
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantification of the ion transport mechanism in protective polymer coatings on lithium metal anodes†
Hongyao
Zhou‡
a,
Haodong
Liu
a,
Xing
Xing
a,
Zijun
Wang
b,
Sicen
Yu
a,
Gabriel M.
Veith
 c and
Ping
Liu
*a
c and
Ping
Liu
*a
aDepartment of NanoEngineering, 9500 Gilman Drive, La Jolla, California 92093, USA. E-mail: piliu@eng.ucsd.edu
bMaterials Science and Engineering Program, University of California San Diego, USA
cMaterial Science and Technology Division, Oak Ridge National Laboratory, Oak Ridge, Tennessee 37831, USA
First published on 12th April 2021
Abstract
Protective Polymer Coatings (PPCs) have been proposed to protect lithium metal anodes in rechargeable batteries to stabilize the Li/electrolyte interface and to extend the cycle life by reducing parasitic reactions and improving the lithium deposition morphology. However, the ion transport mechanism in PPCs remains unclear. Specifically, the degree of polymer swelling in the electrolyte and the influence of polymer/solvent/ion interactions are never quantified. Here we use poly(acrylonitrile-co-butadiene) (PAN–PBD) with controlled cross-link densities to quantify how the swelling ratio of the PPC affects conductivity, Li+ ion selectivity, activation energy, and rheological properties. The large difference in polarities between PAN (polar) and PBD (non-polar) segments allows the comparison of PPC properties when swollen in carbonate (high polarity) and ether (low polarity) electrolytes, which are the two most common classes of electrolytes. We find that a low swelling ratio of the PPC increases the transference number of Li+ ions while decreasing the conductivity. The activation energy only increases when the PPC is swollen in the carbonate electrolyte because of the strong ion–dipole interaction in the PAN phase, which is absent in the non-polar PBD phase. Theoretical models using Hansen solubility parameters and a percolation model have been shown to be effective in predicting the swelling behavior of PPCs in organic solvents and to estimate the conductivity. The trade-off between conductivity and the transference number is the primary challenge for PPCs. Our study provides general guidelines for PPC design, which favors the use of non-polar polymers with low polarity organic electrolytes.
Introduction
Lithium (Li) metal is an ideal anode for increasing the energy density of Li-based batteries, because of the high gravimetric (3860 mA h g−1) and volumetric (2060 mA h cm−3) capacities compared to graphite.1 The current bottleneck for Li metal anodes is, however, the high reactivity of Li metal with liquid electrolyte (LE) and non-uniform deposition of Li metal.1,2 The reaction between Li metal and the LE produces a solid electrolyte interface (SEI) layer with a thickness of c.a. 10 nm on the Li surface.3 Although the SEI layer can impede further side reactions with the LE, the large volume change of Li metal during repeated deposition/dissolution processes continuously destroys the SEI layer and gradually consumes the LE.4Protective Polymer Coatings (PPCs) are an artificial SEI layer preformed on the surface of Li metal to slow down the rate of side reactions with the LE.5 Compared to their inorganic counterpart, PPCs are easier to deposit as a uniform coating at a lower cost and greater scalability. In addition, the stretchable nature of polymer materials can accommodate the volume change of the Li metal anode during repeated cycling. There are four primary functions for PPCs: (1) to reduce the permeation rate of the organic solvent and decrease the rate of side reactions;6 (2) to increase the transference number of the Li+ ion (t+) and mitigate the buildup of the concentration polarization of Li+ at the anode surface;7,8 (3) to increase the flowability of the interface between Li metal and the electrolyte, promoting a uniform and non-dendritic deposition of Li metal;9 and (4) to reinforce the stability of the SEI layer through the designed reaction between the PPC and Li metal.10
Various types of PPCs have been developed meeting one or more of the above four criteria, with a primary focus on demonstrating their effectiveness in a lithium metal battery cell. In contrast, there have been very few fundamental studies searching for the descriptors of the protective functions of PPCs. For example, Lopez used the dielectric constant and the surface energy of PPCs and made a correlation with the particle size of the electrodeposited Li metal.11 More recently, we categorized the PPC materials based on polarities (i.e. Hildebrand solubility parameters), which vary from non-polar to strongly polarized polymers.5 Typically, the polarities between PPC materials and LEs are matched, because the resultant high swelling ratio of polymers increases the ionic conductivity (σ).12 On the other hand, a lower swelling ratio is expected to reduce the permeation rate of the LE (function 1). Furthermore, the interaction between the polymer and Li+ ion is expected to become more significant at lower swelling ratios (i.e. increasing volume fractions of the polymer), which may influence the Li+ ion selectivity (function 2) and activation energy (Ea) for ion transport. In terms of the polarity of PPCs, there is no clear experimental evidence showing the advantage between a polar polymer which actively interacts with the ions or a non-polar polymer which only exerts weak interactions. Therefore, a quantitative analysis of the ion transport properties in PPCs as a function of the swelling ratio, and a comparison in polar or non-polar ion transporting media are highly desired.
The focus of this study is to: (1) establish a platform to systematically vary cross-linking densities of a PPC to control the swelling ratio; (2) carry out a quantitative analysis of the ion transport and the rheology of the bulk PPC material swollen in either ether-based (low polarity) or carbonate-based (high polarity) LEs; and (3) examine the influence of the swelling ratio on Li metal deposition underneath the PPC. Therefore, the selection of the PPC material in this study specifically prioritizes the quantitative analysis to obtain a clear and unambiguous comparison between the properties of polar and non-polar PPC materials. In this regard, we selected polyacrylonitrile-co-butadiene (PAN–PBD) with which the degree of self-cross-linking reaction of the PAN segment is precisely controlled by the concentration of lithium trisulfide (Li2S3) which acts as the cross-linking agent (Fig. 1). Theoretical prediction by using Hansen solubility parameters (HSPs) shows that the large difference in the polarities between PAN (polar) and PBD (non-polar) segments results in the selective swelling of the PAN segment in carbonate and the PBD segment in ether LEs, respectively. Deswelling of the polymer segment with the opposite polarity to the LE serves as the mechanical support and enables fabrication of a thin self-standing PPC with a thickness of only 10 μm. We confirm the decrease of σ value and the increase of t+ value with decreasing swelling ratio when either the PAN segment or the PBD segment is the swollen phase. Strong coupling between the nitrile group in the polar PAN segment and Li+ increases the Ea value in carbonate LE (Fig. 1); in contrast, such behavior is not observed with the non-polar PBD segments swollen in the ether LE. A minimum conductivity of 10−4 S cm−1 is required to deposit Li metal without breaking the 10 μm-thick PPC layers. In addition, a mechanically weak but viscous PPC layer promotes the deposition of spherical rather than dendritic lithium metal particles. By comparing the ion transport and rheological properties of the bipolar PAN–PBD PPC with controlled swelling ratio in ether or carbonate LEs, we provide design guidelines for the development of advanced PPC materials for Li metal anodes.
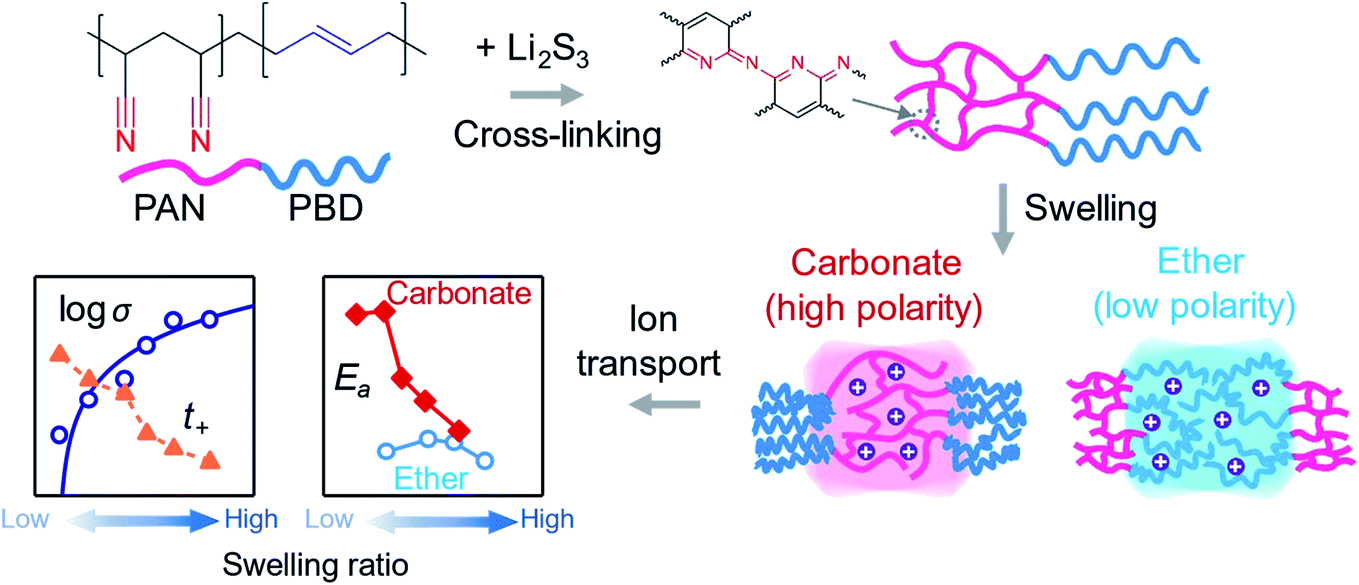 | ||
| Fig. 1 Cross-linking reaction of PAN–PBD and the swelling in ether or carbonate electrolytes. The cross-link density and the swelling ratio are controlled by the concentration of Li2S3 mixed in the polymer. The PAN (polar) and PBD (non-polar) segments selectively swell in carbonate (high polarity) and ether (low polarity) electrolytes, respectively. Ion transport takes place at the swollen polymer phase, and the difference in polymer/solvent polarities results in distinctive ion transport properties as a function of the swelling ratio. Deswelling of the polymer segment with the opposite polarity to the electrolyte enhances the mechanical strength of the PAN–PBD thin film. | ||
Results and discussion
Characterization
Self-cyclization of PAN is an attractive approach to form chemical cross-linking between the PAN segments.13,14 Conventionally, self-cyclization of PAN is initiated at temperatures above 200 °C,15–17 and the resulting cross-link density only varies in a narrow range (e.g. 1.5–2.5 × 10−4 mol cm−3) regardless of the heating time.14 The difficulty in varying the cross-link density of PAN can be attributed to the high activation energy (149 kJ mol−1) for cyclization, and the spontaneous propagation of cyclization after the activation.18 Lithium trisulfide (Li2S3 = Li2S + 2S) in dimethyl formamide (DMF) solution was known to catalyze PAN cyclization at low temperatures.19,20Here we carry out the cross-linking of PAN–PBD in a mixed solution of DMF and tetrahydrofuran (THF) with varied concentrations of Li2S3 (Li2S3/AN = 0–1.0 in mol mol−1) at 100 °C to vary the cross-linking density of PAN segments (Fig. 1). After the cross-linking, residual DMF and Li2S3 are removed by extraction in DME, which has a high solubility for polysulfide species (Sx−) (Fig. S1, ESI†). To carry out a quantitative analysis of the cross-link density, the peak at 970 cm−1 (trans C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H of PBD) is selected as the reference for intensity analysis of the other peaks (Fig. 2). The constant absorbance at 1440 cm−1 (saturated C–H bending of PBD), independent of the Li2S3/AN ratio, indicates that the PBD segment is unreacted during the cross-linking reaction and justifies the use of 970 cm−1 as the reference peak. Increasing absorbance at 1600 cm−1 (CC/CN stretching) and decreasing absorbance at 2240 cm−1 (C
C–H of PBD) is selected as the reference for intensity analysis of the other peaks (Fig. 2). The constant absorbance at 1440 cm−1 (saturated C–H bending of PBD), independent of the Li2S3/AN ratio, indicates that the PBD segment is unreacted during the cross-linking reaction and justifies the use of 970 cm−1 as the reference peak. Increasing absorbance at 1600 cm−1 (CC/CN stretching) and decreasing absorbance at 2240 cm−1 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretching of PAN) suggest that the cyclization of CN into the conjugate CC/CN is catalyzed by Li2S3. The appearance of the peaks at 2190 cm−1 (conjugate CN) and 790 cm−1 (1,2,3-trisubstituted = C–H bending) suggests that dehydrogenation of α- and β-hydrogens of PAN takes place and forms a CC double bond in the PAN main chain. The red shift of the CC/CN peak position from 1620 to 1612 cm−1 with increasing Li2S3/AN ratio indicates an extending length of the CC/CN conjugation (Fig. S2, ESI†). X-ray photoelectron spectroscopy (XPS) shows increasing intensity of the S 2p peak at 163.3 eV with increasing Li2S3/AN ratio, revealing the possible presence of both S–S and S–C bonds21 (Fig. S3, ESI†).
N stretching of PAN) suggest that the cyclization of CN into the conjugate CC/CN is catalyzed by Li2S3. The appearance of the peaks at 2190 cm−1 (conjugate CN) and 790 cm−1 (1,2,3-trisubstituted = C–H bending) suggests that dehydrogenation of α- and β-hydrogens of PAN takes place and forms a CC double bond in the PAN main chain. The red shift of the CC/CN peak position from 1620 to 1612 cm−1 with increasing Li2S3/AN ratio indicates an extending length of the CC/CN conjugation (Fig. S2, ESI†). X-ray photoelectron spectroscopy (XPS) shows increasing intensity of the S 2p peak at 163.3 eV with increasing Li2S3/AN ratio, revealing the possible presence of both S–S and S–C bonds21 (Fig. S3, ESI†).
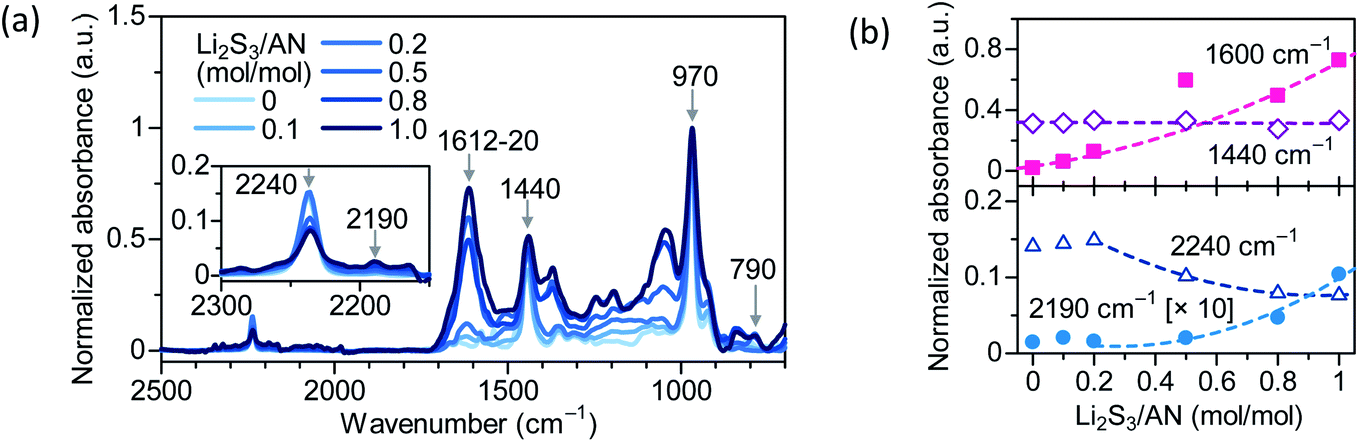 | ||
| Fig. 2 (a) FTIR spectra of PAN–PBD cross-linked under varied Li2S3/AN mole ratios (0–1.0). The inset shows the magnified view of the CN stretching peaks. The absorbance is normalized to the peak at 970 cm−1 (trans CC–H of PBD). (b) Changes of the IR absorbance at the selected peaks: 1600 cm−1, CC/CN; 1440 cm−1, CH2 of PBD; 2240 cm−1, CN; 2190 cm−1 (the absorbance magnified by 10), conjugate CN. The result shows the cyclization of CN into a fused pyridine-ring structure. The dashed lines represent the least-squares fittings to the experimental results. | ||
Based on the above analysis, we propose the following reaction scheme: (1) Li2S3 catalyzes dehydrogenation at the PAN backbone (possibly releasing H2S gas) and forms conjugated CC with CN. (2) Nucleophilic attack of Sx− on CN produces the Sx–CN− anion and initiates intramolecular self-cyclization between adjacent CN groups. The cyclization reaction between the conjugate CN groups is sterically favored (formation of a six-membered ring), compared to between nonconjugated CN groups. (3) The cyclization reaction propagates between different PAN segments and results in intermolecular cross-linking. (Fig. S4, ESI†)
Polymer swelling
We study the swelling behavior of PAN–PBD in two prototypical battery electrolytes: 1 M lithium bis(fluorosulfonyl)imide (LiFSI) in DME as a low-polarity LE, and 1 M lithium hexafluoro phosphate (LiPF6) in a mixed solvent of EC and dimethyl carbonate (DMC) in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 weight ratio as a high-polarity LE.5,22 The PAN–PBD copolymer is known to undergo phase segregation to form nanodomains of PAN and PBD, respectively.23 DME and EC/DMC solvents are expected to be absorbed into the different polymer phases. The swelling ratio of the cross-linked PAN–PBD is expressed as a volume ratio of the swollen polymer (V) to the volume of dry polymer (V0) and plotted as a function of Li2S3/AN ratio (Fig. 3a and b). The swelling ratio in DME solvent decreases with increasing ratio of Li2S3/AN because of the increasing number of the intermolecular cross-linking formed between the PAN segments. The swelling ratio in pure DMC is lower than the value in DME, and the addition of EC in DMC increases the swelling ratio. The addition of LiPF6 in DMC/EC mixed solvent reduces the swelling ratio of PAN–PBD (Fig. 3b). We confirmed that the addition of lithium perchlorate (LiClO4) also reduces the swelling ratio to the same degree as LiPF6 (Fig. S5, ESI†). In contrast, addition of LiFSI causes no change in the swelling ratio in DME (Fig. 3a). FTIR analysis revealed a stronger absorbance of the CN–Li+ dipole–ion pair in DMC/EC/LiPF6, compared with DME/LiFSI (Fig. S6, ESI†).
:1 weight ratio as a high-polarity LE.5,22 The PAN–PBD copolymer is known to undergo phase segregation to form nanodomains of PAN and PBD, respectively.23 DME and EC/DMC solvents are expected to be absorbed into the different polymer phases. The swelling ratio of the cross-linked PAN–PBD is expressed as a volume ratio of the swollen polymer (V) to the volume of dry polymer (V0) and plotted as a function of Li2S3/AN ratio (Fig. 3a and b). The swelling ratio in DME solvent decreases with increasing ratio of Li2S3/AN because of the increasing number of the intermolecular cross-linking formed between the PAN segments. The swelling ratio in pure DMC is lower than the value in DME, and the addition of EC in DMC increases the swelling ratio. The addition of LiPF6 in DMC/EC mixed solvent reduces the swelling ratio of PAN–PBD (Fig. 3b). We confirmed that the addition of lithium perchlorate (LiClO4) also reduces the swelling ratio to the same degree as LiPF6 (Fig. S5, ESI†). In contrast, addition of LiFSI causes no change in the swelling ratio in DME (Fig. 3a). FTIR analysis revealed a stronger absorbance of the CN–Li+ dipole–ion pair in DMC/EC/LiPF6, compared with DME/LiFSI (Fig. S6, ESI†).
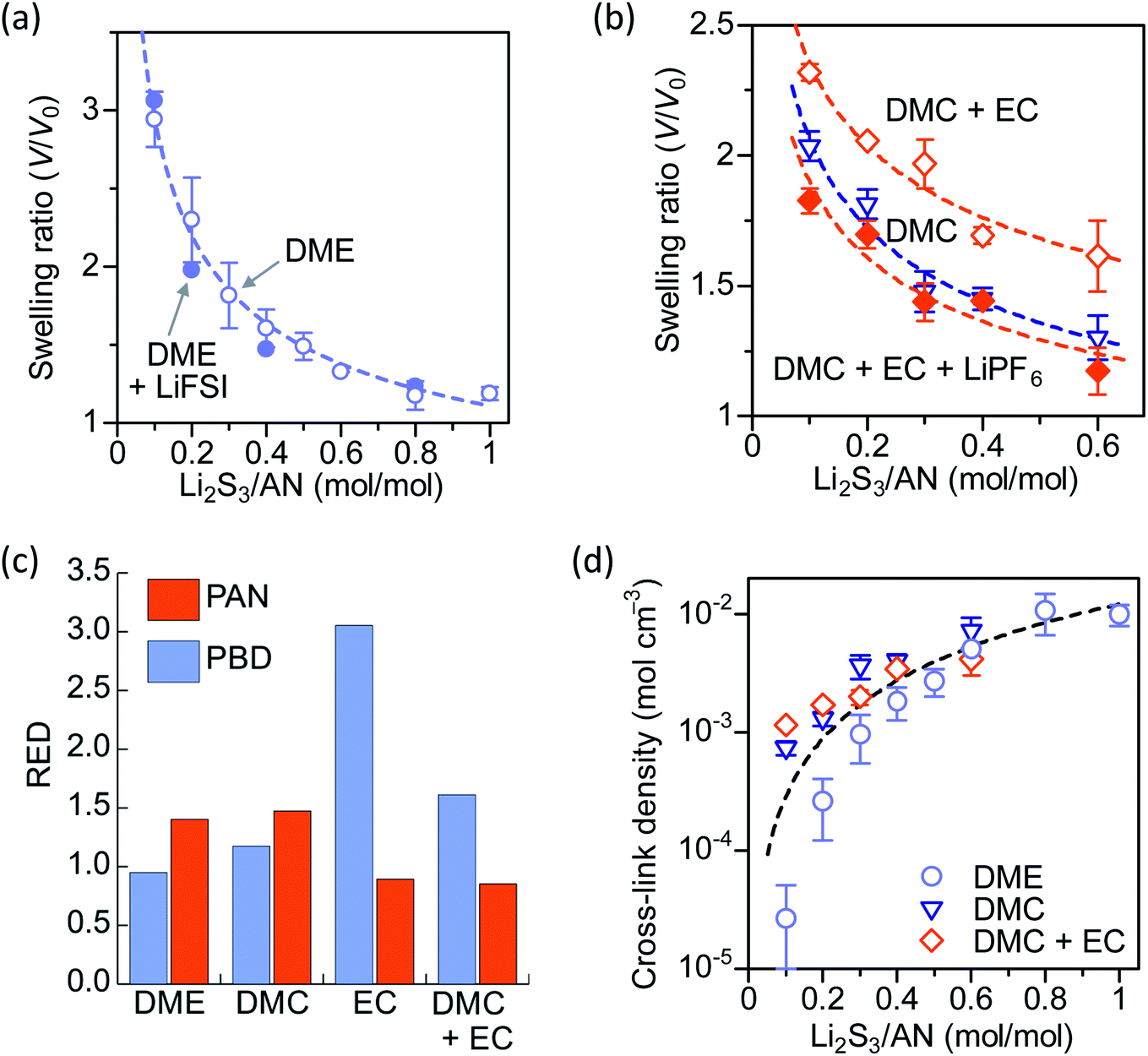 | ||
| Fig. 3 Swelling ratio of PAN–PBD in (a) DME (+LiFSI, 1 ml kg−1), and (b) DMC, DMC + EC (1:1) (+LiPF6, 1 mol kg−1). Formation of CN–Li+ pairs in DMC + EC decreases the swelling ratio when LiPF6 is added. (c) Relative Energy Difference (RED) between the polymer (PAN and PBD) and the solvents (DME, DMC, EC, and DMC + EC). A good solvent for a polymer generally shows RED < 1. (d) Cross-link density of PAN–PBD calculated from the Flory–Rehner equation by using the results from (a) and (b). All the dashed curves represent the least-square fittings to the experimental data. | ||
To obtain a theoretical basis of the swelling behavior, we used HSPs and quantified the degree of interaction between the polymers (PAN, and PBN) and the solvents (DME, DMC, EC, and DMC + EC). The relative energy difference (RED) is a useful parameter to evaluate the solubility of polymers in solvents,24 which is calculated from the difference of HSPs between the polymer and the solvent (Appendix A, ESI†). Typically, a good solvent shows RED < 1 (i.e. the polymer and the solvent have a similar polarity), while a poor solvent shows RED > 1. DME shows a low RED with PBD (0.95) and a high RED with PAN (1.40), indicating preferential solvation of the PBD phase (Fig. 3c). DMC shows high REDs with both PBD (1.17) and PAN (1.47), which explains the low swelling ratio of PAN–PBD in DMC. In contrast, pure EC shows a low RED with PAN (0.89) and a significantly higher RED with PBD (3.05), suggesting that only the PAN phase can swell in EC. In a mixed solvent of DMC and EC (1:1 in weight ratio), RED with PAN (0.85) shows a similar value to pure EC. Despite that the RED with PBD (1.61) is lower in the DMC/EC mixed solvent than in pure EC, RED > 1 suggests that the solvation of the PBD phase is still unlikely in DMC/EC. Therefore, the higher swelling ratio in the DMC/EC mixed solvent than pure DMC is explained by the reduced RED value with the PAN phase. The favorable solvation of PAN in DMC/EC also explains that the decrease of the swelling ratio by the addition of salts (LiPF6 and LiClO4) is caused by the formation of the CN–Li+ pair, which behaves as a cross-linker.
The cross-link density (N) of PAN–PBD can be calculated from the swelling ratio by using the Flory–Rehner equation:25,26
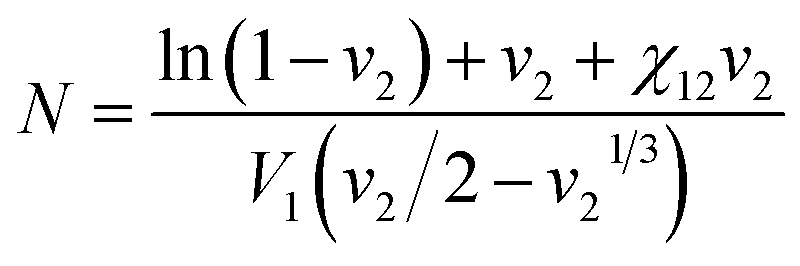 | (1) |
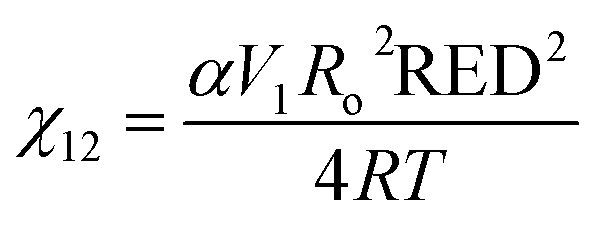 | (2) |
:PBD = 32:68, vol/vol) and, therefore, N is also an averaged value of the entire copolymer structure. Typically, a lower swelling ratio (larger volume fraction of polymer, v2 in eqn (1)) results in greater values of N, because polymer swelling is less limited by chemical cross-linking. As a result, the calculated N value from the swelling ratio in DMC and DMC/EC is greater than the corresponding values in DME at low Li2S3/AN ratios (Fig. 3d). This result may be due to the following reasons: (1) pure DMC is a poor solvent for both PAN and PBD and thus results in a low swelling ratio. (2) the DMC/EC mixture is a good solvent for the PAN segments only. The chemical cross-linking and the strong dipole–dipole interaction28 between the nitrile groups make dissociation of the PAN segments more difficult and reduce the swelling ratio. (3) In contrast, the swelling of non-polar PBD segments in DME is more facilitated thanks to the lower degree of self-association between the PBD segments and thus results in higher swelling ratios.
Ion transport mechanism
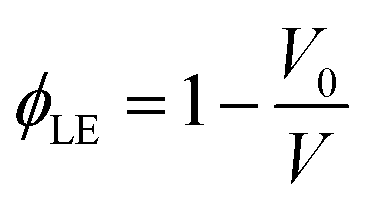 | (3) |
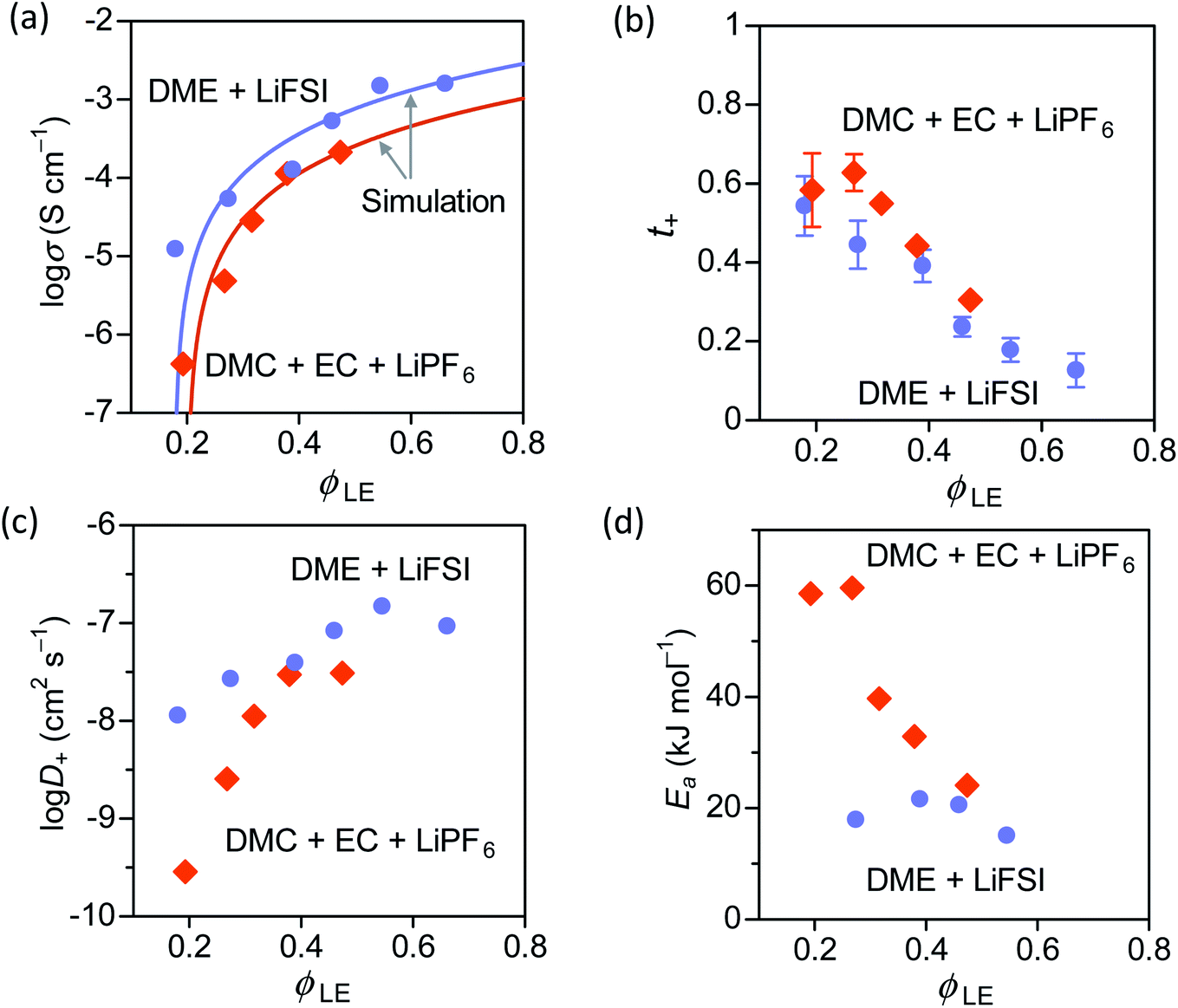 | ||
| Fig. 4 (a) Ionic conductivity, (b) transference number of Li+ ion, (c) diffusion coefficient of Li+ ion, and (d) activation energy of PAN–PBD swollen in DME + LiFSI (1 mol kg−1), and in DMC + EC (1:1) + LiPF6 (1 mol kg−1). The volume fraction of the liquid electrolyte (ϕLE) in the swollen polymer is controlled by the cross-link density of PAN–PBD. | ||
In both carbonate and ether electrolytes, the change of σ values at ϕLE > 0.4 is gradual, and then the σ value sharply decreases as ϕLE drops to 0.2. A similar change of the σ values with decreasing ϕLE was also observed in different gel polymer electrolytes produced from a polymer blend of nitrile-butadiene/styrene-butadiene rubbers (NBR/SBR),29 PEO,12,30 and poly(methyl methacrylate) (PMMA).31 However, no theoretical model has been proposed to explain the relation between σ and ϕLE.
Here we demonstrate the applicability of a percolation model for the first time in gel polymer electrolyte swollen in organic solvents. The percolation model was initially proposed by Hsu to explain an insulator-to-conductor transition of a proton-conductive membrane (e.g. Nafion) as a function of the water content:32
| σ = σ0(ϕLE − ϕ0)n | (4) |
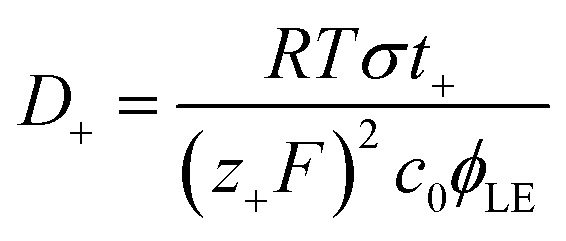 | (5) |
N–Li+ interaction at lower ϕLE values. This explanation is further supported by the increase of Ea only observed when PAN–PBD is swollen in DMC/EC (Fig. 4d, the Arrhenius plot in Fig. S10, ESI†). In contrast, the Ea of the PAN–PBD swollen in DME electrolyte (where PBD is the ion-conduction phase) remains constant even at low ϕLE, indicating that Li+ transport is decoupled from the interaction with the polymer host.
Mechanical and rheological properties
Tensile strain is applied to dry PAN–PBD to study the influence of cross-link density on mechanical properties (Fig. 5a). At the lowest cross-link density (Li2S3/AN = 0.1), PAN–PBD shows a completely elastic elongation without any plastic deformation until fracture. At higher cross-link densities, the transition from elastic to plastic deformation appears after approximately 3% of the strain is applied (inset of Fig. 5a), and the yield strength increases with increasing cross-link density. Young's modulus (E) is calculated from the slope of the stress–strain curve in the elastic region and plotted with the cross-link density estimated from the swelling ratio in DME (Fig. 5b). The value of E increases by a factor of 102 when the cross-link density is increased from 3 × 10−5 to 1 × 10−3 mol cm−3, while the elongation at break remains at 200%, showing the increasing toughness of the material. Further increase of the cross-link density to 1 × 10−2 mol cm−3 increases E by a factor of four, while the elongation at break decreases to 50%, transforming into a relatively rigid and brittle material.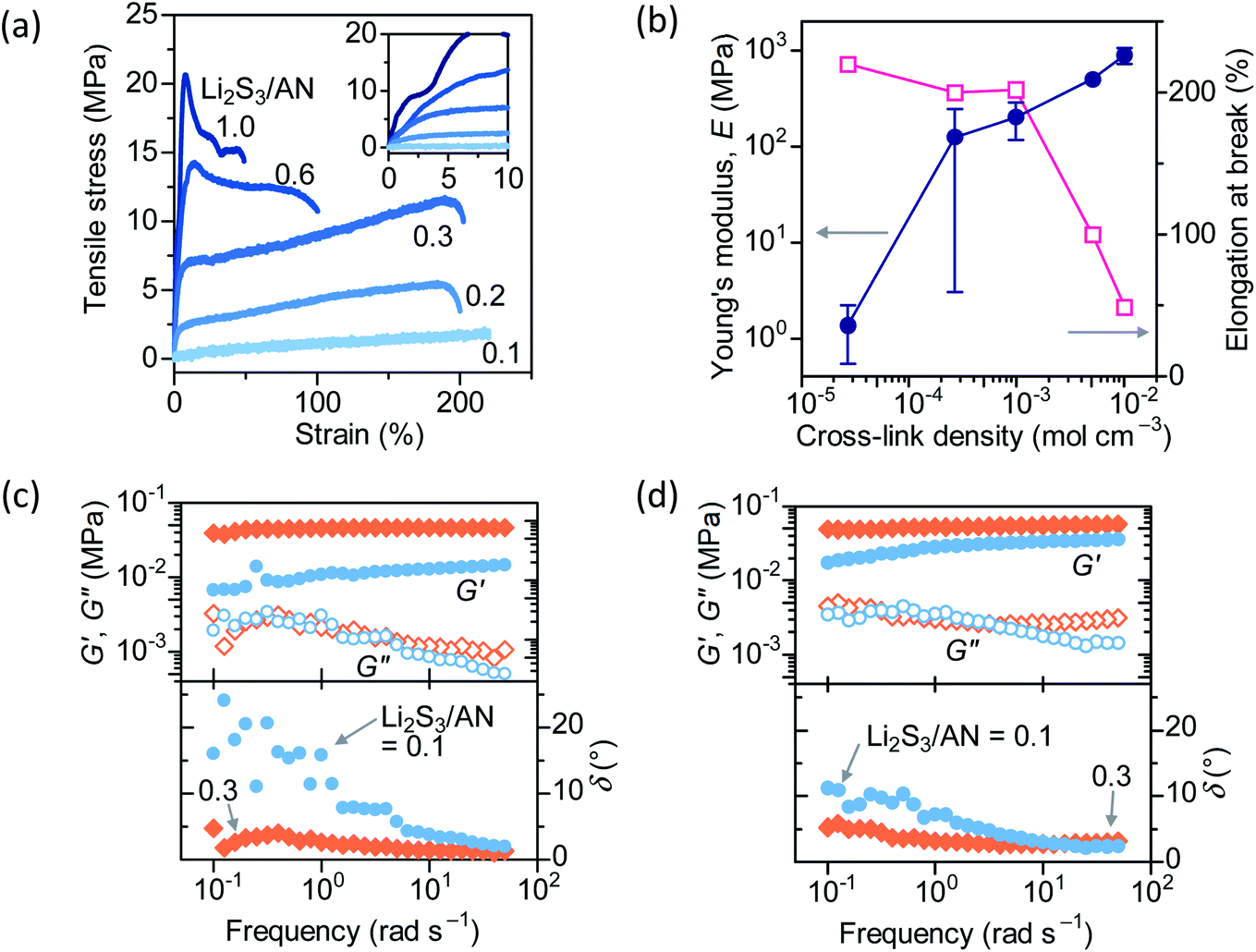 | ||
| Fig. 5 (a) Stress–strain curve of the dry PAN–PBD. (b) Young's modulus and elongation at break evaluated from (a), plotted with the cross-link density. (c and d) Frequency dependence of the storage and loss modulus (G′ and G′′), and the phase angle (δ) of PAN–PBD swollen in (c) DME + LiClO4, and in (d) DMC + EC (1:1) + LiClO4, evaluated under a constant strain of 0.1%. | ||
Oscillatory shear strain is applied to PAN–PBD swollen in DME/LiClO4 or DMC/EC/LiClO4 to evaluate the rheological properties. LiClO4 is used instead of LiFSI or LiPF6 because of the better stability in the ambient environment. The linear viscoelastic regime is evaluated to be below 1% of the strain (Fig. S11, ESI†). Frequency dependence of storage (G′) and loss (G′′) modulus, and the phase angle (δ = tan−1(G′′/G′)) are evaluated at a constant strain of 0.1% (under the linear viscoelastic regime) (Fig. 5c and d). In both DME and DMC/EC electrolytes, the higher cross-link density (Li2S3/AN = 0.3) results in larger G′ values while G′′ shows little difference from the lower cross-link density (Li2S3/AN = 0.1). When the oscillation frequency is reduced from 10 to 0.1 rad s−1, G′ of Li2S3/AN = 0.1 starts to decrease, and δ increases from nearly 0° to 20° in DME and to 10° in DMC/EC electrolytes, respectively. The increase of δ indicates the transition from a purely elastic body to a more viscous material.
Morphology of Li metal
Previously, both PAN6 and PBD43 were used as a PPC5 for Li metal anodes. PAN was reported to reduce the reactivity of EC with Li metal through the dipole–dipole interaction,6 while the PBD copolymer was used as the elastic binder for inorganic nanofillers.43 To use cross-linked PAN–PBD as the PPC, we reduced the thickness to 10 μm and placed it between the battery separator and the copper (Cu) current collector (Fig. S12, ESI†). The PPC is swollen to the equilibrium in the carbonate LE (DMC + EC (1:1) + LiPF6 (1 mol kg−1) + fluoroethylene carbonate, FEC (5 wt%) as an additive) before cell assembly. The additional FEC does not change the swelling behavior of the PPC. During the first deposition of Li metal on Cu, voltage polarization increases with the increasing cross-link density of PPC (Fig. S13, ESI†) because of the reduced σ (Fig. 4a). After Li deposition (capacity = 1 mA h cm−2), we observed that the Li metal breaks the PPC and deposits on the surface when the Li2S3/AN ratio is equal to or greater than 0.4, while all the Li metal is plated underneath the PPC below the ratio of 0.4 (Fig. 6a, see Fig. S14 for the optical images underneath the PPC, ESI†). The observation agrees with the previous study where a thick Nafion PPC (50 μm) with low σ (<10−5 S cm−1) resulted in the non-uniform Li deposition concentrated at the local defects in the PPC.7 Conductivity measurements (Fig. 4a) indicate that a σ value higher than at least 10−5 S cm−1 is required to deposit Li metal underneath the 10 μm-thick PPCs at the initial deposition, and σ > 10−4 S cm−1 is desired for the long-term cycling stability of the PPC (see below).
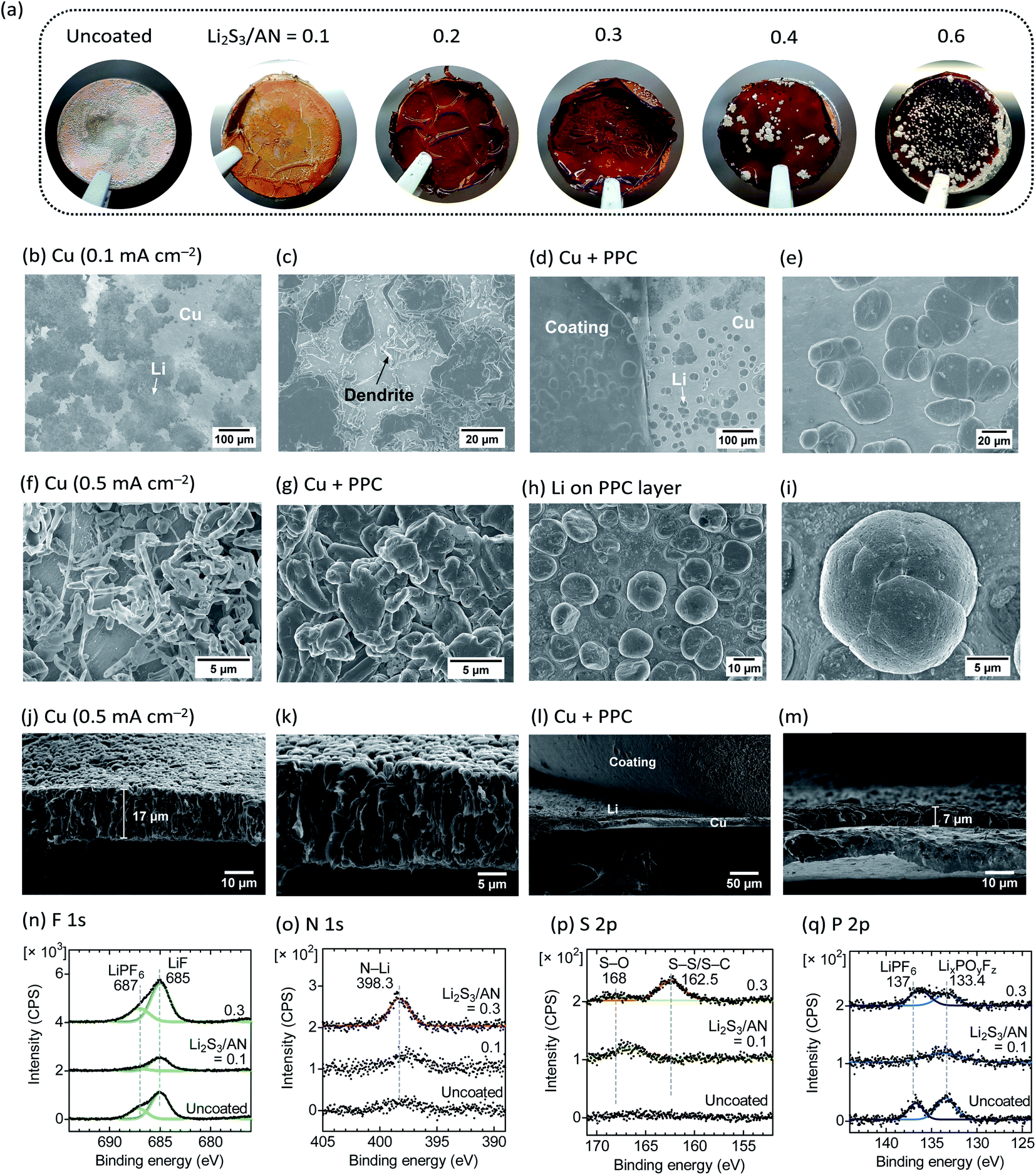 | ||
| Fig. 6 (a) Optical images after the first deposition of Li metal on Cu foil with/without PAN–PBD PPC cross-linked at Li2S3/AN = 0.1–0.6. (b–g) Surface SEM images after the first deposition of Li metal on (b, c and f) uncoated Cu, and on (d, e and g) Cu with the PPC (cross-linked at Li2S3/AN = 0.1). (h and i) Li particles adhered on the viscous PPC layer, showing a spherical morphology resembling (e). (j–m) Cross-sectional SEM images of Li metal deposited on (j and k) uncoated Cu, and on (l and m) Cu with the PPC layer. (n–q) XPS spectra of Li metal after the first deposition on Cu with/without the PPC (Li2S3/AN = 0.1 or 0.3). Current density = 0.1 (a, b–e, n–q), and 0.5 (f–m) mA cm−2. Charge capacity = 1 mA h cm−2 (a–q). Electrolyte = DMC + EC (1:1) + LiPF6 (1 mol kg−1) + FEC (5 wt%). | ||
On the uncoated Cu (current density = 0.1 mA cm−2), two different morphologies of the Li metal are observed: a flat Li deposit where the Li is deformed by the compressive pressure from the battery separator, and a dendritic shape where the Li is less compressed (Fig. 6b and c). In contrast, the Li metal deposited underneath the PPC uniformly shows a spherical morphology with a diameter of 20 μm, and no dendritic Li is observed (Fig. 6d and e). The spherical Li deposits are also observed underneath the PPC with slightly higher cross-link density (Li2S3/AN = 0.2, 0.3). However, when the Li breaks the PPC and plates on the surface (Li2S3/AN = 0.4), only flat and compressed Li deposits (like the morphology on the uncoated Cu) are observed (Fig. S15, ESI†).
When Li is deposited at higher current density (0.5 mA cm−2), the Li on the uncoated Cu shows a smaller particle size (<2 μm), and more pronounced dendritic morphology (Fig. 6f, g, see Fig. S16 for the optical images, and Fig. S17 for the SEM images at different magnifications, ESI†). In contrast, the Li deposited underneath the PPC shows a granular morphology with a larger particle size (≈5 μm) than the Li on the uncoated Cu (Fig. 6g). Furthermore, the Li particles adhered on the least-cross-linked PPC (Li2S3/AN = 0.1) show a large spherical morphology (diameter ≈ 15 μm, Fig. 6h and i) which resembles the Li deposited underneath the PPC at 0.1 mA cm−2 (Fig. 6d and e). No spherical Li particle is observed on the PPC at Li2S3/AN = 0.3. The higher viscosity (δ ≈ 10° at the oscillation frequency of 0.1 rad s−1, Fig. 5d) of the PPC at Li2S3/AN = 0.1 appears to be beneficial for the better adhesion with the Li particles. A recent report on the morphological improvement of Li deposits in the viscous LE further supports the merit of using the viscous PPC.44 Contrary to the common belief that a high shear modulus is required to suppress Li dendrites,45,46 the swollen PPC (with a significantly lower shear modulus of G ∼ 10−2 MPa than the dry SPE, for example, G(PEO) = 26.2 MPa)45 is proved to be useful as the protective coating for Li metal.
The cross-sectional SEM images of Li metal deposited on the uncoated Cu foil (Fig. 6j and k) show the porous structure composed of Li metal filaments. The thickness of the Li metal is 17 μm. In contrast, the Li metal deposited under the PPC layer (cross-linked at Li2S3/AN = 0.1, Fig. 6l and m) is much denser, and the thickness is only 7 μm. Note that this thickness is close to the value of a fully dense Li metal layer (4.85 μm when 1 mA h cm−2 of Li is deposited).5 The denser structure is advantageous for reducing the surface area of Li metal in contact with the LE and preventing the formation of isolated Li metal.5
We also observed the morphology of Li metal under the PPC layer in the ether LE (DME + 1 mol kg−1 LiFSI) (Fig. S23, ESI†). Because the ether LE forms a more stable SEI layer and has lower reactivity with Li metal than the carbonate LE,22 no Li dendrite is observed even without the PPC layer. Interestingly, the application of the PPC layer results in the spherical shape of Li metal, which resembles the morphology observed in the carbonate LE. This result indicates that the spherical morphology is likely a result of the viscoelastic properties of the PPC layer and independent from the choice of the electrolyte.
The combination of the non-polar polymer film (PBD) with the low-polarity electrolyte (DME) shows the advantage of the lower Ea value for Li+ ion transport (Fig. 4d). In the previous studies by Archer and Bao, PPCs with lower surface energy values (i.e. lower polarity) were found to promote the nucleation of larger Li deposits.11,47 The low Ea values associated with the low surface energy of PPCs may have a great impact on the size of Li deposits.
Composition of the SEI layer
XPS spectra in the F 1s and P 2p region reveal that LiF and phosphates (LixPOyFz)48 are invariably present in the SEI layer of the Li deposited on both uncoated and PAN–PBD PPC-coated Cu (Fig. 6n and q). The LiF and phosphates are the decomposition products of LiPF6 or FEC in the LE phase. The N 1s and S 2p spectra respectively show the increasing peak intensity of the N–Li bond,49,50 sulfoxide (S–O), S–S, and possibly the S–C bond21 with increasing Li2S3/AN ratio (Fig. 6o and p). Because the LE contains no N or S species, the N and S compounds must be produced from the decomposition of the PAN–PBD PPC. The concentration of PAN segments in the swollen state is higher at higher cross-link densities (i.e. low LE content), and therefore, more N–Li bond (likely from Li3N)50 is formed from the reduction of CN on the Li surface. The S–O bond is probably formed from the reaction of EC/DMC solvent with residual polysulfide species in the PAN–PBD PPC, and the presence of the S–S bond indicates that the polysulfide species is partially transferred onto the Li surface. XPS analysis points out the importance of tuning the cross-link density (the swelling ratio) of the PPC to balance the ratio between the SEI products derived from the LE, or from the PPC.
Long-term stability of the PPC layer
The long-term stability of PAN–PBD PPC is evaluated by the repetitive deposition/dissolution of Li metal for 50 cycles (Fig. S18, ESI†). All Li is deposited underneath the PPC at Li2S3/AN < 0.3, while a minor amount of Li is deposited on top of the PPC surface at Li2S3/AN = 0.3. The low values of ϕLE (≈0.3), σ (<10−4 S cm−1), and also the low viscosity (δ < 5°) are likely to cause a tip-driven growth of the Li filament through the local defect, which is commonly observed in more ionically resistive and brittle solid state electrolytes.51 Despite the morphological improvement of the Li deposits, the average coulombic efficiency (CE) from 10 to 50 cycles of the Li metal deposition/dissolution with the PAN–PBD PPC shows comparable values to the CE on the uncoated Cu (Fig. S19, ESI†). The morphology of Li deposited after the 50 cycles shows aggregation of fine Li filaments with the size of less than 1 μm (Fig. S20, ESI†). The morphological change during the long-term cycling is possibly caused by the build-up of inactive Li deposits,52 which reduces the effective surface area of Li directly in contact with the PPC, and also the effective pressure exerted on the Li deposits. Optimization of the current collector (e.g. 3D structure),10 and a rigorous control on the cell stack pressure could be a promising approach to enhance the benefits of PPCs.Conclusions
In summary, we employed the cross-linked PAN–PBD copolymer as a useful platform to quantify and model the ion transport and rheological properties in carbonate and ether LEs. The bulk properties of the PAN–PBD PPC are correlated with the protective functions and the morphology of Li metal underneath the PPC layer (Fig. 7). Low cross-link density results in high ϕLE and σ values (which can be simulated by the percolation model), and low Ea values (∼20 kJ mol−1). The high viscosity (δ > 10°) of the PPC leads to better adhesion with the Li deposits and improves the Li morphology from the dendritic shape to the large spherical particles (15–20 μm). In contrast, the high cross-link density results in low ϕLE, σ, and δ values, which promote tip-driven Li growth and penetration through the PPC. Once an electronically conductive path is formed inside the PPC, Li deposition initiates from the PPC surface, leading to the failure of the protective function. To avoid the failure of the PPC over long cycles, a σ value higher than 10−4 S cm−1 is required. On the other hand, we must satisfy the requirement for the σ value at the lowest possible ϕLE values to effectively block LE permeation through the PPC layer and to increase the t+ value. In this regard, none of the reported PPCs meet these requirements. As a general design rule, a non-polar PPC is more advantageous than a polar PPC because the weaker interaction between Li+ and a non-polar polymer segment allows ion transport at lower Ea values. The high chemical stability of the non-polar alkyl segments with Li metal, addition of thermodynamically stable conductive inorganic fillers, and the combination with the state-of-the-art ether electrolyte are expected to break the present upper limit of the CE of Li metal anodes.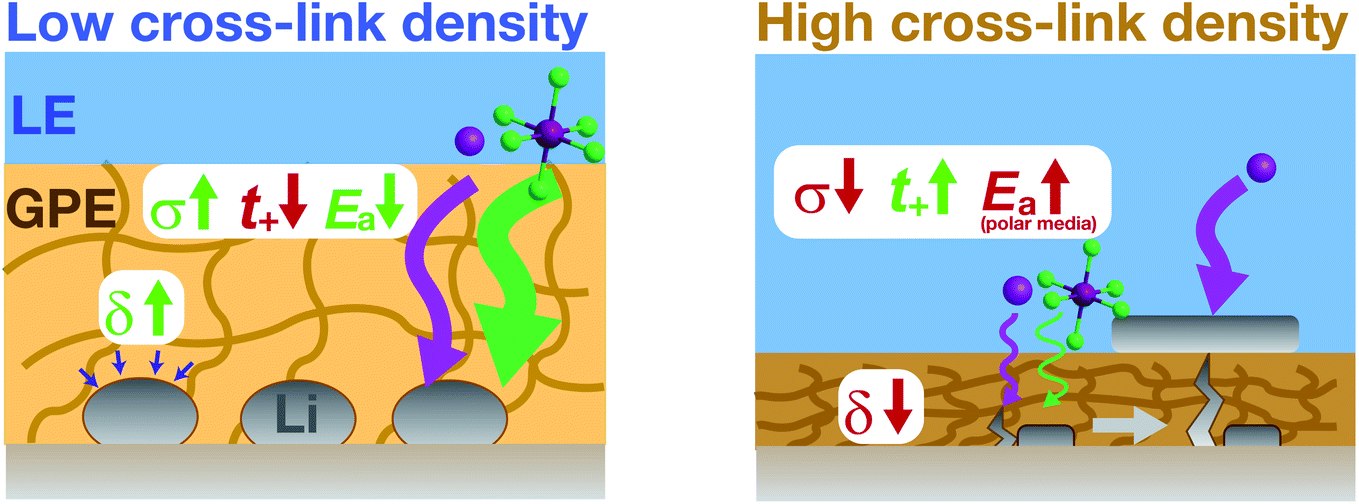 | ||
| Fig. 7 Schematic representation of the ion transport properties (σ, t+, and Ea), and rheology (phase angle, δ) of the PPC at low/high cross-link densities, and the Li deposition process through the PPC layer. At a low cross-link density, the large swelling ratio increases σ but decreases t+ (i.e. lower relative mobility of Li+ ion than the anion). The high viscosity (large δ value) at the low cross-link density results in the spherical morphology of Li deposition. At a high cross-link density, the low swelling ratio decreases σ but increases t+. The increasing degree of dipole–Li+ ion interaction also increases Ea in a polar polymer swollen in a polar LE. The low values of σ and δ promote the tip-driven Li growth through the local defect, eventually leading to Li plating on top of the PPC layer. | ||
Author contributions
P. L. conceived the idea and supervised the study. H. Z. planned the experiment, analyzed all the data, and wrote the manuscript. H. L. and X. X. carried out the preliminary experiment on the cross-linking of PAN–PBD. Z. W. performed the rheological measurements. S. Y. performed the tensile test and XPS on the cross-linked PAN–PBD. G. V. performed XPS on Li metal. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Office of Vehicle Technologies of the U.S. Department of Energy through the Advanced Battery Materials Research (BMR) Program (Battery500 Consortium) under Contract No. DE-EE0007764.References
- J. Liu, Z. Bao, Y. Cui, E. J. Dufek, J. B. Goodenough, P. Khalifah, Q. Li, B. Y. Liaw, P. Liu, A. Manthiram, Y. S. Meng, V. R. Subramanian, M. F. Toney, V. V Viswanathan, M. S. Whittingham, J. Xiao, W. Xu, J. Yang, X.-Q. Yang and J.-G. Zhang, Nat. Energy, 2019, 4, 180–186 CrossRef CAS.
- C. Niu, H. Lee, S. Chen, Q. Li, J. Du, W. Xu, J.-G. Zhang, M. S. Whittingham, J. Xiao and J. Liu, Nat. Energy, 2019, 4, 551–559 CrossRef CAS.
- X. Cao, X. Ren, L. Zou, M. H. Engelhard, W. Huang, H. Wang, B. E. Matthews, H. Lee, C. Niu, B. W. Arey, Y. Cui, C. Wang, J. Xiao, J. Liu, W. Xu and J.-G. Zhang, Nat. Energy, 2019, 4, 796–805 CrossRef CAS.
- X. Cheng, R. Zhang, C. Zhao and Q. Zhang, Chem. Rev., 2017, 117, 10403–10473 CrossRef CAS PubMed.
- H. Zhou, S. Yu, H. Liu and P. Liu, J. Power Sources, 2020, 450, 227632 CrossRef CAS.
- J. Bae, Y. Qian, Y. Li, X. Zhou, J. B. Goodenough and G. Yu, Energy Environ. Sci., 2019, 12, 3319–3327 RSC.
- J. Song, H. Lee, M.-J. Choo, J.-K. Park and H.-T. Kim, Sci. Rep., 2015, 5, 14458 CrossRef CAS PubMed.
- Z. Tu, S. Choudhury, M. J. Zachman, S. Wei, K. Zhang, L. F. Kourkoutis and L. A. Archer, Joule, 2017, 1, 394–406 CrossRef CAS.
- Z. Yu, D. G. Mackanic, W. Michaels, M. Lee, A. Pei, D. Feng, Q. Zhang, Y. Tsao, C. V. Amanchukwu, X. Yan, H. Wang, S. Chen, K. Liu, J. Kang, J. Qin, Y. Cui and Z. Bao, Joule, 2019, 3, 2761–2776 CrossRef CAS.
- Y. Gao, Z. Yan, J. L. Gray, X. He, D. Wang, T. Chen, Q. Huang, Y. C. Li, H. Wang, S. H. Kim, T. E. Mallouk and D. Wang, Nat. Mater., 2019, 18, 384–389 CrossRef CAS PubMed.
- J. Lopez, A. Pei, J. Y. Oh, G.-J. J. N. Wang, Y. Cui and Z. Bao, J. Am. Chem. Soc., 2018, 140, 11735–11744 CrossRef CAS PubMed.
- G. B. Appetecchi, Y. Aihara and B. Scrosati, Solid State Ionics, 2004, 170, 63–72 CrossRef CAS.
- N. Verdier, D. Lepage, R. Zidani, A. Prébé, D. Aymé-Perrot, C. Pellerin, M. Dollé and D. Rochefort, ACS Appl. Energy Mater., 2020, 3, 1099–1110 CrossRef CAS.
- N. Verdier, D. Lepage, A. Prébé, D. Aymé-Perrot, M. Dollé and D. Rochefort, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 1825–1833 CrossRef CAS.
- Z. Fu, B. Liu, Y. Liu, B. Li and H. Zhang, Ind. Eng. Chem. Res., 2018, 57, 8348–8359 CrossRef CAS.
- E. Cipriani, M. Zanetti, P. Bracco, V. Brunella, M. P. Luda and L. Costa, Polym. Degrad. Stab., 2016, 123, 178–188 CrossRef CAS.
- S. Dalton, F. Heatley and P. M. Budd, Polymer, 1999, 40, 5531–5543 CrossRef CAS.
- B. Barua and M. C. Saha, Polym. Eng. Sci., 2018, 58, 1315–1321 CrossRef CAS.
- J. Guo, Z. Yang, Y. Yu, H. D. Abruña and L. A. Archer, J. Am. Chem. Soc., 2013, 135, 763–767 CrossRef CAS PubMed.
- B. S. Lee, S. Cui, X. Xing, H. Liu, X. Yue, V. Petrova, H. D. Lim, R. Chen and P. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 38928–38935 CrossRef CAS PubMed.
- Z. Wang, Y. Dong, H. Li, Z. Zhao, H. Bin Wu, C. Hao, S. Liu, J. Qiu and X. W. Lou, Nat. Commun., 2014, 5, 5002 CrossRef CAS PubMed.
- J. Qian, W. A. Henderson, W. Xu, P. Bhattacharya, M. Engelhard, O. Borodin and J.-G. Zhang, Nat. Commun., 2015, 6, 6362 CrossRef CAS PubMed.
- V. Hervio, B. Bresson, A. Brûlet, I. J. Paredes, A. Sahu, V. Briand, C. Creton and G. E. Sanoja, Macromolecules, 2021, 54, 2828–2834 CrossRef CAS.
- C. M. Hansen, Hansen Solubility Parameters, CRC Press, 2nd edn, 2007 Search PubMed.
- S. G. Croll, J. Coat. Technol. Res., 2010, 7, 49–55 CrossRef CAS.
- J. L. Valentín, J. Carretero-González, I. Mora-Barrantes, W. Chassé and K. Saalwächter, Macromolecules, 2008, 41, 4717–4729 CrossRef.
- T. Lindvig, M. L. Michelsen and G. M. Kontogeorgis, Fluid Phase Equilib., 2002, 203, 247–260 CrossRef CAS.
- J. M. Alía, H. G. M. Edwards, W. R. Fawcett and T. G. Smagala, J. Phys. Chem. A, 2007, 111, 793–804 CrossRef PubMed.
- M. Matsumoto, J. E. Soc, M. Matsumoto, T. Ichino, J. S. Rutt and S. Nishi, J. Electrochem. Soc., 1993, 140, L151 CrossRef CAS.
- M. Kono, E. Hayashi and M. Watanabe, J. Electrochem. Soc., 1999, 146, 1626–1632 CrossRef CAS.
- S. Kuwabata and M. Tomiyori, J. Electrochem. Soc., 2002, 149, A988 CrossRef CAS.
- W. Y. Hsu, J. R. Barkley and P. Meakin, Macromolecules, 1980, 13, 198–200 CrossRef CAS.
- D. R. Morris and X. Sun, J. Appl. Polym. Sci., 1993, 50, 1445–1452 CrossRef CAS.
- T. Thampan, S. Malhotra, H. Tang and R. Datta, J. Electrochem. Soc., 2000, 147, 3242–3250 CrossRef CAS.
- P. Knauth, L. Pasquini, R. Narducci, E. Sgreccia, R.-A. Becerra-Arciniegas and M. L. Di Vona, J. Membr. Sci., 2020, 617, 118622 CrossRef.
- D. M. Pesko, S. Sawhney, J. Newman and N. P. Balsara, J. Electrochem. Soc., 2018, 165, A3014–A3021 CrossRef CAS.
- T. P. Liyana-Arachchi, J. B. Haskins, C. M. Burke, K. M. Diederichsen, B. D. McCloskey and J. W. Lawson, J. Phys. Chem. B, 2018, 122, 8548–8559 CrossRef CAS PubMed.
- S. A. Krachkovskiy, J. D. Bazak, S. Fraser, I. C. Halalay and G. R. Goward, J. Electrochem. Soc., 2017, 164, A912–A916 CrossRef CAS.
- J. Popovic, D. Höfler, J. P. Melchior, A. Münchinger, B. List and J. Maier, J. Phys. Chem. Lett., 2018, 9, 5116–5120 CrossRef CAS PubMed.
- G. M. Geise, M. A. Hickner and B. E. Logan, ACS Appl. Mater. Interfaces, 2013, 5, 10294–10301 CrossRef CAS PubMed.
- S. T. Russell, R. Pereira, J. T. Vardner, G. N. Jones, C. Dimarco, A. C. West and S. K. Kumar, Macromolecules, 2020, 53, 1014–1023 CrossRef CAS.
- J. Newman and K. E. Thomas, Electrochemical Systems, John Wiley & Sons, Inc., Hoboken, New Jersey, 3rd edn, 2004 Search PubMed.
- Y. Liu, D. Lin, P. Y. Yuen, K. Liu, J. Xie, R. H. Dauskardt and Y. Cui, Adv. Mater., 2017, 29, 1605531 CrossRef PubMed.
- S. Wei, Z. Cheng, P. Nath, M. D. Tikekar, G. Li and L. A. Archer, Sci. Adv., 2018, 4, eaao6243 CrossRef PubMed.
- P. Barai, K. Higa and V. Srinivasan, Phys. Chem. Chem. Phys., 2017, 19, 20493–20505 RSC.
- C. Monroe and J. Newman, J. Electrochem. Soc., 2005, 152, A396 CrossRef CAS.
- S. Stalin, M. Tikekar, P. Biswal, G. Li, H. E. N. Johnson, Y. Deng, Q. Zhao, D. Vu, G. W. Coates and L. A. Archer, Nano Lett., 2020, 20, 5749–5758 CrossRef CAS PubMed.
- B. Philippe, R. Dedryvère, M. Gorgoi, H. Rensmo, D. Gonbeau and K. Edström, Chem. Mater., 2013, 25, 394–404 CrossRef CAS.
- L. Yu, N. L. Canfield, S. Chen, H. Lee, X. Ren, M. H. Engelhard, Q. Li, J. Liu, W. Xu and J.-G. Zhang, ChemElectroChem, 2018, 5, 761–769 CrossRef CAS.
- C. Yan, Y.-X. Yao, X. Chen, X.-B. Cheng, X.-Q. Zhang, J.-Q. Huang and Q. Zhang, Angew. Chem., 2018, 130, 14251–14255 CrossRef.
- K. B. Hatzell, X. C. Chen, C. L. Cobb, N. P. Dasgupta, M. B. Dixit, L. E. Marbella, M. T. McDowell, P. P. Mukherjee, A. Verma, V. Viswanathan, A. S. Westover and W. G. Zeier, ACS Energy Lett., 2020, 5, 922–934 CrossRef CAS.
- C. Fang, J. Li, M. Zhang, Y. Zhang, F. Yang, J. Z. Lee, M.-H. Lee, J. Alvarado, M. A. Schroeder, Y. Yang, B. Lu, N. Williams, M. Ceja, L. Yang, M. Cai, J. Gu, K. Xu, X. Wang and Y. S. Meng, Nature, 2019, 572, 511–515 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc06651f |
| ‡ Present address: Department of Chemistry, Graduate School of Science, The University of Tokyo, Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. |
| This journal is © The Royal Society of Chemistry 2021 |