
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInterstitial and substitutional light elements in transition metals for heterogeneous catalysis
Tianyi
Chen
,
Christopher
Foo
 and
Shik Chi
Edman Tsang
*
and
Shik Chi
Edman Tsang
*
Wolfson Catalysis Centre, Department of Chemistry, University of Oxford, Oxford OX1 3QR, UK. E-mail: edman.tsang@chem.ox.ac.uk
First published on 18th December 2020
Abstract
The addition of foreign element dopants to monometallic nanoparticle catalysts is of great importance in industrial applications. Both substitutional and interstitial doping of pure metallic phases can give profound effects such as altering electronic and transport properties, lattice parameters, phase transitions, and consequently various physicochemical properties. For transition metal catalysts, this often leads to changes in catalytic activity and selectivity. This article provides an overview of the recent developments regarding the catalytic properties and characterisation of such systems. In particular, the structure–activity relationship for a number of important chemical reactions is summarised and the future prospects of this area are also explored.
Tianyi Chen obtained her BSc (Chemistry) and MRes (Catalysis: Chemistry and Engineering) from Imperial College London (UK) in 2015 and 2016, respectively. In 2020, she completed her PhD thesis at the University of Oxford (UK), under the supervision of Prof. Edman Tsang and Prof. Peter Nellist. Her interests include synthesis and characterisation of light elements doped transition metals and their use as catalysts in the conversion of biomass-derived substrates to value-added chemicals. |
 Christopher Foo | Christopher Foo obtained his MSci degree from the University of Bristol in 2017, and is now a DPhil candidate at the University of Oxford, UK under the supervision of Prof. Edman Tsang. His current research focuses on the crystallographic characterisation of various catalysis and energy storage systems, in particular the identification and siting of dopants in parent frameworks, ranging from zeolites and protonic conductors to lithium ion intercalation for battery applications. Being partly funded and supervised by the nearby Diamond Light Source, he specialises in the collection and refinement of high quality diffraction data in order to correlate emergent properties with atomic structure. |
 Edman Tsang | Edman Tsang is a Professor of Chemistry and Head of Wolfson Catalysis Centre at the University of Oxford, UK. His main research interests are on nanomaterials and catalysis concerning energy and environment which include developments of catalytic, photocatalytic and electrocatalytic technologies for fine chemicals, cleaner combustion, green chemistry, energy storages, processes and production, etc. Particular expertise is in design and architecture of nanocatalysts, which can lead to understanding of catalytic surfaces and interfaces. He has about 350 referred research publications including Nature, Science and Nature sister journals. He has delivered over 200 plenary and invited presentations at conferences, universities and companies. |
Introduction
Transition metals are often used in heterogeneous catalysis because of their characteristic unfilled d-bands.1 It has been long known that their catalytic performance can be altered by adding impurities, either deliberately or otherwise. The resulting doping can be broadly classified into substitutional doping2 and interstitial doping.3 In substitutional doping, atoms of the host metal are replaced by foreign atoms (Fig. 1a); meanwhile in interstitial doping, foreign atoms, usually light elements, occupy the interstitial sites (Fig. 1b).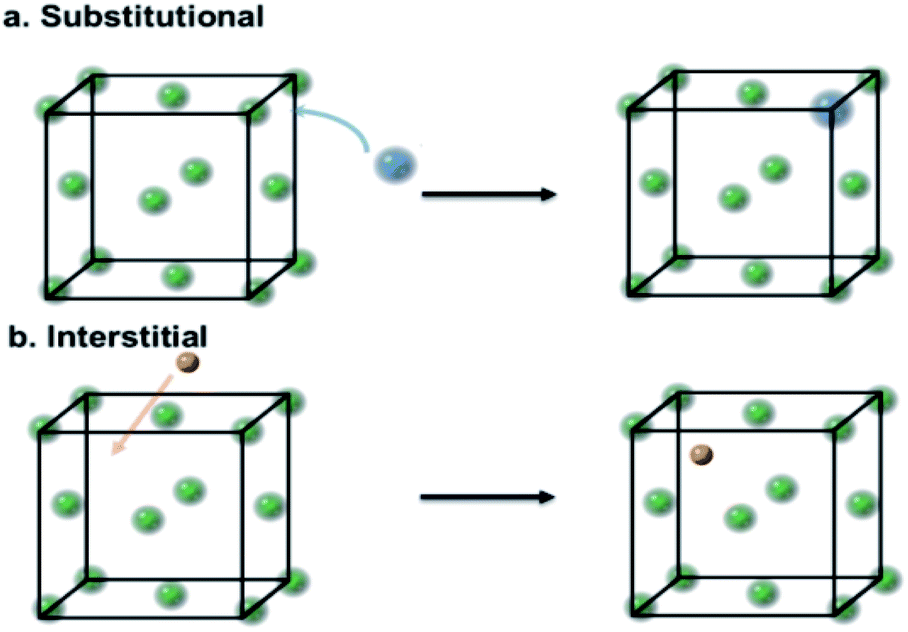 | ||
| Fig. 1 A schematic of substitutional and interstitial atomic modification by light elements to face-centered-cubic (FCC) a host metal lattice (green). (a) Substitutional atomic displacement by foreign light atoms (blue). (b) Interstitial atomic modification by foreign light atoms (brown). | ||
The history of interstitial light-element modification of transition metals for catalysts can be dated back to the modification of tungsten by alloying with carbon atoms forming tungsten carbide, which was used in the isomerisation of neo-pentane to isopentane,4 and displayed a similar catalytic behavior to platinum metal. Some kinds of geometric and electronic modifications of the host structure by the carbon ‘impurities’ critically affect surface and/or bulk structure to give significant improvement in catalytic conversion and selectivity.5 However, despite the dramatic alteration of the catalytic properties by such light-element doping, there is a lack of in-depth understanding of the structure–activity relationship. Recent advances in analytical techniques such as electron ptychography in transmission electron microscopy, and high-resolution synchrotron and neutron diffraction have made strong contributions to the understanding of these catalysts, and consequently the correlations between doping, structure and activity, allowing the rational design of this class of catalysts.6 It is timely to summarise some recent breakthroughs in this area.
Here, in this mini-review, we will outline some of the latest developments in this type of doped transition-metal catalysts, which have been modified with light elements, including hydrogen (H), lithium (Li), boron (B), carbon (C), and nitrogen (N). To provide a more comprehensive understanding, we will also highlight some recent advances in their characterisation with respect to (1) structural aspects: phase identification, local disorder, chemical environment and site location identification; (2) electronic modifications of local and band structure of the host transition metal by DFT calculations; (3) identification and disentanglement of the effects of structural and electronic changes on the observed catalysis phenomena, and give a rational guidance on how modifications of the fundamental physicochemical properties affect catalytic properties; and finally (4) the evaluation of future prospects on the current research field and suggestion of challenges to be overcome.
Terminology: intermetallic compounds versus solid-solution alloys and doping
It is well known that the architecture of the nanocrystal, after alloying with foreign atoms or additives, are modified in terms of crystallinity and disorder, local atomic arrangement and symmetry (space group), various types of structural or electronic defects, long/short-range atomic order and distribution, crystal morphology, and secondary architecture properties including sintering/agglomeration behaviour and support distribution.Regarding the atomic ordering, mixed nano-alloys can further be categorised as either intermetallic compounds or solid-solution alloys (Fig. 2).7 The term ‘intermetallic compound’ is used for systems that have long-range atomic order and well-defined stoichiometry. The space group and atomic arrangements of the intermetallic phase are different to that of the pure parent metal. In contrast, the term ‘solid-solution alloy’ is used to describe a system of two or more components in which the atomic structure of the parent metal is largely unchanged and the dopant is accommodated non-stoichiometrically and distributed non-homogeneously (mixed), i.e. the dopant is dissolved in an unchanged structure. This type of doping is usually characterised by gradual changes in lattice parameter with dopant concentration,8 and may precede a discrete phase transition to an intermetallic compound,9 which can be demonstrated by the boron–palladium binary system.8,9
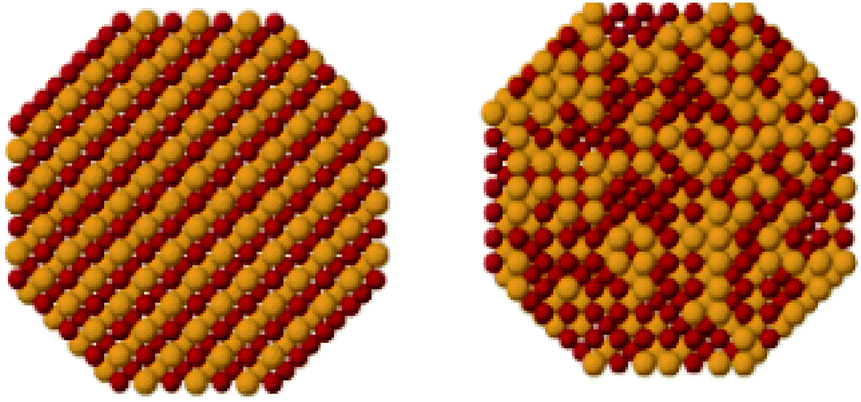 | ||
| Fig. 2 A schematic representation of intermetallic compounds (left) and solid-solution alloys (right). This figure has been reproduced from ref. 12 with permission from American Chemical Society, copyright 2008. | ||
The formation of solid-solution alloys or intermetallic compounds can be explained from a thermodynamic perspective (eqn (1)):10
| ΔGf = ΔHf − TΔSf | (1) |
Therefore, the driving force in the formation of solid-solution alloys (entropy-driven) and intermetallic compounds (enthalpy-driven) are different. However, it is worth noting that this equation fails to consider the factors of strain, defects etc.10 Overall, this classification is essential because solid-solution alloys and interatomic compounds behave differently even if the elemental composition and stoichiometry are the same.
In literature, the term ‘doping’ is used most frequently to describe the addition of a small quantities of a foreign element as a promoter or additive to a catalyst or semiconductor. This type of ‘doping’ is usually described in a general, non-specific way. In this work, this term will also be used to describe large concentrations of additive; for example in Y-doped ZrO2, in which the Y content is above 10%. As admitted by the literature,13 as long as the modification is mild and the parent structure remains approximately unchanged, this term is still applicable. Therefore, it is more suitable to use the term ‘doping’ when describing solid-solution alloys and use other words when describing intermetallic compounds.
Solid-solution alloy: substitutional versus interstitial systems
Substitution systems
For typical substitutional solid-solution alloys, the atomic size and electronic properties of the foreign element are similar or identical to that of the parent metal. Normally, the difference in atomic radius should be less than 15%, as described by the Hume–Rothery rule14 (eqn (2)).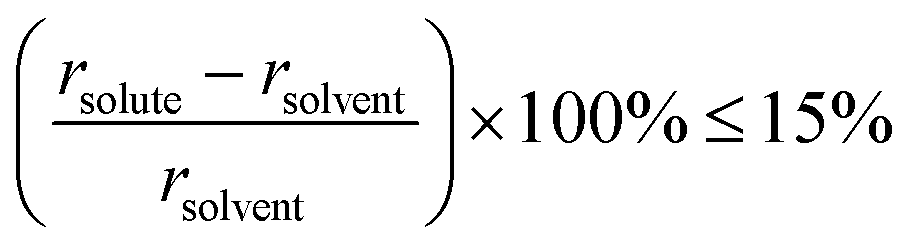 | (2) |
The formation of substitutional alloys can affect the local electronic structure, thereby modifying the chemisorption properties of the metal surface.15 This occurs in both zero valent and metal oxide systems. For example, Cu can be alloyed into Ni, leading to the formation of a substitutional alloy.16 The presence of Cu in the substitutional alloy system is anticipated to fill the d-band vacancies of Ni and suppress the formation of γ-NiOOH, which is a low-activity byproduct formed during electrocatalytic methanol oxidation. In the case of substitutional alloy formation in metal oxides, a small fraction of the metal cations of a host oxide can be substituted by foreign cations,17 and/or the anions replaced by foreign anions such as halides.18 Though, as known in the literature, the stability of substitutional alloys is a problem because the dopant atom often does not have strong location preference with a low mobility barrier resulting in diffusion and instability at elevated temperatures. Hence, segregation or even loss of dopant can occur, resulting in a mixture of the pure constituents.19
Interstitial systems
Unlike substitutional alloys, in order for the formation of interstitial alloys to occur, the size of the foreign species needs to be sufficiently small to diffuse into the interstitial sites. The conventional interstitial alloys are metal hydrides,20 carbides,21 borides22 and nitrides,23 among others. These light elements, from the first period of the p-block and below, often have small atomic radii and dominant s–p orbitals, and often occupy the largest available interstitial site: the octahedral hole in face-centre-cubic (FCC) and hexagonal-close-packed (HCP) structures, and the trigonal prismatic hole in hexagonal structures. The crystal structure is governed by geometric and electronic factors. The geometric factor is based on Hägg's rule,24 which uses the ratio of dopant atomic radius (rX) to that of the host metal atomic radius (rM) for the interstitial alloy formation. When the ratio (rX/rM) is less than 0.59, the structure of the interstitial alloy will be simple, such as the aforementioned FCC, HCP and hexagonal structures. When the ratio (rX/rM) is greater than 0.59, an interstitial alloy with a more complicated structure will be obtained, for instance chromium carbide, Cr3C2.25The second factor that governs crystal structure is the electronic effect. The nature of metallic bonding in interstitial solid-solution transition-metal alloys (d-orbital dominated) is remarkably different to the pristine metal due to orbital mixing between the foreign atoms and host metal system, such as the s–p orbitals of the dopant element and the s–p–d orbitals of the metal elements. In light of the Engel–Brewer theory of metals,26,27 it was suggested that the structure adopted by a metal or interstitial solid-solution alloy is dependent on the s–p counts as follows:26
BCC: sp0.5
HCP: sp0.7–1.1
FCC: sp1.5–2
It can be found that increasing p-orbital mixing can induce the structural transformation from body-centre-cubic (BCC) to HCP to FCC. Additionally, accommodating foreign atoms in a metal structure can result in lattice expansion of the host structure, which is indicative of interstitial solid-solution alloy formation due to repulsive electrostatic interactions.28
The size of solute is a main contribution for the formation of the interstitial solid-solution alloy. As depicted in Fig. 3, the atomic radii of hydrogen, boron, carbon and nitrogen are rather small. However, the atomic radius of Li is relatively large, comparable to some transition metals such as silver (165 pm). Hence, lithium atoms are observed to substitute silver atoms instead of inhabiting interstitial sites in the structure.128 Hence, it is expected that incorporating light elements with smaller atomic radii into transition metals are more likely to form interstitial solid-solution alloys. In addition to the effect of size, the similarity of the electropositivity and crystal structure of the two components needs to be taken into account when considering interstitial solid-solution systems.29
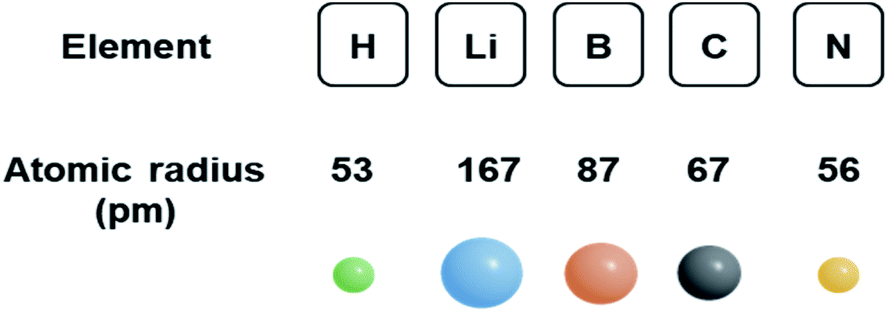 | ||
| Fig. 3 The atomic radii of elemental hydrogen, lithium, boron, carbon and nitrogen. | ||
Since each research group has its own notation for solid-solution alloy systems, this mini-review will express binary systems with the host metal (M) first, and the dopant element (N) second. Substitutional doping and interstitial doping will be expressed in superscript as follows: M–subN and M–intN. Solid-solution phases that are reported as a specified composition are expressed as MmNn. Unclear solid-solution phases are expressed as MmNx, where x denotes that the atomic ratio remains unclear. In bimetallic systems that have been doped with a light element, the two host metals will be separated by a dash. Note that some material labels in this mini-review differ from the original ones in the literature.
Characterisation
As emphasised, once the foreign element either substitutes an atom from the metal framework or occupies an interstitial site, even at very low concentration, profound effects on the physiochemical properties of the host metal system can be altered, such as adjusting the electronic and transport properties, lattice parameter changes, phase transitions, thermal stability and chemical properties, etc. However, regardless of synthetic method used, it is difficult to discern whether the foreign element has been deposited on the surface, at interstitial sites or vertices of the host metal system. Therefore, if the catalytic performance is promoted after addition of the foreign additives, it is anticipated that confirming the location and the degree of occupancy of the foreign elements will significantly contribute to the understanding of the relationship between the dopant-host metal system and catalytic performance.As reflected from literature,17 the employment of appropriate characterisation techniques can help answer these questions:
What drives the accommodation?
What is the chemical state/properties of the foreign elements?
What sites do the foreign elements occupy?
Is the resulting structure crystalline, or only ordered at a short length-scale?
Is the foreign element homogeneously distributed throughout a particle, i.e. surface/bulk distribution?
Can we visualise the dopants directly?
Following advances in the sensitivity of detection and the speed of digital processing, many more state-of-the-art techniques for characterisation have been developed (Table 1). In the following section, the methods used to help answering the above questions will be evaluated.
| Incident particle | Analytical technique | Main information | Ref. |
|---|---|---|---|
| Photons | XRD | Atomic positions and occupancies of crystalline structures | 30 |
| EXAFS/XANES | Interatomic distances, chemical environments/oxidation states | 31 | |
| XPS | Electronic structure, oxidation states | 32 | |
| XPDF | Distorted local structure | 6 | |
| FTIR | Vibrational frequency of intermediates on metallic surface | 33 | |
| Electron | S/TEM | Crystal structure of nanoparticles | 34 and 35 |
| EELS/EDX | Chemical composition/oxidation states | 36 and 37 | |
| Ptychography, DPC | Imaging of light elements | 38 | |
| Other | ssNMR | Chemical environment of light elements | 6 |
| TPR | Oxidation states, oxidative and thermal stability | 30 | |
| DSC/TGA | Thermal stability | 30 | |
| ICP/MS | Elemental composition | 32 |
X-ray diffraction (XRD)
The accommodation of light elements induces structural distortion of the host metal system, for example the presence of carbon in the interstitial site of palladium.39 Normally, the diffraction patterns of the modified system are compared with that of the unmodified system to observe whether the reflections are shifted or new phases are formed. It should be noted that not every case will obey this rule. In recent work on the Au–intC system, doping does not induce any appreciable change in diffraction.40 Expansion or contraction due to substitutional modification is usually interpreted using Vegard's rule,17 which states that the concentration of a substitutional dopant is linearly correlated with the variation in the lattice parameter. However, it should be noted that Vegard's rule does not have a sound theoretical basis, as is often admitted by literature, with many exceptions reported.41Compared to laboratory-source X-rays, those from a synchrotron source have many improved properties, for example brightness, focus area, divergence, and energy bandwidth. Additionally, much higher energies can be achieved. Highly collimated and intense X-rays allow the collection of high-resolution X-ray data, particularly for diffraction. For example, in our recent work, the refinement of high-resolution synchrotron X-ray powder diffraction (SXRD or SXPD) patterns demonstrated the coexistence of FCC and HCP phases in Pd–intB NPs.6
Other characterisation by synchrotron radiation
Other experiments carried out at synchrotron-radiation facilities include the following. Use of the X-ray pair distribution function (XPDF) and the corresponding refinement can probe the local structure, distortion and site occupancy for disordered systems.6 Also useful in characterising non-crystalline systems, extended X-ray absorption fine structure (EXAFS) and X-ray absorption near-edge structure (XANES) can determine the chemical composition, electronic state, environment and local structure, regardless of long-range order.31 For example, the use of XANES spectra is capable of detecting the Pd–C bonds of carbon-containing molecules adsorbed at the surface of Pd NPs. In studies of the Pd–intH phase, Lamberti et al.42 used in situ and in operando X-ray absorption spectroscopy (XAS) to demonstrate the differences in structural and electronic configuration between the core and shell regions of the NP, due to hydriding at different temperatures and hydrogen pressures.X-ray photoelectron spectroscopy (XPS)
XPS is a conventional technique that is used to probe the surface composition and chemical state of solid catalysts, due to the very low penetration depth of the photoelectrons. It is often used to determine the oxidation states and concentrations of sub/surface atoms, which provides complementary information to XRD. Liu et al. synthesised mesoporous Pd–intB nanospheres (MNSs) by using amphiphilic dioctadecyldimethylammonium chloride (DODAC) as the surfactant template, and confirmed the synthesis of the phase by XRD and XPS. Notably, partial electron transfer to the metal framework was observed by the Pd 3d5/2 binding energy in Pd–intB shifting to significantly higher energy (336.1 eV vs. 335.1 eV), which is indicative of partial band filling.Fourier-transform infrared spectroscopy (FTIR)
FTIR spectroscopy is used to probe the variations in vibrational frequencies of metal–adsorbate bonds. Bowker et al.43 compared the CO absorption on doped and undoped Pd NPs using diffuse-reflectance infrared Fourier-transform spectroscopy (DRIFTS). For the reduced undoped Pd NPs, a band located at ∼1990 cm−1 was observed when exposed to CO, which was assigned to the bridge-bonded CO on Pd. However, this band of ∼1990 cm−1 was not seen in the carbidised sample, which indicates that the presence of carbon at the interstitial sites can alter the surface structure and block the bridge site adsorption, alleviating poisoning.Additionally, FTIR can be used to give insight into the catalytic mechanism under typical reaction conditions. Cai et al.44 studied the decomposition of formic acid over Pd–intB/C and undoped Pd/C. The Pd/C exhibited a distinctive CO band from adsorbed formic acid. In contrast, for the Pd–intB/C, there was negligible signal in the CO region (1800–1900 cm−1), and the predominant band was indicative of adsorbed CO2. This result indicates the subtle change in the reaction pathway for the formic acid dehydrogenation over Pd when doped with B.
Probing interstitial light elements with electrons
It is well known that electron microscopy can provide structural and morphological information of doped metallic nanoparticles.45 Benefiting from development in aberration correctors and computational processing, scanning transmission-electron microscope annular dark-field imaging (STEM-ADF) is becoming an important analytical technique to study this class of materials. However, it is rather challenging to directly visualise light elements such as hydrogen, carbon, boron, and nitrogen within the metal lattice due to their low atomic number and hence low electron-scattering ability, which is in stark contrast with highly scattering metal atoms.46 Furthermore, for light-element dopants, additional care must be taken to avoid beam damage when using intense electron beams. In an investigation of an interstitially boron-doped palladium catalyst, Pd–intB, our research group used the emerging electron microscopy technique of ptychography to visualise boron at the octahedral sites of the Pd metal framework.6 Along with other supporting techniques and simulations, certain atomic columns observed by microscopy were interpreted as interstitial boron atoms. In 2020, Gan et al.47 reported that the use of a differentiated and integrated differential phase contrast technique (dDPC and iDPC) in aberration-corrected STEM can even directly visualise atomic columns of hydrogen that correspond to interstitial tetrahedral sites in the Pd–H system, which was also supported by the simulated dDPC images. On the basis of the result, they claimed that hydrogen can also occupy tetrahedral interstitial sites, which is in agreement with previous NPD studies48 and theoretical calculations.49Electron microscopy is often associated with two spectroscopic techniques that yield elemental mapping information. Energy-dispersive X-ray (EDX) spectroscopy can yield elemental composition information, and specialises in the detection of heavy elements. Meanwhile electron energy-loss spectroscopy (EELS) is additionally able to probe the local chemical environment and oxidation state, and specialises in the detection of light elements. Recently, our research group used EELS to map the boron distribution within Pd NPs and estimated the site occupancy of boron at octahedral sites to be approximately 0.2.6 However, the PDF refinement proposed that the average site occupancy across the whole sample was much lower at ∼0.05; hence it was shown that interstitial boron is distributed unevenly across the sample. The dopant was shown to be present as atomic boron within the lattice by the observation of the B K-edge at 188 eV, rather than an energy of 194 eV and 203 eV which is indicative of boron oxides (Fig. 4).6
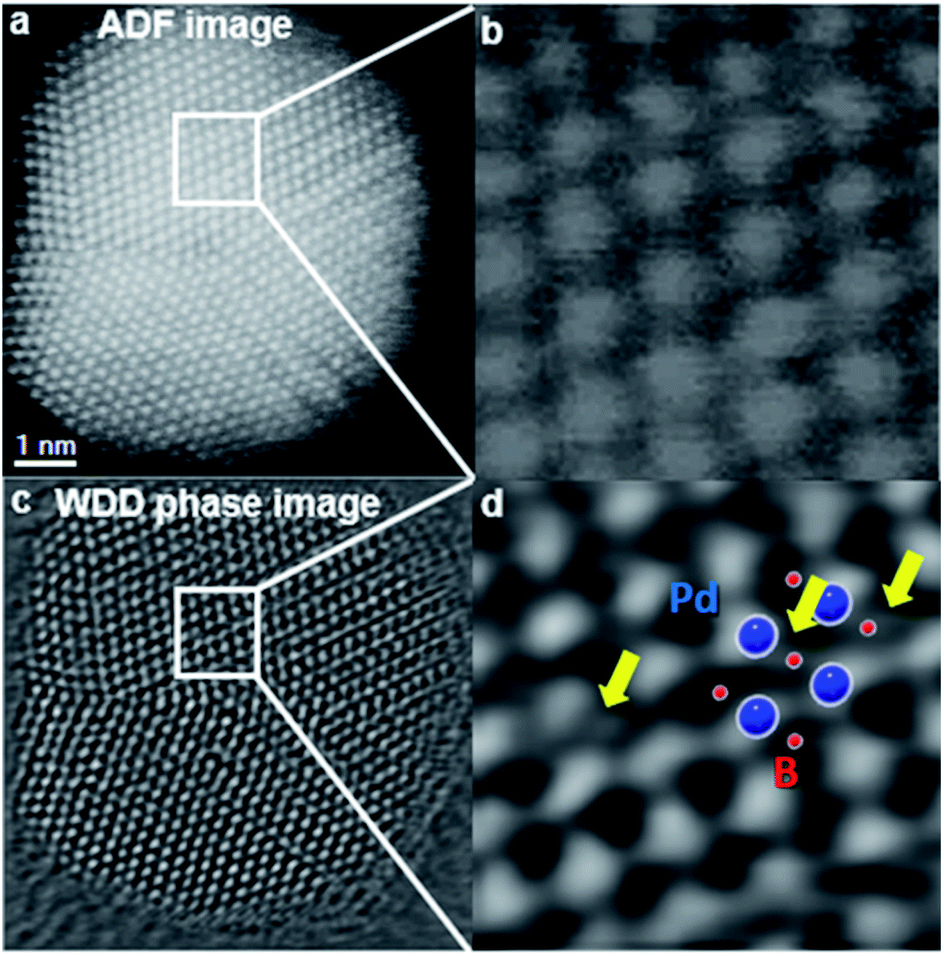 | ||
| Fig. 4 Simultaneously acquired (a) Z-contrast STEM-ADF (b) inset, and (c) ptychographic phase images of a Pd–intB/C NP oriented in the [110] zone axis. The ptychographic phase image is reconstructed using a Wigner distribution deconvolution (WDD) method after aberration correction. (d) Shows an enlarged area in which some scattering between the Pd columns can be superimposed by the model predicted by XPDF. This figure has been reproduced from ref. 6 with permission from American Chemical Society, copyright 2019. | ||
ICP-MS and ssNMR
Inductively coupled plasma mass spectrometry (ICP-MS) is a powerful analytical technique that can provide elemental composition to ppb sensitivity. For instance, Sato et al. quantified the boron composition of Pd–intB NPs synthesised by an electroless deposition method.50 However, ICP-MS is unable to give information about the location or environment of species. Our research group used 11B magic-angle spinning solid-state NMR (ssNMR) to determine the chemical environment of B in Pd–intB.6 Deconvolution of the spectra can show the presence of at least two chemical environments, of which one was attributed to the interstitial site and one to surface boron species. Additionally, comparison of the spin-lattice relaxation time T1 identified which peak corresponded to the interstitial B, as it has a faster relaxation due to its closer proximity to other atoms.Thermal gravimetric analysis (TGA)
TGA is a conventional analytical technique used to study the stability of interstitially-doped metal catalysts. Takanabe et al. used TGA and XANES simultaneously to evaluate the oxidation behaviour of cobalt NPs with and without interstitial boron dopants in CO2. On the basis of the weight change, the result indicated that the oxidation occurs at a lower temperature in the B-doped system in comparison with the fresh catalyst (boron oxidation on the surface encourages the formation of volatile boron oxide).51Temperature programmed reaction (TPR)
TPR is often used to find the most efficient reduction temperature for catalysts. Additionally, TPR is becoming a quick preliminary method for determining whether a dopant resides at the interstitial sites or not. Owing to the formation of interstitial hydride in palladium at sub-ambient temperatures, our research group showed that if interstitial sites are not blocked by foreign small-elements dopants, TPR yields a strong negative peak at approximately 70–80 °C which is attributed to β-PdHx decomposition. In contrast, the disappearance of this peak is due to the occupancy of carbon dopants at the interstitial sites.39Computational investigation
Unlike homogeneous catalysis, only surface atoms of close packed heterogeneous catalyst can participate in the chemical reaction. Therefore, is it crucially important to understand how the foreign light elements that occupy interstitial sites alter the surface atomic and electronic structure of a catalyst. In recent decades, computational simulations have become an important tool since they can disentangle complex phenomena and provide information that experiments cannot provide. Hence theoretical simulations are needed for the correlation of structure with chemical properties, which has repeatedly supported experimental testing and characterisation.Nørskov et al. conducted pioneering research into the role of subsurface carbon in palladium during selective hydrogenation by conducting DFT calculations.52 They identified the absorption energies of carbon at different sites in the Pd metal framework, and showed that the adsorption energy of a methyl group varies as a function of carbon coverage. It was found that the binding of methyl groups is substantially weakened if the carbon resides at the subsurface layer of Pd (111). As indicated, the presence of light elements can expand the unit cell, which can lead to lattice strain. Yang et al.53 compared the electronic effect induced by lattice strain and electronic interaction in the presence of boron atoms during their computational analysis of the hydrogenation of CO to methanol. As demonstrated in Fig. 5, the lattice-strain effect can shift the d-band of palladium to higher energy;54εd shifts to εd′ with an increment of εd′′. Meanwhile the alloying effect can broaden the d-band of palladium. Due to the orbital mixing of p-states of the boron, the d-band centre of the palladium is shifted down, hence weakening its chemisorption.
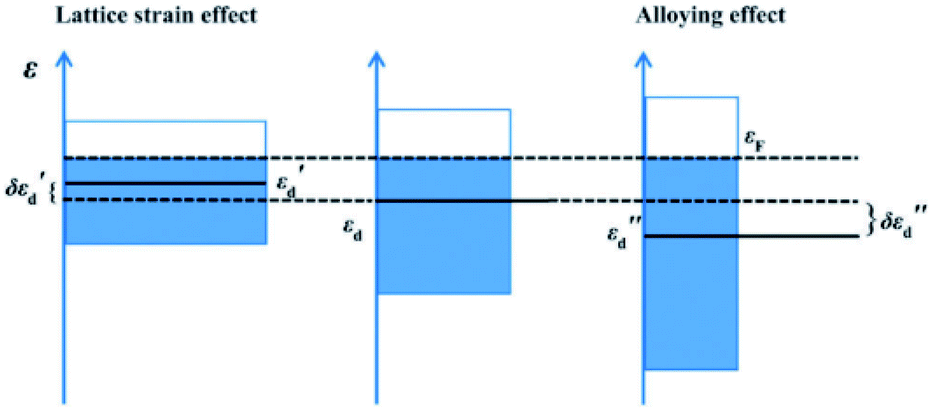 | ||
| Fig. 5 A schematic illustration of the electronic effect provided by lattice strain (left) and electronic interaction (right) in the presence of boron atoms, compared to pure Pd is (middle). εF is the Fermi energy; εd′(εd′′) is the energy of the d-band centre and δεd′(δεd′′) is the shift of the d-band centre, where δεd′ = εd′ − εd and δεd′′ = εd′′ − εd. This figure has been reproduced from ref. 53 with permission from Royal Society of Chemistry, copyright 2016. | ||
In addition to being used for assessing the surface–adsorbate interaction, and strain-alloying effect, DFT has also been used to predict the propensity of light-element accommodation to be interstitial or substitutional, thereby allowing prediction of new chemical and catalytic properties. For example, the experimental report by Ma et al.55 showed that doping boron into copper improves catalytic stability for the hydrogenation of dimethyl oxalate; however the experimental structure still remains uncharacterised. Mushrif et al. used DFT calculations to correlate the stable microstructure of Cu–B with catalytic performance.56
It was found by calculating the binding energy of boron at different sites on the Cu(111) surface that the most stable site is the octahedral site of the first subsurface layer (−486 kJ mol−1). As previously indicated, the role of light elements such as boron or carbon in the palladium subsurface can significantly promote catalytic performance. DFT was also used by Illas et al. to investigate C incorporation in subsurface sites of Cu, Ag and Au by using a supercell slab model and a well-shaped nanoparticle model.57 They reported that the most stable carbon site for the (111) surfaces of Cu and Ag is at the interstitial site, which is also true for their nanoparticle. In contrast, for the Au(111) slab surface, carbon is most stable chemisorbed on the surface, rather than integrated into the structure. For Au NPs however, carbon becomes more stable at the interstitial sites, which is partially due to the low coordination of the site in this morphology.
Catalytic applications
So far, we have demonstrated that incorporating light elements with metal NPs, either at interstitial or substitutional sites, may lead to structural distortion that modifies the surface structure and electronic properties of the host metal system, hence offering a way to tune the surface interactions between reactants and catalyst. In particular, interstitial doping may provide a new method for electronic modification of the metal surface. Herein, the recent studies of H-, Li-, B-, C-, and N-metal nanocatalysts will be summarised. As mentioned before, if the size of the dopant permits, it is energetically preferable for it to occupy interstitial sites rather than substitutional sites. However, examples of substitutional doping have been also reported for catalytic applications. We will highlight their unique catalytic properties accordingly.Boron doping
It is well known that boron is an effective promotor for tuning the catalytic activity of a wide range of metal and non-metal systems.58,59 In general, boron occupies the interstitial site of a host metal, but substitutional doping has also been reported in literature.60 Pd–intB is the most reported interstitial solid-solution system and has been used in electrochemistry and selective hydrogenation. Benefitting from the recent progress in synthesis strategies and characterisation, the Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :B atomic ratio can be precisely controlled, approaching a boron mole fraction of 0.18.61 In addition, Ni–intB and Co–intB systems are attracting attention as new catalysts for some important chemical conversions.
:B atomic ratio can be precisely controlled, approaching a boron mole fraction of 0.18.61 In addition, Ni–intB and Co–intB systems are attracting attention as new catalysts for some important chemical conversions.
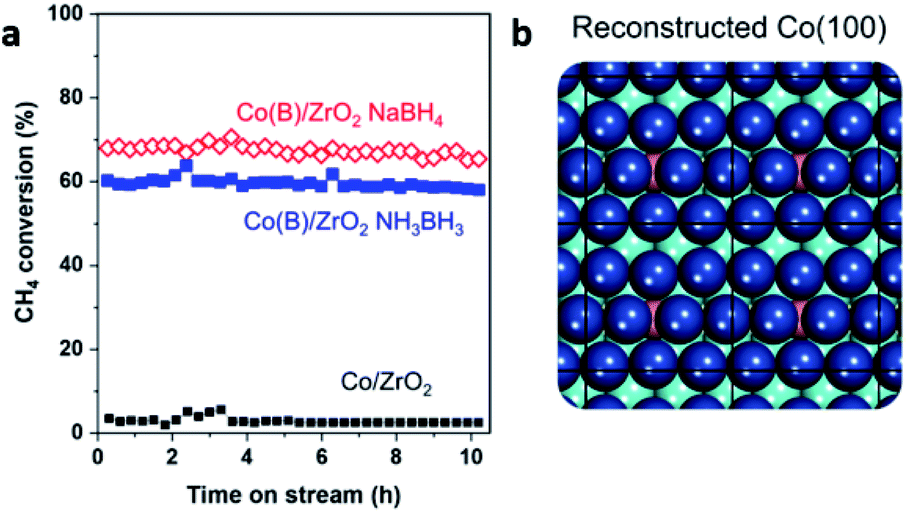 | ||
| Fig. 6 (a) The reaction stream of methane conversion for Co/ZrO2 and two Co–intB/ZrO2 catalysts prepared from NaBH4 and NH3BH3. (b) Simulated surface reorganisation of Co (blue) induced by the presence of interstitial B (pink). This figure has been reproduced from ref. 51 with permission from Elsevier, copyright 2020. | ||
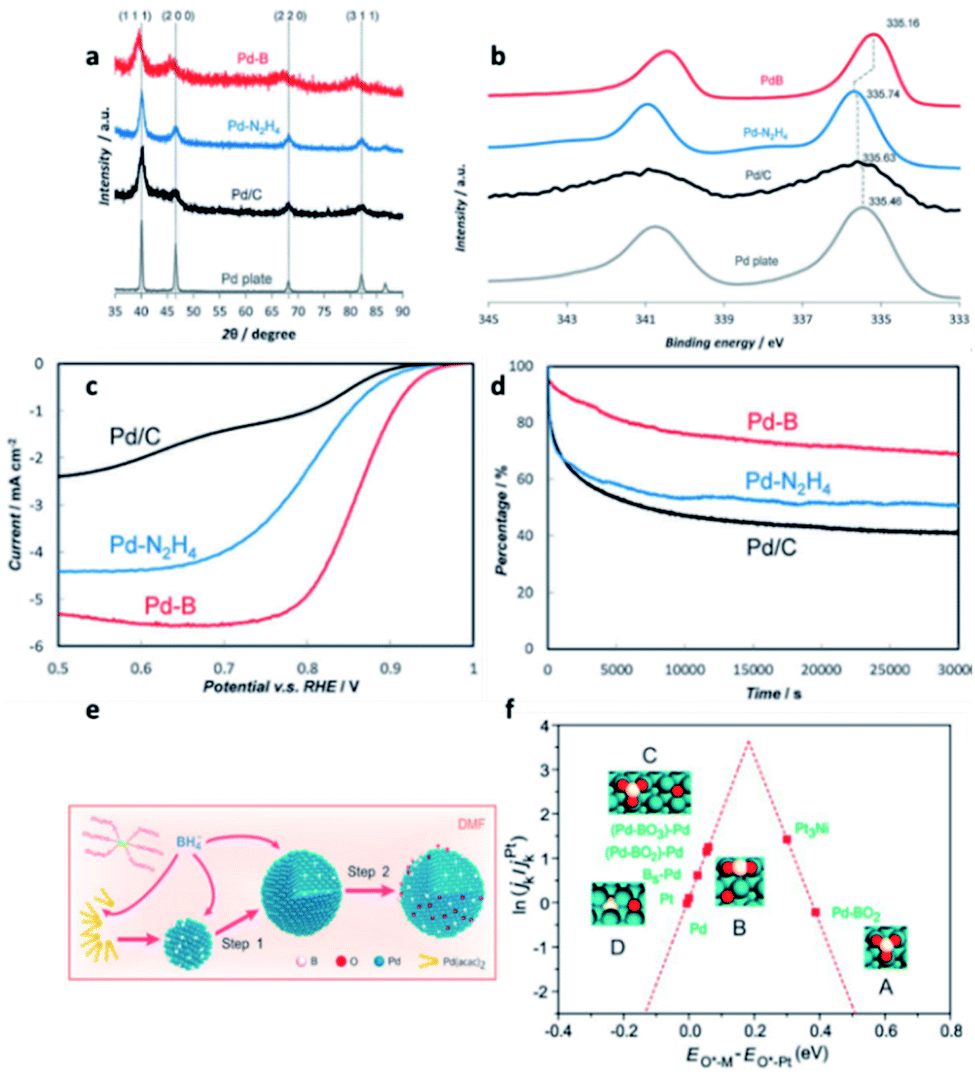 | ||
| Fig. 7 (a) XRD, (b) XPS, (c) ORR polarisation curves, and (d) chronoamperometry responses (as percentage of initial current density) of Pd–intB, Pd–N2H4, Pd/C and a Pd metal plate. This figure has been reproduced from ref. 32 with permission from Wiley-VCH, copyright 2016. (e) Illustration of the synthesis procedure for Pd–intB/C. (f) ORR activity of different surface sites on the Pd–intB surface, in comparison with platinum, with respect to the calculated O adsorption energies; This figure has been reproduced from ref. 74 with permission from American Chemical Society, copyright 2017. | ||
Compared with commercial undoped Pd and Pt catalysts, crystalline Pd–intB demonstrated 14-times and 35-times higher catalytic activity, respectively, as well as improved catalytic stability (Fig. 7c and d). The computational studies demonstrated that the presence of boron can lower the surface core level binding energy of Pd, indicating an increase in the density of states related to the Pd–O anti-bonding interaction with excess electrons from boron. The Pd–Oadsorbate interaction is therefore weakened and hence the catalytic activity is boosted.
In the subsequent years, Cai et al.73 and Wang et al.74 reported that Pd–intB catalysts can be prepared by different synthetic strategies which also exhibited high catalytic activity in the ORR. In the case of Wang et al. work, the concentration of boron can be controlled by adjusting the reaction temperature and precursor concentration (Fig. 7e). According to the DFT calculations reported by Wang et al., they concluded that Pd–BO2 assemblies and neighbouring palladium atoms are responsible for the superior catalytic activity; this effect is in addition to the effect of subsurface boron atoms. They also noted that other boron oxide species might exhibit good catalytic activity, even at low concentration (Fig. 7f).
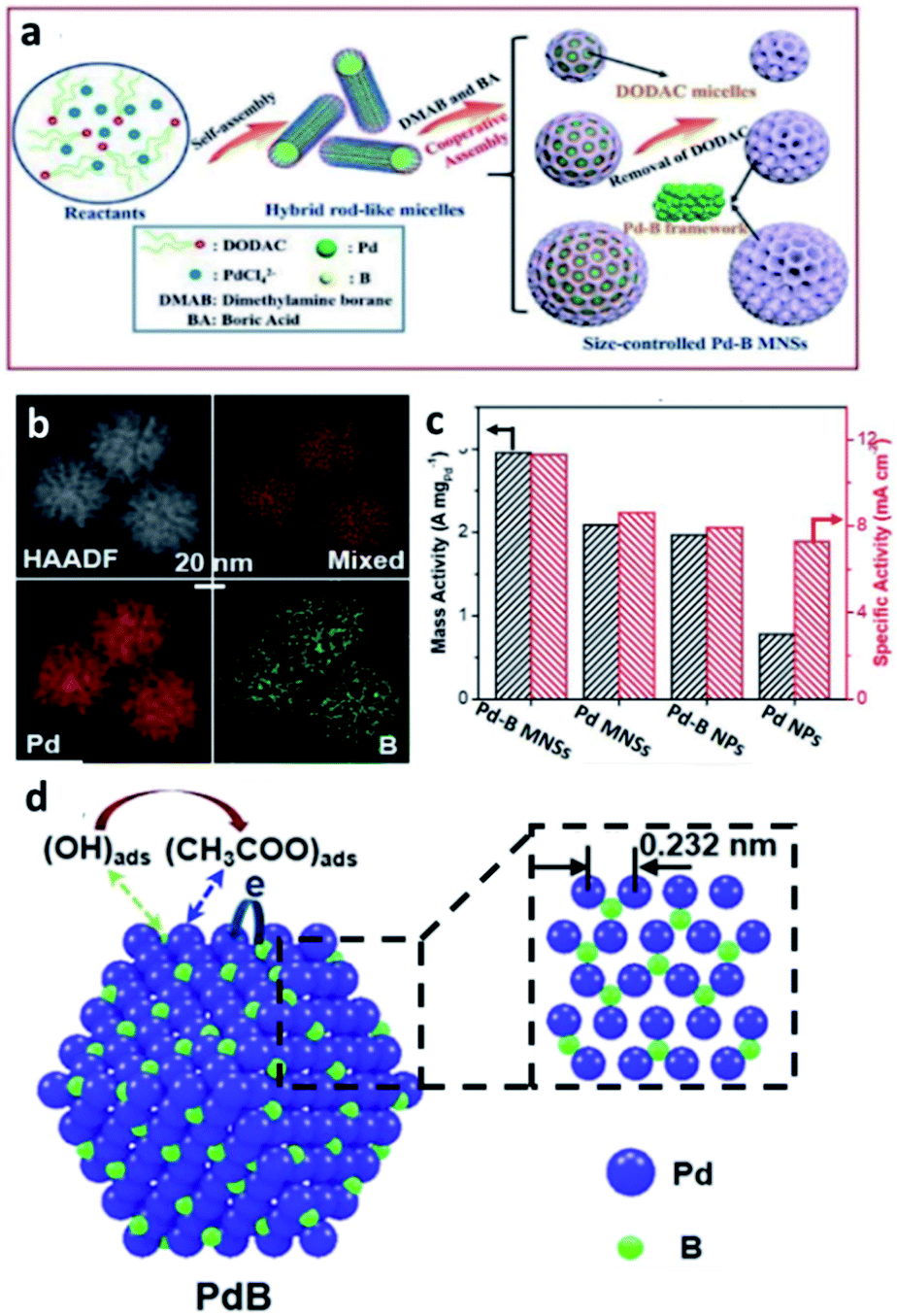 | ||
| Fig. 8 (a) A scheme for the formation of Pd–intB MNSs. (b) EDX elemental mapping; Pd (red), B (green). (c) Mass and specific activities of Pd–intB MNSs, Pd MNSs, Pd–intB NPs and commercial Pd NPs collected in 1.0 M KOH and 1.0 M ethanol. This figure has been reproduced from ref. 77 with permission from Royal Society of Chemistry, copyright 2019. (d) A illustration of electrocatalytic EOR on the surfaces of Pd–intB. The inset shows the crystal structure of Pd–intB: Pd (purple), B (green). This figure has been reproduced from ref. 78 with permission from American Chemical Society, copyright 2020. | ||
In 2020, the same group has continued to investigate the Pd–intB binary system for EOR.78 They suggested that the presence of boron can overcome charge-transfer resistances, and hence increase the rate of charge-transfer during the reaction. Furthermore, they attempted to rationalise the effect of boron on the Pd metal system in terms of electronic and bifunctional promotion (Fig. 8d). Incorporation of boron into metal lattices can not only modify the surface electronic properties of Pd by weakening the strength of adsorption of poisoning ethyoxy intermediates that are generated during the electrocatalytic EOR, but also facilitate the adsorption of OH–(B–OHads) which can accelerate the oxidation of the ethoxy intermediate and the associated diffusion away from the catalyst surface.
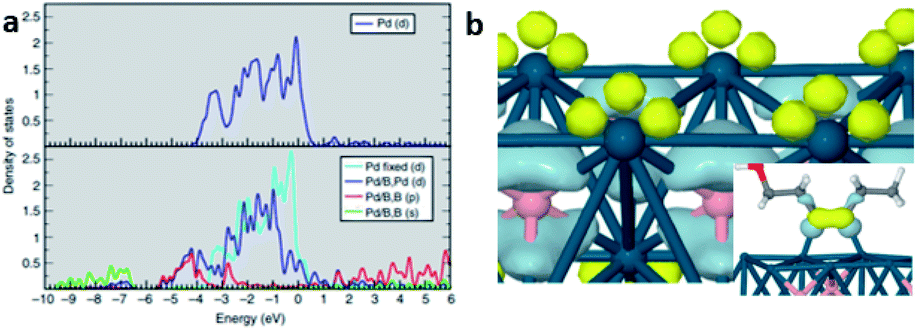 | ||
| Fig. 9 DFT study of Pd–intB system. (a) DOS of pure Pd (top) and Pd–intB (bottom). (b) Electron-density difference isosurfaces show local electron-rich (light blue) and electron-poor regions (yellow) of Pd atoms (blue) with intB atoms (pink) due to states-mixing. Inset shows isolated HOMO/LUMO overlap of the adsorbate/surface interaction. This figure has been reproduced from ref. 37 with permission from Nature, copyright 2014. | ||
Nitrogen doping
Nitrogen is one of the most investigated elements for modifying nanomaterials.83 The research of intermetallic nitride-based compounds has been summarised by Dongil32 and Fan84et al. Unlike boron modifications, nitrogen requires the extreme conditions of high pressure to be alloyed with Pd metal.85 In contrast, cobalt-based catalysts are often alloyed with nitrogen to enhance their catalytic activity. Monometallic cobalt NPs exhibit one of two possible phases, namely HCP and FCC, which are dependent on their size and the temperature employed in the preparation.86 The cobalt HCP–FCC phase transition occurs at around 700 K.87 Cobalt NPs with FCC phase are most commonly reported, which can be mainly attributed to the particle size effect. As illustrated by the work of Shimada et al.,88 particles with FCC structure are energetically more stable below 20 nm.89 Treatment with ammonia gas or other N-containing sources can incorporate nitrogen at the octahedral sites of cobalt, which can allow the formation of Co4N; though to a lesser degree, nitrogen can also reside at the tetrahedral sites, leading to the formation of Co5.47N and CoN. The type of interstitial hole that nitrogen occupies is governed by the synthetic method.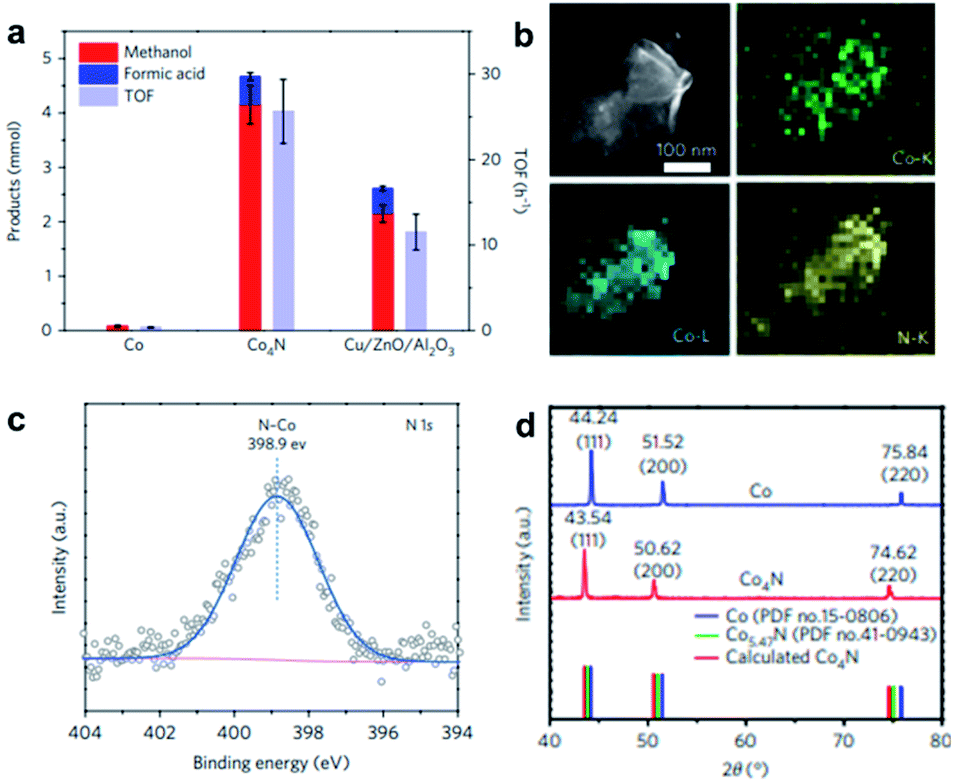 | ||
| Fig. 10 (a) Product selectivity and TOF of Co nanosheets, Co4N nanosheets and Cu/ZnO/Al2O3 in CO2 hydrogenation under 32 bar of 1:3 CO2/H2 at 150 °C for 3 hours. (b) EDX elemental mapping of the Co K-, Co L- and N K-edges of Co4N nanosheets. (c) N 1s XPS spectrum for Co4N sheets. (d) XRD patterns of Co and Co4N sheets. This figure has been reproduced from ref. 90 with permission from Nature, copyright 2017. | ||
In the same year, Wang et al. evaluated Co5.47N supported on porous nitrogen-doped carbon for the application of ORR. This system can exhibit an acceptable catalytic activity, with an onset potential of −42 mV vs. SCE.96 Huang et al. later reported that conducting a single-step preparation increased the activity of the catalyst.97 Sun et al.98 and Wu et al.99 synthesised Co5.47N in complex architectures by adding a third component. They concluded that the interaction between N and Co can enhance the catalytic activity. Additionally, Hou et al. reported that FeN (zinc blende structure) catalysts can be bound onto nitrogen-doped graphene through π–π stacking and oxygen-containing functional groups. Interaction between the support and the Fe–intN catalyst can result in a synergistic effect which enhances the catalytic activity and stability.100
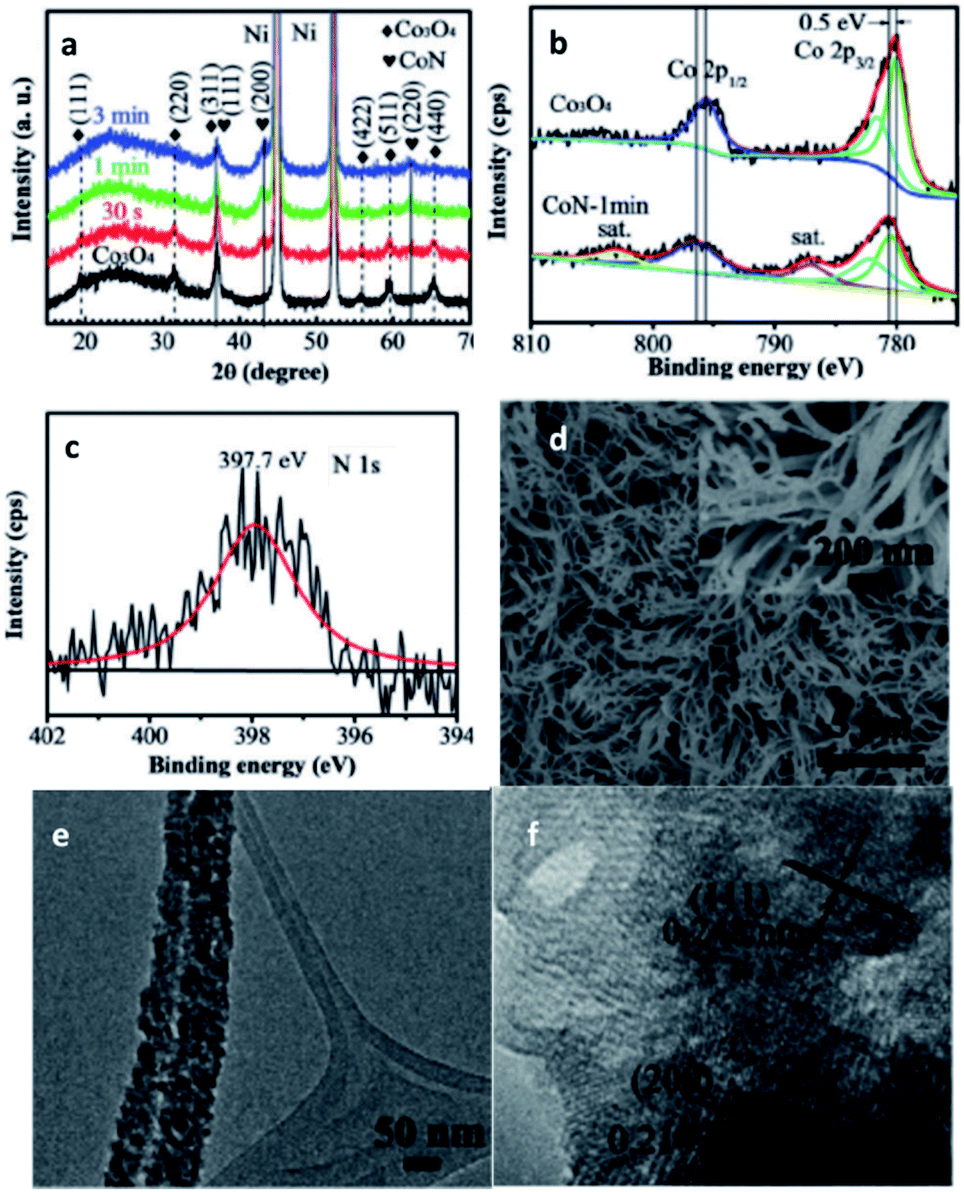 | ||
| Fig. 11 Characterisation of CoN. (a) XRD of Co3O4 and CoN. XPS spectra: (b) Co 2p of Co3O4 and CoN-1 min, (c) N 1s of CoN. (d) SEM, (e) TEM, (f) HR-TEM micrograph of CoN. This figure has been reproduced from ref. 95 with permission from Wiley-VCH, copyright 2016. | ||
Carbon doping
In the field of ferrous metallurgy, carbon is an essential element that can be alloyed into octahedral holes in the Fe bcc lattice, making the steel harder and stronger.29 In contrast, carbonaceous species are often considered poisonous in catalytic applications due to active site blocking. However, recent investigations have demonstrated that carbon can also play a promoting role in catalysis, namely when it resides in the subsurface of the metal catalyst. The subsurface modification of Pd by carbon (the maximum molar fraction is up to 0.13)81 has been the subject of intense research within the field, and will be summarised in the section below. Recent works also report that this strategy has been applied to other mono-40 and bi-metallic104 systems.In comparison with the Lindlar type catalyst, the subsurface modified Pd–intC NPs can suppress undesirable isomerisation and over-hydrogenation, which is attributed to the effects from the hybridisation of the Pd d-orbital with the C sp-orbital.30
In 2019, Jin et al. reported the successful preparation of a Pd–intC nanocrystal based on our synthesis strategy and widened the application to other substrates.106 This strategy has also been applied to other transition-metal catalysts, such as Au. Recently, Wan et al. have reported that carbon can be interstitially doped into Au for selective hydrogenation of alkyne-based substrates.40 Though the XRD did not reveal any lattice variation, they speculated that the location of carbon is at the interstitial sites, as reflected by the XPS and XANES (Fig. 12a and b). The TOF value of the Au–intC NPs was found to be higher than undoped Au/TiO2 with a similar particle size (Fig. 12c). A linear relationship between the activation energy and entropy of activation was observed (Fig. 12d). Their DFT calculations indicated that the activation barrier of hydrogen dissociation can be reduced to 0.59 eV on Au–intC(111), and showed that doping affects the adsorption modes of 3-nitrosytrene (Fig. 12e and f).
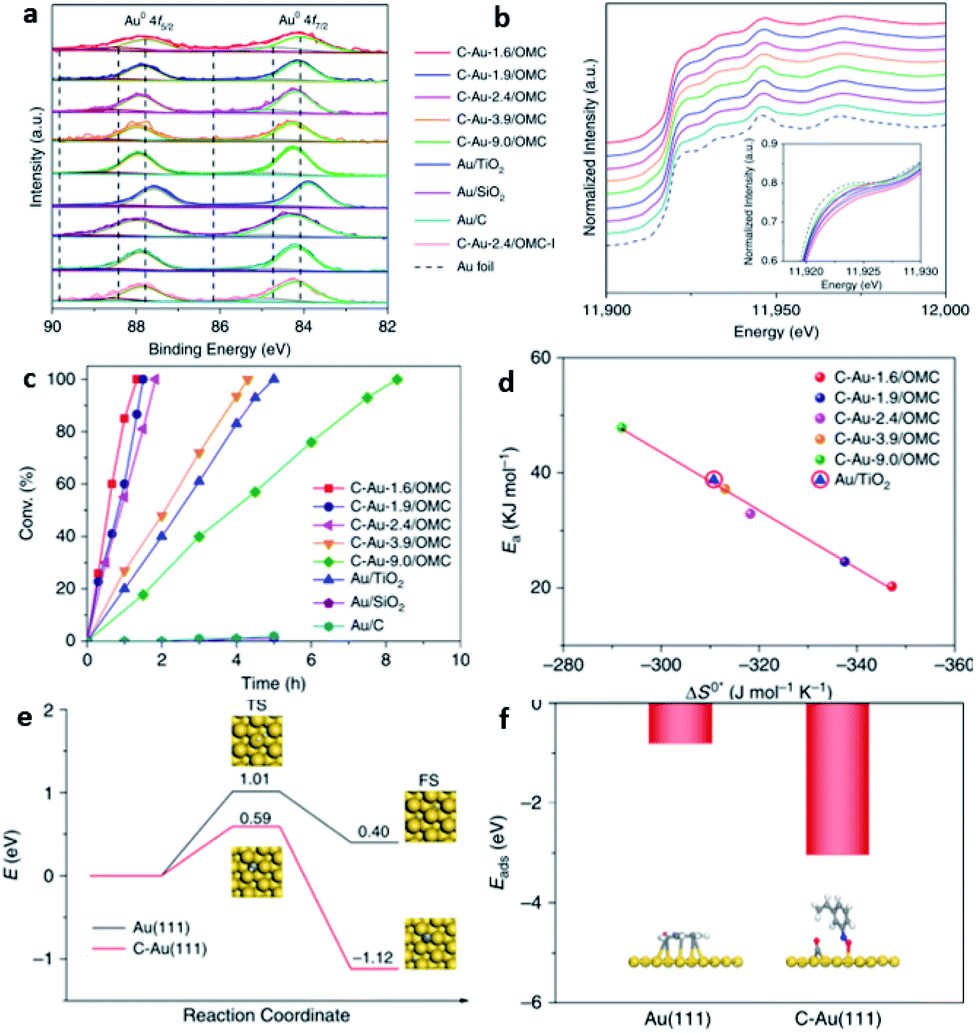 | ||
| Fig. 12 (a) Au 4f XPS and (b) Au L3-edge XANES spectra for various Au–intC NPs decorated on ordered mesoporous carbon. (c) The conversion of 3-nitrostyrene over different ctatalysts. (d) The resulting correlation between the activation energy (Ea) and entropy of activation (ΔS°*). (e) DFT calculations of energy profiles for the dissociation of H2 on pristine Au(111) and Au–intC(111), and (f) corresponding absorption modes. This figure has been reproduced from ref. 40 with permission from Nature, copyright 2020. | ||
Hydrogen doping
Historically, the study of metal hydrides has focussed on their application in hydrogen storage. However, recently hydrogen has also been found to be a promising element to incorporate into metal systems to tune their catalytic activity. Recently, palladium hydride (Pd–intH), an interstitial metal hydride,107 has increasingly received attention for a wide range of catalytic applications. There are two well-known structures of Pd–intH: the α-phase that has a composition of approximately PdH0<x≤0.03 and exhibits a slightly higher lattice parameter than pure palladium (3.889 Å), and the β-phase that has a composition of approximately PdHx≥0.58, whose formation is observed by a sudden increase in lattice parameter to 4.018 Å.108 Sometimes, α-PdHx and β-PdHx phases can coexist in the region of the phase diagram(0.03 ≤ x ≤ 0.58).109,110 The distribution between the octahedral and tetrahedral sites has been shown to be dependent on the depth from the surface, and hence highly dependent on particle size and shape.48 Elsewhere it has been found by NPD that the occupancy of tetrahedral sites in both the α- and β-phases increases when the host Pd framework is alloyed with other metals.111 Recent work on the utilisation of Pd/C in the electro-hydrogenation of dinitrogen demonstrated that the in situ formation of Pd–intH can catalyse the hydrogenation.112 Herein, other recent catalytic applications are detailed.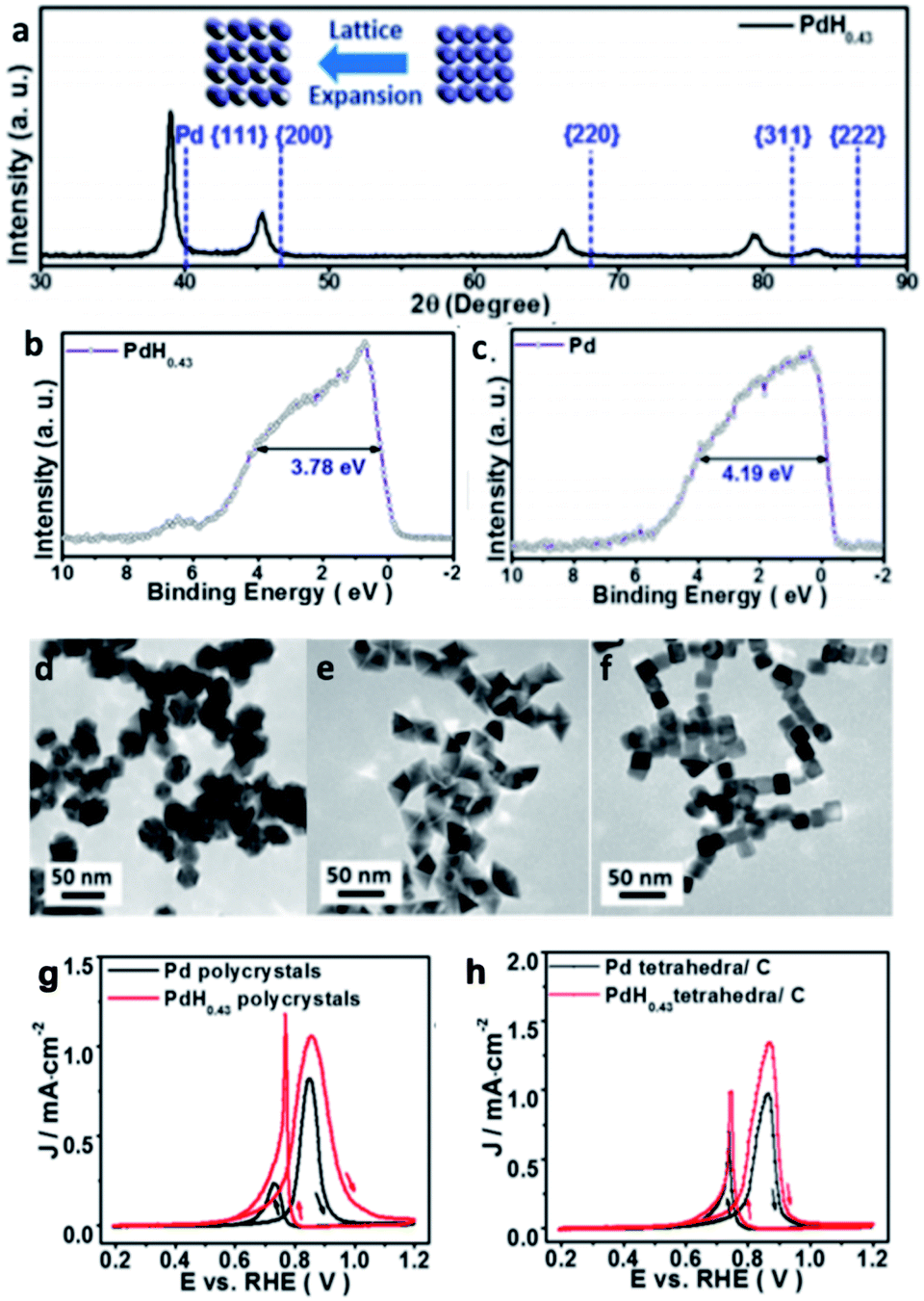 | ||
| Fig. 13 (a) XRD of PdH0.43. Valence band structure of (b) PdH0.43 and (c) Pd. (d–f) TEM micrographs of PdH0.43 nano-polycrystals, nano-tetrahedra and nanotubes. Cyclic voltammetry of Pd and PdH0.43 (g) polycrystals and (h) tetrahedra in N2 saturated, 0.1 M KOH with 0.1 M methanol, scan rate of 10 mV s−1. This figure has been reproduced from ref. 113 with permission from American Chemical Society, copyright 2015. | ||
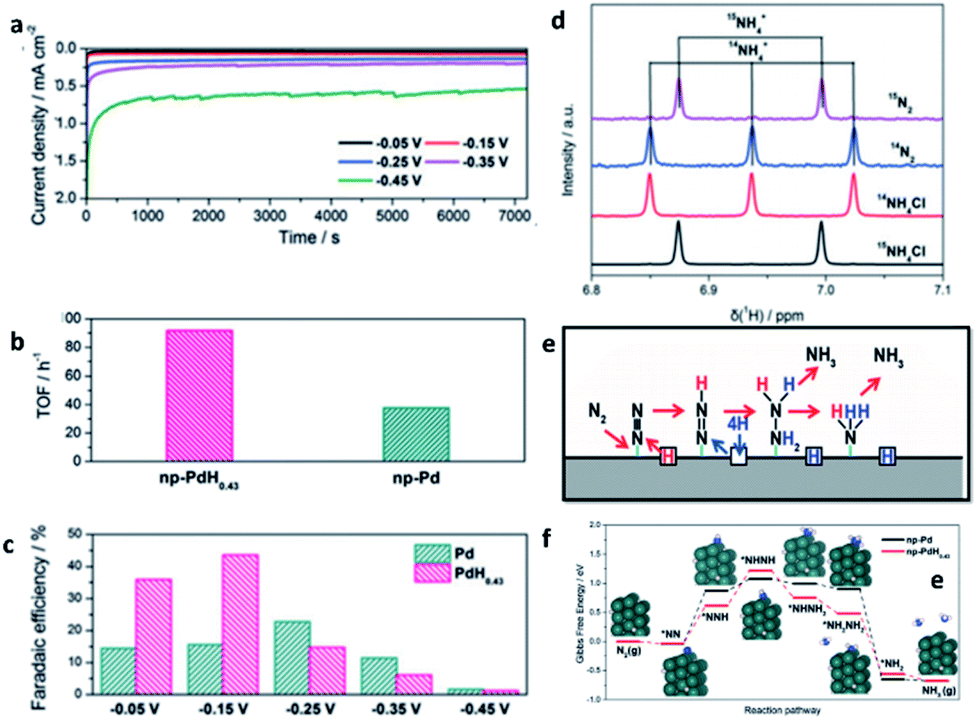 | ||
| Fig. 14 (a) Potentiostatic curves of np-PdH0.43 during NRR. (b) TOF of np-Pd and np-PdH0.43 at −0.15 V. (c) Faradaic efficiencies at selected potentials. (d) Baseline-subtracted 1H-NMR spectra of the post-mortem electrolyte with 15N2 and 14N2 and the reference 14NH4Cl and 15NH4Cl. (e) Proposed reaction pathway for nitrogen reduction. (f) Gibbs free-energy profiles of the NRR on np-Pd and np-PdH0.43. This figure has been reproduced from ref. 118 with permission from Wiley-VCH, copyright 2020. | ||
Lithium doping
Lithium doping is well-known to significantly modify the catalytic properties of metal oxide systems. A typical example, is the use of Li–MgO in oxidative methane coupling (OMC), in which C2 products are produced at high yields by the coupling of methyl radicals under high temperatures (943–1223 K);122 without Li no such catalysis is seen. However, there have been very limited works shown from recent open literature on using lithium doping in transition metal systems catalysis under reducing environments. Though the recent review on the catalytic reactions of light-element doped palladium by Rioux et al.123 was largely comprehensive, they did not include any mention of lithium-doped Pd. Despite limited examples, highlight of these reports are presented below.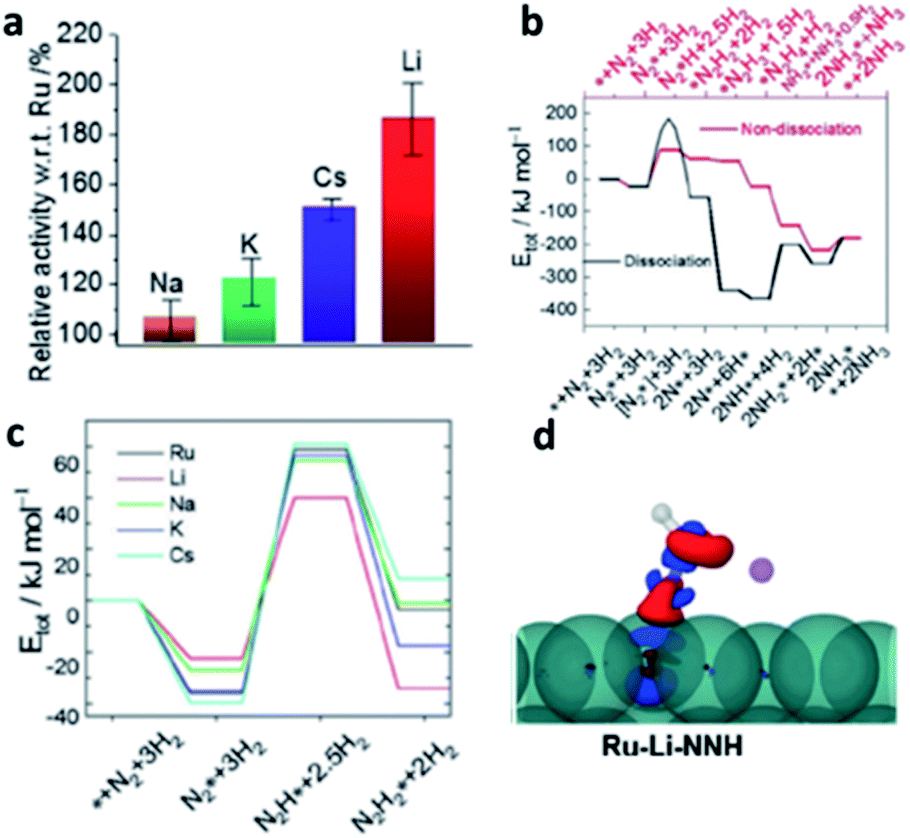 | ||
| Fig. 15 (a) Ammonia synthesis activity of various alkali-metal ion promoted Ba–Ru/AC with respect to total Ru content. (b) Calculated energy diagrams for ammonia synthesis via dissociative and associative pathways on Ru{0001} surface; the binding energy, Etot, is defined relative to the energies for the free Ru{0001} surface (*). Structures of the most stable intermediates are shown above and below the energy diagram for dissociative pathway and associative pathway. (c) Calculations for the most energetic step of adsorbed N2* to N2H* on Ru{0001} terrace with various dopants. (d) Electron-density difference plot (ρtotal − ρRu − ρNNH, where ρ is the electron density of the subscript component); negative (blue), positive (red), which shows the asymmetrical surface polarisation and stabilisation of NNH species by Li+ (purple) to induce electron back donation to π* region from Ru. This figure has been reproduced from ref. 126 with permission from Wiley-VCH, copyright 2020. | ||
DFT modelling was also used to further investigate the stabilisation of adsorbed N2 and hydrogenated species by surface alkali atoms by the associative pathway on terrace Ru sites. There is a particularly strong structural stabilisation for Li to stay closer to δ-N2*H than for any other alkali dopant (Fig. 15c). It is interesting to note from the calculated electron-density difference plots in Fig. 15d that the polarisation of the surface by Li can clearly break the symmetry of N2H* and greatly promote the back donation of Ru to N2H* (to give δ− charge) compared to the unpromoted Ru. This step facilities the associative mechanism for ammonia synthesis.
Conclusions
Overall, we have summarised the recent contributions to several light-element-modified transition-metal systems, discussing their synthesis, characterisation and the relationship between structure and catalytic applications and performance. In contrast to substitutional systems, interstitial light-element modified systems have wide-reaching application across catalysis and hold tremendous appeal across the community.There is no doubt that the chemistry of light-element-modified transition-metal systems will continue to flourish and pave an avenue to replace precious, expensive, and often more toxic transition-metal based catalysts. However, the research about the construction of such nanomaterials is still limited. For instance, the syntheses of nanoscale materials are challenging by conventional strategies. Incorporation of light-elements using recent synthetic methodologies such as forming multi-component alloy nanoparticles by fast non-equilibrium heating of their aerosols could be an exciting direction.128 On the other hand, some advanced structural characterisation techniques are still in the early stages of development and are difficult to access. These issues are being addressed in the continued development of advanced techniques and more facile synthesis procedures, which opens the way for light-element-modified transition-metal systems to play an increasingly important role in heterogeneous catalysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The first and second author thank for the care from their families and friends during the pandemic period of COVID-19.Notes and references
- G. Bond, Heterogeneous catalysis: principles and applications, Oxford: Clarendon Press, Oxford, 1982, vol. 27 Search PubMed.
- B. Huang, H. Kobayashi, T. Yamamoto, T. Toriyama, S. Matsumura, Y. Nishida, K. Sato, K. Nagaoka, M. Haneda, W. Xie, Y. Nanba, M. Koyama, F. Wang, S. Kawaguchi, Y. Kubota and H. Kitagawa, Angew. Chem., Int. Ed., 2019, 58, 2230–2235 CrossRef CAS.
- A. M. Alexander and J. S. J. Hargreaves, Chem. Soc. Rev., 2010, 39, 4388–4401 RSC.
- R. B. Levy and M. Boudart, Science, 1973, 181, 547–549 CrossRef CAS.
- E. W. McFarland and H. Metiu, Chem. Rev., 2013, 113, 4391–4427 CrossRef CAS.
- T. Chen, I. Ellis, T. J. N. Hooper, E. Liberti, L. Ye, B. T. W. Lo, C. O'Leary, A. A. Sheader, G. T. Martinez, L. Jones, P.-L. Ho, P. Zhao, J. Cookson, P. T. Bishop, P. Chater, J. V. Hanna, P. Nellist and S. C. E. Tsang, J. Am. Chem. Soc., 2019, 141, 19616–19624 CrossRef CAS.
- K. D. Gilroy, A. Ruditskiy, H.-C. Peng, D. Qin and Y. Xia, Chem. Rev., 2016, 116, 10414–10472 CrossRef CAS.
- M. Beck, M. Ellner and E. J. Mittemeijer, Mater. Sci. Forum, 2000, 321–324, 604–609 CAS.
- P. K. Liao, K. E. Spear and M. E. Schlesinger, J. Phase Equilib., 1996, 17, 340–345 CrossRef CAS.
- Z. Peng and H. Yang, Nano Today, 2009, 4, 143–164 CrossRef CAS.
- S. Furukawa and T. Komatsu, ACS Catal., 2017, 7, 735–765 CrossRef CAS.
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845–910 CrossRef CAS.
- J. Maier, J. Inorg. Gen. Chem., 2017, 643, 2083–2087 CAS.
- Y. M. Zhang, J. R. G. Evans and S. Yang, J. Cryst. Phys. Chem., 2013, 44, 81–97 Search PubMed.
- M. Armbrüster, Encyclopedia of Catalysis, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2011 Search PubMed.
- Y. An, H. Ijaz, M. Huang, J. Qu and S. Hu, Dalton Trans., 2020, 49, 1646–1651 RSC.
- E. W. McFarland and H. Metiu, Chem. Rev., 2013, 113, 4391–4427 CrossRef CAS.
- A. Machocki and R. Jezior, Chem. Eng. J., 2008, 137, 643–652 CrossRef CAS.
- M. Armbrüster, R. Schlögl and Y. Grin, Sci. Technol. Adv. Mater., 2014, 15, 034803 CrossRef.
- P. Chen, E. Akiba, S. Orimo, A. Zuettel and L. Schlapbach, Hydrogen Science and Engineering: Materials, Processes, Systems and Technology, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2016, pp. 763–790 Search PubMed.
- Y. Xiao, J.-Y. Hwang and Y.-K. Sun, J. Mater. Chem. A, 2016, 4, 10379–10393 RSC.
- S. Carenco, D. Portehault, C. Boissière, N. Mézailles and C. Sanchez, Chem. Rev., 2013, 113, 7981–8065 CrossRef CAS.
- A. B. Dongil, Nanomaterials, 2019, 9, 1111 CrossRef CAS.
- G. Hägg, Phys. Chem. B, 1931, 12, 33 Search PubMed.
- L. H. Bennett, A. J. McAlister and R. E. Watson, Phys. Today, 1977, 30, 34–41 CrossRef CAS.
- L. Brewer, Science, 1968, 161, 115–122 CrossRef CAS.
- S. T. Oyama, Catal. Today, 1992, 15, 179–200 CrossRef CAS.
- J. S. Galsin, in Impurity Scattering in Metallic Alloys, Springer US, Boston, MA, 2002, pp. 93–123 Search PubMed.
- P. Atkins, F. Armstrong, T. Overton, J. Rourke and M. Weller, Inorganic Chemistry, OUP, Oxford, 5th edn, 2009. Search PubMed.
- C. W. A. Chan, K. Y. Tam, J. Cookson, P. Bishop and S. C. Tsang, Catal. Sci. Technol., 2011, 1, 1584 RSC.
- A. L. Bugaev, A. A. Guda, I. A. Pankin, E. Groppo, R. Pellegrini, A. Longo, A. V. Soldatov and C. Lamberti, Catal. Today, 2019, 336, 40–44 CrossRef CAS.
- T. T. Vo Doan, J. Wang, K. C. Poon, D. C. L. Tan, B. Khezri, R. D. Webster, H. Su and H. Sato, Angew. Chem., Int. Ed., 2016, 55, 6842–6847 CrossRef.
- M. Nesselberger and M. Arenz, ChemCatChem, 2016, 8, 1125–1131 CrossRef CAS.
- F. M. Ross, Science of Microscopy, Springer, New York, NY, 2007, pp. 445–534. Search PubMed.
- Z. Wang, D. Santhanagopalan, W. Zhang, F. Wang, H. L. Xin, K. He, J. Li, N. Dudney and Y. S. Meng, Nano Lett., 2016, 16, 3760–3767 CrossRef CAS.
- K. Kobayashi, H. Kobayashi, M. Maesato, M. Hayashi, T. Yamamoto, S. Yoshioka, S. Matsumura, T. Sugiyama, S. Kawaguchi, Y. Kubota, H. Nakanishi and H. Kitagawa, Angew. Chem., Int. Ed., 2017, 56, 6578–6582 CrossRef CAS.
- C. W. A. Chan, A. H. Mahadi, M. M.-J. Li, E. C. Corbos, C. Tang, G. Jones, W. C. H. Kuo, J. Cookson, C. M. Brown, P. T. Bishop and S. C. E. Tsang, Nat. Commun., 2014, 5, 5787 CrossRef CAS.
- H. Yang, R. N. Rutte, L. Jones, M. Simson, R. Sagawa, H. Ryll, M. Huth, T. J. Pennycook, M. L. H. Green, H. Soltau, Y. Kondo, B. G. Davis and P. D. Nellist, Nat. Commun., 2016, 7, 12532 CrossRef CAS.
- K. Okitsu, Y. Mizukoshi, H. Bandow, T. A. Yamamoto, Y. Nagata and Y. Maeda, J. Phys. Chem. B, 1997, 101, 5470–5472 CrossRef CAS.
- Y. Sun, Y. Cao, L. Wang, X. Mu, Q. Zhao, R. Si, X. Zhu, S. Chen, B. Zhang, D. Chen and Y. Wan, Nat. Commun., 2020, 11, 4600 CrossRef CAS.
- T. Fujii, Am. Mineral., 1960, 45, 370–382 CAS.
- A. L. Bugaev, O. A. Usoltsev, A. Lazzarini, K. A. Lomachenko, A. A. Guda, R. Pellegrini, M. Carosso, J. G. Vitillo, E. Groppo, J. A. van Bokhoven, A. V. Soldatov and C. Lamberti, Faraday Discuss., 2018, 208, 187–205 RSC.
- W. Jones, P. P. Wells, E. K. Gibson, A. Chutia, I. P. Silverwood, C. R. A. Catlow and M. Bowker, ChemCatChem, 2019, 11, 4334–4339 CrossRef CAS.
- K. Jiang, J. Chang, H. Wang, S. Brimaud, W. Xing, R. J. Behm and W.-B. Cai, ACS Appl. Mater. Interfaces, 2016, 8, 7133–7138 CrossRef CAS.
- D. S. Su, B. Zhang and R. Schlögl, Chem. Rev., 2015, 115, 2818–2882 CrossRef CAS.
- S. Yamashita, J. Kikkawa, K. Yanagisawa, T. Nagai, K. Ishizuka and K. Kimoto, Sci. Rep., 2018, 8, 12325 CrossRef.
- B. Lin, X. Wu, L. Xie, Y. Kang, H. Du, F. Kang, J. Li and L. Gan, Angew. Chem., Int. Ed., 2020, 59, 20348–20352 CrossRef CAS.
- H. Akiba, M. Kofu, H. Kobayashi, H. Kitagawa, K. Ikeda, T. Otomo and O. Yamamuro, J. Am. Chem. Soc., 2016, 138, 10238–10243 CrossRef CAS.
- T. Ishimoto and M. Koyama, J. Chem. Phys., 2018, 148, 034705 CrossRef.
- K. C. Poon, D. C. L. Tan, T. D. T. Vo, B. Khezri, H. Su, R. D. Webster and H. Sato, J. Am. Chem. Soc., 2014, 136, 5217–5220 CrossRef CAS.
- A. J. Al Abdulghani, J. Park, S. M. Kozlov, D.-C. Kang, B. AlSabban, S. Pedireddy, A. Aguilar-Tapia, S. Ould-Chikh, J.-L. Hazemann, J.-M. Basset, L. Cavallo and K. Takanabe, J. Catal., 2020, 392, 126–134 CrossRef CAS.
- F. Studt, F. Abild-Pedersen, T. Bligaard, R. Z. Sørensen, C. H. Christensen and J. K. Nørskov, Angew. Chem., Int. Ed., 2008, 47, 9299–9302 CrossRef CAS.
- P. Wu and B. Yang, Phys. Chem. Chem. Phys., 2016, 18, 21720–21729 RSC.
- M. Mavrikakis, B. Hammer and J. K. Nørskov, Phys. Rev. Lett., 1998, 81, 2819–2822 CrossRef.
- S. Zhao, H. Yue, Y. Zhao, B. Wang, Y. Geng, J. Lv, S. Wang, J. Gong and X. Ma, J. Catal., 2013, 297, 142–150 CrossRef CAS.
- Q. T. Trinh, A. Banerjee, Y. Yang and S. H. Mushrif, J. Phys. Chem. C, 2017, 121, 1099–1112 CrossRef CAS.
- O. Piqué, I. Z. Koleva, F. Viñes, H. A. Aleksandrov, G. N. Vayssilov and F. Illas, Angew. Chem., Int. Ed., 2019, 58, 1744–1748 CrossRef.
- T. Koretsune and S. Saito, Phys. Rev. B, 2008, 77, 165417 CrossRef.
- S. Agnoli and M. Favaro, J. Mater. Chem. A, 2016, 4, 5002–5025 RSC.
- H. Brodowsky and H. Sagunski, Berichte der Bunsengesellschaft für physikalische Chemie, 1983, 87, 803–805 CrossRef CAS.
- M. Beck, M. Ellner and E. J. Mittemeijer, Mater. Sci. Forum, 2000, 321–324, 604–609 CAS.
- M. Akri, S. Zhao, X. Li, K. Zang, A. F. Lee, M. A. Isaacs, W. Xi, Y. Gangarajula, J. Luo, Y. Ren, Y.-T. Cui, L. Li, Y. Su, X. Pan, W. Wen, Y. Pan, K. Wilson, L. Li, B. Qiao, H. Ishii, Y.-F. Liao, A. Wang, X. Wang and T. Zhang, Nat. Commun., 2019, 10, 5181 CrossRef.
- J. Xu and M. Saeys, J. Catal., 2006, 242, 217–226 CrossRef CAS.
- J. Xu and M. Saeys, Chem. Eng. Sci., 2007, 62, 5039–5041 CrossRef CAS.
- J. Xu and M. Saeys, J. Phys. Chem. C, 2009, 113, 4099–4106 CrossRef CAS.
- A. Fouskas, M. Kollia, A. Kambolis, C. Papadopoulou and H. Matralis, Appl. Catal., A, 2014, 474, 125–134 CrossRef CAS.
- K. F. Tan, J. Chang, A. Borgna and M. Saeys, J. Catal., 2011, 280, 50–59 CrossRef CAS.
- H.-X. Zhang, S.-H. Wang, K. Jiang, T. André and W.-B. Cai, J. Power Sources, 2012, 199, 165–169 CrossRef CAS.
- K. Jiang, K. Xu, S. Zou and W. Bin Cai, J. Am. Chem. Soc., 2014, 136, 4861–4864 CrossRef CAS.
- J. S. Yoo, Z.-J. Zhao, J. K. Nørskov and F. Studt, ACS Catal., 2015, 5, 6579–6586 CrossRef CAS.
- T. Murata, K. Kotsuki, H. Murayama, R. Tsuji and Y. Morita, Commun. Chem., 2019, 2, 46 CrossRef.
- T. T. Vo Doan, J. Wang, K. C. Poon, D. C. L. Tan, B. Khezri, R. D. Webster, H. Su and H. Sato, Angew. Chem., Int. Ed., 2016, 55, 6842–6847 CrossRef.
- M. Wang, X. Qin, K. Jiang, Y. Dong, M. Shao and W. Bin Cai, J. Phys. Chem. C, 2017, 121, 3416–3423 CrossRef CAS.
- J. Li, J. Chen, Q. Wang, W.-B. Cai and S. Chen, Chem. Mater., 2017, 29, 10060–10067 CrossRef CAS.
- L. Chen, L. Lu, H. Zhu, Y. Chen, Y. Huang, Y. Li and L. Wang, Nat. Commun., 2017, 8, 14136 CrossRef CAS.
- R. Jiang, D. T. Tran, J. P. McClure and D. Chu, ACS Catal., 2014, 4, 2577–2586 CrossRef CAS.
- H. Lv, L. Sun, D. Xu, J. Henzie, Y. Yamauchi and B. Liu, J. Mater. Chem. A, 2019, 7, 24877–24883 RSC.
- L. Sun, H. Lv, Y. Wang, D. Xu and B. Liu, J. Phys. Chem. Lett., 2020, 11, 6632–6639 CrossRef CAS.
- M. Crespo-Quesada, A. Yarulin, M. Jin, Y. Xia and L. Kiwi-Minsker, J. Am. Chem. Soc., 2011, 133, 12787–12794 CrossRef CAS.
- M.-K. Kang and J. Nielsen, J. Ind. Microbiol. Biotechnol., 2017, 44, 613–622 CrossRef CAS.
- A. Borodziński and G. C. Bond, Catal. Rev., 2006, 48, 91–144 CrossRef.
- D. Teschner, J. Borsodi, A. Wootsch, Z. Revay, M. Havecker, A. Knop-Gericke, S. D. Jackson and R. Schlogl, Science, 2008, 320, 86–89 CrossRef CAS.
- M. Inagaki, M. Toyoda, Y. Soneda and T. Morishita, Carbon, 2018, 132, 104–140 CrossRef CAS.
- Y. Zhong, X. Xia, F. Shi, J. Zhan, J. Tu and H. J. Fan, Adv. Sci., 2016, 3, 1500286 CrossRef.
- J. C. Crowhurst, A. F. Goncharov, B. Sadigh, J. M. Zaug, D. Aberg, Y. Meng and V. B. Prakapenka, J. Mater. Res., 2008, 23, 1–5 CrossRef CAS.
- V. A. de la Peña O′Shea, P. R. de la Piscina, N. Homs, G. Aromí and J. L. G. Fierro, Chem. Mater., 2009, 21, 5637–5643 CrossRef.
- R. Lizárraga, F. Pan, L. Bergqvist, E. Holmström, Z. Gercsi and L. Vitos, Sci. Rep., 2017, 7, 3778 CrossRef.
- O. Kitakami, H. Sato, Y. Shimada, F. Sato and M. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 13849–13854 CrossRef CAS.
- P. van Helden, I. M. Ciobîcă and R. L. J. Coetzer, Catal. Today, 2016, 261, 48–59 CrossRef CAS.
- L. Wang, W. Zhang, X. Zheng, Y. Chen, W. Wu, J. Qiu, X. Zhao, X. Zhao, Y. Dai and J. Zeng, Nat. Energy, 2017, 2, 869–876 CrossRef CAS.
- R. Razzaq, C. Li, M. Usman, K. Suzuki and S. Zhang, Chem. Eng. J., 2015, 262, 1090–1098 CrossRef CAS.
- L. An, W. Huang, N. Zhang, X. Chen and D. Xia, J. Mater. Chem. A, 2014, 2, 62–65 RSC.
- H. Abroshan, P. Bothra, S. Back, A. Kulkarni, J. K. Nørskov and S. Siahrostami, J. Phys. Chem. C, 2018, 122, 4783–4791 CrossRef CAS.
- Y. Yao, X. Lin and X. Zhang, J. Mater. Sci., 2018, 53, 7691–7702 CrossRef CAS.
- Y. Zhang, B. Ouyang, J. Xu, G. Jia, S. Chen, R. S. Rawat and H. J. Fan, Angew. Chem., Int. Ed., 2016, 55, 8670–8674 CrossRef CAS.
- Y. Zhou, Z. Zhou, R. Shen, R. Ma, Q. Liu, G. Cao and J. Wang, Energy Storage Mater., 2018, 13, 189–198 CrossRef.
- Z. Rong, C. Dong, S. Zhang, W. Dong and F. Huang, Nanoscale, 2020, 12, 6089–6095 RSC.
- M. Jiang, C. Fu, R. Cheng, W. Zhang, T. Liu, R. Wang, J. Zhang and B. Sun, Adv. Sci., 2020, 7, 2000747 CrossRef.
- Z. Chen, Y. Ha, Y. Liu, H. Wang, H. Yang, H. Xu, Y. Li and R. Wu, ACS Appl. Mater. Interfaces, 2018, 10, 7134–7144 CrossRef CAS.
- H. Yin, C. Zhang, F. Liu and Y. Hou, Adv. Funct. Mater., 2014, 24, 2930–2937 CrossRef CAS.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS.
- C. Guan, A. Sumboja, W. Zang, Y. Qian, H. Zhang, X. Liu, Z. Liu, D. Zhao, S. J. Pennycook and J. Wang, Energy Storage Mater., 2019, 16, 243–250 CrossRef.
- R. Xu, F. Luo, M. Li and Z. Yang, Chem. Commun., 2019, 55, 13394–13397 RSC.
- Y. Niu, X. Huang, Y. Wang, M. Xu, J. Chen, S. Xu, M. Willinger, W. Zhang, M. Wei and B. Zhang, Nat. Commun., 2020, 11, 1–9 Search PubMed.
- M. Bauer, R. Schoch, L. Shao, B. Zhang, A. Knop-Gericke, M. Willinger, R. Schlögl and D. Teschner, J. Phys. Chem. C, 2012, 116, 22375–22385 CrossRef CAS.
- R. Guo, Q. Chen, X. Li, Y. Liu, C. Wang, W. Bi, C. Zhao, Y. Guo and M. Jin, J. Mater. Chem. A, 2019, 7, 4714–4720 RSC.
- Q. Lai, T. Wang, Y. Sun and K.-F. Aguey-Zinsou, Adv. Mater. Technol., 2018, 3, 1700298 CrossRef.
- A. J. Maeland and T. R. P. Gibb, J. Phys. Chem., 1961, 65, 1270–1272 CrossRef CAS.
- B. D. Adams and A. Chen, Mater. Today, 2011, 14, 282–289 CrossRef CAS.
- Y. Fukai, The Metal-Hydrogen System, Springer, Berlin, Heidelberg, 1993, vol. 21 Search PubMed.
- D. E. Nanu, W. J. Legerstee, S. W. H. Eijt, W. G. Haije, J. F. Vente, M. G. Tucker and A. J. Böttger, Acta Mater., 2008, 56, 6132–6140 CrossRef CAS.
- J. Wang, L. Yu, L. Hu, G. Chen, H. Xin and X. Feng, Nat. Commun., 2018, 9, 1795 CrossRef.
- Z. Zhao, X. Huang, M. Li, G. Wang, C. Lee, E. Zhu, X. Duan and Y. Huang, J. Am. Chem. Soc., 2015, 137, 15672–15675 CrossRef CAS.
- D. Gao, H. Zhou, F. Cai, D. Wang, Y. Hu, B. Jiang, W.-B. Cai, X. Chen, R. Si, F. Yang, S. Miao, J. Wang, G. Wang and X. Bao, Nano Res., 2017, 10, 2181–2191 CrossRef CAS.
- M. K. Kabiraz, J. Kim, W. J. Lee, B. Ruqia, H. C. Kim, S. U. Lee, J. R. Kim, S. M. Paek, J. W. Hong and S. Il Choi, Chem. Mater., 2019, 31, 5663–5673 CrossRef CAS.
- X.-T. Li, L. Chen, G.-F. Wei, C. Shang and Z.-P. Liu, ACS Catal., 2020, 10, 9694–9705 CrossRef CAS.
- J. Zhang, M. Chen, H. Li, Y. Li, J. Ye, Z. Cao, M. Fang, Q. Kuang, J. Zheng and Z. Xie, Nano Energy, 2018, 44, 127–134 CrossRef CAS.
- W. Xu, G. Fan, J. Chen, J. Li, L. Zhang, S. Zhu, X. Su, F. Cheng and J. Chen, Angew. Chem., 2020, 132, 3539–3544 CrossRef.
- Y. Lu, J. Wang, Y. Peng, A. Fisher and X. Wang, Adv. Energy Mater., 2017, 7, 1700919 CrossRef.
- S. Wang, D. Tian, X. Wang, J. Qin, Y. Tang, J. Zhu, Y. Cong, H. Liu, Y. Lv, C. Qiu, Z. Gao and Y. Song, Electrochem. Commun., 2019, 102, 67–71 CrossRef CAS.
- L. Yan, X.-X. Liu and Y. Fu, RSC Adv., 2016, 6, 109702–109705 RSC.
- B. L. Farrell, V. O. Igenegbai and S. Linic, ACS Catal., 2016, 6, 4340–4346 CrossRef CAS.
- T. Xie and R. M. Rioux, Catal. Today DOI:10.1016/j.cattod.2020.07.062.
- I. T. Ellis, E. H. Wolf, G. Jones, B. Lo, M. Meng-Jung Li, A. P. E. York and S. C. Edman Tsang, Chem. Commun., 2017, 53, 601–604 RSC.
- D. E. Brown, T. Edmonds, R. W. Joyner, J. J. McCarroll and S. R. Tennison, Catal. Lett., 2014, 144, 545–552 CrossRef CAS.
- J. Zheng, F. Liao, S. Wu, G. Jones, T. Chen, J. Fellowes, T. Sudmeier, I. J. McPherson, I. Wilkinson and S. C. E. Tsang, Angew. Chem., Int. Ed., 2019, 58, 17335–17341 CrossRef CAS.
- Y. Toda, H. Yanagi, E. Ikenaga, J. J. Kim, M. Kobata, S. Ueda, T. Kamiya, M. Hirano, K. Kobayashi and H. Hosono, Adv. Mater., 2007, 19, 3564–3569 CrossRef CAS.
- A. D. Pelton, Bull. Alloy Phase Diagrams, 1986, 7, 223–228 Search PubMed.
- C. Autthanit, W. Chatkaew, P. Praserthdam and B. Jongsomjit, J. Environ. Chem. Eng., 2020, 8, 103547 CrossRef CAS.
- C. Krutpijit, W. Tian, B. Jongsomjit, D. Pjontek and J. E. Herrera, Mol. Catal., 2020, 483, 110717 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |