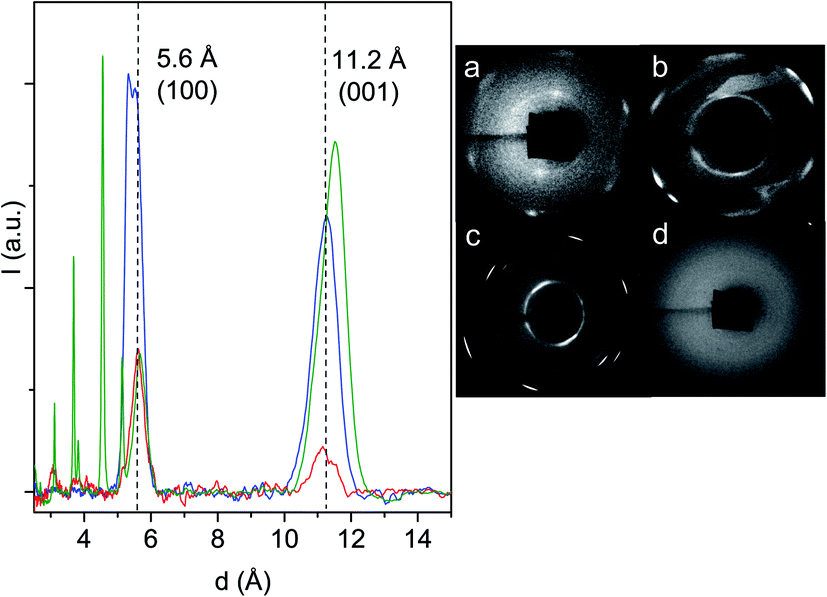 | ||
| Fig. 5 Left: 1D azimuthally integrated diffraction patterns reported as a function of the d spacing recorded with synchrotron light on the samples recovered from the ambient temperature reactions performed at 21.7 (blue), 25.0 (red) and 31.5 GPa (green). The baseline is the result of the subtraction of the Compton background. The additional narrow peaks observed at lower d in the green trace are relative to the unreacted azobenzene crystal. The two dashed lines and the relative d values (angstrom) indicate the position of the (100) and (001) diffraction peaks of the monoclinic cell in which the nanothreads are arranged (see the following discussion). Right: 2D images of the samples recovered from the reactions performed at 300 K and 25.0 GPa (a); 300 K and 21.7 GPa (b); 300 K and 31.5 GPa (c); 530 K and 10.0 GPa (d). | ||
The thread formation is confirmed by the high resolution transmission electron microscopy (HRTEM) analysis of the recovered materials. In Fig. 6, we report selected images collected at 60 keV primary electron beam energy on the samples recovered from the ambient temperature reactions performed at 25.0 and 31.5 GPa. The fragments analyzed have been obtained by breaking with a needle the sample contained in the gasket and selecting the thinner ones which are likely obtained from the peripheral areas of the recovered product. In these fragments are encountered both areas dominated by the presence of amorphous hydrogenated carbon (see ESI6†) and large portions showing perfectly parallel nanothreads extending for tens of nanometers. The occurrence of two products is in line with the competing mechanisms characterizing this kind of reaction. In some regions, these threads are bent (see the upper left panel of Fig. 6) producing curved bundles. A similar occurrence is observed in the micrographs obtained by Nobrega et al. on the nanothreads recovered by the compression of aniline26 and could likely be ascribed to the polycrystalline nature of the starting samples, which nevertheless allows the propagation of the thread through differently oriented adjacent crystals. The distance between the threads is very reproducible varying between 3.3 and 3.5 Å, a distance remarkably lower, about 30%, than those reported for benzene,10,16 pyridine24,25 and aniline,26 thus evidencing a denser and more compact material.
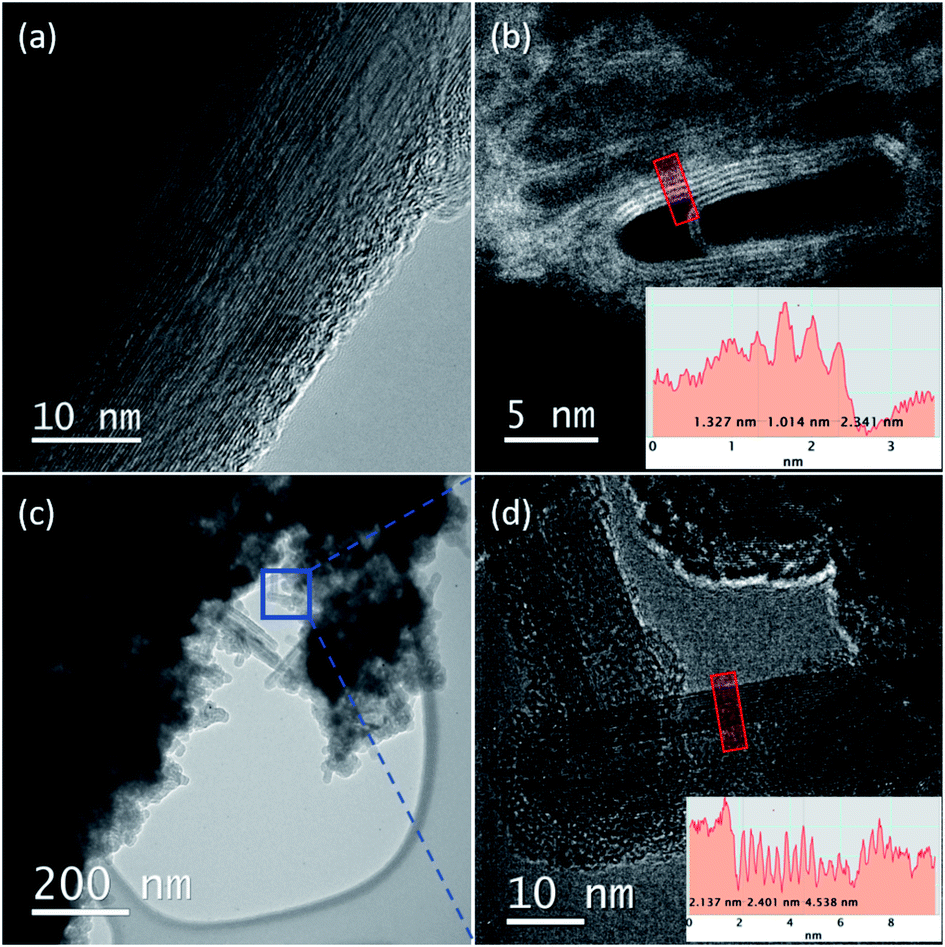 | ||
| Fig. 6 TEM micrographs recorded at different magnifications on the samples recovered from the ambient temperature reactions performed at 25.0 (a, b) and 31.5 (c, d) GPa. For the sake of completeness, micrographs are acquired both in bright-field (a, c, and d) and in Z-contrast HAADF STEM (b) mode. The micrographs have been acquired using an accelerating voltage of 60 kV, to avoid the knock-on damage. Nanothreads are clearly visible in all the highest magnification images. In (b) and (d), the insets report the line profiles obtained along the red boxes in the selected areas of the two recovered samples (separation between the threads ranging between 3.3 and 3.5 Å). In (a), the amazing long-range ordering of the formed nanothreads is evidenced. Bundles of nanothreads are perfectly visible in the lower magnification image (c), where the blue box corresponds to the particular area shown in (d). | ||
The XRD and HRTEM data define the constrains for the identification of the 2D crystalline arrangement of the synthesized nanothreads. XRD data unambiguously indicate a pseudo-hexagonal 2D arrangement with an interlayer distance of 5.6 Å corresponding to a lattice parameter a = 6.46 Å. However, the observation of the higher d peak provides additional constrains to identify the molecular arrangement in the 2D unit cell, which must also be reconciled with the small separation between the nanothreads, which develop as usual in the direction perpendicular to this plane, identified by HRTEM (3.3–3.5 Å). A monoclinic cell (a = 6.46 Å; c = 13.05 Å; β = 120°) as that reported in Fig. 7, with the molecule's dimensions as determined by X-ray diffraction,54 well accounts for all the experimental observations exhibiting diffraction peaks at 5.6 Å (100) and 11.2 Å (001) (see Fig. 5). The distance between the threads, obtained by piling up the phenyl rings along the direction perpendicular to the 2D lattice plane, depends on the viewing direction. In Fig. 7, we have highlighted how planes separated by 3.3 Å can be obtained rotating the threads by 90° around axes lying in the ac plane. This side view is also the most probable in the TEM experiment because the thin sample portions prepared for transmission experiments derive from a reproducible cleavage of the sample occurring along the thread direction due to the large Young's modulus of the nanothreads.12 Therefore, XRD experiments performed on the bulk sample contained in the gasket and TEM analysis performed on fragments obtained by breaking the sample provide complementary information related to the nearly orthogonal observation directions.
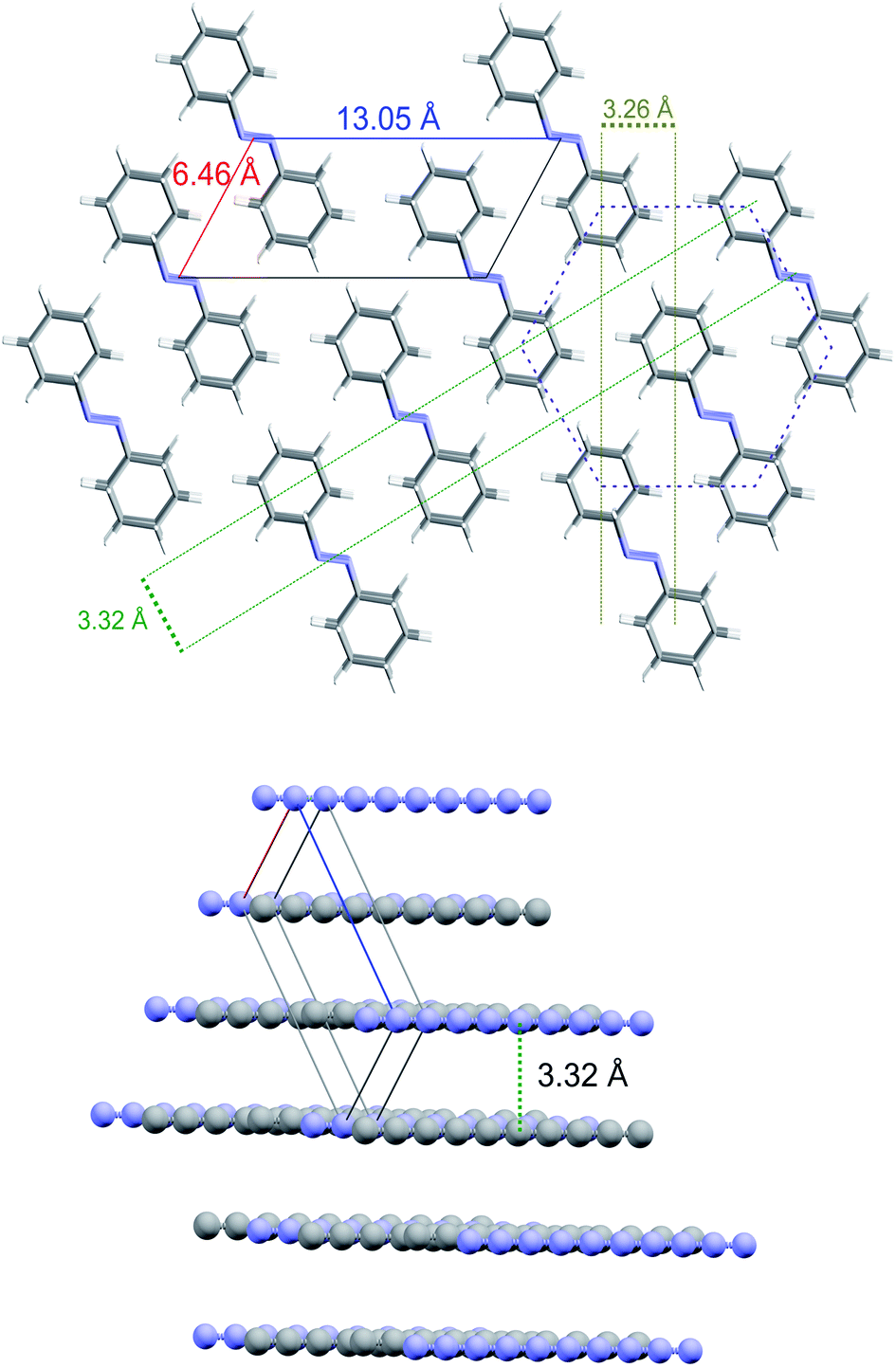 | ||
| Fig. 7 (Top) Schematic arrangement of the nanothreads in the plane perpendicular to the direction of formation. The monoclinic cell and the corresponding lattice parameters are evidenced as well as the hexagonal arrangement of the threads (dashed purple line). The two pairs of dashed parallel lines indicate side views identifying a nanothread separation of 3.29(3) Å in excellent agreement with the value extracted from the HRTEM images. (Bottom) Example of one of such side views where for the sake of clarity grey and blue balls represent the center of mass of phenyl rings and azo groups, respectively. | ||
Our experimental data unambiguously indicate the obtainment of nanothreads, where the tubular structures connected by untouched azo groups are formed through the bonding among phenyl rings arranged in stacks along the azobenzene b crystal axis. In spite of this evidence, we do not have a clue of the nanothread structure. In the attempt of bridging such a gap, we considered the four polymeric structures recognized so far as the most favorable:15,55 tube (3,0), polymer I, polytwistane and zipper. Among these four structures, we can immediately rule out the polytwistane since the formation of this 1D helical structure along the stack direction would require a relative rotation of the neighbouring molecules, thereby becoming incompatible with the formation of double core nanothreads and the preservation of the azo groups. Also the formation of the zipper polymer is unlikely because a relative rotation of neighbouring molecules is required, thus increasing the activation energy of the reaction, and in spite of the possibility of realizing a double core polymer (see Fig. 8), a considerable strain characterizes the thread, thus making the internal energy considerably larger than that of the two remaining structures. According to these preliminary considerations, we have computed, using density functional theory (DFT), the energetics of isolated fully sp3 nanothread fragments composed of six molecular units having the tube (3, 0) and polymer I structures. In Fig. 8, we show the optimized structures of these nanothreads and their Gibbs energies after subtracting the electronic contribution. The detailed results of the calculation are reported in ESI7.† The energy of the polymer I structure is about 2.4 eV lower than that of the tube (3, 0) but it must be considered that in the latter polymorph 6 more hydrogen atoms are required to saturate the terminations thus increasing of 18 units the number of vibrational modes contributing to the zero point energy. By suitably subdivide these vibrations between C–H stretching (6) and bending (12) modes and assuming an average frequency for these modes of 2900 and 1200 cm−1, respectively, an additional contribution to the zero point energy of 1.97 eV occurs. Therefore, although the polymer 1 structure still presents an energy lower by approximately 0.5 eV, the small difference does not allow a certain selection of the thread structure. For this purpose, we have computed the IR spectra of these two nanothread fragments in order to compare them with those recorded on the recovered materials. After the structural optimization, no imaginary vibrational frequencies were obtained, thus indicating the achievement of the minimum of the potential surface for the resulting geometries. The comparison between computed and experimental spectra is shown in Fig. 8. The similarities between the computed polymer 1 spectrum and the experimental ones are impressive also in view of the reduced chain length considered. All the following characteristic features are nicely reproduced: the doublet below 800 cm−1, the reduced intensity between 900 and 1000 cm−1, the absorption density between 1000 and 1400 cm−1 and the other characteristic doublet between 1500 and 1600 cm−1. For the latter feature, the discrepancy in the intensity of the higher frequency component can be due to the presence of unsaturation in the recovered materials. The strong band around 1450 cm−1 present in the computed spectrum of the tube (3, 0) deserves a comment: it is ascribable to the –CH2 scissoring modes given the large number of such groups present in the simulated tube terminations. A schematic drawing of the possible reaction path leading to the polymer 1 thread, assuming an isotropic compression of the ambient pressure crystal structure and considering the monomer-thread structural relationships, is reported in the ESI8.† However, accurate XRD studies of the pressure evolution of the azobenzene crystal structure up to the reaction threshold pressure are mandatory to elucidate the reaction mechanism.
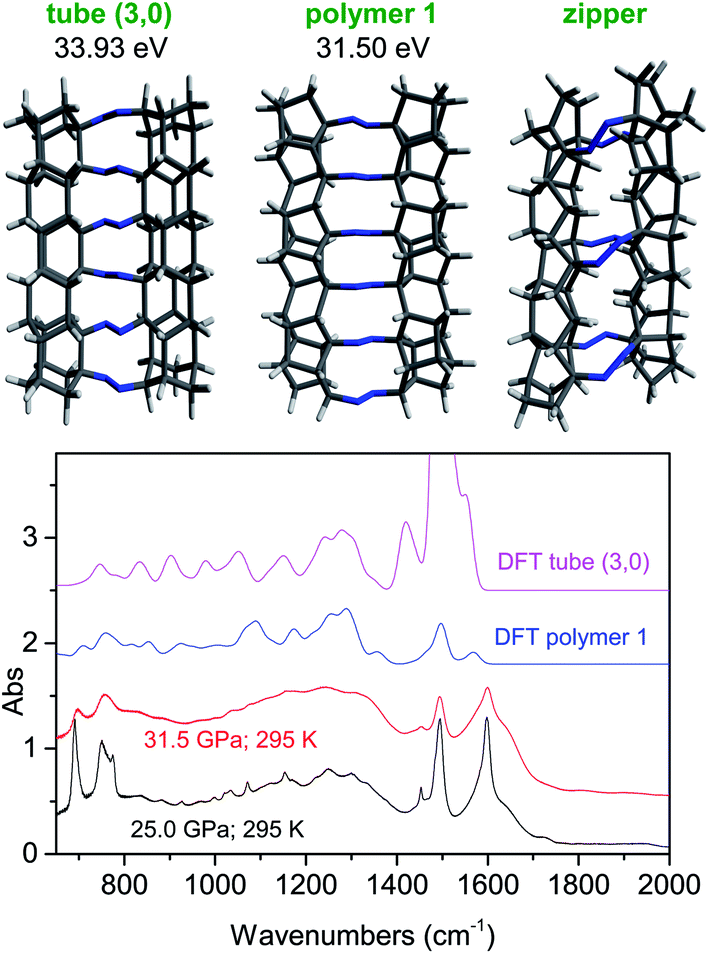 | ||
| Fig. 8 (Upper panel) Optimized structures of nanothread fragments composed of six azobenzene units and the relative Gibbs energies after the subtraction of the electronic contribution. (Lower panel) Comparison among the IR spectra of the materials recovered by different ambient temperature reactions (P–T conditions of the reactions are indicated in the Figure) and those computed for polymer 1 and tube (3, 0) nanothread structures composed of six molecular units. | ||
Conclusions
Since the discovery of carbon nanothreads, one dimensional fully saturated diamond-like wires, considerable progress has been made in the understanding of the reaction mechanisms and the crystal structure–reactivity relationship. A few successful examples, mainly involving aromatic molecules, provided evidence of the possible insertion of heteroatoms or simple substituents along the chain, prerequisite for the synthesis of nanothreads with targeted properties. The present knowledge can be therefore exploited for designing ultrahard fibers with specific photophysical and photochemical properties realized by inserting and preserving chromophores bound, but not incorporated, to the saturated thread structure in order to preserve the mechanical properties. We have successfully accomplished such a synthesis by compressing azobenzene in a DAC at 20 GPa and room temperature, the archetype of a large family of dyes. The stacking formed by the phenyl rings along the b axis of the azobenzene crystal allows the formation of C–C interring bonds leading to the formation of high quality double core nanothreads arranged, according to DFT calculations, in a polymer 1 geometry. Importantly, the azo group (–NN–) is preserved during the reaction and in the quenched material, which exhibits a pseudohexagonal 2D ordered structure with a distance between the planes identified by the centres of the threads of 5.6 Å. Another remarkable feature of the recovered material is the extremely high density of the packed threads because of the reduced distance between neighbouring double threads (3.3–3.5 Å) much lower than that of any other analogous materials synthesized so far. The preservation of the azo group is of fundamental importance for tuning the optical properties of this material by changing, for example, the azobenzene concentration in a starting doped or co-crystal. The results presented in this work represent therefore an important milestone in the challenging search of systems and methodologies directed to control and exploit the electronic properties of these mechanically superior materials.
Methods
Crystalline azobenzene (from Sigma Aldrich with a purity >99%) was loaded without any compression medium into a membrane diamond anvil cell (MDAC) equipped with IIa type diamonds together with a ruby chip for pressure calibration by the ruby fluorescence method.56 Samples were laterally contained by rhenium gaskets drilled to have an initial sample diameter of 150 μm and a thickness of about 50 μm. High temperature was obtained by resistive heating, and the sample temperature was measured with a K-type thermocouple placed close to the diamonds with a 0.1 K accuracy.Angle-dispersive synchrotron X-ray diffraction measurements were performed on the recovered products at the Xpress beamline at the Elettra Italian synchrotron with a beam energy of 25.0 KeV, a focal spot diameter of about 80 μm (FWHM) and an image plate detector (MAR345). In-house X-ray diffraction measurements were performed with a custom made, laboratory diffractometer, equipped with a focused Xenocs-GeniX Mo Small Spot microsource with a wavelength of 0.7107 Å and a beam diameter on the focal plane of 150 μm. A PI-SCX 4300 CCD was employed as the detector.
Fourier transform infrared absorption spectra were recorded using a Bruker-IFS 120 HR spectrometer suitably modified for experiments in a diamond anvil cell, with an instrumental resolution set to 1 cm−1.57 The sample pressure was measured by the ruby fluorescence method using few mW of a 532 nm laser line from a doubled Nd:YAG laser source.
Transmission electron microscopy (TEM) investigation was performed with a JEOL ARM200F Cs-corrected microscope, equipped with a cold-field emission gun having an energy spread of 0.3 eV and operating at 60 keV. In conventional TEM (CTEM) mode, micrographs have been acquired in bright field (BF), while in STEM mode imaging has also been performed in Z-contrast mode using a high-angle annular dark field (HAADF) detector, with a probe size of 1.0 Å. The recovered material was mechanically removed from the gasket with a needle and placed directly onto the surface of a standard TEM lacey carbon film, covering a 300-mesh copper grid.
The structural optimization and the infrared spectrum calculation of the nanothread fragment composed of six molecular units have been performed using density functional theory approximation (DFT) with the Orca program,58 with the REVPBE density functional, the def2-TZVPP basis set, and the def2-TZVPP/C auxiliary basis set. The infrared frequencies are obtained by numerical Hessian computation.
Author contributions
S. F. and R. B. conceived and designed the experiments. S. F. and S. R. conducted the experiments, analyzed the data and performed the calculations. S. F., S. R., F. A. and A. M. M. characterized the recovered materials. R. B. prepared the manuscript with the help and contribution of all the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the European Laboratory for Nonlinear Spectroscopy (LENS) for hosting the research, and the Deep Carbon Observatory and the “Fondazione CR Firenze” for strong support. The research has been supported by the following grants: Extreme Physics and Chemistry of Carbon: Forms, Transformations, and Movements in Planetary Interiors funded by the Alfred P. Sloan Foundation; Fondazione Cassa di Risparmio di Firenze under the project “Utilizzo dell'anisotropia strutturale nella sintesi di nanofili di carbonio diamond-like ad alta pressione”. The TEM analyses were performed at the Beyond-Nano laboratory of the CNR-IMM, which is supported by the Italian Ministry of Education and Research (MIUR) under the “Beyond-Nano” project (PON a3_00363). We also thank MIUR-Italy (“Progetto Dipartimenti di Eccellenza 2018–2022′′ allocated to the Department of Chemistry “Ugo Schiff”) and the Elettra Italian synchrotron for hosting our experiment under the proposal number 20200230.References
- A. C. Ferrari, et al., Science and Technology Roadmap for Graphene, related Two-Dimensional Crystals, and Hybrid Systems, Nanoscale, 2015, 7, 4587–5062 RSC.
- V. N. Mochalin, O. Shenderova, D. Ho and Y. Gogotsi, The Properties and Applications of Nanodiamonds, Nat. Nanotechnol., 2011, 7, 11–23 CrossRef PubMed.
- P. Nemeth, K. McColl, L. A. J. Garvie, C. G. Salzmann, M. Murri and P. F. McMillan, Complex Nanostructures in Diamond, Nat. Mater., 2020, 19, 1126–1131 CrossRef CAS PubMed.
- J. L. Blackburn, A. J. Ferguson, C. Cho and J. C. Grunlan, Carbon-Nanotube-Based Thermoelectric Materials and Devices, Adv. Mater., 2018, 30, 1704386 CrossRef PubMed.
- T. Lv, M. Liu, D. Zhu, L. Gan and T. Chen, Nanocarbon-Based Materials for Flexible All-Solid-State Supercapacitors, Adv. Mater., 2018, 30, 1705489 CrossRef PubMed.
- K. P. Loh, D. Ho, G. Ngar, C. Chiu, D. T. Leong, G. Pastorin and E. K.-H. Chow, Clinical Applications of Carbon Nanomaterials in Diagnostics and Therapy, Adv. Mater., 2018, 30, 1802368 CrossRef PubMed.
- D. Stojkovic, P. Zhang and V. H. Crespi, Smallest Nanotube: Breaking the Symmetry of sp3 Bonds in Tubular Geometries, Phys. Rev. Lett., 2001, 87, 125502 CrossRef CAS PubMed.
- X.-D. Wen, R. Hoffmann and N. W. Ashcroft, Benzene under High Pressure: a Story of Molecular Crystals Transforming to Saturated Networks, with a Possible Intermediate Metallic Phase, J. Am. Chem. Soc., 2011, 133, 9023–9035 CrossRef CAS PubMed.
- S. R. Barua, H. Quanz, M. Olbrich, P. R. Schreiner, D. Trauner and W. D. Allen, Polytwistane, Chem. – Eur. J., 2014, 20, 1638–1645 CrossRef CAS PubMed.
- T. C. Fitzgibbons, M. Guthrie, E. S. Xu, V. H. Crespi, S. K. Davidowski, G. D. Cody, N. Alem and J. V. Badding, Benzene-Derived Carbon Nanothreads, Nat. Mater., 2015, 14, 43–47 CrossRef CAS PubMed.
- R. E. Roman, K. Kwan and S. W. Cranford, Mechanical Properties and Defect Sensitivity of Diamond Nanothreads, Nano Lett., 2015, 15, 1585–1590 CrossRef CAS PubMed.
- H. Zhan, G. Zhang, V. B. C. Tan, Y. Cheng, J. M. Bell, Y.-W. Zhang and Y. Gu, From Brittle to Ductile: a Structure Dependent Ductility of Diamond Nanothread, Nanoscale, 2016, 8, 11177–11184 RSC.
- H. Zhan, G. Zhang, J. M. Bell, V. B. C. Tan and Y. Gu, High Density Mechanical Energy Storage with Carbon Nanothread Bundle, Nat. Commun., 2020, 11, 1905 CrossRef CAS PubMed.
- H. Zhan, G. Zhang, G. Zhuang, R. Timon and Y. Gu, Low Interfacial Thermal Resistance between Crossed Ultra-Thin Carbon Nanothreads, Carbon, 2020, 165, 216–224 CrossRef CAS.
- B. Chen, R. Hoffmann, N. W. Ashcroft, J. Badding, E. S. Xu and V. Crespi, Linearly Polymerized Benzene Arrays As Intermediates, Tracing Pathways to Carbon Nanothreads, J. Am. Chem. Soc., 2015, 137, 14373–14386 CrossRef CAS PubMed.
- X. Li, M. Baldini, T. Wang, B. Chen, E.-s. Xu, B. Vermilyea, V. H. Crespi, R. Hoffmann, J. J. Molaison, C. A. Tulk, M. Guthrie, S. Sinogeikin and J. V. Badding, Mechanochemical Synthesis of Carbon Nanothread Single Crystals, J. Am. Chem. Soc., 2017, 139, 16343–16349 CrossRef CAS PubMed.
- P. Duan, X. Li, T. Wang, B. Chen, S. J. Juhl, D. Koeplinger, V. H. Crespi, J. V. Badding and K. Schmidt-Rohr, The Chemical Structure of Carbon Nanothreads Analyzed by Advanced Solid-State NMR, J. Am. Chem. Soc., 2018, 140, 7658–7666 CrossRef CAS PubMed.
- S. J. Juhl, T. Wang, B. Vermilyea, X. Li, V. H. Crespi, J. V. Badding and N. Alem, Local Structure and Bonding of Carbon Nanothreads Probed by High-Resolution Transmission Electron Microscopy, J. Am. Chem. Soc., 2019, 141, 6937–6945 CrossRef CAS PubMed.
- L. Ciabini, M. Santoro, F. A. Gorelli, R. Bini, V. Schettino and S. Raugei, Triggering Dynamics of the High-Pressure Benzene Amorphization, Nat. Mater., 2007, 6, 39–43 CrossRef CAS PubMed.
- L. Ciabini, M. Santoro, R. Bini and V. Schettino, High Pressure Reactivity of Solid Benzene Probed by Infrared Spectroscopy, J. Chem. Phys., 2002, 116, 2928–2935 CrossRef CAS.
- P. G. Demingos and A. R. Muniz, Carbon Nanothreads from Polycyclic Aromatic Hydrocarbon Molecules, Carbon, 2018, 140, 644–652 CrossRef CAS.
- J. F. R. V. Silveira and A. R. Muniz, Functionalized Diamond Nanothreads from Benzene Derivatives, Phys. Chem. Chem. Phys., 2017, 19, 7132–7137 RSC.
- P. G. Demingos and A. R. Muniz, Electronic and Mechanical Properties of Partially Saturated Carbon and Carbon Nitride Nanothreads, J. Phys. Chem. C, 2019, 123, 3886–3891 CrossRef CAS.
- X. Li, T. Wang, P. Duan, M. Baldini, H.-T. Huang, B. Chen, S. J. Juhl, D. Koeplinger, V. H. Crespi, K. Schmidt-Rohr, R. Hoffmann, N. Alem, M. Guthrie, X. Zhang and J. V. Badding, Carbon Nitride Nanothread Crystals Derived from Pyridine, J. Am. Chem. Soc., 2018, 140, 4969–4972 CrossRef CAS PubMed.
- S. Fanetti, M. Santoro, F. Alabarse, E. Berretti and R. Bini, Modulating the H-Bond Strength by Varying the Temperature for the High Pressure Synthesis of Nitrogen Rich Carbon Nanothreads, Nanoscale, 2020, 12, 5233–5242 RSC.
- M. M. Nobrega, E. Teixeira-Neto, A. B. Cairns, M. L. A. Temperini and R. Bini, One-Dimensional Diamondoid Polyaniline-like Nanothreads from Compressed Crystal Aniline, Chem. Sci., 2018, 9, 254–260 RSC.
- A. Biswas, M. D. Ward, T. Wang, L. Zhu, H.-T. Huang, J. V. Badding, V. H. Crespi and T. A. Strobel, Evidence for Orientational Order in Nanothreads Derived from Thiophene, J. Phys. Chem. Lett., 2019, 10, 7164–7171 CrossRef CAS PubMed.
- M. D. Ward, W. S. Tang, L. Zhu, D. Popov, G. D. Cody and T. A. Strobel, Controlled Single-Crystalline Polymerization of C10H8·C10F8 under Pressure, Macromolecules, 2019, 52, 7557–7563 CrossRef CAS.
- A. Friedrich, I. E. Collings, K. Dziubek, S. Fanetti, K. Radacki, J. Ruiz-Fuertes, J. Pellicer-Porres, M. Hanfland, D. Sieh, R. Bini, S. J. Clark and T. B. Marder, Pressure-Induced Polymerization of Polycyclic Arene-Perfluoroarene co-Crystals: Single Crystal X-Ray Diffraction Studies, Reaction Kinetics, and Design of Columnar Hydrofluorocarbons, J. Am. Chem. Soc., 2020, 142(44), 18907–18923 CrossRef CAS PubMed.
- H.-T. Huang, L. Zhu, M. D. Ward, T. Wang, B. Chen, B. L. Chaloux, Q. Wang, A. Biswas, J. L. Gray, B. Kuei, G. D. Cody, A. Epshteyn, V. H. Crespi, J. V. Badding and T. A. Strobel, Nanoarchitecture through Strained Molecules: Cubane-Derived Scaffolds and the Smallest Carbon Nanothreads, J. Am. Chem. Soc., 2020, 142, 17944–17955 CrossRef CAS PubMed.
- M. Citroni, S. Fanetti, C. Bazzicalupi, K. Dziubek, M. Pagliai, M. M. Nobrega, M. Mezouar and R. Bini, Structural and Electronic Competing Mechanisms in the Formation of Amorphous Carbon Nitride by Compressing s-Triazine, J. Phys. Chem. C, 2015, 119, 28560–28569 CrossRef CAS.
- M. M. Nobrega, M. L. A. Temperini and R. Bini, Probing the Chemical Stability of Aniline under High Pressure, J. Phys. Chem. C, 2017, 121, 7495–7501 CrossRef CAS.
- S. Fanetti, M. Citroni, K. Dziubek, M. M. Nobrega and R. Bini, The Role of H-Bond in the High-Pressure Chemistry of Model Molecules, Phys. Rev. B, 2018, 30, 094001 CrossRef PubMed.
- S. Fanetti, M. M. Nobrega, E. Teixeira-Neto, M. L. A. Temperini and R. Bini, Effect of Structural Anisotropy in High-Pressure Reaction of Aniline, J. Phys. Chem. C, 2018, 122, 29158–29164 CrossRef CAS.
- R. Bini and V. Schettino, Materials Under Extreme Conditions: Molecular Crystals at High Pressure, Imperial College Press, London, 2014 Search PubMed.
- H. M. Dhammika Bandarab and S. C. Burdette, Photoisomerization in Different Classes of Azobenzene, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- E. Wagner-Wysiecka, N. Łukasik, J. F. Biernat and E. Luboch, Azo Group(s) in Selected Macrocyclic Compounds, J. Inclusion Phenom. Macrocyclic Chem., 2018, 90, 189–257 CrossRef CAS PubMed.
- D. Bléger and S. Hecht, Visible-Light-Activated Molecular Switches, Angew. Chem., Int. Ed., 2015, 54, 11338–11349 CrossRef PubMed.
- S. Crespi, N. A. Simeth and B. König, Heteroaryl Azo Dyes as Molecular Photoswitches, Nat. Rev. Chem., 2019, 3, 133–146 CrossRef CAS.
- M. Dong, A. Babalhavaeji, S. Samanta, A. A. Beharry and G. A. Woolley, Red-Shifting Azobenzene Photoswitches for in Vivo Use, Acc. Chem. Res., 2016, 7, 11090 Search PubMed.
- M. Döbbelin, et al., Light-Enhanced Liquid-Phase Exfoliation and Current Photoswitching in Graphene-Azobenzene Composites, Nat. Commun., 2015, 48, 2662–2670 CrossRef PubMed.
- C. J. A. Brown, Refinement of the Crystal Structure of Azobenzene, Acta Crystallogr., 1966, 21, 146–152 CrossRef CAS.
- J. Harada and K. Ogawa, X-ray Diffraction Analysis of Nonequilibrium States in Crystals: Observation of an Unstable Conformer in Flash-Cooled Crystals, J. Am. Chem. Soc., 2004, 126, 3539–3544 CrossRef CAS PubMed.
- A. Li, C. Bi, S. Xu, H. Cui and W. Xu, Structural Change of Trans-Azobenzene Crystal and Powder under High Pressure, J. Mol. Struct., 2020, 1206, 127745 CrossRef CAS.
- Z. Dong, N. M. Seemann, N. Lu and Y. Song, Effects of High Pressure on Azobenzene and Hydrazobenzene Probed by Raman Spectroscopy, J. Phys. Chem. B, 2011, 115, 14912–14918 CrossRef CAS PubMed.
- Y. Hao, S. Huang, Y. Guo, L. Zhou, H. Hao, C. J. Barrett and H. Yu, Photoinduced Multi-Directional Deformation of Azobenzene Molecular Crystals, J. Mater. Chem. C, 2019, 7, 503–508 RSC.
- T. Taniguchi, T. Asahi and H. Koshima, Photomechanical Azobenzene Crystals, Crystals, 2019, 9, 437 CrossRef CAS.
- M. Avrami, Kinetics of Phase Change. I General Theory, J. Chem. Phys., 1939, 7, 1103–1112 CrossRef CAS.
- M. Avrami, Kinetics of Phase Change. II Transformation-Time Relations for Random Distribution of Nuclei, J. Chem. Phys., 1940, 8, 212–224 CrossRef CAS.
- M. G. Avrami, Phase Change, and Microstructure Kinetics of Phase Change. III, J. Chem. Phys., 1941, 9, 177–184 CrossRef CAS.
- S. F. Hulbert, Models for Solid-State Reactions in Powdered Compacts: A Review, J. Br. Ceram. Soc., 1969, 6, 11–20 CAS.
- D. Chelazzi, M. Ceppatelli, M. Santoro, R. Bini and V. Schettino, Pressure-Induced Polymerization in Solid Ethylene, J. Phys. Chem. B, 2005, 109, 21658–21663 CrossRef CAS PubMed.
- D. R. Armostrong, J. Clarkson and W. E. Smith, Vibrational Analysis of Trans-Azobenzene, J. Phys. Chem., 1995, 99, 17825–17831 CrossRef.
- J. A. Bouwstra, A. Schouten and J. Kroon, Structural Studies of the System Trans-Azobenzene/Trans-Stilbene. I. A Reinvestigation of the Disorder in the Crystal Structure of Trans-Azobenzene, C12H10N2, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1983, C39, 1121–1123 CrossRef CAS.
- B. Chen, T. Wang, V. H. Crespi, X. Li, J. Badding and R. Hoffmann, All the Ways To Have Substituted Nanothreads, J. Chem. Theory Comput., 2018, 14, 1131–1140 CrossRef CAS PubMed.
- H. K. Mao, P. M. Bell, J. V. Shaner and D. J. Steinberg, Specific Volume Measurements of Cu, Mo, Pd, and Ag and Calibration of the Ruby R1 Fluorescence Pressure Gauge from 0.06 to 1 Mbar, J. Appl. Phys., 1978, 49, 3276 CrossRef CAS.
- R. Bini, R. Ballerini, G. Pratesi and H. J. Jodl, Experimental Setup for Fourier Transform Infrared Spectroscopy Studies in Condensed Matter at High Pressure and Low Temperatures, Rev. Sci. Instrum., 1997, 68, 3154–3160 CrossRef CAS.
- F. Neese, The ORCA Program System, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc06968j |
| This journal is © The Royal Society of Chemistry 2021 |