
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards the stable chelation of radium for biomedical applications with an 18-membered macrocyclic ligand†
Diane S.
Abou‡
 abc,
Nikki A.
Thiele‡
de,
Nicholas T.
Gutsche
f,
Alexandria
Villmer
ab,
Hanwen
Zhang
ab,
Joshua J.
Woods
dg,
Kwamena E.
Baidoo
f,
Freddy E.
Escorcia
f,
Justin J.
Wilson
*d and
Daniel L. J.
Thorek
*abhi
abc,
Nikki A.
Thiele‡
de,
Nicholas T.
Gutsche
f,
Alexandria
Villmer
ab,
Hanwen
Zhang
ab,
Joshua J.
Woods
dg,
Kwamena E.
Baidoo
f,
Freddy E.
Escorcia
f,
Justin J.
Wilson
*d and
Daniel L. J.
Thorek
*abhi
aDepartment of Radiology, Washington University in St. Louis School of Medicine, St. Louis, MO 63110, USA. E-mail: thorek.lab@wustl.edu
bProgram in Quantitative Molecular Therapeutics, Washington University in St. Louis School of Medicine, St. Louis, MO 63110, USA
cRadiology Cyclotron Facility, Mallinckrodt Institute of Radiology, Washington University in St. Louis, St. Louis, MO 63110, USA
dDepartment of Chemistry and Chemical Biology, Cornell University, Ithaca, NY 14853, USA. E-mail: jjw275@cornell.edu
eChemical Sciences Division, Oak Ridge National Laboratory, Oak Ridge, TN 37830, USA
fMolecular Imaging Program, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD 20892, USA
gRobert F. Smith School for Chemical and Biomolecular Engineering, Cornell University, Ithaca, NY 14853, USA
hDepartment of Biomedical Engineering, Washington University in St. Louis, St. Louis, MO 63110, USA
iOncologic Imaging Program, Siteman Cancer Center, Washington University in St. Louis School of Medicine, St. Louis, MO 63110, USA
First published on 29th January 2021
Abstract
Targeted alpha therapy is an emerging strategy for the treatment of disseminated cancer. [223Ra]RaCl2 is the only clinically approved alpha particle-emitting drug, and it is used to treat castrate-resistant prostate cancer bone metastases, to which [223Ra]Ra2+ localizes. To specifically direct [223Ra]Ra2+ to non-osseous disease sites, chelation and conjugation to a cancer-targeting moiety is necessary. Although previous efforts to stably chelate [223Ra]Ra2+ for this purpose have had limited success, here we report a biologically stable radiocomplex with the 18-membered macrocyclic chelator macropa. Quantitative labeling of macropa with [223Ra]Ra2+ was accomplished within 5 min at room temperature with a radiolabeling efficiency of >95%, representing a significant advancement over conventional chelators such as DOTA and EDTA, which were unable to completely complex [223Ra]Ra2+ under these conditions. [223Ra][Ra(macropa)] was highly stable in human serum and exhibited dramatically reduced bone and spleen uptake in mice in comparison to bone-targeted [223Ra]RaCl2, signifying that [223Ra][Ra(macropa)] remains intact in vivo. Upon conjugation of macropa to a single amino acid β-alanine as well as to the prostate-specific membrane antigen-targeting peptide DUPA, both constructs retained high affinity for 223Ra, complexing >95% of Ra2+ in solution. Furthermore, [223Ra][Ra(macropa-β-alanine)] was rapidly cleared from mice and showed low 223Ra bone absorption, indicating that this conjugate is stable under biological conditions. Unexpectedly, this stability was lost upon conjugation of macropa to DUPA, which suggests a role of targeting vectors in complex stability in vivo for this system. Nonetheless, our successful demonstration of efficient radiolabeling of the β-alanine conjugate with 223Ra and its subsequent stability in vivo establishes for the first time the possibility of delivering [223Ra]Ra2+ to metastases outside of the bone using functionalized chelators, marking a significant expansion of the therapeutic utility of this radiometal in the clinic.
Introduction
[223Ra]RaCl2 is the first and currently only approved α particle-emitting radiopharmaceutical, with an indication for men with bone-metastatic castrate-resistant prostate cancer.1 Since its approval in 2013, [223Ra]RaCl2 has been used to treat over 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 patients, substantially improving the quality of life for those suffering from bone pain, reducing fracture risk, and extending overall survival.2–4 This radiometal is administered as a chloride salt in aqueous citrate buffer without a biological targeting vector or chelating agent. As a bone seeker, [223Ra]Ra2+ is readily incorporated at sites of bone turnover,5–7 including sites of osseous metastases, where it subsequently decays to irradiate the surrounding malignant tissue. Through its decay to stable 207Pb, the 4 high-energy, short-range α particles of 223Ra and its daughters annihilate disseminated bone metastatic prostate cancer cells, while largely sparing neighboring healthy tissues.
000 patients, substantially improving the quality of life for those suffering from bone pain, reducing fracture risk, and extending overall survival.2–4 This radiometal is administered as a chloride salt in aqueous citrate buffer without a biological targeting vector or chelating agent. As a bone seeker, [223Ra]Ra2+ is readily incorporated at sites of bone turnover,5–7 including sites of osseous metastases, where it subsequently decays to irradiate the surrounding malignant tissue. Through its decay to stable 207Pb, the 4 high-energy, short-range α particles of 223Ra and its daughters annihilate disseminated bone metastatic prostate cancer cells, while largely sparing neighboring healthy tissues.
The cytotoxic potential of α particles, evinced by the clinical success of [223Ra]RaCl2, has motivated efforts to expand this form of therapy. To achieve this goal, researchers have coupled α particle emitters, such as 225Ac, 213Bi, 227Th, and 211At, to biological targeting vectors, including peptides and antibodies, that bind with high specificity to receptors overexpressed on cancer cells. This combination, referred to as targeted α-particle therapy (TAT), allows for these radionuclides to be used more broadly in a capacity that is independent of their inherent biodistribution properties.8–12 The success of TAT is reflected by ongoing clinical trials of 225Ac-PSMA-617 for the treatment of castrate-resistant prostate cancer.13,14 This radiopharmaceutical agent delivers 225Ac to prostate cancer cells that overexpress the prostate-specific membrane antigen (PSMA), and has shown remarkable efficacy in prolonging and improving patient lifespans.15 Despite the promising clinical applications of 225Ac-PSMA-617, the limited availability of 225Ac and the challenges in scaling up its production remain significant hurdles that need to be overcome before this therapeutic isotope can be applied universally.16,17
Given the established clinical efficacy, safety, and availability of 223Ra in comparison to other α particle emitters such as 225Ac, the implementation of this radiometal in TAT is very attractive. To effectively use 223Ra for this purpose, however, this radiometal must be stably conjugated to tumor-targeting vectors via a bifunctional chelating agent (BFC). An effective BFC must match the coordination chemistry of the radiometal of interest such that the ion remains stably bound to the targeting vector in vivo.18 Despite decades of interest in Ra2+ for biomedical and environmental applications, no effective chelator for 223Ra has been identified that is suitable for biological applications.19–21
The challenge in finding an efficient chelator for the stable chelation of this ion arises from its chemical properties. As an s-block ion, its interactions with ligands are predominantly electrostatic. Furthermore, Ra2+ is the largest +2 ion in the periodic table (eight-coordinate ionic radius = 1.48 Å (ref. 22)), and thus possesses a low charge-to-ionic radius ratio that results in electrostatic metal–ligand interactions that are substantially weaker than those of the smaller alkaline earth ions.23 Previous efforts to investigate the aqueous coordination chemistry of Ra2+ have focused on linear polyaminocarboxylate ligands, such as ethylenediamine(tetraacetic) acid (EDTA).24–28 These studies, which employed either long-lived 226Ra or tracer 228Ra, demonstrate that complexes of Ra2+ with these ligands form, albeit with lower stability constants than the lighter alkaline earth ions. Competition extraction studies have also shown that macrocycles such as DOTA and Kryptofix 2.2.2 can bind to 223Ra, although the biological stability of these complexes has not been evaluated.29 A further body of work in this field has been directed towards the development of organic-soluble extractants that partition this ion to the organic phase in biphasic systems.29–38 Of these extractants, the most common structural motif explored has been that containing a calixarene core. Although calixarene-based ligands have demonstrated efficiency for the selective extraction of Ra2+ in the context of environmental remediation,32–35 attempts to use these ligands for Ra2+ complexation for nuclear medicine applications have been unsuccessful due to either failed radiolabeling36 or poor kinetic stability of the resulting complex.29 These unsuccessful efforts have led many in the radiopharmaceutical community to the conclusion that stable chelation of 223Ra sufficient for its in vivo use cannot be achieved. In turn, significant work has been directed towards the incorporation of this radionuclide into various nanoparticle constructs.39,40 Doping of the Ra2+ ion into solid-state nanoparticles, such as those of BaSO4, LaPO4, Fe2O3, TiO2, hydroxyapatite, and nanozeolites, has been shown to be an effective means of stabilizing this radionuclide and altering its biodistribution properties.41–46 However, the complementary use of molecularly-targeted constructs using a BFC would enable more possibilities for radiopharmaceutical optimization via appropriate chemical modifications.
In this work, we show that macropa, an 18-membered bis-picolinate diazacrown macrocycle (Fig. 1a), is an effective chelator of [223Ra]Ra2+, demonstrating rapid complexation kinetics and profound in vivo stability. This ligand is the first chelator, to the best of our knowledge, that can stabilize this large ion in vivo. Further, we investigated Ra2+ chelation utilizing a bifunctional derivative of macropa conjugated to a single amino acid, β-alanine, or a prostate cancer-targeting agent, DUPA (Fig. 1b), to probe the effects of chelator functionalization on Ra2+–ligand complex stability. Ultimately, this work garners a better understanding of the elusive coordination chemistry of the Ra2+ ion and opens the path for the generation of targeted 223Ra therapeutics for novel biomedical applications.
 | ||
| Fig. 1 (a) Structure of macropa and its coordination to radium. (b) Structures of DOTA and bifunctional constructs of macropa explored in this work for 223Ra chelation. | ||
Results and discussion
223Ra labeling and in vitro characterization
Macropa is an expanded 18-membered macrocyclic ligand that contains two pendent picolinate donor arms appended to a diaza-18-crown-6 core (Fig. 1a).47 We and others have previously shown that ligands based on this macrocycle exhibit an unusual preference for binding to large over small ions.47–50 This property has made macropa useful for chelating the large radiometals 132/135La and 225Ac for therapeutic nuclear medicine applications.51–54 Furthermore, we have shown that this ligand forms a complex of high thermodynamic stability with Ba2+, the largest non-radioactive +2 ion (eight-coordinate ionic radius (ref. 22) = 1.42 Å),55 prompting its recent investigation as a chelator for the diagnostic isotope 131Ba.56 In aqueous solution at pH 7.4, the conditional stability constant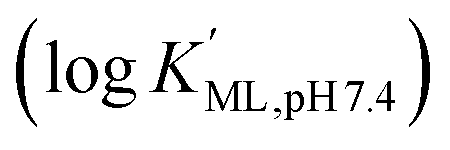 of the [Ba(macropa)] complex is 10.74 (I = 0.1 M KCl, 25 °C), indicating that macropa is the highest-affinity chelator for Ba2+ at physiological pH reported to date. Based on these properties, we hypothesized that macropa might also be suitable for binding to Ra2+, the heavier alkaline earth ion congener of Ba2+, for TAT applications (Fig. 1a). The ionic radius of Ra2+ is only marginally larger than that of Ba2+, and as members of the same group in the periodic table, their donor atom preferences are expected to be similar. Density functional theory calculations (Fig. S1 and Tables S1–S3, ESI†) support this assertion, as both Ba2+ and Ra2+ are computed to form structurally analogous complexes with macropa.
of the [Ba(macropa)] complex is 10.74 (I = 0.1 M KCl, 25 °C), indicating that macropa is the highest-affinity chelator for Ba2+ at physiological pH reported to date. Based on these properties, we hypothesized that macropa might also be suitable for binding to Ra2+, the heavier alkaline earth ion congener of Ba2+, for TAT applications (Fig. 1a). The ionic radius of Ra2+ is only marginally larger than that of Ba2+, and as members of the same group in the periodic table, their donor atom preferences are expected to be similar. Density functional theory calculations (Fig. S1 and Tables S1–S3, ESI†) support this assertion, as both Ba2+ and Ra2+ are computed to form structurally analogous complexes with macropa.
To probe the suitability of macropa for the stable chelation of Ra2+, we first sought to establish conditions for radiolabeling macropa with [223Ra]Ra2+. The reaction was carried out at room temperature by mixing a solution of macropa with 3.7 kBq (0.1 μCi) of [223Ra]Ra2+, obtained from an 227Ac/227Th generator (Section 1.2 of the ESI†).57 The complexation was conducted at room temperature and buffered in metal-free ammonium acetate (0.1 M) at pH 6. Aliquots from these radiolabeling reactions were removed at different time points and analyzed by radio-instant thin layer chromatography (radioTLC) to assess complex formation. Under these TLC conditions, which used diglycolamide (DGA)-impregnated silica as the solid support and 0.1 M NaOH as the mobile phase, free [223Ra]Ra2+ remained at the baseline (Rf = 0), whereas the complexed species migrated with the solvent front (Rf = 1) (Fig. 2a). The TLC strips were visualized using a phosphorimager after radioactive equilibrium was reached to quantify the extent of 223Ra complexation. Within 5 min at room temperature, [223Ra][Ra(macropa)] was formed with a radiolabeling efficiency (RL% ± SD) of 95 ± 1.05% (n = 5) (Fig. 2a). These mild radiolabeling conditions are highly favorable for use with temperature-sensitive biological targeting vectors like antibodies, which degrade at elevated temperatures. By contrast, the most widely used chelator in nuclear medicine, 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA, Fig. 1b), fails to completely coordinate [223Ra]Ra2+ under similar conditions, as does the common polyaminocarboxylate ligand EDTA (see Section 1.3 and Fig. S2 in the ESI†). These results indicate that macropa provides an effective coordination environment for Ra2+.
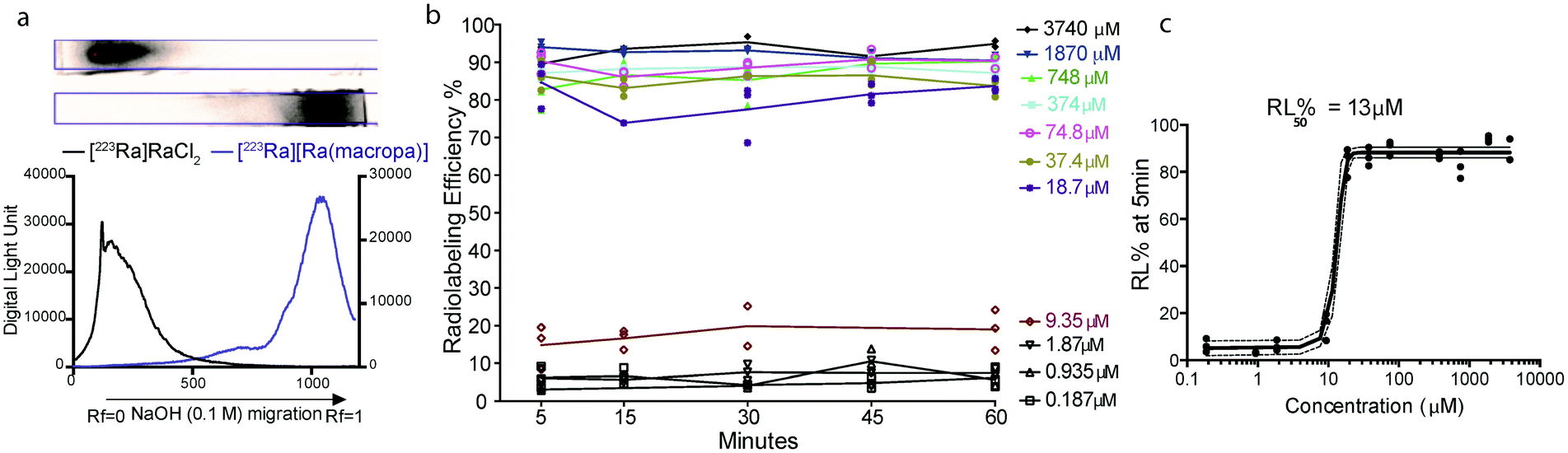 | ||
| Fig. 2 Formation of [223Ra][Ra(macropa)] under different conditions. (a) Migration of [223Ra]RaCl2 (top) and [223Ra][Ra(macropa)] (bottom) on DGA-coated chromatographic strips, which were developed using a mobile phase of 0.1 M NaOH and visualized using autoradiographic quantification profiles. (b) RL% measured as a function of time at room temperature, pH 6, with varying concentration of macropa. (c) RL% as a function of macropa concentration 5 min after co-mixing; the ligand concentration required for 50% RL% (RL50%) was found to be 13 μM, and >80% RL% was achieved at a concentration of 18 μM. | ||
Next, the concentration of macropa was varied over a range of 187 nM to 3.74 mM with a fixed amount of 223Ra activity (3.7 kBq) to determine the maximum apparent molar activity that we could obtain with this system. Above a ligand concentration of 18 μM, the RL%'s were greater than 80% with no change over the 1 h duration (Fig. 2b). At a ligand concentration of less than 18 μM, however, the RL% dropped significantly to values of <20%. A plot of the RL% versus ligand concentration as determined at 5 min is shown in Fig. 2c. Interpolation of these data indicate that 50% RL% is achieved with a ligand concentration of 13 μM. Radiolabeling data obtained at the 1 h time point also gave the same ligand concentration required for 50% RL% (Fig. S3†), further showing that complexation efficiencies remain constant after the initial 5 min. Collectively, these data show that [223Ra][Ra(macropa)] is rapidly and quantitatively formed with molar activities ranging from 0.26 to 55.5 Ci mol−1 or 2.05 to 9.62 × 106 MBq mol−1, utilizing very low activity amounts (<5 μCi or <185 kBq) with radiochemical purity >95%. Although these molar activities are generally lower than those obtained for 225Ac-DOTA constructs,58,59 the high purity of [223Ra][Ra(macropa)] precludes the needs for additional chromatography of this radiocomplex prior to in vivo administration.
Having established that macropa can rapidly bind 223Ra under mild conditions, we next sought to determine the stability of the complex in conditions that are encountered in vivo using human serum at 37 °C over 12 days. Although radioTLC proved useful for assessing RL% in aqueous buffer, this method (silica or DGA-coated TLCs) failed when analyzing 223Ra speciation in the complex media of serum. Specifically, TLC plates analyzed from serum gave rise to multiple peaks and streaking, which prevented unambiguous assignment of free versus complexed [223Ra]Ra2+ (Fig. S4†). To overcome this limitation, we turned to size exclusion chromatography (SEC) using dual detection of absorbance (at 280 nm UV) and gamma counting of collected fractions at radioactive equilibrium, a method that we have validated to be sufficient for unambiguously distinguishing between free and complexed [223Ra]Ra2+ (Section 1.4 and Fig. S5 in ESI†). Upon incubation of [223Ra]RaCl2 in human serum at 37 °C (Fig. 3a), the UV chromatogram of this mixture shows several peaks below 20 mL, corresponding to serum proteins. The radiochromatogram shows only a broad peak at 30 mL, which is indicative of free Ra2+. Importantly, no activity is observed to coincide with the protein fractions, indicating that free Ra2+ does not strongly interact with other components in serum. Having validated this method for evaluating serum stability, we incubated [223Ra][Ra(macropa)] in human serum at 37 °C and analyzed aliquots of this mixture by SEC. A representative chromatogram of this mixture after 2 h is shown in Fig. 3b. At this time point, the radiochromatogram shows only a single peak at 20 mL, which matches that of [Ba(macropa)] and [223Ra][Ra(macropa)] (Fig. S5†). The absence of the broad peak near 30 mL confirms that the [223Ra]Ra2+ ion remains bound within the macrocycle over this time period and that the complex is stable. After 12 days in serum, the span of approximately one half-life of 223Ra, [223Ra][Ra(macropa)] remains approximately 90% intact (Fig. 3c). This high degree of stability was also found for the non-radioactive [Ba(macropa)] complex, which remained intact in the presence of hydroxyapatite, a major constituent of bone matrix that also binds these metals (Section 1.5 and Fig. S6 in ESI†). The robustness of [223Ra][Ra(macropa)] compares favorably with other α particle-emitting radiopharmaceuticals and their respective chelation platforms (e.g.225Ac-DOTA-antibody).58
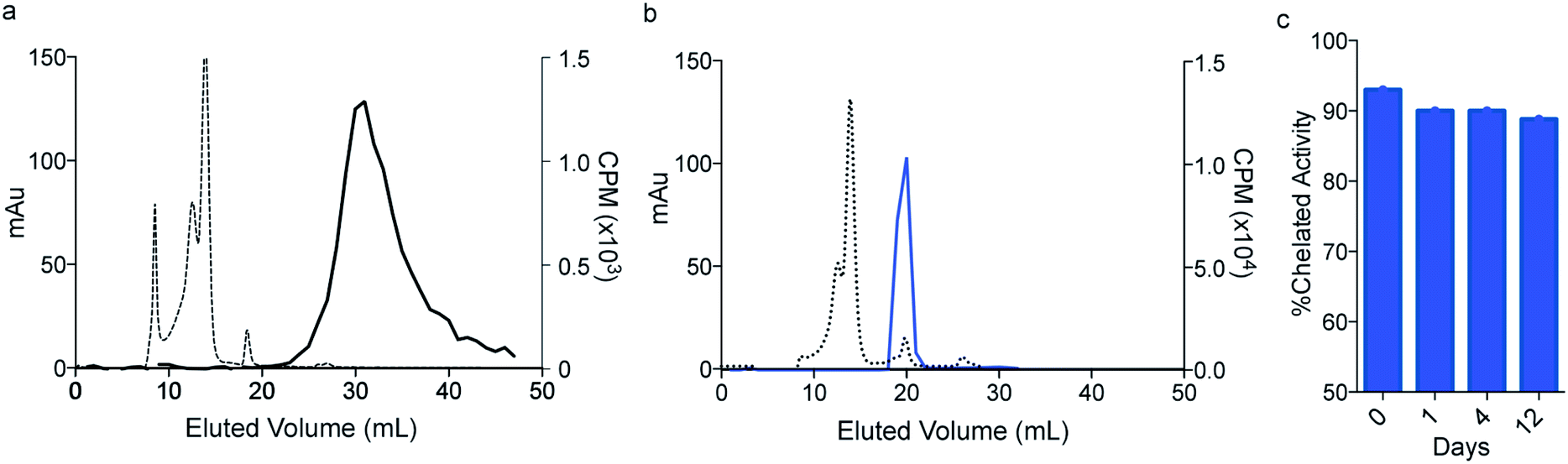 | ||
| Fig. 3 In vitro stability of [223Ra][Ra(macropa)]. Size exclusion chromatography (SEC) of (a) [223Ra]RaCl2 mixed in serum, detected at 280 nm (dashed line) and by gamma counting (solid line); (b) [223Ra][Ra(macropa)] after 2 h in human serum; (c) percent intact complex following human serum challenge over 12 days. | ||
In vivo biodistribution of [223Ra][Ra(macropa)]
Based upon the promising in vitro stability of [223Ra][Ra(macropa)], we proceeded to evaluate the biodistribution of this complex. Previously, we have shown that [223Ra]RaCl2 distribution in mice closely resembles that observed in humans, with rapid blood clearance, localization to sites of active bone remodeling, and excretion predominantly occurring through the small bowel and kidneys.6 To evaluate the in vivo stability of [223Ra][Ra(macropa)], its biodistribution was compared to the FDA/EMA approved formulation of [223Ra]RaCl2 administered in citrate buffer. We utilized healthy, skeletally mature rodent models combined with quantitative gamma counting measurements of organ activity normalized to weight. Organs were excised at 15 min and 24 h post-injection (p.i.) (Fig. 4). Consistent with its bone-seeking properties, a significant amount of [223Ra]Ra2+ was detected in bones (>10% IA per g) for the control [223Ra]RaCl2 citrate-administered groups at 15 min and 24 h p.i. (Fig. 4a). By contrast, an order of magnitude decrease in bone uptake was observed for animals injected with [223Ra][Ra(macropa)]. The latter demonstrated a bone uptake of 1.6 ± 0.2% IA per g versus 22 ± 1% IA per g for the control group at 24 h (****p < 0.0001; Fig. 4b and c). | ||
| Fig. 4 In vivo evaluation of [223Ra][Ra(macropa)]. (a) [223Ra]RaCl2 and (b) [223Ra][Ra(macropa)] radioactive organ distribution (% injected activity normalized to weight; % IA per g) was assessed utilizing healthy, skeletally mature mice sacrificed at 15 min and 24 h p.i. Differences in splenic, renal, and bone uptake were observed between the two groups. (c) Mice administered [223Ra][Ra(macropa)] exhibited 10 to 25-fold lower osseous uptake than that of free 223Ra at 15 min and 24 h p.i. (**p = 0.0096; ****p < 0.0001). | ||
In addition to decreasing the bone uptake, macropa markedly altered the excretion profile of this radiometal. The majority of [223Ra][Ra(macropa)] is rapidly cleared from the blood (2.55 ± 0.85% IA per g at 15 min to 0.007 ± 0.006 at 24 h) through renal excretion. Gut and spleen uptake were reduced (1.67 ± 1.5 and 0.7 ± 0.14% IA per g, respectively) as compared to the [223Ra]RaCl2 control (4.08 ± 1.9 and 11.6 ± 6.1% IA per g). Preventing splenic accumulation (previously noted as a site of uptake of [223Ra]RaCl2 in the rodent5) and gut accumulation are meaningful improvements as gastrointestinal distress is a commonly reported symptom from [223Ra]RaCl2 treatment in patients.2,3 At 15 min p.i., we measured a 300-fold higher activity in the urine, sampled from the bladder, in mice treated with [223Ra][Ra(macropa)] compared to those treated with [223Ra]RaCl2 (Fig. S7a†). At 24 h, all other organs showed negligible counts (<0.37 Bq or 0.01% IA).
The lower kidney activity combined with increased bladder accumulation of [223Ra][Ra(macropa)] compared to [223Ra]RaCl2 (Fig. S7a†) indicates that the complex is rapidly cleared, thus providing further support that macropa retains this radiometal in vivo. As a final confirmation of the in vivo stability of [223Ra][Ra(macropa)], we analyzed urine samples of mice treated with this compound by SEC. The resulting radiochromatogram shows that the excreted species has the same retention time (20 mL) as [223Ra][Ra(macropa)], with no evidence of free or protein-bound activity (Fig. S7b†). In conjunction with the biodistribution data, this chromatogram shows conclusively that the [223Ra][Ra(macropa)] complex is stable in vivo, marking the first observation of a stable Ra2+ coordination complex in this setting.
Radiolabeling and testing of macropa conjugates
Following our demonstration of [223Ra][Ra(macropa)] in vivo stability, the next objective was to explore conjugated forms of this chelator. We have previously reported macropa-NCS, a BFC version of macropa containing an amine-reactive isothiocyanate group appended to one of the picolinate pendant arms.51 Using this bifunctional ligand, we first prepared a simple amino acid conjugate, macropa-β-alanine (see Section 1.7 and Fig. S8–S14 in the ESI†). In this construct, β-alanine is linked to macropa via a thiourea bond. This compound provides a straightforward starting point to evaluate the effects of the thiourea conjugation strategy on Ra2+ complex stability. As a first means of evaluating the effects of this group, we carried out pH-potentiometric titrations to determine both the pKa values and metal stability constants of this ligand for comparison to unfunctionalized macropa. The results of these titrations are compiled in Tables 1, S4 and Fig. S15, S16.† The pKa values of macropa-β-alanine are very similar to those previously reported for macropa (Table 1). As expected, two additional protonation constants for the deprotonation of the NH thiourea and β-alanine carboxylic acid are present. The similarity of the other pKa values, however, suggests that the donor properties of macropa-β-alanine are largely unchanged from those of macropa. The stability constants of macropa-β-alanine for Ca2+, Sr2+, and Ba2+ confirm this assertion. These values are scarcely perturbed from those measured for macropa. For example, we measured logKBaL for macropa-β-alanine to be 11.06(0.10), whereas this value is 11.11 for macropa. Based on these values, both [Ba(macropa)] and [Ba(macropa-β-alanine)] are completely complexed at physiological pH (Fig. 5a). Thus, these thermodynamic data show that the addition of the thiourea group has little effect on the binding affinity of macropa for the alkaline earth ions.
| Macropa-β-ala4−f | Macropa2− | |
|---|---|---|
| a Data provided for comparison. b From combined UV spectrophotometric-potentiometric titrations. c Ref. 55, I = 0.1 M KCl. d Ref. 47, I = 0.1 M KCl. e Ref. 48, I = 0.1 M KNO3. f Values in parentheses correspond to one standard deviation. | ||
| logKa1 |
11.25(0.11), 11.36(0.01)b | 7.41c, 7.41d |
| logKa2 |
7.49(0.03) | 6.90c, 6.85d |
| logKa3 |
6.90(0.02) | 3.23c, 3.32d |
| logKa4 |
4.32(0.03) | 2.45c, 2.36d |
| logKa5 |
3.19(0.01) | 1.69d |
| logKa6 |
2.51(0.05) | |
| logKBaL |
11.06(0.10) | 11.11c |
| logKBaHL |
11.18(0.05) | 3.76c |
| logKBaH2L |
4.54(0.03) | 2.49c |
| logKBaH3L |
3.67(0.03) | |
| logKSrL |
9.58(0.13) | 9.44c, 9.57e |
| logKSrHL |
10.97(0.12) | 3.35c, 4.16e |
| logKSrH2L |
4.59(0.03) | |
| logKCaL |
6.15(0.05) | 5.79c, 5.25e |
| logKCaHL |
10.78(0.06) | |
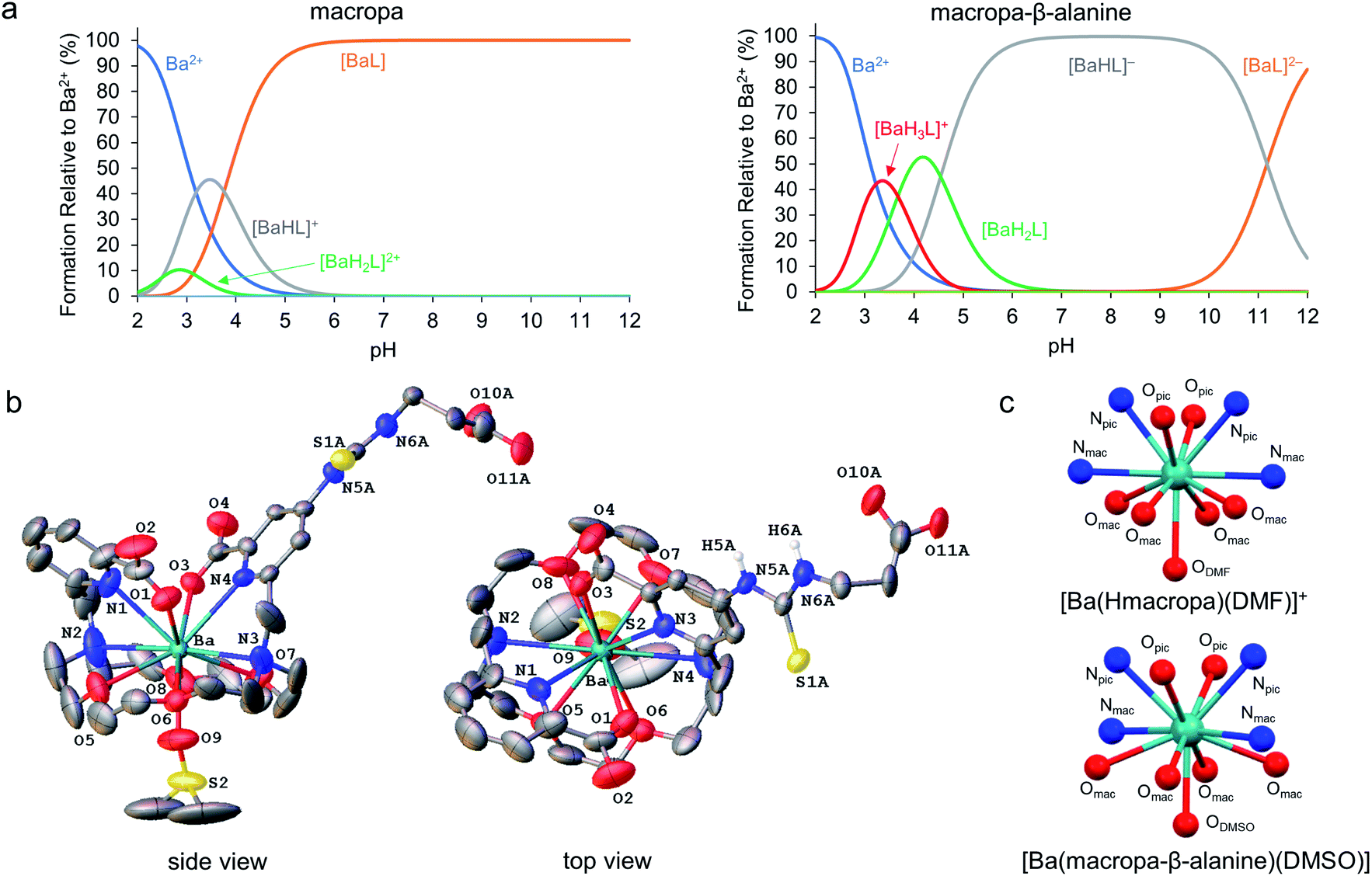 | ||
| Fig. 5 Alkaline earth metal complexes of macropa-β-alanine. (a) Species distribution diagrams of macropa (left) and macropa-β-alanine (right) in the presence of Ba2+ at [Ba2+]tot = [L]tot = 1.0 mM, I = 0.1 M KCl, and 25 °C. (b) X-ray crystal structure of [Ba(macropa-β-alanine)(DMSO)]. Ellipsoids are drawn at the 50% probability level. Nonacidic hydrogen atoms are omitted for clarity. A full discussion of the crystallographic disorder of the structure is provided in the ESI.† (c) Comparison of the immediate coordination sphere of the Ba2+ center in the crystal structures of [Ba(Hmacropa)(DMF)]+ and [Ba(macropa-β-alanine)(DMSO)]. | ||
To further confirm that the thiourea group has no negative effects on alkaline earth coordination, the Ba2+ complex of macropa-β-alanine was synthesized (see Section 1.7 and Fig. S17–S19 in the ESI†). To evaluate its kinetic stability, this complex was challenged with hydroxyapatite, a model of the major constituent of bone matrix. These challenge studies showed that Ba2+ was well retained by macropa-β-alanine, verifying that this complex has a high degree of kinetic inertness (Fig. S6†). Furthermore, single crystals of this complex were subsequently grown from dimethyl sulfoxide (DMSO) and analyzed by X-ray diffraction (Fig. 5b and Tables S5–S10†). Although this structure suffers from significant crystallographic disorder and was weakly diffracting, the overall atomic connectivity and coordination geometry of Ba2+ could be discerned. The Ba2+ center is 11-coordinate with 10 donors provided by macropa and an 11th afforded by a disordered DMSO solvent molecule that interpenetrates the macrocyclic base. In general, this structure resembles closely that of the unfunctionalized [Ba(macropa)] complex, which we previously reported.55 The conformation of the macrocycle about the Ba2+ center is similar for both structures (Fig. 5c). The thiourea and β-alanine moieties were heavily disordered in the [Ba(macropa-β-alanine)(DMSO)] crystal structure, but in both possible configurations these groups were positioned far from the direct coordination sphere of the Ba2+ ion. These structural data further support the potentiometric titrations by showing that thiourea conjugation has no significant effects on the Ba2+ coordination properties of this ligand, suggesting that this BFC will retain high affinity for the Ra2+ ion.
To further probe the utility of this BFC for [223Ra]Ra2+ chelation, we evaluated its ability to bind this radiometal. Using conditions identical to those employed for macropa, RL%'s of >90% were obtained for macropa-β-alanine with [223Ra]Ra2+ (n = 5), and the resulting complex was characterized with SEC (Fig. S20a†). We tested the in vitro stability of the radiolabeled conjugate in human serum by SEC. These studies revealed that >70% of the complex remained intact after 12 days (Fig. 6a and S20b†). Although this stability is slightly lower than what we observed for unfunctionalized [223Ra][Ra(macropa)], these data show that this ion can be kinetically stabilized in different macropa-like ligands. In the case of [223Ra][Ra(macropa-β-alanine)], the SEC chromatogram revealed a small amount of activity (≃5% of initial content) in the elution volume that is characteristic of proteins (10–15 mL). Because we had shown that free [223Ra]Ra2+ does not bind directly to proteins as determined by SEC, we hypothesize that the presence of activity in the protein fractions for [223Ra][Ra(macropa-β-alanine)] is due to outer sphere interactions of this intact complex with proteins and not due to an inherent instability of the Ra2+ coordination sphere.
 | ||
| Fig. 6 In vitro and in vivo evaluation of [223Ra][Ra(macropa-β-alanine)]. (a) Stability of [223Ra][Ra(macropa-β-alanine)] over the course of 12 days in human serum at 37 °C. (b) Organ distribution of [223Ra][Ra(macropa-β-alanine)] at 24 h p.i. Significant differences in osseous (*p < 0.005), splenic, and renal uptake were observed in comparison to the control [223Ra]RaCl2. | ||
To assess the in vivo stability of [223Ra][Ra(macropa-β-alanine)], its biodistribution was investigated in mice. Similarly to [223Ra][Ra(macropa)], major differences in the organ distribution were observed as compared to [223Ra]RaCl2. Notably, at 24 h p.i., it showed very low bone uptake (2.69 ± 0.24% IA per g) in comparison to [223Ra]RaCl2 (9.7 ± 1.66%, *p = 0.0027) (Fig. 6b). Furthermore, [223Ra][Ra(macropa-β-alanine)] displayed approximately 5-fold lower splenic uptake (0.95 ± 0.5% IA per g vs. 5.52 ± 2.3% IA per g) and ∼7-fold lower kidney uptake compared to [223Ra]RaCl2, results that can be explained by the faster and more favorable renal clearance of the β-alanine conjugate. At 24 h, significantly less radioactivity is present in all tissues for [223Ra][Ra(macropa-β-alanine)] in comparison to mice treated with [223Ra]RaCl2, for which significant activity was detected in the bone, spleen, kidney and intestine. Thus, these data show that conjugates of macropa have the potential to be highly stable in vivo. Furthermore, these results suggest that with the choice of an appropriate biological targeting vector, targeted therapy employing 223Ra is feasible.
To probe the potential of TAT using 223Ra, macropa-NCS was conjugated to a glutamate–urea–glutamate (DUPA) targeting vector.54 This DUPA targeting vector binds with high affinity to the PSMA, which is overexpressed on prostate cancer adenocarcinoma cells. Macropa-DUPA was found to chelate Ba2+ effectively (Fig. S21–S23†) and prevent this ion from adsorbing onto hydroxyapatite, albeit to a slightly lower extent than macropa-β-alanine (Fig. S6†). Having confirmed its ability to complex Ba2+, radiolabeling with [223Ra]Ra2+ was carried out. Incorporation of this radiometal proceeded efficiently, giving RL% of >95% (Fig. S24a†). Furthermore, the radiolabeled conjugate remained >90% intact upon incubation in serum at 37 °C for 12 days (Fig. S24b†).
Based on these promising in vitro data, we evaluated the biodistribution of this conjugate in mice. Unexpectedly, the macropa-DUPA conjugate exhibited no difference in biodistribution in comparison to free [223Ra]RaCl2 (Fig. S25†). Notably, high levels of bone uptake consistent with the release of free [223Ra]Ra2+ were detected. Thus, despite the promising stability of macropa and macropa-β-alanine with 223Ra, this stability is lost upon attachment to the DUPA PSMA-targeting vector. This observation contrasts with our previous report of stable chelation with radioactive lanthanum utilizing macropa-DUPA,54 highlighting the unique requirements for successful chelation of radium. Although the reasons for the lack of stability in the DUPA conjugate, in comparison to the β-alanine conjugate, are not fully understood, these data show that targeting vectors can affect metal-chelate stabilities. This phenomenon warrants further investigation, as it may have significant implications in the development of new metal-based radiopharmaceutical agents.
Conclusions
We have demonstrated the robust, rapid, and stable chelation of radium in aqueous solution using the macrocyclic ligand macropa. Upon complexation at room temperature, the [223Ra(macropa)] complex displays a high degree of long-term stability both in vitro under physiological conditions and in vivo in murine models. Although our efforts to target 223Ra to soft-tissue prostate cancers using the PSMA-binding ligand DUPA were unsuccessful, the in vitro and in vivo success of both [223Ra][Ra(macropa)] and [223Ra][Ra(macropa-β-alanine)] opens the path forward for possible cancer-targeted α-particle radiotherapy with 223Ra. Considering that within the radiopharmaceutical community, targeted forms of 223Ra2+ were considered inaccessible due to the lack of biologically stable chelator complexes, our demonstration that macropa and macropa-β-alanine can retain this ion in vivo will motivate continued efforts towards applying this radionuclide for new applications in TAT. We are currently focusing on efforts to develop TAT constructs that are sufficiently stable in vivo for such applications.Conflicts of interest
Wilson and Thiele hold intellectual property rights on macropa; Abou and Thorek have filed provisional patent protection for radium-related production and utilization through WUSTL. Justin Wilson holds equity in Noria Therapeutics, Inc., which has licensed this technology.Acknowledgements
In vivo experiments were conducted following a laboratory protocol approved by IACUC animal welfare of Washington University (DCM), conforming to the recommendations published in Animal Welfare Act and Animal Welfare Regulations (the “Blue Book”) by the USDA animal care. Research reported in this publication was supported by the National Cancer Institute and National Institute of Biomedical Imaging and Bioengineering of the National Institutes of Health under Award Numbers ZIA BC 011800 (FEE), NCI R01CA229893 (DLJT), R01CA201035 (DLJT), R01CA240711 (DLJT), R21EB027282 (JJW), and R01EB029259 (JJW). This research was also supported by a Cottrell Research Scholar Award from the Research Corporation for Science Advancement to JJW. This research made use of the NMR facility at Cornell University, which is supported, in part, by the National Science Foundation under Award CHE-1531632. Isotopes were provided in part through the Department of Energy Isotope Program. We thank the support of the WUSTL EH&S team.Notes and references
- P. G. Kluetz, W. Pierce, V. E. Maher, H. Zhang, S. Tang, P. Song, Q. Liu, M. T. Haber, E. E. Leutzinger, A. Al-Hakim, W. Chen, T. Palmby, E. Alebachew, R. Sridhara, A. Ibrahim, R. Justice and R. Pazdur, Clin. Cancer Res., 2014, 20, 9–14 CrossRef CAS PubMed.
- C. Parker, S. Nilsson D Heinrich, S. I. Helle, J. M. O'Sullivan, S. D. Fosså, A. Chodacki, P. Wiechno, J. Logue, M. Seke, A. Widmark, D. C. Johannessen, P. Hoskin, D. Bottomley, N. D. James, A. Solberg, I. Syndikus, J. Kliment, S. Wedel, S. Boehmer, M. Dall'Oglio, L. Franzén, R. Coleman, N. J. Vogelzang, C. G. O'Bryan-Tear, K. Staudacher, J. Garcia-Vargas, M. Shan, S. Bruland and O. Sartor, N. Engl. J. Med., 2013, 369, 213–223 CrossRef CAS PubMed.
- O. Sartor, R. Coleman, S. Nilsson, D. Heinrich, S. I. Helle, J. M. O'Sullivan, S. D. Fosså, A. Chodacki, P. Wiechno, J. Logue, A. Widmark, D. C. Johannessen, P. Hoskin, N. D. James, A. Solberg, I. Syndikus, N. J. Vogelzang, C. G. O'Bryan-Tear, M. Shan, Ø. S. Bruland and C. Parker, Lancet Oncol., 2014, 15, 738–746 CrossRef CAS PubMed.
- H. Jadvar, S. Challa, D. I. Quinn and P. S. Conti, Cancer Biother. Radiopharm., 2015, 30, 195–199 CrossRef CAS PubMed.
- G. Henriksen, D. R. Fisher, J. C. Roeske, Ø. S. Bruland and R. H. Larsen, J. Nucl. Med., 2003, 44, 252–259 CAS.
- D. S. Abou, D. Ulmert, M. Doucet, R. F. Hobbs, R. C. Riddle and D. L. J. Thorek, J. Natl. Cancer Inst., 2016, 108, djv380 CrossRef PubMed.
- W. Jiang, D. Ulmert, B. W. Simons, D. S. Abou and D. L. J. Thorek, Nucl. Med. Biol., 2018, 62–63, 1–8 CrossRef CAS PubMed.
- Y.-S. Kim and M. W. Brechbiel, Tumor Biol., 2012, 33, 573–590 CrossRef CAS PubMed.
- Y. Dekempeneer, M. Keyaerts, A. Krasniqi, J. Puttemans, S. Muyldermans, T. Lahoutte, M. D'huyvetter and N. Devoogdt, Expert Opin. Biol. Ther., 2016, 16, 1035–1047 CrossRef CAS PubMed.
- M. Makvandi, E. Dupis, J. W. Engle, F. M. Nortier, M. E. Fassbender, S. Simon, E. R. Birnbaum, R. W. Atcher, K. D. John, O. Rixe and J. P. Norenberg, Target. Oncol., 2018, 13, 189–203 CrossRef PubMed.
- A. Morgenstern, C. Apostolidis, C. Kratochwil, M. Sathekge, L. Krolicki and F. Bruchertseifer, Curr. Radiopharm., 2018, 11, 200–208 CrossRef CAS PubMed.
- J. G. Jurcic, Semin. Nucl. Med., 2020, 50, 152–161 CrossRef PubMed.
- C. Kratochwil, F. Bruchertseifer, F. L. Giesel, M. Weis, F. A. Verburg, F. Mottaghy, K. Kopka, C. Apostolidis, U. Haberkorn and A. Morgenstern, J. Nucl. Med., 2016, 57, 1941–1944 CrossRef CAS PubMed.
- C. Kratochwil, U. Haberkorn and F. L. Giesel, Semin. Nucl. Med., 2020, 50, 133–140 CrossRef PubMed.
- M. J. van der Doelen, N. Mehra, I. M. van Oort, M. G. Looijen-Salamon, M. J. R. Janssen, J. A. E. Custers, P. H. J. Slootbeek, L. I. Kroeze, F. Bruchertseifer, A. Morgenstern, U. Haberkorn, C. Kratochwil, J. Nagarajah and W. R. Gerritsen, Urol. Oncol.: Semin. Orig. Invest., 2020 DOI:10.1016/j.urolonc.2020.12.002.
- J. Engle, Curr. Radiopharm., 2018, 11, 173–179 CrossRef CAS PubMed.
- A. Morgenstern, C. Apostolidis and F. Bruchertseifer, Semin. Nucl. Med., 2020, 50, 119–123 CrossRef PubMed.
- E. W. Price and C. Orvig, Chem. Soc. Rev., 2014, 43, 260–290 RSC.
- K. B. Gunn and K. B. Mistry, Plant Soil, 1970, 33, 7–16 CrossRef CAS.
- J.-C. Chao, A. Hong, R. W. Okey and R. W. Peters, Proc. Conf. Hazard. Waste Res.: Bridging Gaps Technol. Cult., 1998, 142–160 CAS.
- M. Gott, J. Steinbach and C. Mamat, Open Chem., 2016, 14, 118–129 CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- N. S. Poonia and A. V. Bajaj, Chem. Rev., 1979, 79, 389–445 CrossRef CAS.
- B. P. Nikolsky, A. M. Trofimov and N. B. Vysokoostrovskaya, Radiochemistry, 1959, 1, 147–154 Search PubMed.
- F. Nelson, R. A. Day and K. A. Kraus, J. Inorg. Nucl. Chem., 1960, 15, 140–150 CrossRef CAS.
- L. Baetslé and E. Bengsch, J. Chromatogr. A, 1962, 8, 265–273 CrossRef.
- T. Sekine, Y. Kawashima, T. Unnai and M. Sakairi, Bull. Chem. Soc. Jpn., 1968, 41, 3013–3015 CrossRef CAS.
- A. V. Matyskin, N. L. Hansson, P. L. Brown and C. Ekberg, J. Solution Chem., 2017, 46, 1951–1969 CrossRef CAS PubMed.
- G. Henriksen, P. Hoff and R. H. Larsen, Appl. Radiat. Isot., 2002, 56, 667–671 CrossRef CAS PubMed.
- X. Chen, M. Ji, D. R. Fisher and C. M. Wai, Inorg. Chem., 1999, 38, 5449–5452 CrossRef CAS PubMed.
- F. W. B. Van Leeuwen, W. Verboom, X. Shi, J. T. Davis and D. N. Reinhoudt, J. Am. Chem. Soc., 2004, 126, 16575–16581 CrossRef CAS PubMed.
- F. W. van Leeuwen, H. Beijleveld, A. H. Velders, J. Huskens, W. Verboom and D. N. Reinhoudt, Org. Biomol. Chem., 2005, 3, 1993–2001 RSC.
- F. van Leeuwen, C. Miermans, H. Beijleveld, T. Tomasberger, J. Davis, W. Verboom and D. Reinhoudt, Environ. Sci. Technol., 2005, 39, 5455–5459 CrossRef CAS PubMed.
- F. W. B. Van Leeuwen, H. Beijleveld, C. J. H. Miermans, J. Huskens, W. Verboom and D. N. Reinhoudt, Anal. Chem., 2005, 77, 4611–4617 CrossRef CAS PubMed.
- F. W. B. van Leeuwen, W. Verboom and D. N. Reinhoudt, Chem. Soc. Rev., 2005, 34, 753–761 RSC.
- M. Freesmeyer, W. Weigand and T. Weisheit, Nuklearmedizin, 2018, 57, 242–246 CrossRef PubMed.
- M. Gott, P. Yang, U. Kortz, H. Stephan, H.-J. Pietzsch and C. Mamat, Chem. Commun., 2019, 55, 7631–7634 RSC.
- D. Bauer, F. Reissig, H.-J. Pietzsch, J. Steinbach and C. Mamat, J. Med. Imaging Radiat. Sci., 2019, 50, S39 CrossRef.
- M. Silindir-Gunay, M. Karpuz and A. Y. Ozer, Cancer Biother. Radiopharm., 2020, 35, 446–458 CrossRef CAS PubMed.
- A. Majkowska-Pilip, W. Gawęda, K. Żelechowska-Matysiak, K. Wawrowicz and A. Bilewicz, Nanomaterials, 2020, 10, 1366 CrossRef CAS PubMed.
- J. V. Rojas, J. D. Woodward, N. Chen, A. J. Rondinone, C. H. Castano and S. Mirzadeh, Nucl. Med. Biol., 2015, 42, 614–620 CrossRef CAS PubMed.
- F. Reissig, R. Hübner, J. Steinbach, H. J. Pietzsch and C. Mamat, Inorg. Chem. Front., 2019, 6, 1341–1349 RSC.
- M. Czerwińska, G. Gracasso, M. Pruszyński, A. Bilewicz, M. Kruszewski, A. Majkowska-Pilip and A. Lankoff, Materials, 2020, 13, 3875 CrossRef PubMed.
- F. Reissig, K. Zarschler, R. Hübner, H. J. Pietzsch, K. Kopka and C. Mamat, ChemistryOpen, 2020, 9, 797–805 CrossRef CAS PubMed.
- W. Gawęda, M. Pruszyński, E. Cędrowska, M. Rodak, A. Majkowska-Pilip, D. Gaweł, F. Bruchertseifer, A. Morgenstern and A. Bilewicz, Nanomaterials, 2020, 10, 2067 CrossRef PubMed.
- P. Suchánková, E. Kukleva, E. Nykl, P. Nykl, M. Sakmár, M. Vlk and J. Kozempel, Nanomaterials, 2020, 10, 1632 CrossRef PubMed.
- A. Roca-Sabio, M. Mato-Iglesias, D. Esteban-Gómez, É. Toth, A. de Blas, C. Platas-Iglesias and T. Rodríguez-Blas, J. Am. Chem. Soc., 2009, 131, 3331–3341 CrossRef CAS PubMed.
- R. Ferreirós-Martínez, D. Esteban-Gómez, É. Tóth, A. de Blas, C. Platas-Iglesias and T. Rodríguez-Blas, Inorg. Chem., 2011, 50, 3772–3784 CrossRef PubMed.
- M. P. Jensen, R. Chiarizia, I. A. Shkrob, J. S. Ulicki, B. D. Spindler, D. J. Murphy, M. Hossain, A. Roca-Sabio, C. Platas-Iglesias, A. de Blas and T. Rodríguez-Blas, Inorg. Chem., 2014, 53, 6003–6012 CrossRef CAS PubMed.
- M. Regueiro-Figueroa, J. L. Barriada, A. Pallier, D. Esteban-Gómez, A. de Blas, T. Rodríguez-Blas, É. Tóth and C. Platas-Iglesias, Inorg. Chem., 2015, 54, 4940–4952 CrossRef CAS PubMed.
- N. A. Thiele, V. Brown, J. M. Kelly, A. Amor-Coarasa, U. Jermilova, S. N. MacMillan, A. Nikolopoulou, S. Ponnala, C. F. Ramogida, A. K. H. Robertson, C. Rodríguez-Rodríguez, P. Schaffer, C. Williams Jr, J. W. Babich, V. Radchenko and J. J. Wilson, Angew. Chem., Int. Ed., 2017, 56, 14712–14717 CrossRef CAS PubMed.
- N. A. Thiele and J. J. Wilson, Cancer Biother. Radiopharm., 2018, 33, 336–348 CrossRef PubMed.
- J. M. Kelly, A. Amor-Coarasa, S. Ponnala, A. Nikolopoulou, C. Williams Jr, N. A. Thiele, D. Schlyer, J. J. Wilson, S. G. DiMagno and J. W. Babich, J. Nucl. Med., 2019, 60, 649–655 CrossRef CAS PubMed.
- E. Aluicio-Sarduy, N. A. Thiele, K. E. Martin, B. A. Vaughn, J. Devaraj, A. P. Olson, T. E. Barnhart, J. J. Wilson, E. Boros and J. W. Engle, Chem.–Eur. J., 2020, 26, 1238–1242 CrossRef CAS PubMed.
- N. A. Thiele, S. N. MacMillan and J. J. Wilson, J. Am. Chem. Soc., 2018, 140, 17071–17078 CrossRef CAS PubMed.
- F. Reissig, D. Bauer, M. Ullrich, M. Kreller, J. Pietzsch, C. Mamat, K. Kopka, H. Pietzsch and M. Walther, Pharmaceuticals, 2020, 13, 272 CrossRef CAS PubMed.
- D. S. Abou, J. Pickett, J. E. Mattson and D. L. J. Thorek, Appl. Radiat. Isot., 2017, 119, 36–42 CrossRef CAS PubMed.
- W. F. Maguire, M. R. McDevitt, P. M. Smith-Jones and D. A. Scheinberg, J. Nucl. Med., 2014, 55, 1492–1498 CrossRef CAS PubMed.
- A. Cortez, A. Josefsson, G. McCarty, A. E. Shtekler, A. Rao, Z. Austin and J. R. Nedrow, Nucl. Med. Biol., 2020, 88–89, 62–72 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Preparation of 223Ra, ligand synthesis and radiolabeling procedures, serum stability and in vivo protocols, spectral characterization of synthesized compounds, details of computational and crystallographic studies, and supporting figures and tables. CCDC 2035004. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc06867e |
| ‡ D. S. A. and N. A. T. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |