
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery and biosynthesis of guanipiperazine from a NRPS-like pathway†
Jing
Shi‡
a,
Xiang
Xu‡
a,
Pei Yi
Liu
a,
Yi Ling
Hu
a,
Bo
Zhang
a,
Rui Hua
Jiao
a,
Ghader
Bashiri
b,
Ren Xiang
Tan
*a and
Hui Ming
Ge
 *a
*a
aState Key Laboratory of Pharmaceutical Biotechnology, School of Life Sciences, Chemistry and Biomedicine Innovation Center (ChemBIC), Institute of Artificial Intelligence Biomedicine, Nanjing University, Nanjing 210023, China. E-mail: rxtan@nju.edu.cn; hmge@nju.edu.cn
bLaboratory of Structural Biology, Maurice Wilkins Centre for Molecular Biodiscovery, School of Biological Sciences, University of Auckland, Auckland 1010, New Zealand
First published on 5th January 2021
Abstract
Nonribosomal peptide synthetases (NRPSs) are modular enzymes that use a thiotemplate mechanism to assemble the peptide backbones of structurally diverse and biologically active natural products in bacteria and fungi. Unlike these canonical multi-modular NRPSs, single-module NRPS-like enzymes, which lack the key condensation (C) domain, are rare in bacteria, and have been largely unexplored to date. Here, we report the discovery of a gene cluster (gup) encoding a NRPS-like megasynthetase through genome mining. Heterologous expression of the gup cluster led to the production of two unprecedented alkaloids, guanipiperazines A and B. The NRPS-like enzyme activates two L-tyrosine molecules, reduces them to the corresponding amino aldehydes, and forms an unstable imine product. The subsequent enzymatic reduction affords piperazine, which can be morphed by a P450 monooxygenase into a highly strained compound through C–O bond formation. Further intermolecular oxidative coupling forming the C–C or C–O bond is catalyzed by another P450 enzyme. This work reveals the huge potential of NRPS-like biosynthetic gene clusters in the discovery of novel natural products.
Introduction
Nonribosomal peptide synthetases (NRPSs) are modular megaenzymes consisting of multiple covalently-linked domains. These megaenzymes catalyze the synthesis of important peptide natural products, including vancomycin, daptomycin, and cyclosporine A.1–3 The biosynthesis of such nonribosomal peptides depends on a minimal domain set for adenylation (A), thiolation (T), and condensation (C).1,4,5 The A domain selects and activates a carboxylic acid substrate through adenylation at the expense of ATP, then transfers the acyl group to the phosphopantetheinyl arm attached to the T domain to form a thioester linkage. The C domain mediates peptide bond formation in two adjacent modules. In comparison, single-module NRPS-like enzymes do not contain a C domain and lack this extension function. These enzymes are functionally diverse in primary and secondary metabolism. Depending the type of downstream releasing domains, NRPS-like enzymes catalyse a broad range of reactions, including but not limited to Dieckmann cyclization,6,7 reduction,8,9 and Claisen condensation.10The carboxylic acid reductases (CARs) belong to the single-module NRPS-like enzymes, and comprise an N-terminal adenylation domain, which is fused to a C-terminal reductase (R) domain via a T domain.11 Mechanistically, the T domain delivers the acyl thioester to the R domain for reduction by NADPH to give an aldehyde product. For example, Lys2 can reduce α-aminoadipate at C-6 to the semialdehyde in the biosynthesis of lysine in Saccharomyces cerevisiae;12 CmlP is involved in the biosynthesis of the antibiotic chloramphenicol (Scheme 1) in Streptomyces;13 PvfC is responsible for the synthesis of a family of pyrazine N-oxides in Pseudomonas virulence;14,15 and HamD is related to valdiazen biosynthesis in Burkholderia cenocepacia (Scheme 1).16 CARs have also recently attracted interest as green biocatalysts due to their abilities to convert a broad range of carboxylic acid substrates to the corresponding aldehydes.17,18 CARs alone or combined with other enzymes have been applied to generate pharmaceuticals, biofuels, and other valuable products.17,19,20
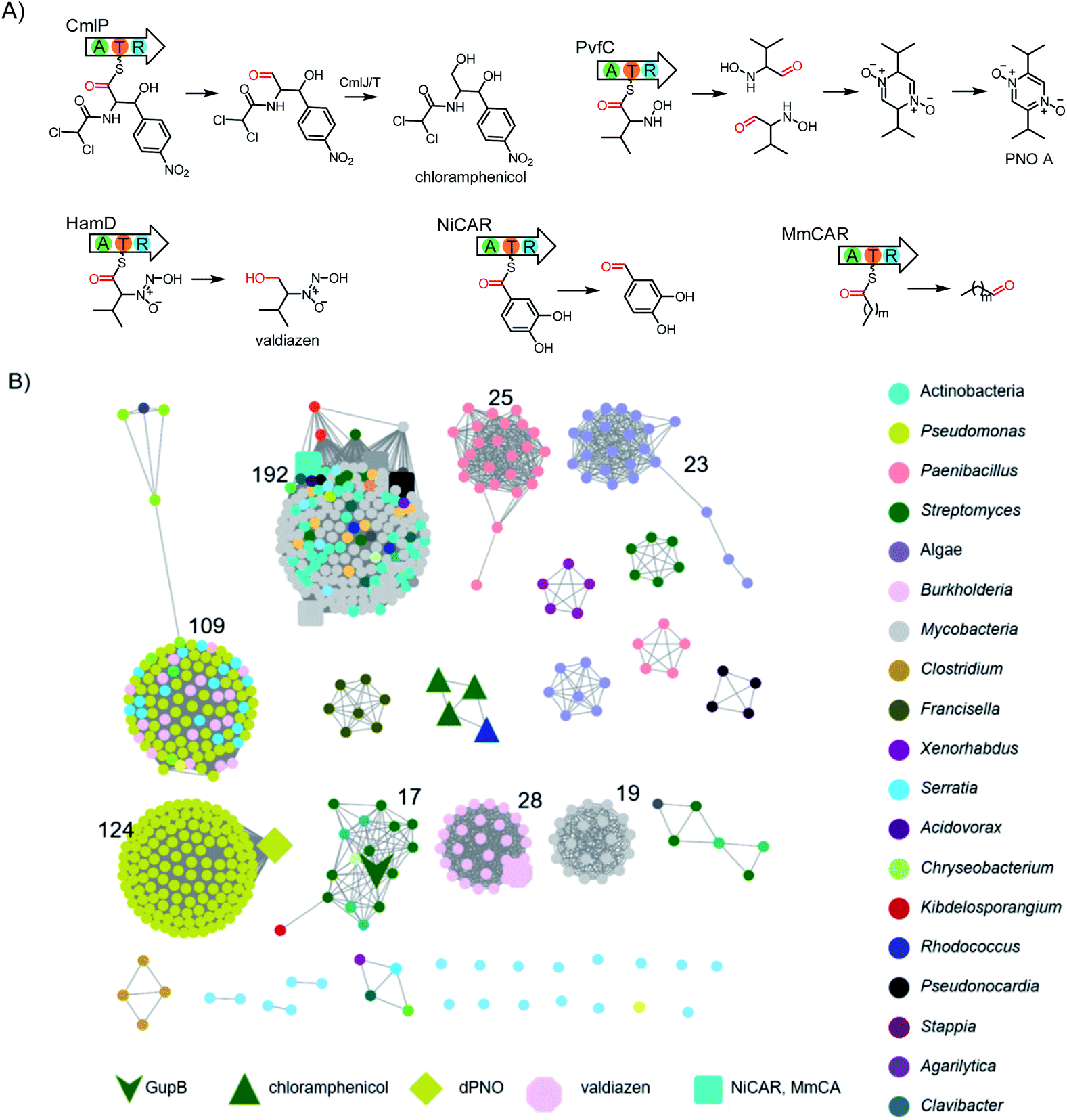 | ||
| Scheme 1 Genome mining of single-module CAR enzymes from bacteria. (A) The enzymatic reactions catalyzed by characterized bacterial CAR enzymes. (B) Sequence similarity network (SSN) analysis of single-module CAR enzymes with a sequence identity of >40% visualized by Cytoscape. | ||
Recent advances of microbial genomics have led to the rapid accumulation of uncharacterized biosynthetic gene clusters (BGCs) in genome database. The diverse functions of CARs and their biocatalytic potential encouraged us to identify new members through genome mining. Here, we report the discovery of a unique four-gene operon encoding an F420H2-dependent reductase, a CAR, and two P450 proteins in Streptomyces. We reconstituted four enzymes that catalyse the production of two unprecedented alkaloids (1 and 2). We demonstrated the detailed biosynthetic steps towards the formation of 1 and 2.
Results and discussion
Genome mining uncovered the gup BGC
To identify single-module NRPS-like CAR encoding genes, we systematically investigated the genomic data in our own and the antiSMASH database,21 which totally contain over 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 bacterial BGCs. When the R domain was used as the search query, 1230 NRPS BGCs containing a terminal R domain were retrieved. After manually removing the multi-modular NRPSs, we were left with 608 single-module NRPS-like BGCs.
000 bacterial BGCs. When the R domain was used as the search query, 1230 NRPS BGCs containing a terminal R domain were retrieved. After manually removing the multi-modular NRPSs, we were left with 608 single-module NRPS-like BGCs.
From these 608 hits, we generated a sequence similarity network (SSN) based on their corresponding R domain sequences.22 We showed that CARs from different organisms may evolve independently as many of them are related to their taxonomies (Scheme 1).23 Nonetheless, the known CAR enzymes with similar functions are clustered together. The largest clade of proteins contains homologs of NiCARs and MmCARs,24 suggesting this clade is most likely involved in the reduction of aromatic or aliphatic carboxylic acids to their corresponding aldehydes. We also excluded homologous enzymes of PvfC, HamD, and CmlP from further investigation (Scheme 1).
We hypothesize that other tailoring enzymes in BGCs are important for the structural complexity of the corresponding natural products, as exemplified by the chloramphenicol BGC where a series of reductase, halogenase, amidase, and oxygenase encoding genes are present.13 Thus, we further submitted the CAR-gene containing loci to antiSMASH for gene cluster analysis.21 We identified an orphan BGC (gup) in S. chrestomyceticus consisting of a four-gene operon encoding a putative F420H2-dependent reductase, a CAR, and two P450s (Fig. 1). A subsequent BLAST homology search in the NCBI and JGI genome databases revealed that many other Streptomyces strains harbour this homologous, yet cryptic, gene cluster (Fig. S1†).
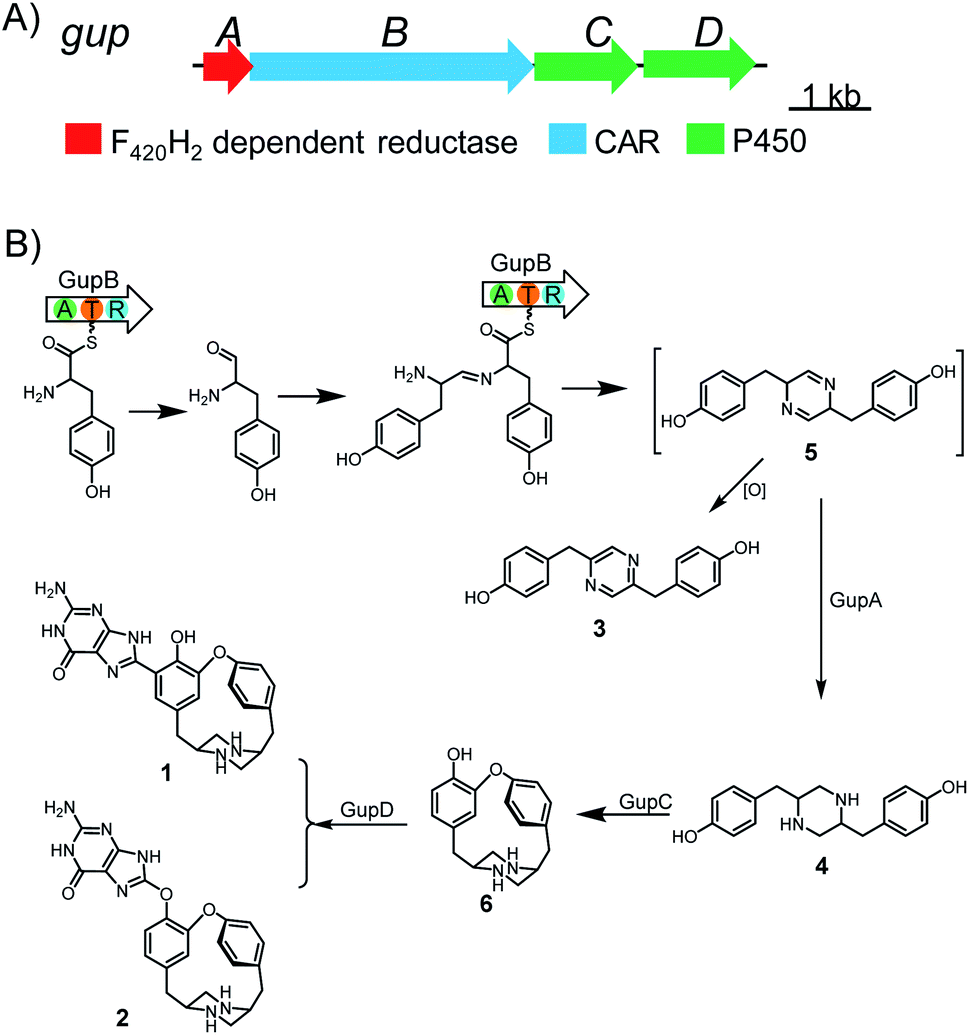 | ||
| Fig. 1 Biosynthesis of guanipiperazines A and B (1 and 2). (A) Biosynthetic gene cluster of gup; (B) the proposed biosynthetic pathway of 1 and 2. | ||
Heterologous production of guanipiperazines
Initial attempts to obtain the natural products encoded by the gup BGC in the S. chrestomyceticus wild-type strain by fermentation and media optimization failed. Therefore, we performed heterologous expression of the gup BGC in S. lividans TK24, a prolific host that is widely used for producing actinobacterial natural products.25 We cloned the intact gup BGC into a Streptomyces integrative plasmid, pSET152, under the control of a strong promoter PkasO*,26 and introduced the plasmid into the S. lividans TK24 strain. After cultivating this strain in ISP4 medium for 10 days, two compounds, 1 and 2 were produced, both with m/z 446.2 [M + H]+. In contrast, these two compounds were not present in the control strain harbouring an empty pSET152 plasmid (Fig. 2). Subsequent large scale fermentation of the S. lividans TK24/gupABCD strain led to the isolation of sufficient amounts of 1 and 2. Their structures were characterized by analyses of the 1D and 2D NMR data, including 1H, 13C, HSQC, HMBC, 1H–1H COSY, and NOESY (Fig. 1, Tables S4 and S5†). Both 1 and 2 showed antimicrobial activities against Saccharomyces cerevisiae with minimum inhibitory concentrations (MICs) of 4.5 and 5.6 μg mL−1, respectively.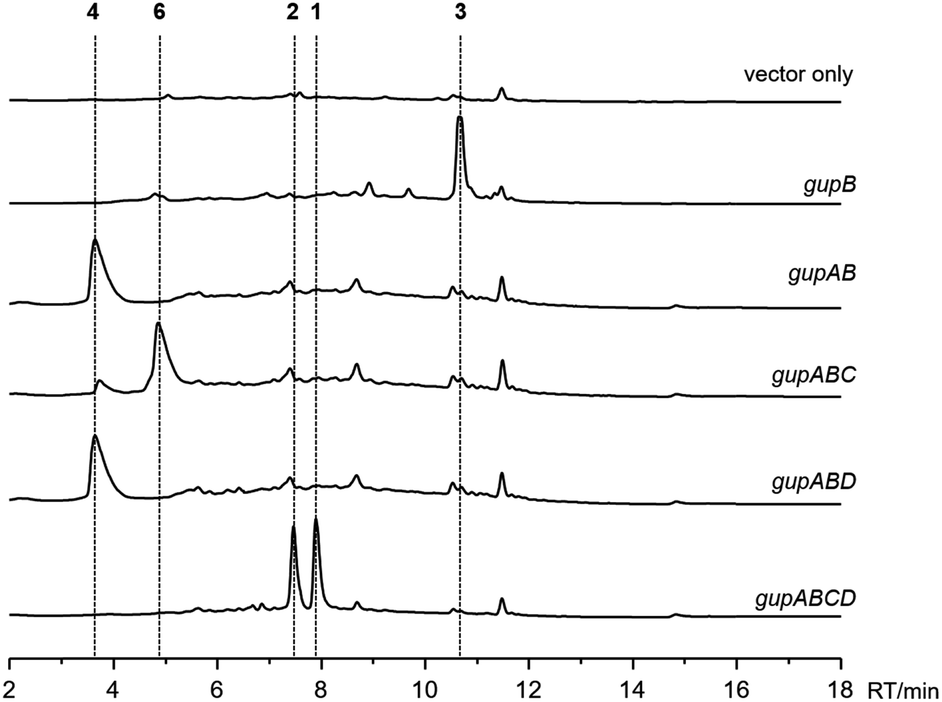 | ||
| Fig. 2 HPLC analysis (monitored at 280 nm) of the heterologous expression of the gup biosynthetic gene cluster in Streptomyces lividans TK24. | ||
In vivo characterization of GupA and GupB
Previous studies have reported the formation of a strained piperazine with a C–C cross-link between the phenol sides in fungus.8,27 To dissect the biosynthesis of 1 and 2, which feature a unique C–O cross-linked piperazine core and a guaninyl modification, we set out to reconstitute the biosynthetic pathway through a bottom-up strategy in S. lividans TK24. Expressing gupB alone in S. lividans TK24 led to the formation of a new product 3 (Fig. 1, Table S6†). Compound 3 has a pyrazine-type core, inconsistent with the structures of 1 and 2. To test if 3 is an on-pathway intermediate, we fed 3 to a S. lividans TK24/gupACD strain, in which the gupB gene was deleted from the gup BGC. The production of 1 or 2 could not be restored (Fig. S2†), indicating 3 could be a shunt metabolite.GupA is annotated as an F420H2-dependent reductase. Co-expression of the gupB and gupA genes yielded a piperazine-type compound 4 (Fig. 1, Table S7†). Feeding 4 to the S. lividans TK24/gupACD strain restored the production of 1 and 2 (Fig. S2†), indicating 4 is an on-pathway intermediate. Hence, the CAR GupB enzyme may successively activate two tyrosine molecules and reduce them to the corresponding amino aldehydes. Condensation between two amino aldehydes followed by dehydration led to the formation of a proposed intermediate 5. In the absence of GupA, 5 is spontaneously overoxidized by air to give 3; alternatively, 5 is reduced to form intermediate 4 in the presence of GupA.
In vitro characterization of GupA and GupB
To investigate the function of GupB in vitro, we overproduced the intact GupB protein in Escherichia coli BL21(DE3) cells and purified the apo-form to homogeneity via Ni-NTA affinity chromatography (Fig. S3†). An in vitro adenylation activity assay was used to monitor the pyrophosphate production in the presence of different amino acids.28L-Tyrosine is the preferred substrate for adenylation of the A domain of GupB, whereas D-tyrosine and other amino acids show relatively low activities (Fig. 3). Thus, we hypothesize that the chiral centers at C-3 and C-13 are retained in 1 and 2, as the downstream modifications would not affect their stereochemistry.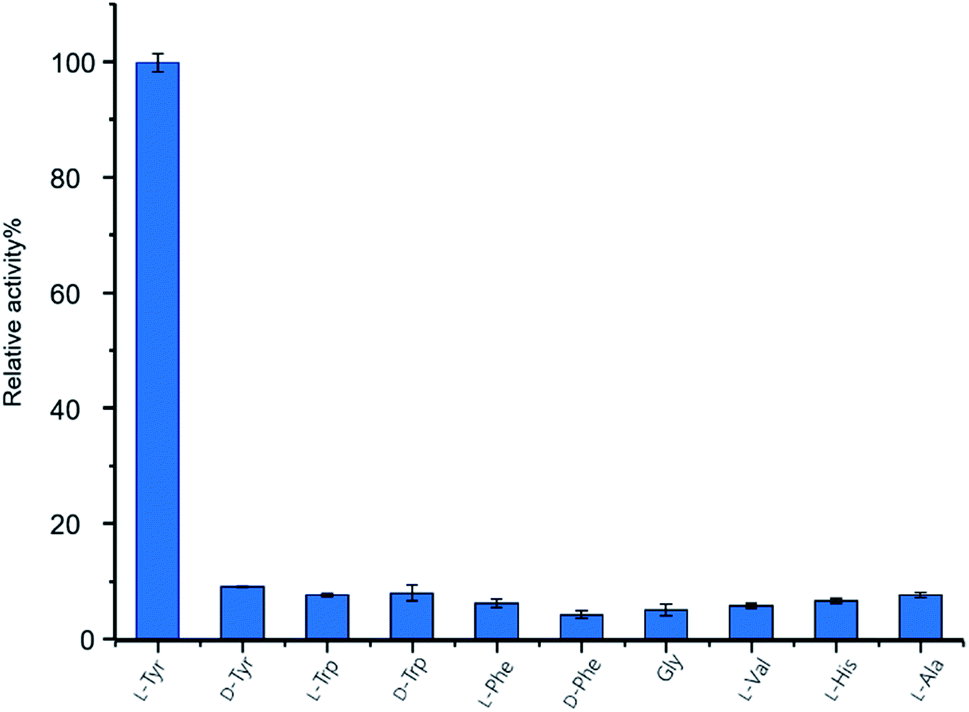 | ||
| Fig. 3 The relative adenylation activities for GupB. | ||
GupA is a small protein (162 amino acids) that belongs to the RV1155-F420 superfamily, and contains a conserved F420-binding domain. However, the functions of members of this protein superfamily are unclear. We tested whether GupA can reduce the proposed intermediate 5. We purified the cofactor F420 from a genetically-engineered strain of Mycobacterium smegmatis.29 F420-dependent glucose-6-phosphate dehydrogenase (FGD) from M. tuberculosis was used for in situ regeneration of the reduced form of F420 (F420H2) (Fig. S5†).30 We reconstituted the holo-GupB by incubating apo-GupB, CoA, and Sfp (a phosphopantetheinyl transferase in Bacillus subtilis).31 Further supplementation of ATP and L-tyrosine in the above reaction system led to the production of 3 (Fig. 4), indicating the reconstituted GupB protein is functional. When we incubated GupA, F420, and the F420H2 regeneration system into the abovementioned enzymatic reaction, production of 4 was observed.
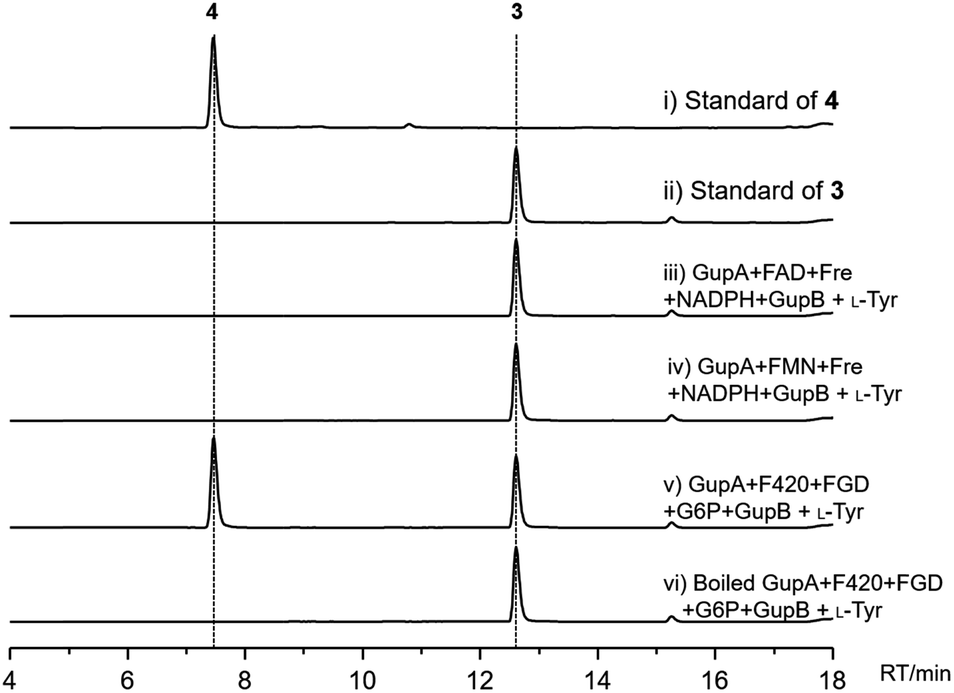 | ||
| Fig. 4 HPLC analysis (monitored at 280 nm) of GupB and GupA enzymatic reactions in vitro. | ||
As cofactor F420 is structurally similar to the flavins (FMN or FAD), we evaluated if GupA could utilize FMNH2/FADH2 as reducing agents. FMNH2/FADH2 can be regenerated by NADPH, FMN/FAD, and flavin reductase (Fre) from E. coli.32 However, the production of 4 was not detected upon the introduction of FMNH2/FADH2 (Fig. S6†). These data reveal that GupB activates and reduces two L-tyrosine molecules and forms the condensed intermediate 5, while GupA is required for the formation of 4. To the best of our knowledge, GupA is the first member of the RV1155-F420 superfamily to be functionally characterized.
Functional characterization of GupC and GupD
Next, we aimed to identify the enzymes responsible for the intramolecular diaryl ether (C–O bond) and intermolecular diaryl (C–C bond) and ether (C–O bond) couplings. The C–C and C–O aryl bonds have been observed in other peptides and alkaloids, and are often introduced by P450 enzymes; for example, OxyC, StaP, and CYP121 mediate the diary C–C bond formation in vancomycin, staurosporine, and mycocyclosin biosynthesis,33–35 respectively; and OxyA, OxyB and OxyE catalyse the diary C–O bond formation in vancomycin biosynthesis.33 Therefore, we postulated the remaining two P450 encoding genes, gupC and gupD, which share 32% sequence identity, are involved in C–C and C–O bond formation.The gupC and gupD genes were deleted individually from the gup BGC through in-frame deletion. LC-MS analysis showed that S. lividans TK24/gupABD did not produce 1 and 2, but accumulated 4 as the dominant product (Fig. 2). Meanwhile, the S. lividans TK24/gupABC strain generated a new compound 6 with m/z 297.2 [M + H]+ (Fig. 2), which is 2 Da less than that of 4. The structure of 6 is the expected strained piperazine compound (Fig. 1, Table S8†). Our in vivo heterologous expression data indicated that GupC catalyzes the intramolecular C–O bond formation, while GupD couples 6 and a guanine to form 1 and 2 through C–C and C–O linkage.
To verify functions of the two P450s in vitro, GupC and GupD were overexpressed in E. coli BL21(DE3) cells and purified to near homogeneity as (His)6-fusion proteins. In addition, P450 enzyme activity requires the supply of electrons to the iron center via the electron transport chain. The bacterial P450 enzyme utilizes a two-component system consisting of ferredoxin (Fdx) and ferredoxin reductase (Fdr). Thus, we overexpressed and purified the native Fdx and Fdr enzymes from S. chrestomyceticus (Fig. S3†). Incubation of GupC and 4 in the presence of the electron transport system and NADPH led to the consumption of 4 with the concomitant appearance of 6. These findings indicate GupC catalyzes the intramolecular C–O bond formation.
When GupD was incubated with 6 and GTP in the presence of the electron transport system, we observed the formation of 1 and 2 (Fig. 5). This indicated that GupD acts as a guaninyl transferase that catalyzes the attachment of a guanine moiety to the piperazine core via a C–C or C–O bond. This function of GupD is reminiscent of the P450 enzyme PcmD (with 37% identity to GupD),36 which transfers a guaninyl moiety to a cyclodipeptide in the biosynthesis of purincyclamide. PcmD uses a guanine as the substrate instead of GTP, whereas GupD only accepts GTP as the substrate, and is inactive to ATP, UTP, and guanine (Fig. S7†).
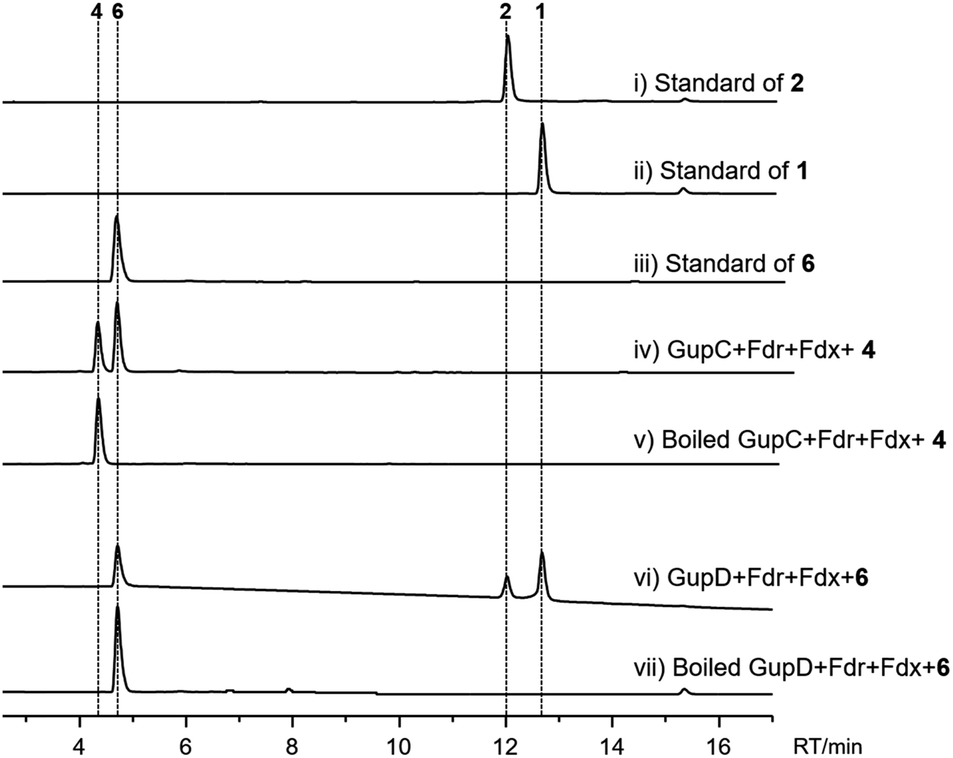 | ||
| Fig. 5 HPLC analysis (monitored at 280 nm) of GupC and GupD enzymatic reactions in vitro. | ||
Conclusions
In conclusion, by genome mining towards the single-module NRPS-like genes in bacteria we discovered a four-gene operon (gup) that is actually abundant and widespread in Streptomyces strains. Heterologous expression of the gup BGC led to the characterization of two novel alkaloids, guanipiperazines A and B, which feature a rare, highly strained piperazine core and a guaninyl modification. GupB is a new CAR enzyme in secondary metabolism that plays a key role in the construction of the piperazine core. An F420H2-dependent reductase and two P450 enzymes subsequently mediate the remaining redox steps to form the guanipiperazines. Our work highlights the diverse functions of CAR enzymes, and paves the way for new natural products to be discovered from other clades of CAR enzymes in the future.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by MOST (2019YFC0312500, 2018YFC1706200 and 2018YFA0902000), NSFC (81925033, 21861142005, 81773591, 81673333, 81803380, 2171101213, and 21661140001), the Fundamental Research Funds for the Central Universities (14380092, 14380113, and 143801389) and a Sir Charles Hercus Fellowship (Health Research Council of New Zealand) to G. B. (17/058).Notes and references
- R. D. Sussmuth and A. Mainz, Angew. Chem., Int. Ed., 2017, 56, 3770–3821 CrossRef.
- M. A. Marahiel, Nat. Prod. Rep., 2016, 33, 136–140 RSC.
- C. T. Walsh, Nat. Prod. Rep., 2016, 33, 127–135 RSC.
- H. D. Mootz, D. Schwarzer and M. A. Marahiel, ChemBioChem, 2002, 3, 490–504 CrossRef CAS.
- M. Strieker, A. Tanovic and M. A. Marahiel, Curr. Opin. Struct. Biol., 2010, 20, 234–240 CrossRef CAS.
- C. J. Balibar, A. R. Howard-Jones and C. T. Walsh, Nat. Chem. Biol., 2007, 3, 584–592 CrossRef CAS.
- M. Klapper, D. Braga, G. Lackner, R. Herbst and P. Stallforth, Cell Chem. Biol., 2018, 25, 659–665 CrossRef CAS.
- X. Yu, F. Liu, Y. Zou, M.-C. Tang, L. Hang, K. N. Houk and Y. Tang, J. Am. Chem. Soc., 2016, 138, 13529–13532 CrossRef CAS.
- A. Tietze, Y. N. Shi, M. Kronenwerth and H. B. Bode, ChemBioChem, 2020, 21, 2750–2754 CrossRef CAS.
- J. A. Baccile, J. E. Spraker, H. H. Le, E. Brandenburger, C. Gomez, J. W. Bok, J. Macheleidt, A. A. Brakhage, D. Hoffmeister, N. P. Keller and F. C. Schroeder, Nat. Chem. Biol., 2016, 12, 419–424 CrossRef CAS.
- H. Stolterfoht, D. Schwendenwein, C. W. Sensen, F. Rudroff and M. Winkler, J. Biotechnol., 2017, 257, 222–232 CrossRef CAS.
- D. E. Ehmann, A. M. Gehring and C. T. Walsh, Biochemistry, 1999, 38, 6171–6177 CrossRef CAS.
- L. T. Fernandez-Martinez, C. Borsetto, J. P. Gomez-Escribano, M. J. Bibb, M. M. Al-Bassam, G. Chandra and M. J. Bibb, Antimicrob. Agents Chemother., 2014, 58, 7441–7450 CrossRef.
- A. M. Kretsch, G. L. Morgan, J. Tyrrell, E. Mevers, I. Vallet-Gely and B. Li, Org. Lett., 2018, 20, 4791–4795 CrossRef CAS.
- G. L. Morgan and B. Li, Angew. Chem., Int. Ed., 2020, 59, 21387–21391 CrossRef CAS.
- C. Jenul, S. Sieber, C. Daeppen, A. Mathew, M. Lardi, G. Pessi, D. Hoepfner, M. Neuburger, A. Linden, K. Gademann and L. Eberl, Nat. Commun., 2018, 9, 1297 CrossRef.
- S. R. Derrington, N. J. Turner and S. P. France, J. Biotechnol., 2019, 304, 78–88 CrossRef CAS.
- K. Napora-Wijata, G. A. Strohmeier and M. Winkler, Biotechnol. J., 2014, 9, 822–843 CrossRef CAS.
- S. P. France, S. Hussain, A. M. Hill, L. J. Hepworth, R. M. Howard, K. R. Mulholland, S. L. Flitsch and N. J. Turner, ACS Catal., 2016, 6, 3753–3759 CrossRef CAS.
- G. Qu, J. Guo, D. Yang and Z. Sun, Green Chem., 2018, 20, 777–792 RSC.
- K. Blin, V. P. Andreu, E. L. C. de los Santos, F. Del Carratore, S. Y. Lee, M. H. Medema and T. Weber, Nucleic Acids Res., 2019, 47, D625–D630 CrossRef CAS.
- J. A. Gerlt, Biochemistry, 2017, 56, 4293–4308 CrossRef CAS.
- D. Kalb, G. Lackner and D. Hoffmeister, Appl. Environ. Microbiol., 2014, 80, 6175–6183 CrossRef.
- S. P. France, S. Hussain, A. M. Hill, L. J. Hepworth, R. M. Howard, K. R. Mulholland, S. L. Flitsch and N. J. Turner, ACS Catal., 2016, 6, 3753–3759 CrossRef CAS.
- Y. Ahmed, Y. Rebets, M. R. Estevez, J. Zapp, M. Myronovskyi and A. Luzhetskyy, Microb. Cell Fact., 2020, 19, 5 CrossRef CAS.
- W. Wang, X. Li, J. Wang, S. Xiang, X. Feng and K. Yang, Appl. Environ. Microbiol., 2013, 79, 4484–4492 CrossRef CAS.
- R. R. Forseth, S. Amaike, D. Schwenk, K. J. Affeldt, D. Hoffmeister, F. C. Schroeder and N. P. Keller, Angew. Chem., Int. Ed., 2013, 52, 1590–1594 CrossRef CAS.
- J. Shi, Y. J. Zeng, B. Zhang, F. L. Shao, Y. C. Chen, X. Xu, Y. Sun, Q. Xu, R. X. Tan and H. M. Ge, Chem. Sci., 2019, 10, 3042–3048 RSC.
- G. Bashiri, A. M. Rehan, D. R. Greenwood, J. M. J. Dickson and E. N. Baker, PLoS One, 2010, 5, e15803 CrossRef CAS.
- G. Bashiri, J. Antoney, E. N. M. Jirgis, M. V. Shah, B. Ney, J. Copp, S. M. Stuteley, S. Sreebhavan, B. Palmer, M. Middleditch, N. Tokuriki, C. Greening, C. Scott, E. N. Baker and C. J. Jackson, Nat. Commun., 2019, 10, 1558 CrossRef.
- L. E. N. Quadri, P. H. Weinreb, M. Lei, M. M. Nakano, P. Zuber and C. T. Walsh, Biochemistry, 1998, 37, 1585–1595 CrossRef CAS.
- Y. S. Wang, B. Zhang, J. P. Zhu, C. L. Yang, Y. Guo, C. L. Liu, F. Liu, H. Q. Huang, S. W. Zhao, Y. Liang, R. H. Jiao, R. X. Tan and H. M. Ge, J. Am. Chem. Soc., 2018, 140, 10909–10914 CrossRef CAS.
- K. Haslinger, M. Peschke, C. Brieke, E. Maximowitsch and M. J. Cryle, Nature, 2015, 521, 105–U271 CrossRef CAS.
- M. Makino, H. Sugimoto, Y. Shiro, S. Asamizu, H. Onaka and S. Nagano, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11591–11596 CrossRef CAS.
- P. Belin, M. H. Le Du, A. Fielding, O. Lequin, M. Jacquet, J. B. Charbonnier, A. Lecoq, R. Thai, M. Courcon, C. Masson, C. Dugave, R. Genet, J. L. Pernodet and M. Gondry, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 7426–7431 CrossRef.
- J. Shi, X. Xu, E. J. Zhao, B. Zhang, W. Li, Y. Zhao, R. H. Jiao, R. X. Tan and H. M. Ge, Org. Lett., 2019, 21, 6825–6829 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc06135b |
| ‡ Contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |