DOI:
10.1039/D0SC06366E
(Edge Article)
Chem. Sci., 2021,
12, 3509-3515
Structural elucidation of a methylenation reagent of esters: synthesis and reactivity of a dinuclear titanium(III) methylene complex†
Received
20th November 2020
, Accepted 19th January 2021
First published on 19th January 2021
Abstract
Transmetallation of a zinc methylene complex [ZnI(tmeda)]2(μ-CH2) with a titanium(III) chloride [TiCl3(tmeda)(thf)] produced a titanium methylene complex. The X-ray diffraction study displayed a dinuclear methylene structure [TiCl(tmeda)]2(μ-CH2)(μ-Cl)2. Treatment of an ester with the titanium methylene complex resulted in methylenation of the ester carbonyl to form a vinyl ether. The titanium methylene complex also reacted with a terminal olefin, resulting in olefin-metathesis and olefin-homologation. Cyclopropanation by methylene transfer from the titanium methylene proceeded by use of a 1,3-diene. The mechanistic study of the cyclopropanation reaction by the density functional theory calculations was also reported.
Introduction
Since the first report of the Tebbe reagent in 1978,1,2 titanium methylene species3 have been widely investigated and utilized for Wittig-type olefination of carbonyls,1,3b,3c,4 C–H activation,5 olefin homologation,1 olefin metathesis6 and ring-opening metathesis polymerization.7 In the same year of Tebbe's report,1 Oshima and Takai also reported a methylenation reagent prepared from CH2Br2, Zn and TiCl4,8 which was modified later by Lombardo.9 In 1994, it turned out that the originally used zinc powder contained lead as an impurity derived from the method of metallurgy, i.e. pyrometallurgy,10 which was what catalyzed generation of the methylenation reagent (vide infra).11 It was also found that addition of N,N,N′,N′-tetramethylethylenediamine (TMEDA) to the original mixture (RCHBr2, Zn, cat. PbCl2 and TiCl4) dramatically changed the functional selectivity; the new reagent undergoes alkylidenation of esters,12,13 which cannot be achieved by the Nysted reagent [Zn3Br2(μ-CH2)2(thf)]14 with titanium chlorides (TiCl4, [TiCl2(OiPr)2], [Cp2TiCl2])15 or the original CH2X2–Zn(Pb)–TiCl4 reagent without TMEDA.8,16
The first key step in preparing the CH2X2–Zn(Pb)–TiCl4 methylenation reagent involves reductive cleavage of C–X bonds by Zn(0) to form a zinc methylene species “CH2(ZnX)2”,11,17 which was trapped as CH2(SnMe3)2 upon treatment with ClSnMe3 (Scheme 1b).8,16a The second reductive cleavage of a C–X bond by Zn(0) to give the zinc methylene “CH2(ZnI)2” is accelerated via transmetallation with a catalytic amount of a lead(II) salt. This catalytic amount of lead crucially affects the generation of the CH2X2–Zn(Pb)–TiCl4 reagent.11 The other key step is reduction of titanium(IV) to titanium(III) by Zn(0) (Scheme 1c),18 which takes place simultaneously with formation of the zinc methylene “CH2(ZnX)2”. Given the idea of a terminally bound mononuclear titanium methylene, known as “methylidene”, generated from the Tebbe and Petasis reagents (Scheme 1a),1,19 we believe that a transmetallation event between the zinc methylene “CH2(ZnI)2” and titanium(III) chloride18b should take place to generate a titanium(III) methylidene species [(L)nTi(=CH2)Cl] due to its powerful methylenation reactivity.12,13 To clarify the reactive species in the methylenation reagent, we have recently synthesized and isolated the zinc methylene “CH2(ZnX)2” as a dinuclear zinc μ-methylene complex, namely [ZnI(L)n]2(μ-CH2) (1a: L = tmeda, n = 1; 1b: L = 2,6-lutidine, n = 2), and methylenation of ester carbonyls proceeded by mixing 1a and [TiCl3(tmeda)(thf)] (2).20 Herein, we report a structural characterization of a titanium methylene complex formed by transmetallation between the zinc methylene and titanium(III) chloride as well as methylene transfer reactions to ether carbonyls and olefins.
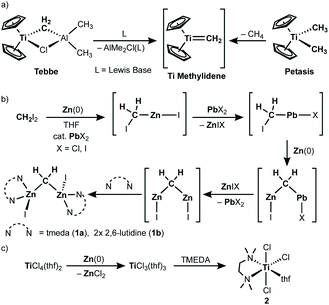 |
| | Scheme 1 (a) Generation of a titanium methylidene from the Tebbe and Petasis reagents; (b) generation of dinuclear zinc methylene species from diiodomethane and zinc(0) catalyzed by a lead(II) salt; (c) reduction of titanium(IV) chloride to titanium(III) by zinc(0). | |
Results and discussion
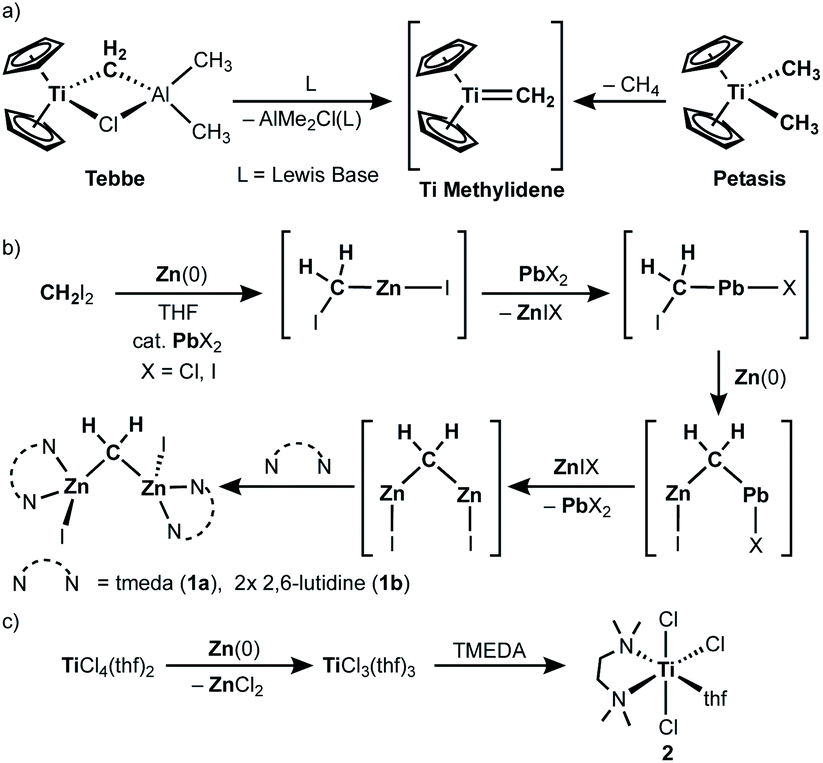
As in our recent report,20 the dinuclear zinc methylene complex (1) was prepared by reaction of Zn(0) with CH2I2 in the presence of lead(II) chloride11 and addition of TMEDA or 2,6-lutidine in THF afforded the corresponding adducts 1a and 1b, respectively. A solid-state structure of the TMEDA adduct 1a (Fig. 1, left) was obtained by X-ray diffraction study of a single crystal grown in THF/hexane. Akin to the recently reported bpyMes adduct [ZnI(bpyMes)]2(μ-CH2) (bpyMes = 6-Mes-2,2′-bipyridyl),20 the methylene ligand is bridging between two zinc iodido centers (Zn–C: 1.969(7) Å, 1.979(7) Å; Zn–C–Zn: 109.4(3)°), along with TMEDA in a bidentate coordination mode. The molecular structure of 1a in solid-state revealed a slightly distorted C2 structure, which gave pseudo-C2 symmetric NMR spectra in solution.
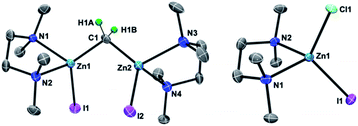 |
| | Fig. 1 POV-ray drawing of 1a (left) and [ZnClI(tmeda)] (right) with thermal ellipsoids at the 50% probability level. Hydrogen atoms with the exception of the methylene ligand of 1a have been omitted for clarity. | |
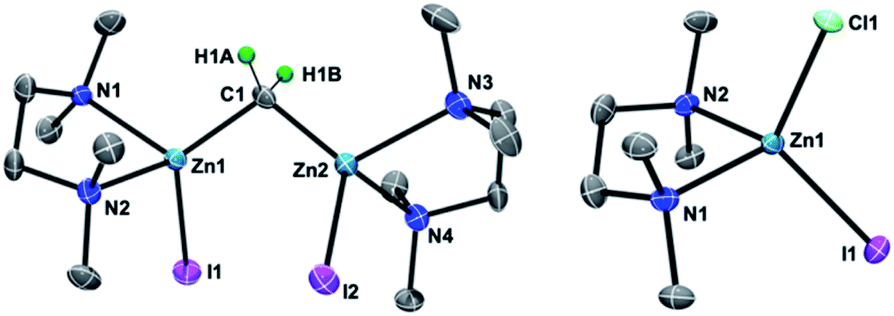
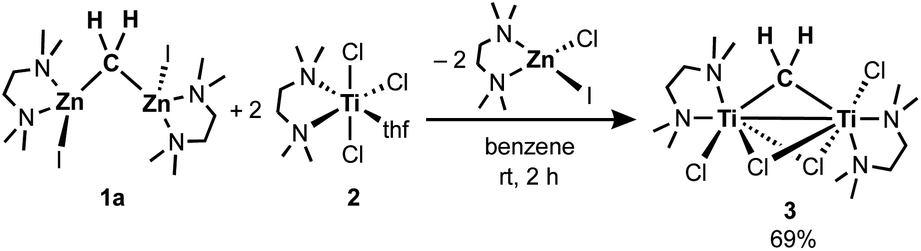
To gain insight into the transmetallation process between the zinc methylene and titanium chloride, multiple combinations of zinc methylene complexes (1a and 1b) and titanium chlorides ([TiCl4(thf)2], [TiCl4(tmeda)], [TiCl3(thf)3], and 2) with and without additional ligands (PR3, pyridine, 4-dimethylaminopyridine, and ethers) were attempted and monitored by NMR spectroscopy. In most cases, 1H NMR spectra revealed consumption of the zinc methylene species around −1 ppm along with formation of CH4 and C2H4 as well as some paramagnetic species. Interestingly, the combination of both TMEDA adducts 1a and 2 exclusively resulted in a clean formation of a new diamagnetic titanium methylene species at 9.94 ppm,21 even though the originally proposed methylidene species should have a single titanium(III) center to be paramagnetic. However, the NMR spectrum still showed a mixture with the remaining zinc methylene species in an approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, which was consumed completely by addition of another equivalent of 2 (Fig. S5, ESI†).21 Accordingly, treatment of 1a with two equivalents of 2 in benzene (Scheme 2) afforded complex 3 in 69% isolated yield as a reddish brown solid after removal of a zinc chlorido–iodido TMEDA complex [ZnClI(tmeda)] (Fig. 1, right), which was structurally characterized by X-ray diffraction. The NMR spectra of the isolated product 3 corroborate the methylene ligand at 1H: 9.45 ppm and 13C: 248.2 ppm (1JCH = 114 Hz) along with inequivalent CH3 and CH2 resonances of TMEDA. The titanium methylene resonances of 3 are slightly down-field shifted from those of di- or trinuclear μ-methylene complexes of titanium (1H: 5.51–8.81 ppm, 13C: 188.5–253.1 ppm),22 but much more up-field shifted from those of mononuclear titanium methylidenes (1H: 11.61–12.12 ppm, 13C: 285.9–295.9 ppm).23 The methylene complex 3 is stable in solid-state at room temperature for weeks, but the 1H NMR resonance of the CH2 ligand at 9.45 ppm gradually diminished in solution at room temperature to form methane CH4, which was not deuterated even in THF-d8 or CD2Cl2.21
:1 ratio, which was consumed completely by addition of another equivalent of 2 (Fig. S5, ESI†).21 Accordingly, treatment of 1a with two equivalents of 2 in benzene (Scheme 2) afforded complex 3 in 69% isolated yield as a reddish brown solid after removal of a zinc chlorido–iodido TMEDA complex [ZnClI(tmeda)] (Fig. 1, right), which was structurally characterized by X-ray diffraction. The NMR spectra of the isolated product 3 corroborate the methylene ligand at 1H: 9.45 ppm and 13C: 248.2 ppm (1JCH = 114 Hz) along with inequivalent CH3 and CH2 resonances of TMEDA. The titanium methylene resonances of 3 are slightly down-field shifted from those of di- or trinuclear μ-methylene complexes of titanium (1H: 5.51–8.81 ppm, 13C: 188.5–253.1 ppm),22 but much more up-field shifted from those of mononuclear titanium methylidenes (1H: 11.61–12.12 ppm, 13C: 285.9–295.9 ppm).23 The methylene complex 3 is stable in solid-state at room temperature for weeks, but the 1H NMR resonance of the CH2 ligand at 9.45 ppm gradually diminished in solution at room temperature to form methane CH4, which was not deuterated even in THF-d8 or CD2Cl2.21
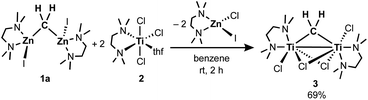 |
| | Scheme 2 Synthesis of the titanium methylene complex 3. | |
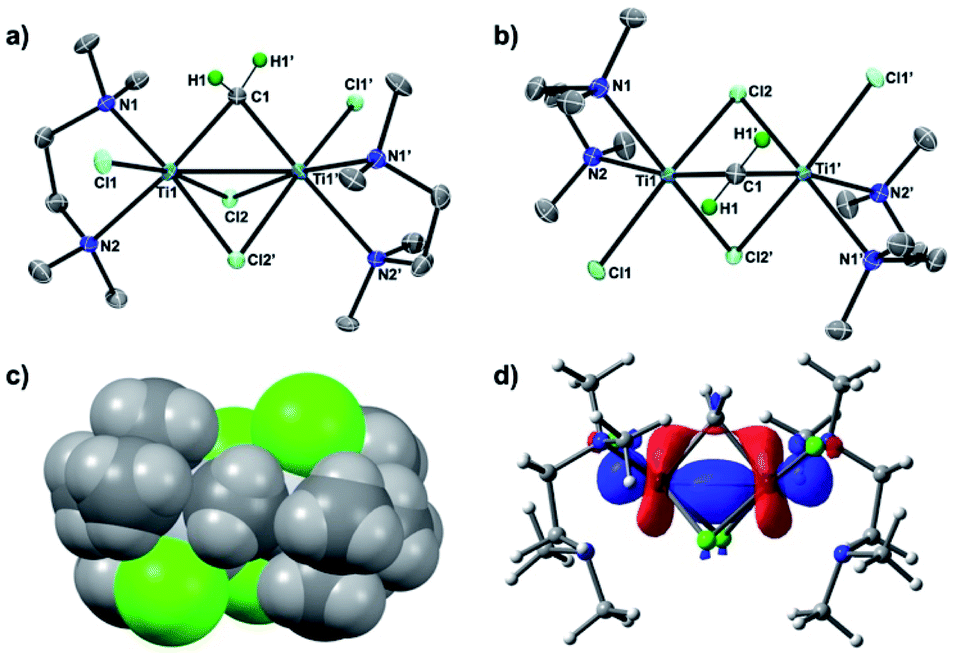
To conclusively establish the connectivity in 3, X-ray diffraction data on a single crystal grown from a concentrated THF solution were collected. As shown in Fig. 2, the solid-state structure of 3 displayed a C2 symmetric dinuclear titanium structure bridged by a methylene ligand (Ti1–C1: 2.084(6) Å; Ti1–C1–Ti1′: 78.4(3)°) and chlorides (Ti1–Cl1: 2.409(2) Å; Ti1–Cl2: 2.405(2) Å, 2.472 (2) Å). The crystal structures of the Tebbe complex [Cp2Ti(μ-CH2)(μ-Cl)AlMe2] have been reported by Mindiola24a and more recently by Anwander,24b and the Ti–CH2 bond length in complex 3 (2.084(6) Å) is comparable to the reported Ti–CH2 distances (2.095(5) Å,24a 2.058(3) Å)24b of the Tebbe complex. The bridging mode of the methylene ligand, where the hydrogen atoms were located from the difference map and refined isotropically, is oriented to avoid the steric repulsion with the methyl groups of TMEDA (Fig. 2c). The dinuclear methylene complex 3 is almost isostructural to the recently reported dinuclear chromium(III) alkylidene complexes with TMEDA ligands, [CrCl(tmeda)]2(μ-CHR)(μ-Cl)2 (R = SiMe3, GeMe3, SnMeCl2), but those dinuclear chromium(III) alkylidene complexes could be enforced to have a Cs symmetry due to the bulky substituents on the bridging alkylidenes.26 In contrast to the reported dinuclear Ti(IV)–Ti(IV) methylene complexes,22 the molecular structure of the Ti(III)–Ti(III) methylene complex 3 showed a short Ti–Ti distance of 2.634(2) Å, which is much shorter than the sum of van der Waals radii.25 The density functional theory (DFT) calculations of 3 in singlet (13) revealed a Ti–Ti bonding interaction (2.637 Å) at the HOMO (Fig. 2d) along with the Wiberg bond order index 0.96, while the optimized structure in triplet (33) has a much longer Ti–Ti distance (3.224 Å).
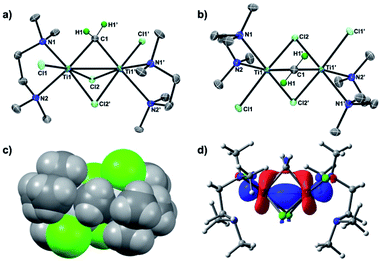 |
| | Fig. 2 (a) Side-view of POV-ray drawing of complex 3 with thermal ellipsoids at the 50% probability level. Hydrogen atoms with the exception of the methylene ligand have been omitted for clarity; (b) top-view; (c) space-filling model of 3; (d) molecular orbital of 3 at HOMO with the 0.04 isovalue. | |
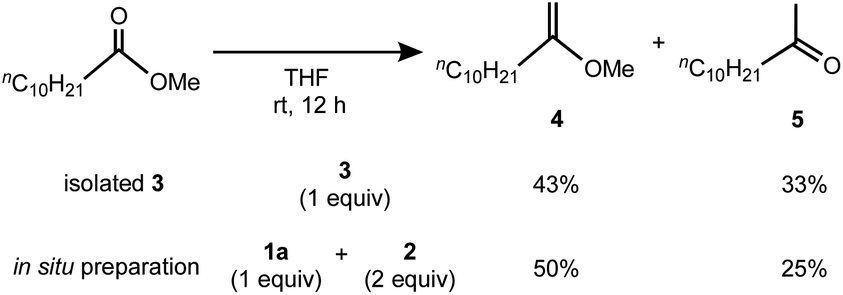
Having the methylene complex 3 in hand, we resorted to demonstrating methylenation of esters,12 which can be achieved by titanium methylidene species such as the Tebbe reagent.4a As shown in Scheme 3, methylenation of methyl undecanoate with one equivalent of 3 in THF smoothly proceeded in 76% yield to give a mixture of 2-methoxy-1-decene (4) and its hydrolysis product 5,27 comparable to that with the reagent prepared in situ from 1a and 2 in a 1:2 ratio (4: 50%; 5: 25%). In contrast, no methylenation products 4 or 5 were observed without TMEDA, e.g. a mixture of the 2,6-lutidine adduct 1b and [TiCl3(thf)3],20 implying the necessity of TMEDA to generate the reactive species for methylenation of esters. In addition, methylenation of cyclic esters with 3 could proceed to afford cyclic vinyl ethers, but some ring-opening and oligomerization products were also formed.21
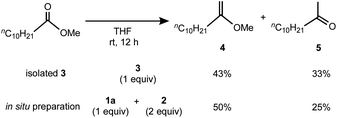 |
| | Scheme 3 Methylenation of methyl undecanoate by isolated 3 and in situ preparation of 3. | |
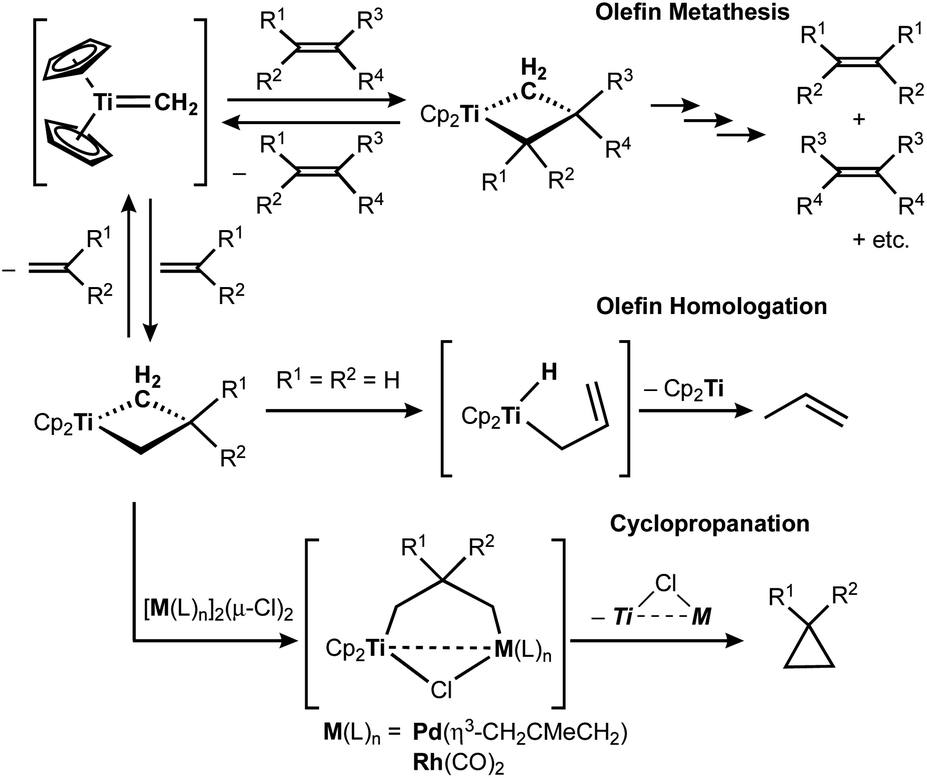
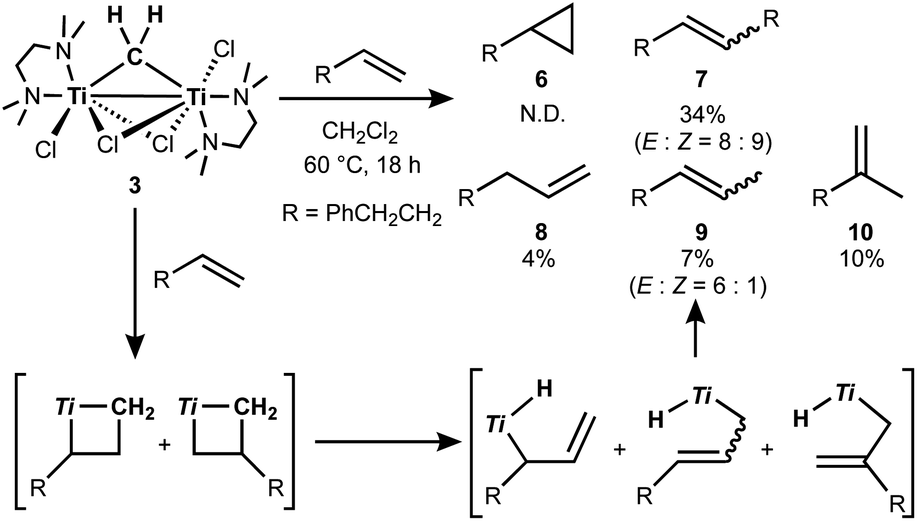
The titanocene methylidene generated from the Tebbe or Petasis reagents reacts with olefins to form titanacyclobutanes reversibly (Scheme 4),1,28 resulting in olefin-metathesis.6,7 Tebbe and Parshall also found that titanacyclobutanes can undergo β-H elimination and formation of olefin-homologation products.1 In contrast to other metallacyclobutanes,29 Grubbs and co-workers pointed out the difficulty in promoting reductive elimination of cyclopropane from the mononuclear titanium(IV) metallacyclobutane due to formation of a thermodynamically unfavored titanium(II) product,30,31 unless assisted by oxidation with I232 or formation of metal–metal interacting heterobimetallic species.33 In fact, formation of the cyclopropanation product (6) by treatment of a terminal olefin, 4-phenyl-1-butene, with our dinuclear titanium methylene 3 was not observed. Instead, NMR and GC analyses revealed formation of the corresponding olefin-metathesis (7: 34%)34 and olefin-homologation products (8: 4%; 9: 7%; 10: 10%).21 As illustrated in Scheme 5, each olefin-homologation product should be formed via the corresponding titanacyclobutane and allyl-hydrido intermediates.
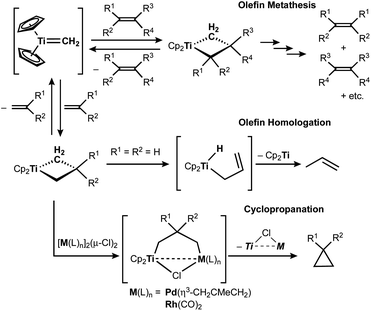 |
| | Scheme 4 Reversible [2+2]-cycloaddition of olefin to titanium methylidene1,28 and olefin-metathesis,6,7 olefin-homologation,1 and formation of cyclopropane from titanacyclobutanes.32,33 | |
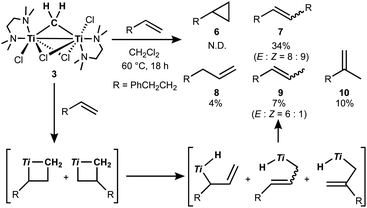 |
| | Scheme 5 Olefin-metathesis and homologation of terminal olefin by 3 and the proposed mechanism (Ti = TiCl(tmeda)). | |
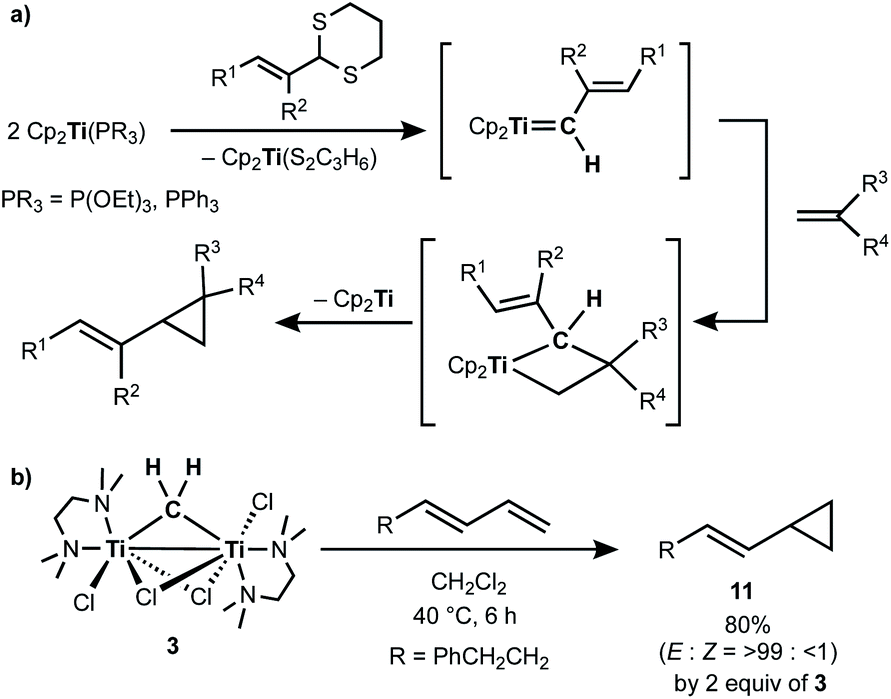
Takeda and co-workers reported a successful example of cyclopropanation by a combination of titanocene allylidenes and terminal olefins (Scheme 6a),35 while olefin-homologation of terminal olefins was observed by use of titanocene alkylidenes without α-vinyl groups.36 Inspired by Takeda's work on the vinylcyclopropanation system, we employed a 1,3-diene for the cyclopropanation reaction by methylene transfer from complex 3. Treatment of a 1,3-diene, (E)-6-phenyl-1,3-hexadiene, with 3 at room temperature in CH2Cl2 resulted in selective formation of an (E)-vinyl cyclopropane (11) in 49% yield (Scheme 6b). More conversion of the 1,3-diene to cyclopropane 11 (80% yield) was achieved by further addition of 3 (2 equiv.) and gentle heating at 40 °C. However, various olefin-metathesis products from the 1,3-diene as well as a small amount of the Z-isomer of 11 were formed by performing the reaction at 80 °C (Fig. S12, ESI†).
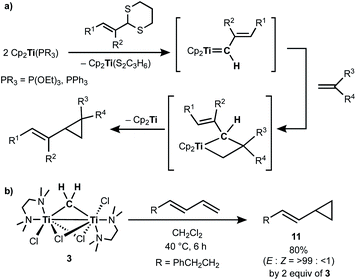 |
| | Scheme 6 (a) Vinylcyclopropanation of terminal olefins by titanocene alkylidenes;35 (b) cyclopropanation of a 1,3-diene by complex 3. | |
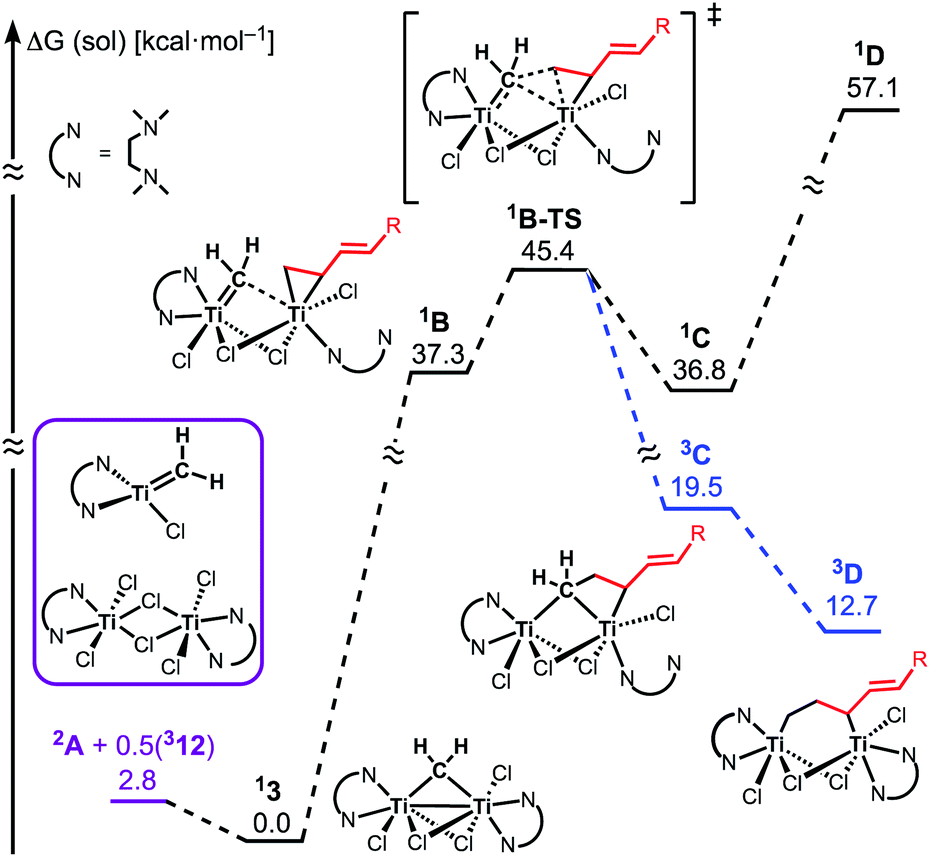
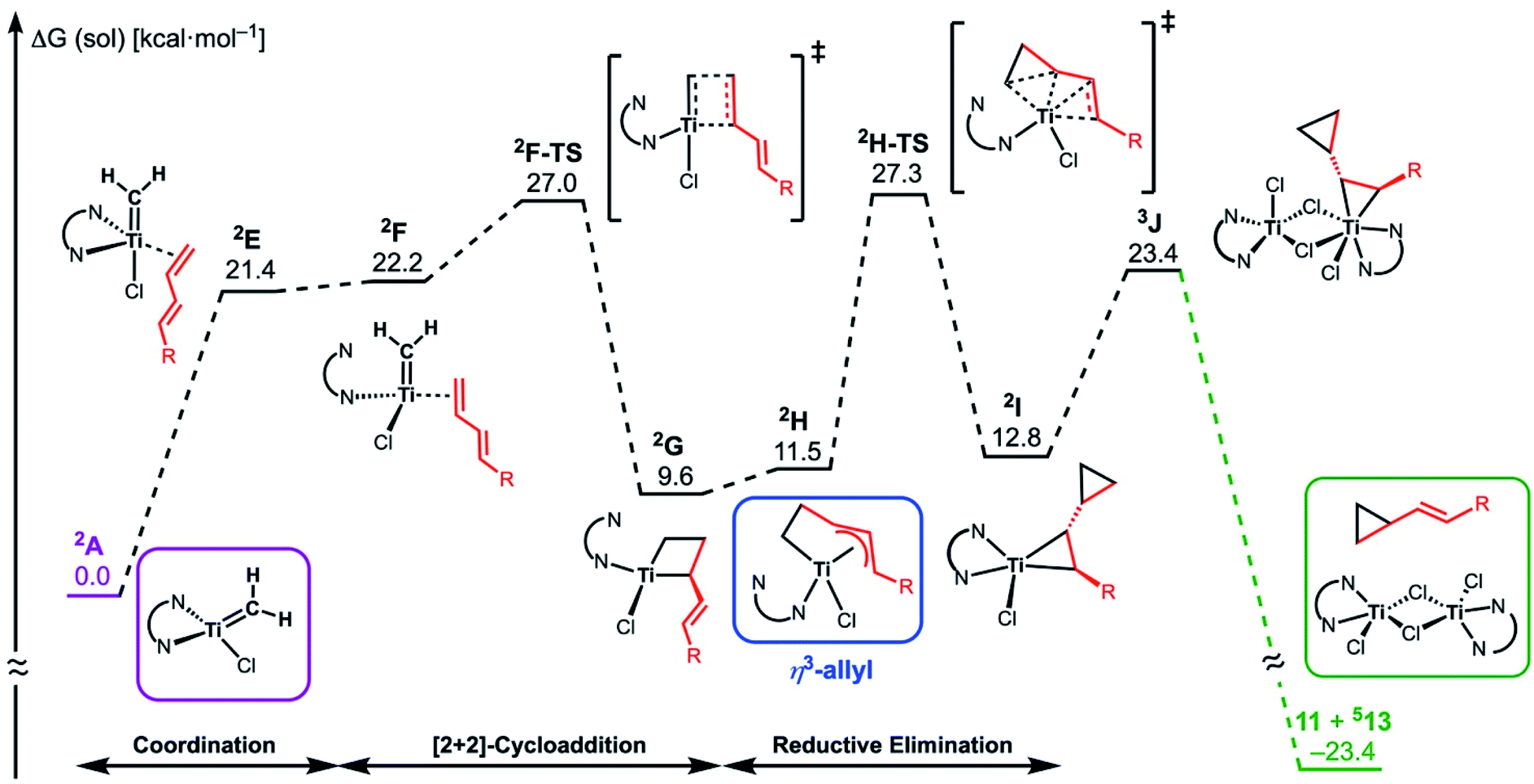
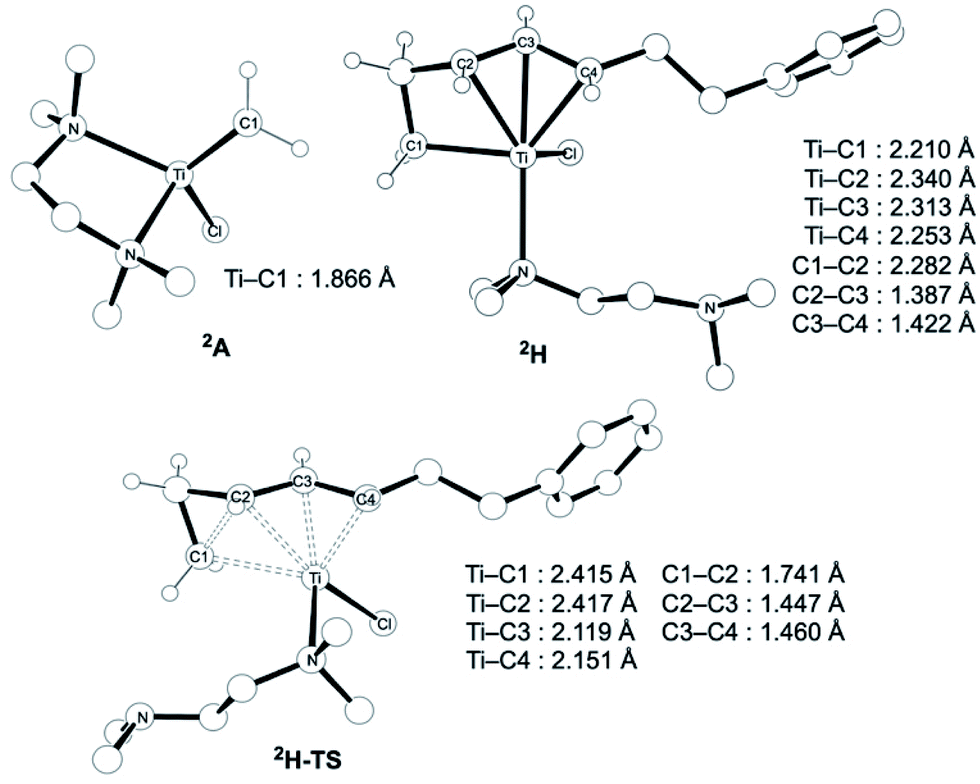
The similar reactivity of our titanium methylene 3 with titanocene alkylidenes raised the question of whether our dinuclear Ti(III)–Ti(III) system remains in the dinuclear structure during the reaction37 or generates a mononuclear titanium methylidene. Unfortunately, experimental observation of the dinuclear or mononuclear titanium methylidene species in the reaction system has not been achieved and we decided to carry out the mechanistic study by quantum chemical calculations based on DFT. The vinylcyclopropanation reaction system by complex 3 and a 1,3-diene was employed to gain insight into the reaction selectivity of cyclopropanation. We first examined whether the 1,3-diene forms a new C–C bond via insertion to the Ti–C bond in the dinuclear complex 3 (Fig. 3) or via [2+2]-cycloaddition with a mononuclear titanium methylidene species [Ti![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2] (A) (Fig. 4). Despite the dinuclear structure of 3 being 2.8 kcal mol−1 lower in relative free energy than generation of A and [TiCl2(tmeda)](μ-Cl)2 (12) (Fig. S14, ESI†), the dinuclear 1,3-diene adduct B, in which the [TiCH2Ti] unit is better described as a semi-bridging titanium methylidene (Ti–CH2: 1.886 Å, 2.546 Å), could be located at 37.3 kcal mol−1 (Fig. 3).38 The transition state of the insertion step (B-TS) was also found with a very high reaction barrier of 45.4 kcal mol−1 leading to dimetallacycle species C and D. In contrast, the mononuclear 1,3-diene adduct E was located at 21.4 kcal mol−1 from A. Upon [2+2]-cycloaddition as illustrated in Fig. 4, the TMEDA ligand changes its coordination mode into a κ1-fashion (F) to traverse the transition state F-TS. The resulted titanacyclobutane intermediate G undergoes hapticity change of a vinyl group on the α-position to transform into an η3-allyl species H. To afford the corresponding cyclopropane 11, reductive elimination from the mononuclear titanium(III) metallacyclobutane should take place, requiring formation of a thermodynamically unfavored titanium(I) species. The π-allylic interaction in H allows reductive ring-closing elimination in H-TS to maintain the titanium(III) character (Fig. 5). As a result, this back-bonding interaction lowers the reaction barrier of the reductive elimination process to give the cyclopropanation product selectively rather than undergoing metathesis or β-H elimination process as in the reaction of terminal olefins.39 Note that a positional isomer of G, β-vinyl titanacyclobutane (G′) given by [2+2]-cycloaddition in the other fashion, is also a conceivable intermediate but cannot form a similar π-allyl configuration due to its strained structure for coordination to the titanium center.40 The mononuclear complex I may bind [TiCl3(tmeda)] to form a dinuclear chloride (J) and then eliminate cyclopropane 11 and a titanium(II) chloride dimer (13) rather than formation of unlikely “TiCl(tmeda)”. Although a mononuclear titanium(II) chloride [TiCl2(tmeda)2] is kinetically stable,30 the titanium(II) chloride dimer [TiCl(tmeda)]2(μ-Cl)2 (13) can readily undergo disproportionation to titanium(III) chloride and some low-valent byproducts.30a,31 In fact, a dinuclear mixed-valent Ti(II)–Ti(III) chloride [TiCl(tmeda)]2(μ-Cl)3 (Fig. S15, ESI†), which has been reported by Gambarotta from thermal decomposition of [TiCl2(tmeda)2],30a was reproducibly observed in our cyclopropanation reaction system.
CH2] (A) (Fig. 4). Despite the dinuclear structure of 3 being 2.8 kcal mol−1 lower in relative free energy than generation of A and [TiCl2(tmeda)](μ-Cl)2 (12) (Fig. S14, ESI†), the dinuclear 1,3-diene adduct B, in which the [TiCH2Ti] unit is better described as a semi-bridging titanium methylidene (Ti–CH2: 1.886 Å, 2.546 Å), could be located at 37.3 kcal mol−1 (Fig. 3).38 The transition state of the insertion step (B-TS) was also found with a very high reaction barrier of 45.4 kcal mol−1 leading to dimetallacycle species C and D. In contrast, the mononuclear 1,3-diene adduct E was located at 21.4 kcal mol−1 from A. Upon [2+2]-cycloaddition as illustrated in Fig. 4, the TMEDA ligand changes its coordination mode into a κ1-fashion (F) to traverse the transition state F-TS. The resulted titanacyclobutane intermediate G undergoes hapticity change of a vinyl group on the α-position to transform into an η3-allyl species H. To afford the corresponding cyclopropane 11, reductive elimination from the mononuclear titanium(III) metallacyclobutane should take place, requiring formation of a thermodynamically unfavored titanium(I) species. The π-allylic interaction in H allows reductive ring-closing elimination in H-TS to maintain the titanium(III) character (Fig. 5). As a result, this back-bonding interaction lowers the reaction barrier of the reductive elimination process to give the cyclopropanation product selectively rather than undergoing metathesis or β-H elimination process as in the reaction of terminal olefins.39 Note that a positional isomer of G, β-vinyl titanacyclobutane (G′) given by [2+2]-cycloaddition in the other fashion, is also a conceivable intermediate but cannot form a similar π-allyl configuration due to its strained structure for coordination to the titanium center.40 The mononuclear complex I may bind [TiCl3(tmeda)] to form a dinuclear chloride (J) and then eliminate cyclopropane 11 and a titanium(II) chloride dimer (13) rather than formation of unlikely “TiCl(tmeda)”. Although a mononuclear titanium(II) chloride [TiCl2(tmeda)2] is kinetically stable,30 the titanium(II) chloride dimer [TiCl(tmeda)]2(μ-Cl)2 (13) can readily undergo disproportionation to titanium(III) chloride and some low-valent byproducts.30a,31 In fact, a dinuclear mixed-valent Ti(II)–Ti(III) chloride [TiCl(tmeda)]2(μ-Cl)3 (Fig. S15, ESI†), which has been reported by Gambarotta from thermal decomposition of [TiCl2(tmeda)2],30a was reproducibly observed in our cyclopropanation reaction system.
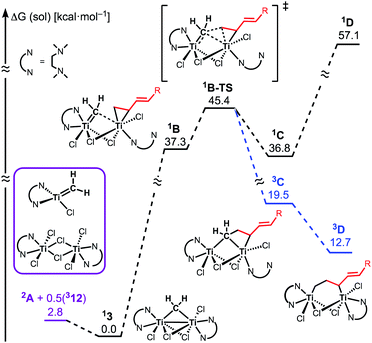 |
| | Fig. 3 Free energy profiles for insertion of 1,3-diene to Ti–CH2. The superscripted number on the left top represents the spin-multiplicity of each titanium species. | |
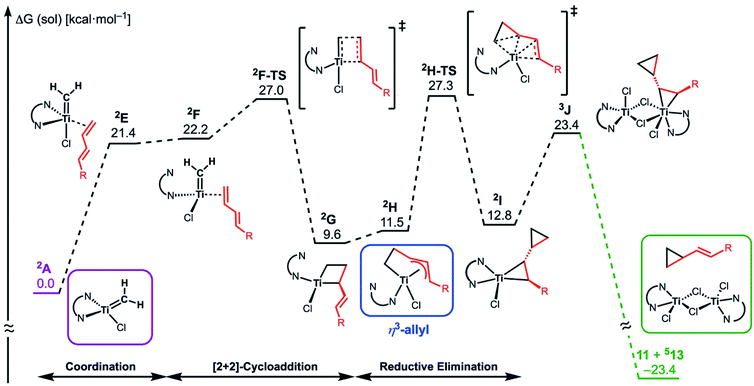 |
| | Fig. 4 Free energy profile for the most plausible pathway of cyclopropanation of 1,3-diene via a mononuclear titanium(III) methylidene (A). The free energy values are presented based on the methylidene species A. The superscripted number on the left top represents the spin-multiplicity of each titanium species. | |
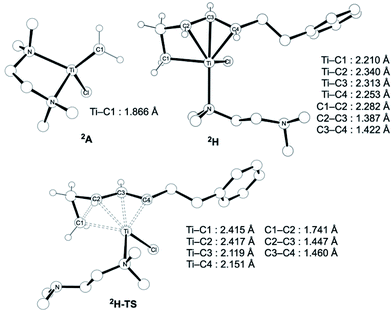 |
| | Fig. 5 Optimized structures of a titanium(III) methylidene species 2A (left top), η3-allyl intermediate 2H, and a transition state of cyclopropanation H-TS (bottom). | |
Conclusions
We have shown a transmetallation pathway between a zinc methylene complex [ZnI(tmeda)]2(μ-CH2) and titanium(III) chloride, resulting in formation of a dinuclear titanium methylene [TiCl(tmeda)]2(μ-CH2)(μ-Cl)2. The solid-state structure showed the first example of a dinuclear Ti(III)–Ti(III) methylene complex. Methylene transfer reactions to ester, terminal olefin and 1,3-diene have been demonstrated. The powerful methylenation reactivity and mechanistic study both propose generation of a titanium(III) methylidene species.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by a Grant-in-Aid for Scientific Research (no. 18H03911) from MEXT, Japan. The DFT calculations were performed at the Research Center for Computational Science, Okazaki, Japan. The authors gratefully thank Prof. Hiroyuki Kawaguchi and Dr Yutaka Ishida (Tokyo Institute of Technology) for analyses of air-sensitive compounds. The authors also thank Ms Nana Yamaji (Okayama University) for work provided on the preliminary study of vinylcyclopropanation.
Notes and references
- F. N. Tebbe, G. W. Parshall and G. S. Reddy, J. Am. Chem. Soc., 1978, 100, 3611–3613 CrossRef CAS.
- J. Scott and D. J. Mindiola, Dalton Trans., 2009, 8463–8472 RSC.
-
(a) R. Beckhaus, Angew. Chem., Int. Et. Engl., 1997, 36, 686–713 CrossRef;
(b) R. C. Hartley and G. J. McKiernan, J. Chem. Soc., Perkin Trans. 1, 2002, 2763–2793 RSC;
(c) R. C. Hartley, J. Li, C. A. Main and G. J. McKiernan, Tetrahedron, 2007, 63, 4825–4864 CrossRef.
-
(a) S. H. Pine, R. Zahler, D. A. Evans and R. H. Grubbs, J. Am. Chem. Soc., 1980, 102, 3270–3272 CrossRef CAS;
(b) L. Clawson, S. L. Buchwald and R. H. Grubbs, Tetrahedron Lett., 1984, 25, 5733–5736 CrossRef CAS;
(c) L. F. Cannizzo and R. H. Grubbs, J. Org. Chem., 1985, 50, 2386–2387 CrossRef CAS;
(d) S. H. Pine, R. J. Pettit, G. D. Geib, S. G. Cruz, C. H. Gallego, T. Tijerina and R. D. Pine, J. Org. Chem., 1985, 50, 1212–1216 CrossRef CAS.
- C. McDade, J. C. Green and J. E. Bercaw, Organometallics, 1982, 1, 1629–1634 CrossRef CAS.
-
(a) F. N. Tebbe, G. W. Parshall and D. W. Ovenall, J. Am. Chem. Soc., 1979, 101, 5074–5075 CrossRef CAS;
(b) T. R. Howard, J. B. Lee and R. H. Grubbs, J. Am. Chem. Soc., 1980, 102, 6876–6878 CrossRef CAS.
-
(a) K. J. Ivin, J. J. Rooney, C. D. Stewart, M. L. H. Green and R. Mehtab, J. Chem. Soc., Chem. Commun., 1978, 604–606 RSC;
(b) L. R. Gilliom and R. H. Grubbs, J. Am. Chem. Soc., 1986, 108, 733–742 CrossRef CAS;
(c) N. A. Petasis and D.-K. Fu, J. Am. Chem. Soc., 1993, 115, 7208–7214 CrossRef CAS.
- K. Takai, Y. Hotta, K. Oshima and H. Nozaki, Tetrahedron Lett., 1978, 27, 2417–2420 CrossRef.
-
(a) L. Lombardo, Tetrahedron Lett., 1982, 23, 4293–4296 CrossRef CAS;
(b) L. Lombardo, Org. Synth., 1987, 65, 81 CrossRef CAS.
- K. Takai, T. Kakiuchi and K. Utimoto, J. Org. Chem., 1994, 59, 2671–2673 CrossRef CAS.
- K. Takai, T. Kakiuchi, Y. Kataoka and K. Utimoto, J. Org. Chem., 1994, 59, 2668–2670 CrossRef CAS.
- T. Okazoe, K. Takai, K. Oshima and K. Utimoto, J. Org. Chem., 1987, 52, 4410–4412 CrossRef CAS.
-
(a) B. M. Johnson and K. P. C. Vollhardt, Synlett, 1990, 209–210 CrossRef CAS;
(b) O. Fujimura, G. C. Fu and R. H. Grubbs, J. Org. Chem., 1994, 59, 4029–4031 CrossRef CAS;
(c) R.-A. Fallahpour and H. J. Hansen, Helv. Chim. Acta, 1994, 77, 2297–2302 CrossRef CAS;
(d) K. A. Tony, R. W. Denton, A. Dilhas, J. Jiménez-Barbero and D. R. Mootoo, Org. Lett., 2007, 9, 1441–1444 CrossRef CAS;
(e) U. Majumder and J. D. Rainier, Tetrahedron Lett., 2005, 46, 7209–7211 CrossRef CAS;
(f) K. Iyer and J. D. Rainier, J. Am. Chem. Soc., 2007, 129, 12604–12605 CrossRef CAS.
-
L. N. Nysted, US Pat., 3865848, Chem. Abstr., 1975, 83, 10406q Search PubMed.
-
(a) J. J. Eisch and A. Piotrowski, Tetrahedron Lett., 1983, 24, 2043–2046 CrossRef CAS;
(b) C. Aïssa, R. Riveiros, J. Ragot and A. Fürstner, J. Am. Chem. Soc., 2003, 125, 15512–15520 CrossRef;
(c) A. Haahr, Z. Rankovic and R. C. Hartley, Tetrahedron Lett., 2011, 52, 3020–3022 CrossRef CAS;
(d) W. E. Noland, C. L. Etienne and N. P. Lanzatella, J. Heterocycl. Chem., 2011, 48, 381–388 CrossRef CAS;
(e) B. Barnych, B. Fenet and J.-M. Vatèle, Tetrahedron, 2013, 69, 334–340 CrossRef CAS;
(f) D. Tymann, U. Bednarzick, L. Iovkova-Berends and M. Hiersemann, Org. Lett., 2018, 20, 4072–4076 CrossRef CAS.
-
(a) K. Takai, Y. Hotta, K. Oshima and H. Nozaki, Bull. Chem. Soc. Jpn., 1980, 53, 1698–1702 CrossRef CAS;
(b) J. Hibino, T. Okazoe, K. Takai and H. Nozaki, Tetrahedron Lett., 1985, 26, 5579–5580 CrossRef CAS;
(c) T. Okazoe, J. Hibino, K. Takai and H. Nozaki, Tetrahedron Lett., 1985, 26, 5581–5584 CrossRef CAS.
-
(a) P. Turnbull, K. Syhora and J. H. Fried, J. Am. Chem. Soc., 1966, 88, 4764–4766 CrossRef CAS;
(b) H. Hashimoto, M. Hida and S. Miyano, J. Organomet. Chem., 1967, 10, 518–520 CrossRef CAS;
(c) I. T. Harrison, R. J. Rawson, P. Turnbull and J. H. Fried, J. Org. Chem., 1971, 36, 3515–3517 CrossRef CAS;
(d) K. Utimoto, N. Toda, T. Mizuno, M. Kobata and S. Matsubara, Angew. Chem., Int. Ed. Engl., 1997, 36, 2804–2805 CrossRef CAS;
(e) S. Matsubara, K. Oshima and K. Utimoto, J. Organomet. Chem., 2001, 617–618, 39–46 CrossRef CAS;
(f) M. Sada, M. Uchiyama and S. Matsubara, Synlett, 2014, 25, 2831–2841 CrossRef CAS and references cited therein.
-
(a) T. Oshiki, T. Kiriyama, K. Tsuchida and K. Takai, Chem. Lett., 2000, 29, 334–335 CrossRef;
(b) V. Bjornstad and K. Undheim, Synthesis, 2007, 235–238 CAS.
- N. A. Petasis and E. I. Bzowej, J. Am. Chem. Soc., 1990, 112, 6392–6394 CrossRef CAS.
- Y. Nishida, N. Hosokawa, M. Murai and K. Takai, J. Am. Chem. Soc., 2015, 137, 114–117 CrossRef CAS.
- See ESI†.
-
(a) L. Scoles, R. Minhas, R. Duchateau, J. Jubb and S. Gambarotta, Organometallics, 1994, 13, 4978–4983 CrossRef CAS;
(b) O. Buitrago, C. R. de Arellano, G. Jiménez and T. Cuenca, Organometallics, 2004, 23, 5873–5876 CrossRef CAS;
(c) J. J. Carbó, O. G. Moral, A. Martín, M. Mena, J.-M. Poblet and C. Satamaría, Chem. Eur. J., 2008, 14, 7930–7938 CrossRef;
(d) J. J. Carbó, O. G. Moral, A. Martín, M. Mena, J.-M. Poblet and C. Satamaría, Eur. J. Inorg. Chem., 2009, 643–653 CrossRef;
(e) S. Liu, A. M. Invergo, J. P. Mclnnis, A. R. Mouat, A. Motta, T. L. Lohr, M. Delferro and T. J. Marks, Organometallics, 2017, 36, 4403–4421 CrossRef CAS.
-
(a) T. Kurogi, P. J. Carroll and D. J. Mindiola, Chem. Commun., 2017, 53, 3412–3414 RSC;
(b) L. N. Grant, S. Ahn, B. C. Manor, M.-H. Baik and D. J. Mindiola, Chem. Commun., 2017, 53, 3415–3417 RSC.
-
(a) R. Thompson, E. Nakamura-Ogiso, C.-H. Chen, M. Pink and D. J. Mindiola, Organometallics, 2014, 33, 429–432 CrossRef CAS;
(b) V. M. Birkelbach, F. Kracht, H. M. Dietrich, C. Stuhl, C. Maichle-Mössmer and R. Anwander, Organometallics, 2020, 39, 3490–3504 CrossRef CAS.
- S. S. Batsanov, Inorg. Mater., 2001, 37, 871–885 CrossRef CAS.
-
(a) M. Murai, R. Taniguchi, N. Hosokawa, Y. Nishida, H. Mimachi, T. Oshiki and K. Takai, J. Am. Chem. Soc., 2017, 139, 13184–13192 CrossRef CAS;
(b) M. Murai, R. Taniguchi, C. Mizuta and K. Takai, Org. Lett., 2019, 21, 2668–2672 CrossRef CAS.
- According to ref. 20, formation of the hydrolysis product 5 was still observed under anhydrous reaction conditions. Ketone 5 could also be formed by β-alkoxo elimination of the oxa-titanacycle intermediate.
-
(a) D. A. Straus and R. H. Grubbs, Organometallics, 1982, 1, 1658–1661 CrossRef CAS;
(b) J. D. Meinhart, E. V. Anslyn and R. H. Grubbs, Organometallics, 1989, 8, 583–589 CrossRef CAS.
-
(a) D. C. L. Perkins, R. J. Puddephatt, M. C. Rendle and C. F. H. Tipper, J. Organomet. Chem., 1980, 195, 105–112 CrossRef CAS;
(b) E. J. Parsons and P. W. Jennings, J. Am. Chem. Soc., 1987, 109, 3973–3977 CrossRef CAS;
(c) T. W. Hanks and P. W. Jennings, J. Am. Chem. Soc., 1987, 109, 5023–5025 CrossRef CAS;
(d) A. Miyashita, M. Ohyoshi, H. Shitara and H. Nohira, J. Organomet. Chem., 1980, 338, 103–111 CrossRef;
(e) E. Lindner, R.-M. Jansen, W. Hiller and R. Fawzi, Chem. Ber., 1989, 122, 1403–1409 CrossRef CAS;
(f) K. D. Kitiachvili, D. J. Mindiola and G. L. Hillhouse, J. Am. Chem. Soc., 2004, 126, 10554–10555 CrossRef CAS.
-
(a) J. J. H. Edema, R. Duchateau, S. Gambarotta, R. Hynes and E. Gabe, Inorg. Chem., 1991, 30, 154–156 CrossRef CAS;
(b) E. M. Zolnhofer, G. B. Wijeratne, T. A. Jackson, S. Fortier, F. W. Heinemann, K. Meyer, J. Krzystek, A. Ozarowski, D. J. Mindiola and J. Telser, Inorg. Chem., 2020, 59, 6187–6201 CrossRef CAS.
- G. B. Wijeratne, E. M. Zolnhofer, S. Fortier, L. N. Grant, P. J. Carroll, C.-H. Chen, K. Meyer, J. Krzystek, A. Ozarowski, T. A. Jackson, D. J. Mindiola and J. Telser, Inorg. Chem., 2015, 54, 10380–10397 CrossRef CAS.
- S. C. H. Ho, D. A. Straus and R. H. Grubbs, J. Am. Chem. Soc., 1984, 106, 1533–1534 CrossRef CAS.
- P. B. Mackenzie, R. J. Coots and R. H. Grubbs, Organometallics, 1989, 8, 8–14 CrossRef CAS.
-
(a) U. Majumder and J. D. Rainier, Tetrahedron Lett., 2005, 46, 7209–7211 CrossRef CAS;
(b) H. W. B. Johnson, U. Majumder and J. D. Rainier, J. Am. Chem. Soc., 2005, 127, 848–849 CrossRef CAS.
- Y. Horikawa, T. Nomura, M. Watanabe, T. Fujiwara and T. Takeda, J. Org. Chem., 1997, 62, 3678–3682 CrossRef CAS.
- A. Tsubouchi, E. Nishio, Y. Kato, T. Fujiwara and T. Takeda, Tetrahedron Lett., 2002, 43, 5755–5758 CrossRef CAS.
-
(a) M. R. Berman, P. B. Comita, C. B. Moore and R. G. Bergman, J. Am. Chem. Soc., 1980, 102, 5694–5695 CrossRef;
(b) M. Cooke, N. J. Forrow and S. A. R. Knox, J. Chem. Soc., Dalton Trans., 1983, 2435–2440 RSC.
- Geometrical optimization of the 1,3-diene adduct based on 1B in triplet (3B) could not be converged. Instead, dissociation of 1,3-diene from the titanium center was observed probably due to a weak interaction of 1,3-diene without back-donation from the titanium center.
- The corresponding transition state in quartet (4H-TS) was located at ΔG = 40.9 kcal·mol–1.
- Geometrical optimization of a modeled π-allylic titanacyclobutane species with the β-vinyl group led to the non-coordinate β-vinyl titanacyclobutane.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2045300–2045305. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc06366e |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *,
Kaito
Kuroki
*,
Kaito
Kuroki