
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceActivation of ammonia and hydrazine by electron rich Fe(II) complexes supported by a dianionic pentadentate ligand platform through a common terminal Fe(III) amido intermediate†‡
Lucie
Nurdin
a,
Yan
Yang
b,
Peter G. N.
Neate
c,
Warren E.
Piers
 *a,
Laurent
Maron
*b,
Michael L.
Neidig
*c,
Jian-Bin
Lin
a and
Benjamin S.
Gelfand
a
*a,
Laurent
Maron
*b,
Michael L.
Neidig
*c,
Jian-Bin
Lin
a and
Benjamin S.
Gelfand
a
aDepartment of Chemistry, University of Calgary, 2500 University Drive NW, Calgary, Alberta T2N 1N4, Canada. E-mail: wpiers@ucalgary.ca
bLPCNO, Université de Toulouse, INSA, UPS, Toulouse, France
cDepartment of Chemistry, University of Rochester, Rochester, New York 14627, USA
First published on 22nd December 2020
Abstract
We report the use of electron rich iron complexes supported by a dianionic diborate pentadentate ligand system, B2Pz4Py, for the coordination and activation of ammonia (NH3) and hydrazine (NH2NH2). For ammonia, coordination to neutral (B2Pz4Py)Fe(II) or cationic [(B2Pz4Py)Fe(III)]+ platforms leads to well characterized ammine complexes from which hydrogen atoms or protons can be removed to generate, fleetingly, a proposed (B2Pz4Py)Fe(III)–NH2 complex (3Ar-NH2). DFT computations suggest a high degree of spin density on the amido ligand, giving it significant aminyl radical character. It rapidly traps the H atom abstracting agent 2,4,6-tri-tert-butylphenoxy radical (ArO˙) to form a C–N bond in a fully characterized product (2Ar), or scavenges hydrogen atoms to return to the ammonia complex (B2Pz4Py)Fe(II)–NH3 (1Ar-NH3). Interestingly, when (B2Pz4Py)Fe(II) is reacted with NH2NH2, a hydrazine bridged dimer, (B2Pz4Py)Fe(II)–NH2NH2–Fe(II)(B2Pz4Py) ((1Ar)2-NH2NH2), is observed at −78 °C and converts to a fully characterized bridging diazene complex, 4Ar, along with ammonia adduct 1Ar-NH3 as it is allowed to warm to room temperature. Experimental and computational evidence is presented to suggest that (B2Pz4Py)Fe(II) induces reductive cleavage of the N–N bond in hydrazine to produce the Fe(III)–NH2 complex 3Ar-NH2, which abstracts H˙ atoms from (1Ar)2-NH2NH2 to generate the observed products. All of these transformations are relevant to proposed steps in the ammonia oxidation reaction, an important process for the use of nitrogen-based fuels enabled by abundant first row transition metals.
Introduction
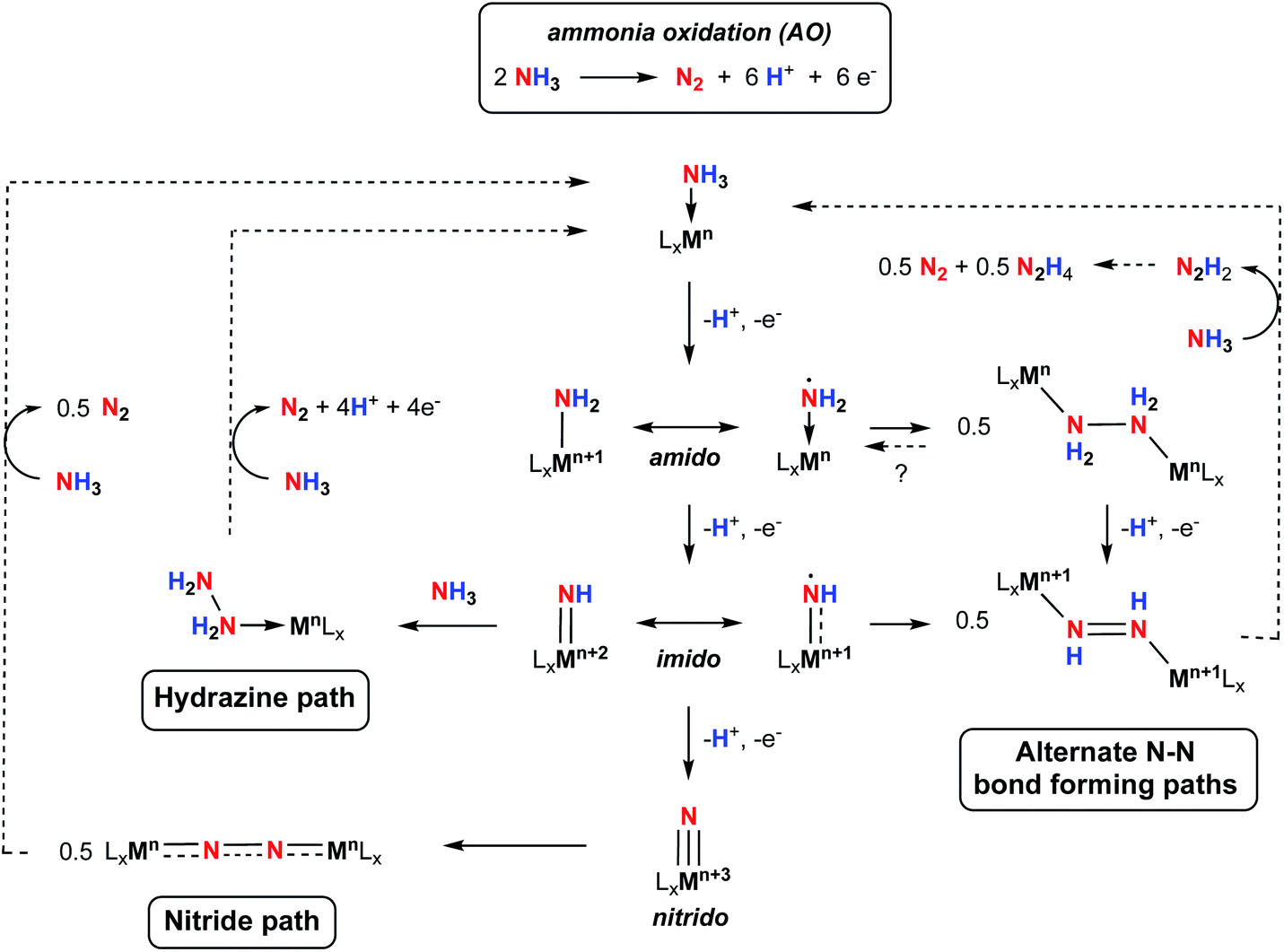
Storing energy in chemical bonds, in particular protons and electrons in nitrogen-based fuels, has become an attractive alternative to supply carbon-free energy carriers on-demand.1,2 Ammonia (NH3) has recently emerged as a promising candidate for carbon-free fuel replacements3 because it is prepared on an industrial scale through the well-established Haber–Bosch (H–B) process, and the infrastructure to store and distribute this chemical are available worldwide.4 With the increasing use of renewable sources to power the H–B process, and with the tremendous effort dedicated to using renewable hydrogen from water electrolysis as an alternative to steam reformation of methane, the potential for use of NH3 as a less carbon-intensive fuel is rising.5Ammonia can be used directly in NH3 fuel cells through the ammonia oxidation reaction (AO, Scheme 1), or can be used as a medium to store hydrogen (H2) resulting from its high energy density.6 However, to mediate this transformation under manageable conditions and reasonable rates and selectivities, catalysts are required. While there have been several reports on heterogeneous catalysts for ammonia oxidation with limited success,7 examples of homogeneous catalysts are attracting increasing attention.8–12 Homogeneous catalysts offer the prospect of greater selectivity, more control over active site steric and electronic properties, and an opportunity to understand the fundamental chemistry involved through detailed spectroscopic and structural studies.13
 | ||
| Scheme 1 Postulated steps and intermediates in homogeneous ammonia oxidation. | ||
The oxidation of NH3 into N2 is an inherently challenging process due to the difficulty in breaking all three of its strong N–H bonds (the first N–H bond dissociation energy, BDE, of NH3 is 107.6 kcal mol−1),14 as well as the multiple electron and proton transfers involved in the formation of N2 (Scheme 1).15 The energy associated with N–H bond breaking can be lowered by coordination of NH3 to a metal center, and subsequent strategies such as N–H oxidative addition,16 heterolytic cleavage through metal–ligand cooperativity,17–21 or hydrogen atom abstraction (HAA)8,9,22–27 have been successful at mediating N–H bond cleavage of NH3. In addition to N–H bond cleavage, N–N bond formation is a key process. Initially, two classes of N–N bond formation were postulated by analogy to water oxidation mechanisms, namely nucleophilic attach by NH3 on electrophilic imido groups (the “hydrazine path”, Scheme 1), or direct homocoupling of two terminal nitride intermediates (the “nitride path”, Scheme 1).10 While the nitride pathway requires a +3 change in the metal oxidation state to yield metal–nitride complexes, which can then undergo N–N homocoupling, the hydrazine pathway only requires a +2 change in the metal oxidation state to yield a metal–imido complex, which can then react further with NH3 to form a hydrazine adduct via N–N coupling. More recently, N–N bond formation via coupling of other intermediates, for example M–NH2 or M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH compounds with significant spin density on the amido/imido nitrogen, have been proposed.28 This path to N–N bond formation requires only a +1 oxidation state change and may be more important for catalysts based on first row transition metals. Thus, despite the recent interest in AO, much remains to be uncovered concerning the mechanistic nuances of this process. The key to advancing our understanding of the fundamental reactivity profiles and thermochemistry associated with the cleavage of the individual N–H bonds, the nature of intermediates involved in this transformation, and the character of N–N bond forming steps lies in detailed mechanistic investigations on a variety of systems.23 Furthermore, to operate at scale, higher stability under typical catalytic conditions is required, as well as catalysts based on sustainable metals. Notably, only one catalyst featuring an earth-abundant, first-row transition metal has been reported but shows low TON due to catalyst decomposition.11 There is therefore room for fundamental research in catalyst design and mechanistic studies to advance this nascent area of catalysis.
NH compounds with significant spin density on the amido/imido nitrogen, have been proposed.28 This path to N–N bond formation requires only a +1 oxidation state change and may be more important for catalysts based on first row transition metals. Thus, despite the recent interest in AO, much remains to be uncovered concerning the mechanistic nuances of this process. The key to advancing our understanding of the fundamental reactivity profiles and thermochemistry associated with the cleavage of the individual N–H bonds, the nature of intermediates involved in this transformation, and the character of N–N bond forming steps lies in detailed mechanistic investigations on a variety of systems.23 Furthermore, to operate at scale, higher stability under typical catalytic conditions is required, as well as catalysts based on sustainable metals. Notably, only one catalyst featuring an earth-abundant, first-row transition metal has been reported but shows low TON due to catalyst decomposition.11 There is therefore room for fundamental research in catalyst design and mechanistic studies to advance this nascent area of catalysis.
In terms of catalyst design, the supporting ligand must be robust enough to support the metal center in multiple oxidation states, in particular when using first-row transition metals. Polypyridyl ligands have been used in three out of the five reported homogeneous catalysts for AO.9–11 These and related tetrapodal pentadentate platforms have been employed across the periodic table and have been successful at supporting a variety of metal centers in various oxidation states, mainly in the context of water oxidation/reduction catalysis.29–33 Bullock and coworkers recently reported a detailed theoretical investigation using the PY5 ligand platform34,35 for AO with M = Cr, Mo, W, Mn, Fe, Ru, Os.36 They observed that the [PY5]Fe(II) complex behaved quite differently to the rest of the metals investigated, notably due to the significant radical character of the Fe–amido and Fe–imido intermediates computed. We therefore thought of using our tetrapodal dianionic pentadentate B2Pz4Py ligand platform, which has been successful at accessing a variety of first-row metal complexes.37–41 The B2Pz4Py ligand features two borate moieties at the linkage position between the four equatorial pyrazole arms and the axial pyridine, rendering the ligand dianionic and slightly more rigid than the PY5 platform.42 The charge modification from the PY5 to the B2Pz4Py platform renders complexes of M(II) ions neutral, and higher oxidation state complexes are generally more accessible with this more electron rich ligand system.37 In addition, the decrease in the overall charge of the complex favors pathways involving dimerization due to lower coulombic repulsion.
Herein we report the synthesis and characterization of Fe complexes bearing the B2Pz4Py ligand platform and their reactivity with ammonia and hydrazine. The reactivity observed is supported by both experimental and theoretical mechanistic studies involving higher-valent, reactive intermediates. In particular, evidence for the involvement of a reactive terminal Fe(III)–NH2 species is presented; as indicated in Scheme 1, such compounds are proposed to be key intermediates of AO but are rarely isolated and characterized43–45 in the absence of Lewis acid stabilization.46,47
Results and discussion
We have previously reported two versions of the [B2Pz4Py]HLi ligand precursor, differing by the aromatic ring bound to the borate group (Ar = Ph,39p-Tol37). Our initial report on Fe was carried out using the phenyl version of the ligand, featuring complexes 1Ph-THF and 1Ph (Scheme 2) but these derivatives were characterized by poor solubility in many solvents. Incorporation of tolyl groups leads to greatly enhanced solubility of the resulting Fe complexes, and the tolyl derivative was employed instead for the majority of the chemistry described herein. An improved synthesis was developed to make complexes 1Ar-THF by treating the monolithium pyridinium salt directly with one equivalent of Fe(HMDS)2 (ref. 48 and 49) in THF (Scheme 2). This synthesis allowed for a greater yield of LiBr free product, and simplification of the purification procedure. Compounds 1Ar are isolated as light green solids in ≈70% yield by heating 1Ar-THF under vacuum for four hours (Scheme 2). While 1Ph is insoluble in aprotic solvents such as toluene or benzene, 1Tol is readily dissolved in these media. The difference in solubility between 1Ph and 1Tol is striking and can serve as a distinguishing feature for the isolation of various key complexes, under the assumption that the difference between the two Ar groups does not significantly influence the course of reactivity at the Fe center. Evans method measurement of the magnetic susceptibility of 1Tol gives a μeff = 4.99, consistent with a high spin (HS) system with 4 unpaired electrons (S = 2) as previously reported for 1Ph.391Ar can coordinate a variety of L type ligands, including THF and MeCN. While THF adducts 1Ar-THF are HS, stronger π-acceptor ligands such as MeCN enforce a low spin (LS) environment (S = 0), noted by a characteristic 1H NMR spectrum of a diamagnetic Fe(II) complex obtained in CD3CN (Fig. S1 and S2‡). This is supported by the data obtained from Mössbauer spectroscopy. A solid sample of 1Tol gives an isomer shift consistent with a HS system (δ = 1.04 mm s−1 and ΔEQ = 0.93 mm s−1, Fig. S3‡). In contrast, when a pale green benzene solution of 1Tol under a dinitrogen atmosphere is frozen to 80 K, it turns orange and gives rise to a Mössbauer spectrum with parameters supporting a LS configuration (δ = 0.44 mm s−1 and ΔEQ = 0.43 mm s−1 Fig. S4‡), suggesting coordination of dinitrogen and a spin state change. Complexes 1Ar and 1Ar-L are extremely air sensitive and can easily be oxidized as shown by a low potential for the Fe(II/III) redox couple at −0.44 V vs. Fc/Fc+ (Fig. S5‡). When exposed to O2, Fe(II) complexes quickly react to form an iron μ-oxo bridge species, Fe(III)–O–Fe(III).50 Therefore, all reactions must be performed under rigorously air free conditions.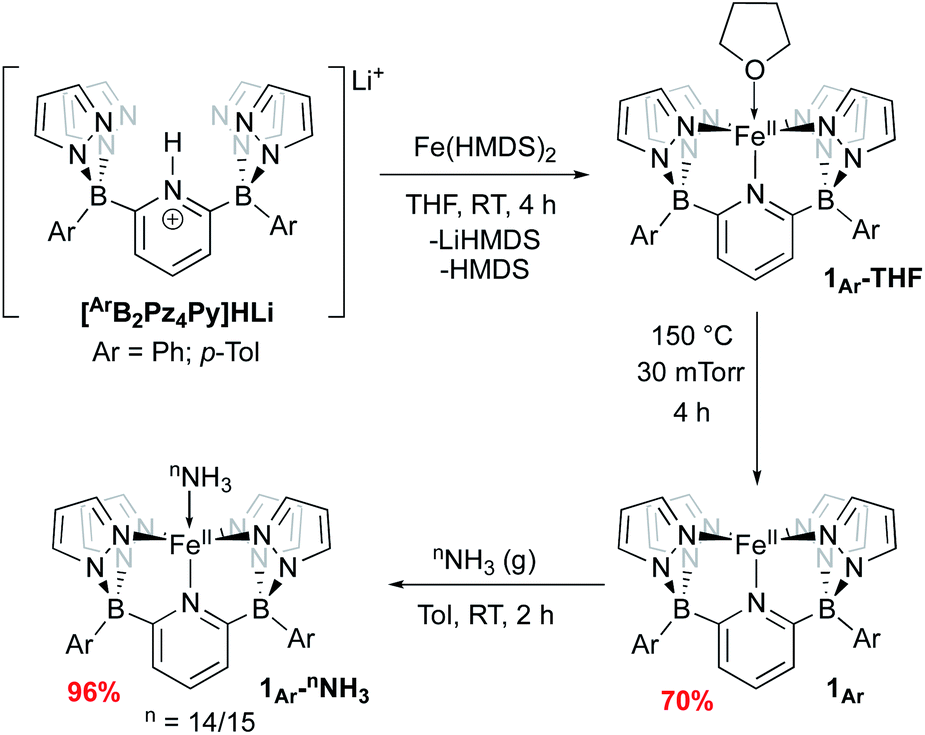 | ||
| Scheme 2 Synthesis of 1Ar and 1Ar-nNH3. | ||
Treatment of 1Ar with 14NH3 (1 atm) results in a clear color change from light green to dark brown forming the NH3 adduct 1Ar-14NH3 (Scheme 2). This is accompanied by a transition to a new 1H NMR spectrum featuring peaks located in the diamagnetic region, although the resonances are quite broadened. When 15NH3 is employed, the singlet at +2.73 ppm becomes a doublet with a 1J15N–1H of 62.4 Hz (Fig. S6‡), consistent with the other 15NH3 adducts previously reported.8,9,26 The 11B chemical spectrum also changes drastically from +38.4 ppm for complex 1Tol to −1.6 ppm for 1Tol-nNH3, which is in the region of diamagnetic metal complexes with this ligand platform.401Tol-nNH3 was isolated in 96% yield as a brown solid (Scheme 2) and was characterized by single-crystal X-ray crystallography (Fig. 1). The Fe–N10 bond of 2.072(2) Å confirms the LS configuration by comparison to similar LS Fe–NH3 adducts.11,44,51,52 In addition, the average bond length of 1.998 Å for the five other Fe–N bonds vs. 2.115 Å for 1Ph-THF (HS)39 also attests to the LS spin structure of this Fe(II)–NH3 complex. The broadening of the chemical shifts in the 1H NMR spectrum may be due to a fast spin flipping between the LS and HS systems.53 Peters and coworkers recently reported that both LS and HS configurations of their polypyridyl Fe(II)–NH3 complex were present in the solid-state structure.11 DFT calculations on the ground state of 1Tol-NH3 show that the LS and HS systems only differ by 5.6 kcal mol−1. Furthermore, NH3 has a field strength located between THF and MeCN, where THF is the weakest field ligand and MeCN the strongest.54 The Mössbauer parameters of 1Tol-NH3 (δ = 0.55 mm s−1 and ΔEQ = 0.22 mm s−1) also suggest that the LS configuration is favored at low temperatures, but some dissociation of the NH3 ligand could not be prevented under an N2 atmosphere and the purported LS dinitrogen Fe(II) adduct is also present in the spectrum in variable amounts (≈20%) in these spectra (Fig. S7‡).
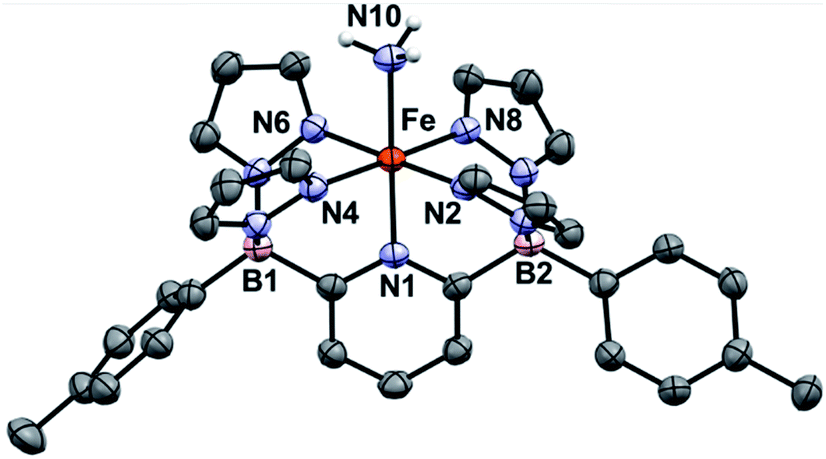 | ||
| Fig. 1 ORTEP diagrams for 1Tol-NH3. Hydrogen, boron, carbon, nitrogen, and iron atoms are white, pink, grey, light blue, and orange respectively. Thermal ellipsoids are shown at the 50% probability level. Calculated hydrogen atoms are omitted for clarity except those on the ammonia ligand (N10). The solvent molecule has been omitted for clarity. Selected bond distances (Å): Fe–N1, 1.969(2); Fe–N2, 2.000(2); Fe–N4, 1.964(2); Fe–N6, 2.000(2); Fe–N8, 2.004(2); Fe–N10, 2.072(2). Selected bond angles (°): N1–Fe–N2, 91.1(1); N2–Fe–N4, 92.8(8); N1–Fe–N10, 179.0(9). Further metrical data are given in Table S1.‡ | ||
An important first step in AO is the loss of “H˙” from amine complexes, so reactions of 1Ar-nNH3 with hydrogen atom abstractors (HAAs) were explored. This strategy has been employed in a variety of systems,22,27 and has been successful for catalytic and stoichiometric AO.8,9 To choose a suitable reagent, the first N–H bond dissociation energy (BDE) of 1Tol-NH3 was computed and found to be 75.5 kcal mol−1, which is significantly lower than the first N–H BDE of NH3 (107.6 kcal mol−1).14 No reaction was observed when TEMPO was used as the HAA (O–H = 70.0 kcal mol−1),14 so the stronger 2,4,6-tri-tert-butylphenoxy radical (ArO˙, BDE of ArO–H = 81.6 kcal mol−1),14 which has proven successful in mediating N–H bond cleavage,8,26 was then tried. To deconvolute this chemistry, both the phenyl and tolyl ligands were employed, exploiting their differing solubility properties as described. Accordingly, treatment of 1Ph-15NH3 with excess ArO˙ for 16 h at room temperature in toluene, gave a new diamagnetic, forest green product, 2Ph-15N (Scheme 3). The 1H NMR spectrum of the product features three key resonances: two different – tBu groups in the alkyl region (1.64 and 0.83 ppm), and one deshielded doublet at +17.82 ppm (J = 66.6 Hz) integrating for 1H and suggesting a 1J15N–1H coupling (Fig. S8‡).55 This doublet becomes a singlet when 1Ph-14NH3 is employed in the reaction, confirming the assignment as a 15N–H proton (Fig. S9‡). The parameters obtained from the Mössbauer spectrum of this species are consistent with a low spin Fe(II) system (δ = 0.29 mm s−1 and ΔEQ = 0.97 mm s−1, Fig. S10‡). Unfortunately, due to the poor solubility of 2Ph, no X-ray quality crystals could be obtained. The same reaction using 1Tol-nNH3 gave a spectroscopically similar product, but 2Tol could not be isolated from the ArOH by product of the reaction due to its greater solubility (Fig. S11‡). Luckily, a pentane solution of this reaction mixture stored in the glove box freezer deposited blue, X-ray quality crystals and the molecular structure of 2Tol was revealed (Fig. 2). This structure is fully consistent with all the spectroscopic and analytical data obtained for 2Ph. Analysis of the metrical data confirmed the depiction of 2Ar as an Fe(II) activated imine complex. In particular, the short N10–C1, 1.330(4); C2–O, 1.240(5); C3–C4 1.353(6); and C5–C6 1.357(5) bond distances (Å) confirm the double bond assignment around the conjugated ring. Interestingly, the short Fe–N10 bond distance, 1.859(3) Å, attests to the stability of compounds 2Ar, and when 2Ar is treated with excess ArO˙ and/or excess NH3, no further reactivity is observed (Fig. S12‡). The stability of 2Ar is most likely enhanced due to the hydrogen bonding interaction between NH–O (2.145(4) Å), see Fig. 2.
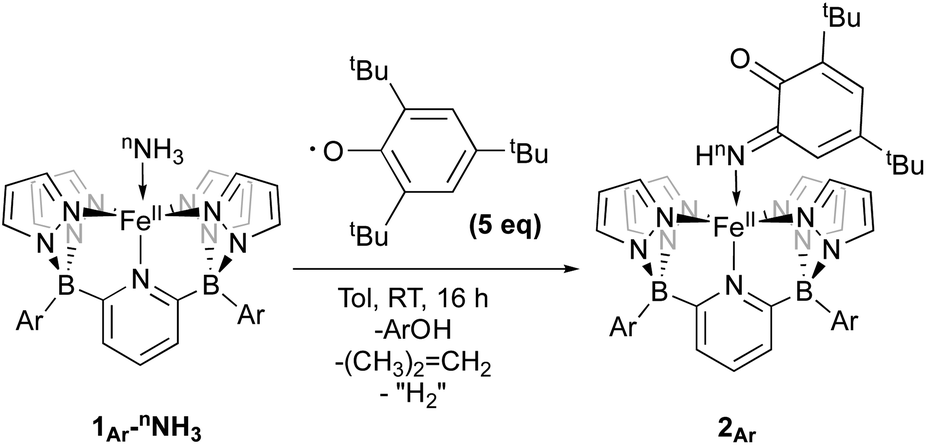 | ||
| Scheme 3 Reactivity of 1Ar-nNH3 with ArO˙ to form 2Ar. | ||
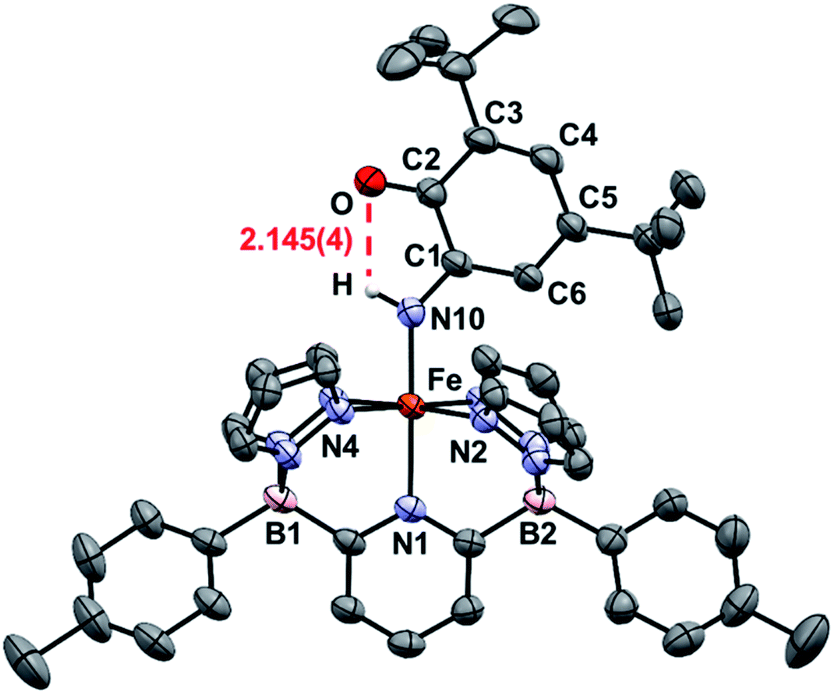 | ||
| Fig. 2 ORTEP diagrams for 2Tol. Hydrogen, boron, carbon, nitrogen, oxygen, and iron atoms are white, pink, grey, light blue, red, and orange respectively. Thermal ellipsoids are shown at the 50% probability level. Calculated hydrogen atoms are omitted for clarity except those on atom N10. The hydrogen bonding interaction is shown in red. Selected bond distances (Å): Fe–N1, 2.036(3); Fe–N2, 1.986(3); Fe–N4, 1.963(3); Fe–N10, 1.859(3); N10–C1, 1.330(4); C2–O, 1.240(5); C1–C2, 1.484(5); C2–C3, 1.477(5); C3–C4, 1.353(6); C4–C5, 1.437(6); C5–C6, 1.357(5); C6–C1, 1.429(5); O–H, 2.145(4). Selected bond angles (°): N1–Fe–N2, 91.6(1); N2–Fe–N4, 92.9(1); N1–Fe–N10, 178.1(1); Fe–N10–C1, 136.2(2). Further metrical data are given in Table S1.‡ | ||
Isolation of products 2Ar suggests that ArO˙ abstracts a hydrogen atom from 1Ar-NH3 to form Fe(III)–NH2(3Ar-NH2) which then is trapped by excess aryloxy radical; the reaction does not proceed unless excess radical is employed. Consistent with this, signals for Ar–OH are observed in the crude reaction mixture (Fig. S13‡). The loss of a tBu group from the aryloxy framework implies that iso-butene is released, along with two hydrogen atoms; the former was detected by GC-MS in the headspace of the reaction (Fig. S14‡). Thevenet and coworkers reported similar reactivity with ArO˙ and aromatic amines, where the loss of isobutene arose from heterocleavage of the C–C bond linked to the – tBu group on the ortho position.56 Notably, we do not detect any H2 but observe that more than one equivalent of ArOH is formed in the reaction suggesting that excess aryloxy radical serves as a hydrogen atom scavenger in the product forming step from C-Northo (Fig. 3). DFT investigation (B3PW91 functional, see ESI‡ for full computational details) of 3Tol-NH2 shows why it would prefer to engage in C–N coupling rather than undergo a second hydrogen atom abstraction. These computations show that the formally Fe(III)–NH2 has significant unpaired spin density on N (22%) (Fig. 3) and has partial aminyl radical character.57 Furthermore, the computed N–H BDE for 3Tol-NH2 is substantially higher (97.1 kcal mol−1) than that of 1Tol-NH3, indicating that a second abstraction is thermodynamically prohibited. Thus, amides 3Ar-NH2 react with the excess ArO˙ via lower barrier, exothermic C–N heterocoupling of 3Ar-NH2 with the aromatic ring of ArO˙ (Fig. 3). According to the calculations performed by Mayer and coworkers on the distribution of the residual spin density in ArO˙, the para position contains the bulk of the unpaired density and is therefore the preferred site for reactivity.58 However, here we observe no products arising from para heterocoupling in the reaction between 1Ar-NH3 and ArO˙. By DFT computations, the C–N bond forming steps to intermediates C-Northo and C-Npara have a similar barrier of ∼26 kcal mol−1, but intermediate C-Northo is more stable than C-Npara by 10 kcal mol−1; indeed, formation of the latter is endothermic, while the former is favorable by −4.2 kcal mol−1 (Fig. 3). This stabilization most likely arises from hydrogen bonding interaction similar to what is seen in the final product, which is absent in the intermediate produced from C–N bond formation at the para position. Assuming that loss of iso-butene is irreversible, the equilibrium between these two isomeric intermediates thus strongly favors the ortho species, leading to the observed products 2Ar.
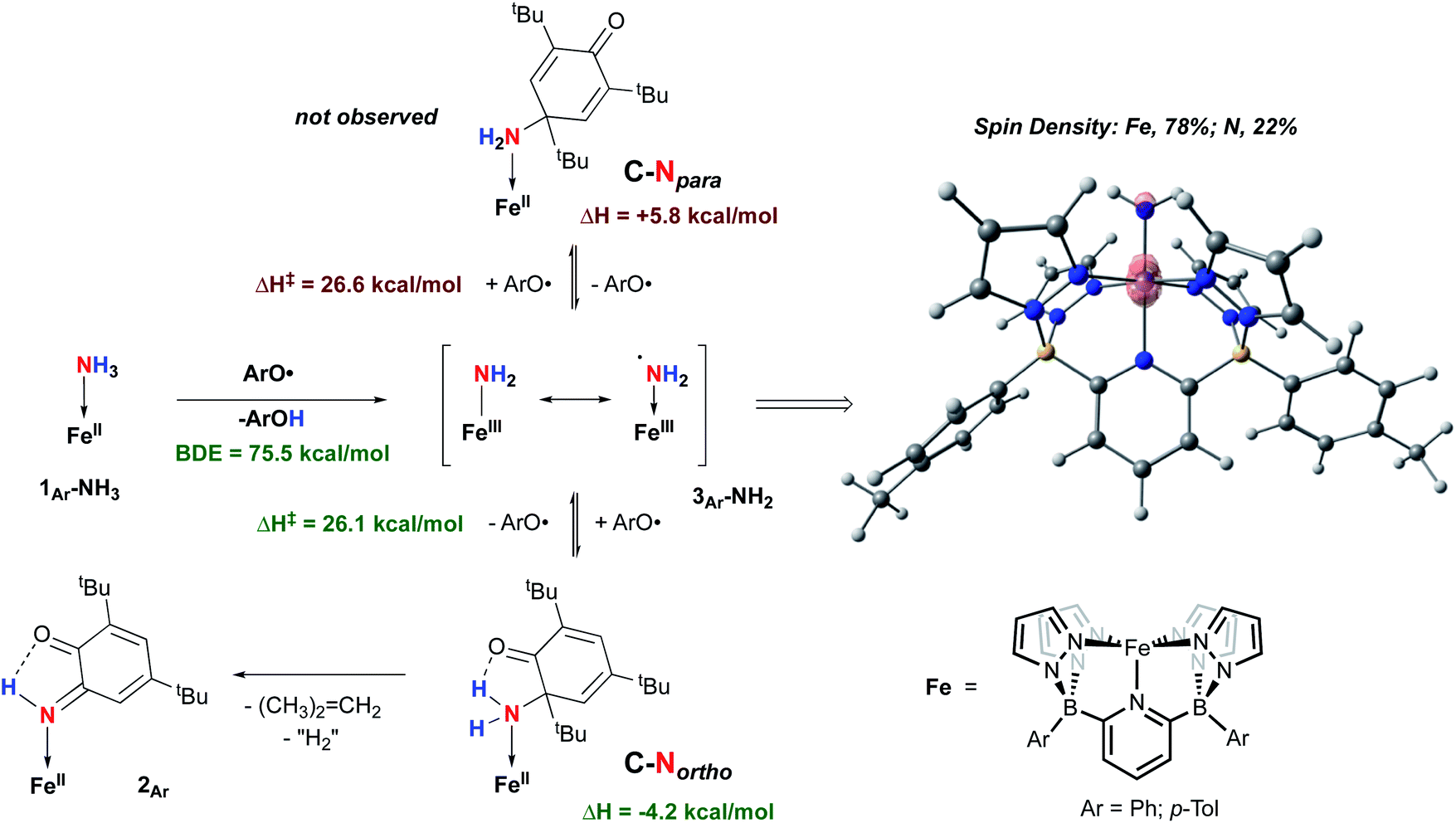 | ||
| Fig. 3 Two mechanisms probed by DFT (B3PW91) calculation for the reactivity of 1Ar-NH3 and ArO˙ at 298 K. Enthalpies are given relative to compound 1Ar-NH3 set to 0.0 kcal mol−1. | ||
The reaction pathway C-Northo, although unusual, complements the recent report by Bullock and coworkers using a Ru porphyrin system.9 They proposed that a Ru(III)NH2 intermediate, akin to complexes 3Ar, reacts with the para position of ArO˙ to form a Ru(II)NH2R complex, analogous to complex C-Npara. In their case, however, the Ru(II)–NH2R bond is labile enough to release NH2R in the presence of excess NH3. Here, we do not observe any such turnover and 2Ar are terminal products in this reaction. The term “catalytic diversion” employed by Bullock can also be used here where the reactivity between ArO˙ and 3Ar-NH2 prevents any catalysis to happen.
Iron amido complexes (Fe–NH2) have been postulated as key intermediates in a variety of reactions.22,59–61 In particular, Peters et al. recently reported a polypyridyl Fe catalyst for AO and proposed an Fe(III)NH2 intermediate as part of their mechanistic analysis.11 We therefore attempted to generate compound 3Tol-NH2via other routes to obtain more direct evidence for it. Treatment of 1Tol-NH3 with other H atom abstractors like trityl radicals (˙CPh3 (ref. 62) or ˙C(p-tBu-Ph)3 (ref. 63)) to prevent C–N coupling were unsuccessful, presumably due to steric effects. Therefore, two other paths (A and B, Scheme 4) to 3Tol-NH2 were explored.
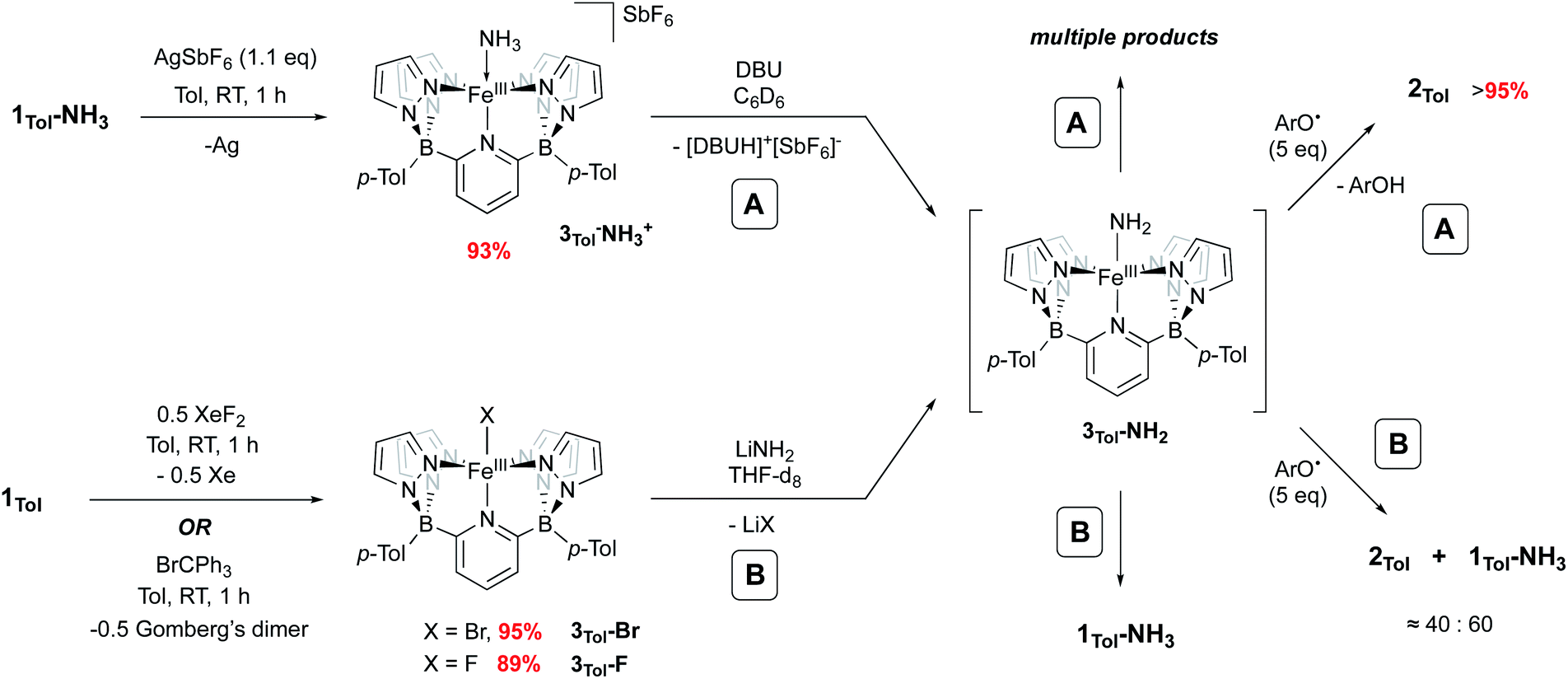 | ||
| Scheme 4 Independent generation and trapping of 3Tol-NH2. (A) Oxidation of 1Tol-NH3 to 3Tol-NH3+ and deprotonation with DBU. (B) Salt metathesis using 3Tol-Br and 3Tol-F. | ||
Going from 1Tol-NH3 to 3Tol-NH2 can be seen as a proton-coupled electron transfer (PCET) by removal of a hydrogen atom (H˙) but can also be broken down into successive steps: oxidation by 1 e− and removal of 1 H+. 1Tol-NH3 is cleanly oxidized by AgSbF6 in toluene, giving a dark red solution and deposited Ag(0). Upon work up, the Fe(III) ammine complex 3Tol-NH3+ is isolated in excellent yield (93%, Scheme 4). 3Tol-NH3+ exhibits a LS (S = 1/2) configuration (μeff = 1.80 by Evans method), and its structure was confirmed by X-ray crystallography (Fig. 4). The shorter Fe–N10 bond of 2.010(2) in 3Tol-NH3+vs. 2.072(2) for 1Tol-NH3 confirmed the oxidation of the Fe center from +II to +III and is a rare example of a Fe–NH3 adduct in the +III oxidation state.64,65 The compound demonstrates the ability of the B2Pz4Py platform to stabilize and access higher oxidation state complexes. Deprotonation of 3Tol-NH3+ with 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU) was carried out at room temperature in C6D6. The reaction goes through a deep purple intermediate which rapidly turns into a gold coloured solution, which contains a mixture of paramagnetic species, including 1Tol-NH3 as the main product (Fig. S15 and S16‡). However, if the deprotonation of 3Tol-15NH3+ is performed in the presence of excess phenoxy radical ArO˙, the reaction proceeds cleanly and affords 2Tol-15N as the sole iron-containing complex, along with ArOH and [DBUH][SbF6] by-products (Fig. S17‡).
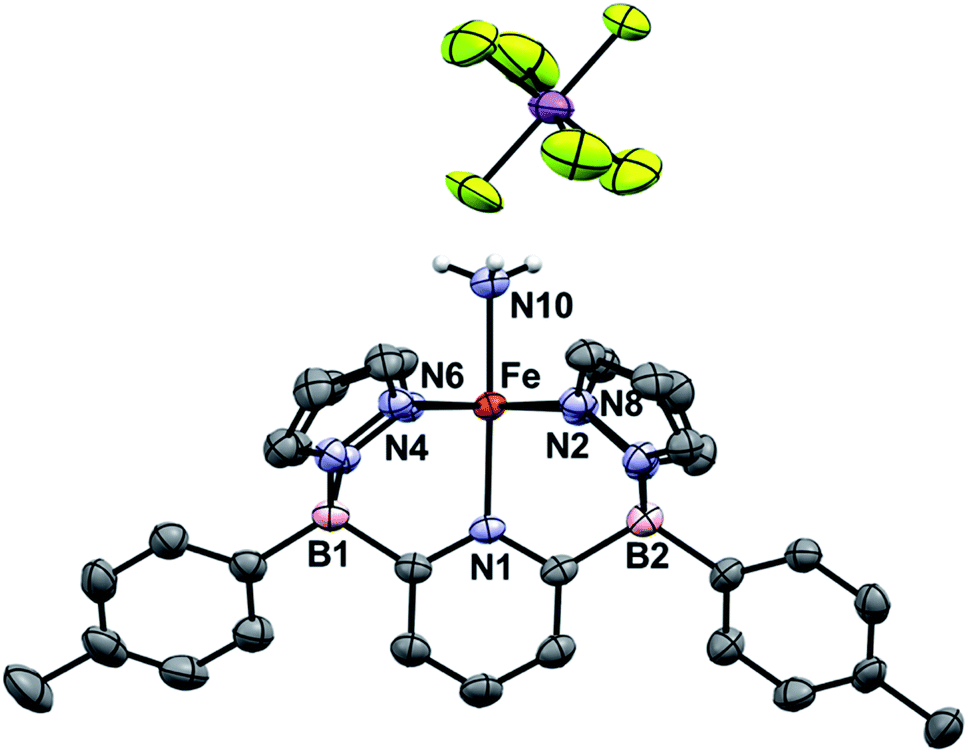 | ||
| Fig. 4 ORTEP diagram for 3Tol-NH3+. Hydrogen, boron, carbon, nitrogen, antimony, fluorine, and iron atoms are white, pink, grey, light blue, purple, yellow, and orange respectively. Thermal ellipsoids are shown at the 50% probability level. Calculated hydrogen atoms are omitted for clarity except those on the ammonia ligand (N10). The solvent molecule has been omitted for clarity. Selected bond distances (Å): Fe–N1, 2.009(3); Fe–N2, 1.999(4); Fe–N4, 1.990(3); Fe–N6, 1.951(3); Fe–N8, 1.942(4); Fe–N10, 2.010(3). Selected bond angles (°): N1–Fe–N2, 91.9(1); N2–Fe–N4, 94.7(1); N1–Fe–N10, 176.8(1). Further metrical data are given in Table S1.‡ | ||
Salt metathesis using LiNH2 and an M–X (X = halogen) complex has been the most successful strategy employed to access and isolate M–NH2 complexes.43,44,66 We therefore attempted this reaction as depicted in Scheme 4, path B. The required Fe(III) halide complexes 3Tol-X (X = F, Br) were prepared by oxidation of 1Tol with XeF2 or trityl bromide, respectively and were obtained in excellent yield. These compounds were fully characterized, including their solid state structures (Fig. S18,‡3Tol-Br; Fig. S19,‡3Tol-F). No reaction between these compounds and LiNH2 was observed in non-polar solvents, even with addition of crown ethers, so THF-d8 was employed. Perhaps unsurprisingly, when 3Tol-Br was treated with LiNH2 in THF, after five days at RT, 1Tol-NH3 was isolated as the sole product of the reaction (Fig. S20 and S21‡). In this reaction, when 3Tol-NH2 is generated by salt metathesis, it acquires a hydrogen atom, most likely coming from the THF solvent44,67 to form 1Tol-NH3.39 A similar result was observed when the iron fluoride 3Tol-F was employed, although the reaction was significantly faster. As with the deprotonation strategy, when these reactions were carried out in the presence of an excess of ArO˙, significant amounts of 2Tol were observed (up to ≈40%, Fig. S22 and S23‡), although here the scavenging of H atoms occurs at a competitive rate, with the balance of iron ending up as 1Tol-NH3, as summarized in Scheme 4.
Taken together, the chemistry described above suggests that, while it is possible to generate 3Tol-NH2 by a variety of methods, it is highly reactive due to a preponderance of spin density on the NH2 nitrogen (Fig. 3). It thus behaves as an Fe(II) aminyl57 and is readily trapped by ArO˙ or scavenges H˙ atoms from appropriate donors (THF coordinated to Li, for example). Our attempts to generate it in the absence of such traps were motivated by the possibility that it might dimerize through N–N bond formation, which has been proposed in a variety of catalytic cycles,68 and in particular for ammonia oxidation catalysts operating through the hydrazine pathway (Scheme 1).9,10 Indeed, such a process has recently been observed in a related d7 Ni(III) system.28 However, no evidence for this was found in any of the experiments used to generate 3Tol-NH2. It occurred to us that, because of the electron rich nature of the B2Pz4Py ligand system, the Fe(II) compound might be a strong enough reductant to induce N–N bond cleavage in hydrazine, that is the reverse of homocoupling of 3Tol-NH2. The reactions of 1Tol with hydrazine were thus explored and, as described below, a reaction path indicative of H2N–NH2 reductive cleavage is implicated by both experiments and computations.
When 1Tol is treated with 0.5 equivalents of hydrazine, a rapid color change from light green to dark forest green was observed and a clean 1H NMR spectrum indicating the presence of two products was obtained (Fig. S24‡). One of the products is complex 1Tol-NH3, which has already been made and characterized independently (Scheme 2 and Fig. 1). The second product, also diamagnetic, was isolated as a deep blue solid by washing the reaction mixture with cold Et2O and was identified as described below to be the diazene dimer 4Tol (Scheme 5). When this reaction was carried out at −78 °C, 1H NMR spectroscopy revealed that upon mixing 1Tol with hydrazine, an intermediate assigned as the dimeric hydrazine adduct (1Tol)2-NH2NH2 is formed (Fig. S25‡). This intermediate is diamagnetic and the equivalent hydrazine protons appear at +3.65 ppm in the spectrum taken at 210 K, integrating to 4 relative to the 12 protons of the tolyl methyl groups and confirming its dimeric nature. This resonance appears as a broad doublet in a sample prepared with isotopically labeled 15N2H4, exhibiting a 1J15N–1H constant of 66 Hz (Fig. S26‡). As samples containing in situ generated (1Tol)2-NH2NH2 are warmed to −15 °C, peaks corresponding to the two products 1Tol-NH3 and 4Tol emerge with no other (diamagnetic) intermediates apparent (Fig. S27‡), until at room temperature the reaction proceeds to completion. Notably, no hydrogen gas was detected by 1H NMR spectroscopy at any temperature during this experiment.
 | ||
| Scheme 5 The reaction of 1Tol with N2H4 and the two isolated products 1Tol-NH3 and 4Tol. | ||
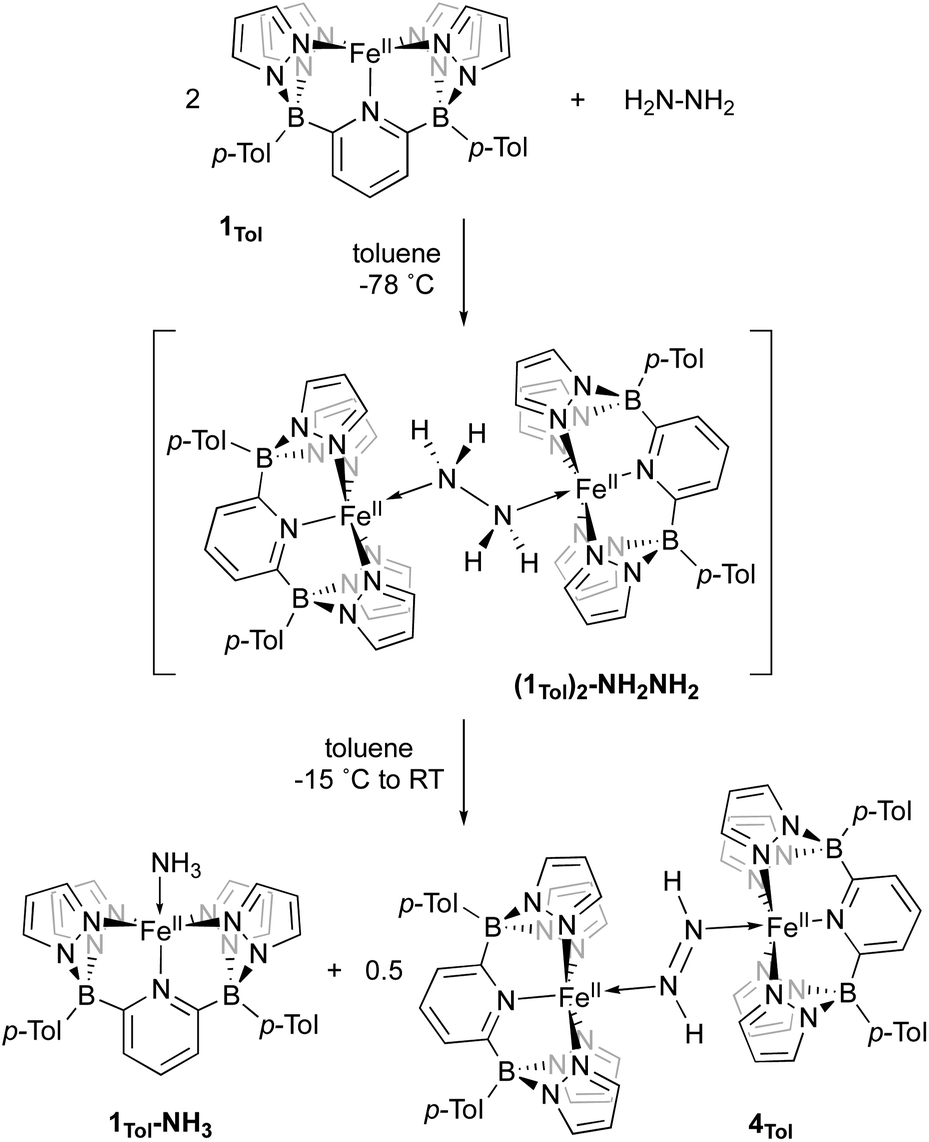
Complex 4Tol is a low spin, diamagnetic, Fe(II) complex and was characterized by a variety of spectroscopic techniques, as well as X-ray crystallography (Fig. 5). The diazene protons in the 1H NMR spectrum appeared as a singlet at +18.66 ppm. When 15N2H4 is used, the singlet becomes a diagnostic AA‘XX’ multiplet (Fig. 5a), confirming the presence of a bridging diazene moiety. From the simulation of the multiplet (red spectrum, Fig. 5a), the different J values can be obtained.55 In particular, the 3JHH of 22.4 Hz is consistent with a trans isomer, and in addition, a N–N bond length of 1.298 Å can be predicted based on the empirical correlation between the 3JHH value and the N–N bond distance of trans diazene complexes.51,69 This is supported by the solid-state structure of 4Tol depicted in Fig. 5b. The N–N bond distance of 1.288(4) Å is consistent with an N–N double bond and with the predicted value, and the average value of 132° for the Fe–NN bond angles is consistent with sp2 hybridized nitrogen atoms. The shorter experimental N–N bond combined with the intense blue color of the species (Fig. S28‡) suggest a strong interaction between the Fe d orbitals and the π system of the diazene moiety. Furthermore, the rRaman spectrum of 4Tol shows an intense stretch at 1321 cm−1 corresponding to the 14N14N bond. This band shifts to 1279 cm−1 for the 4Tol-(15N)2 isotopologue, which is in good agreement with the calculated shift (14N/15NΔcalc. = 45 cm−1; 14N/15NΔexpt. = 42 cm−1). Finally, the 80 K Mössbauer spectrum of 4Tol gives parameters δ = 0.41 mm s−1 and ΔEQ = 0.49 mm s−1 which are in agreement with expectations for an Fe(II), LS complex (Fig. S29‡). Overall, the characteristics of complex 4Tol are similar to other Fe diazene complexes found in the literature but remains a rare example of a trans diazene Fe(II) complex.51,70–72 Notably, complex 4Tol is highly thermally stable (heated at 110 °C in toluene for a week) and does not dissociate when subjected to vacuum (30 mTorr). This is reflective of the substitutionally inert low spin Fe(II) octahedral centre and attests to the robustness of Fe complexes supported by the B2Pz4Py ligand in comparison to other systems where the trans diazene complex is much more reactive.51,73
 | ||
| Fig. 5 (A) 1H NMR spectra of the diazene multiplet of 4Tol-(15N)2 in C6D6 indicating an AA‘XX’ system (experimental, black; simulation, red). The experimental spectrum was simulated with TopSpin using the following parameters: δ = 18.66 ppm; 1JNH = 69.4 Hz; 2JNH = 0.45 Hz; 3JHH = 22.4 Hz (trans); 1JNN = 10.6 Hz, and line width = 3.0 Hz. (B) ORTEP diagrams for 4Tol. Thermal ellipsoids are shown at the 50% probability level. Calculated hydrogen atoms are omitted for clarity except those on the diazene ligand (N10 and N11). The molecule of solvent has been omitted for clarity. Selected bond distances (Å): Fe1–N1, 1.979(3); Fe1–N2, 2.001(3); Fe1–N4, 1.994(3); Fe1–N10, 1.923(3); Fe2–N11, 1.887(4); Fe2–N12, 2.031(4); Fe2–N14, 2.008(3); Fe2–N16, 2.026(2); N10–N11, 1.288(4). Selected bond angles (°): N2–Fe–N4, 91.9(1); N1–Fe–N10, 177.2(1); Fe1–N10–N11, 131.8(2); N14–Fe2–N16, 94.9(1); N12–Fe2–N11, 176.9(1); Fe2–N11–N10, 132.1(3). Further metrical data are given in Table S2.‡ | ||
Even though the homogeneously catalysed disproportionation of hydrazine to ammonia and diazene has previously been reported in a few studies,74–76 the mechanism of this transformation has yet to be studied in great detail.60,68,77,78 We therefore sought to study the mechanism of the reaction between 1Tol and hydrazine, both experimentally and theoretically. Two main mechanisms are postulated to explain the formation of 1Tol-NH3 and 4Tol from hydrazine disproportionation mediated by 1Tol. They were probed by DFT calculations using the B3PW91 functional and are summarized in Fig. 6.
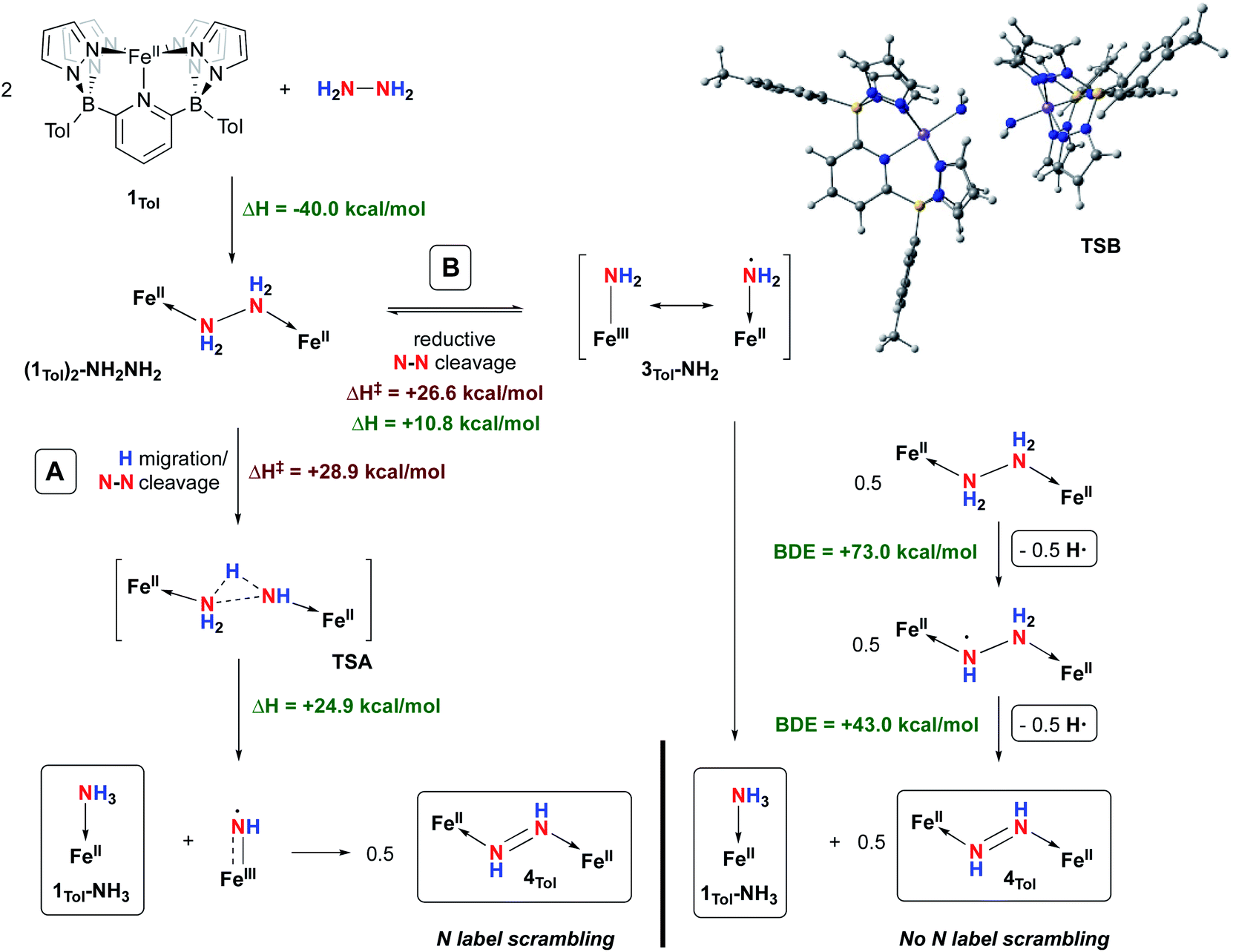 | ||
| Fig. 6 Two potential mechanisms for the disproportionation of hydrazine using complex 1Tol at 298 K. Enthalpies are given relative to compound 1Tol set to 0.0 kcal mol−1. The final products are shown in the boxes. | ||
The coordination of hydrazine to 1Tol to form the hydrazine adduct (1Tol)2-NH2NH2 is favored enthalpically by 40.0 kcal mol−1, consistent with experimental observation. From this intermediate, two pathways for N–N bond cleavage can be envisioned. The first (path A) is reminiscent of what we proposed for O–O bond cleavage in the isoelectronic cobalt hydrogen peroxide dication, formed upon double protonation of the Co(III)–Co(III) peroxo complex supported by the B2Pz4Py framework.37 Here, migration of a proton from one nitrogen to another is coupled with N–N cleavage in TSA, and this path leads directly to one of the observed products, 1Tol-NH3, along with the formally Fe(III) imidyl species shown with a barrier of 28.9 kcal mol−1. This barrier is considerably higher than what we found for the same step in the [Co(III)–(H)O–O(H)–Co(III)]2+ dication;37 this is likely due to the lower acidity associated with the N–H protons in neutral (1Tol)2-NH2NH2. Furthermore, in contrast to the cobalt/hydrogen peroxide chemistry, the step through TSA is significantly endothermic (24.9 kcal mol−1). However, computational analysis of the iron “imido” species shows that, like 3Tol-NH2, it is accurately described as an Fe(III) imidyl radical because the majority of spin density (54%) is found on the nitrogen as opposed to the iron (46%). This compound is therefore highly reactive and dimerizes in an exothermic reaction (more than 90 kcal mol−1) to the observed diazene product 4Tol, providing the main driving force for this path. It is likely that the trans-diazene is strongly favored for steric reasons, and we did not explore the path to formation of the cis-isomer. While some structurally characterized cis-diazene complexes exist,79,80 they are favored by constraints imposed by the ligand systems employed and most adopt trans structures for both steric51,70–72 and electronic81,82 reasons.
The mechanism depicted in path A to the diazene product 4Tol should lead to scrambling of 14N/15N labeling if a mixture of (1Tol)2-14NH214NH2 and (1Tol)2-15NH215NH2 were allowed to proceed to formation of the products. The isotopic distribution within the NN bond in the product 4Tol can be determined by rRaman spectroscopy. The reaction of 1Tol with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 15N2H4 and 14N2H4 at 210 K as described above generates the required 1:1 mixture of isotopologues. As shown in Fig. 7, analysis of the rRaman spectrum of the product mixture clearly shows that only unscrambled 4Tol-(14N)2 and 4Tol-(15N)2 were formed, with no evidence of the mixed isotopologue 4Tol-14N–15N. This experiment prompted us to consider a mechanism in which isotopic scrambling would not be predicted, namely path B in Fig. 6.
:1 mixture of 15N2H4 and 14N2H4 at 210 K as described above generates the required 1:1 mixture of isotopologues. As shown in Fig. 7, analysis of the rRaman spectrum of the product mixture clearly shows that only unscrambled 4Tol-(14N)2 and 4Tol-(15N)2 were formed, with no evidence of the mixed isotopologue 4Tol-14N–15N. This experiment prompted us to consider a mechanism in which isotopic scrambling would not be predicted, namely path B in Fig. 6.
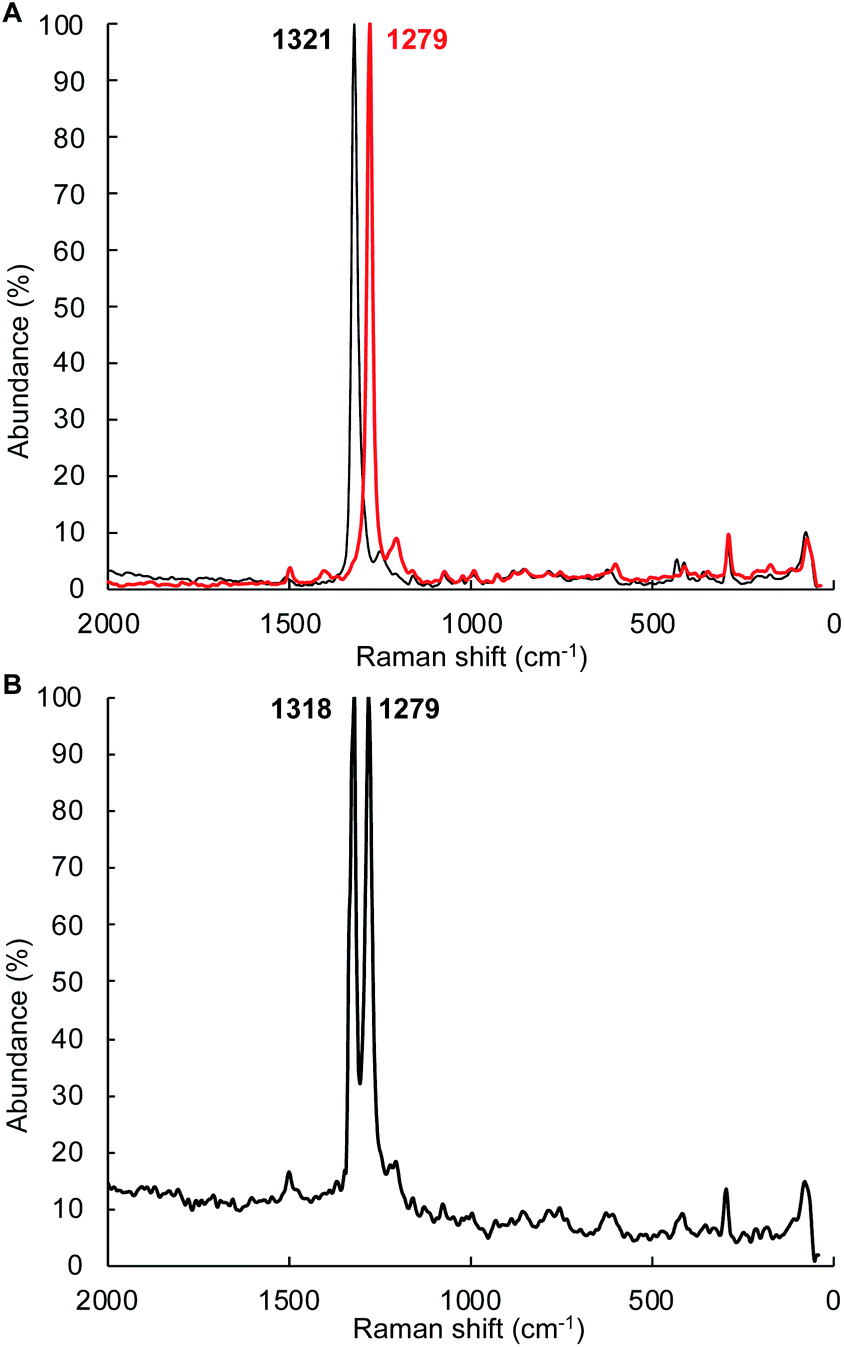 | ||
| Fig. 7 Scrambling of nN label in the disproportionation of a 1:1 mixture of 15N2H4 and 14N2H4 with 1Tol as determined by resonance Raman spectroscopy. The rRaman spectra were obtained with a 1064 nm laser excitation wavelength at room temperature. A. rRaman spectra of 4Tol-(14N)2 (black trace) and 4Tol-(15N)2 (red trace). (B) rRaman spectrum of the 4Tol-(nN)2 isotopologues obtained by reaction of nN2H4 with 1Tol. | ||
Pathway B is based on an equilibrium between the hydrazine adduct (1Tol)2-NH2NH2 and its monomeric form, complex 3Tol-NH2 (Fig. 6). Computational analysis of this step reveals that it is endothermic by 10.8 kcal mol−1, but proceeds viaTSB, which is lower in energy than TSA by 2.3 kcal mol−1. The N–N distance in TSB is 2.332 Å, indicating that most of the barrier is due to homolytic cleavage of the N–N bond in coordinated hydrazine. While dimerization back to (1Tol)2-NH2NH2 has a low barrier (≈15 kcal mol−1), if 3Tol-NH2 finds another equivalent of (1Tol)2-NH2NH2 it may abstract a relatively weak N–H bond (73.0 kcal mol−1, cf. that of 75.5 kcal mol−1 in 1Tol-NH3). The resulting complex has an even weaker N–H bond of 43.0 kcal mol−1, and undergoes further H atom abstraction to yield the diazene product 4Tol. This path is consistent with the lack of scrambling of nitrogen label in the experiment described above. Because of the endothermic nature of equilibrium of path B, the concentration of highly reactive 3Tol-NH2 is expected to be very small and would scavenge an H atom from the weakest and most abundant source in the medium, in this case (1Tol)2-NH2NH2. Unfortunately, attempts to detect the LS (S = 1/2) iron amido/amidyl species 3Tol-NH2 by following the reaction of 1Tol with hydrazine by low temperature EPR spectroscopy were not successful, indicating it must be an extremely short-lived species.
Conclusions
Iron(II) complexes supported by the dianionic, pentadentate ligand platform B2Pz4Py39 were used to coordinate and activate ammonia and hydrazine. Several lines of experimental evidence, supported by DFT computations, implicate the generation of a highly reactive aminyl species, 3Ar-NH2 through abstraction of H˙ or H+ from suitable precursors. Such compounds are understood to be important intermediates in ammonia oxidation processes as mediated by transition metal-based catalysts. While 3Ar-NH2 might formally be regarded as an Fe(III) amido derivative, the electron richness of the diborate ligand framework favors the Fe(II) aminyl formulation, accounting for its high propensity to react with other radical species (e.g. ArO˙) or H˙ sources. Interestingly, this compound also appears to be accessible via hydrazine activation through a reductive cleavage of the N–N bond in hydrazine. This is the reverse of a potentially important N–N bond forming path in AO. These transformations show that the tetrapodal pentadentate platform furnished by the B2Pz4Py ligand has promise not only for studying new stoichiometric reactions of archetypical nitrogen containing fuel sources, but also for catalyst development.Conflicts of interest
There are no conflicts to declare.Acknowledgements
W. E. P. thanks the Canada Research Chair secretariat for a Tier I CRC (2013–2027). Funding for the experimental work described was provided by the Natural Sciences and Engineering Research Council of Canada in the form of a Discovery Grant to W. E. P. Funding for the computational work described was provided by a French CNRS PICS project. L. N. thanks Alberta Innovates Technology Futures and the Vanier Canada Graduate Scholarships for support. LM is a senior member of the Institut Universitaire de France. Y. Y. thanks the Chinese Scholarship Council (CSC) for financial support. CalMip is acknowledged for a generous grant of computing time. M. L. N. acknowledges support for this work from the National Institutes of Health under grant R01GM111480.Notes and references
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS.
- H. B. Gray, Nat. Chem., 2009, 1, 7 CrossRef CAS.
- J. G. Chen, R. M. Crooks, L. C. Seefeldt, K. L. Bren, R. M. Bullock, M. Y. Darensbourg, P. L. Holland, B. Hoffman, M. J. Janik, A. K. Jones, M. G. Kanatzidis, P. King, K. M. Lancaster, V. S. Lymar, P. Pfromm, W. F. Schneider and R. R. Schrock, Science, 2018, 360, eaar6611 CrossRef.
- P. H. Pfromm, J. Renewable Sustainable Energy, 2017, 9, 034702 CrossRef.
- G. Soloveichik, Nat. Catal., 2019, 2, 377–380 CrossRef CAS.
- N. M. Adli, H. Zhang, S. Mukherjee and G. Wu, J. Electrochem. Soc., 2018, 165, 3130–3147 CrossRef.
- S. T. Hatscher, T. Fetzer, E. Wagner and H.-J. Kneuper, in Handbook of Heterogeneous Catalysis, 2008, pp. 2575–2592, DOI:10.1002/9783527610044.hetcat0130.
- P. Bhattacharya, Z. M. Heiden, G. M. Chambers, S. I. Johnson, R. M. Bullock and M. T. Mock, Angew. Chem., Int. Ed., 2019, 58, 11618–11624 CrossRef CAS.
- P. L. Dunn, S. I. Johnson, W. Kaminsky and R. M. Bullock, J. Am. Chem. Soc., 2020, 142, 3361–3365 CrossRef CAS.
- F. Habibzadeh, S. L. Miller, T. W. Hamann and M. R. Smith, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 2849–2853 CrossRef CAS.
- M. D. Zott, P. Garrido-Barros and J. C. Peters, ACS Catal., 2019, 9, 10101–10108 CrossRef CAS.
- K. Nakajima, H. Toda, K. Sakata and Y. Nishibayashi, Nat. Chem., 2019, 11, 702–709 CrossRef CAS.
- P. L. Dunn, B. J. Cook, S. I. Johnson, A. M. Appel and R. M. Bullock, J. Am. Chem. Soc., 2020, 142, 17845–17858 CrossRef CAS.
- J. J. Warren, T. A. Tronic and J. M. Mayer, Chem. Rev., 2010, 110, 6961–7001 CrossRef CAS.
- D. Cheddie, in Hydrogen Energy – Challenges and Perspectives, 2012, DOI:10.5772/47759.
- J. Zhao, Science, 2005, 307, 1080–1082 CrossRef CAS.
- V. D. Gutsulyak, W. E. Piers, J. Borau-Garcia and M. Parvez, J. Am. Chem. Soc., 2013, 135, 11776–11779 CrossRef.
- G. W. Margulieux, Z. R. Turner and P. J. Chirik, Angew. Chem., Int. Ed., 2014, 53, 14211–14215 CrossRef CAS.
- E. Khaskin, M. A. Iron, L. J. W. Shimon, J. Zhang and D. Milstein, J. Am. Chem. Soc., 2010, 132, 8542–8543 CrossRef CAS.
- Y.-H. Chang, Y. Nakajima, H. Tanaka, K. Yoshizawa and F. Ozawa, J. Am. Chem. Soc., 2013, 135, 11791–11794 CrossRef CAS.
- R. M. Brown, J. Borau-Garcia, J. Valjus, C. J. Roberts, H. M. Tuononen, M. Parvez and R. Roesler, Angew. Chem., Int. Ed., 2015, 54, 6274–6277 CrossRef CAS.
- M. G. Scheibel, J. Abbenseth, M. Kinauer, F. W. Heinemann, C. Würtele, B. De Bruin and S. Schneider, Inorg. Chem., 2015, 54, 9290–9302 CrossRef CAS.
- M. J. Bezdek, S. Guo and P. J. Chirik, Science, 2016, 354, 730–733 CrossRef CAS.
- G. W. Margulieux, M. J. Bezdek, Z. R. Turner and P. J. Chirik, J. Am. Chem. Soc., 2017, 139, 6110–6113 CrossRef CAS.
- M. J. Bezdek and P. J. Chirik, Angew. Chem., Int. Ed., 2018, 130, 2246–2250 CrossRef.
- P. Bhattacharya, Z. M. Heiden, E. S. Wiedner, S. Raugei, N. A. Piro, W. S. Kassel, R. M. Bullock and M. T. Mock, J. Am. Chem. Soc., 2017, 139, 2916–2919 CrossRef CAS.
- B. J. Cook, S. I. Johnson, G. M. Chambers, W. Kaminsky and R. M. Bullock, Chem. Commun., 2019, 55, 14058–14061 RSC.
- G. Nina, O. H. Paul and P. Jonas, Hydrazine Formation via NiIII-NH2 Radical Coupling in Ni-Mediated Ammonia Oxidation, 2020 Search PubMed.
- J. Y. Shen, M. Wang, P. L. Zhang, J. Jiang and L. C. Sun, Chem. Commun., 2017, 53, 4374–4377 RSC.
- D. J. Wasylenko, R. D. Palmer and C. P. Berlinguette, Chem. Commun., 2013, 49, 218–227 RSC.
- D. M. Ekanayake, K. M. Kulesa, J. Singh, K. K. Kpogo, S. Mazumder, H. B. Schlegel, C. N. Verani, H. Bernhard Schlegel and C. N. Verani, Dalton Trans., 2017, 46, 16812–16820 RSC.
- W. T. Eckenhoff, Coord. Chem. Rev., 2018, 373, 295–316 CrossRef CAS.
- L. Z. Chen, A. Khadivi, M. Singh and J. W. Jurss, Inorg. Chem. Front., 2017, 4, 1649–1653 RSC.
- R. T. Jonas and T. D. P. Stack, J. Am. Chem. Soc., 1997, 119, 8566–8567 CrossRef CAS.
- M. E. de Vries, R. M. La Crois, G. Roelfes, H. Kooijman, A. L. Spek, R. Hage and B. L. Feringa, Chem. Commun., 1997, 1549–1550 RSC.
- S. I. Johnson, S. P. Heins, C. M. Klug, E. S. Wiedner, R. M. Bullock and S. Raugei, Chem. Commun., 2019, 55, 5083–5086 RSC.
- L. Nurdin, D. M. Spasyuk, L. Fairburn, W. E. Piers and L. Maron, J. Am. Chem. Soc., 2018, 140, 16094–16105 CrossRef CAS.
- D. W. Beh, W. E. Piers, B. S. Gelfand and J.-B. Lin, Dalton Trans., 2020, 49, 95–101 RSC.
- D. M. Spasyuk, S. H. Carpenter, C. E. Kefalidis, W. E. Piers, M. L. Neidig and L. Maron, Chem. Sci., 2016, 7, 5939–5944 RSC.
- D. W. Beh, W. E. Piers, I. del Rosal, L. Maron, B. S. Gelfand, C. Gendy and J.-B. Lin, Dalton Trans., 2018, 47, 13680–13688 RSC.
- L. Nurdin, D. M. Spasyuk, W. E. Piers and L. Maron, Inorg. Chem., 2017, 56, 4157–4168 CrossRef CAS.
- T. J. Morin, B. Bennett, V. S. Lindeman and J. R. Gardinier, Inorg. Chem., 2008, 47, 7468–7470 CrossRef CAS.
- D. J. Fox and R. G. Bergman, J. Am. Chem. Soc., 2003, 125, 8984–8985 CrossRef CAS.
- S. E. Creutz and J. C. Peters, Chem. Sci., 2017, 8, 2321–2328 RSC.
- J. S. Anderson, M.-E. Moret and J. C. Peters, J. Am. Chem. Soc., 2013, 135, 534–537 CrossRef CAS.
- J. J. Kiernicki, E. E. Norwine, M. Zeller and N. K. Szymczak, Chem. Commun., 2019, 55, 11896–11899 RSC.
- A. J. Mountford, W. Clegg, S. J. Coles, R. W. Harrington, P. N. Horton, S. M. Humphrey, M. B. Hursthouse, J. A. Wright and S. J. Lancaster, Chem.–Eur. J., 2007, 13, 4535–4547 CrossRef CAS.
- R. A. Andersen, K. Faegri, J. C. Green, A. Haaland, M. F. Lappert, W. P. Leung and K. Rypdal, Inorg. Chem., 1988, 27, 1782–1786 CrossRef CAS.
- D. L. J. Broere, I. Čorić, A. Brosnahan and P. L. Holland, Inorg. Chem., 2017, 56, 3140–3143 CrossRef CAS.
- L. Nurdin, Ph.D. thesis, University of Calgary, 2020.
- C. T. Saouma, C. E. Moore, A. L. Rheingold and J. C. Peters, Inorg. Chem., 2011, 50, 11285–11287 CrossRef CAS.
- Y. Lee, N. P. Mankad and J. C. Peters, Nat. Chem., 2010, 2, 558–565 CrossRef CAS.
- D. N. Bowman and E. Jakubikova, Inorg. Chem., 2012, 51, 6011–6019 CrossRef CAS.
- Y. Shimura, Bull. Chem. Soc. Jpn., 1988, 61, 693–698 CrossRef CAS.
- J. Mason, in Patai's Chemistry of Functional Groups, 2009, DOI:10.1002/9780470682531.pat0148.
- M. Hedoyatullah and F. Thevenet, Bull. Soc. Chim. Belg., 2010, 96, 311–323 CrossRef.
- A. I. O. Suarez, V. Lyaskovskyy, J. N. H. Reek, J. I. van der Vlugt, B. de Bruin, J. I. van der Vlugt and B. de Bruin, Angew. Chem., Int. Ed., 2013, 52, 12510–12529 CrossRef CAS.
- V. W. Manner, T. F. Markle, J. H. Freudenthal, J. P. Roth and J. M. Mayer, Chem. Commun., 2008, 256–258 RSC.
- R. L. Lucas, D. R. Powell and A. S. Borovik, J. Am. Chem. Soc., 2005, 127, 11596–11597 CrossRef CAS.
- K. Umehara, S. Kuwata and T. Ikariya, J. Am. Chem. Soc., 2013, 135, 6754–6757 CrossRef CAS.
- Y.-H. Chang, P.-M. Chan, Y.-F. Tsai, G.-H. Lee and H.-F. Hsu, Inorg. Chem., 2014, 53, 664–666 CrossRef CAS.
- M. Gomberg, J. Am. Chem. Soc., 1900, 22, 757–771 CrossRef.
- T. H. Colle and E. S. Lewis, J. Am. Chem. Soc., 1979, 101, 1810–1814 CrossRef CAS.
- N. S. Sickerman, S. M. Peterson, J. W. Ziller and A. S. Borovik, Chem. Commun., 2014, 50, 2515–2517 RSC.
- M. M. Olmstead, G. Sigel, H. Hope, X. Xu and P. P. Power, J. Am. Chem. Soc., 1985, 107, 8087–8091 CrossRef CAS.
- J. S. Anderson, J. Rittle and J. C. Peters, Nature, 2013, 501, 84–87 CrossRef CAS.
- G. T. Sazama and T. A. Betley, Inorg. Chem., 2010, 49, 2512–2524 CrossRef CAS.
- C. Ferousi, S. H. Majer, I. M. DiMucci and K. M. Lancaster, Chem. Rev., 2020, 120, 5252–5307 CAS.
- M. A. Cooper and S. L. Manatt, J. Am. Chem. Soc., 1969, 91, 6325–6333 CAS.
- D. Sellmann and J. Sutter, Acc. Chem. Res., 1997, 30, 460–469 CAS.
- D. Sellmann, W. Soglowek, F. Knoch and M. Moll, Angew. Chem., Int. Ed., 1989, 28, 1271–1272 Search PubMed.
- D. Sellmann, D. C. F. Blum and F. W. Heinemann, Inorg. Chim. Acta, 2002, 337, 1–10 CAS.
- C. G. Balesdent, J. L. Crossland, D. T. Regan, C. T. López and D. R. Tyler, Inorg. Chem., 2013, 52, 14178–14187 CrossRef CAS.
- N. X. Gu, G. Ung and J. C. Peters, Chem. Commun., 2019, 55, 5363–5366 CAS.
- P. B. Hitchcock, D. L. Hughes, M. J. Maguire, K. Marjani and R. L. Richards, J. Chem. Soc., Dalton Trans., 1997, 24, 4747–4752 Search PubMed.
- L. D. Field, H. L. Li and A. M. Magill, Inorg. Chem., 2009, 48, 5–7 CrossRef CAS.
- H. Tanaka, S. Hitaoka, K. Umehara, K. Yoshizawa and S. Kuwata, Eur. J. Inorg. Chem., 2020, 2020, 1472–1482 CrossRef CAS.
- Y. Zheng, W. Zheng, J. Wang, H. Chang and D. Zhu, J. Phys. Chem. A, 2018, 122, 2764–2780 CAS.
- Y. Chen, L. Liu, Y. Peng, P. Chen, Y. Luo and J. Qu, J. Am. Chem. Soc., 2011, 133, 1147–1149 CrossRef CAS.
- C. T. Saouma, P. Müller and J. C. Peters, J. Am. Chem. Soc., 2009, 131, 10358–10359 CrossRef CAS.
- J. A. Pople and L. A. Curtiss, J. Chem. Phys., 1991, 95, 4385–4388 CrossRef CAS.
- A. Sindhu, R. Pradhan, U. Lourderaj and M. Paranjothy, Phys. Chem. Chem. Phys., 2019, 21, 15678–15685 RSC.
Footnotes |
| † The authors would like to dedicate this manuscript to Prof. Robert H. Morris, University of Toronto, scholar, gentleman and organometallic chemist extraordinaire. |
| ‡ Electronic supplementary information (ESI) available: Experimental and characterization details for all new compounds, including spectroscopic data, X-ray crystallographic data and computational details with the cartesian coordinates for calculated structures. CCDC codes 2044409–2044414 contain the crystallographic data for compounds 1Tol-NH3, 2Tol, 3Tol-NH3+, 3Tol-Br, 3Tol-F, and 4Tol, respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc06466a |
| This journal is © The Royal Society of Chemistry 2021 |