
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergizing the potential of bacterial genomics and metabolomics to find novel antibiotics
Fabian
Panter
abc,
Chantal D.
Bader
ab and
Rolf
Müller
*abc
aDepartment of Microbial Natural Products, Helmholtz-Institute for Pharmaceutical Research Saarland (HIPS), Helmholtz Centre for Infection Research (HZI), Department of Pharmacy, Saarland University, Campus E8 1, 66123 Saarbrücken, Germany. E-mail: Rolf.Mueller@helmholtz-hips.de
bGerman Centre for Infection Research (DZIF), Partner Site Hannover–Braunschweig, Germany
cHelmholtz International Lab for Anti-infectives, Campus E8 1, 66123 Saarbrücken, Germany
First published on 29th March 2021
Abstract
Antibiotic development based on natural products has faced a long lasting decline since the 1970s, while both the speed and the extent of antimicrobial resistance (AMR) development have been severely underestimated. The discovery of antimicrobial natural products of bacterial and fungal origin featuring new chemistry and previously unknown mode of actions is increasingly challenged by rediscovery issues. Natural products that are abundantly produced by the corresponding wild type organisms often featuring strong UV signals have been extensively characterized, especially the ones produced by extensively screened microbial genera such as streptomycetes. Purely synthetic chemistry approaches aiming to replace the declining supply from natural products as starting materials to develop novel antibiotics largely failed to provide significant numbers of antibiotic drug leads. To cope with this fundamental issue, microbial natural products science is being transformed from a ‘grind-and-find’ study to an integrated approach based on bacterial genomics and metabolomics. Novel technologies in instrumental analytics are increasingly employed to lower detection limits and expand the space of detectable substance classes, while broadening the scope of accessible and potentially bioactive natural products. Furthermore, the almost exponential increase in publicly available bacterial genome data has shown that the biosynthetic potential of the investigated strains by far exceeds the amount of detected metabolites. This can be judged by the discrepancy between the number of biosynthetic gene clusters (BGC) encoded in the genome of each microbial strain and the number of secondary metabolites actually detected, even when considering the increased sensitivity provided by novel analytical instrumentation. In silico annotation tools for biosynthetic gene cluster classification and analysis allow fast prioritization in BGC-to-compound workflows, which is highly important to be able to process the enormous underlying data volumes. BGC prioritization is currently accompanied by novel molecular biology-based approaches to access the so-called orphan BGCs not yet correlated with a secondary metabolite. Integration of metabolomics, in silico genomics and molecular biology approaches into the mainstream of natural product research will critically influence future success and impact the natural product field in pharmaceutical, nutritional and agrochemical applications and especially in anti-infective research.
Introduction
Antibiotics research has come a long way since the discoveries of first penicillin and later streptomycin in the 30s and 40s of the last century.1 These discoveries marked the start of the so-called golden era of antibiotics by introducing antimicrobial substances that would become some of the most successful small molecule drugs in human history.1,2 The golden era of antibiotics discovery lasted from the 1930s until the 1960s, a time that saw numerous discoveries and rapid development of novel antibiotic substances.3 Most of the antibiotic drugs developed early on, such as aminoglycosides or tetracyclines were derived from actinomycetes and were provided by so-called bioactivity-guided antibiotics isolation approaches.4 The development of sulfonamides was an exception at that time, as they represent purely synthetic antibacterial agents.5 Natural products are preselected structures in the search for drug leads in antibiotics research as they are produced by the respective microbes or plants to defend themselves against predators or competitors in their ecological niches.6 Even though plants have been used for the longest time for medicinal purposes among the natural product producers, fungi and bacteria are more suitable when it comes to antibiotics discovery, as survival in their microbial community forces them to compete with other microbes and defence against higher developed animals is assumed less important.7Not long after the antibiotic discovery boom, the general sentiment that bacterial infections are generally under control emerged among both natural product scientists as well as pharmacists and clinicians (see Fig. 1). Infectious diseases were no longer considered to rank among the biggest health risks to humans, which inevitably led to shifting priorities towards other topics such as anti-cancer research.8,9 However, bacterial and fungal pathogens adapted much more quickly than predicted to the more and more widespread use of antibiotics by developing and sharing antibiotic resistance genes, which is why infectious diseases are seen as one of the major global health threats again.10
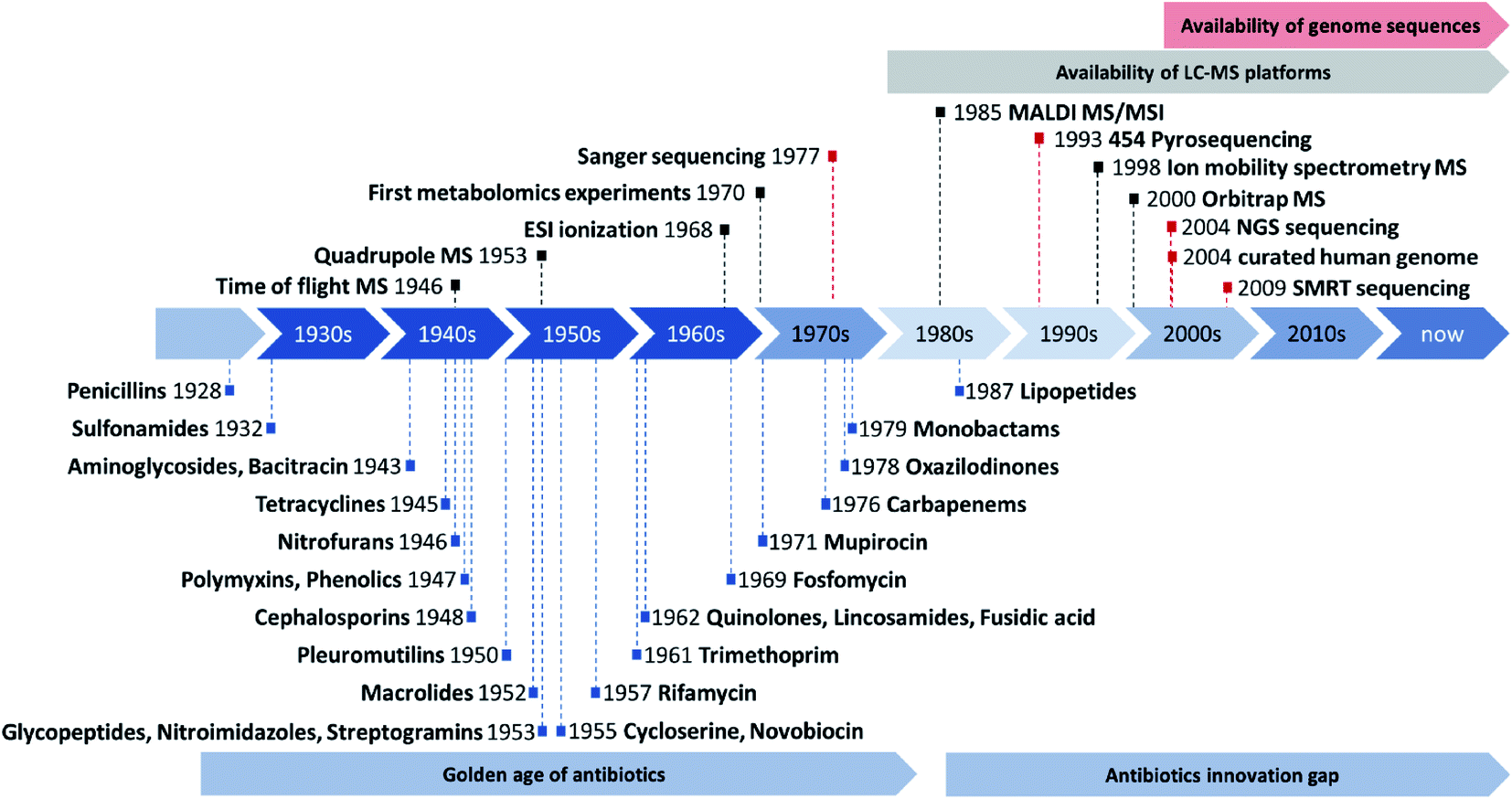 | ||
| Fig. 1 Overview about the discovery of novel antibiotics during the golden age of antibiotics discovery and the subsequent innovation gap adapted from Silver L. (blue).11 Overview about key milestones in the development (not commercialization) of mass spectrometry instrumentation (black) and genome sequencing technology (red) as well as reasonable timeframes for the availability of LC-MS and genome sequencing technology in natural products laboratories. Sequencing milestone abbreviations are next generation sequencing (NGS) or illumina technology and single molecule real time (SMRT) sequencing or PacBio technology. | ||
To date, most infectious diseases caused by bacteria remain controllable due to semisynthetic improvement of clinically used antibiotics derived from natural products in several iterations.12,13 Many antibiotic classes, especially the ones discovered early on during the golden age of antibiotics have already reached the 5th generation of synthetic derivatives, limiting further chemical modifications.13 The increasing number of antibiotics resistance adaptation cycles followed by semisynthetic optimization aimed at breaking this pathogen resistance development leaves many well-known antibiotics without much room for synthetic improvement. There have been significant efforts to tackle this challenge with de novo synthesized small molecules based on the molecular understanding of bacterial growth and metabolism in the pharmaceutical industry, but the antibiotic drug lead output did not warrant the investment costs.14 Therefore, the combination of scientific difficulties associated with antibiotic discovery and development, as well as a number of economic challenges related to antibiotics marketing, led to most big pharmaceutical companies leaving this research field.15
Natural product based antibiotics research has met additional challenges, as antibiotic drug leads, produced by well-known strains in high yields, as well as those that feature strong mass spectrometry (MS) or ultra-violet (UV) spectroscopy signals, commonly referred to as the ‘low hanging fruits’ of antibiotics research, have predominantly been discovered and characterized. Thus, current research has to refocus on isolation, cultivation and analysis of lesser-investigated bacterial and fungal species, as these have higher chances of producing novel natural products.16 For such bacterial strains, it is thought that the majority of the produced natural products are not described yet. Since the golden age of antibiotics, many new microbial strains as potential producers of novel secondary metabolites have been isolated, which greatly expands the taxonomical diversity available in the laboratory. Furthermore, most of the already investigated strains have only been used to isolate a single natural product class. However, according to in silico predictions almost all of them are capable of producing many more than just single natural products classes, leaving a huge chemical space still untapped.17 Furthermore, metagenomics analyses suggest that the majority of microorganisms have not been cultivated in laboratory environments yet.18 Additionally, there is a strong need to implement improved analytics systems or a wider variety of different detection techniques to explore the metabolites that are difficult to detect.19 That way, metabolites displaying irregular polarity or ionization properties can be accessed as well as metabolites produced in low amounts. The ability of the natural products discovery field to adapt to this new situation and extract value out of what is commonly referred to as the ‘hidden metabolome’ of bacteria will ultimately determine the future role of bacterial natural product research, not only in antibiotics discovery.
In this review, we will discuss recent developments of the fields of instrumental analytics, microbial in silico genomics and molecular biology approaches, which are used in various combinations to improve the outcome of natural product research (see Fig. 2). Furthermore, we shed light on the evolution of natural products discovery workflows and uncover promising current modifications. We classify emerging approaches in natural products discovery regarding current challenges and their potential to advance discoveries of novel antimicrobials from microorganisms when applied efficiently. In particular, this review focuses on the evaluation of techniques that successfully entered the natural products laboratory and contributed to the discovery of novel secondary metabolites, thus proving their wider applicability in natural products research.
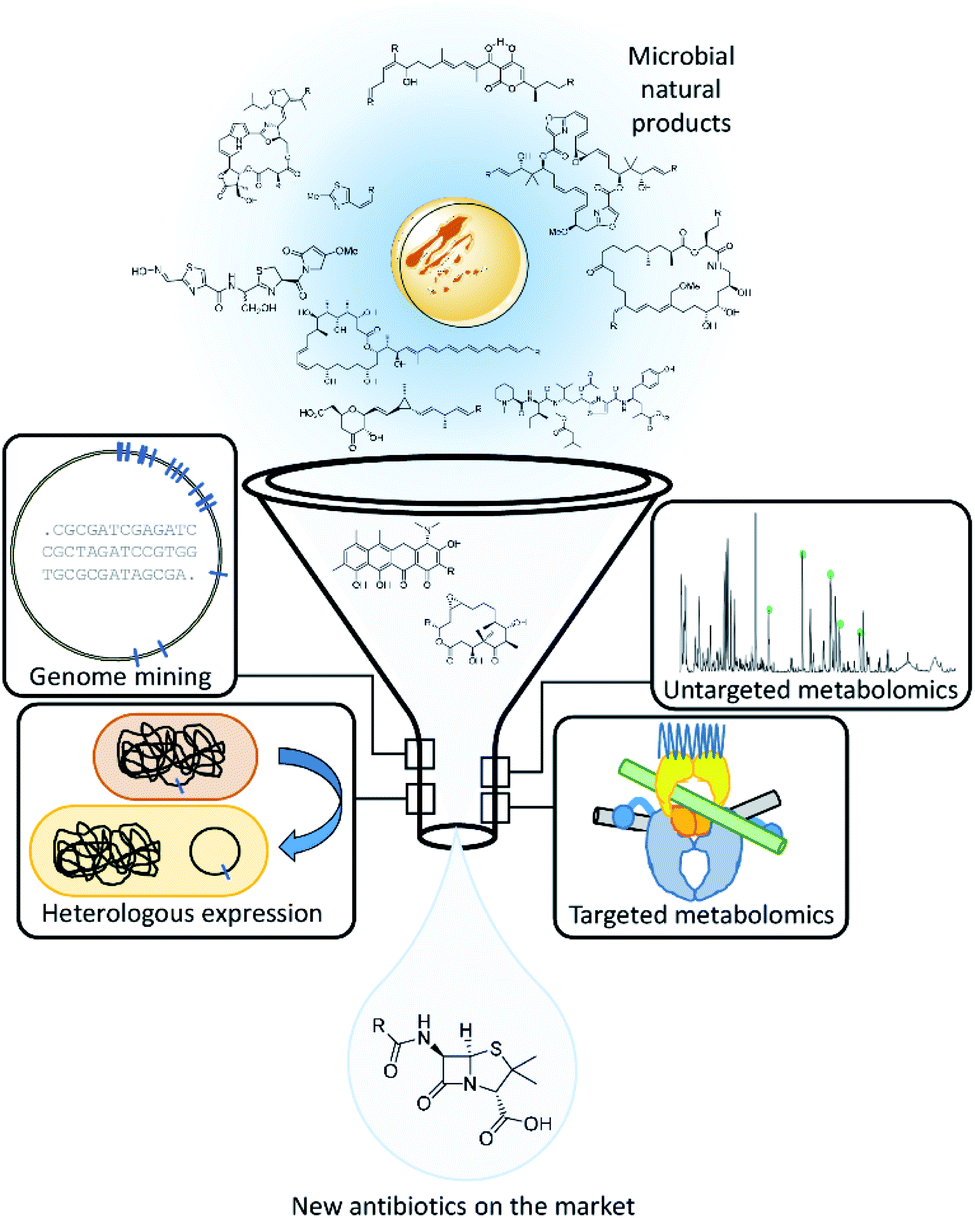 | ||
| Fig. 2 Schematic overview over the antibiotics discovery pipeline based on natural products featuring the approaches we shed light on in this review. | ||
The analytical challenge: untargeted bacterial secondary metabolomics, efficient data mining and prioritization
Natural products scientists have always dealt with extracts from natural sources as a means to find novel biologically active molecules. These extracts are complex mixtures of primary and secondary metabolites of the extracted organism, whose components stretch all across the polarity spectrum and comprise a significant range in molecular mass.20 In microbial natural product research, these extracts additionally contain complex matrices composed of co-extracted components stemming from cultivation media or additives that may lead to UV and MS signals with orders of magnitude more intense than the analytes of interest. The analysis of these extracts has always been inherently challenging and no single analysis method will be able to separate and annotate its entire content.21The following two sections highlight challenges and novel approaches in targeted and untargeted metabolomics. The respective methods and instrumentations were developed to overcome these obstacles in non-targeted metabolomics workflows focusing on expanding the range of detectable metabolites. Compounds that are detected and isolated in non-targeted metabolomics or via in silico genomics and molecular biology-based campaigns for antibiotics discovery are often tested for in vitro bioactivity against a variety of pathogens or pathogen-like organisms and evaluated for their primary cytotoxicity in parallel.9 Unfortunately, after confirming bioactivity in an untargeted metabolomics workflow, it is often difficult to elucidate the mode of action and molecular target of these newly found antibiotics. In targeted metabolomics workflows, which aim to identify antibiotics with a specific target, the methods mainly aim at improving the detectability of the compound to protein interactions for example, as well as the incorporation of a more diverse set of analytical methods.19 Target-based metabolomics workflows or those relying on a functional assay, limit the detectable natural products to specific binders showing activity in said assay and are therefore a step ahead in the process of developing a natural product-based antibiotic. However, as already mentioned above, these techniques are limited to known molecular targets, wherefore the application of non-targeted metabolomics workflows are still of high importance.
Non-targeted microbial metabolomics to expand the detectable secondary metabolite space
Early separation and detection methods in natural product research, such as thin layer chromatography (TLC) with staining agents, fail in analysing complexity due to limited resolving power and detection restrictions regarding specific functional groups. The introduction of liquid chromatography in combination with UV detectors, a technique available in almost every natural products laboratory nowadays, has drastically improved these two issues.22 However, UV detection heavily biased the analysis towards molecules with chromophores, which is reflected in the types of natural products that were discovered using this technology.23The introduction of robust and relatively cheap mass spectrometry devices (MS) and significant advances in nuclear magnetic resonance spectroscopy (NMR) have been game changers for analysis of natural products containing extracts.19 The chemical space detectable by (high-resolution) MS measurements combined with NMR data represented a quantum leap compared to staining reagents and UV data and provided much more useful information about the elemental composition and chemical structure of the analytes.24
The following sections will therefore focus on latest developments in MS and NMR-based metabolomics investigation for the discovery of novel antimicrobial natural products, as well as on challenges in data treatment, arising from the immense amount of data produced in non-targeted metabolomics workflows.
Analytical setups for extract analysis
Current MS-based secondary metabolomics analysis mainly focuses on liquid chromatography electrospray ionization (LC-ESI) MS-based techniques.22 While these techniques offer a very broad detection coverage of natural products scaffolds combined with a relative ease of use and cost efficiency, there are a variety of natural products classes that are difficult-to-impossible to detect due to polarity or ionization constraints.17 Detectability problems may arise in each step of this process, wherefore we present alternatives for LC, ESI and MS in the following sections. During the LC process, extremely polar and water-soluble substances such as the nucleoside antibiotic pseudouridimycin portray almost no retention in reversed phase high performance liquid chromatography (RP-HPLC), whereas strongly nonpolar substances such as terpenes show too much affinity to an RP-HPLC column and may not be eluted from the column in a standardized gradient.25 Furthermore, common eluents used for LC include a small amount of organic acids to increase separation and subsequent ionization with ESI, which may degrade acid-sensitive natural products. Natural products not detectable due to incompatibility with the LC system may, however, be detectable when directly infusing the bacterial crude extracts, without prior separation by LC, which becomes feasible by increasing the resolving power of the mass spectrometer.21ESI, as a soft ionization technique, provides intact masses even for sensitive molecules, but fails to ionize poorly oxygenated natural products, such as terpene scaffolds or steroids.25,26 These natural products however, may be detectable when changing the ionization technique to matrix assisted laser desorption ionization (MALDI) or atmospheric pressure chemical ionization (APCI).27 Mass spectrometry in general only provides very limited information about the structural composition of a molecule. This issue can be tackled when introducing NMR to metabolomics workflows, either as an alternative to or as hyphenation with MS.19 In order to broaden the natural products space and to facilitate the detection of novel antibiotics from the bacterial origin, we have to diversify analytical techniques for both natural product separation and detection (see Fig. 3).
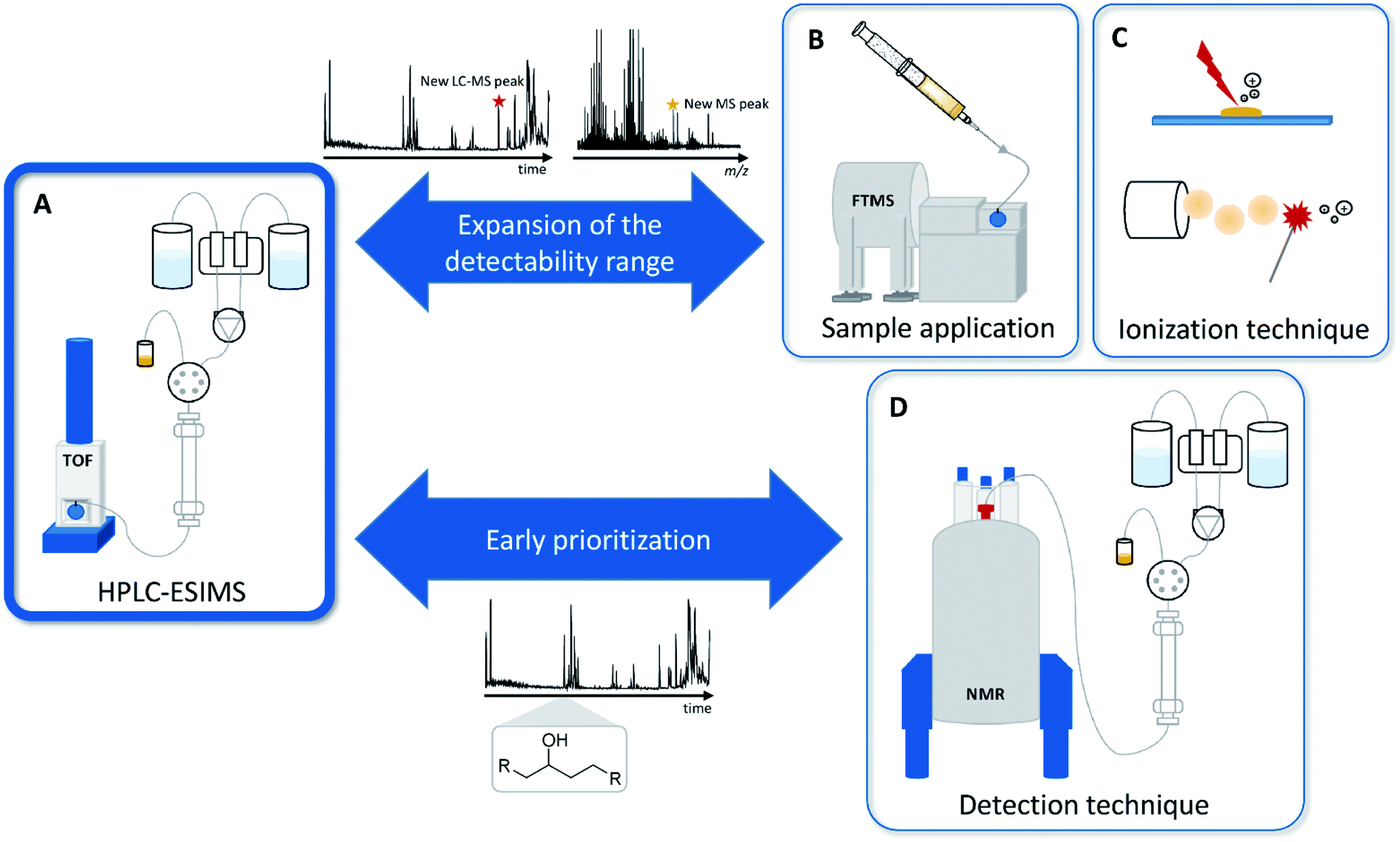 | ||
| Fig. 3 (A) The commonly used RP-HPLC-MS setup mainly used for natural products analysis. (B) Variation of the sample application method to direct infusion. (C) Alternative ionization techniques such as MALDI and APCI. (D) Alternative detection and early prioritization techniques such as LC-NMR instead of LC-MS. | ||
DI-Fourier transform mass spectrometry in secondary metabolomics
One of the mass spectrometry techniques emerging in recent years, which is increasingly integrating into the mainstream of analytics procedures for bacterial secondary metabolite analysis, is direct infusion (DI) mass spectrometry on ultrahigh resolution (UHR) mass spectrometers such as Fourier transform ion cyclotron resonance (FTICR) and Orbitrap mass spectrometers.28 This technique can be an excellent addition to LC-MS based bacterial secondary metabolite discovery, as it removes the polarity constraints imposed by LC-columns.21Plant derived secondary metabolomics have successfully applied DI-FTICR as an add-on to LC-MS based secondary metabolomics approaches as exemplified by Park et al.29 However, for bacterial secondary metabolomics DI Fourier transform-based MS is only rarely used, even though it was shown to extend the chemical space detectable in natural product extracts.21 While there are studies that successfully employed FTICR data to unravel interactions between organisms of different kingdoms of life, such as the interaction of Rhizoctonia strains with Solanum tuberosum or the interaction between Pseudomonas aeruginosa and Caenorhabditis elegans, the technique remains underused as a screening tool to overcome the LC-ESI-MS detectability bias.30
While FTICR has proven to be slightly advantageous when it comes to determination of molecular formulae from complex mixtures in a direct comparison with Orbitrap MS, its high costs and the extensive expertise required for operation strongly limit its application.31 Orbitrap MS, which became available around 2004 as an affordable alternative, may therefore increasingly be used for DI Fourier transform-based natural product discovery in the future. Both FTICR and Orbitrap technologies often unambiguously provide the molecular composition of an ionized molecule based on mass accuracy and the isotopic fine structure. Additionally, they may be used in MSn experiments to provide further insights into the structural composition. They both however lack any additional structural information as for example polarity and the surface as assessed by LC and IMS.
Ion separation according to collision cross-section – ion mobility spectrometry in natural product research
The development of ion mobility spectrometry (IMS) coupled MS systems that became commercially available and thus increasingly available to natural products laboratories already started in 1998.32 The big advantage in coupling IMS to MS relies on the separation principle behind ion mobility. Ion mobility cells separate ions in an electric field set in opposing direction to a stream of collision gas molecules, mainly nitrogen gas. In this arrangement, ions are separated according to a collision cross section (CCS) per charge parameter, which is semi-orthogonal to m/z values and thus offers an additional ion separation dimension. For that reason, IMS applications have been developed by several suppliers of MS systems including the travelling wave systems commercialized by Waters, drift tube systems commercialized by Agilent and trapped ion systems commercialized by Bruker. These IMS systems are linked to high-resolution TOF systems and achieve high resolving power due to combined acquisition of CCS and m/z values. These latest developments shifted IMS-MS applications from a niche application for specialized applications to a technique that becomes more and more mainstream in natural products laboratories.33 The key strength of IMS-MS lies in its ability to discern structural isomers that often occur in natural products mixtures, as most structure isomers have different CCS values due to their different geometry. A prominent example for such a case is the detection of streptorubin B from S. coelicolor.34 The utility of IMS-MS as a screening tool was further demonstrated by the principal component analysis based comparison of Nocardiopsis sp. mutants that revealed the effect of antibiotic resistance on secondary metabolite production.35Secondary metabolite identification by alternative ionization types – shifting away from ESI
As mentioned in the first section on metabolomics, bacterial secondary metabolomics suffers from a detection bias introduced by the almost exclusive use of ESI as the only ionization technique employed in many natural products laboratories.36 When trying to expand the molecular space detectable in mass spectrometry experiments, it is of vital importance to broaden the ionization methods used, as every ionization method, such as ESI, MALDI, atmospheric pressure photoionization (APPI) or APCI, only ionizes a section of the chemical space.27 APCI ionization as a rather harsh ionization method is predominantly used to ionize more non-polar metabolites e.g. terpenes or hydrophobic halogenated small molecules.37 APPI on the other hand, shows significant overlap with ESI ionization but seems to provide better signal to noise ratios that might enable the discovery of additional secondary metabolites.38MALDI ionization for example, often employed in imaging workflows, has shown its potential to expand the detectable secondary metabolome both in terms of visualizing ions with an alternative ionization source but also in terms of observing microorganisms growing on a solid support.39 This ionization technique is extremely useful for tapping into the bacterial secondary metabolome of bacteria, which are hard to ferment in liquid media. MALDI imaging was employed in metabolomics experiments in Frankia sp., a group of actinobacteria difficult to cultivate in liquid media, making fermentation for isolation of any novel natural product a particularly challenging task.40
Mass spectrometry imaging in natural product research
But other than just analysing bacteria only growing on solid surfaces, mass spectrometry imaging (MSI) secondary metabolite identification also allows spatial resolution of mass spectrometry data on a 2D surface.27,41,42 Spatial resolution of mass spectrometry observations can be used to observe a variety of different phenomena linked to the identification of novel bioactive natural products. It allows, for example, pinpointing secondary metabolites linked to bacterial life cycle stages such as sporulation as it was shown for the myxobacterium Myxococcus xanthus.43 Co-cultivation experiments followed up by MALDI-MSI with termite gut-associated Streptomyces strains and a Pleosporales fungus showed spatially resolved appearance of a novel lanthipeptide that only appears in the co-cultivation contact zone.44 These secondary metabolites appearing in the contact zone of a co-cultivation experiment are of particular interest as these molecules are often secreted as defence secondary metabolites that are predestined to become initial lead structures in antibiotics development. Another example for employing MALDI-MSI as a means to discover and isolate novel antibiotics is the discovery of arylomycin from Streptomyces roseosporus.45 In this case, the MS signals stemming from arylomycin were shown to be linked to the growth inhibition zone that made the arylomycin antibiotics visible by MALDI-MSI. A similar study involving MALDI-MSI of the growth inhibition zone between two fungal species led to the discovery of an antifungal peptide called leucinostatin Z.46However, one of the main drawbacks of MALDI usage for the discovery of new antibiotics is that abundant ions heavily suppress the ionization of analytes produced in low amounts or are less prone to ionization.41,43 Furthermore, co-cultivation experiments as the ones described in this paragraph are challenging to upscale for production and isolation of the respective antibiotics, wherefore co-cultivation-based MALDI-MSI experiments often remain limited to an analytical scale. To overcome ionizability limitations arising from ionization via MALDI, a technique to combine the advantages of ESI and MSI was developed. This technique, which is called desorptive electrospray ionization (DESI) uses an ESI spray to perform desorptive ionization from surfaces, which enables an ESI based MSI.47 As described by Parrot et al. DESI-MSI is more readily able to ionize compounds of higher polarity than MALDI-MSI that more predominantly ionizes lipid type metabolites and is therefore more tailored towards the detection of most secondary metabolite classes.47 Still, while the potential of DESI-MSI has been shown in the MSI based analysis of clinical samples especially when combined with metabolite annotation platforms such as METASPACE, so far the technique seems to be widely underused in the natural products discovery process.48 A study that involves future utility of DESI-MSI for natural product research shows antibiotics production during predation by Myxococcus xanthus bacteria.49 Although the study pinpoints a number of molecular features belonging to natural products associated with bacterial predation, isolation of those compounds remains a very challenging task due to the limited scalability of 2D cultivations on surfaces.
Nuclear magnetic resonance spectroscopy applications in bacterial secondary metabolomics
NMR spectra provide more detailed structural information about a compound than high-resolution mass spectra, which typically only provide the exact mass and the molecular formula of a compound, along with the molecule's MS fragment masses. NMR spectroscopy is widely used in structure elucidation of natural products, but a central challenge to its application as a metabolomics tool is its limited applicability on compound mixtures.50 In addition to that, due to inherent sensitivity limitations of NMR spectroscopy, the technique is only able to detect the major components of a complex mixture in stark contrast to DI-FTICR approaches for example. NMR spectroscopy, however, can already pinpoint key structural elements at an early stage in the secondary metabolite under investigation.51 A recently developed method relies on identifying interesting compounds by their individual and characteristic unique NMR signals, instead of focusing on the entire NMR spectrum of a complex metabolite mixture.52 This technique is one of the increasingly needed approaches to apply NMR-based secondary metabolomics at an early stage of the natural products detection process, in order to obtain preliminary structural information.A technique that combines advantages of HPLC based separation of a complex biological extract with the structural information provided by NMR spectroscopy is the solid phase extraction (SPE) based method LC-SPE-NMR.53 This technique allows for harnessing the structural information of a chromatographic peak in an extract that was collected on an SPE cartridge by subsequent NMR analysis, which provides fast insight into structural features of a peak in a complex natural product mixture.54 SPE enrichment of an eluting LC peak is necessary to obtain concentrations sufficient for NMR analysis and exchanging the LC solvent to a deuterated solvent. Still, as for the aforementioned techniques, NMR-based metabolomics techniques play a niche role in supplementing the widely used RP-HPLC-MS platforms for natural product detection, even though it is more and more used in primary metabolomics studies.
Apart from NMR applications in metabolomics, multi-dimensional NMR spectroscopy remains the gold standard for downstream natural products structure elucidation based on the purified compound.55 Novel NMR techniques that aim at increasing sensitivity and resolution in multidimensional NMR to increase spectra quality as well as downsizing the necessary sample amount are increasingly integrated in natural products science.56 Combined with stronger NMR magnets, these techniques help push down the minimal amounts necessary for the acquisition of interpretable NMR spectra. This represents a much-needed improvement in natural products sciences, as interesting natural products can oftentimes only be isolated in very small amounts.
Sorting, classifying and prioritizing metabolomics data using automated data treatment and chemometrics
The sheer complexity and data size of mass spectrometry data created in high-throughput MS or NMR-based state-of-the-art natural products detection workflows clearly exceed manual data analysis capabilities. For unbiased analysis of the MS data of biological samples, such as natural products extracts, some degree of automated background or matrix filtering combined with MS signal prioritization has to be performed to mine natural products mass spectrometry data more efficiently.57 This analysis that aims at reducing the complexity of unbiased analysis of natural products samples by application of mathematical and statistical methods is called chemometrics.58In mass spectrometry, complex biological matrices in secondary metabolomics often hamper efficient data mining due to an abundance in culture medium or primary metabolism derived MS signals. Statistics tools, separating blank culture associated molecular features and subtract these features before selective application of tandem mass spectrometry, help resolve these issues.59 Many of these approaches rely on principal component analysis (PCA) based on LC-MS features. PCA can be used to objectively discern LC-MS features associated with microbial growth from matrix-derived LC-MS features.59–61 This can be used to ensure optimal acquisition of MS2 spectra by directly targeting only relevant precursor ions for the MS2 experiments and thus reserving the duty cycle for acquiring a maximum of meaningful tandem MS spectra.61
Novel MS2 analysis tools provide structural insights based on comparison of fragmentation trees and allow early prediction of interesting structural elements in natural products.62 Combined with either MS2 spectral networking tools such as GNPS or molecular fingerprinting tools such as NC-MFP, MAP4 or Sirius 4.0, this technology can speed up the analysis and detection process in microbial secondary metabolomics by several orders of magnitude.62–64 In this case, the user obtains a filtered, clustered and unbiased overview of all bacterial secondary metabolites present in a microbial extract. One big advantage of repositories such as GNPS and NPAtlas is that they are community curated.63,65 Keeping these repositories up-to-date benefits the whole community and on the other side – maybe more importantly – they will most likely not become limited due to commercialization and connected access issues. Hiding crucial reference data behind paywalls is counterproductive for the natural products community in general. Open access natural product databases prevent rediscoveries and thus increase the efficiency of natural product research altogether.66 Efficient prioritization of mass spectrometry data from a microbial origin needs both automated and unbiased analysis of mass spectrometry data and comprehensive natural product repositories that are easy to access.
Integration of taxonomic information into metabolomics experiments
It is widely known among natural product research laboratories, that the further apart two species are taxonomically, the more likely it is that their secondary metabolite profile is distinct and provides the best chances for the isolation of structurally novel natural products. Consequently, there has been a focus in natural product research on the discovery of novel and phylogenetically distant species for antibiotics discovery as described in the first section. Until very recently, this intuitive assumption has not been evaluated quantitatively. Hoffmann et al. showed an efficient method to utilize statistics-based filtering of large amounts of mass spectrometry data across taxonomic boundaries and pinpoint secondary metabolites correlating with a specific bacterial strain or genus.16 Dissecting increasingly complex secondary metabolomics data according to taxonomy will increase natural products science's focus on specialized secondary metabolites evolved to give a specific genus a competitive edge in its ecological niche. Analysing this specialized secondary metabolome is likely to uncover secondary metabolites exhibiting promising biological activities.Both the use of the novel analysis techniques discussed in this section and the analysis of the secondary metabolome of microbes that are genetically distant from well-known laboratory type strains provide higher chances for the discovery of bioactive natural products based on novel core structures. These approaches can therefore help to increase the output of the microbial natural product discovery field.
Targeted metabolomics approaches based on known molecular targets as prioritization tools
In contrast to the aforementioned non-targeted metabolomics workflows, which are not limited to any specific molecular target, approaches focusing on specific binders for molecular targets offer the possibility of obtaining a biologically active secondary metabolite with the information about its target and mode of action early in the process.67 Molecular target in this case means a binding site or binding pocket on a macromolecular entity, often a protein, protein complex or nucleic acid macromolecule. This molecular target can be addressed by a small molecule binder to affect the corresponding macromolecular entity.68 Once the assay is set up, it is a very elegant way of discovering novel antibiotic lead structures. This approach, however, is limited to known, well-described molecular target structures such as topoisomerase proteins like GyrA, parts of the transcription machinery like RnaP, proteins involved in DNA replication like DnaN, the protein biosynthesis or targets involved in cell wall biosynthesis like lipid II.69As these targets are already well established, this approach runs the risk for rediscoveries. Furthermore, strongly active natural products produced by a variety of producer organisms reoccur in these screenings. As a number of the identified small molecule binders show overlapping binding sites, problems with cross-resistance may limit the effectiveness of the approach.70 Molecular target-based approaches furthermore require the purified target structure to be physically available. Because protein expressions are often straightforward, most of these approaches tend to involve protein targets in these experiments.71 The protein is subsequently incubated with bacterial extracts or purified compounds from libraries and potential binders are analysed and subsequently confirmed by diverse analytical techniques. To the best of our knowledge, native protein MS is the only detection method used for high-throughput screening of extracts, whereas methods such as co-crystallization X-ray crystallography, surface plasmon resonance (SPR) or microscale thermophoresis (MST) are mainly used for screening of pre-purified extract fractions or libraries of purified natural products.72–74 The following sections summarize how these technologies are implemented in screening workflows for novel antimicrobials.
Protein-based inhibitor fishing in crude extracts using native protein MS
Significant advances in mass accuracy, mainly fuelled by advances in high-resolution mass spectrometry instrumentation, allow the acquisition of extremely accurate mass spectra of proteins in their natively folded form by direct infusion ESI-MS.75 In contrast to many MALDI- or LC-based techniques that can only analyse digested or denatured protein, protein DI-MS techniques transfer the protein with its intact tertiary structure into the gas phase and charge it for MS analysis. The ability to determine the exact mass of multi-charged protein complexes in their native form led to development of high throughput binding assays using these protein complexes. Thereby one can identify the mass of a protein with and without incubation with a non-covalently bound inhibitor.72Besides determining whether the inhibitor is binding or not, this approach also allows (with the help of deconvolution software) determining the inhibitor's exact mass from the mass deviation of the protein complexes.73 Native protein MS therefore is well suited for screening complex mixtures such as crude extracts, as it does not only show whether protein binding occurs, but also indicates which component of the mixture did bind to the protein of interest.73 While Yang et al. successfully applied this technique for the identification of an anti-malarial agent out of natural products extracts, access to and application of this technique have been so far limited.76 Despite its tremendous potential in molecular target-directed natural product discovery, native protein MS therefore remains a niche research field. Increased access to hrMS platforms may encourage further spread of this technique in the field of natural product research. A more specialized case in the protein based inhibitor fishing is the detection and analysis of covalently bound protein inhibitors. While these proteins have to be in their active native folding state during binding of the corresponding inhibitor, they will not lose the inhibitor upon denaturing, as it would be the case for non-covalently bound protein inhibitors. Inhibitor screens can therefore be performed by LC-MS under denaturing conditions. An example for the usage of LC-MS based identification of a covalently bound inhibitor to an – in this case – digested protein is the evaluation of thioredoxin inhibition by suicide inhibitors.77 Intact protein LC-MS analysis as a means to identify covalent inhibitors is exemplified by the work on blocking the biosynthesis of the microbial pathogenicity factor Tillivalin by molecules binding covalently to the acyl carrier protein involved in Tillivalin biosynthesis.78
Protein-based natural product library screens using X-ray crystallography, cryogenic electron microscopy, SPR or MST
The screening of pre-purified natural product libraries majorly benefits from reduced complexity of the sample, even though for high-throughput workflows, samples are commonly pooled in the first evaluation rounds. The detection methods applied here derive from molecular target-based medicinal chemistry approaches.79A valuable tool developed in recent years is the structure determination of enzymes using cryogenic electron microscopy (cryo-EM). Recent improvements in resolution towards low, single-digit angstrom values on one hand allows this technique to be harnessed in structure elucidation of enzyme–inhibitor complexes.80 On the other hand, if the target to analyse is crystallizing well, target-directed secondary metabolite discovery by X-ray crystallography is becoming more and more popular.81 Still, the majority of natural product research laboratories neither have direct access to cryo-EM platforms nor to X-ray crystal structure analysis platforms, which makes better implementation of these techniques in natural product research a challenge to be solved in inter-laboratorial cooperation.
Two benchtop devices that are more commonly available are SPR and MST. These techniques are used to not only determine binding, but also binding affinities of pure natural products to their targets or to validate hits from virtual screening campaigns, but also have proven useful screening tools for natural products libraries.82 Non-specific binding may complicate screening of bacterial crude extracts by MST or SPR as these techniques are unable to discriminate specific from non-specific binding. Therefore, a combination of screening technologies may still prove their usefulness for screening of extracts.
In summary, new instrumentation and analysis techniques are available that have already shown to be useable for antibiotic natural product discovery and prioritization for isolation based on a specific protein target. Major improvements have been achieved in the field of mass spectrometry, which influenced many of the non-targeted and targeted metabolomics workflows. Subsequent data treatment is more easily performed with increasing availability of computing power, as well as processing tools streamlining high-throughput screenings. However, all metabolomics-based workflows can obviously only detect natural products that are produced by the microorganisms studied under laboratory conditions. With more and more microbial genome sequences available, it became clear that the genome inscribed biosynthetic potential of bacteria and fungi far exceeds what is detectable even when expanding the analytical methods applied.83 The following section evaluates information obtained from microbial genome sequences and how it can be used to harness and valorise a larger part of the genome-encoded microbial natural product potential.
The biosynthetic genomics challenge: prediction and prioritization of secondary metabolite biosynthetic gene clusters
Natural product research has undergone a revolution, triggered by the dramatic drop in the genome sequencing cost as well as the increased reliability of modern genome sequencing techniques.84 This allowed the evolution of natural product science from a research field mainly extracting microbial cultures and performing bio testing in so-called ‘grind-and-find’ approaches towards a research field guided by predicting the theoretical secondary metabolite production potential based on genomic information.85 For the first time, researchers were able to increase the chances for identifying novel secondary metabolite structures by specifically looking at microbial species that exhibit a large number of secondary metabolite biosynthetic pathways encoded in their genomes. Even the established, most proficient producers turned out to harbour a much larger biosynthetic potential per genome than anticipated by the number of compounds isolated from these strains.17Shortly after the genome sequencing revolution, a variety of bioinformatics tools were developed that allow fast and efficient prediction and annotation of biosynthetic genes. This particularly holds true for bacteria, where secondary metabolite biosynthesis pathways are mostly clustered as so-called biosynthetic gene clusters (BGCs). Tools, such as antiSMASH or PRISM, not only allow prediction of a BGC's location, but nowadays also provide first insights into the putative structure of the encoded secondary metabolite.86 BGC annotation principles have been transferred to other organisms such as fungi and even plants, although BGC prediction in these organisms still lags behind the predictive quality reached in bacteria.87 In addition to the genomic analysis of cultured species, the detectability of BGCs in complex environmental metagenome DNA samples is of special interest for bacteria that so far are not cultivatable in laboratories, an aspect that will be discussed in more detail below.
The advances in this field led to a dramatic uptick in the number of detected BGCs across all bacterial genera coupled with information about microbial genera that contain gene cluster-rich genomes.88,89 These so-called BGC-rich genomes cluster among several ‘highly productive’ microbial phyla all across the tree of life.88 However, a significant proportion of BGCs are recurring among bacteria, especially among closely related strains, which makes efficient BGC dereplication a necessity. Community-curated open access repositories such as MiBiG are extremely helpful tools to prevent natural product rediscoveries stemming from the distribution of the same biosynthetic gene cluster across several microbes.90 Even though there is a clear trend that secondary metabolite BGC distribution follows taxonomy, there can be exceptions to this rule. Horizontal gene transfer events for entire BGCs may lead to rediscoveries of natural products from phylogenetically distant bacteria. BGC databases like the aforementioned MiBiG can prevent repetition of work that has already been performed and thus streamline natural product research.91
In the following, we highlight principles for prioritization of one BGC for subsequent follow up studies to decipher the encoded chemistry. For most bacteria, the number of detected ‘orphan’ BGCs exceeds the number of BGCs correlated with a known natural product.17 An orphan gene cluster in this context means that the genetic data combined with BGC annotation tools clearly show the presence of this BGC in the host strains genome, while no corresponding secondary metabolite can be associated with the applied instrumental analytics and under the cultivation conditions tested.92 Even for bacterial reference stains such as Streptomyces coelicolor A3 (2) or Streptomyces albus J1074, bacterial strains that have been known for a long time and extensively analysed, the BGC content exceeds the number of known secondary metabolites by far.93 Outside of the realm of actinobacteria, the discrepancy between genome inscribed secondary metabolite potential and secondary metabolites actually detected remains equally high, as exemplified by the myxobacterial model strain Myxococcus xanthus DK1622.94 A study on Streptomyces griseus describes in detail how such biosynthetic potential based mining approaches can lead to the discovery of novel natural products by selection of ‘high biosynthetic potential strains’ that are subsequently investigated in depth.95 These examples show that even well-known and extensively used bacterial strains still have enormous biosynthetic potential that has not been harnessed yet. How genomics data can be linked to MS data at an early stage to correlate these orphan gene clusters to their corresponding products which leads to integration of genomics and metabolomics data at an early point in the analysis will be discussed in the following sections in detail.
Prioritization of BGCs and their activation
The emergence of readily available and cheap genome sequencing technology, and reliable gene cluster prediction, as well as the availability of molecular biology technologies for the capture and transfer of BGCs for gene cluster heterologous expression untapped an enormous potential for the activation of BGCs.96,97 However, not all of these BGCs can be analysed in the laborious and time-consuming process required for in-depth analysis of one BGC and its corresponding natural product family. This holds especially true, if this work involves genetic engineering to activate the corresponding BGC or increase its production, which makes concise gene cluster prioritization very important.98A rather obvious approach towards BGC prioritization for in-depth investigation is based on gene cluster complexity. Many BGCs, especially modular ones belonging to non-ribosomal peptide synthetase (NRPS) or polyketide synthase (PKS) families, indicate the complexity of the produced natural product in the sequence size and domain complexity of the corresponding BGC.99 Secondary metabolism is an energy intensive process and large secondary metabolite BGCs consume a disproportionally high amount of cellular resources. Investigating these large BGCs is therefore assumed to hold high chances to reveal encoded biologically active substances, as this energy investment has to be counter balanced by a survival benefit for the producing organism (Fig. 4).100
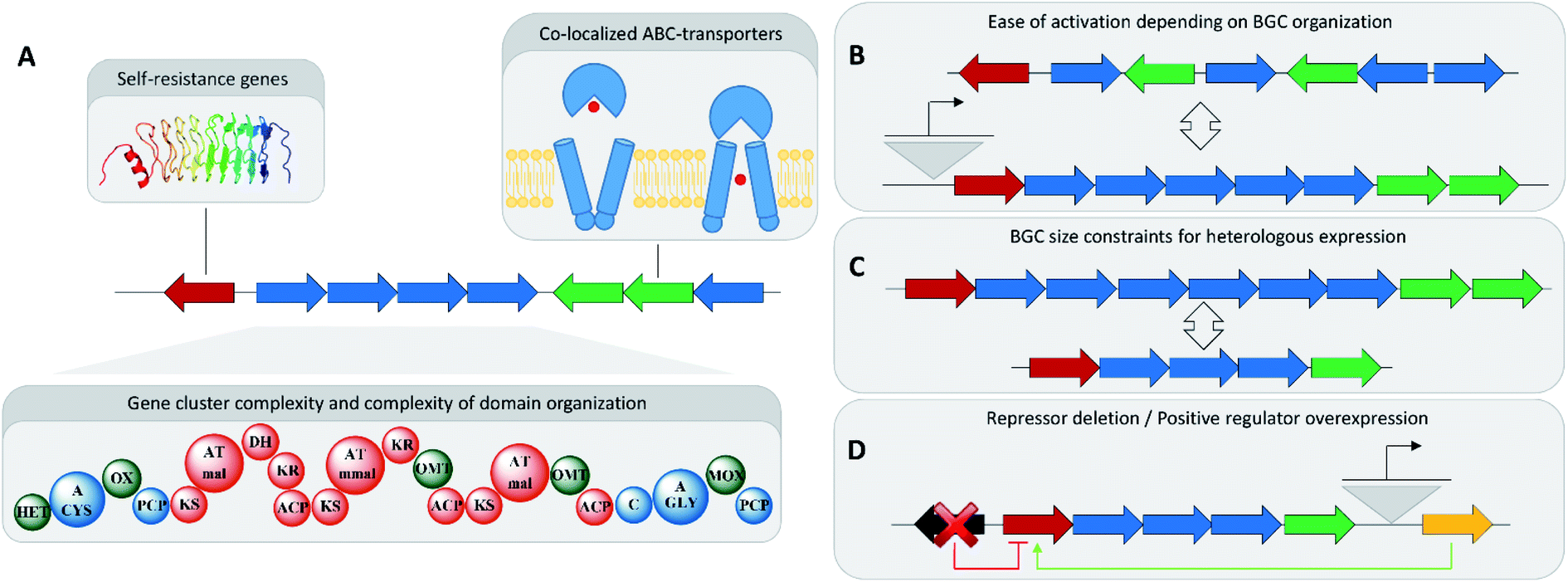 | ||
| Fig. 4 (A) Gene cluster prioritization techniques. (B) Comparison of well-organized BGCs to BGCs with a complex operon and regulatory structure regarding ease of activation. (C) BGC size limitations in BGC overexpression attempts. (D) Ability to engineer the regulatory environment of a BGC. | ||
In antibiotics research based on bacterial secondary metabolomics, mining of self-resistance markers in close proximity to said BGCs has shown great success in uncovering bioactive natural products. Self-resistance is important for antibiotics producing microorganisms, as these microorganisms have to develop means to survive their own antibiotic production.101 Thus, co-localization of an ‘orphan’ BGC with a potential resistance gene indicates the formation of a novel natural product with antibiotic activities.101 Identification, isolation and structure elucidation of pyxidicyclines from myxobacteria, and taromycin from terrestrial, as well as thiotetronic acid from marine actinomycetes are prime examples of how potential self-resistance mechanisms can be harnessed to predict and identify the target of the small molecule product of an ‘orphan’ BGC.102,103
Automated annotation of self-resistance mechanisms, as it is available in the ARTS tool, can furnish additional information. If this is combined with antiSMASH-based gene cluster identification it allows efficient prioritization of potential antibiotic BGCs.104 Another key indicator for spotting biologically active BGCs would be co-localization of so-called ABC-transporters responsible for efficient export of the natural product out of the bacterial cell. This correlation indicates that the corresponding natural product binds to targets in other organisms or strong auto-toxicity in the producing organism, both of which are characteristics of natural product antibiotics.
Activation of BGCs in wild type strains
A traditional straightforward strategy used to activate BGCs in wild type microorganisms is to challenge them with exogenous compounds such as (heavy) metal ions, hormones of plant or fungal origin and bacterial quorum sensing molecules.105 This approach, which is referred to as the one strain many compounds (OSMAC) approach, targets to activate the regulatory mechanisms of microbial biosynthetic gene clusters.106 For the most part, such an approach is necessary in standard settings as microorganisms have little-to-no incentive to defend themselves against competitors under the optimal growth conditions in axenic cultures fermented in the laboratory with no competitors or predators present.A more targeted approach directed at activating specific biosynthetic gene clusters can be genetic engineering based activation of ‘orphan’ BGCs in a wild type microorganism. This is commonly performed by exchange of the wild type promoter(s), controlling the BGC expression, against inducible promoters that will transcriptionally activate the BGC and remove it from its native regulatory network on the DNA level.107 This can be achieved either by homologous recombination in the producer strain as in the case of pyxidicyclines or by using the CRISPR/Cas9 system as performed in the case of the Streptomyces viridochromogenes pigment.102,108 Applicability of this method critically relies on the availability of genetic tools, including DNA transfer technologies for the corresponding strain, which can be very laborious to develop. It is possibly for this reason that self-resistance based prioritization and activation of BGCs have not been attempted in high-throughput and therefore not delivered an extensive amount of novel bioactive natural products.
An alternative strategy to remove a BGC from the regulatory network in the corresponding wild type organism is to delete BGC-specific repressors or overexpress BGC-specific transcriptional activators. These BGC-specific regulators are often co-localized with the BGC, especially in actinomycetes.109 Overexpression of positive regulators can lead to production of natural products as it was shown for Aspergillus species.110 In streptomycetes, knockout experiments of repressors close to BGCs have shown to be a valuable tool in the activation of ‘cryptic’ natural products.111 These principles can be expanded to other microorganisms to increase the accessible natural product space by activating ‘cryptic’ BGCs.
Cloning and heterologous expression of BGCs
For many wild type bacteria, genetic engineering tools do not exist, which hinders direct activation of an orphan BGC in said organisms. Therefore, there has been a drive to develop techniques to clone BGCs onto target vectors followed by heterologous expression in well-characterized model strains, which are ideally phylogenetically closely related to the organism of origin of the corresponding BGC. The only way to clone and engineer the biosynthetic machinery of this BGC, in order to activate the production of the corresponding natural product, is extracting the BGC sequence and refactoring it on a genetic element (mainly done in E. coli) followed by transfer of the refactored or activated BGC in a production strain of choice. As this heterologous expression process is time intensive, heterologous expression of interesting cryptic BGCs is preceded by screening the strains' metabolome and genome with large natural products and gene cluster databases such as NPAtlas and MiBiG.90,112 This process limits the number of heterologous expression efforts that result in rediscoveries of previously described natural products.Techniques that have been successfully used to transfer BGCs from the native host organism to a more easily handled model organism include Rec/ET-based direct cloning, transformation-associated recombination (TAR)-based direct cloning, assembly of polymerase chain reaction (PCR) products by TAR, Golden gate cloning or Gibson assembly.96,113,114 These techniques will become even more important with the increased availability of cheap synthetic DNA that is more and more used as an alternative source for assembly of putative antibiotics BGCs (Fig. 5). Recent examples of such heterologous expressions are cloning and heterologous expression of argyrins, kocurin, daptomycin, corallopyronins or pyxidicyclines.102,115 Taken together these techniques can help overcome accessibility limitations that arise from slow or insufficient growth of the wild type organism, a lack of genetic tools to access its cryptic BGCs and yield limitations in the wild type strain.116
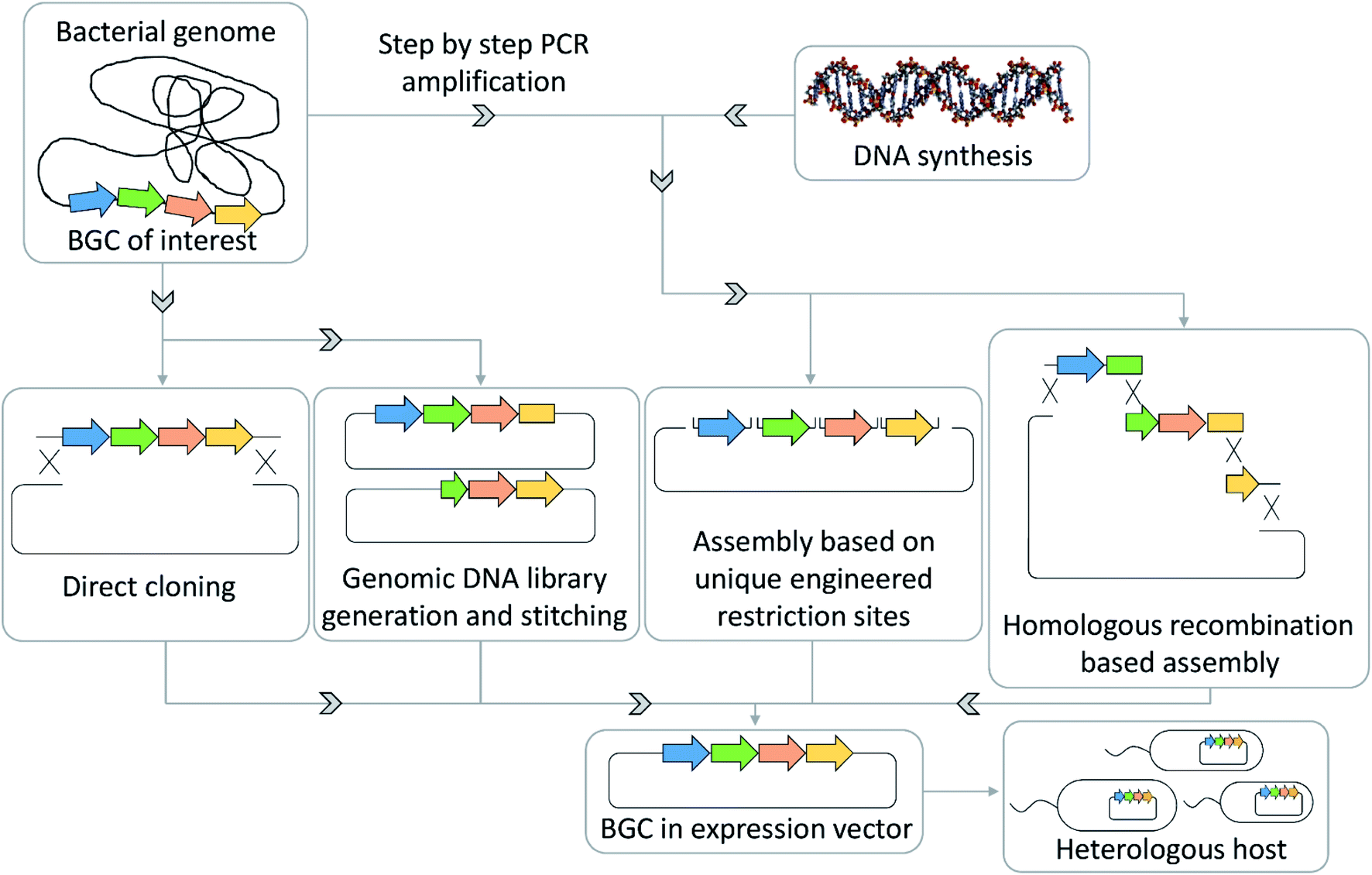 | ||
| Fig. 5 BGC assembly strategies for the heterologous expression of secondary metabolite pathways. | ||
All these strategies for activation of a BGC or its heterologous expression are driven by the attempt to produce novel antibiotics. If successfully applied, they all strongly rely on appropriate chemical analytics applied to the developed host organism, where wild type metabolome profiles are compared with those of the genetically engineered strain.19 Typically, initial production titres in heterologous expression are low and need to be improved by systematic studies in an iterative fashion. Thus, it is imperative to apply one of the analytical detection methods described in the first paragraph highlighting the importance of the interconnection between genomics and metabolomics data in natural product science.
Linking genomics to metabolomics data at the earliest stage
In many instances, secondary metabolomics and microbial genomics are still treated as processes that are mostly performed separately or consecutively. Bringing together these domains, e.g. by associative statistics to link BGC appearances to MS signals, can speed up natural product discoveries and the detection of the corresponding biosynthetic machinery.117 As described in the paragraph on taxonomic diversity, many of these novel cryptic BGCs of interest are found in newly isolated wild type strains that are taxonomically distant from described type strains for which genetic engineering tools do not exist yet.88 Genomics analysis has been supported by varying technologies making investigation of strains not suitable for metabolomics studies (e.g. as they are hard to culture under laboratory conditions) accessible. With their genome sequence available, their BGCs can still be investigated with bioinformatics tools and prioritized BGCs can be expressed in heterologous hosts, as soon as entire BGCs are assembled by one of the aforementioned BGC assembly techniques.118 Using these technologies, the amount of genomics data already exceeds the metabolome data available. Looking forward, the imbalance between genomics and metabolomics is likely to increase rather than decrease, as the overall speed of genome sequencing, generation of publicly available genome sequence information and BGC prediction currently exceeds the speed of natural product isolation and structure elucidation by orders of magnitude. Furthermore, there is currently no new purification and structure elucidation technique in sight to inverse this development.Mining metagenomes to access BGCs of yet uncultured bacteria
A large detriment to efficient mining of BGCs is that a significant number of newly observed microorganisms, especially those from uncommon ecological niches, are difficult to culture and thus uncultured under laboratory conditions.119 Thus, for many of these organisms, isolation of secondary metabolites from their wild type cultures remains out of reach unless an efficient approach of culturing these organisms is found. This holds especially true for symbionts, microorganisms that are often thought to be not viable in axenic culture.120 Metagenomics analyses indicate that large parts of microbial communities do not even have a single congener cultivated in the laboratory so far.18 Therefore, BGCs from underexploited or even uncultured microbial genera have a larger probability to be responsible for production of a structurally distinct natural product.To access the biosynthetic potential of these organisms, metagenome BGC prediction is of great importance. As a note, one limitation might be that not all BGCs turn out to be co-localized in one genomic region. Such non-clustered BGCs of unknown function provide additional challenges for which currently no methodology of prediction and association is available. Mining tools such as eSNaPD, that streamline BGC detection and prediction from metagenomes, can help accessing the secondary metabolite space hidden in metagenomes.121 In addition to that, BGC cloning and heterologous expression techniques, such as recombineering (homologous recombination based genetic engineering) tools described earlier, furnish tools to capture these gene clusters and express them in taxonomically closely related laboratory strains to harness the encoded secondary metabolites (Fig. 5).113,122
Conclusions
In summary, current developments in secondary metabolite identification processes boosted by innovations in novel instrumental analytics equipment (especially in the realm of mass spectrometry) are pushing detection limits and unlock a large, previously unseen part of the bacterial secondary metabolome. Software solutions are increasingly used in natural product laboratories to sift through metabolomics data helping with automated detection of interesting mass spectrometry signals based on objective criteria.123 In addition to that, better access to larger computing power resources put the unbiased processing of large-scale metabolomics data into the mainstream of natural product research. Improvements in BGC prediction as well as the staggering increase in publicly available bacterial genome data led to an enormous boom in BGCs available for analysis while computing power currently available allows implementing, sorting and prioritization algorithms to quickly point at BGCs of interest.123Contrary to these developments – and even though natural product isolation and structure elucidation processes have sped up due to more widespread availability of innovative technologies such as semi-preparative HPLC – isolation and structure elucidation of natural products remain a one-structure-at-a-time process. Therefore, this process does not currently scale with the aforementioned improvements in genomics and metabolomics. Even though tandem mass-spectrometry based prediction of structural relatedness has improved greatly over the last years, as it is well exemplified by the aforementioned GNPS platform, de novo structure elucidation solely based on fragment spectra seems out of reach for now.63 Novel software frameworks such as SIRIUS 4.0 are able to match tandem MS spectra against libraries of predefined structure formulae, which in itself can be considered a big leap forward, but are still unable to predict unknown structures with accuracy.62 This process is therefore likely to remain the bottleneck in natural product science, unless there is a yet hardly imaginable ground breaking innovation as for example small molecule structure elucidation from mixtures by cryo-EM that would make isolation and structure elucidation scalable with the generation of genomics and metabolomics data. Discovery of biologically active bacterial secondary metabolites (here more specifically of antibiotics) in our opinion seems to neither be limited by the availability of BGCs as their numbers explode, even when only counting the ones found in the public domain, nor the amount of detectable mass spectrometry features provided from yet uncharacterized natural products.
Increasing sensitivity in mass spectrometers that are more commonly used in natural product laboratories, such as TOF, Orbitrap or FT-ICR systems is revealing an ever-increasing number of natural products in biological samples from an analytical chemistry point of view. Previously discussed alternatives to the most commonly employed LC-ESI-MS techniques were introduced by variations of ionization techniques, as well as the increasing introduction of direct infusion based metabolomics approaches in natural product laboratories, to broaden the scope of detectable natural product classes per extract. Thus, the fundamentals for a bright future for natural product based antibiotics research, meaning abundant metabolome data and a plethora of ‘orphan’ BGCs that are not yet correlated with any known natural product, are available.
The question of whether future antibiotics research based on bacterial secondary metabolomics will continue to thrive critically relies on innovative ways of BGC prioritization or efficient sorting, classification and prioritization of mass spectrometry data of microbial extracts and more importantly, the ability of the field to incorporate these novel developments into the mainstream of natural product research. In antibiotics research, solutions for prioritization of BGCs currently rely on prediction of self-resistance mechanisms, a technique that suffers from its inability to predict novel targets as the corresponding BGC encoded self-resistance mechanisms would necessarily also be unknown and thus not be predictable from genome sequence data alone. Selective activation of BGCs prioritized by architectural complexity is an efficient method to unlock larger structural diversity of secondary metabolites, but structural complexity of a bacterial secondary metabolite is not inherently correlated with strong biological activity. On the metabolomics side, pushing down detection limits has created an additional problem. The LC-MS data of microbial extracts created with state-of-the-art high-resolution mass spectrometers generate too much data for manual analysis. For objective unbiased interpretation of such analyses, automated data treatment is of utmost importance. In recent years, the field has seen progress as evidenced in the section on statistical data treatment, that helps classify the data created by a modern mass spectrometer into categories of potentially interesting secondary metabolites and matrix LC-MS signals such as medium derived LC-MS peaks. Secondary metabolite spectral networking has succeeded at extracting preliminary structural information at an early stage, but its usability depends on community curation of the corresponding open access repositories, such as spectral libraries and structure databases.
As a conclusion, we can determine that a wide variety of different methods is available to natural product scientists to harness the biosynthetic potential of microorganisms with the aforementioned technologies from an analytical chemistry perspective, as well as from an in silico genomics and molecular biology standpoint. Many laboratories still only focus on a limited set of natural product discovery methods and workflows, which have proven to be successful in the past, but tend to introduce heavy discovery biases. As we believe that this focus also strongly limits the detectable space of natural products, we suggest the evaluation of new technologies for antibiotics discovery continuously, while combining these efforts with in silico prediction methods to uncover the most promising secondary metabolites and secondary metabolite producers. Advances in detection and prioritization of structurally novel natural products and promising candidate BGCs will be crucial to increase the success rate of the downstream natural product discovery workflow parts with limited scalability such as heterologous expression of biosynthetic gene clusters and structure elucidation. Feeding these currently non-scalable, labour-intensive parts of the natural product detection processes with well-curated metabolomics and/or genomics data – in the sense of carefully evaluated and interpreted MS data and well-prioritized orphan BGCs will ultimately determine success or failure of natural product sciences, as it critically influences its output of biologically active natural products.
Author contributions
F. P. contributed to conception and definition of scope and focus of this study. F. P. and C. D. B. contributed to literature research, conceiving, writing and editing of the manuscript. R. M. contributed to the conception of the article and performed editing and proofreading of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors thank D. Krug and F. P. Jake Haeckl for carefully revising the manuscript.References
- R. Gaynes, Emerging Infect. Dis., 2017, 23, 849 CrossRef.
- S. A. Waksman, Science, 1953, 118, 259 CrossRef CAS PubMed.
- M. I. Hutchings, A. W. Truman and B. Wilkinson, Curr. Opin. Microbiol., 2019, 51, 72 CrossRef CAS PubMed.
- D. Lyddiard, G. L. Jones and B. W. Greatrex, FEMS Microbiol. Lett., 2016, 363(8), fnw084 CrossRef PubMed.
- O. C. Nunes, C. M. Manaia, B. A. Kolvenbach and P. F. X. Corvini, Appl. Microbiol. Biotechnol., 2020, 104, 10389–10408 CrossRef CAS PubMed.
- R. C. G. Corrêa, S. A. Heleno, M. J. Alves and I. C. F. R. Ferreira, Curr. Pharm. Des., 2020, 26, 815 CrossRef PubMed.
- J. J. Hug, D. Krug and R. Müller, Nat. Rev. Chem., 2020, 4, 172 CrossRef CAS.
- T. Saga and K. Yamaguchi, JMAJ, 2009, 52, 103 Search PubMed.
- B. Ribeiro da Cunha, L. P. Fonseca and C. R. C. Calado, Antibiotics, 2019, 8, 45 CrossRef PubMed.
- (a) H. W. Boucher, G. H. Talbot, J. S. Bradley, J. E. Edwards, D. Gilbert, L. B. Rice, M. Scheld, B. Spellberg and J. Bartlett, Clin. Infect. Dis., 2009, 48, 1 CrossRef PubMed; (b) A. Brenciani, G. Morroni, S. Pollini, E. Tiberi, M. Mingoia, P. E. Varaldo, G. M. Rossolini and E. Giovanetti, J. Antimicrob. Chemother., 2016, 71, 307 CrossRef CAS PubMed; (c) Interagency coordination group on antimicrobial resistance, No time to wait: securing the future from drug-resistant infections. Report to the secretary general of the united nations, 2019 Search PubMed.
- L. L. Silver, Clin. Microbiol. Rev., 2011, 24, 71 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770 CrossRef CAS PubMed.
- P. M. Wright, I. B. Seiple and A. G. Myers, Angew. Chem., Int. Ed., 2014, 53, 8840 CrossRef CAS PubMed.
- (a) A. Towse, C. K. Hoyle, J. Goodall, M. Hirsch, J. Mestre-Ferrandiz and J. H. Rex, Health Policy, 2017, 121, 1025 CrossRef PubMed; (b) D. J. Payne, M. N. Gwynn, D. J. Holmes and D. L. Pompliano, Nat. Rev. Drug Discovery, 2007, 6, 29 CrossRef CAS PubMed.
- B. Plackett, Nature, 2020, 586, S50–S52 CrossRef CAS.
- T. Hoffmann, D. Krug, N. Bozkurt, S. Duddela, R. Jansen, R. Garcia, K. Gerth, H. Steinmetz and R. Müller, Nat. Commun., 2018, 9, 803 CrossRef PubMed.
- C. D. Bader, F. Panter and R. Müller, Biotechnol. Adv., 2020, 39, 107480 CrossRef CAS PubMed.
- D. Nichols, N. Cahoon, E. M. Trakhtenberg, L. Pham, A. Mehta, A. Belanger, T. Kanigan, K. Lewis and S. S. Epstein, Appl. Environ. Microbiol., 2010, 76, 2445 CrossRef CAS PubMed.
- J. J. Hug, C. D. Bader, M. Remškar, K. Cirnski and R. Müller, Antibiotics, 2018, 7, 44 CrossRef PubMed.
- O. Sticher, Nat. Prod. Rep., 2008, 25, 517 RSC.
- C. D. Bader, P. A. Haack, F. Panter, D. Krug and R. Müller, J. Nat. Prod., 2021, 84(2), 268–277 CrossRef CAS PubMed.
- F. Chen, M. Wang and K.-W. Cheng, in Bioactive natural products. Detection, isolation, and structural determination, ed. S. M. Colegate and R. J. Molyneux, CRC Press, Boca Raton, 2nd edn, 2008, pp. 245–265 Search PubMed.
- J.-L. Wolfender, Planta Med., 2009, 75, 719 CrossRef CAS PubMed.
- (a) V. Exarchou, M. Krucker, T. A. van Beek, J. Vervoort, I. P. Gerothanassis and K. Albert, Magn. Reson. Chem., 2005, 43, 681 CrossRef CAS PubMed; (b) A. Bouslimani, L. M. Sanchez, N. Garg and P. C. Dorrestein, Nat. Prod. Rep., 2014, 31, 718 RSC.
- M. F. Chellat and R. Riedl, Angew. Chem., Int. Ed., 2017, 56, 13184 CrossRef CAS PubMed.
- W. Gao, T. Stalder, P. Foley, M. Rauh, H. Deng and C. Kirschbaum, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2013, 928, 1 CrossRef CAS PubMed.
- J. E. Spraker, G. T. Luu and L. M. Sanchez, Nat. Prod. Rep., 2019, 37, 150–162 RSC.
- M. Ghaste, R. Mistrik and V. Shulaev, Int. J. Mol. Sci., 2016, 17(6), 816 CrossRef PubMed.
- K. H. Park, M. S. Kim, S. J. Baek, I. H. Bae, S.-W. Seo, J. Kim, Y. K. Shin, Y.-M. Lee and H. S. Kim, Plant Methods, 2013, 9, 15 CrossRef CAS PubMed.
- (a) K. A. Aliferis and S. Jabaji, PLoS One, 2012, 7, e42576 CrossRef CAS PubMed; (b) M. Witting, M. Lucio, D. Tziotis, B. Wägele, K. Suhre, R. Voulhoux, S. Garvis and P. Schmitt-Kopplin, Anal. Bioanal. Chem., 2015, 407, 1059 CrossRef CAS PubMed.
- J. A. Hawkes, T. Dittmar, C. Patriarca, L. Tranvik and J. Bergquist, Anal. Chem., 2016, 88, 7698 CrossRef CAS PubMed.
- C. S. Hoaglund, S. J. Valentine, C. R. Sporleder, J. P. Reilly and D. E. Clemmer, Anal. Chem., 1998, 70, 2236 CrossRef CAS PubMed.
- (a) K. Masike, M. A. Stander and A. de Villiers, J. Pharm. Biomed. Anal., 2021, 195, 113846 CrossRef CAS PubMed; (b) A. C. Schrimpe-Rutledge, S. D. Sherrod and J. A. McLean, Curr. Opin. Chem. Biol., 2018, 42, 160 CrossRef CAS PubMed.
- A. P. Marshall, A. R. Johnson, M. M. Vega, R. J. Thomson and E. E. Carlson, J. Nat. Prod., 2020, 83, 159 CrossRef CAS PubMed.
- D. K. Derewacz, C. R. Goodwin, C. R. Mcnees, J. A. McLean and B. O. Bachmann, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2336 CrossRef CAS PubMed.
- C. Seger, S. Sturm and H. Stuppner, Nat. Prod. Rep., 2013, 30, 970 RSC.
- K. Haraguchi, Y. Kato, K. Atobe, S. Okada, T. Endo, F. Matsubara and T. Oguma, Anal. Chem., 2008, 80(24), 9748–9755 CrossRef CAS PubMed.
- T. Hebra, V. Eparvier and D. Touboul, J. Chromatogr. A, 2020, 1630, 461533 CrossRef CAS PubMed.
- M. Z. Israr, D. Bernieh, A. Salzano, S. Cassambai, Y. Yazaki and T. Suzuki, Clin. Chem. Lab. Med., 2020, 58(6), 883–896 CrossRef CAS PubMed.
- D. W. Udwary, E. A. Gontang, A. C. Jones, C. S. Jones, A. W. Schultz, J. M. Winter, J. Y. Yang, N. Beauchemin, T. L. Capson, B. R. Clark, E. Esquenazi, A. S. Eustáquio, K. Freel, L. Gerwick, W. H. Gerwick, D. Gonzalez, W.-T. Liu, K. L. Malloy, K. N. Maloney, M. Nett, J. K. Nunnery, K. Penn, A. Prieto-Davo, T. L. Simmons, S. Weitz, M. C. Wilson, L. S. Tisa, P. C. Dorrestein and B. S. Moore, Appl. Environ. Microbiol., 2011, 77, 3617 CrossRef CAS PubMed.
- J. D. Watrous and P. C. Dorrestein, Nat. Rev. Microbiol., 2011, 9, 683 CrossRef CAS PubMed.
- A. R. Buchberger, K. DeLaney, J. Johnson and L. Li, Anal. Chem., 2018, 90, 240 CrossRef CAS PubMed.
- M. Hoffmann, D. Auerbach, F. Panter, T. Hoffmann, P. C. Dorrestein and R. Müller, ACS Chem. Biol., 2018, 13, 273 CrossRef CAS PubMed.
- R. Benndorf, H. Guo, E. Sommerwerk, C. Weigel, M. Garcia-Altares, K. Martin, H. Hu, M. Küfner, Z. W. de Beer, M. Poulsen and C. Beemelmanns, Antibiotics, 2018, 7(3), 83 CrossRef CAS PubMed.
- W.-T. Liu, R. D. Kersten, Y.-L. Yang, B. S. Moore and P. C. Dorrestein, J. Am. Chem. Soc., 2011, 133, 18010 CrossRef CAS PubMed.
- R. Liu, R. A. A. Khan, Q. Yue, Y. Jiao, Y. Yang, Y. Li and B. Xie, Biochem. Biophys. Res. Commun., 2020, 527, 689 CrossRef CAS PubMed.
- D. Parrot, S. Papazian, D. Foil and D. Tasdemir, Planta Med., 2018, 84, 584 CrossRef CAS PubMed.
- (a) G. Zhang, J. Zhang, R. J. DeHoog, S. Pennathur, C. R. Anderton, M. A. Venkatachalam, T. Alexandrov, L. S. Eberlin and K. Sharma, Metabolomics, 2020, 16, 11 CrossRef CAS PubMed; (b) A. Palmer, P. Phapale, I. Chernyavsky, R. Lavigne, D. Fay, A. Tarasov, V. Kovalev, J. Fuchser, S. Nikolenko, C. Pineau, M. Becker and T. Alexandrov, Nat. Methods, 2017, 14, 57 CrossRef CAS PubMed.
- B. M. Ellis, C. N. Fischer, L. B. Martin, B. O. Bachmann and J. A. McLean, Anal. Chem., 2019, 91, 13703–13711 CrossRef CAS PubMed.
- T. Sugiki, K. Furuita, T. Fujiwara and C. Kojima, Molecules, 2018, 23(1), 148 CrossRef PubMed.
- X. Li, H. Luo, T. Huang, L. Xu, X. Shi and K. Hu, Anal. Bioanal. Chem., 2019, 411, 1301 CrossRef CAS PubMed.
- B. M. Duggan, R. Cullum, W. Fenical, L. A. Amador, A. D. Rodríguez and J. J. La Clair, Angew. Chem., 2020, 132, 1160 CrossRef.
- Y. -L. Yang, W. -Y. Liao, W. -Y. Liu, C. -C. Liaw, C. -N. Shen, Z. -Y. Huang and S. -H. Wu, Chem.–Eur. J., 2009, 15, 11573 CrossRef CAS PubMed.
- (a) L. Keller, A. Plaza, C. Dubiella, M. Groll, M. Kaiser and R. Müller, J. Am. Chem. Soc., 2015, 137, 8121 CrossRef CAS PubMed; (b) L. Etzbach, A. Plaza, R. Garcia, S. Baumann and R. Müller, Org. Lett., 2014, 16, 2414 CrossRef CAS PubMed.
- R. M. Boiteau, D. W. Hoyt, C. D. Nicora, H. A. Kinmonth-Schultz, J. K. Ward and K. Bingol, Metabolites, 2018, 8(1), 8 CrossRef PubMed.
- M. Foroozandeh, R. W. Adams, M. Nilsson and G. A. Morris, J. Am. Chem. Soc., 2014, 136, 11867 CrossRef CAS PubMed.
- T. Ito and M. Masubuchi, J. Antibiot., 2014, 67, 353 CrossRef CAS PubMed.
- L. Yi, N. Dong, Y. Yun, B. Deng, D. Ren, S. Liu and Y. Liang, Anal. Chim. Acta, 2016, 914, 17 CrossRef CAS PubMed.
- T. Hoffmann, D. Krug, S. Hüttel and R. Müller, Anal. Chem., 2014, 86, 10780 CrossRef CAS PubMed.
- Y. Hou, D. R. Braun, C. R. Michel, J. L. Klassen, N. Adnani, T. P. Wyche and T. S. Bugni, Anal. Chem., 2012, 84, 4277 CrossRef CAS PubMed.
- F. Panter, D. Krug and R. Müller, ACS Chem. Biol., 2019, 14, 88 CrossRef CAS PubMed.
- K. Dührkop, M. Fleischauer, M. Ludwig, A. A. Aksenov, A. V. Melnik, M. Meusel, P. C. Dorrestein, J. Rousu and S. Böcker, Nat. Methods, 2019, 16, 299 CrossRef PubMed.
- M. Wang, J. J. Carver, V. V. Phelan, L. M. Sanchez, N. Garg, Y. Peng, D. D. Nguyen, J. Watrous, C. A. Kapono, T. Luzzatto-Knaan, C. Porto, A. Bouslimani, A. V. Melnik, M. J. Meehan, W.-T. Liu, M. Crusemann, P. D. Boudreau, E. Esquenazi, M. Sandoval-Calderon, R. D. Kersten, L. A. Pace, R. A. Quinn, K. R. Duncan, C.-C. Hsu, D. J. Floros, R. G. Gavilan, K. Kleigrewe, T. Northen, R. J. Dutton, D. Parrot, E. E. Carlson, B. Aigle, C. F. Michelsen, L. Jelsbak, C. Sohlenkamp, P. Pevzner, A. Edlund, J. McLean, J. Piel, B. T. Murphy, L. Gerwick, C.-C. Liaw, Y.-L. Yang, H.-U. Humpf, M. Maansson, R. A. Keyzers, A. C. Sims, A. R. Johnson, A. M. Sidebottom, B. E. Sedio, A. Klitgaard, C. B. Larson, C. A. Boya P, D. Torres-Mendoza, D. J. Gonzalez, D. B. Silva, L. M. Marques, D. P. Demarque, E. Pociute, E. C. O'Neill, E. Briand, E. J. N. Helfrich, E. A. Granatosky, E. Glukhov, F. Ryffel, H. Houson, H. Mohimani, J. J. Kharbush, Y. Zeng, J. A. Vorholt, K. L. Kurita, P. Charusanti, K. L. McPhail, K. F. Nielsen, L. Vuong, M. Elfeki, M. F. Traxler, N. Engene, N. Koyama, O. B. Vining, R. Baric, R. R. Silva, S. J. Mascuch, S. Tomasi, S. Jenkins, V. Macherla, T. Hoffman, V. Agarwal, P. G. Williams, J. Dai, R. Neupane, J. Gurr, A. M. C. Rodriguez, A. Lamsa, C. Zhang, K. Dorrestein, B. M. Duggan, J. Almaliti, P.-M. Allard, P. Phapale, L.-F. Nothias, T. Alexandrov, M. Litaudon, J.-L. Wolfender, J. E. Kyle, T. O. Metz, T. Peryea, D.-T. Nguyen, D. VanLeer, P. Shinn, A. Jadhav, R. Muller, K. M. Waters, W. Shi, X. Liu, L. Zhang, R. Knight, P. R. Jensen, B. O. Palsson, K. Pogliano, R. G. Linington, M. Gutierrez, N. P. Lopes, W. H. Gerwick, B. S. Moore, P. C. Dorrestein and N. Bandeira, Nat. Biotechnol., 2016, 34, 828 CrossRef CAS PubMed.
- (a) M. Seo, H. K. Shin, Y. Myung, S. Hwang and K. T. No, J. Cheminf., 2020, 12, 6 CrossRef CAS PubMed; (b) A. Capecchi, D. Probst and J.-L. Reymond, J. Cheminf., 2020, 12, 43 CrossRef CAS PubMed.
- J. A. van Santen, G. Jacob, A. L. Singh, V. Aniebok, M. J. Balunas, D. Bunsko, F. C. Neto, L. Castaño-Espriu, C. Chang, T. N. Clark, J. L. Cleary Little, D. A. Delgadillo, P. C. Dorrestein, K. R. Duncan, J. M. Egan, M. M. Galey, F. J. Haeckl, A. Hua, A. H. Hughes, D. Iskakova, A. Khadilkar, J.-H. Lee, S. Lee, N. LeGrow, D. Y. Liu, J. M. Macho, C. S. McCaughey, M. H. Medema, R. P. Neupane, T. J. O’Donnell, J. S. Paula, L. M. Sanchez, A. F. Shaikh, S. Soldatou, B. R. Terlouw, T. A. Tran, M. Valentine, J. J. J. van der Hooft, D. A. Vo, M. Wang, D. Wilson, K. E. Zink and R. G. Linington, ACS Cent. Sci., 2019, 5(11), 1824–1833 CrossRef CAS PubMed.
- J. A. van Santen, S. A. Kautsar, M. H. Medema and R. G. Linington, Nat. Prod. Rep., 2021, 38, 264–278 RSC.
- F. E. Koehn, Prog. Drug Res., 2008, 65, 175 CAS.
- Y. Futamura, M. Muroi and H. Osada, Mol. BioSyst., 2013, 9, 897 RSC.
- (a) G. A. Jacoby, M. A. Corcoran and D. C. Hooper, Antimicrob. Agents Chemother., 2015, 59(11), 6689–6695 CrossRef CAS PubMed; (b) A. Kling, P. Lukat, D. V. Almeida, A. Bauer, E. Fontaine, S. Sordello, N. Zaburannyi, J. Herrmann, S. C. Wenzel, C. König, N. C. Ammerman, M. B. Barrio, K. Borchers, F. Bordon-Pallier, M. Brönstrup, G. Courtemanche, M. Gerlitz, M. Geslin, P. Hammann, D. W. Heinz, H. Hoffmann, S. Klieber, M. Kohlmann, M. Kurz, C. Lair, H. Matter, E. Nuermberger, S. Tyagi, L. Fraisse, J. H. Grosset, S. Lagrange and R. Müller, Science, 2015, 348, 1106 CrossRef CAS PubMed; (c) A. Müller, A. Klöckner and T. Schneider, Nat. Prod. Rep., 2017, 34, 909 RSC.
- M. Tulp and L. Bohlin, Bioorg. Med. Chem., 2005, 13, 5274 CrossRef CAS PubMed.
- T. M. Bakheet and A. J. Doig, BMC Bioinf., 2010, 11, 1 CrossRef PubMed.
- L. Pedro and R. J. Quinn, Molecules, 2016, 21, 984 CrossRef PubMed.
- H. Vu, N. B. Pham and R. J. Quinn, J. Biomol. Screening, 2008, 13, 265 CrossRef CAS PubMed.
- (a) Z. Chilingaryan, Z. Yin and A. J. Oakley, Int. J. Mol. Sci., 2012, 13, 12857 CrossRef CAS PubMed; (b) D. Bruno, A. Grondin, P. Schambel, M. Vitorino and D. Zeyer, Phytochem. Rev., 2020, 19, 1141 CrossRef; (c) J. M. Rainard, G. C. Pandarakalam and S. P. McElroy, SLAS Discovery, 2018, 23, 225 CAS.
- A. Leitner, T. Walzthoeni, A. Kahraman, F. Herzog, O. Rinner, M. Beck and R. Aebersold, Mol. Cell. Proteomics, 2010, 9, 1634 CrossRef CAS PubMed.
- B. Yang, Y. J. Feng, H. Vu, B. McCormick, J. Rowley, L. Pedro, G. J. Crowther, W. C. van Voorhis, P. I. Forster and R. J. Quinn, J. Biomol. Screening, 2016, 21, 194 CrossRef CAS PubMed.
- H. Seki, S. Xue, S. Pellett, P. Šilhár, E. A. Johnson and K. D. Janda, J. Am. Chem. Soc., 2016, 138, 5568 CrossRef CAS PubMed.
- A. von Tesmar, M. Hoffmann, A. Abou Fayad, S. Hüttel, V. Schmitt, J. Herrmann and R. Müller, ACS Chem. Biol., 2018, 13, 812 CrossRef CAS PubMed.
- R. M. Kainkaryam and P. J. Woolf, Curr. Opin. Drug Discovery Dev., 2009, 12, 339 CAS.
- A. Merk, A. Bartesaghi, S. Banerjee, V. Falconieri, P. Rao, M. I. Davis, R. Pragani, M. B. Boxer, L. A. Earl, J. L. S. Milne and S. Subramaniam, Cell, 2016, 165, 1698 CrossRef CAS PubMed.
- (a) D. R. Cooper, P. J. Porebski, M. Chruszcz and W. Minor, Expert Opin. Drug Discovery, 2011, 6, 771 CrossRef CAS; (b) T. L. Blundell, H. Jhoti and C. Abell, Nat. Rev. Drug Discovery, 2002, 1, 45 CrossRef CAS PubMed.
- (a) C. J. Henrich and J. A. Beutler, Nat. Prod. Rep., 2013, 30, 1284 RSC; (b) V. Ravichandran, L. Zhong, H. Wang, G. Yu, Y. Zhang and A. Li, Front. Cell. Infect. Microbiol., 2018, 8, 292 CrossRef PubMed.
- M. R. Seyedsayamdost, J. Ind. Microbiol. Biotechnol., 2019, 46, 301 CrossRef CAS PubMed.
- J. M. Heather and B. Chain, Genomics, 2016, 107, 1 CrossRef CAS PubMed.
- A. L. Harvey, R. Edrada-Ebel and R. J. Quinn, Nat. Rev. Drug Discovery, 2015, 14, 111 CrossRef CAS.
- (a) K. Blin, T. Wolf, M. G. Chevrette, X. Lu, C. J. Schwalen, S. A. Kautsar, H. G. Suarez Duran, E. L. C. de Los Santos, H. U. Kim, M. Nave, J. S. Dickschat, D. A. Mitchell, E. Shelest, R. Breitling, E. Takano, S. Y. Lee, T. Weber and M. H. Medema, Nucleic Acids Res., 2017, 45, W36–W41 CrossRef CAS PubMed; (b) K. Blin, H. U. Kim, M. H. Medema and T. Weber, Briefings Bioinf., 2017, 20(4), 1103–1113 CrossRef PubMed; (c) M. A. Skinnider, N. J. Merwin, C. W. Johnston and N. A. Magarvey, Nucleic Acids Res., 2017, 45, W49–W54 CrossRef CAS PubMed; (d) M. A. Skinnider, C. W. Johnston, M. Gunabalasingam, N. J. Merwin, A. M. Kieliszek, R. J. MacLellan, H. Li, M. R. M. Ranieri, A. L. H. Webster, M. P. T. Cao, A. Pfeifle, N. Spencer, Q. H. To, D. P. Wallace, C. A. Dejong and N. A. Magarvey, Nat. Commun., 2020, 11, 6058 CrossRef CAS PubMed.
- (a) T. A. J. van der Lee and M. H. Medema, Fungal Genet. Biol., 2016, 89, 29 CrossRef CAS PubMed; (b) S. A. Kautsar, H. G. Suarez Duran, K. Blin, A. Osbourn and M. H. Medema, Nucleic Acids Res., 2017, 45, W55–W63 CrossRef CAS PubMed.
- P. Cimermancic, M. H. Medema, J. Claesen, K. Kurita, L. C. Wieland Brown, K. Mavrommatis, A. Pati, P. A. Godfrey, M. Koehrsen, J. Clardy, B. W. Birren, E. Takano, A. Sali, R. G. Linington and M. A. Fischbach, Cell, 2014, 158, 412 CrossRef CAS PubMed.
- Z. Charlop-Powers, A. Milshteyn and S. F. Brady, Curr. Opin. Microbiol., 2014, 70 CrossRef CAS PubMed.
- S. A. Kautsar, K. Blin, S. Shaw, J. C. Navarro-Muñoz, B. R. Terlouw, van der Hooft, J. J. Justin, J. A. van Santen, V. Tracanna, H. G. Suarez Duran, V. Pascal Andreu, N. Selem-Mojica, M. Alanjary, S. L. Robinson, G. Lund, S. C. Epstein, A. C. Sisto, L. K. Charkoudian, J. Collemare, R. G. Linington, T. Weber and M. H. Medema, Nucleic Acids Res., 2019, 48(D1), D454–D458 Search PubMed.
- M. H. Medema, R. Kottmann, P. Yilmaz, M. Cummings, J. B. Biggins, K. Blin, I. de Bruijn, Y. H. Chooi, J. Claesen, R. C. Coates, P. Cruz-Morales, S. Duddela, S. Düsterhus, D. J. Edwards, D. P. Fewer, N. Garg, C. Geiger, J. P. Gomez-Escribano, A. Greule, M. Hadjithomas, A. S. Haines, E. J. Helfrich, M. L. Hillwig, K. Ishida, A. C. Jones, C. S. Jones, K. Jungmann, C. Kegler, H. U. Kim, P. Kötter, D. Krug, J. Masschelein, A. V. Melnik, s. M. Mantovani, E. A. Monroe, M. Moore, N. Moss, H.-W. Nützmann, G. Pan, A. Pati, D. Petras, F. J. Reen, F. Rosconi, Z. Rui, Z. Tian, N. J. Tobias, Y. Tsunematsu, P. Wiemann, E. Wyckoff, X. Yan, G. Yim, F. Yu, Y. Xie, B. Aigle, A. K. Apel, C. J. Balibar, E. P. Balskus, F. Barona-Gómez, A. Bechthold, H. B. Bode, R. Borriss, S. F. Brady, A. A. Brakhage, P. Caffrey, Y.-Q. Cheng, J. Clardy, R. J. Cox, R. de Mot, S. Donadio, M. S. Donia, W. van der Donk, P. C. Dorrestein, S. Doyle, A. J. Driessen, M. Ehling-Schulz, K.-D. Entian, M. A. Fischbach, L. Gerwick, W. H. Gerwick, H. Gross, B. Gust, C. Hertweck, M. Höfte, S. E. Jensen, J. Ju, L. Katz, L. Kaysser, J. L. Klassen, N. P. Keller, J. Kormanec, O. P. Kuipers, T. Kuzuyama, N. C. Kyrpides, H.-J. Kwon, S. Lautru, R. Lavigne, C. Y. Lee, B. Linquan, X. Liu, W. Liu, A. Luzhetskyy, T. Mahmud, Y. Mast, C. Méndez, M. Metsä-Ketelä, J. Micklefield, D. A. Mitchell, B. S. Moore, L. M. Moreira, R. Müller, B. A. Neilan, M. Nett, J. Nielsen, F. O'Gara, H. Oikawa, A. Osbourn, M. S. Osburne, B. Ostash, S. M. Payne, J.-L. Pernodet, M. Petricek, J. Piel, O. Ploux, J. M. Raaijmakers, J. A. Salas, E. K. Schmitt, B. Scott, R. F. Seipke, B. Shen, D. H. Sherman, K. Sivonen, M. J. Smanski, M. Sosio, E. Stegmann, R. D. Süssmuth, K. Tahlan, C. M. Thomas, Y. Tang, A. W. Truman, M. Viaud, J. D. Walton, C. T. Walsh, T. Weber, G. P. van Wezel, B. Wilkinson, J. M. Willey, W. Wohlleben, G. D. Wright, N. Ziemert, C. Zhang, S. B. Zotchev, R. Breitling, E. Takano and F. O. Glöckner, Nat. Chem. Biol., 2015, 11, 625 CrossRef CAS.
- Y.-M. Chiang, S.-L. Chang, B. R. Oakley and C. C. C. Wang, Curr. Opin. Chem. Biol., 2011, 15, 137 CrossRef CAS.
- (a) G. L. Challis, J. Ind. Microbiol. Biotechnol., 2014, 41, 219 CrossRef CAS PubMed; (b) R. F. Seipke, PLoS One, 2015, 10, e0116457 CrossRef PubMed.
- (a) S. C. Wenzel and R. Müller, Mol. BioSyst., 2009, 5, 567 RSC; (b) A. D. Steele, C. N. Teijaro, D. Yang and B. Shen, J. Biol. Chem., 2019, 294(45), 16567–16576 CrossRef CAS.
- P. Xie, M. Ma, M. E. Rateb, K. A. Shaaban, Z. Yu, S.-X. Huang, L.-X. Zhao, X. Zhu, Y. Yan, R. M. Peterson, J. R. Lohman, D. Yang, M. Yin, J. D. Rudolf, Y. Jiang, Y. Duan and B. Shen, J. Nat. Prod., 2014, 77, 377 CrossRef CAS PubMed.
- J. J. Zhang, K. Yamanaka, X. Tang and B. S. Moore, Methods Enzymol., 2019, 621, 87 CAS.
- (a) U. Galm and B. Shen, Expert Opin. Drug Discovery, 2006, 1, 409 CrossRef CAS PubMed; (b) Y. Luo, B. Enghiad and H. Zhao, Nat. Prod. Rep., 2016, 33, 174 RSC.
- P. N. Tran, M.-R. Yen, C.-Y. Chiang, H.-C. Lin and P.-Y. Chen, Appl. Microbiol. Biotechnol., 2019, 103, 3277 CrossRef CAS PubMed.
- C. T. Walsh, Nat. Prod. Rep., 2016, 33, 127 RSC.
- H. B. Bode and R. Müller, in Myxobacteria: Multicellularity and differentiation, ed. D. Whitworth, ASM Press, Chicago, 2007, pp. 259–282 Search PubMed.
- Y. Yan, N. Liu and Y. Tang, Nat. Prod. Rep., 2020, 37, 879 RSC.
- F. Panter, D. Krug, S. Baumann and R. Müller, Chem. Sci., 2018, 9, 4898 RSC.
- (a) X. Tang, J. Li, N. Millán-Aguiñaga, J. J. Zhang, E. C. O'Neill, J. A. Ugalde, P. R. Jensen, s. M. Mantovani and B. S. Moore, ACS Chem. Biol., 2015, 10, 2841 CrossRef CAS PubMed; (b) K. Yamanaka, K. A. Reynolds, R. D. Kersten, K. S. Ryan, D. J. Gonzalez, V. Nizet, P. C. Dorrestein and B. S. Moore, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 1957 CrossRef CAS PubMed.
- M. D. Mungan, M. Alanjary, K. Blin, T. Weber, M. H. Medema and N. Ziemert, Nucleic Acids Res., 2020, 48(W1), W546–W552 CrossRef CAS PubMed.
- K. Ishida, T. Lincke, S. Behnken and C. Hertweck, J. Am. Chem. Soc., 2010, 132, 13966 CrossRef CAS PubMed.
- S. Romano, S. A. Jackson, S. Patry and A. D. W. Dobson, Mar. Drugs, 2018, 16(7), 244 CrossRef PubMed.
- P. J. Rutledge and G. L. Challis, Nat. Rev. Microbiol., 2015, 13, 509 CrossRef CAS PubMed.
- M. M. Zhang, F. T. Wong, Y. Wang, S. Luo, Y. H. Lim, E. Heng, W. L. Yeo, R. E. Cobb, B. Enghiad, E. L. Ang and H. Zhao, Nat. Chem. Biol., 2017, 13, 607–609 CrossRef CAS PubMed.
- J. Wei, L. He and G. Niu, Synth. Syst. Biotechnol., 2018, 3, 229 CrossRef PubMed.
- T. Nakazawa, K. 'i. Ishiuchi, A. Praseuth, H. Noguchi, K. Hotta and K. Watanabe, ChemBioChem, 2012, 13, 855 CrossRef CAS PubMed.
- R. Chen, Q. Zhang, B. Tan, L. Zheng, H. Li, Y. Zhu and C. Zhang, Org. Lett., 2017, 19, 5697 CrossRef CAS PubMed.
- npatlas – The Natural Products Atlas, https://www.npatlas.org.
- J. Fu, X. Bian, S. Hu, H. Wang, F. Huang, P. M. Seibert, A. Plaza, L. Xia, R. Müller, A. F. Stewart and Y. Zhang, Nat. Biotechnol., 2012, 30, 440 CrossRef CAS.
- (a) C. Engler, R. Kandzia and S. Marillonnet, PLoS One, 2008, 3, e3647 CrossRef; (b) D. G. Gibson, L. Young, R. Y. Chuang, J. C. Venter, C. A. Hutchison III and H. O. Smith, Nat. Methods, 2009, 6, 343 CrossRef CAS; (c) H.-J. Nah, H.-R. Pyeon, S.-H. Kang, S.-S. Choi and E.-S. Kim, Front. Microbiol., 2017, 8, 394 Search PubMed.
- (a) D. Pogorevc, Y. Tang, M. Hoffmann, G. Zipf, H. S. Bernauer, A. Popoff, H. Steinmetz and S. C. Wenzel, ACS Synth. Biol., 2019, 8, 1121 CrossRef CAS PubMed; (b) L. Linares-Otoya, V. Linares-Otoya, L. Armas-Mantilla, C. Blanco-Olano, M. Crüsemann, M. L. Ganoza-Yupanqui, J. Campos-Florian, G. M. König and T. F. Schäberle, Microbiology, 2017, 163, 1409 CrossRef CAS PubMed; (c) D. Pogorevc, F. Panter, C. Schillinger, R. Jansen, S. C. Wenzel and R. Müller, Metab. Eng., 2019, 55, 201 CrossRef CAS PubMed; (d) S. Choi, H.-J. Nah, S. Choi and E.-S. Kim, J. Microbiol. Biotechnol., 2019, 29, 1931 CrossRef CAS PubMed.
- Y. Ahmed, Y. Rebets, M. R. Estévez, J. Zapp, M. Myronovskyi and A. Luzhetskyy, Microb. Cell Fact., 2020, 19, 1 CrossRef PubMed.
- J. R. Doroghazi, J. C. Albright, A. W. Goering, K.-S. Ju, R. R. Haines, K. A. Tchalukov, D. P. Labeda, N. L. Kelleher and W. W. Metcalf, Nat. Chem. Biol., 2014, 10, 963 CrossRef CAS PubMed.
- J. Handelsman, Microbiol. Mol. Biol. Rev., 2004, 68, 669 CrossRef CAS.
- E. J. Stewart, J. Bacteriol., 2012, 194, 4151 CrossRef CAS PubMed.
- Y. Hongoh, Biosci., Biotechnol., Biochem., 2010, 74(6), 1145–1151 CrossRef CAS PubMed.
- B. V. B. Reddy, A. Milshteyn, Z. Charlop-Powers and S. F. Brady, Chem. Biol., 2014, 21, 1023 CrossRef CAS.
- (a) H. Wang, Z. Li, R. Jia, Y. Hou, J. Yin, X. Bian, A. Li, R. Muller, A. F. Stewart, J. Fu and Y. Zhang, Nat. Protoc., 2016, 11, 1175 CrossRef CAS PubMed; (b) J. H. Kim, Z. Feng, J. D. Bauer, D. Kallifidas, P. Y. Calle and S. F. Brady, Biopolymers, 2010, 93, 833 CrossRef CAS PubMed.
- M. H. Medema, Nat. Prod. Rep., 2021, 38, 301 RSC.
| This journal is © The Royal Society of Chemistry 2021 |