
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn orthogonal and reactivity-based one-pot glycosylation strategy for both glycan and nucleoside synthesis: access to TMG-chitotriomycin, lipochitooligosaccharides and capuramycin†
Haiqing
He‡
,
Lili
Xu‡
,
Roujing
Sun
,
Yunqin
Zhang
,
Yingying
Huang
,
Zixi
Chen
,
Penghua
Li
,
Rui
Yang
and
Guozhi
Xiao
 *
*
State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, University of Chinese Academy of Sciences, Chinese Academy of Sciences, Kunming 650201, China. E-mail: xiaoguozhi@mail.kib.ac.cn
First published on 23rd February 2021
Abstract
Both glycans (O-glycosides) and nucleosides (N-glycosides) play important roles in numerous biological processes. Chemical synthesis is a reliable and effective means to solve the attainability issues of these essential biomolecules. However, due to the stereo- and regiochemical issues during glycan assembly, together with problems including the poor solubility and nucleophilicity of nucleobases in nucleoside synthesis, the development of one-pot glycosylation strategies toward efficient synthesis of both glycans and nucleosides remains poor and challenging. Here, we report the first orthogonal and reactivity-based one-pot glycosylation strategy suitable for both glycan and nucleoside synthesis on the basis of glycosyl ortho-(1-phenylvinyl)benzoates. This one-pot glycosylation strategy not only inherits the advantages including no aglycon transfers, no undesired interference of departing species, and no unpleasant odors associated with the previously developed orthogonal one-pot glycosylation strategy based on glycosyl ortho-alkynylbenzoates, but also highly expands the scope (glycans and nucleosides) and increases the number of leaving groups that could be employed for the multistep one-pot synthesis (up to the formation of four different glycosidic bonds). In particular, the current one-pot glycosylation strategy is successfully applied to the total synthesis of a promising tuberculosis drug lead capuramycin and the divergent and formal synthesis of TMG-chitotriomycin with potent and specific inhibition activities toward β-N-acetylglucosaminidases and important endosymbiotic lipochitooligosaccharides including the Nod factor and the Myc factor, which represents one of the most efficient and straightforward synthetic routes toward these biologically salient molecules.
Introduction
Both glycans (O-glycosides) and nucleosides (N-glycosides) are essential biomolecules with a variety of important roles in numerous biological processes such as viral and bacterial infection, DNA and RNA synthesis, cell growth, proliferation and signaling, immune response, as well as enzyme metabolism and regulation.1–3 Due to the issues of the micro-heterogeneity, it is extremely difficult to isolate pure and homogeneous glycans and nucleosides from natural resources. Chemical synthesis of both glycans and nucleosides is an effective, reliable and scalable method to solve the issues of the attainability, which can highly facilitate the development of new therapeutic agents and the decipherment of their functions.3–11 During the past three decades, many strategies including automated chemical synthesis,12 one-pot glycosylation strategies,13 orthogonal glycosylation14 and latent-active synthesis15 have been developed to achieve the efficient construction of glycosidic bonds (C–O bonds), among which one-pot glycosylation strategies are highly efficient and attractive. In comparison with traditional glycosylation methods, which need tedious deprotection and protection manipulations during glycan assembly, one-pot glycosylation strategies not only avoid the purifications of intermediates and workups of the glycosylation interval, but also highly accelerate glycan synthesis and reduce chemical wastes. However, one-pot glycosylation strategies for nucleoside synthesis (C–N bonds) are much less investigated. The development of one-pot glycosylation strategies suitable for both glycan and nucleoside synthesis remain poor and challenging due to the stereo- and regiochemical issues during glycan assembly,13,16 together with other problems including the poor solubility and nucleophilicity of nucleobases in nucleoside synthesis.3,11In 2019, we reported that the orthogonal one-pot glycosylation strategy based on glycosyl ortho-alkynylbenzoates6b (ABz) can be applied to the efficient synthesis of a variety of glycans, which solves the shortcomings including aglycon transfers, the interference of departing species and unpleasant odor inherent to orthogonal one-pot glycosylation based on thioglycosides13b (Scheme 1). This orthogonal one-pot glycosylation strategy has recently been successfully used in the modular synthesis of a nona-decasaccharide motif from Psidium guajava polysaccharides with potent α-glucosidase inhibitory activity.13c Notwithstanding these advances, challenges still remain in this field. For instance, the number of leaving groups that can be utilized in multi-step orthogonal one-pot synthesis is still limited.12b,13 Furthermore, one-pot glycosylation strategies suitable for the construction of both C–O and C–N bonds remain undeveloped. Recently, we developed a new, versatile and efficient glycosylation method for both glycan and nucleoside synthesis, which uses glycosyl ortho-(1-phenylvinyl)benzoates (PVB) as novel glycosyl donors.11a We wondered whether the utilization of orthogonalities and reactivities of glycosyl PVB could be explored to achieve the efficient one-pot synthesis of both glycans and nucleosides.
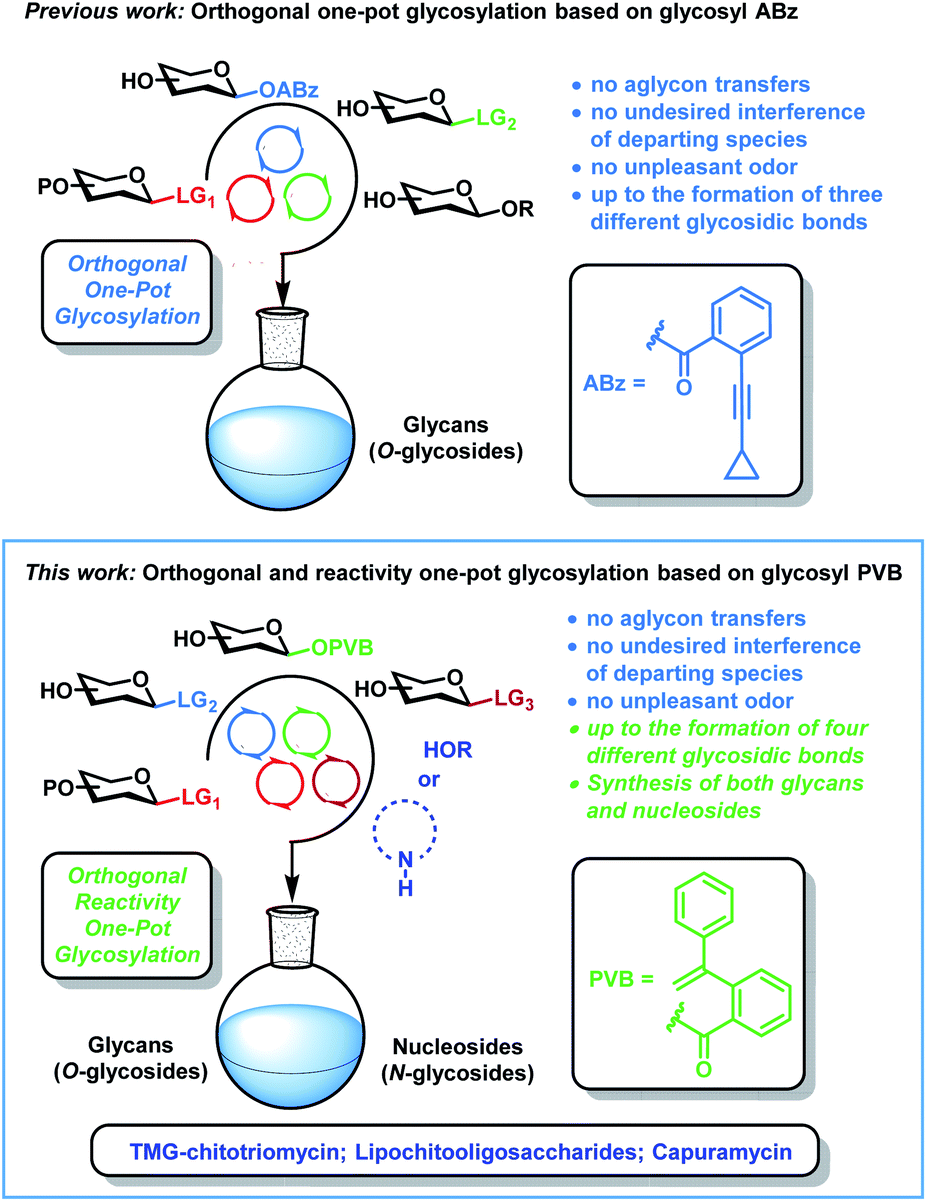 | ||
| Scheme 1 Strategies of one-pot glycosylation based on glycosyl ABz and glycosyl PVB. | ||
Herein, we report the first orthogonal and reactivity-based one-pot glycosylation strategy suitable for both glycan and nucleoside synthesis on the basis of glycosyl PVB. This one-pot glycosylation strategy not only inherits advantages including no aglycon transfers, no undesired interference of departing species, and no unpleasant odors associated with the previously developed orthogonal one-pot glycosylation strategy based on glycosyl ABz, but also highly expands the scope (glycans and nucleosides) and increases the number of leaving groups that could be employed for the multistep one-pot synthesis (up to the formation of four different glycosidic bonds). A wide range of nucleosides and glycans including α-Gal epitope, isoGb3 trisaccharide and phytoalexin elicitor β-glucan heptasaccharide have been efficiently synthesized with this strategy. Furthermore, the synthetic advantages and utilities of this strategy have been successfully demonstrated by the divergent and formal synthesis of TMG-chitotriomycin with potent and specific inhibition activities toward β-N-acetylglucosaminidases and important endosymbiotic lipochitooligosaccharides including the Nod factor and the Myc factor, as well as the total synthesis of nucleoside antibiotic capuramycin with promising anti-tuberculosis activities (Scheme 1).
Results and discussion
In order to develop one-pot glycosylation strategies (Scheme 1), two criteria should be met: (1) when the leaving group LGn is activated, the next leaving group LGn+1 must be stable and unaffected; (2) sideproducts produced during the glycosylation should not interfere with the next glycosylations. To this end, we commenced with the investigation of the orthogonalities of glycosyl PVB with the other glycosyl donors. First, glycosyl PVB was fixed as the bifunctional acceptor with the activatable leaving group and the free hydroxyl group. Several different glycosyl donors were studied, including glycosyl trichloroacetimidate (TCAI),17 glycosyl N-phenyltrifluoroacetimidate (PTFAI),18S-benzoxazolyl (SBox) glycoside,19S-thiazolinyl (STaz) glycoside20 and glycosyl ortho-alkynylbenzoate (ABz).6b The screening results showed that the bifunctional acceptor glycosyl PVB 2 could be successfully coupled with a variety of glycosyl donors under the activation conditions for TCAI 1a and PTFAI 1b (cat. TMSOTf, −20 °C), SBox 1c (AgOTf, rt), STaz 1d (MeOTf, rt) and ABz 1e (PPh3AuOTf, rt), affording the corresponding PVB disaccharides 3a–3b in excellent yields (Scheme 2). Self-condensation products were not detected.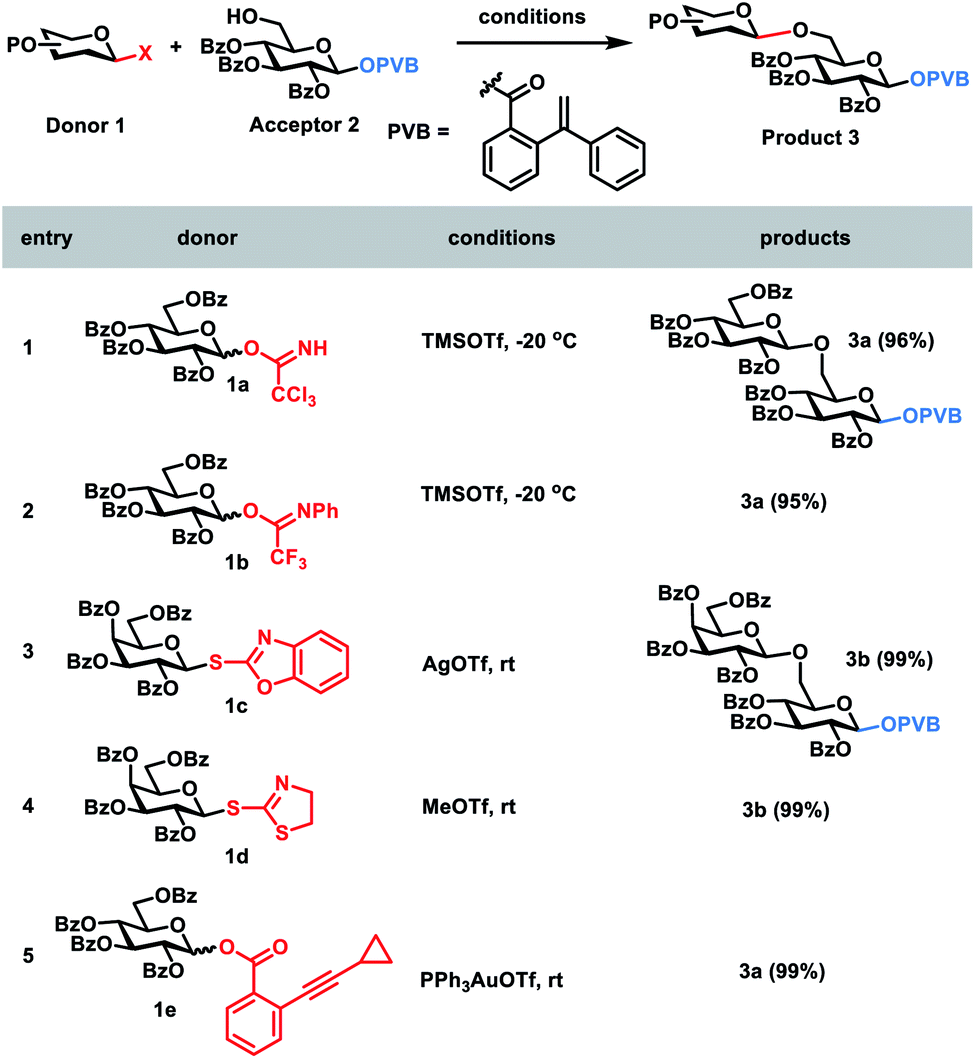 | ||
| Scheme 2 The investigation of the orthogonalities of glycosyl PVB with other glycosyl donors. | ||
Next, we went on to study whether glycosyl PVB could be selectively coupled with other bifunctional acceptors in the activation of NIS and TMSOTf. Due to the much higher reactivities of glycosyl PVB than thioglycosides,11a the desired disaccharide 6a was indeed obtained in an excellent yield (90%) during glycosylation between the disarmed glucosyl PVB 4 and the disarmed thioglycoside215a in the presence of NIS and TMSOTf at −15 °C. Similarly, PVB 4 was selectively coupled with the disarmed acceptor n-Pen225b, providing the desired disaccharide 6b in 90% yield (Scheme 3).
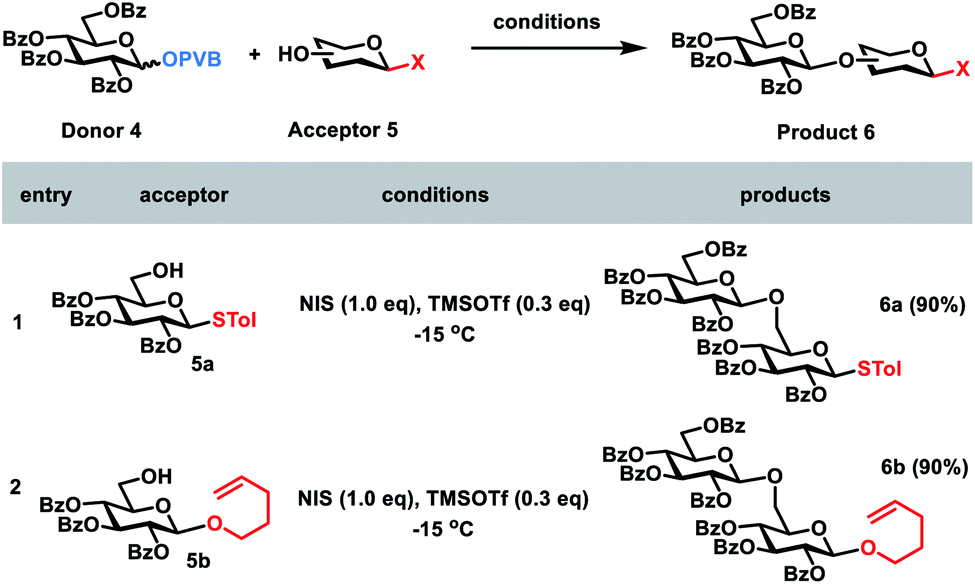 | ||
| Scheme 3 The investigation of the reactivities of glycosyl PVB with other glycosyl donors. | ||
Having investigated orthogonalities and reactivities of glycosyl PVB with other glycosyl donors, we next focused on the one-pot synthesis of glycans (Scheme 4). For the glycosyl TCAI and PVB pair, TCAI 1a (1.2 equiv.) was glycosylated with PVB 2 (1.0 equiv.) in the catalysis of TMSOTf at −20 °C, furnishing the PVB disaccharide intermediate. The above intermediate was further coupled with the disarmed thioglycoside 5a (0.9 equiv.) under the activation of NIS and TMSOTf at −15 °C, providing the trisaccharide 25 in 87% yield in one pot. Similarly, for the glycosyl PTFAI and PVB pair, the successive coupling of Gla PTFAI 7, Gla PVB 8 and the poor 4-OH glucosaminyl thioglycoside acceptor 9 produced the trisaccharide 26 in 54% yield in a one-pot manner. This trisaccharide motif was discovered as an α-Gal epitope that could induce xenograft rejection.23 Replacing the above acceptor 9 with the poor 4-OH glucosyl acceptor 10a also generated trisaccharide 27 in the same flask in 81% yield, the motif of which is the glycosphingolipid analogue of isoGb3 that could serve as a key endogenous human NKT cell antigen.24 It is noted that the 4,6-di-tert-butylsilyene group ensures the highly stereoselective α-galactosylation during the synthesis of 26–27.25 For the SBox glycoside and glycosyl PVB pair, coupling of SBox 1c (1.3 equiv.) with PVB 2 (1.0 equiv.) in the activation of AgOTf at room temperature produced the disaccharide intermediate, which was further glycosylated with the acceptor 11 (0.8 equiv.) in the promotion of NIS and TMSOTf, efficiently generating trisaccharide 28 in 91% yield in one pot. Similarly, for the STaz glycoside and glycosyl PVB pair, MeOTf was used to activate STaz 1d over PVB 2, followed by the addition of acceptor 10b and the activators NIS and TMSOTf, successfully furnishing trisaccharide 29 in 90% yield in one pot. For the glycosyl ABz and PVB pair, Yu glycosylation of ABz 12 (1.3 equiv.) with 3-OH glucosyl PVB 13 (1.0 equiv.) under the catalysis of PPh3AuOTf afforded the PVB disaccharide, which was followed by coupling with the acceptor 11 (1.0 equiv.) in the presence of NIS and TMSOTf, generating trisaccharide 30 in a one-pot manner in 88% yield. Using the above similar procedures, sequential glycosylation of ABz 1e, PVB 2 and 3-OH glucosyl acceptor 14 provided trisaccharide 32 in the same flask in 89% yield. For the glycosyl PVB and thioglycoside pair, the disarmed PVB 4 (1.0 equiv.) was coupled with the disarmed STol 5a (1.0 equiv.) in the presence of NIS (1.0 equiv.) and TMSOTf (0.3 equiv.) at −15 °C, generating the STol disaccharide intermediate. The above intermediate was further glycosylated with acceptor 15 (0.8 equiv.) under the activation of NIS and TMSOTf at a higher temperature (−15 °C to rt), producing trisaccharide 33 efficiently in 88% yield via reactivity-based one-pot glycosylation. For the glycosyl PVB and n-Pen glycoside pair, reactivity-based one-pot glycosylation of PVB 4 (1.1 equiv.), n-Pen 5b (1.0 equiv.) and acceptor 16 (0.8 equiv.) using the above similar procedures also successfully afforded trisaccharide 34 in 81% yield.
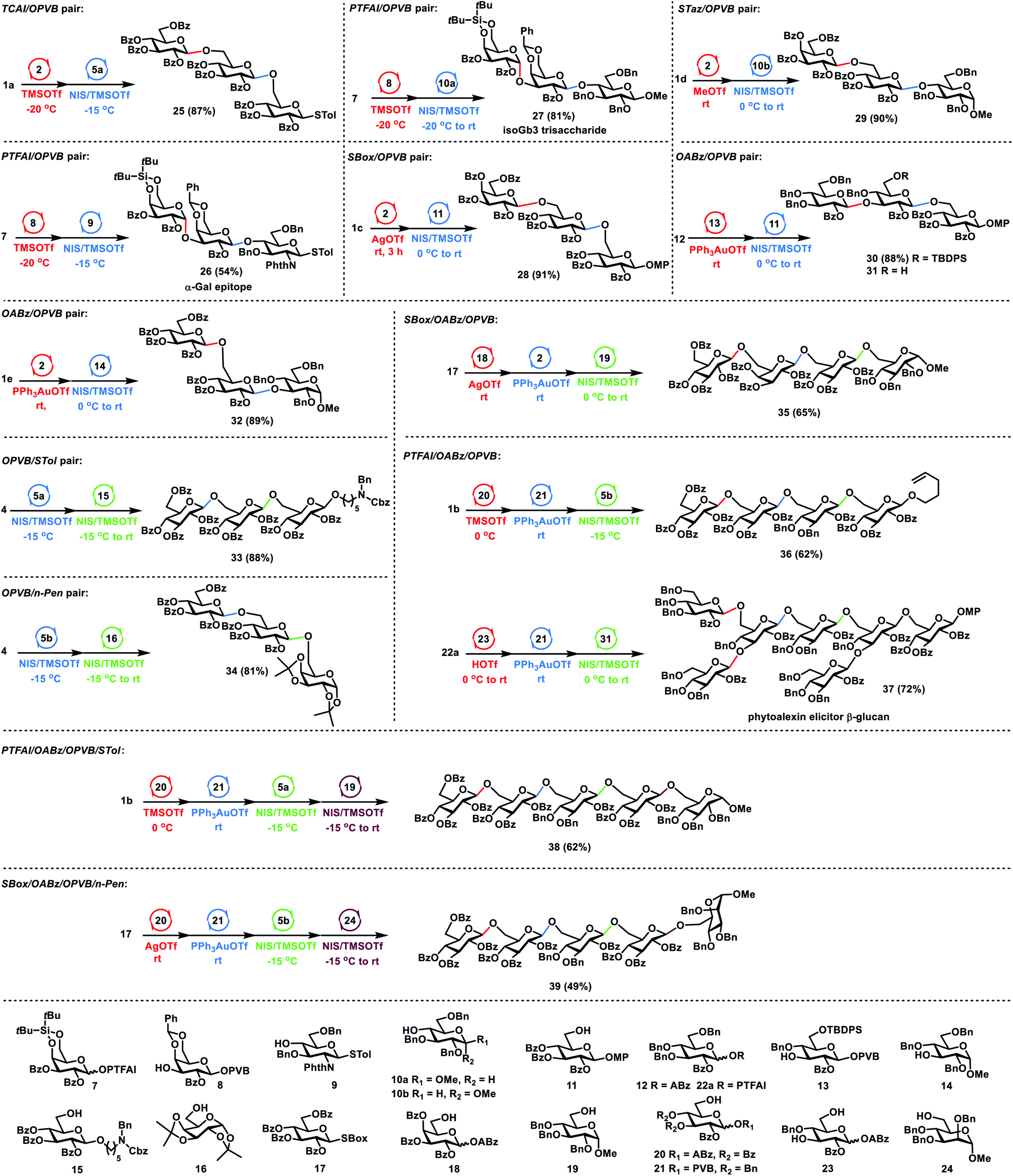 | ||
| Scheme 4 The orthogonal and reactivity-based one-pot glycosylation strategy for glycan synthesis. | ||
Then, we investigated the multistep one-pot synthesis of oligosaccharides. For the SBox glycoside, glycosyl ABz and PVB triplet, the orthogonal one-pot glycosylation of SBox 17 (1.2 equiv.), ABz 18 (1.0 equiv.), PVB 2 (0.9 equiv.) and the acceptor 19 (0.8 equiv.) successfully provided the tetrasaccharide 35 in 65% yield. For the glycosyl PTFAI, ABz and PVB triplet, the sequential glycosylation of PTFAI 1b (1.3 equiv.), ABz 20 (1.1 equiv.), PVB 21 (1.0 equiv.), and the acceptor 5b (1.2 equiv.) also proceeded smoothly, generating the tetrasaccharide 36 in the same flask with 62% yield, whose sulfated derivatives exhibit strong proangiogenic activity.26 Fungal β-glucan oligosaccharides possess the structure of the branched heptasaccharide motif 37, which could elicit phytoalexin production in soybean plant.27 Orthogonal double glycosylation of PTFAI 22a (2.5 equiv.) with 3-OH, 6-OH-glucosyl ABz 23 (1.0 equiv.) in the catalysis of HOTf provided the ABz trisaccharide, which was further coupled with PVB 21 (0.9 equiv.) via Yu glycosylation, generating the PVB tetrasaccharide. The above intermediate was further glycosylated with the trisaccharide acceptor 31 (0.9 equiv.) derived from 30, producing the branched heptasaccharide 37 with the formation of four new glycosidic linkages in the same flask in 72% yield. For a group of glycosyl PTFAI, ABz, PVB and thioglycoside, orthogonal one-pot glycosylation of PTFAI 1b (1.3 equiv.), ABz 20 (1.1 equiv.) and PVB 21 (1.0 equiv.) efficiently furnished the PVB trisaccharide intermediate, which was further coupled with the disarmed STol 5a (1.0 equiv.) in the presence of NIS and TMSOTf at −15 °C, producing the tetrasaccharide. The above tetrasaccharide intermediate was further coupled with acceptor 19 (0.9 equiv.) in the activation of NIS and TMSOTf at a higher reaction temperature (−15 °C to rt), affording the pentasaccharide 38 in 62% yield with the production of four new linkages via an orthogonal and reactivity-based one-pot glycosylation strategy. For a group of SBox glycoside, glycosyl ABz and PVB, as well as n-Pen glycoside, the orthogonal and reactivity-based one-pot glycosylation of SBox 17 (1.3 equiv.), ABz 20 (1.1 equiv.), PVB 21 (1.0 equiv.), n-Pen 5b (1.2 equiv.) and the acceptor 24 (0.9 equiv.) also successfully provided the desired pentasaccharide 39 in 49% yield with the formation of four new different glycosidic bonds.
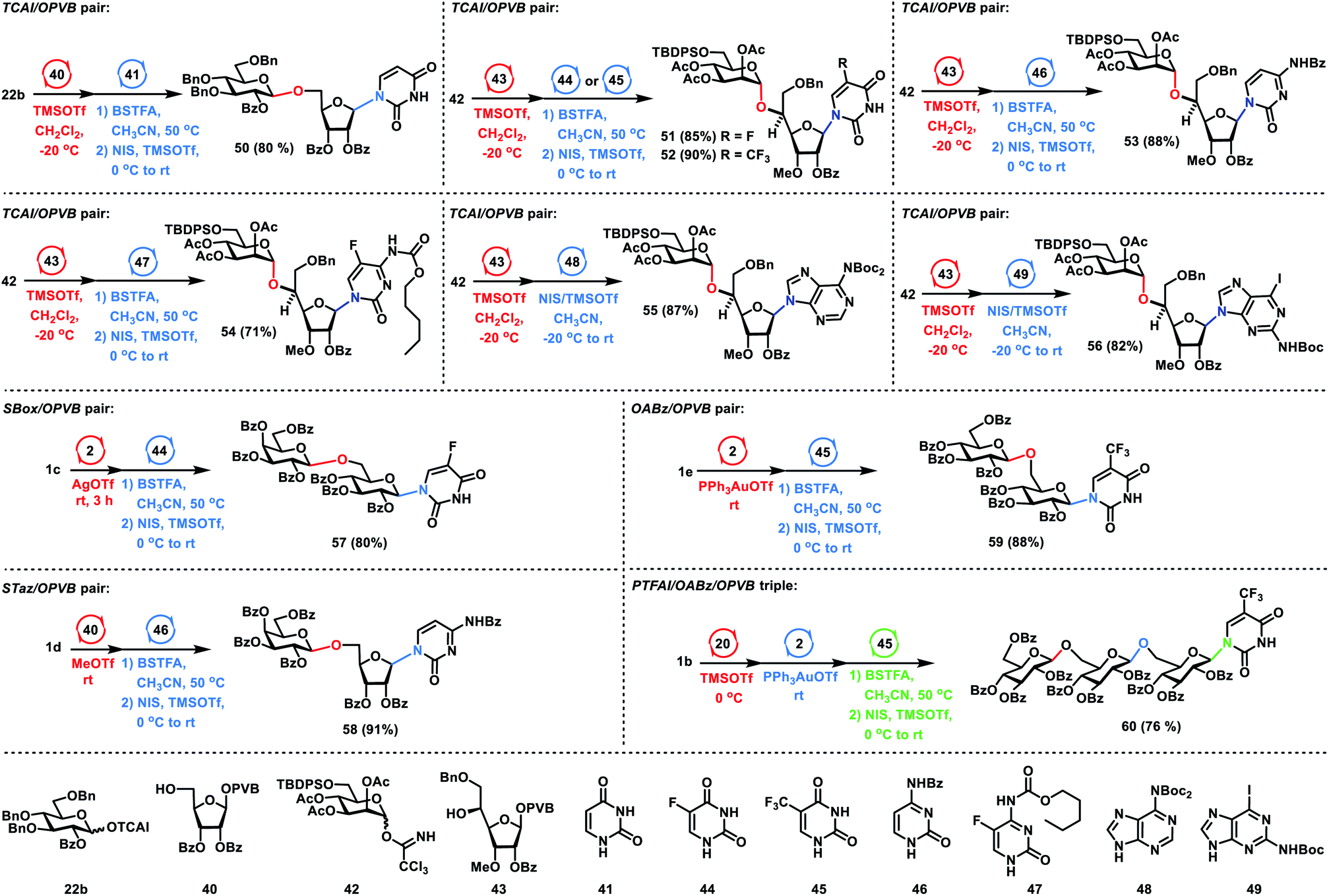
Next, we focused on the one-pot synthesis of nucleosides (Scheme 5). For the glycosyl TCAI and PVB pair, coupling of Glc TCAI 22b (1.2 equiv.) with Rib PVB 40 (1.0 equiv.) in the activation of TMSOTf at −20 °C generated the disaccharide PVB intermediate, which underwent further N-glycosylation with the silylated uracil intermediate derived from silylating uracil 41 (2.0 equiv.) with N,O-bis(trimethylsilyl)trifluoroacetamide (BSTFA) in CH3CN, providing the desired nucleoside 50 in 80% yield in a one-pot manner. Under similar reaction conditions, a catalytic amount of TMSOTf was used to couple Man TCAI 42 with Rib PVB 43, affording the disaccharide PVB intermediate. The above PVB intermediate was further glycosylated with silylated nucleobases derived from 5-fluorouridine 44, trifluorothymine 45, N4-benzoylcytosine 46 and 5-fluorocytosine derivative 47 with BSTFA in CH3CN, furnishing the desired nucleosides 51–54 in 85%, 90%, 88%, and 71% yields respectively in one-pot. It is noteworthy that the current protocol of N-glycosylation of pyrimidines with glycosyl PVB tolerates the carbamate functionality in pyrimidines. When a famous Vorbrüggen type reaction28 was utilized to couple the silylated 5-fluorocytosine derivative 47 under the action of pyridinium triflate salts,29 significant decomposition of the carbamate functionality and moderate coupling yield (50% yield) were observed.30 Besides pyrimidines, purines are also viable substrates. It was noted that N-glycosylation of purines was a challenging task due to the N9/N7 regioselectivity issues of purines in the glycosylation reactions. Nevertheless, nucleoside 55 was prepared successfully in an excellent 87% yield in one-pot by successive coupling of TCAI 42, PVB 43, and N6-bis(tert-butoxycarbonyl)adenine 48. It was noted that the tert-butoxycarbonyl (Boc) group installed in purine 48 served two purposes, namely, solving the solubility issues of purine nucleobases and blocking the glycosylation of N7 position. Similarly, sequential glycosylation of TCAI 42, PVB 43, and N2-tert-butoxycarbonyl-2-amino-6-iodopurine 49 gave the desired nucleoside 56 in 82% yield in a one-pot manner, which is a useful intermediate to the guanine nucleoside or 6-substituted analogs. For the SBox glycoside and glycosyl PVB pair, the coupling of Gla SBox 1c (1.3 equiv.) with Glc PVB 2 (1.0 equiv.) in the activation of AgOTf produced the disaccharide intermediate, which was further glycosylated with the silylated nucleobase derived from 5-fluorouridine 44 (2.0 equiv.), furnishing the desired nucleoside 57 in a one-pot manner with 80% yield. For the STaz glycoside and glycosyl PVB pair, MeOTf was used to activate Gla STaz 1d (1.3 equiv.) over Rib PVB 40 (1.0 equiv.), followed by N-glycosylation with silylated N4-benzoylcytosine 46 (2.0 equiv.) in the presence of NIS and TMSOTf, providing nucleoside 58 in 91% yield in the same flask. For the glycosyl ABz and PVB pair, Yu glycosylation6b between ABz 1e (1.2 equiv.) and PVB 2 (1.0 equiv.) produced the disaccharide intermediate, which further underwent N-glycosylation with silylated trifluorothymine 45 (2.0 equiv.), affording the desired nucleoside 59 in 88% yield in a one-pot manner. For a group of glycosyl PTFAI, ABz and PVB, orthogonal one-pot glycosylation of PTFAI 1b (1.3 equiv.), ABz 20 (1.1 equiv.), PVB 2 (1.0 equiv.) and silylated trifluorothymine 45 (2.0 equiv.) generated the desired nucleoside 60 in 76% yield.
 | ||
| Scheme 5 The orthogonal one-pot glycosylation for nucleoside synthesis. | ||
After the orthogonal and reactivity-based one-pot synthesis of both glycans and nucleosides was successfully investigated, we further applied this one-pot glycosylation strategy to the formal synthesis of TMG-chitotriomycin and lipochitooligosaccharides (Scheme 6). TMG-chitotriomycin is an enzyme inhibitor specific for insect and fungal β-N-acetylglucosaminidases (GlcNAcase) with no inhibition of mammalian and plant GlcNAcases.31 Especially because the unique inhibitory character of TMG-chitotriomycin can be very useful for the development of selective pesticides or fungicides and chitinolytic systems studies, TMG-chitotriomycin has stimulated broad synthetic interest.32 Structurally, TMG-chitotriomycin is very close to important glycolipids and lipochitooligosaccharides including the Nod factors33 and the Myc factors.34 They are important signal molecules involved in two ecologically crucial endosymbioses, nitrogen-fixing rhizobia and legume endosymbiosis and arbuscular mycorrhiza endosymbiosis between fungi and terrestrial plants. Due to their important actions and natural scarcity, these molecules also intrigued chemists for chemical synthesis.35 However, because of the poor solubility of glucosamine units and the notoriously poor nucleophilicity of the 4-hydroxyl group of glucosamine derivatives, the efficient and stereoselective one-pot synthesis of these bioactive molecules still remains challenging.
 | ||
| Scheme 6 Divergent and formal synthesis of TMG-chitotriomycin and lipochitooligosaccharides via the orthogonal one-pot glycosylation strategy based on glycosyl PVB. | ||
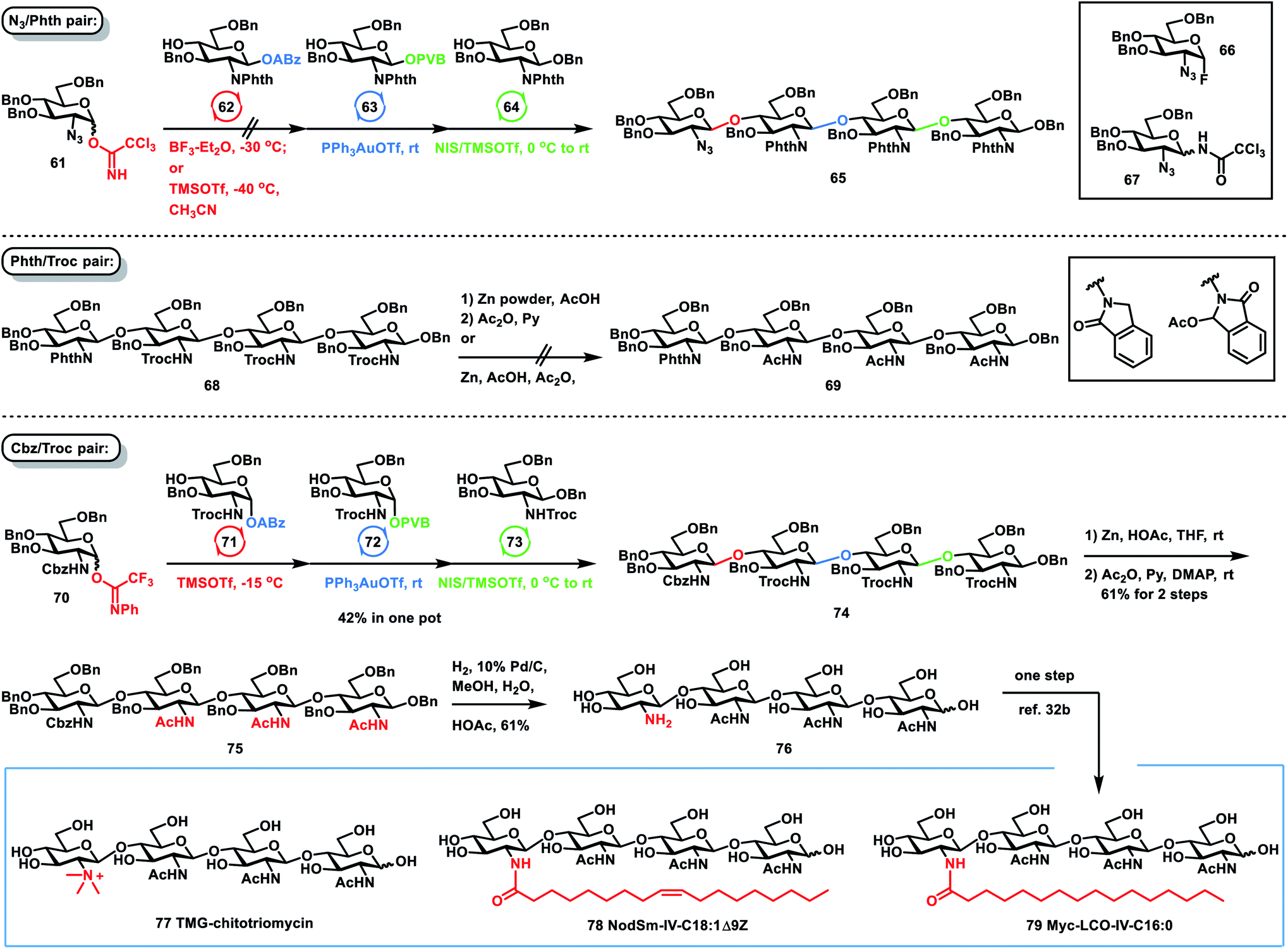
The correct selection of protecting groups for the differentiation of the amino groups between the nonreducing units and reducing unit is required to achieve the successful, efficient and stereoselective one-pot synthesis of TMG-chitotriomycin 77, the Nod factor 78 and the Myc factor 79. At first, we chose a pair of azido (N3) and phthalimido (Phth) groups as amino protecting groups. However, it was unsuccessful in preparing tetrasaccharide 65via orthogonal one-pot glycosylation of 2-N3 GlcN TCAI 61, 2-N-Phth GlcN ABz 62, 2-N-Phth GlcN PVB 63 and GlcN 64. The issue is the first glycosylation between 61 and 62. When BF3–Et2O was used to activate 2-N3 GlcN 61 over 2-N-Phth GlcN 62 at −30 °C, considerable amounts of side products glycosyl fluoride 66 and N-glycosyl trichloroacetamide 67via rearrangement were formed, which was detrimental to the successful one-pot assembly of 65. While TMSOTf was employed as a catalyst for coupling of 61 and 62 in the presence of CH3CN at −40 °C, no glycosyl fluoride byproduct 66 was formed, but the stereoselectivity of this glycosylation was low (β/α = 2/1). Later, we switched to Phth and 2,2,2-trichloroethoxycarbonyl (Troc) groups as the pair of amino protecting groups. Although one-pot assembly of tetrasaccharide 68 was successfully carried out on the basis of a group of glycosyl PTFAI, ABz and PVB, the conversion of 68 to the desired tetrasaccharide 69 was not successful via selective deprotection of three Troc groups over one Phth group. While zinc powder and acetic acid were used to remove three Troc groups, the Phth group in 68 was also reduced, generating an isoindolin-1-one derivative or a 3-oxoisoindolin-1-yl acetate derivative. Finally, we used a benzyloxycarbonyl (Cbz) amino protecting group instead of the Phth group. To our delight, orthogonal one-pot glycosylation of 2-N-Cbz PTFAI 70 (1.4 equiv.), 2-N-Troc ABz 71 (1.0 equiv.), 2-N-Troc PVB 72 (0.8 equiv.) and the acceptor 2-N-Troc GlcN 73 (0.7 equiv.) proceeded smoothly and stereoselectively, affording the desired tetrasaccharide 74 in a satisfactory 42% yield. Furthermore, selective removal of three Troc groups over one Cbz group in 74 was successfully carried out with zinc power and acetic acid, which was followed by acetylation of the resulting amine group, furnishing the desired tetrasaccharide 75 in 61% yield over two steps. Finally, global hydrogenolysis of 10 Bn groups and one Cbz group in the presence of 10% Pd/C successfully provided the desired tetrasaccharide 76 in 61% yield. The analytical data of synthetic tetrasaccharide 76 are consistent with those reported for 76, from which the synthesis of TMG-chitotriomycin 77, the Nod factor 78 and the Myc factor 79 was reported by Jean-Marie Beau and co-workers via one-step protocol of methylation and acylation, respectively.32b Compared with the previous synthetic approaches toward lipochitooligosaccharides and TMG-chitotriomycin,32,35 the current one-pot and divergent synthetic strategy represents one of the most efficient and straightforward synthetic routes toward these important and bioactive molecules.
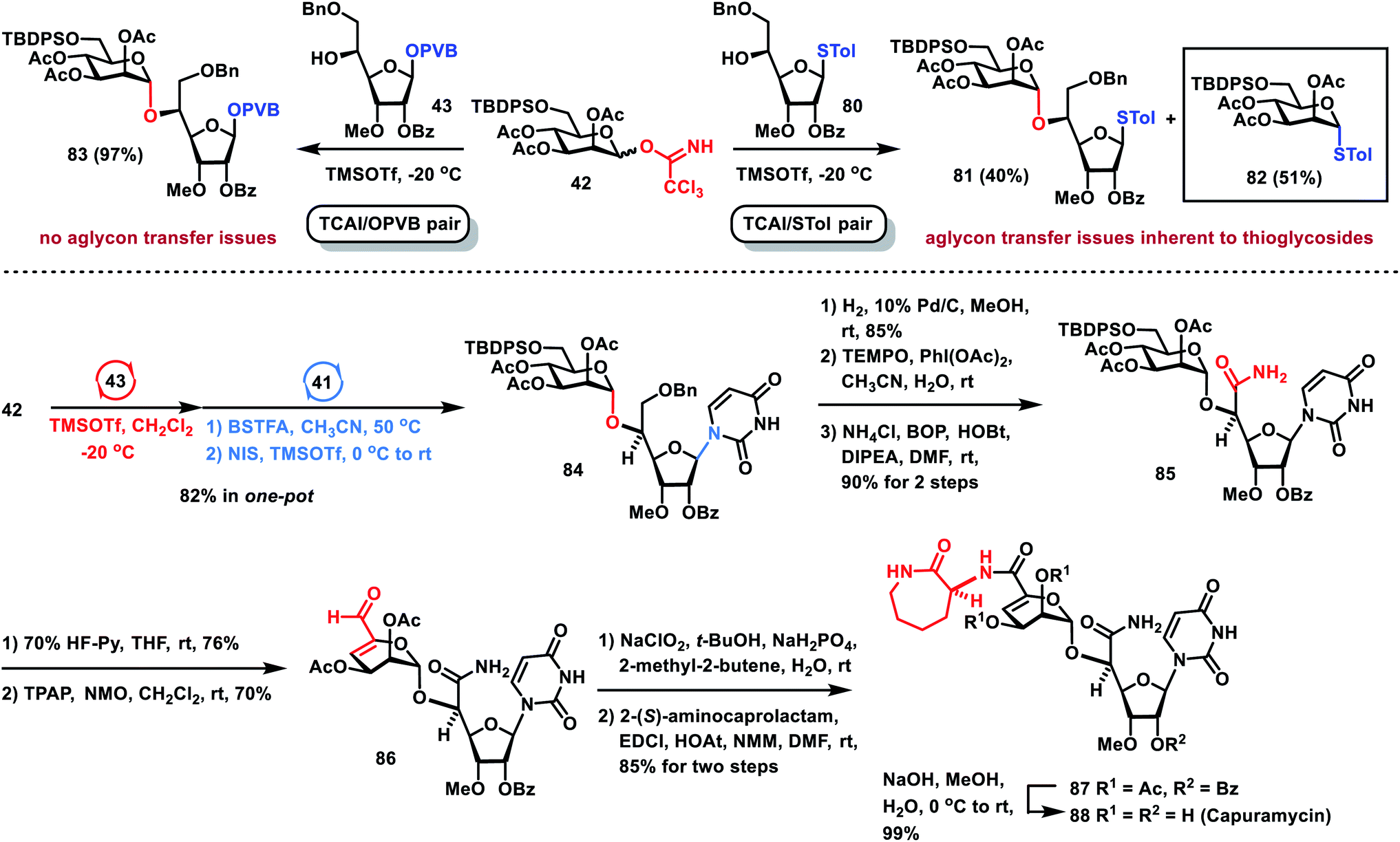
After the one-pot and formal synthesis of TMG-chitotriomycin 77, the Nod factor 78 and the Myc factor 79 has been successfully carried out, we further demonstrated that the one-pot glycosylation strategy based on glycosyl PVB can be successfully applied to the efficient total synthesis of the nucleoside antibiotic capuramycin 88 (Scheme 7), which is a promising tuberculosis drug lead.36,37 When Man TCAI 42 was coupled with thioglycoside 80 under the catalysis of TMSOTf, besides the desired disaccharide 81 (40% yield), a significant amount of aglycone transfer byproduct 82 (51% yield) was obtained, which exposed the aglycone transfer issue inherent to one-pot glycosylation based on thioglycosides.38 Instead, under the same reaction conditions, glycosylation of TCAI 42 and PVB 43 proceeded smoothly, generating the desired disaccharide PVB 83 in an excellent 97% yield, which precludes the aglycone transfer issue. Furthermore, orthogonal one-pot glycosylation of TCAI 42 (1.2 equiv.), PVB 43 (1.0 equiv.) and silylated uracil 41 (2.0 equiv.) furnished the desired nucleoside 84 in 82% yield. Hydrogenolytic removal of the benzyl group in 84 produced an alcohol, which was oxidized to carboxylic acid under TEMPO oxidation conditions,39 followed by condensation with NH4Cl to give the corresponding primary amide 85. The removal of the TBDPS group in 85 with 70% HF–Py afforded the alcohol intermediate, which was subjected to the same TEMPO oxidation conditions, producing complex mixtures. Modified Parikh–Doering oxidation of the alcohol intermediate also gave unsuccessful results.40 However, Ley oxidation of the resulting alcohol (TPAP, NMO) resulted in the successful oxidation–elimination reaction, providing the desired α,β-unsaturated aldehyde 86 in 70% yield.41 Pinnick oxidation of aldehyde 86 (NaClO2, t-BuOH, NaH2PO4, 2-methyl-2-butene, H2O) furnished a carboxylic acid, which was condensed with 2-(S)-aminocaprolactam (EDCI, HOAt, NMM), producing protected capuramycin 87 in 85% yield over two steps. Saponification of 87 by using NaOH in aq. MeOH provided capuramycin 88 in quantitative yield. The analytical data of synthetic capuramycin 88 are in good agreement with those reported for nucleoside antibiotic capuramycin.37
 | ||
| Scheme 7 Total synthesis of capuramycin via the orthogonal one-pot glycosylation strategy based on glycosyl PVB. | ||
Conclusions
In summary, the orthogonalities and reactivities of glycosyl PVB with the other leaving groups have been systematically investigated, and the first orthogonal and reactivity-based one-pot glycosylation strategy suitable for both glycan and nucleoside synthesis based on glycosyl PVB has been successfully developed, which has advantages such as no aglycon transfers, no undesired interference of departing species, and no unpleasant odors. Furthermore, the scope (glycans and nucleosides) and the number of leaving groups that could be employed for the multistep one-pot synthesis has also been highly expanded with this strategy. A variety of nucleoside and glycan motifs including α-Gal epitope, isoGb3 trisaccharide and phytoalexin elicitor β-glucan heptasaccharide had been efficiently prepared with this strategy. In particular, this one-pot glycosylation strategy has been successfully applied to the divergent and formal synthesis of TMG-chitotriomycin with the potent and specific inhibition of GlcNAcase activities and important plant endosymbiotic glycolipids. The correct selection of Cbz and Troc orthogonal amino protecting groups is crucial for the divergent, stereoselective and formal one-pot synthesis of TMG-chitotriomycin and lipochitooligosaccharides, which represents one of the most straightforward synthetic routes toward these biologically salient molecules. Furthermore, the total synthesis of nucleoside antibiotic capuramycin with promising anti-tuberculosis activities has also been successfully demonstrated with the current one-pot glycosylation strategy, which can highly streamline the chemical synthesis of both glycans and nucleosides with important biological functions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Key Project of the Natural Science Foundation of Yunnan Province, the CAS Pioneer Hundred Talents Program (No. 2017-128), the State Key Laboratory of Phytochemistry and Plant Resources in West China (P2020-ZZ05, P2018-ZZ02), and the Start-up funding of Kunming Institute of Botany is greatly acknowledged.Notes and references
- C. R. Bertozzi and L. L. Kiessling, Science, 2001, 291, 2357–2364 CrossRef CAS.
- A. Varki, Glycobiology, 2017, 27, 3–49 CrossRef CAS.
- (a) L. P. Jordheim, D. Durantel, F. Zoulim and C. Dumontet, Nat. Rev. Drug Discovery, 2013, 12, 447–464 CrossRef CAS; (b) J. Shelton, X. Lu, J. A. Hollenbaugh, J. H. Cho, F. Amblard and R. F. Schinazi, Chem. Rev., 2016, 116, 14379–14455 CrossRef CAS; (c) C. M. Galmarini, J. R. Mackey and C. Dumontet, Lancet Oncol., 2002, 3, 415–424 CrossRef CAS; (d) E. De Clercq, Med. Res. Rev., 2013, 33, 1215–1248 CrossRef CAS; (e) P. Merino, Chemical Synthesis of Nucleosides Analogues, Wiley, 2013 CrossRef.
- (a) C.-H. Wong, Carbohydrate-Based Drug Discovery, Wiley, 2003 CrossRef; (b) P. H. Seeberger and D. B. Werz, Nature, 2007, 446, 1046–1051 CrossRef CAS; (c) B. Ernst and J. Magnani, Nat. Rev. Drug Discovery, 2009, 8, 661–677 CrossRef CAS; (d) T. J. Boltje, T. Buskas and G. J. Boons, Nat. Chem., 2009, 1, 611–622 CrossRef CAS; (e) S. J. Danishefsky, Y.-K. Shue, M. N. Chang and C.-H. Wong, Acc. Chem. Res., 2015, 48, 643–652 CrossRef CAS.
- L. Krasnova and C.-H. Wong, J. Am. Chem. Soc., 2019, 141, 3735–3754 CrossRef CAS.
- (a) G. Xiao, X. Shao, D. Zhu and B. Yu, Nat. Prod. Rep., 2019, 36, 769–787 RSC; (b) B. Yu, Acc. Chem. Res., 2018, 51, 507–516 CrossRef CAS; (c) D. Zhu and B. Yu, Chin. J. Chem., 2018, 36, 681–691 CrossRef CAS; (d) Y. Yang, X. Zhang and B. Yu, Nat. Prod. Rep., 2015, 32, 1331–1355 RSC.
- (a) W.-L. Leng, H. Yao, J.-X. He and X.-W. Liu, Acc. Chem. Res., 2018, 51, 628–639 CrossRef CAS; (b) P. Peng and R. R. Schmidt, Acc. Chem. Res., 2017, 50, 1171–1183 CrossRef CAS.
- (a) C. S. Bennett and M. C. Galan, Chem. Rev., 2018, 18, 7931–7985 CrossRef; (b) P. O. Adero, H. Amarasekara, P. Wen, L. Bohé and D. Crich, Chem. Rev., 2018, 118, 8242–8284 CrossRef CAS; (c) M. M. Nielsen and C. M. Pedersen, Chem. Rev., 2018, 118, 8285–8358 CrossRef CAS.
- For selective reviews, see: (a) J. Stagg and M. J. Smyth, Oncogene, 2010, 29, 5346–5358 CrossRef CAS; (b) E. De Clercq, Med. Res. Rev., 2010, 30, 667–707 CAS; (c) A. J. A. Cobb, Org. Biomol. Chem., 2007, 5, 3260–3275 RSC; (d) I. Luyten and P. Herdewijn, Eur. J. Med. Chem., 1998, 33, 515–576 CrossRef CAS; (e) M. Egli, Angew. Chem., Int. Ed. Engl., 1996, 35, 1894–1909 CrossRef.
- P. Herdwijn, Modified Nucleosides in Biochemistry, Biotechnology and Medicine, Wiley-VCH, Weinheim, 2008 Search PubMed.
- (a) P. Li, H. He, Y. Zhang, R. Yang, L. Xu, Z. Chen, Y. Huang, L. Bao and G. Xiao, Nat. Commun., 2020, 11, 405 CrossRef CAS; (b) P. Li, H. He, L. Xu, Y. Huang, Z. Chen, Y. Zhang, R. Yang and G. Xiao, Green Synth. Catal., 2020, 1, 160–166 CrossRef; (c) M. Meanwell, S. M. Silverman, J. Lehmann, B. Adluri, Y. Wang and R. Cohen, Science, 2020, 369, 725–730 CrossRef CAS; (d) Q. Zhang, J. Sun, Y. Zhu, F. Zhang and B. Yu, Angew. Chem., Int. Ed., 2011, 50, 4933–4936 CrossRef CAS; (e) S. Wang, Q. Zhang, Y. Zhao, J. Sun, W. Kang, F. Wang, H. Pan, G. Tang and B. Yu, Angew. Chem., Int. Ed., 2019, 58, 10558–10562 CrossRef CAS; (f) S. Wang, J. Sun, Q. Zhang, X. Cao, Y. Zhao, G. Tang and B. Yu, Angew. Chem., Int. Ed., 2018, 57, 2884–2888 CrossRef CAS; (g) J. Li and B. Yu, Angew. Chem., Int. Ed., 2015, 54, 6618–6621 CrossRef CAS; (h) S. Nie, W. Li and B. Yu, J. Am. Chem. Soc., 2014, 136, 4157–4160 CrossRef CAS.
- For selective reviews, see: (a) M. Guberman and P. H. Seeberger, J. Am. Chem. Soc., 2019, 141, 5581–5592 CrossRef CAS; (b) M. Panza, S. G. Pistorio, K. J. Stine and A. V. Demchenko, Chem. Rev., 2018, 118, 8105–8150 CrossRef CAS; (c) P. H. Seeberger, Acc. Chem. Res., 2015, 48, 1450–1463 CrossRef CAS.
- (a) S. S. Kulkarni, C.-C. Wang, N. M. Sabbavarapu, A. R. Podilapu, P.-H. Liao and S.-C. Hung, Chem. Rev., 2018, 118, 8025–8104 CrossRef CAS; (b) Y. Zhang, G. Xiang, S. He, Y. Hu, Y. Liu, L. Xu and G. Xiao, Org. Lett., 2019, 21, 2335–2339 CrossRef CAS; (c) Y. Zhang, Z. Chen, Y. Huang, S. He, X. Yang, Z. Wu, X. Wang and G. Xiao, Angew. Chem., Int. Ed., 2020, 59, 7576–7584 CrossRef CAS.
- O. Kanie, Y. Ito and T. Ogawa, J. Am. Chem. Soc., 1994, 116, 12073–12074 CrossRef CAS.
- T. C. Shiao and R. Roy, Top. Curr. Chem., 2011, 301, 69–108 CrossRef CAS.
- (a) H.-Y. Wang, S. A. Blaszczyk, G. Xiao and W. Tang, Chem. Soc. Rev., 2018, 47, 681–701 RSC; (b) G. Xiao, G. A. C. Rosado, D. A. Glazier, B. Xi, C. Liu, P. Liu and W. Tang, J. Am. Chem. Soc., 2017, 139, 4346–4349 CrossRef CAS; (c) C.-H. Hsu, S.-C. Hung, C.-Y. Wu and C.-H. Wong, Angew. Chem., Int. Ed., 2011, 50, 11872–11923 CrossRef CAS.
- (a) R. R. Schmidt and W. Kinzy, Adv. Carbohydr. Chem. Biochem., 1994, 50, 21–123 CrossRef CAS; (b) R. R. Schmidt, Angew. Chem., Int. Ed. Engl., 1986, 25, 212–235 CrossRef.
- (a) B. Yu and J. Sun, Chem. Commun., 2010, 46, 4668–4679 RSC; (b) B. Yu and H. Tao, Tetrahedron Lett., 2001, 42, 2405–2407 CrossRef CAS.
- A. V. Demchenko, N. N. Malysheva and C. De Meo, Org. Lett., 2003, 5, 455–458 CrossRef CAS.
- A. V. Demchenko, P. Pornsuriyasak, C. De Meo and N. N. Malysheva, Angew. Chem., Int. Ed., 2004, 43, 3069–3072 CrossRef CAS.
- G. Lian, X. Zhang and B. Yu, Carbohydr. Res., 2015, 403, 13–22 CrossRef CAS.
- B. Fraser-Reid, U. E. Udodong, Z. Wu, H. Ottosson, J. R. Merritt, C. S. Rao, C. Roberts and R. Madsen, Synlett, 1992, 1992, 927–942 CrossRef.
- U. Galili, Immunol. Today, 1993, 14, 480–482 CrossRef CAS.
- D. Zhou, J. Mattner, C. N. Schrantz, N. Yin, Y. Gao, Y. Sagiv, K. Hudspeth, Y.-P. Wu, T. Yamashita, S. Teneberg, D. Wang, R. L. Proia, S. B. Levery, P. B. Savage, L. Teyton and A. Bendelac, Science, 2004, 306, 1786–1789 CrossRef CAS.
- A. Imamura, H. Ando, S. Korogi, G. Tanabe, O. Muraoka, H. Ishida and M. Kiso, Tetrahedron Lett., 2003, 44, 6725–6728 CrossRef CAS.
- S. A. Mousa, X. Feng, J. Xie, Y. Du, Y. Hua, H. He, L. OQConnor and R. J. Linhardt, J. Cardiovasc. Pharmacol., 2006, 48, 6–13 CrossRef CAS.
- A. Darvill, C. Augur, C. Bergmann, R. W. Carlson, J. Cheong, S. Eberhard, M. G. Hahn, V. M. Lo, V. Marfa, B. Meyer, D. Mohnen, M. Neill, M. Spiro, H. Halbeek, W. York and P. Albersheim, Glycobiology, 1992, 2, 181–198 CrossRef CAS.
- (a) H. Vorbruggen, Acc. Chem. Res., 1995, 28, 509–520 CrossRef; (b) H. Vorbruggen and K. Krolikiewicz, Angew. Chem., Int. Ed. Engl., 1975, 14, 421–422 CrossRef; (c) U. Niedballa and H. Vorbruggen, Angew. Chem., Int. Ed. Engl., 1970, 9, 461–462 CrossRef CAS.
- A. Sniady, M. W. Bedore and T. F. Jamison, Angew. Chem., Int. Ed., 2011, 50, 2155–2158 CrossRef CAS.
- B. Shen and T. F. Jamison, Org. Lett., 2012, 14, 3348–3351 CrossRef CAS.
- H. Usuki, T. Nitoda, M. Ichikawa, N. Yamaji, T. Iwashita, H. Komura and H. Kanzaki, J. Am. Chem. Soc., 2008, 130, 4146–4152 CrossRef CAS.
- (a) Y. Yang, Y. Li and B. Yu, J. Am. Chem. Soc., 2009, 131, 12076–12077 CrossRef CAS; (b) G. Despras, A. Alix, D. Urban, B. Vauzeilles and J. M. Beau, Angew. Chem., Int. Ed., 2014, 53, 11912–11916 CrossRef CAS; (c) T. Nokami, Y. Isoda, N. Sasaki, A. Takaiso, S. Hayase, T. Itoh, R. Hayashi, A. Shimizu and J. Yoshida, Org. Lett., 2015, 17, 1525–1528 CrossRef CAS; (d) Y. Isoda, N. Sasaki, K. Kitamura, S. Takahashi, S. Manmode, N. T. Okuda, J. Tamura, T. Nokami and T. Itoh, Beilstein J. Org. Chem., 2017, 13, 919–924 CrossRef CAS.
- (a) P. Lerouge, P. Roche, C. Faucher, F. Maillet, G. Truchet, J.-C. Prome and J. Denarie, Nature, 1990, 344, 781–784 CrossRef CAS; (b) G. Truchet, P. Roche, P. Lerouge, J. Vasse, S. Camut, F. de Billy, J.-C. Prome and J. Denarie, Nature, 1991, 351, 670–673 CrossRef CAS.
- F. Maillet, V. Poinsot, O. Andre, V. Puech-Pages, A. Haouy, M. Gueunier, L. Cromer, D. Giraudet, D. Formey, A. Niebel, E. A. Martinez, H. Driguez, G. Becard and J. Denarie, Nature, 2011, 469, 58–63 CrossRef CAS.
- For the recent review, see: Y. Yang and B. Yu, Tetrahedron, 2014, 70, 1023–1046 CrossRef CAS.
- (a) A. L. Biecker, X. Liu, J. S. Thorson, Z. Yang and S. G. V. Lanen, Molecules, 2019, 24, 433 CrossRef; (b) M. Serpi, V. Ferrari and F. Pertusati, J. Med. Chem., 2016, 59, 10343–10382 CrossRef CAS.
- (a) S. Knapp and S. R. Nandan, J. Org. Chem., 1994, 59, 281 CrossRef CAS; (b) M. Kurosu, K. Li and D. C. Crick, Org. Lett., 2009, 11, 2393 CrossRef CAS; (c) Y. Wang, S. Siricilla, B. A. Aleiwi and M. Kurosu, Chem.–Eur. J., 2013, 19, 13847 CrossRef CAS.
- (a) J. D. C. Codee, B. Stubba, M. Schiattarella, H. S. Overkleeft, C. A. A. van Boeckel, J. H. van Boom and G. A. van der Marel, J. Am. Chem. Soc., 2005, 127, 3767–3773 CrossRef CAS; (b) H. Tanaka, M. Adachi and T. Takahashi, Tetrahedron Lett., 2004, 45, 1433–1436 CrossRef CAS; (c) H. Yu, B. Yu, X. Wu, Y. Hui and X. Han, J. Chem. Soc., Perkin Trans. 1, 2000, 1445–1453 RSC.
- A. De Mico, R. Margarita, L. Parlanti, A. Vescovi and G. Piancatelli, J. Org. Chem., 1997, 62, 6974–6977 CrossRef CAS.
- J. R. Parikh and W. E. Doering, J. Am. Chem. Soc., 1967, 89, 5505–5507 CrossRef CAS.
- W. P. Griffith and S. V. Ley, J. Chem. Soc., Chem. Commun., 1987, 1625–1627 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc06815b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |