
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient and selective DNA modification on bacterial membranes†
Qian
Tian
,
Yousef
Bagheri
 ,
Puspam
Keshri
,
Rigumula
Wu
,
Kewei
Ren
,
Qikun
Yu
,
Bin
Zhao
and
Mingxu
You
*
,
Puspam
Keshri
,
Rigumula
Wu
,
Kewei
Ren
,
Qikun
Yu
,
Bin
Zhao
and
Mingxu
You
*
Department of Chemistry, University of Massachusetts, Amherst, Massachusetts 01003, USA. E-mail: mingxuyou@chem.umass.edu
First published on 28th December 2020
Abstract
With highly precise self-assembly and programmability, DNA has been widely used as a versatile material in nanotechnology and synthetic biology. Recently, DNA-based nanostructures and devices have been engineered onto eukaryotic cell membranes for various exciting applications in the detection and regulation of cell functions. While in contrast, the potential of applying DNA nanotechnology for bacterial membrane studies is still largely underexplored, which is mainly due to the lack of tools to modify DNA on bacterial membranes. Herein, using lipid–DNA conjugates, we have developed a simple, fast, and highly efficient system to engineer bacterial membranes with designer DNA molecules. We have constructed a small library of synthetic lipids, conjugated with DNA oligonucleotides, and characterized their membrane insertion properties on various Gram-negative and Gram-positive bacteria. Simply after incubation, these lipid–DNA conjugates can be rapidly and efficiently inserted onto target bacterial membranes. Based on the membrane selectivity of these conjugates, we have further demonstrated their applications in differentiating bacterial strains and potentially in pathogen detection. These lipid–DNA conjugates are promising tools to facilitate the possibly broad usage of DNA nanotechnology for bacterial membrane analysis, functionalization, and therapy.
Introduction
Bacterial membranes play a critical role in cellular communication, survival, and topology. As a highly complex and dynamic system, the membrane structures help protect bacteria against different hostile environments.1 By engineering bacterial membranes with artificial functional moieties, various bioanalytical and medical applications have been achieved, including membrane imaging, photodynamic therapy, immunotherapy, and regulating host–pathogen interactions.2–4 Different types of fluorophores, photosensitizers, antibiotics, peptides, and synthetic polymers have been used as the functional moieties in these membrane modifications.4–7As another promising functional unit, surprisingly, DNA has rarely been used for bacterial membrane engineering.8 DNA can form highly precise and programmable self-assembly and well-defined nanostructures. A large variety of DNA nanodevices have been developed for nanomedicine, diagnostics, computing, biophysical characterization, and structural biology.9–11 More recently, these DNA nanodevices have been modified onto mammalian cell membranes to generate artificial membrane channels, detect membrane signaling, monitor membrane biophysical phenomena, regulate cell-surface and intercellular interactions, and deliver genes and various cargos.12–23
Our goal in this study is to provide a simple and efficient approach to functionalize DNAs onto bacterial cell membranes, with the hope of achieving similar level of versatile decoration, biosensing, and therapy as that on the mammalian cell membranes. Different methods have been developed to engineer bacterial membranes, including metabolic labeling, chemical cross-linking, and hydrophobic insertion.3,24 For example, a number of DNA aptamer molecules have been identified to selectively recognize specific bacterial membrane targets and further used for the pathogen detection.25–28 However, the membrane modification efficiency and affinity of these aptamers are often limited. In addition, the identification of aptamers for many bacterial strains is still challenging with the current time-consuming and labor-intensive screening process. Considering the straightforward procedure of hydrophobic insertion, i.e., simply by incubation, we wondered if it is possible to insert DNA oligonucleotides onto bacterial membranes just by adding a hydrophobic moiety, such as lipids.
Lipid–DNA conjugates have emerged as a potent tool for the modification of mammalian cell membranes.16,17,29–31 These amphiphilic conjugates have attracted great interest due to their simple procedure, fast insertion, and high efficiency.32,33 Nevertheless, considering the inherent differences in the membrane composition of bacterial and mammalian cells, lipid–DNA conjugates that have been tested on mammalian cell membranes may not function similarly on bacterial membranes. We hope to demonstrate here that by fine-tuning the hydrophobic lipid moieties, these lipid–DNA conjugates could be also potentially used for bacterial membrane engineering.
In this study, we constructed a library of lipids, with different structure and hydrophobicity, to conjugate with DNA oligonucleotides. Interestingly, the membrane insertion efficiency of these lipid–DNA conjugates is highly dependent on the bacterial species, and even the strains. Selective bacterial membrane modification can be achieved based on the choice of lipid moieties. We have further demonstrated that these lipid–DNA conjugates can be used for the detection of various target bacteria, including methicillin-resistant Staphylococcus aureus (MRSA). With selective, rapid, and efficient modification, these lipid–DNA moieties will pave the way for the potential versatile applications of DNA nanostructures and devices for bacterial membrane analysis and regulation.
Result and discussion
Design and bacterial membrane insertion of lipid–DNA conjugates
To study the effect of lipid structures on the bacterial membrane insertion efficiency, we first synthesized a library of lipids containing cholesterol and five other lipids of different fatty acid chain number, length, and degree of saturation (Fig. 1a). Cholesterol was chosen because it is one of the most popular lipids used for modifying DNAs onto mammalian cells.17,29 Each of these lipids was conjugated with a fluorescein amidite (FAM)-labeled 20-nucleotide (nt)-long DNA strand through standard phosphoramidite chemistry. This DNA strand has been designed to have no secondary structure to minimize potential interactions with the bacterial membranes (Table S1†).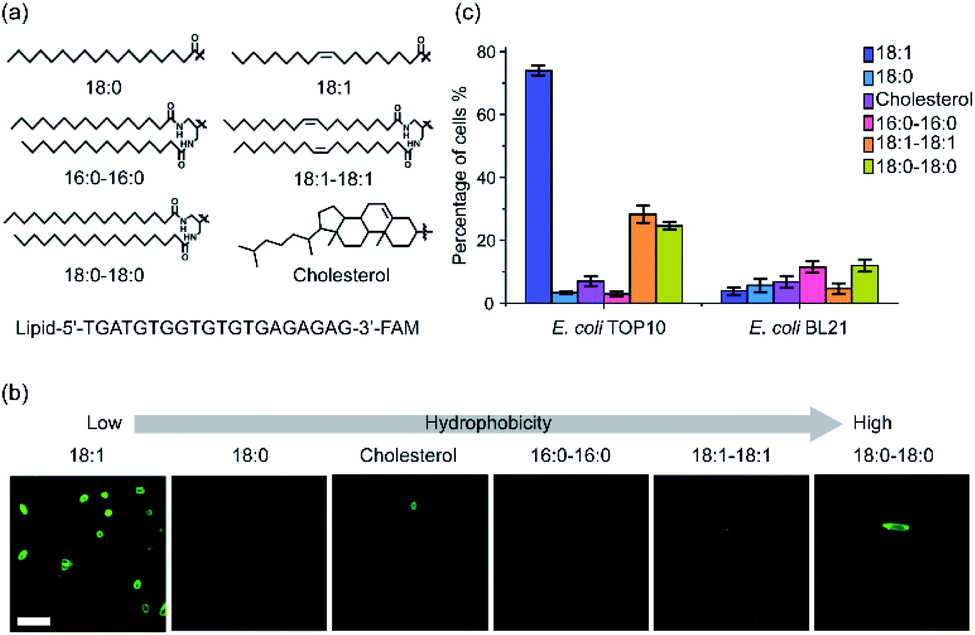 | ||
| Fig. 1 The structures of lipid–DNA conjugates and their insertion onto E. coli cell membranes. (a) Chemical structures of the lipids and sequence of the lipid–DNA conjugates. (b) Fluorescence imaging of the lipid–DNA insertion onto the membranes of E. coli TOP10 cells. Images were taken after 1 μM conjugate was incubated with TOP10 cells for 1 h at 37 °C. Scale bar, 5 μm. (c) Modification efficiency of 1 μM each lipid–DNA conjugate on the TOP10 and BL21 cells after 1 h incubation at 37 °C. Shown was the percentage of cells exhibited fluorescence intensity larger than two-fold of cellular autofluorescence background. At least 100 cells were analyzed in each case from different imaging regions. | ||
After purification and validation, we first asked if these lipid–DNA conjugates can be inserted onto the membranes of Gram-negative Escherichia coli (E. coli) cells. We chose two commonly used E. coli strains: a K-12 strain, TOP10, and a B strain, BL21. After incubating 1 μM of each lipid–DNA conjugate with the cells for 1 h at 37 °C and washing away free conjugates, cell membrane fluorescence signal was imaged with a confocal microscope. As shown in Fig. 1b, among these conjugates, the 18:1-DNA exhibited obvious fluorescence signals on most (74%) TOP10 cell membranes (Fig. 1c and Table S2†). While the 18:0–18:0 and 18:1–18:1-based conjugates can modify 25% and 28% of the TOP10 cells, respectively. There are some clear cell-to-cell variations in the membrane modification efficiency. These variations among individual cells can be due to their differences in the membrane compositions, phases of growth, aggregation status, etc. All other lipid–DNA conjugates have minimal modification (3–12%) on both TOP10 and BL21 cell membranes (Fig. 1c and S1†). As a control, the DNA oligonucleotide itself will not be inserted onto the bacterial membranes (Fig. S1†).
We also used super-resolution structured illumination microscopy to further confirm that the observed fluorescence signal was indeed from the bacterial cell surfaces (Fig. S2a†). To study if these lipid–DNA conjugates are located in the outer or inner membranes, we added 30% sucrose-containing M9 medium to induce plasmolysis of these DNA-modified E. coli cells. Plasmolysis results in the shrinkage of bacterial cytoplasm, which further leads to the separation of the inner and outer membranes. Indeed, using DiI-C12 to specifically label the inner cytoplasmic membrane of E. coli,34 we can clearly visualize the cellular shrinkage and internalized Dil-C12 fluorescence signal after treating with 30% sucrose (Fig. S2b†). In contrast, under the same condition, the fluorescence of 18:1-DNA conjugate stayed on the membranes, indicating these lipid–DNA conjugates are located on the outer membranes of E. coli cells (Fig. S2b†).
We also studied the membrane insertion of these lipid–DNA conjugates onto another type of Gram-negative bacteria, Pseudomonas aeruginosa (P. aeruginosa). However, none of these conjugates can be modified on the P. aeruginosa membranes (<1%, Table S2†). Our data indicated that the membrane insertion of lipid–DNA conjugates has specific preference on the bacterial species.
Next, we also asked if these lipid–DNA conjugates can be inserted onto the membranes of Gram-positive bacteria, such as Corynebacterium glutamicum (C. glutamicum), Staphylococcus aureus (S. aureus), and Micrococcus luteus (M. luteus). After 1 h incubation, three types of lipid–DNA conjugates, 18:0–18:0, 18:1–18:1, and 16:0–16:0, can efficiently modify the membranes (80–94%, Table S2†) of both C. glutamicum and S. aureus (Fig. 2). In contrast, the cholesterol–DNA conjugate can be selectively inserted onto C. glutamicum membranes, while the 18:1-DNA conjugate prefers S. aureus. There is no lipid–DNA conjugate in the library that can modify M. luteus cells though (Table S2†). Indeed, different Gram-positive bacterial species can also be modified distinctly with these lipid–DNA conjugates (Fig. 2b and c).
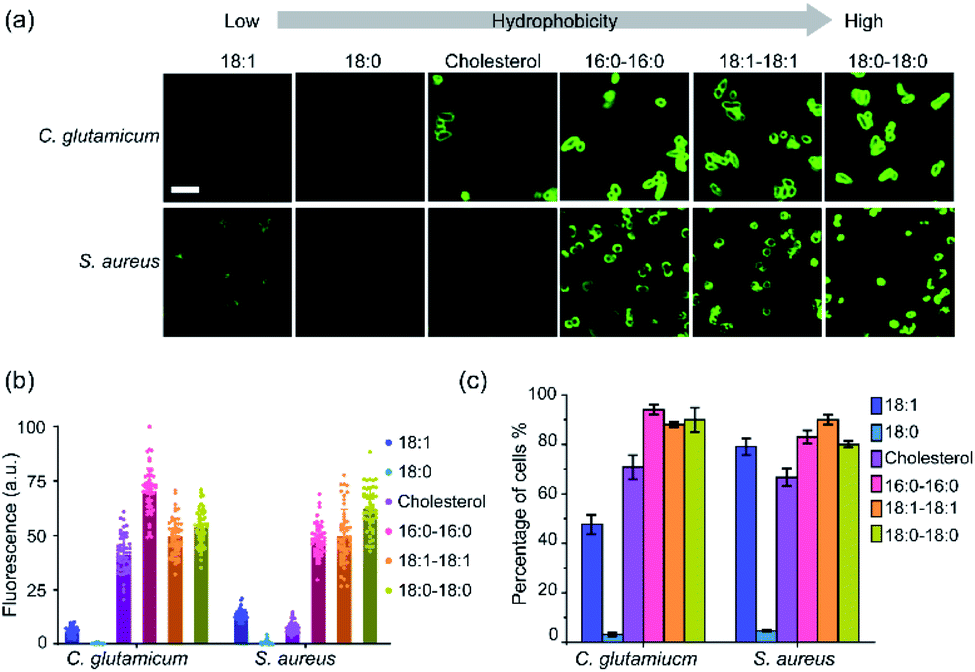 | ||
| Fig. 2 Membrane insertion of lipid–DNA conjugates onto Gram-positive cells. (a) Fluorescence imaging of the lipid–DNA insertion onto the membranes of C. glutamicum and S. aureus cells. Images were taken after 1 μM conjugate was incubated with cells for 1 h at 37 °C. Scale bar, 5 μm. (b) Fluorescence distributions on individual C. glutamicum and S. aureus cell membranes after 1 h incubation at 37 °C with 1 μM of each lipid–DNA conjugate. These fluorescence intensities were normalized to the maximum cellular fluorescence observed. At least 50 cells were analyzed in each case from different regions of imaging. (c) Modification efficiency of 1 μM each lipid–DNA conjugate on the C. glutamicum and S. aureus cells after 1 h incubation at 37 °C. Shown was the percentage of cells exhibited fluorescence intensity larger than two-fold of cellular autofluorescence background. At least 100 cells were analyzed in each case from different imaging regions. | ||
Membrane insertion kinetics & persistence of lipid–DNA conjugates
After demonstrating the bacterial membrane insertion of these lipid–DNA conjugates, we wanted to further characterize and understand these modification behaviors. First, we studied the membrane insertion kinetics of these lipid–DNA conjugates. We chose to measure the insertion kinetics of 18:1-DNA onto E. coli TOP10 cells and that of 16:0–16:0-DNA onto S. aureus and C. glutamicum cells. This choice of lipid–DNA conjugates is based on their high modification efficiency (Fig. 3 and S3–S5†). Our results indicated that within 5 min, clear membrane fluorescence signals can be observed on all these three types of bacteria (Fig. S3–S5†). When incubated at 37 °C, half maximum fluorescence intensity was shown at ∼5 min, 17 min, and 20 min for the TOP10, S. aureus and C. glutamicum cells, respectively; 90% of the maximum signal was reached after 22–60 min incubation (Fig. 3a–c). As supported by these data, lipid–DNA conjugates can rapidly insert onto bacterial membranes.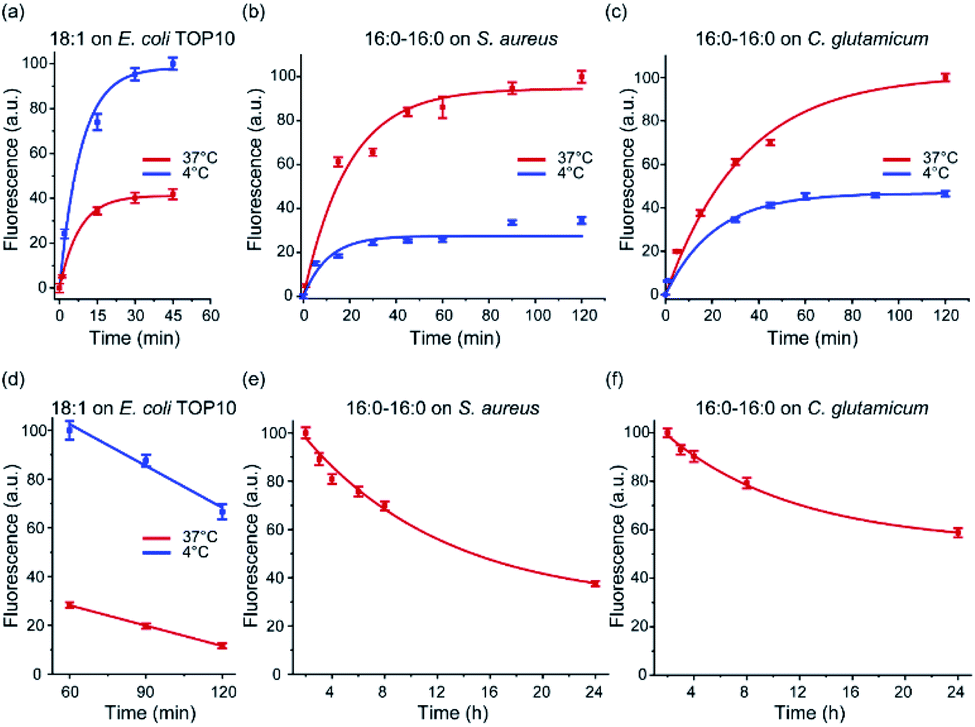 | ||
| Fig. 3 Membrane insertion kinetics and persistence of lipid–DNA conjugates. (a) Membrane insertion kinetics of the 18:1-DNA conjugate on E. coli TOP10 cells. At 0 min, 1 μM of the conjugate was added and incubated with the cells at either 37 °C or 4 °C. (b, c) Membrane insertion kinetics of the 16:0–16:0-DNA conjugate on the S. aureus or C. glutamicum cells. At 0 min, 1 μM of the conjugate was added and incubated with the cells at either 37 °C or 4 °C. (d) Membrane persistence of the 18:1-DNA conjugate on the TOP10 cells. These cells were pre-incubated with 1 μM 18:1-DNA conjugate for 1 h and then imaged for another 1 h at either 37 °C or 4 °C. (e, f) Membrane persistence of the 16:0–16:0-DNA on the S. aureus or C. glutamicum cells. These cells were pre-incubated with 1 μM 16:0–16:0-DNA conjugate for 2 h and then tracked for another 22 h at 37 °C. Error bars represent the standard error of the mean values as analyzed from at least 50 cells in each case from different imaging regions. | ||
We next asked if the membrane insertion kinetics is influenced by the incubation temperature. To test this, we repeated the above-mentioned kinetic measurement at 4 °C. Similar as that of 37 °C, very fast membrane insertion was observed, with half maximum fluorescence shown at 7–20 min and 90% of maximum signal exhibited at 25–60 min. Interestingly, for the E. coli TOP10 cells, a faster membrane insertion and higher maximum fluorescence signal was shown at 4 °C (Fig. 3a). While in contrast, for S. aureus and C. glutamicum, a lower insertion kinetics and efficiency was observed at 4 °C than 37 °C (Fig. 3b and c). As shown in the following section, we believe this difference in the bacteria-specific temperature effect is likely due to different hydrophobicities of these membranes. Temperature can affect the membrane insertion of the lipid–DNA conjugates.
We also wondered if these lipid–DNA conjugates can stay on these bacterial membranes for a long time. By elongating the incubation time, on the TOP10 cell membranes, the fluorescence signal of 18:1-DNA was shown to be decreased by ∼50% after ∼90 min incubation at 37 °C. While at 4 °C, a much higher membrane probe density and persistence was observed, with only a ∼30% reduction in fluorescence after 2 h incubation (Fig. 3d). Interestingly, the 16:0–16:0-DNA conjugate was highly stable on both S. aureus and C. glutamicum membranes. Even after 24 h incubation at 37 °C, ∼40–50% of the conjugate was still on these cell membranes (Fig. 3e and f). These highly stable modifications can be potentially useful for long-term membrane analysis and regulations.
Membrane modification efficiency of lipid–DNA conjugates
Our next goal is to quantify the bacterial membrane insertion efficiency of these lipid–DNA conjugates. We realized that the membrane densities of lipid–DNA conjugates can be tuned based on their initial concentrations during the incubation. By incubating 0.1–2 μM of lipid–DNA conjugates with the same density of E. coli TOP10, S. aureus, and C. glutamicum cells, brighter bacterial membrane signal was generally induced with a higher initial concentration of the conjugates (Fig. S6†). When adding 16:0–16:0-DNA onto S. aureus cells, there was even a linear correlation (Fig. S6b†). The maximum membrane insertion was normally observed after adding 0.5–1 μM of lipid–DNA conjugates.To further study the correlation between the membrane density of lipid–DNA and the observed fluorescence intensity, we prepared 1,2-dilauroyl-sn-glycero-3-phosphocholine (DLPC)-based supported lipid bilayers containing different concentrations of lipid–DNA conjugates. Lipid–DNA conjugates can be homogenously distributed on these supported lipid bilayers. Under the same imaging condition as that for the bacterial membrane studies, a linear correlation between the membrane fluorescence intensity and lipid–DNA density was observed (Fig. S7b†). Based on this standard calibration curve, we can now quantify the bacterial membrane insertion efficiency of each lipid–DNA conjugate.
On C. glutamicum membranes, up to 0.7 DNA per nm2 area can be inserted at 37 °C with the help of the 16:0–16:0-DNA conjugate. Similarly, the highest DNA density on S. aureus membranes was 0.6 nm−2 when 1 μM of 18:0–18:0-DNA was added for 1 h at 37 °C (Table S3†). In comparison, the maximum DNA modification on E. coli TOP10 membranes was achieved at 4 °C (0.4 nm−2), rather than 37 °C (0.2 nm−2), with the addition of 1 μM 18:1-DNA conjugate (Table S3†). Indeed, the lipid–DNA conjugates can be efficiently modified onto bacterial membranes. Meanwhile, on each type of these bacterial membranes, there are some clear variations in the modification efficiency among different lipid–DNA conjugates. After 1 h incubation with 1 μM of each conjugate, up to 13.5-fold difference in the membrane DNA density was shown. For the same lipid–DNA conjugate, a 1.8–7.4-fold variation in the maximum modification efficiency was observed on these E. coli TOP10, S. aureus, and C. glutamicum cells.
Effect of lipid–DNA hydrophobicity on the membrane insertion
Our next goal is to study how these lipid–DNA conjugates can modify bacterial membranes with different selectivity. We wondered if the difference in these membrane insertion efficiency is due to different hydrophobicities of the lipid–DNA conjugates. The hydrophobicity of each lipid–DNA conjugate has been quantified using an HPLC assay.33 We realized that more hydrophobic lipid–DNA conjugates, such as 18:1–18:1-DNA and 18:0–18:0-DNA, tend to insert onto S. aureus and C. glutamicum membranes with larger densities (Fig. 4). This result may be attributed to the highly hydrophobic environment in these bacterial membranes. Indeed, it has been reported that the membranes of S. aureus and C. glutamicum contains a large number of highly hydrophobic branched chain amino acids and fatty acids.35–38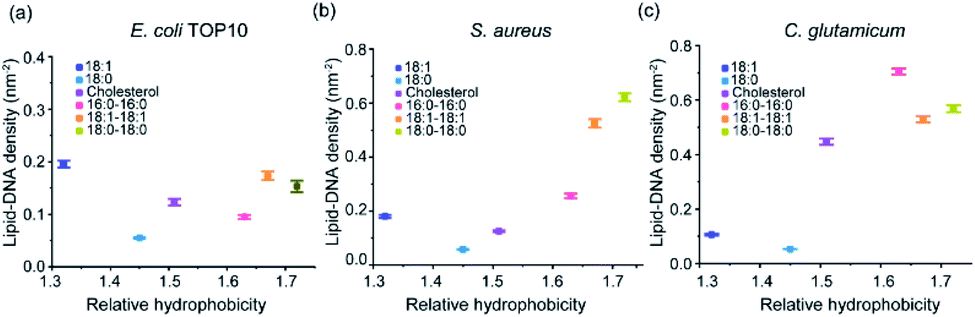 | ||
| Fig. 4 The relationships between the hydrophobicity of each lipid–DNA conjugate and their corresponding membrane densities. These membrane densities were measured after 1 h incubation of each lipid–DNA conjugate with (a) E. coli TOP10, (b) S. aureus, and (c) C. glutamicum cells at 37 °C. The relative hydrophobicity was determined from an HPLC assay.28 Error bars represent the standard error of the mean values as analyzed from at least 50 cells in each case from different imaging regions. | ||
While in contrast, for the E. coli TOP10 cells, both the least hydrophobic (18:1-DNA) and most hydrophobic (18:1–18:1-DNA and 18:0–18:0-DNA) conjugates exhibited high membrane modification efficiency (Fig. 4a). There seems to be no clear correlation between the lipid–DNA hydrophobicity and the TOP10 membrane modification efficiency. Compared to S. aureus and C. glutamicum, these E. coli K12 cells are known to be more hydrophilic due to the existence of lipopolysaccharide chains on the outer membranes.38–40 The membrane hydrophobicity of these E. coli cells can be further reduced at low temperatures due to the increased content of unsaturated fatty acids.41 This fact may also explain the above-mentioned higher TOP10 membrane modification of 18:1-DNA at 4 °C compared to 37 °C (Fig. 3a). Bacterial membranes may indeed prefer the insertion of lipid–DNA conjugates of similar hydrophobicity.
We also wanted to study the effect of DNA length on the bacterial membrane modification. For this purpose, we synthesized a cholesterol–DNA conjugate based on an 80 nt-long DNA oligonucleotide. After incubating this conjugate with E. coli TOP10 and S. aureus cells, respectively, at 37 °C for 1 h, the observed cell membrane fluorescence signal was quite similar as that of the 20 nt cholesterol–DNA conjugate (Fig. S8†). These data indicated that the effect of DNA length on the membrane insertion efficiency may not be as dramatic as that of the lipid moiety. Efficient membrane insertion of both short and long oligonucleotides can be achieved with the help of these lipid–DNA conjugates.
Selective targeting and detection of bacteria
To potentially apply these lipid–DNA conjugates for membrane analysis and regulation, we asked if these membrane-anchored DNAs remain to be accessible for hybridization. To test this, after inserting FAM-labelled lipid–DNA conjugates onto S. aureus and C. glutamicum cell membranes, we added a Cy5-labelled complementary DNA strand (Cy5-cDNA). Without a lipid tail, these Cy5-cDNA cannot modify bacterial membranes by themselves (Fig. S9†). While by hybridizing with membrane-anchored DNAs, indeed, highly colocalized FAM and Cy5 fluorescence signals can be clearly visualized on the cell membranes of C. glutamicum and S. aureus (Fig. S9 and S10†). As a control, when non-complementary DNA was added, only the FAM signal can be observed on the cell membranes of C. glutamicum (Fig. S9†). These results indicated that membrane-anchored DNAs are still available to hybridize with the corresponding complementary DNA strands.We would like to also mention that in these Gram-positive C. glutamicum and S. aureus bacteria, there are some cell walls located outside of the plasma membranes. The thickness and composition of these cell walls will likely affect the membrane insertion efficiency of these lipid–DNA conjugates. Some of these lipid–DNA may actually anchor directly into the cell walls. Indeed, compared to cell-wall-embedded Gram-negative cells, these Gram-positive bacteria exhibit overall higher lipid–DNA modification efficiency (Fig. 4).
To further demonstrate other potential applications of these membrane-anchored lipid–DNA conjugates, we asked if these lipid–DNA conjugates can be used for the selective detection of bacteria from a mixture. We chose to study two bacterial mixtures, E. coli BL21 + S. aureus, and E. coli BL21 + TOP10. These BL21 cells have been transformed with a red fluorescent protein, RFP670, for easy distinction. Considering the bacterial membrane selectivity of these lipid–DNA conjugates (Tables S2 and S3†), a FAM-labelled 18:1–18:1-DNA and a 18:1-DNA conjugate was respectively used to target S. aureus and TOP10 cells in the mixture. Indeed as expected, these conjugates can selectively modify S. aureus and TOP10 cell membranes. 96% and 92% of the labeled cells were correct target bacteria in the presence of E. coli BL21 (Fig. 5a).
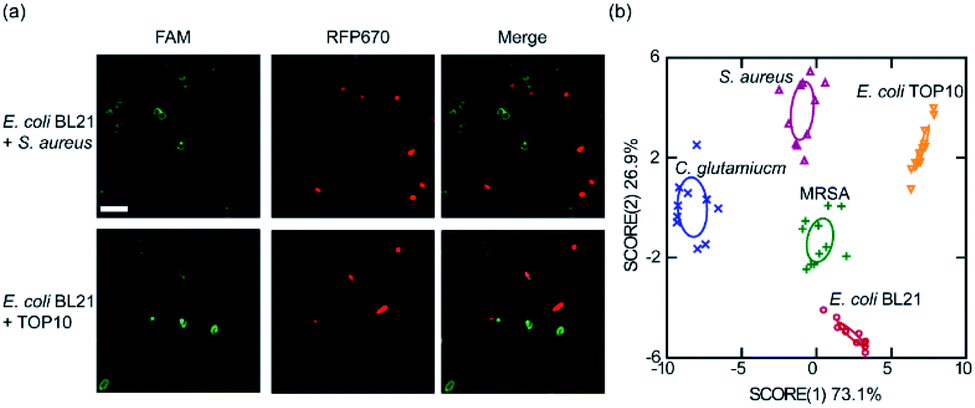 | ||
| Fig. 5 Bacterial differentiation and detection with the lipid–DNA conjugates. (a) (Top) FAM-labeled 18:1–18:1-DNA conjugate can be used to distinguish S. aureus cells from a mixture with RFP670-expressing E. coli BL21 cells. (Bottom) Similarly, FAM-labeled 18:1-DNA conjugate was used to distinguish E. coli TOP10 cells from a mixture with BL21. Here, 1 μM of the lipid–DNA conjugate was incubated with the cell mixture for 1 h at 37 °C. Scale bar, 5 μm. (b) Linear discriminant analysis based on the fluorescence response pattern of the 18:1-DNA and 16:0–16:0-DNA conjugates on five types of bacterial strains. The transformed canonical scores were plotted with 95% confidence ellipses around the centroid of each group. | ||
We finally asked if it is possible to use these lipid–DNA conjugates to distinguish target bacteria in a more complex cell system. We realized that it is still difficult in using just a single lipid–DNA conjugate for this purpose, instead, a pattern-based bacterial detection using a simple array of lipid–DNA conjugates may be feasible. To test this idea, we wondered if a pair of two lipid–DNA conjugates can be enough to distinguish E. coli TOP10, E. coli BL21, C. glutamicum, and S. aureus. Indeed, based on the specific recognition pattern of 18:1-DNA and 16:0–16:0-DNA, all these strains can be categorized into separate clusters in a linear discriminant analysis (Fig. 5b). Not only this pair of 18:1-DNA/16:0–16:0-DNA conjugates, other pairs of lipid–DNA conjugates, such as cholesterol–DNA/18:1–18:1-DNA, can also be used to differentiate each of these bacterial strains (Fig. S11†). More interestingly, these simple lipid–DNA arrays are able to not only distinguish bacteria from mammalian cells, but also bacteria of minor differences, for example, S. aureus vs. methicillin-resistant S. aureus (MRSA), a clinically important health-threating bacterial pathogen (Fig. S11†).42 Indeed, these lipid–DNA conjugates can be potentially used for the selective detection of various bacterial species, including antibiotic-resistant superbugs.
Conclusions
In this study, we have developed a simple, rapid, and effective method to engineer bacterial membranes with DNA oligonucleotides. After several minutes of incubation, a large number of DNA strands can be readily modified onto these bacterial membranes. The membrane density of DNA can be rationally tuned based on the choice of lipid–DNA conjugates and their initial concentrations. As high as 0.4–0.7 DNA insertion per nm2 membrane area can be achieved on various Gram-negative and Gram-positive bacterial species. These membrane-anchored DNAs are still available for hybridization and can stay on the membranes for a long period of time. In addition, these lipid-mediated DNA modifications have interesting selectivity on the bacterial membranes. By fine-tuning the hydrophobicity of the lipid moieties, targeted bacterial membrane engineering can be achieved for potential diagnostic and biomedical applications.This study can potentially largely extend the applications of DNA nanotechnology in the field of microbiology. With the help of these lipid–DNA conjugates, versatile DNA scaffolds, structures, and devices can now be functionalized onto various types of bacterial membranes. These membrane-anchored functional DNA nanodevices can be likely used for the generation of artificial signaling pathways, analytical and biophysical characterization of bacterial membranes, structural regulation, and therapy.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the support of NIH R35GM133507, Sloan Research Fellowship, and a start-up grant from UMass Amherst to M. You. We are grateful to Dr James Chambers for the assistance in fluorescence imaging, and Dr Sloan Siegrist for comments and suggestions. C. glutamicum and S. aureus were gifted from Dr Sloan Siegrist. We thank Dr Vincent Rotello for assistance on linear discriminant analysis and gifting M. luteus and MRSA. We also thank other members of the You Lab for useful discussion.References
- T. J. Silhavy, D. Kahne and S. Walker, Cold Spring Harbor Perspect. Biol., 2010, 2, a000414 CrossRef
.
- S. Gautam, T. J. Gniadek, T. Kim and D. A. Spiegel, Trends Biotechnol., 2013, 31, 258–267 CrossRef CAS
- M. S. Siegrist, B. M. Swarts, D. M. Fox, S. A. Lim and C. R. Bertozzi, FEMS Microbiol. Rev., 2015, 39, 184–202 CrossRef
- H. Jia, Y. Zhu, Z. Chen and F. Wu, ACS Appl. Mater. Interfaces, 2017, 9, 15943–15951 CrossRef CAS
- P. Shieh, M. S. Siegrist, A. J. Cullen and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 5456–5461 CrossRef CAS
- H. Etayash, L. Norman, T. Thundat, M. Stiles and K. Kaur, ACS Appl. Mater. Interfaces, 2014, 6, 1131–1138 CrossRef CAS
- A. Parthasarathy, H. C. Pappas, E. H. Hill, Y. Huang, D. G. Whitten and K. S. Schanze, ACS Appl. Mater. Interfaces, 2015, 7, 28027–28034 CrossRef CAS
- N. Lahav-Mankovski, P. K. Prasad, N. Oppenheimer-Low, G. Raviv, T. Dadosh, T. Unger, T. M. Salame, L. Motiei and D. Margulies, Nat. Commun., 2020, 11, 1299 CrossRef CAS
- Y. Krishnan and M. Bathe, Trends Cell Biol., 2012, 22, 624–633 CrossRef CAS
- N. C. Seeman and H. F. Sleiman, Nat. Rev. Mater., 2018, 3, 17068 CrossRef CAS
- Y. Chen, B. Groves, R. A. Muscat and G. Seelig, Nat. Nanotechnol., 2015, 10, 748–760 CrossRef CAS
- G. Feng, X. Luo, X. Lu, S. Xie, L. Deng, W. Kang, F. He, J. Zhang, C. Lei, B. Lin, Y. Huang, Z. Nie and S. Yao, Angew. Chem., Int. Ed., 2019, 58, 6590–6594 CrossRef CAS
- L. Qiu, T. Zhang, J. Jiang, C. Wu, G. Zhu, M. You, X. Chen, L. Q. Zhang, C. Cui, R. Yu and W. Tan, J. Am. Chem. Soc., 2014, 136, 13090–13093 CrossRef CAS
- R. Peng, X. Zheng, Y. Lyu, L. Xu, X. Zhang, G. Ke, Q. Liu, C. You, S. Huan and W. Tan, J. Am. Chem. Soc., 2018, 140, 9793–9796 CrossRef CAS
- W. Zhao, S. Schafer, J. Choi, Y. J. Yamanaka, M. L. Lombardi, S. Bose, A. L. Carlson, J. A. Phillips, W. Teo, I. A. Droujinine, C. Cui, R. K. Jain, J. Lammerding, J. C. Love, C. Lin, D. Sarkar, R. Karnik and J. M. Karp, Nat. Nanotechnol., 2011, 6, 524–531 CrossRef CAS
- P. Shi and Y. Wang, Angew. Chem., Int. Ed., 2021 DOI:10.1002/anie.202010278
- S. Huo, H. Li, A. Boersma and A. Herrmann, Adv. Sci., 2019, 6, 1–17 Search PubMed
- B. Zhao, C. O'Brien, A. P. K. K. K. Mudiyanselage, N. Li, Y. Bagheri, R. Wu, Y. Sun and M. You, J. Am. Chem. Soc., 2017, 139, 18182–18185 CrossRef CAS
- M. You, Y. Lyu, D. Han, L. Qiu, Q. Liu, T. Chen, C. Wu, L. Peng, L. Zhang, G. Bao and W. Tan, Nat. Nanotechnol., 2017, 12, 453–459 CrossRef CAS
- B. Zhao, N. Li, T. Xie, Y. Bagheri, C. Liang, P. Keshri, Y. Sun and M. You, Chem. Sci., 2020, 11, 8558–8566 RSC
- A. Saminathan, J. Devany, A. T. Veetil, B. Suresh, K. S. Pillai, M. Schwake and Y. Krishnan, Nat. Nanotechnol., 2021 DOI:10.1038/s41565-020-00784-1
- Z. Ge, J. Liu, L. Guo, G. Yao, Q. Li, L. Wang, J. Li and C. Fan, J. Am. Chem. Soc., 2020, 142, 8800–8808 CrossRef
- X. Xiong, H. Liu, Z. Zhao, M. B. Altman, D. Lopez-Colon, C. Yang, L. Chang, C. Liu and W. Tan, Angew. Chem., Int. Ed., 2013, 52, 1472–1476 CrossRef CAS
- R. M. Epand, C. Walker, R. F. Epand and N. A. Magarvey, Biochim. Biophys. Acta, Biomembr., 2016, 1858, 980–987 CrossRef CAS
- Y. K. Huang, X. J. Chen, Y. Xia, S. J. Wu, N. Duan, X. Y. Ma and Z. P. Wang, Analytical Methods, 2014, 6, 690–697 RSC
- N. E. Trunzo and K. L. Hong, Int. J. Mol. Sci., 2020, 21, 5074 CrossRef CAS
- N. Alizadeh, M. Y. Memar, S. R. Moaddab and H. S. Kafil, Biomed. Pharmacother., 2017, 93, 737–745 CrossRef CAS
- S. Marton, F. Cleto, M. A. Krieger and J. Cardoso, PLoS One, 2016, 11, e0153637 CrossRef
- B. Zhao, Q. Tian, Y. Bagheri and M. You, Current Opinion in Biomedical Engineering, 2020, 13, 76–83 CrossRef
- Q. Shen, M. W. Grome, Y. Yang and C. Lin, Adv. Biosyst., 2020, 4, 1900215 CrossRef CAS
- A. Lopez and J. W. Liu, Langmuir, 2018, 34, 15000–15013 CrossRef CAS
- Y. Bagheri, F. Shafiei, S. Chedid, B. Zhao and M. You, Supramol. Chem., 2019, 31, 532–544 CrossRef CAS
- Y. Bagheri, S. Chedid, F. Shafiei, B. Zhao and M. You, Chem. Sci., 2019, 10, 11030–11040 RSC
- F. Oswald, H. Varadarajan, H. Lill, E. J. Peterman and Y. J. Bollen, Biophys. J., 2016, 110, 1139–1149 CrossRef CAS
- F. Reifsteck, S. Wee and B. J. Wilkinson, J. Med. Microbiol., 1987, 24, 65–73 CrossRef CAS
- R. Takeshita, H. Ito and M. Wachi, Biosci., Biotechnol., Biochem., 2010, 74, 1617–1623 CrossRef CAS
- J. Marienhagen, N. Kennerknecht, H. Sahm and L. Eggeling, J. Bacteriol., 2005, 187, 7639–7646 CrossRef CAS
- M. Rosenberg, D. Gutnick and E. Rosenberg, FEMS Microbiol. Lett., 1980, 9, 29–33 CrossRef CAS
- A. Zita and M. Hermansson, FEMS Microbiol. Lett., 1997, 152, 299–306 CrossRef CAS
- F. Hamadi, H. Latrache, H. Zahir, A. Elghmari, M. Timinouni and M. Ellouali, Braz. J. Microbiol., 2008, 39, 10–15 CrossRef
- M. M. Suutari and S. Laakso, Crit. Rev. Microbiol., 1994, 20, 285–328 CrossRef CAS
- G. J. Moran, A. Krishnadasan, R. J. Gorwitz, G. E. Fosheim, L. K. McDougal, R. B. Carey and D. A. Talan, N. Engl. J. Med., 2006, 355, 666–674 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, Fig. S1–S11, Table S1–S6. See DOI: 10.1039/d0sc06630c |
| This journal is © The Royal Society of Chemistry 2021 |