
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA tailored phosphoaspartate probe unravels CprR as a response regulator in Pseudomonas aeruginosa interkingdom signaling†
Patrick W. A.
Allihn
 a,
Mathias W.
Hackl
a,
Christina
Ludwig
b,
Stephan M.
Hacker
*c and
Stephan A.
Sieber
*a
a,
Mathias W.
Hackl
a,
Christina
Ludwig
b,
Stephan M.
Hacker
*c and
Stephan A.
Sieber
*a
aTUM Center for Functional Protein Assemblies (CPA), Department of Chemistry and Chair of Organic Chemistry II, Technical University of Munich, Lichtenbergstraße 4, 85748 Garching, Germany. E-mail: stephan.sieber@tum.de
bBavarian Center for Biomolecular Mass Spectrometry (BayBioMS), Technical University of Munich, 85354 Freising, Germany. E-mail: tina.ludwig@tum.de
cDepartment of Chemistry, Technical University of Munich, 85748 Garching, Germany. E-mail: stephan.m.hacker@tum.de
First published on 8th February 2021
Abstract
Pseudomonas aeruginosa is a difficult-to-treat Gram-negative bacterial pathogen causing life-threatening infections. Adaptive resistance (AR) to cationic peptide antibiotics such as polymyxin B impairs the therapeutic success. This self-protection is mediated by two component systems (TCSs) consisting of a membrane-bound histidine kinase and an intracellular response regulator (RR). As phosphorylation of the key RR aspartate residue is transient during signaling and hydrolytically unstable, the study of these systems is challenging. Here, we apply a tailored reverse polarity chemical proteomic strategy to capture this transient modification and read-out RR phosphorylation in complex proteomes using a nucleophilic probe. In-depth mechanistic insights into an ideal trapping strategy were performed with a recombinant RR demonstrating the importance of fine-tuned acidic pH values to facilitate the attack on the aspartate carbonyl C-atom and prevent unproductive hydrolysis. Analysis of Bacillus subtilis and P. aeruginosa proteomes revealed the detection of multiple annotated phosphoaspartate (pAsp) sites of known RRs in addition to many new potential pAsp sites. With this validated strategy we dissected the signaling of dynorphin A, a human peptide stress hormone, which is sensed by P. aeruginosa to prepare AR. Intriguingly, our methodology identified CprR as an unprecedented RR in dynorphin A interkingdom signaling.
Introduction
Pseudomonas aeruginosa, a Gram-negative bacterial pathogen, is listed by the WHO as a critical priority for the development of novel antibiotics to fight multiresistance.1 Its intricate pathogenesis mechanisms cause severe diseases such as pneumonia in cystic fibrosis patients.2,3 These mechanisms are controlled by elaborate signaling mechanisms, which coordinate population density (quorum sensing), virulence and antibiotic defense.4,5P. aeruginosa was recently shown to sense the human peptide hormone dynorphin, which is secreted by eukaryotic cells upon stress e.g. induced by bacterial infections.6 While dynorphin only exhibits moderate antibiotic activity, other cationic antimicrobial peptides, produced in response to the infection, are able to destroy the intruding pathogen.7 Recently, chemical proteomics revealed that dynorphin A is sensed by the P. aeruginosa two-component system ParRS, which leads to upregulation of the antimicrobial peptide response mediated by the ArnBCADTEF system.8 This interkingdom signaling between a human stress hormone and P. aeruginosa confers a competitive advantage to the pathogen, which listens in on the human defense mechanism and thereby prepares itself for a forthcoming attack.
ParR and ParS belong to the prevalent group of two-component gene regulatory systems (TCS) with crucial roles for the physiology and pathogenicity of bacteria (Fig. 1a).9 They are composed of a membrane-bound histidine kinase (HK) and a response regulator (RR). Signal transduction occurs via sensing of external stimuli and signal relay to the kinase domain, which induces autophosphorylation of a histidine residue. In a second step the phosphate group is shuttled from the histidine to a conserved aspartate of the RR yielding phosphoaspartate (pAsp) and inducing further downstream signaling.
 | ||
| Fig. 1 Architecture of two-component system signaling and chemical trapping of pAsp. (a) Upon an external signal, the membrane-bound histidine kinase is autophosphorylated on a conserved His residue (1), followed by phosphotransfer to a conserved Asp residue of its cognate response regulator (2). Upon a conformational change, the response regulator binds to target genes and regulates their expression (3). (b) Hydroxylamine-based probes attack either the carbonyl-C (pH = 4) or the phosphate-P-atom (pH = 7) of pAsp. (c) Probes used for RP-ABPP for the detection of pAsp by Chang et al. (DBHA)14 and in this study (HA-yne). | ||
TCSs facilitate rapid signaling events such as driving motility changes in chemotaxis,10 expression of virulence factors in quorum sensing6 and triggering adaptive resistance.11 In contrast to the chemically stable phosphorylation of serine, threonine and tyrosine in eukaryotic cells, the prokaryotic pAsp is a mixed carboxylic acid–phosphoric acid anhydride (acyl phosphate), which has limited half-life under neutral, acidic and alkaline conditions12 impeding its detection via common analytical methods such as immobilized metal affinity chromatography (IMAC) followed by liquid chromatography coupled to tandem MS (LC-MS/MS). Thus, a chemical methodology is required, which rapidly transforms the labile acyl phosphate into a stable modification that can be detected using standard proteomic workflows. Here, α-effect nucleophiles such as hydroxylamine capture the labile modification yielding stable N-hydroxy-asparagine derivatives (Fig. 1b).13 This reaction requires slightly acidic conditions in order to promote a selective attack at the carbonyl C-atom, while neutral conditions favor phosphate cleavage by attack on the P-atom.12,15 To expand the chemical proteomic toolbox for monitoring pAsp modifications, Chang et al. recently introduced a desthiobiotin containing hydroxylamine (DBHA) probe facilitating the profiling of pAsp in Escherichia coli proteomes under neutral pH (Fig. 1c).14
We here provide in-depth molecular insights into the preferred nucleophilic attack of hydroxylamine probes under different pH values and demonstrate the importance of acidic conditions along with solubilizing detergents for selective modification of the aspartate carboxylate during the reverse polarity activity based profiling (RP-ABPP) approach.16 Moreover, our study accounts for the reactivity toward other electrophilic residues and elucidates their overall impact on model proteins. The tailored conditions of these model studies were integrated into an advanced chemical proteomic platform featuring a minimal clickable17 hydroxylamine alkyne probe (HA-yne) with access to sterically demanding protein pockets (Fig. 1c) as well as the application of isotopically labeled desthiobiotin azide (isoDTB) tags18 for the detection and quantification of modified sites. This methodology revealed 123 HA-yne modified aspartate sites with high fidelity. These fine-tuned conditions enabled the surprising discovery of dynorphin A-mediated phosphorylation of the RR CprR, a so far unknown target in interkingdom signaling.
Results
Acyl phosphates are trapped as stable hydroxamates under optimized acidic conditions
In this work, we aimed to decipher the role of dynorphin A in interkingdom signaling by unraveling its RRs in P. aeruginosa. Prior to these studies, we refined the nucleophilic profiling platform to become most efficient for this need. As previous work did not focus on the molecular basis of the nucleophile reaction with acyl phosphates within proteins, we initiated this study with a well-established model RR from E. coli termed PhoB. PhoB is a key regulator in E. coli phosphate metabolism, which is phosphorylated at Asp53 by the sensor histidine kinase PhoR to initiate transcription of its client genes.19–21E. coli PhoB was recombinantly overexpressed, purified and phosphorylated at Asp53 in vitro with acetyl phosphate as a phosphor donor.21,22 Intact protein MS (IPMS) confirmed the addition of one phosphate group to PhoB, while a corresponding aspartate-to-asparagine (D53N) mutation prevented this modification demonstrating site specificity (Fig. 2 and S1, ESI†). To probe the conversion of phosphorylated D53 with a strong α-effect nucleophile, we treated phosphorylated PhoB with sterically unhindered hydroxylamine at previously established pH = 7 (final concentration of NH2OH HCl = 0.5 M) in a time-dependent intact protein MS assay. However, this experiment resulted in a lack of hydroxylamine labeling in line with the favored attack at the phosphorous atom eventually leading to the unmodified aspartate residue. (Fig. S2, ESI†).15,23 In order to mediate an attack at the carbonyl C-atom, we conducted the same experiment at acidic pH (pH = 4)15,23 and a rapid modification to N4-hydroxyasparagine was observed within minutes with quantitative conversion after 30 min (Fig. 2b and S3a, ESI†). As expected, unphosphorylated PhoB did not show any mass change (Fig. S4, ESI†). We thus conclude that acidic pH is crucial for productive acyl phosphate trapping and maximizing labeling efficiency. | ||
| Fig. 2 Intact-protein mass spectrometric (IPMS) assay for the in vitro phosphorylation and chemical trapping of pAsp of model response regulator PhoB of E. coli. (a) PhoB (green) was phosphorylated in vitro with acetyl phosphate (red) and converted with 500 mM hydroxylamine or HA-yne at pH = 4 (blue). (b and c) IPMS assay results using hydroxylamine (b) and HA-yne (c). In vitro phosphorylation yielded around 50% pPhoB, which was quantitatively converted with both nucleophiles. | ||
Prior to the analysis of pAsp modifications in complex biological samples, the sensitivity of detection needed to be enhanced by application of an enrichment strategy. In order to minimize undesired steric clashes within protein pockets, we synthesized a minimal hydroxylamine probe linked to an alkyne handle (HA-yne), which was previously used for oxime ligations (Fig. 1c).24 Once its reaction with pAsp is complete, desthiobiotin azide can be clicked to the alkyne functional group facilitating subsequent affinity enrichment. Furthermore, the modular nature of the alkyne handle has the advantage that it allows attachment of various labels for different detection methods like fluorophores for gel-based analysis or isoDTB tags18 for quantitative proteomics. Similar to hydroxylamine, the HA-yne probe led to rapid conversion of phosphorylated PhoB under the set conditions of pH = 4 (Fig. 2c and S3b, ESI†).
For the application of the probe in whole proteomes, it is crucial to trap the transient acyl phosphate modifications rapidly upon cell lysis. We thus added the probe directly to the lysis buffer (pH = 4). Labeling of B. subtilis revealed an almost quantitative protein precipitation attributed to the high probe concentration at acidic labeling conditions (Fig. S5, ESI†). A screen of diverse detergents demonstrated that the addition of 1% (w/v) lauryldimethylamine oxide (LDAO) maintains protein solubility (Fig. S6, ESI†). Labeling under these conditions, acidic pH and LDAO, resulted in the detection of strong fluorescent bands upon clicking the probe to rhodamine azide and fluorescent SDS-PAGE analysis. Sufficient labeling intensity was observed at high concentrations starting at 125 mM HA-yne (Fig. S7, ESI†), which we set as ideal parameters for subsequent LC-MS/MS studies. We next spiked HA-yne labeled PhoB into the labeled proteome, clicked to commercially available desthiobiotin azide and upon streptavidin bead enrichment, proteolytic digestion and elution from the beads, the peptides were subjected to LC-MS/MS analysis. Our chemoproteomic workflow successfully identified the HA-yne modification at the expected residue D53 of PhoB (Fig. 3).
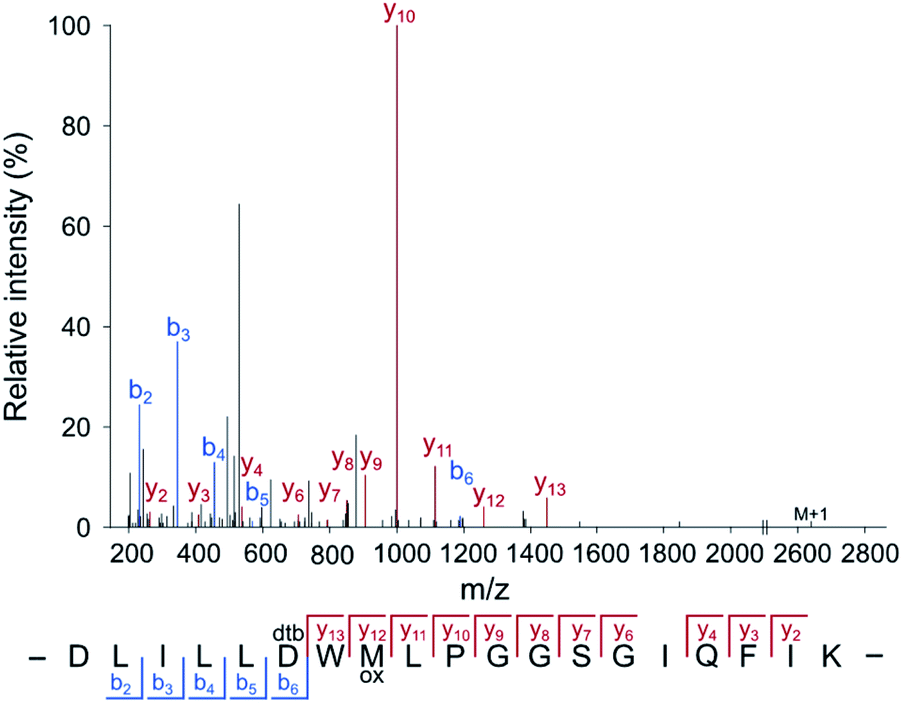 | ||
| Fig. 3 Annotated MS2 spectrum of the HA-yne-modified PhoB peptide. For RP-ABPP method development, HA-yne modified E. coli PhoB (Fig. 2c) was spiked into a labeled bacterial proteome, clicked to desthiobiotin-azide, enriched, digested and subjected to LC-MS/MS analysis. The predicted pAsp peptide was identified with the modification at the annotated position (D53) using MaxQuant software.25 | ||
A hydroxylamine-alkyne probe reveals modified aspartate sites at conserved response regulator motifs in bacterial proteomes
With optimized conditions for proteome labeling in hand, we commenced with the analysis of modification sites in B. subtilis and P. aeruginosa proteomes. In brief, following HA-yne treatment during cell lysis, the proteome was clicked to desthiobiotin azide, digested and peptides were enriched on streptavidin beads followed by LC-MS/MS analysis (Fig. 4a and S8, ESI†). Overall, we detected 141 and 198 modified Asp sites with high fidelity in B. subtilis and P. aeruginosa, respectively, which includes 15% (6/41) and 19% (18/93) of known pAsp sites from UniProt (Fig. 4b, d and Table S1, ESI†).28 Moreover, within pAsp annotated proteins the expected pAsp residue was modified almost exclusively (24/25). Within this dataset, so far only poorly characterized proteins, like the probable response regulators PA2798 and PA1243, were detected with modification at the expected residues confirming these pAsp sites. Hence, our method verifies many phosphoaspartate sites experimentally and represents a complementary strategy to the annotation by sequence similarity. Furthermore, many new potential pAsp sites were detected. An overlay of all identified sequences in P. aeruginosa revealed distinct similarities with the pAsp motif of known RRs (Fig. 4c). Nevertheless, the motifs were not identical and for B. subtilis (Fig. 4e) the resemblance was rather weak pointing to the fact that we also detect many modifications that do not reside in previously known sequence motifs. In P. aeruginosa, positions −5 (D), +3 (M), +4 (P), +6 (M) and +8 (G) show a distinct similarity. Accordingly, signal transduction RR domains were among the top enriched protein domains (Fig. S9, ESI†). The sequence motif enrichment of this refined methodology is more pronounced compared to the previous study demonstrating the general utility of the approach in acyl phosphate trapping.14 As we cannot a priori exclude the capture of other electrophilic modifications such as ADP-ribosylation,29 glutamate methylation30 and transamidation31 we extended our analysis to all amino acids as possible modification sites. To our surprise we obtained significant labeling at Glu, Asn and Gln which cumulated in about 6000 additional sites, foremost on Gln, in both proteomes (Fig. S10 and Table S1, ESI†). Regarding Glu modified peptides in both organisms, positions −1 (V), +1 (V) and +2 (D) show significant preferences. Interestingly, in P. aeruginosa, the sequence around the modified residue for Glu, Asn, Gln shows a significant enrichment of lysine residues. Furthermore, Asn and Gln modified sequences also show a preference for acidic residues, which is even more pronounced in B. subtilis (Fig. S11, ESI†). As previous work did not account for modification sites other than Asp, we further investigated if the applied labeling conditions could be responsible for this effect. It is known that Asn and Gln containing peptides can form succinimide as well as glutarimide intermediates which could be attacked by nucleophiles resulting in ring opening and covalent bond formation (Fig. S12, ESI†).32–35 Although this deamidation reaction may only occur to a minor extent, the MS workflow would still be sensitive enough for its detection. We thus performed a quantitative analysis on test proteins, α-Casein, BSA and phosphorylated PhoB, which we treated with the probe under labeling conditions and determined the extent of probe addition on each protein. Interestingly, while phosphorylated D53 on PhoB was labeled with near quantitative conversion, modification of unphosphorylated proteins was not detected (Fig. S13, ESI†). As we do not detect strong modification of any of the unmodified proteins, we can at this stage not fully explain the reactivity at Glu, Asn and Gln in the proteome. As we identified a specific sequence motif for these sites (Fig. S11, ESI†), it might be that this protein sequence leads to an unidentified electrophilic acyl modification. Nevertheless, it is encouraging that, while in general modified sites of predominantly high abundance proteins are detected (Fig. S14, ESI†), annotated pAsp sites are also detected in low abundance proteins. In line with our initial goal, we thus recommend utilizing this strategy for detection of Asp phosphorylation. | ||
| Fig. 4 RP-ABPP workflow and binding site analysis. (a) Bacterial cells of P. aeruginosa and B. subtilis were lysed in a buffer containing the nucleophilic HA-yne probe (125 mM) at pH = 4 and clicked to desthiobiotin-azide (DTB-PEG3-N3). Modified proteins were tryptically digested, enriched on streptavidin beads, eluted from the beads and subjected to LC-MS/MS analysis. (b and d) Table of HA-yne modified sites in P. aeruginosa and B. subtilis, that also have UniProt annotated pAsp sites. Additionally, the corresponding genes, TCSs and their implications are listed. For the complete list of modified sites see Table S1 (ESI†). Only sites with an Andromeda26 localization probability exceeding 75% for the relative HA-yne modified residue were included in the analysis using MaxQuant software.25 (c and e) Comparison of pAsp annotated and HA-yne modified sequence motifs in P. aeruginosa and B. subtilis using pLogo.27 Residues at positions ranging from −10 to +10 next to the modification site were included in the analysis. pAsp annotated and HA-yne modified sequences (fg) were compared with the complete proteomic background (bg) in P. aeruginosa or B. subtilis from the UniProt database.28 Red horizontal bars indicate the Bonferroni-corrected statistical significance (p = 0.05). | ||
Dynorphin A exposure uncovers CprR as a response regulator in P. aeruginosa interkingdom signaling
The refined and validated workflow was finally utilized for the investigation of downstream dynorphin A signaling in P. aeruginosa. Dynorphin A treatment was performed applying conditions of our previously established AfBPP method for target identification of a dynorphin A photoprobe.8 Cells were treated with either dynorphin A or DMSO for either 1, 5 or 15 min followed by HA-yne addition during lysis. The shorter dynorphin A treatment durations compared to our AfBPP method (30 min) were used in order to enable the capture of transient phosphoaspartate modifications for fast dynorphin A induced signaling events. Proteomes were clicked to desthiobiotin azide, digested and peptides enriched on streptavidin beads followed by LC-MS/MS analysis to unravel their identity and site of modification. Overall, 103 vs. 113 modified Asp sites were detected in dynorphin A treated and untreated samples, respectively (Table S1, ESI†). Importantly, among the annotated pAsp sites only one residue, D53 in the protein assigned as cationic peptide response regulator (CprR), was solely present in the dynorphin A treated sample at all time points. While we cannot directly prove phosphorylation of D53 of CprR using our method as other electrophilic modifications would lead to the same readout, the detected peptide was modified at the predicted position of the well-known motif and, therefore, phosphorylation of CprR at D53 is firmly assumed to be the modification providing the electrophilic acyl group for HA-yne conversion. CprR and its cognate sensor kinase CprS belong to a family of TCSs, which sense environmental signals such as antibiotics to activate the expression of the arn lipopolysaccharide (LPS) modification operon.36 The corresponding modification of negatively charged LPS with positively charged arabinosamine reduces cationic peptide binding and facilitates adaptive resistance. Similar to the ParRS mediated arn activation via dynorphin A sensing, CprRS has been shown to respond to cationic peptide antibiotics such as polymyxin B.36To further boost sensitivity and enable direct quantification of sites, we applied the recently introduced isoDTB tags,18 utilizing heavy and light isotopes incorporated in the linker of the desthiobiotin azide. Their use allows a direct, quantitative comparison of pooled samples treated with dynorphin A or the control, respectively (Fig. 5a). Intriguingly, this experiment revealed D53 of CprR with the highest intensity ratio (dynorphin A treated vs. untreated = 16.2) among all detected modification sites (Fig. 5b, S15 and Table S1, ESI†). To directly compare our findings to those from our previous AfBPP study, we repeated the experiment with 30 min dynorphin A treatment, which resulted in a comparable CprR ratio of 12.0. (Fig. S16a, b and Table S1, ESI†).
 | ||
| Fig. 5 RP-isoDTB workflow for quantitative MS1 and MS2 (PRM) analysis. (a) Intact cells of P. aeruginosa were treated with DMSO or 10 μM dynorphin A (Dyn), lysed in a buffer containing the nucleophilic HA-yne probe (125 mM) at pH 4 and clicked to the isoDTB tags. Modified proteins were combined, tryptically digested, enriched on streptavidin beads, eluted from the beads and subjected to LC-MS/MS analysis. (b) Waterfall plot representing the ratio between dynorphin A (light) and DMSO (heavy) treated HA-yne modified Asp and Glu residues. Red dots indicate sites, that are also annotated as pAsp sites in UniProt. (c) PRM transitions (Dyn/light vs. DMSO/heavy) of pAsp annotated and HA-yne modified peptides of response regulators CprR and ParR. Data was analyzed using the Skyline software.37 MS2 ratios of 20.8 and 2.0 were obtained for CprR and ParR, respectively, unraveling CprR as the only protein with highly enhanced modification. | ||
ParR, the cognate RR of the previously identified dynorphin A target sensor histidine kinase ParS, was solely detected upon dynorphin A treatment so that no ratio could be obtained. For a closer inspection of the ratios we performed parallel reaction monitoring (PRM) measurements with precursors for peptides containing D53 of CprR and D57 of ParR in the inclusion list for MS2 fragmentation during the whole chromatographic run (Table S2, ESI†). This method enabled the detection of the two corresponding modified peptides and revealed a ratio of 2.0 for ParR and a higher ratio for CprR of 20.8 (Fig. 5c and Table S2, ESI†). Again, 30 min dynorphin A treatment yielded a similar outcome with ratios of 19.4 and 2.8 for CprR and ParR, respectively (Fig. S16c and Table S2, ESI†). These results suggest, that under the given conditions, dynorphin A elicits a much stronger response via CprR as compared to ParR, highlighting the importance to study multiple bacterial sensory systems via tailored chemical tools in order to fully decipher the multiple facets, sensor kinase binding and corresponding RR phosphorylation, of the antimicrobial peptide response.
Conclusions
Given the impressive success of phosphoproteomics to study signaling networks in eukaryotic cells, the exploration of prokaryotic phosphorylation signals lacks behind. One reason is the prevalent but transient modification of aspartate resulting in a labile acyl phosphate group, which escapes detection by conventional methods. The capture of this modification by strong nucleophiles is a promising approach, however, care has to be taken in the selection of appropriate labeling conditions in order to maximize the yield and fidelity. Our study revealed that the addition of a minimal hydroxylamine-alkyne probe at pH = 4 supplemented with a solubilizing detergent favors the desired attack on the aspartate carbonyl C-atom and maximizes the readout of conserved RR sites in whole proteomes. This approach was validated by the detection of about 20% of all known RRs with modification at the known pAsp sites but also a large number of so far unknown sites. Interestingly, an alignment of all detected aspartate-modified peptides in P. aeruginosa displayed similarity to the conserved RR consensus motif suggesting that the method not only reports known but also putative unknown signaling systems. Given the prevalence of open reading frames encoding regulator proteins in P. aeruginosa, these findings will be subject to further functional studies.The tailored alkyne probe (HA-yne) is readily accessible and the reported procedures should allow straightforward implementation for the detection of this important post-translational modification in other laboratories. HA-yne for the first time allows phosphoaspartate monitoring through downstream application of click chemistry, which largely increases the flexibility of the approach. Besides the possibility of attaching desthiobiotin azide for enrichment, this modular nature of the technology enabled straightforward implementation of fluorescence gel-based assays using a fluorescent azide and application of the probe in the isoDTB-ABPP platform, which allowed quantitatively understanding of TCS signaling processes.
The main goal of implementing this refined prokaryotic phosphoproteomic platform was the application in deciphering molecular details of P. aeruginosa cationic peptide signaling. While ParS has been detected as a direct target of dynorphin A previously via a complementary affinity-based protein profiling (AfBPP) approach to identify sensor histidine kinases, we here show that under the selected conditions its cognate RR ParR is only slightly stronger modified in response to dynorphin A treatment. Unexpectedly, we here identify CprR as a modified and presumably phosphorylated RR, whose native histidine kinase CprS was not discovered by the previous AfBPP method. Thus, the two approaches (AfBPP and pAsp-trapping) unravel a high degree of complementarity with useful applications in deciphering bacterial signaling.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was funded by the European Research Council (ERC) and the European Union's Horizon 2020 research and innovation programme (grant agreement no. 725085, CHEMMINE, ERC consolidator grant). SMH acknowledges funding by the Fonds der Chemischen Industrie (Liebig Fellowship) and the TUM Junior Fellow Fund. The authors gratefully acknowledge M. Wolff, K. Bäuml and K. Gliesche for technical assistance.References
- E. Tacconelli, E. Carrara, A. Savoldi, S. Harbarth, M. Mendelson, D. L. Monnet, C. Pulcini, G. Kahlmeter, J. Kluytmans, Y. Carmeli, M. Ouellette, K. Outterson, J. Patel, M. Cavaleri, E. M. Cox, C. R. Houchens, M. L. Grayson, P. Hansen, N. Singh, U. Theuretzbacher, N. Magrini and W. H. O. P. P. L. W. Group, Lancet Infect. Dis.c, 2018, 18, 318–327 CrossRef.
- J. B. Lyczak, C. L. Cannon and G. B. Pier, Clin. Microbiol. Rev., 2002, 15, 194–222 CrossRef CAS.
- A. Y. Bhagirath, Y. Li, D. Somayajula, M. Dadashi, S. Badr and K. Duan, BMC Pulm. Med., 2016, 16, 174 CrossRef.
- P. K. Singh, A. L. Schaefer, M. R. Parsek, T. O. Moninger, M. J. Welsh and E. P. Greenberg, Nature, 2000, 407, 762–764 CrossRef CAS.
- J. Lee and L. Zhang, Protein Cell, 2015, 6, 26–41 CrossRef CAS.
- O. Zaborina, F. Lepine, G. Xiao, V. Valuckaite, Y. Chen, T. Li, M. Ciancio, A. Zaborin, E. O. Petrof, J. R. Turner, L. G. Rahme, E. Chang and J. C. Alverdy, PLoS Pathog., 2007, 3, e35 CrossRef.
- G. Wang, Pharmaceuticals, 2014, 7, 545–594 CrossRef CAS.
- M. H. Wright, C. Fetzer and S. A. Sieber, J. Am. Chem. Soc., 2017, 139, 6152–6159 CrossRef CAS.
- A. M. Stock, V. L. Robinson and P. N. Goudreau, Annu. Rev. Biochem., 2000, 69, 183–215 Search PubMed.
- J. J. Falke, R. B. Bass, S. L. Butler, S. A. Chervitz and M. A. Danielson, Annu. Rev. Cell Dev. Biol., 1997, 13, 457–512 CrossRef CAS.
- L. Fernandez, W. J. Gooderham, M. Bains, J. B. McPhee, I. Wiegand and R. E. Hancock, Antimicrob. Agents Chemother., 2010, 54, 3372–3382 CrossRef CAS.
- P. V. Attwood, P. G. Besant and M. J. Piggott, Amino Acids, 2011, 40, 1035–1051 CrossRef CAS.
- S. Black and N. G. Wright, J. Biol. Chem., 1955, 213, 27–38 CrossRef CAS.
- J. W. Chang, J. E. Montgomery, G. Lee and R. E. Moellering, Angew. Chem., Int. Ed., 2018, 57, 15712–15716 CrossRef CAS.
- D. E. Koshland, J. Am. Chem. Soc., 1952, 74, 2286–2292 CrossRef CAS.
- M. L. Matthews, L. He, B. D. Horning, E. J. Olson, B. E. Correia, J. R. Yates 3rd, P. E. Dawson and B. F. Cravatt, Nat. Chem., 2017, 9, 234–243 CrossRef CAS.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- P. R. A. Zanon, L. Lewald and S. M. Hacker, Angew. Chem., Int. Ed., 2020, 59, 2829–2836 CrossRef CAS.
- Y. J. Hsieh and B. L. Wanner, Curr. Opin. Microbiol., 2010, 13, 198–203 CrossRef CAS.
- R. A. VanBogelen, E. R. Olson, B. L. Wanner and F. C. Neidhardt, J. Bacteriol., 1996, 178, 4344–4366 CrossRef CAS.
- R. L. Creager-Allen, R. E. Silversmith and R. B. Bourret, J. Biol. Chem., 2013, 288 Search PubMed.
- W. R. McCleary, Mol. Microbiol., 1996, 20, 1155–1163 CrossRef CAS.
- D. R. Phillips and T. H. Fife, J. Am. Chem. Soc., 1968, 90, 6803–6809 CrossRef CAS.
- Y. Chen, Y. Cong, B. Quan, T. Lan, X. Chu, Z. Ye, X. Hou and C. Wang, Redox Biol., 2017, 12, 712–718 CrossRef CAS.
- J. Cox and M. Mann, Nat. Biotechnol., 2008, 26, 1367–1372 CrossRef CAS.
- J. Cox, N. Neuhauser, A. Michalski, R. A. Scheltema, J. V. Olsen and M. Mann, J. Proteome Res., 2011, 10, 1794–1805 CrossRef CAS.
- J. P. O'Shea, M. F. Chou, S. A. Quader, J. K. Ryan, G. M. Church and D. Schwartz, Nat. Methods, 2013, 10, 1211–1212 CrossRef.
- The UniProt Consortium, Nucleic Acids Res., 2019, 47, D506–D515 CrossRef.
- Y. Zhang, J. Wang, M. Ding and Y. Yu, Nat. Methods, 2013, 10, 981–984 CrossRef CAS.
- D. R. Ortega, A. D. Fleetwood, T. Krell, C. S. Harwood, G. J. Jensen and I. B. Zhulin, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 12809–12814 CrossRef CAS.
- T. I. Chio, B. R. Demestichas, B. M. Brems, S. L. Bane and L. N. Tumey, Angew. Chem., Int. Ed., 2020, 59, 13814–13820 CrossRef CAS.
- A. B. Robinson and C. J. Rudd, Curr. Top. Cell. Regul., 1974, 8, 247–295 CAS.
- T. Geiger and S. Clarke, J. Biol. Chem., 1987, 262, 785–794 CrossRef CAS.
- J. X. Zhu and D. W. Aswad, Anal. Biochem., 2007, 364, 1–7 CrossRef CAS.
- W. P. Jencks and M. Gilchrist, J. Am. Chem. Soc., 1964, 86, 5616–5620 CrossRef CAS.
- L. Fernandez, H. Jenssen, M. Bains, I. Wiegand, W. J. Gooderham and R. E. Hancock, Antimicrob. Agents Chemother., 2012, 56, 6212–6222 CrossRef CAS.
- B. MacLean, D. M. Tomazela, N. Shulman, M. Chambers, G. L. Finney, B. Frewen, R. Kern, D. L. Tabb, D. C. Liebler and M. J. MacCoss, Bioinformatics, 2010, 26, 966–968 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, supporting figures and compound characterization. See DOI: 10.1039/d0sc06226j |
| This journal is © The Royal Society of Chemistry 2021 |