
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSymmetry-related residues as promising hotspots for the evolution of de novo oligomeric enzymes†
Jaeseung
Yu
,
Jinsol
Yang
,
Chaok
Seok
and
Woon Ju
Song
 *
*
Department of Chemistry, College of Natural Sciences, Seoul National University, Seoul 08826, Republic of Korea. E-mail: woonjusong@snu.ac.kr
First published on 17th February 2021
Abstract
Directed evolution has provided us with great opportunities and prospects in the synthesis of tailor-made proteins. It, however, often requires at least mid to high throughput screening, necessitating more effective strategies for laboratory evolution. We herein demonstrate that protein symmetry can be a versatile criterion for searching for promising hotspots for the directed evolution of de novo oligomeric enzymes. The randomization of symmetry-related residues located at the rotational axes of artificial metallo-β-lactamase yields drastic effects on catalytic activities, whereas that of non-symmetry-related, yet, proximal residues to the active site results in negligible perturbations. Structural and biochemical analysis of the positive hits indicates that seemingly trivial mutations at symmetry-related spots yield significant alterations in overall structures, metal-coordination geometry, and chemical environments of active sites. Our work implicates that numerous artificially designed and natural oligomeric proteins might have evolutionary advantages of propagating beneficial mutations using their global symmetry.
Introduction
Natural enzymes have evolved throughout numerous rounds of selection.1,2 Accumulating evidence indicates that proteins have adapted to altered chemical environments by sequence modification. For example, it is proposed that the Great Oxygenation Event (GOE) is correlated with the elevated frequency of metalloenzymes with air-stable metal ions.3,4 The introduction of natural and synthetic herbicides and antibiotics also led to the appearance of enzymes that react with these substances, resulting in resistance to these substances.5,6 Analogously, specific chemical pressures can be applied to randomized mutant libraries in a laboratory. Numerous novel proteins have been created, yielding novel catalytic activity,7–10 altered substrate selectivity,11–13 and/or elevated thermal stability.14,15Despite significant progress in protein design and evolution,16–18 there is no standard rule prioritizing the residues for sequence optimization. The amino acid residues in the vicinity of active sites are often the first and primary candidates. B-Factor and sequence conservation analyses have also been carried out to generate focused mutant libraries.19,20 However, our current state-of-the-art understanding of the protein sequence-structure–function relationship is incomplete in that seemingly trivial or distant mutations are essential to induce the desired properties,21,22 while even proximal residues to the active site may cause no detectable changes in structure and function.23 In addition, multiple mutations often exhibit non-additive effects so that iterative mutations and screenings are necessary,24 requiring at least mid to high throughput screening.25–30 Therefore, an alternative guideline to pinpoint key residues for sequence optimization would be advantageous to facilitate enzyme design and to elucidate intertwined protein sequence networks.
In the exploration of this complicated yet, essential question, de novo enzymes could be a versatile model system. Their sequence and structure are not strongly correlated with the nascent function, and thus, enzyme evolution may occur with little underlying interrelation to the intrinsic nature of proteins. Previously, numerous protein scaffolds have been transformed into artificial enzymes, particularly metalloenzymes.31 A large portion of these examples is found as homomeric proteins, having catalytic metal-sites on the protein–protein interfaces.32–34 One of these examples is an artificial metallo-β-lactamase, AB5, where a catalytic center, Zn-OH2/OH unit, was created on the protein–protein interfaces of an α-helical homo-tetramer (Fig. 1a).35 Intriguingly, we found that a seemingly trivial mutation (C96T) yielded substantial structural alterations (Fig. 1b), resulting in the conversion of a tetramer with a large void space into a closely packed one. Besides, hydrolytic activities with ampicillin were enhanced. In contrast, several attempts to optimize K85, E92, Q103, and K104 residues, close to the Zn-active sites (Fig. 1a), exerted no significant enhancement in the hydrolytic activities. Based on these observations, we surmised that C96 residue is a more effective hotspot than four proximal residues (K85, E92, Q103, and K104) because of its location on the C2-rotation axis. If the mutations of residues related to symmetry operations such as rotation may produce significant impacts on overall protein structure and function, possibly using the fluxional protein–protein interface, we further inquired whether other residues on rotational axes can be efficiently targeted for directed evolution of de novo designed oligomeric enzymes, despite being distantly located from active sites and seemingly unrelated to enzyme catalysis.
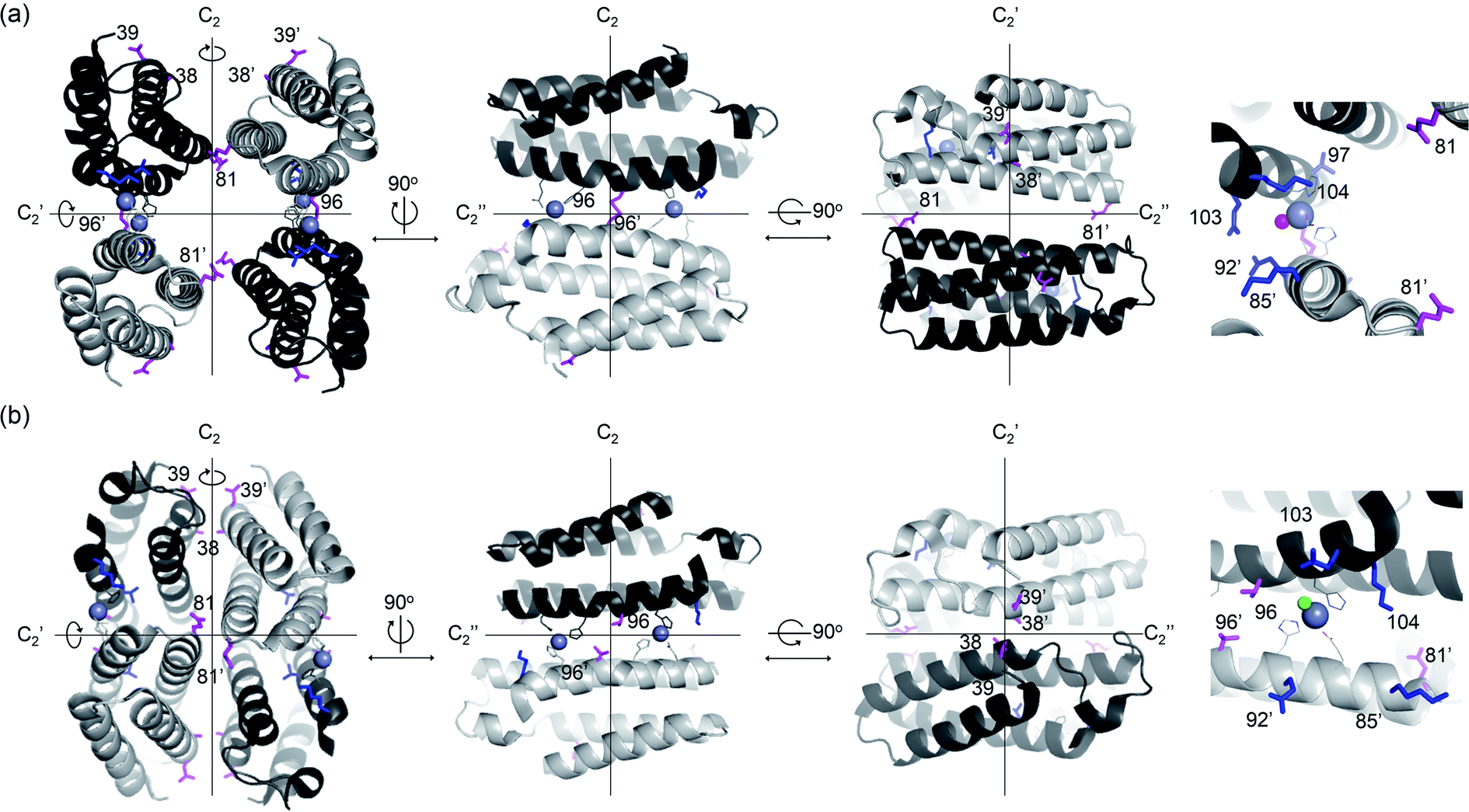 | ||
| Fig. 1 X-ray crystal structures of de novo Zn-dependent β-lactamases related to this study. (a) AB5 protein with C96 residue (PDB 5XZI). (b) C96T variant (PDB 5XZJ). Residues located at the C2 rotational axes, such as 96, 38, 39, and 81, are shown with magenta sticks. Residues located at proximal to the active sites, such as 85, 92, 97, 103, and 104, are shown with blue sticks. Zn ions located at the catalytic sites and metal-ligating residues are shown with grey spheres and lines, respectively. The metal-bound water or chloride ions in catalytic sites are depicted with red and green spheres, respectively. The Zn ions at the structural sites and crystal packing interactions are omitted for clarity. | ||
Results
Construction and screening of symmetry versus proximity-related libraries
To demonstrate whether symmetry-guided residues are potential hotspots for the construction of mutant libraries and screening, we selected three residues located on the C2-rotation axes, A38, D39, and E81 (Fig. 1a). They are located 17.2–24.1 Å from the active site (Table S1a in the ESI†), and even the closest symmetry-related residue to the active site, C96, is not in direct contact with the catalytic unit, a Zn-OH2/OH species. As a parallel study, we also selected two non-symmetry-related but proximal residues to the active site, T97 and K104, which lie at 9.6 and 6.4 Å, respectively. We then individually randomized the symmetry-related and proximal residues by saturated mutagenesis using the NNK codon (Table S2 and Fig. S1 in the ESI†).Both symmetry and proximity-related mutant libraries were screened for the presence of whole-cell hydrolytic activities with an antibiotic β-lactam substrate, ampicillin. For full coverage with a 95% confidence level,36 greater than 94 colonies for each single-site saturated mutant library and 1953 colonies in total were screened (Fig. 2a). For more efficient and quantitative cell-based screening, we slightly modified the method from previous studies35 so that we measured the cell-growth rates upon the addition of a high concentration (10–15 mg L−1) of ampicillin instead of using LB/agar plates containing a relatively low concentration of ampicillin. Then, mutations detrimental to β-lactamase activity would lead to zero or negative cell-growth rates upon the addition of antibiotic substrates, if the cells no longer grow or get ruptured by the cell wall-targeting antibiotics, respectively. In contrast, beneficial mutations would lead to the rates faster than that of the parent protein. When the individual cells of each mutant library were sorted in increasing order of cell-growth rate relative to that of the parent protein, the fitness effect, which can be estimated by the magnitude of alterations upon mutations, irrespective of the occurrence of beneficial or detrimental effects upon mutation, followed the order T97X ≈ K104X ≪ E81X ≤ A38X ≈ C96X < D39X (Fig. 2b and c), where X indicates the 20 randomized amino acid. Notably, the four libraries comprising mutants randomized at the symmetry-related residues, 96, 38, 39, and 81 (Fig. 2b), exhibited considerably greater magnitudes of alterations in cell-growth rate than those of the two libraries saturated at residues 97 and 104 (Fig. 2c), indicating that symmetry-related residues exert greater degrees of perturbation to the protein than proximal residues. The distribution of fitness effects37–39 was also depicted by box charts (Fig. 2c inset), where the mutations at symmetry-related residues exhibit much more outspread cell-growth rates than those of proximal residues. The fastest-growing cells from each library were sequenced, resulting in T97T (parent), K104S, E81G, A38D, C96I, C96K, and D38E mutants.
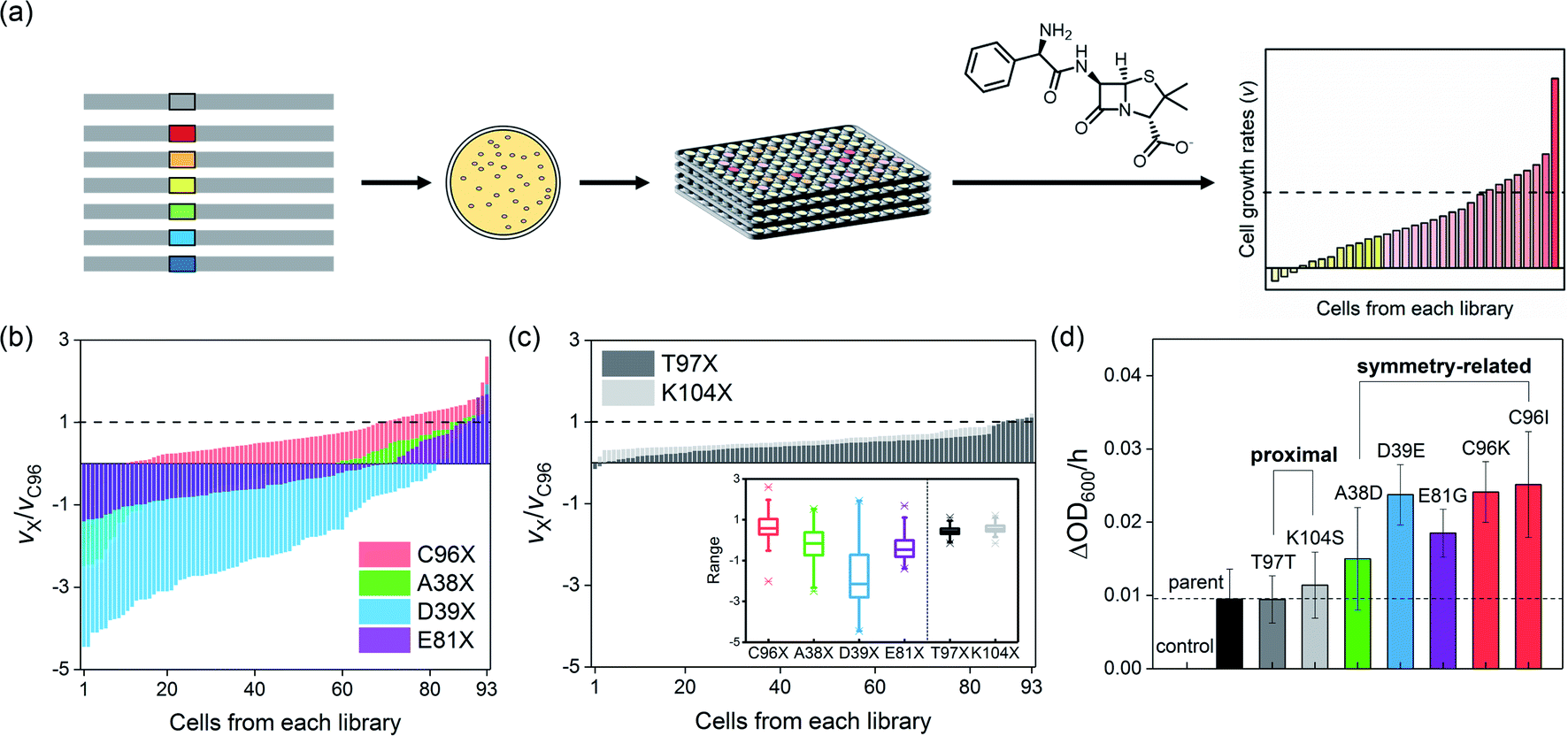 | ||
| Fig. 2 The screening of the first-generation libraries. (a) A scheme for β-lactamase activity-based screening of single-site randomized mutant libraries with ampicillin. Representative screening results of (b) symmetry- and (c) proximity-related mutants. The relative cell-growth rates of 93 colonies to the AB5 protein (C96) are plotted in the increasing order. In the (c) inset, a box chart per each mutant library is included, where the boundaries indicate the 25 and the 75 percentiles, and the lines in the box represent the median values. The caps represent the 1 and 99 percentiles, and whiskers indicate the minimum (below) and maximum (above) cell-growth rates. (d) The cell-growth rates of the best one or two hits from each mutant library. Control and parent indicate C96RIDC1 and AB5 proteins, respectively, where the former exhibits no catalytic Zn-site. The error bars in (d) indicate the standard deviations of three runs of the experiments. | ||
Characterization of the screened single variants
To validate whether the accelerated cell-growth rates were indeed derived from enhanced β-lactamase activities, we measured the cell-growth rates of the sequenced ones, resulting in the order C96 ≈ T97 ≈ K104S ≤ A38D ≤ E81G ≤ D39E ≈ C96K ≈ C96I (Fig. 2d). The order was similar to that of the degrees of perturbations described above, implying that a mutant library with a higher fitness effect is likely to possess more beneficial mutations. Therefore, our results indicate that residues located at symmetry-related positions are promising hotspots for enzyme evolution, potentially exhibiting higher evolvability. These data are consistent with the recent studies of insertion and deletion mutagenesis, where both desirable and strongly deleterious mutations were co-isolated,38 suggesting that a well-focused library might exhibit drastic fitness effects in either directions. In contrast, no hit was observed from the proximity-related library with substantially exceeded cell-growth rates than that of the parent protein; one of the best mutants was indeed the parent (T97), and the other was K104S, and the latter was previously characterized to be only marginally better than the parent protein in terms of catalytic activities.35For more accurate kinetic measurements of the single-site mutants, we carried out in vitro steady-state kinetic analysis of the purified Zn-complexed β-lactamase AB5 variants. By applying various substrate concentrations, the Michaelis–Menten kinetic parameters of the C96K and C96I variants, the best hits from the first-round screening, were obtained (Fig. 3a and b). These variants indeed exhibited up to 2.9- and 2.7-fold increased kcat and kcat/KM values, respectively relative to those of the parent AB5 protein (Table S4 in the ESI†). The results contrasted with the previous attempts to evolve the protein by randomizing the residues at the active sites, 85, 92, 103, and 104.35
 | ||
| Fig. 3 Characterization of the positive hits from the first-round screening. (a) Michaelis–Menten plots and (b) kinetic parameters with ampicillin as a substrate. (c) Michaelis–Menten plots and (d) kinetic parameters with carbenicillin as an alternative β-lactam substrate. The error bars indicate the standard deviations of three runs of the experiments. | ||
Intriguingly, we noted that the enhanced β-lactamase activities were primarily derived from the elevated kcat rather than lowered KM, where the latter value is related to the substrate-binding affinity or dissociation constant (Kd). These results implicate that the single mutations selectively accelerated turnover rates rather than increasing substrate affinity. The molecular origin of acceleration contrasted previous work with an analogous protein, AB3, where enhanced catalytic activities were derived from elevated substrate-binding affinity.40 To identify whether the discrete enhancement mechanism was related to the modified screening conditions using higher concentrations of ampicillin, we reduced the concentration of ampicillin (1.8 mg L−1) for the screening of the C96X mutant library (Fig. S4b in the ESI†). While C96I is still a positive readout, the parent (C96) and C96V became the dominant hits. When we further modified the screening conditions by replacing ampicillin with carbenicillin, another β-lactam antibiotic, at low concentration (Fig. S4d in the ESI†), cells containing C96V and C96L grew relatively fast, implying that screening conditions, such as substrate concentrations and structures, alter the selected readouts.
More importantly, the kinetic parameters of the newly detected variants, C96V and C96L, exhibited substantially lower KM values for ampicillin and/or carbenicillin, respectively, than did the parent protein or previously detected active variants, C96I and C96K (Fig. 3b and d). These data indicated that the tighter substrate-binding affinity obtained by C96V and C96L mutations is likely to be beneficial when low concentrations of β-lactams are applied for activity-based screening. In contrast, C96I and C96K variants with higher turnover rates and catalytic efficiency are likely to be more advantageous when the substrate concentration and binding affinity are no longer limited.
The correlation between the kinetic parameters and the screening conditions was further supported by measuring the substrate-binding affinity or dissociation constant (Kd) directly from intrinsic fluorescence assays (Fig. S5 and Table S5 in the ESI†). The Stern–Volmer plots41,42 of the C96 variant demonstrate that the thermodynamic parameters were consistent with the KM values from kinetic analysis, suggesting that the screening conditions and the chemical properties of the selected products might have promoted specific evolutionary trajectories beneficial for the applied chemical pressures.
To further monitor the effects of single mutations in symmetry-related residues, X-ray crystal structures of three variants from the first-round screening, C96I, C96K, and C96V, were determined (Fig. 4 and Table S6 in the ESI†). The C96I, C96K, and C96V proteins were isolated as tetramers, consistent with the oligomeric states determined in solution (Fig. S6a in ESI†). They were also similar in that all tetramers possess two sets of Zn-binding sites, structural and catalytic ones (Fig. S7 and S8 in the ESI†). Notably, N-terminal residues, surface-exposed acidic residues, and/or H59 and E81 also formed Zn-binding sites, although they are likely to be catalytically irrelevant and generated due to the crystal packing interactions in the presence of excess Zn ions for crystallization (Fig. S9 in the ESI†).
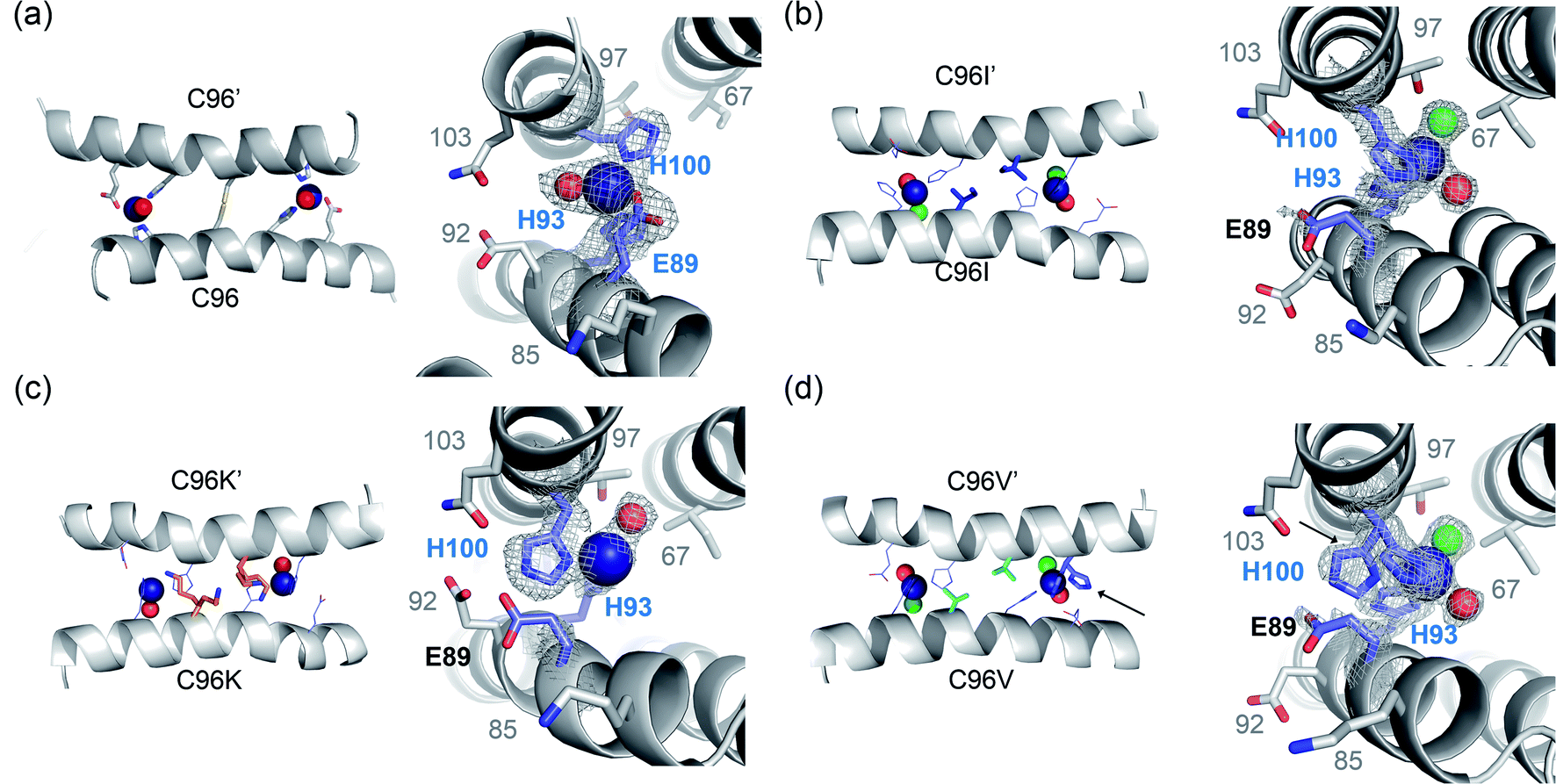 | ||
| Fig. 4 X-ray crystal structures of C96 variants from the first-round screening. The catalytic Zn sites of (a) the parent protein, AB5 (PDB 5XZI) (b) C96I (c) C96K and (d) C96V variants. Zn ion and metal-binding residues are shown with navy spheres and navy sticks, respectively. Metal-bound water molecules and chloride ions are shown with red and green spheres, respectively. Two rotamers of H100 residue in C96V protein are highlighted with black arrows. The first metal-coordination sites are shown with 2Fo–Fc electron density contoured at 1.0σ overlaid. | ||
All three variants were closely packed tetramers, similar to the C96T mutant (Fig. 1b) and different from the parent protein with the C96 residue (Fig. 1a). Consequently, both the structural and catalytic Zn-binding sites were drastically altered by the single mutations. In particular, the directionality of the catalytic Zn-OH2/OH species in all C96 variants was flipped towards I67 and T97, instead of E92 and Q103 (Fig. 4). In addition, the first coordination sphere of the Zn center was greatly altered. Whereas the C96 parent protein exhibits a catalytic Zn ion ligated by 2His/1Glu (E89, H93, H100) and a buffer-derived molecule/ion (Fig. 4a), E89 was no longer coordinated to the catalytic Zn ions in the C96I variant (Fig. 4b, S8a and b in the ESI†). Instead, solvent-derived anions or molecules were bound to Zn, creating the distinct first metal-coordination spheres, the secondary microenvironments, and substrate-binding pockets, which would have enhanced metal-dependent enzyme catalysis. Notably, significantly shorter distances between Zn ions and ligating N atoms of H93 and H100 were observed in C96I protein (1.79–2.03 Å) relative to the parent protein (2.17–2.34 Å) (Table S7 in the ESI†). In addition, two discrete angles of εN (H93)–Zn–δN (H100), 91.3° and 122.7°, were observed, one of which significantly deviated from the parent protein (92.8°), creating two asymmetric bis-His motifs for hydrolytically active Zn-centers.
The C96K mutation also altered the first coordination spheres of the catalytic Zn-binding sites (Fig. 4c). The coordination spheres were composed of H93 and H100 residues with one water molecule, and Zn-OH2/OH was pointed towards I67 and T97. E89 was also dissociated from the Zn ion and was hydrogen-bonded to E92, Q103, and one ordered water molecule near the catalytic Zn-binding sites (Fig. S8c and d in the ESI†). Two discrete angles of two histidine residues and zinc ion were again observed as 103.3 and 125.1°, even further deviated from those in the parent protein. In addition, two conformations of the C96K side chains were observed, forming hydrogen bonds with H93, possibly tuning the chemical properties of the Zn site, such as the pKa value of catalytically critical residues, the nucleophilicity of Zn-OH species, and Zn-binding affinity.
In the C96V protein, Zn ions were ligated by H93 and H100, and E89 was again no longer directly coordinated to Zn upon C96 mutation (Fig. 4d). Instead, E89 was hydrogen-bonded to a metal-bound water molecule or was pointed away from the metal-coordination site. Consequently, two solvent-derived molecules, such as Cl− and H2O, were coordinated to the Zn ion in a tetrahedral geometry. In addition, one of the two H100 residues in the asymmetric unit was in two discrete rotameric states, which was not observed in other variants (Fig. S8e and f in the ESI†). As a result, the angle of εN (H93)–Zn–δN (H100) became 95.3° on one side and 90.2° and 148.6° on the other side, resulting in the mutant possessing either nearly symmetric or the most asymmetric Zn sites on the α-helical protein–protein interface.
To explore whether these geometric perturbations are related to catalytic activities, we carried out substrate-docking simulations using Galaxydock43,44 (Fig. S10 in the ESI†). The data suggest that the discrete Zn-binding sites induced by C96 mutations may favor alternative modes in substrate-binding. Because C96I, C96K, and C96V variants exhibit the unique kinetic parameters or substrate-binding affinity, even the slightly modified Zn-coordination sites might not be an artifact of the crystalline packing interactions but related to the dynamic snapshots of the protein structure in solution. Then, these results indicate that seemingly trivial single mutations on the rotational axis can modify the chemical properties of catalytically active Zn sites.
After structural characterization, we further measured the pH-dependent hydrolytic activities to estimate the pKa value of catalytically critical residues or Zn-OH2 species (Table S8 and Fig. S11 in the ESI†). The parent protein exhibited a pKa value of 8.4(1),35 whereas those of C96I, C96K, and C96V were estimated to be 8.6(1), 9.3(1), and 9.2(2). These data indicate that a single mutation at a symmetry-related position alters the net concentration of catalytically active species by tuning the chemical environments of catalytic sites.
Construction and screening of second-, third-, and fourth-round libraries
Because even a single mutation at a symmetry-related site gave rise to a significant modification in catalytic activities, protein structures, and biochemical properties, we iteratively constructed second-round libraries with C96I as the template. Again, we constructed two groups of single-site mutant libraries, one by altering symmetry-related residues, such as A38, D39, and E81, and the other by randomizing proximity-related residues, such as T97 and K104. For full coverage with a 95% confidence level,36 greater than 94 colonies for each single-site saturated mutant library and 1395 colonies in total were screened. The variation in the relative cell-growth rate upon single mutation, or fitness effect, was observed to follow the order of K104X ≤ T97X ≪ D39X ≤ E81X < A38X (Fig. 5a and b), as also represented by box charts (Fig. 5b inset), again demonstrating that mutations of residues located on the C2 rotational axes, such as A38, D39, and E81, give rise to more drastic impacts on cell-growth rate than do T97 and K104 mutations. When one or two cells with the highest growth rates from each library were sequenced, A38S, A38I, D39E, E81N, T97M, and T97S mutants were obtained, while in the K104X library, only the template, C96I variant having K104 residue, was the most active cell. The measured specific whole-cell activities of the screened cells were in the following order: T97M ≈ T97S ≈ C96I ≪ E81N ≤ D39E ≤ A38I ≤ A38S (Fig. 5c and S12 in the ESI†).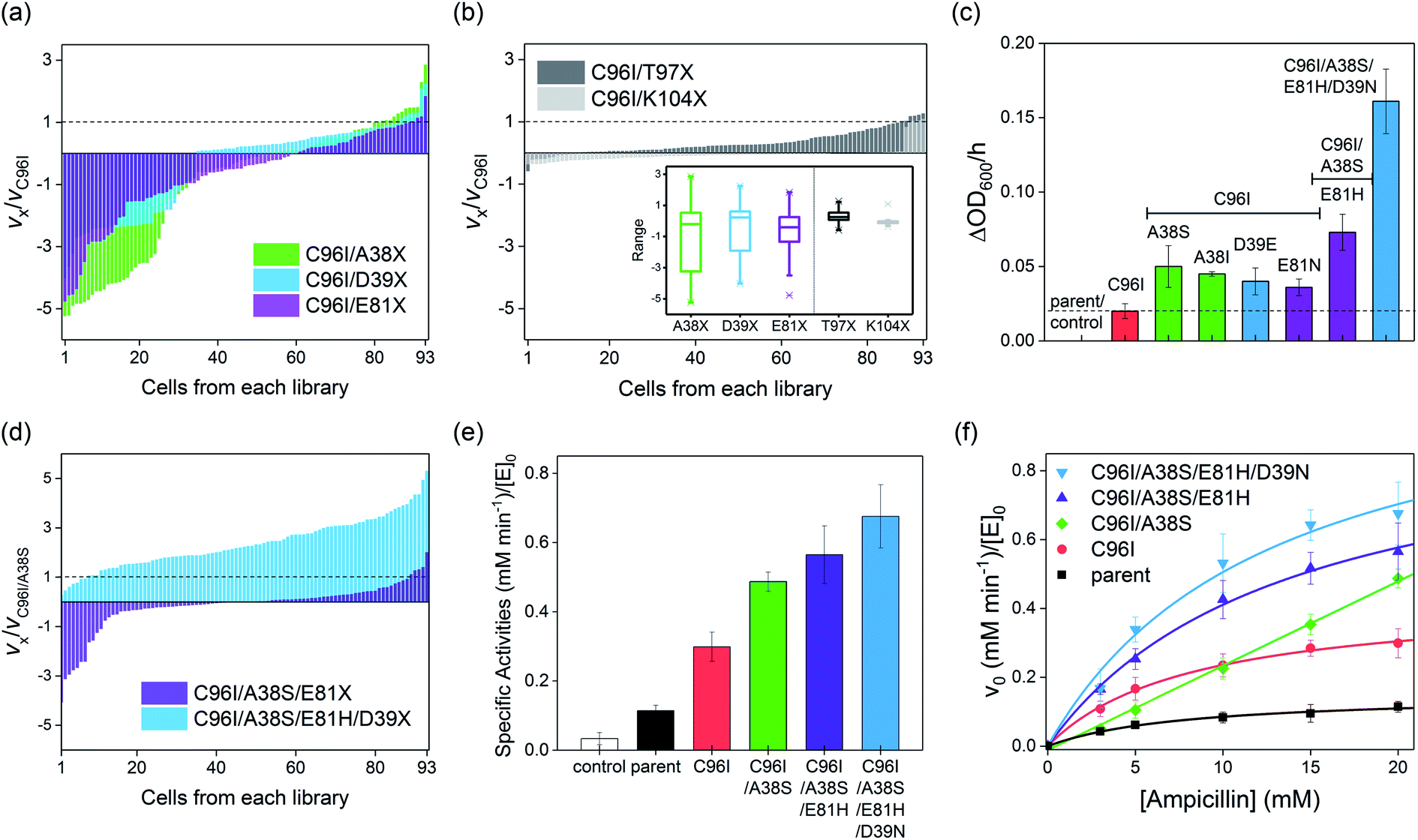 | ||
| Fig. 5 The screening and characterization of the hits from the second to fourth rounds of screening. The second round of representative results with (a) symmetry- and (b) proximity-related mutant libraries. The relative cell-growth rates to C96I variant are plotted for each mutant library. In the (b) inset, box chart per each mutant library was included. (c) The cell-growth rates of the best hits from the second-round library in (a). (d) The third and fourth rounds of the screening with symmetry-related mutant libraries. (e) Specific in vitro activities of the selected variants. (f) Michaelis–Menten kinetic analysis of the best hits. The error bars in (c), (e), and (f) indicate the standard deviations of three runs of the experiments. | ||
Because C96I/A38S double variants yielded the highest catalytic efficiency, we constructed a third round of the mutant library by randomizing the E81 residue of C96I/A38S as the template (Fig. 5d). The outputs exhibit variations in cell-growth rates by ca. 3- and 5-fold in positive and negative directions, respectively, and the best-hit was sequenced to be E81H, resulting in a triple variant, C96I/A38S/E81H (Fig. 5c). Then, we further randomized and screened the last symmetry-related residue, D39. The randomized single-mutant library yielded significantly altered cell-growth rates by up to ca. 5-fold relative to that of the C96I/A38S double mutant (Fig. 5d). The sequencing results indicate that the quadruple variant, C96I/A38S/E81H/D39N, displayed the fastest cell-growth rate, resulting in 8.1-, 3.2-, and 2.2-fold enhancements relative to that of the best hit from the first, second, and third round of screening, C96I, C96I/A38S, and C96I/A38S/E81H, respectively. These data suggest that the iterative sequence optimizations were carried out efficiently by targeting symmetry-related residues.
Characterization of the second-, third-, and fourth-round mutants
To demonstrate that the increased cell-growth rates in the presence of antibiotic substrates are derived from enhanced β-lactamase activities, we isolated the best hits from the second, third, and fourth rounds of screening. Then, we measured the steady-state specific activities with 20 mM ampicillin (Fig. 5e). The specific β-lactamase activities consecutively increased, yielding a 5.9-fold increase relative to that of the parent (AB5 protein) upon quadruple mutations. In contrast, proximity-related mutations attempted in the first and second rounds of optimizations gave rise to negligible enhancements in hydrolytic activities, consistent with the in vivo cell-based activities.The Michaelis–Menten kinetic analysis of the best hits from each round was also carried out (Fig. 5f; Table S8 and Fig. S14 in the ESI†). The overall activities were consecutively elevated throughout every round of evolution, resulting in higher kcat and kcat/KM values than those of the variants from the previous rounds of screening. Relative to the uncatalyzed rate (kuncat) of 3.0(1) × 10−6 min−1, the activities of the quadruple mutant accounted for 4.0(7) × 105 and 3.3(3) × 107 M−1 in terms of rate enhancement (kcat/kuncat) and catalytic proficiency (kcat/KM/kuncat), respectively (Table S8 and Fig. S15 in the ESI†). Notably, no improvement in KM was observed throughout the evolution, and C96I/A38S even lost a saturation behavior with increasing substrate concentration, resulting in a second-order rate constant (k2), instead of kcat/KM. The weaker substrate-binding affinities of the screened mutants might be attributed to the screening conditions, where substantially high concentrations of antibiotics were applied. Then, the microscopic catalytic properties of the evolved variants might be the result of β-lactamases discretely evolved to elevate turnover rates rather than to lower substrate-binding affinity.
In addition, we measured the pH-dependent hydrolytic activities of the quadruple variant. The pKa value was 8.4(1) (Table S9 and Fig. S16 in the ESI†), which differs by 0.6 from that of C96I/A38S. Notably, we observed an inverse correlation between the pKa values and kinetic parameters (such as kcat and kcat/KM) of the mutants having compact tetrameric structures, C96T before the screening, C96I, C96I/A38S, C96I/A38S/E81H, and C96I/A38S/E81H/D39N from evolution, implying that distant mutations increased the net concentration of catalytically essential species, such as Zn-OH species, over that of Zn-OH2.
The structural features of the variants were evaluated by size exclusion chromatography. The parent protein exhibits two disulfide bonds formed via four C96 residues in tetramer form,35 which enables the formation of a tetramer even in the absence of Zn ions at the protein–protein interfaces. Upon the removal of the disulfide bond, Zn-free apo-protein was isolated as a mixture of monomers and dimers, similar to C96T35 (Fig. S17 in the ESI†). The isolated mutants from the screening, C96V, C96K, C96I/A38S, C96I/A38S/E81H, and C96I/A38S/E81H/D39N, exhibit analogous Zn-dependent oligomerization, exclusively forming tetramers even after a series of mutations on the protein–protein interfaces. Intriguingly, exceptions were detected for the C96I and C96L variants from the first-round of screening. They form nearly exclusively tetramers even in the absence of Zn ions. The hydrophobic residues in the two opposing α-helical domains are analogous to those in leucine zippers,45,46 where repeated hydrophobic amino acids form noncovalent interactions, inducing oligomerization. Although it is unclear whether the enhanced protein–protein interfaces kinetically promote the in vivo activity of C96I and C96L by forming tetramers prior to metal binding and increasing the effective concentrations of catalytically active species inside the cells, the results indicate that the impact of single-site mutations at symmetry-related spots is indeed substantial in both structural and functional aspects, and can therefore give rise to significant impacts on protein evolution.
Finally, one of the evolved variants was characterized by X-ray crystallography (Fig. 6). The C96I/A38S variant from the second-round screening was a compact tetramer, similar to the C96I, C96K, and C96V variants. The catalytic Zn-binding sites were also similar in that they are composed of Zn ions coordinated to H93, H100 (Zn–N = 2.0–2.3 Å), and one or two water molecules. Notably, the angle of εN (H93)–Zn–δN (H100) was measured to be 92.2° and 94.5°, resulting in more symmetric metal-centers located at the α-helical domains than in the C96I single variant. The subtle yet significant geometric perturbation at the first metal-coordination sphere, induced by A38S mutation, might be associated with the enhanced catalytic activities, possibly by adjusting the nucleophilicity of the Zn-OH species, optimizing noncovalent interactions with the secondary coordination spheres, and/or shaping the internal active site pocket. Notably, A38S is 17 Å distant from the catalytic Zn center. Therefore, the mutation effect exerted at the rotational axis is likely to be transferred through fluxional protein–protein interfaces, suggesting that distant and beneficial mutations can be created efficiently by targeting symmetry-related residues for protein evolution.
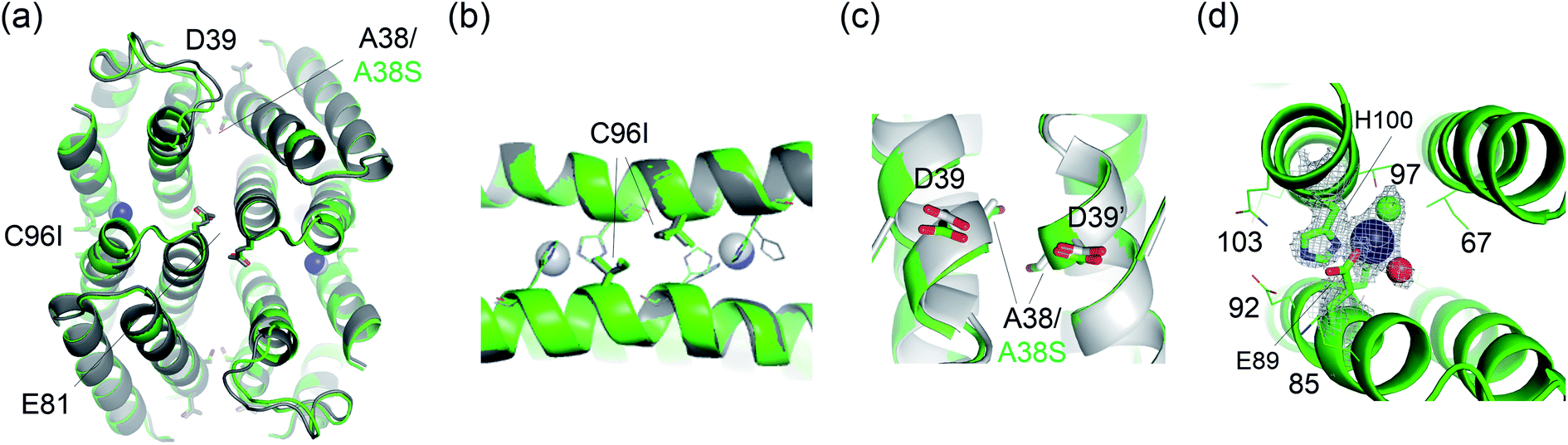 | ||
| Fig. 6 X-ray crystal structure of C96I/A38S protein. (a–c) The superimposed structures of C96I/A38S protein with C96I variant, colored in green and grey, respectively. Zn ions in the catalytic sites are shown with navy and grey spheres, respectively. Symmetry-related residues located at the C2 rotational axes, A38 or A38S, D39, E81, and C96I, are shown with sticks. (d) The enlarged catalytic Zn site in C96I/A38S double variant. A Zn ion, a metal-bound water molecule, and a chloride ion are shown with navy, red, and green spheres, respectively. Metal-binding (H93 and H100) and weakly interacting residue (E89) are shown with sticks. The first metal-coordination site in (d) is shown with 2Fo–Fc electron density contoured at 1.0σ overlaid. | ||
Conclusions
We demonstrated that mutation of residues located on rotational axes can give rise to substantial alterations in the structure and function of a de novo metallo-β-lactamase. Because numerous artificial metalloenzymes and even natural proteins are homo-oligomers possessing rotational axes, targeting symmetry-related residues can be a novel strategy in the construction of well-focused mutant libraries. In addition, this approach is orthogonal to the commonly adapted criteria used in the design of targeted libraries, such as distance from active sites, B-factor, and sequence conservation, therefore providing diverse routes to explore protein sequence networks. Our results might be related to the evolutionary advantages of protein oligomers that might multiply and propagate mutation effects as well.47–50 In addition, the in vitro characterization of the outputs indicates that discrete evolutionary chemical pressures lead to the emergence and divergence of proteins with discrete catalytic properties. The chemical pressure-dependent evolution suggests that artificial enzymes or whole-cell biocatalysts can be created to aim for specific kinetic and/or thermodynamic properties at a microscopic level, increasing the accuracy and predictability of directed evolution. Therefore, a more efficient design strategy for mutant libraries and screening might expedite enzyme evolution and our exploration of protein sequence–structure–function relations with greater accuracy.Experimental
Construction of mutant libraries
Saturated mutagenesis was carried out using primers containing the NNK degenerate codon as described previously.51 PCR was performed using custom-designed primers (Table S2†). After Dpn I (Enzynomics) digestion for 1.5 h at 37 °C, the PCR mixtures were transformed to DH5α E. coli competent cells. Greater than 100 colonies were selected for each single-site randomized library, and they were inoculated to LB medium containing 50 mg L−1 kanamycin. After the overnight cell-growth at 37 °C, plasmids were extracted using a mini-prep kit and sent out for sequencing (Macrogen or Bionics). When the selected position was not fully randomized, additional PCRs with the redesigned primers were performed. Representative sequencing chromatograms for each library are shown in Fig. S1 in the ESI.†Next-generation-sequencing of C96X libraries
To validate the quality of the randomized mutant libraries, we carried out next-generation-sequencing (NGS) of the C96X library as a representative (Macrogen). Amplicon samples were prepared by attaching sequencing primers and the pre-adaptor sequences (Table S3 and Fig. S2a in the ESI†). The size of the PCR products was validated by 1% agarose gel electrophoresis, prior to the sequencing (Fig. S2b in the ESI†). The predicted and NGS results are shown in Fig. S2c.†Screening of the mutant libraries
The plasmid mixtures were transformed to BL21 (DE3) E. coli competent cells containing the cytochrome c maturation cassette (ccm)52 and were grown overnight on LB/agar containing 50 mg L−1 kanamycin and 30 mg L−1 chloramphenicol. Over 100 colonies for each single-site randomized library were inoculated in a 96-well microplate containing 200 μL of LB medium with the antibiotics described above. After overnight growth at 37 °C with constant shaking at 200 rpm, the cultures were diluted 10-fold with 200 μL of LB medium containing 50 mg L−1 kanamycin, 35 mg L−1 chloramphenicol, and 50 μM ZnCl2. Increasing concentrations of ampicillin (10 mg L−1 in the first round, 15 mg L−1 in the second round, and 17.5 mg L−1 in the third and fourth round of the screening) were added throughout the screening when the optical density at 600 nm (OD600) reached approximately 0.4. Notably, the initial cell density was critical in the determination of relative cell-growth rates as illustrated by the Pearson correlation coefficient,53 and the value of our screening condition (−0.08) was sufficiently lower than the reported threshold, −0.3 (Fig. S3 in the ESI†). After monitoring the cell-growth rates at 37 °C for 3 h, the fastest-growing cells were selected for sequencing (Fig. S4 and S12 in the ESI†). At least 2–4 sets of a 96-well plate with the colonies of 93 mutants and 3 parent or template proteins were screened for each library and representative results are included in Fig. 2 and 5.Protein expression, purification, and biochemical characterization
Protein expression and purification of the positive hits were carried out as described previously.35 In short, pET20b(+) plasmid for encoding the ab5 gene was transformed to BL21 (DE3) competent cells. The cells were grown in LB medium with 150 rpm shaking at 37 °C for 18 h. Cell pellets were harvested by centrifugation at 5000 rpm (4715 g) at 7 °C for 10 min. After sonication of the cell pellets in 10 mM sodium phosphate (NaPi) pH 8.0 buffer with pulse on/off = 9/9 s for 40 min in an iced bath, HCl (25%) solution was added to the cell lysates up to pH 5.0. After centrifugation at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm (18800g) at 7 °C for 30 min, the supernatants were adjusted to pH 8 by adding 2 M NaOH solution. Then, the solutions were manually loaded onto a Q-sepharose column pre-equilibrated with 10 mM NaPi pH 8.0 buffer. By applying the step gradients of 0–1 M NaCl in NaPi buffer, red fractions due to the presence of heme cofactor were collected and concentrated using stirred cells (Amicon) with 10 kDa cutoff membranes. Then, the samples were loaded onto a HiTrap Q HP anion exchange column (GE Healthcare Life Sciences) pre-equilibrated with 10 mM NaPi pH 8.0 buffer at 4 °C using FPLC (ÄKTA pure). A linear gradient of 0–1 M NaCl was applied, and the fractions with A415/A280 ≥ 4 measured using a UV-Vis spectrophotometer (Agilent Cary 8454) were collected using and concentrated.
000 rpm (18800g) at 7 °C for 30 min, the supernatants were adjusted to pH 8 by adding 2 M NaOH solution. Then, the solutions were manually loaded onto a Q-sepharose column pre-equilibrated with 10 mM NaPi pH 8.0 buffer. By applying the step gradients of 0–1 M NaCl in NaPi buffer, red fractions due to the presence of heme cofactor were collected and concentrated using stirred cells (Amicon) with 10 kDa cutoff membranes. Then, the samples were loaded onto a HiTrap Q HP anion exchange column (GE Healthcare Life Sciences) pre-equilibrated with 10 mM NaPi pH 8.0 buffer at 4 °C using FPLC (ÄKTA pure). A linear gradient of 0–1 M NaCl was applied, and the fractions with A415/A280 ≥ 4 measured using a UV-Vis spectrophotometer (Agilent Cary 8454) were collected using and concentrated.
Then, the protein sample was loaded onto a HiLoad 16/600 Superdex 75 pg column (GE Healthcare Life Sciences) pre-equilibrated with 20 mM Tris/HCl pH 7.0 buffer with 150 mM NaCl. After elution, pure fractions were collected and concentrated. To prepare the metal-free, apo protein, 10-fold EDTA to the protein was treated for 1 h at 4 °C, and the excess EDTA was removed by 10DG desalting column (Biorad) chromatography. Metal content was measured by a colorimetric assay using 4-(2-pyridylazo) resorcinol (PAR) as described previously.54,55 The purified metal-free, apo-protein was concentrated up to 1.0 mM and stored at −80 °C until further use. The protein concentration was determined by using ε415 nm = 148000 cm−1 M−1.
In vitro hydrolytic activity assay with ampicillin
In vitro hydrolytic activities of Zn-complexed positive hits were measured by time-dependent HPLC analysis as reported previously.35,40 The reaction was initiated by the addition of either ampicillin or carbenicillin into Zn2+-bound protein (7 μM in 100 μL of reaction volume) in 100 mM MOPS pH 7.0 at 25 °C. The reaction mixture (2 μL) was injected onto the C18 column in HPLC (Agilent Infinity 1260) and eluted with a linear gradient, starting from 90% H2O/10% CH3CN to 10% H2O/90% CH3CN for 20 min. Trifluoroacetic acid (TFA) was added to the elution solvents, H2O and CH3CN, at 0.05% and 0.1% (v/v) as final concentrations, respectively. The substrate consumption rates were measured by monitoring the absorbance changes at 220 nm. The initial rates were determined from the reactions with various concentrations of the applied substrate. Michaelis–Menten parameters such as kcat and kcat/KM or second-order rate constants, k2, were determined from iterative non-linear or linear plots, respectively, using Origin 2016 software (Fig. 3, 5f; Tables S4, S8 and Fig. S15 in the ESI†).The pH-dependent hydrolytic activities of Zn-complexed positive hits were monitored by measuring the specific activity with 3 mM ampicillin under various pH conditions; 100 mM MOPS buffer for pH 7.0–7.5 and 100 mM sodium borate buffer for pH 8.0–10.0 ranges (Table S9, Fig. S11 and S16 in the ESI†). Then, the following equation was used for iterative non-linear fitting using Origin 2016 software; y = (kmax × 10(pH−pKa))/(1 + 10(pH−pKa)).
Determination of substrate-binding affinities with intrinsic fluorescence
Intrinsic tryptophan and tyrosine fluorescence changes were monitored using a microplate reader (Biotek Synergy H1) at 25 °C. The parent protein (AB5 with C96 residue) possesses a tryptophan (W66) and a tyrosine (Y105) in the vicinity of the active site (Fig. S5a in the ESI†). Various concentrations of ampicillin or carbenicillin (0–10 mM) were added to Zn2+-bound protein complexes and were incubated for 3 min either in 100 mM MOPS pH 7.0. Notably, ampicillin and carbenicillin have no considerable absorption at 260 and 295 nm, and the inner filter effect can be negligible up to 4 mM for tyrosine and 10 mM for tryptophan residues (Fig. S5b in the ESI†). The intrinsic tyrosine and tryptophan fluorescence changes were observed at 315 nm and 350 nm, respectively, upon the addition of ampicillin when 260 nm and 290 nm excitation were applied, respectively (Fig. S5c in the ESI†). The relative fluorescence changes from tyrosine were fit to a linear Stern–Volmer equation, F0/F = 1 + (Ka × [substrate]). For tryptophan fluorescence changes, a non-linear modified Stern–Volmer equation, F0/(−ΔF) = 1/(Ka × fa × [substrate]) + (1/fa) was applied to account for the presence of multiple tryptophan residues, where Ka and fa values represent the Stern–Volmer constant and accessible fraction of tryptophan, respectively (Table S5, Fig. 5d and e in the ESI†).Determination of oligomeric states of the proteins
The oligomerization states of the parent protein and the evolved variants were determined by size exclusion chromatography. The proteins (150 μM) were loaded on a HiLoad 16/600 Superdex 75 pg column (GE Healthcare and Life Sciences). The retention volume for the monomer, dimer, and tetramer was determined by a linear fit analysis (Fig. S6a in the ESI†). The oligomeric states of the best hits from each round of screening, C96I/A38S, C96I/A38S/E81H, and C96I/A38S/E81H/D39N variants, were also similarly determined by size exclusion chromatography (Fig. S17 in the ESI†).Crystallization and determination of X-ray structures
For crystallization, additional ZnCl2 (0.2 equiv. per monomer) was added to the Zn-complexed tetrameric proteins. Single crystals of Zn2+-bound C96X variants (X = I, K, and V) and C96I/A38S double mutants were obtained by sitting-drop vapor diffusion at room temperature using 1 μL of protein stock (1.0 mM) and 0.5 μL of precipitants listed in Table S6.† The data were collected in the Pohang Accelerator Laboratory (PAL) using either 7A or 11C beamline. Diffraction data were processed with HKL 200056 and CCP4i.57 Molecular replacements were performed with Molrep58 by using a structure of C96RIDC1 monomer (PDB 3IQ6) as a search model. Rigid-body and restrained refinements were carried out using REFMAC559 along with manual inspection and refinements with COOT60 (Table S6†). The resolutions of C96I, C96K, C96V, and C96I/A38S protein structures were determined as 1.98, 2.35, 2.00, and 2.45 Å, respectively. Structural and catalytic Zn-binding sites in C96X variants (X = I, K, and V) are shown in Fig. S7–S9.† The geometric parameters of the Zn-binding sites are summarized in Table S7 in the ESI.†
Docking simulation using GalaxyDock
A flexible protein–ligand docking program GalaxyDock43 was used to predict the binding poses and energies of ampicillin and carbenicillin to the C96X variants. To simulate catalytic binding modes, a close distance between the catalytic water oxygen and the β-lactam ring carbonyl carbon of the substrate was favored during docking by adding a harmonic penalty for a large distance. Side-chains in the active site were allowed to move except for the Zn-coordinating side chains. After filtering out poses with oxygen-carbon distances longer than 3.5 Å, binding stabilities of the predicted binding poses were estimated using the GalaxyDock BP2 score.44 Predicted binding poses are displayed in Fig. S10 in the ESI.†Author contributions
J. Y. and W. J. S. conceptualized the project, J. Y. performed biochemical experiments, J. Y. and C. S. carried out docking simulations, and J. Y. and W. J. S. wrote the original draft, reviewed, and edited the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Creative-pioneering researchers program from Seoul National University (SNU), the National Research Foundation from Korea government (NRF-2019R1C1C1003863), and the Collaborative Genome Program of the Korea Institute of Marine Science and Technology Promotion (KIMST) funded by the Ministry of Oceans and Fisheries (MOF) (No. 20180430).References
- D. Davidi, L. M. Longo, J. Jablonska, R. Milo and D. S. Tawfik, Chem. Rev., 2018, 118, 8786–8797 CrossRef CAS
.
- N. Tokuriki and D. S. Tawfik, Science, 2009, 324, 203–207 CrossRef CAS
- A. D. Anbar, Science, 2008, 322, 1481 CrossRef CAS
- C. L. Dupont, A. Butcher, R. E. Valas, P. E. Bourne and G. Caetano-Anollés, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 10567 CrossRef CAS
- J. L. Seffernick and L. P. Wackett, Biochemistry, 2001, 40, 12747–12753 CrossRef CAS
- M. C. Orencia, J. S. Yoon, J. E. Ness, W. P. C. Stemmer and R. C. Stevens, Nat. Struct. Biol., 2001, 8, 238–242 CrossRef CAS
- B. Seelig and J. W. Szostak, Nature, 2007, 448, 828 CrossRef CAS
- D. Rothlisberger, O. Khersonsky, A. M. Wollacott, L. Jiang, J. DeChancie, J. Betker, J. L. Gallaher, E. A. Althoff, A. Zanghellini, O. Dym, S. Albeck, K. N. Houk, D. S. Tawfik and D. Baker, Nature, 2008, 453, 190–195 CrossRef
- S. Studer, D. A. Hansen, Z. L. Pianowski, P. R. E. Mittl, A. Debon, S. L. Guffy, B. S. Der, B. Kuhlman and D. Hilvert, Science, 2018, 362, 1285 CrossRef CAS
- M. Jeschek, R. Reuter, T. Heinisch, C. Trindler, J. Klehr, S. Panke and T. R. Ward, Nature, 2016, 537, 661–665 CrossRef CAS
- M. A. Siddiq, G. K. A. Hochberg and J. W. Thornton, Curr. Opin. Struct. Biol., 2017, 47, 113–122 CrossRef CAS
- O. May, P. T. Nguyen and F. H. Arnold, Nat. Biotechnol., 2000, 18, 317–320 CrossRef CAS
- J.-b. Wang, G. Li and M. T. Reetz, Chem. Commun., 2017, 53, 3916–3928 RSC
- L. Giver, A. Gershenson, P.-O. Freskgard and F. H. Arnold, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 12809–12813 CrossRef CAS
- H. Minagawa, Y. Yoshida, N. Kenmochi, M. Furuichi, J. Shimada and H. Kaneko, Cell. Mol. Life Sci., 2007, 64, 77–81 CrossRef CAS
- C. Zeymer and D. Hilvert, Annu. Rev. Biochem., 2018, 87, 131–157 CrossRef CAS
- F. H. Arnold, P. L. Wintrode, K. Miyazaki and A. Gershenson, Trends Biochem. Sci., 2001, 26, 100–106 CrossRef CAS
- M. S. Packer and D. R. Liu, Nat. Rev. Genet., 2015, 16, 379–394 CrossRef CAS
- M. T. Reetz, J. D. Carballeira and A. Vogel, Angew. Chem., Int. Ed., 2006, 45, 7745–7751 CrossRef CAS
- L. Sumbalova, J. Stourac, T. Martinek, D. Bednar and J. Damborsky, Nucleic Acids Res., 2018, 46, W356–W362 CrossRef CAS
- S. Wu, J. P. Acevedo and M. T. Reetz, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 2775–2780 CrossRef CAS
- E. Campbell, M. Kaltenbach, G. J. Correy, P. D. Carr, B. T. Porebski, E. K. Livingstone, L. Afriat-Jurnou, A. M. Buckle, M. Weik, F. Hollfelder, N. Tokuriki and C. J. Jackson, Nat. Chem. Biol., 2016, 12, 944–950 CrossRef CAS
- A. Wagner, Nat. Rev. Genet., 2008, 9, 965–974 CrossRef CAS
- C. M. Miton and N. Tokuriki, Protein Sci., 2016, 25, 1260–1272 CrossRef CAS
- M. S. Newton, V. L. Arcus, M. L. Gerth and W. M. Patrick, Curr. Opin. Struct. Biol., 2018, 48, 110–116 CrossRef CAS
- P. A. Romero and F. H. Arnold, Nat. Rev. Mol. Cell Biol., 2009, 10, 866–876 CrossRef CAS
- J. D. Bloom and F. H. Arnold, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 9995 CrossRef CAS
- K. M. Esvelt, J. C. Carlson and D. R. Liu, Nature, 2011, 472, 499–503 CrossRef CAS
- C. L. Moore, L. J. Papa and M. D. Shoulders, J. Am. Chem. Soc., 2018, 140, 11560–11564 CrossRef CAS
-
P. C. Cirino, K. M. Mayer and D. Umeno, Directed evolution library creation, Humana Press, 2003 Search PubMed
- W. J. Jeong, J. Yu and W. J. Song, Chem. Commun., 2020, 56, 9586–9599 RSC
- A. J. Burton, A. R. Thomson, W. M. Dawson, R. L. Brady and D. N. Woolfson, Nat. Chem., 2016, 8, 837–844 CrossRef CAS
- M. L. Zastrow, A. F. A. Peacock, J. A. Stuckey and V. L. Pecoraro, Nat. Chem., 2012, 4, 118–123 CrossRef CAS
- C. M. Rufo, Y. S. Moroz, O. V. Moroz, J. Stöhr, T. A. Smith, X. Hu, W. F. DeGrado and I. V. Korendovych, Nat. Chem., 2014, 6, 303–309 CrossRef CAS
- W. J. Song, J. Yu and F. A. Tezcan, J. Am. Chem. Soc., 2017, 139, 16772–16779 CrossRef CAS
- M. T. Reetz and J. D. Carballeira, Nat. Protoc., 2007, 2, 891–903 CrossRef CAS
- J. I. Boucher, D. N. Bolon and D. S. Tawfik, Protein Sci., 2016, 25, 1219–1226 CrossRef CAS
- S. Emond, M. Petek, E. J. Kay, B. Heames, S. R. A. Devenish, N. Tokuriki and F. Hollfelder, Nat. Commun., 2020, 11, 3469 CrossRef CAS
- F. Santiago, E. Doscher, J. Kim, M. Camps, J. Meza, S. Sindi and M. Barlow, PLoS One, 2020, 15, e0228240 CrossRef CAS
- W. J. Song and F. A. Tezcan, Science, 2014, 346, 1525 CrossRef CAS
-
J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer, Boston, MA, 3rd edn, 2006 Search PubMed
- J. A. Poveda, M. Prieto, J. A. Encinar, J. M. González-Ros and C. R. Mateo, Biochemistry, 2003, 42, 7124–7132 CrossRef CAS
- W.-H. Shin and C. Seok, J. Chem. Inf. Model., 2012, 52, 3225–3232 CrossRef CAS
- M. Baek, W.-H. Shin, H. W. Chung and C. Seok, J. Comput.-Aided Mol. Des., 2017, 31, 653–666 CrossRef CAS
- W. H. Landschulz, P. F. Johnson and S. L. McKnight, Science, 1988, 240, 1759 CrossRef CAS
- T. Alber, Curr. Opin. Genet. Dev., 1992, 2, 205–210 CrossRef CAS
- N. J. Marianayagam, M. Sunde and J. M. Matthews, Trends Biochem. Sci., 2004, 29, 618–625 CrossRef CAS
- M. H. Ali and B. Imperiali, Bioorg. Med. Chem., 2005, 13, 5013–5020 CrossRef CAS
- I. André, C. E. M. Strauss, D. B. Kaplan, P. Bradley and D. Baker, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 16148–16152 CrossRef
- J. A. Marsh and S. A. Teichmann, Annu. Rev. Biochem., 2015, 84, 551–575 CrossRef CAS
- M. T. Reetz and J. D. Carballeira, Nat. Protoc., 2007, 2, 891 CrossRef CAS
- M. Braun and L. Thöny-Meyer, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12830 CrossRef CAS
-
J. Wang, in Encyclopedia of Systems Biology, ed. W. Dubitzky, O. Wolkenhauer, K.-H. Cho and H. Yokota, Springer New York, New York, NY, 2013, pp. 1671–1671, DOI:10.1007/978-1-4419-9863-7_372
- J. D. Brodin, A. Medina-Morales, T. Ni, E. N. Salgado, X. I. Ambroggio and F. A. Tezcan, J. Am. Chem. Soc., 2010, 132, 8610–8617 CrossRef CAS
- K. A. McCall and C. A. Fierke, Anal. Biochem., 2000, 284, 307–315 CrossRef CAS
-
Z. Otwinowski and W. Minor, in Methods in Enzymology, Academic Press, 1997, vol. 276, pp. 307–326 Search PubMed
- Collaborative Computational Project Number 4, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1994, 50, 760–763 CrossRef
- A. Vagin and A. Teplyakov, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 22–25 CrossRef CAS
- G. N. Murshudov, A. A. Vagin and E. J. Dodson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1997, 53, 240–255 CrossRef CAS
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486–501 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Supporting figures, supporting tables, and X-ray diffraction analysis data are included. See DOI: 10.1039/d0sc06823c |
| This journal is © The Royal Society of Chemistry 2021 |