
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNickel-catalyzed C–O/N–H, C–S/N–H, and C–CN/N–H annulation of aromatic amides with alkynes: C–O, C–S, and C–CN activation†
Yasuaki
Iyori
,
Rina
Ueno
,
Aoi
Morishige
and
Naoto
Chatani
 *
*
Department of Applied Chemistry, Faculty of Engineering, Osaka University, Suita, Osaka 565-0871, Japan. E-mail: chatani@chem.eng.osaka-u.ac.jp
First published on 9th December 2020
Abstract
The Ni-catalyzed reaction of ortho-phenoxy-substituted aromatic amides with alkynes in the presence of LiOtBu as a base results in C–O/N–H annulation with the formation of 1(2H)-isoquinolinones. The use of a base is essential for the reaction to proceed. The reaction proceeds, even in the absence of a ligand, and under mild reaction conditions (40 °C). An electron-donating group on the aromatic ring facilitates the reaction. The reaction was also applicable to carbamate (C–O bond activation), methylthio (C–S bond activation), and cyano (C–CN bond activation) groups as leaving groups.
Introduction
The cleavage of C(aryl)–O bonds mediated by transition metal complexes is of significant interest for the development of cross-coupling reactions and biomass utilization.1 A variety of cross-coupling reactions that utilize phenol-derived electrophiles, such as pivalates, carbamates, sulfonates, and anisoles as an alternative to aryl halide coupling partners has been developed, because these are readily available and no halogen-containing waste products are generated during the reaction. The most broadly used catalysts in cross-coupling reactions of oxygen-based electrophiles are Ni complexes. The cleavage of inert C(aryl)–O bonds by a Ni complex generally requires a high reaction temperature along with the addition of a strong donor ligand, such as trialkylphosphines or an N-heterocyclic carbene (NHC) ligand, which leads to a decrease in functional group tolerance. While transformations that involve C(aryl)–O bond activation as a key step have been extensively studied, most of the reactions reported thus far are largely limited to cross-coupling reactions.2 In this context, C(aryl)–O bond activation continues to be a relatively undeveloped area of research. We recently reported that the Ni-catalyzed C–F/N–H annulation of aromatic secondary amides with alkynes proceeds even in the absence of a ligand and at low reaction temperatures (40–100 °C).3 A key to the success of this reaction is the use of a base, which functions to abstract a proton from an aromatic amide resulting in the formation of an amidate species. Because one equivalent of a base is used, the amidate is a bona fide substrate, which reacts with a Ni(0) complex to give a highly active nickel ate complex. We were interested in whether this methodology would be applicable to the activation of other unreactive bonds.We herein report that the C–O/N–H annulation of aromatic amides with alkynes results in the production of isoquinolinones (Scheme 1).4,5 Remarkably, the reaction was also applicable to C–S/N–H and C–CN/N–H annulation, which proceed via C–S and C–CN bond activation.
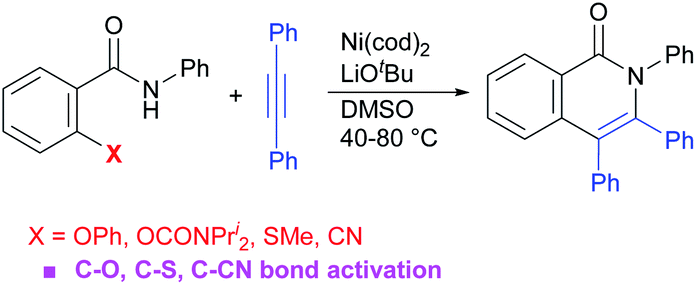 | ||
| Scheme 1 Nickel-catalyzed C–O/N–H, C–S/N–H, C–CN/N–H annulation of aromatic amides with alkynes. | ||
Results and discussion
We began our studies by examining 2-phenoxy-N-phenylbenzamide (1a) as a model substrate and diphenylacetylene (2a) as a coupling partner with Ni(cod)2 as a catalyst to evaluate the optimal reaction conditions for such a reaction (Table 1). The reaction of 1a with 2a (1.5 equiv.) in the presence of 10 mol% of Ni(cod)2 and KOtBu (1 equiv.) in DMF (0.5 mL) at 40 °C for 2 h gave the expected product 3aa in 69% yield, along with 30% of 1a being recovered and a trace amount of amide 4a, which appears to be formed via the transfer of a phenyl group from an oxygen atom in 1a to a nitrogen atom (Table 1, entry 1). After screening a series of solvents, it was found that the product yield could be improved to 85% when DMSO was used as a solvent (entries 2–4). Curiously, the presence of ligands, such as PPh3, dppe, and dtbbpy in the reaction mixture resulted in a decreased product yield (entries 5–7). When the reaction was carried out using NaOtBu or LiOtBu, the yield of the product was improved and the formation of byproduct 4a was suppressed (entries 8 and 9). When the reaction was carried out using LiOtBu for 5 h, the starting amide was completely consumed and the desired product was obtained in 88% isolated yield (entry 10). The reaction did not proceed in the absence of the Ni catalyst (entry 11). Finally, we determined the conditions for the reaction as shown in entry 10 as the standard conditions.|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Ligand | Base | NMR yieldsb3aa/4a/1a |
| a Reaction conditions: 1a (0.2 mmol), 2a (0.3 mmol), Ni(cod)2 (0.02 mmol), and base (0.2 mmol) in solvent (0.5 mL) at 40 °C for 2 h. b NMR yields were determined from 1H NMR with 1,1,1,2-tetrachloroethane as the internal standard. c 0.4 mmol scale, for 5 h. d Without Ni(cod)2. e Isolated yield. | ||||
| 1 | DMF | None | KOtBu | 69%/trace/30% |
| 2 | Toluene | None | KOtBu | 10%/none/>99% |
| 3 | 1,4-Dioxane | None | KOtBu | 48%/none/56% |
| 4 | DMSO | None | KOtBu | 85%/2%/7% |
| 5 | DMSO | PPh3 | KOtBu | 57%/3%/47% |
| 6 | DMSO | dppe | KOtBu | 33%/3%/70% |
| 7 | DMSO | dtbbpy | KOtBu | 12%/5%/89% |
| 8 | DMSO | None | NaOtBu | 93%/none/trace |
| 9 | DMSO | None | LiOtBu | 93%/none/4% |
| 10c | DMSO | None | LiOtBu | >99% (88%)/none/nonee |
| 11d | DMSO | None | LiOtBu | None/3%/>99% |
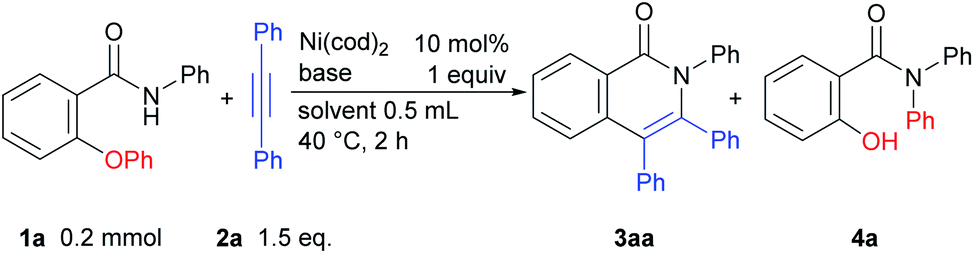
The results of a survey of substrate scope are shown in Scheme 2. First, the effect of the substituent group on the amide nitrogen atom was examined (3ba and 3ca). An electron-withdrawing group on the nitrogen atom caused a slight decrease in the product yield. Various functional groups on the aromatic ring, such as methoxy, trifluoromethyl, fluoro, cyano, diethylamino, and even aldehyde groups were tolerated in the reaction. It was also found that when a substrate bearing an electron-withdrawing group was used, a longer reaction time or a higher reaction temperature was needed, as in 3ea, 3fa, 3ga, and 3ia suggesting that the cleavage of the C–O bond is not the rate-determining step.6,7 While it is well known that Ni complexes can be used to activate C–OMe,1 C–F,8 and CN bonds,9 these bonds remained intact, as in 3ba, 3da, 3fa, 3ka, and 3ma. Substrates bearing a substituent at the ortho position of the phenoxy group were also applicable to this reaction to give 3ka in spite of the steric hinderance imposed by the phenoxy group. An amide bearing both C–OPh and C–F bonds at the ortho position 1l reacted with 2 to give a mixture of two products, 3la and 3la′ in favor of 3la′, indicating that C–F bond activation predominated over C–O bond activation under the reaction conditions used. In the reaction of amide 1m, which contains both OMe and OPh groups at the ortho-position, only the C–OPh bond was selectively cleaved to give 3ma in 75% yield.
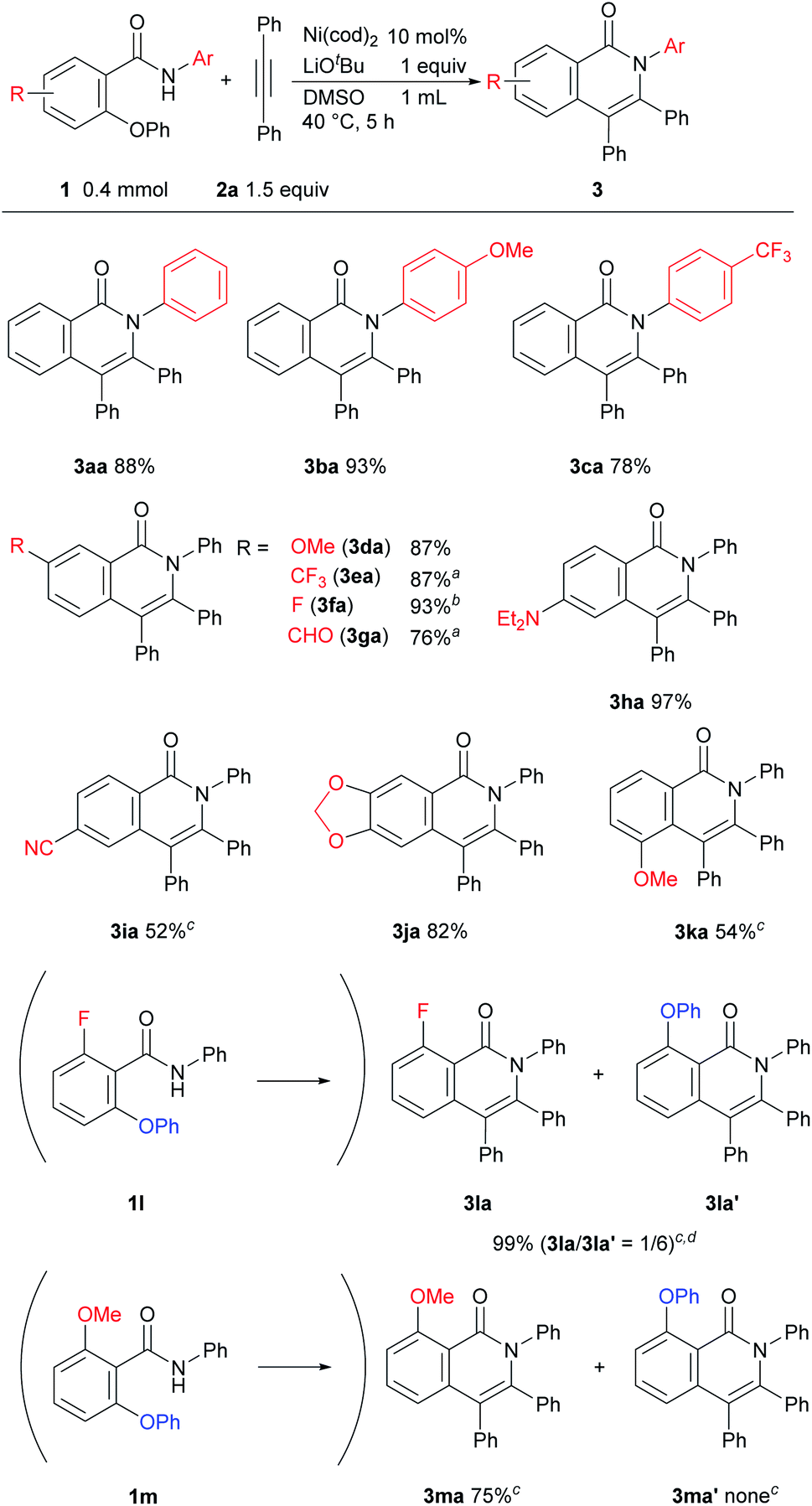 | ||
| Scheme 2 Scope of amides for the reaction. Reaction conditions: amide (0.4 mmol), 2a (0.6 mmol), Ni(cod)2 (0.04 mmol), LiOtBu (0.4 mmol) in DMSO (1 mL) at 40 °C for 5 h. Yields shown are isolated yields. a60 °C. b24 h. c80 °C. dThe product yield and the ratio of products were determined by 1H NMR. | ||
The scope of the reaction with respect to alkynes was also examined (Scheme 3). An alkyne bearing an electron-donating group 2b reacted efficiently to give 3ab in 92% isolated yield. This reaction was carried out in DMF as a solvent due to the insolubility of 2b to DMSO. The reaction of electron-deficient alkynes required higher temperatures or longer reaction times (3ac and 3ad). Alkynes with a thiophene ring and an aliphatic alkyne were also applicable for this reaction (3ae and 3af). When an unsymmetrical alkyne 2g was used, the product 3ag was obtained in a regioselective manner.
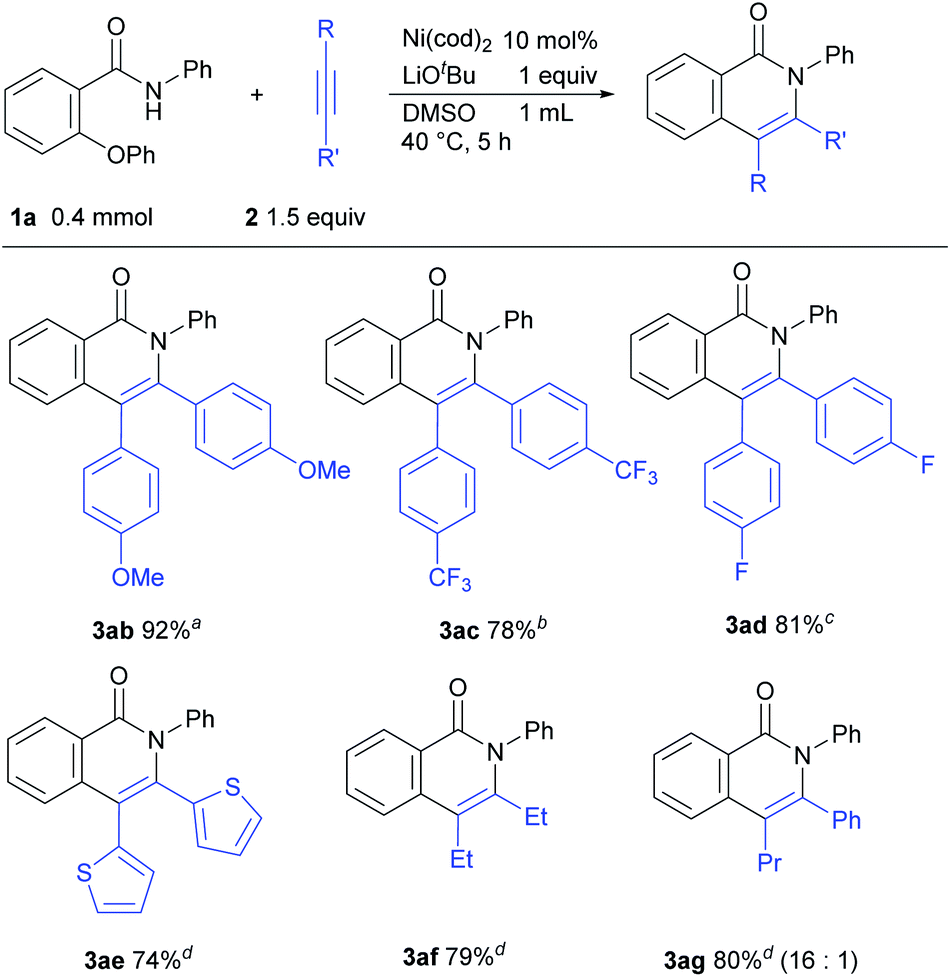 | ||
| Scheme 3 Scope of alkynes. Reaction conditions: 1a (0.4 mmol), alkyne (0.6 mmol), Ni(cod)2 (0.04 mmol), LiOtBu (0.4 mmol) in DMSO (1 mL) at 40 °C for 5 h. Yields shown are the isolated yields. aDMF was used instead of DMSO as a solvent. b80 °C. c24 h. d60 °C. | ||
In the course of our examination of other oxygen-based leaving groups, carbamates 5 were also found to participate in the reaction. Under the same reaction conditions as those used in the reaction of the phenoxy substrate 1a, the expected products 3aa were formed, but the undesired by-product 6aa was also produced. After the brief optimization of the reaction conditions using 5c as a substrate, toluene was determined to be the solvent of choice at 100 °C.10 The effect of substituents on the nitrogen-atom was next examined (Table 2). A carbamate having two methyl groups on the nitrogen atom 5a gave the desired product 3aa in 52% yield, but the chromone derivative 6aa was also obtained in 2% yield as a by-product (entry 1). The chromone 6aa appears to be produced by acyl transfer from the oxygen atom to the nitrogen atom (see ESI†). When a carbamate with two isopropyl groups 5d was used in the reaction, the desired product 3aa was obtained in high yield and the formation of 6aa was not observed (entry 4). The use of 5e gave 3aa in only 3% yield, although the starting material was completely consumed (entry 5).
|
|
||
|---|---|---|
| Entrya | R, R′ | NMR yieldsb3aa/6aa |
| a Reaction conditions: 5 (0.2 mmol), 2a (0.3 mmol), Ni(cod)2 (0.02 mmol), and KOtBu (0.2 mmol) in toluene (1 mL) at 100 °C for 3 h. b Yields were determined from 1H NMR with 1,1,2,2-tetrachloroethane as the internal standard. | ||
| 1 | Me, Me (5a) | 52%/2% |
| 2 | Me, Et (5b) | 74%/6% |
| 3 | Et, Et (5c) | 90%/4% |
| 4 | iPr, iPr (5d) | 98%/none |
| 5 | Ph, Ph (5e) | 3%/none |
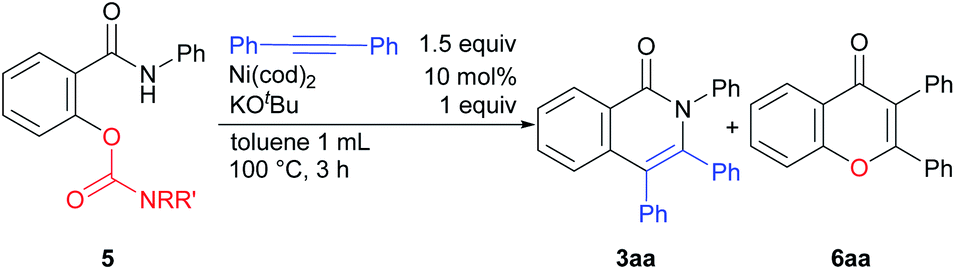
The results of reactions in which a carbamate is used as the leaving group are shown in Scheme 4. The reaction of substrates bearing an electron-donating group proceeded efficiently to give good yields of the desired products 3da and 3na. The reaction of carbamates with an electron-withdrawing fluoro group required a higher reaction temperature (120 °C) for good yields of 3oa to be obtained. The electronic effects of substituents on the alkyne had no effect on product yields (3ab and 3ac). In the reaction of heteroaromatic acetylene and aliphatic acetylene derivatives, a longer reaction time was necessary (3ae and 3af). When pivalate was used as a leaving group instead of a carbamate, in 5i, the desired product 3aa was obtained in lower yield and N-phenyl salicylamide was also produced in 38% NMR yield, which shows that the pivalate is unstable under basic conditions and C(O)–O bond is cleaved.
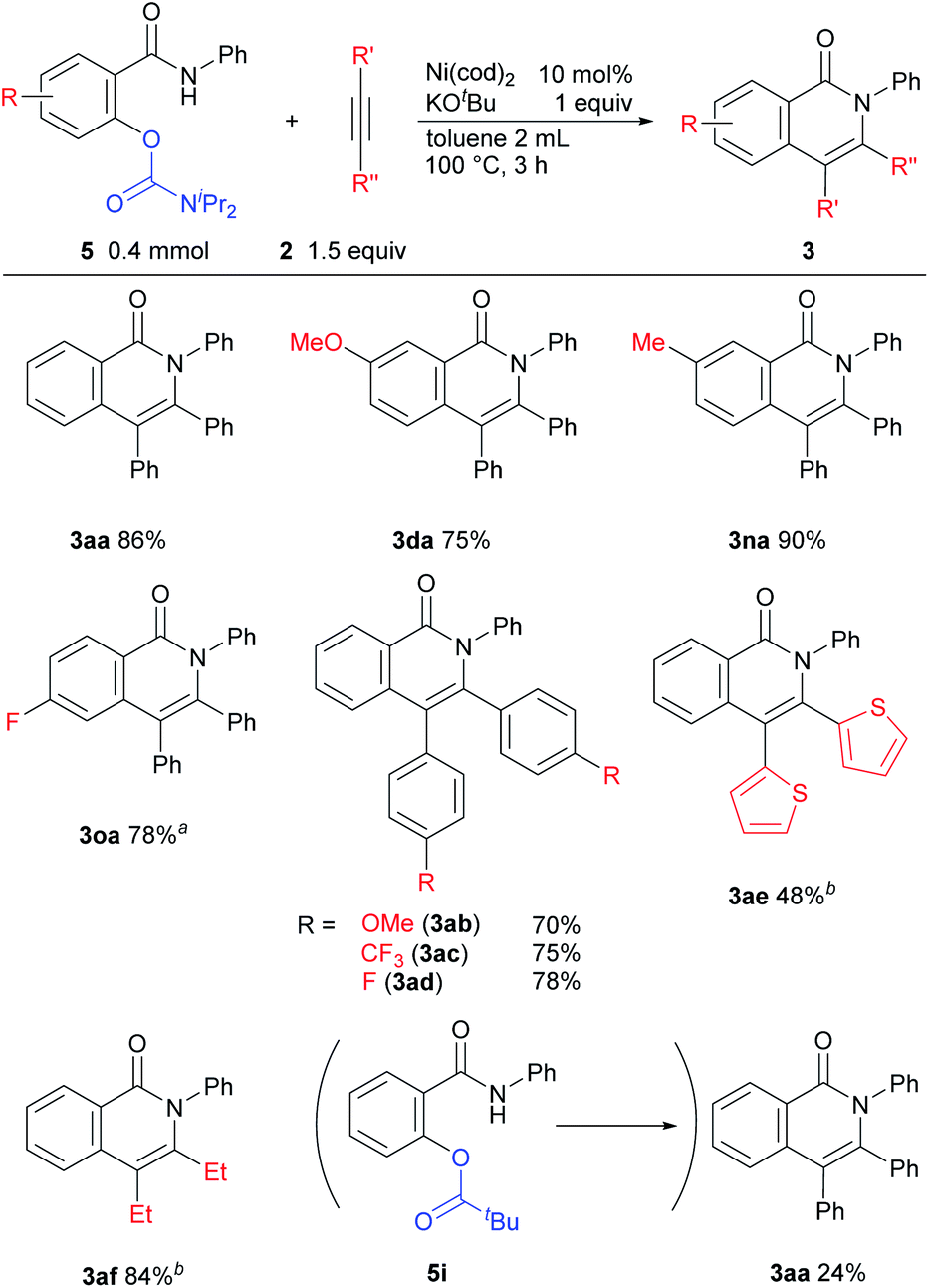 | ||
| Scheme 4 Substrate scope for the reaction of carbamates. Reaction conditions: amide (0.4 mmol), alkyne (0.6 mmol), Ni(cod)2 (0.04 mmol), KOtBu (0.4 mmol) in toluene (2 mL) for 3 h at 100 °C. Yields shown are isolated yields. a120 °C. b22 h. | ||
Some mechanistic studies were conducted to gain insights into the reaction mechanism (Scheme 5). Only a negligible effect was observed in the case of 1a, 1n, and 1o (Scheme 1a). In sharp contrast, the product 3aa was not formed, but, rather, an aryl group transfer product 4p was produced as a sole product in the reaction of aromatic amide 1p containing a substituted trifluoromethyl group, suggesting that a trifluoromethyl group promotes an aryl group transfer from an oxygen atom to a nitrogen atom. The reaction of the deuterium labeled substrate 1a-d was also examined (Scheme 1b). In this case, no H/D exchange was observed in both the starting material and the product. This result indicates that C–H bond activation did not occur under the reaction conditions employed11 and that only C–O bond activation took place in the reaction.
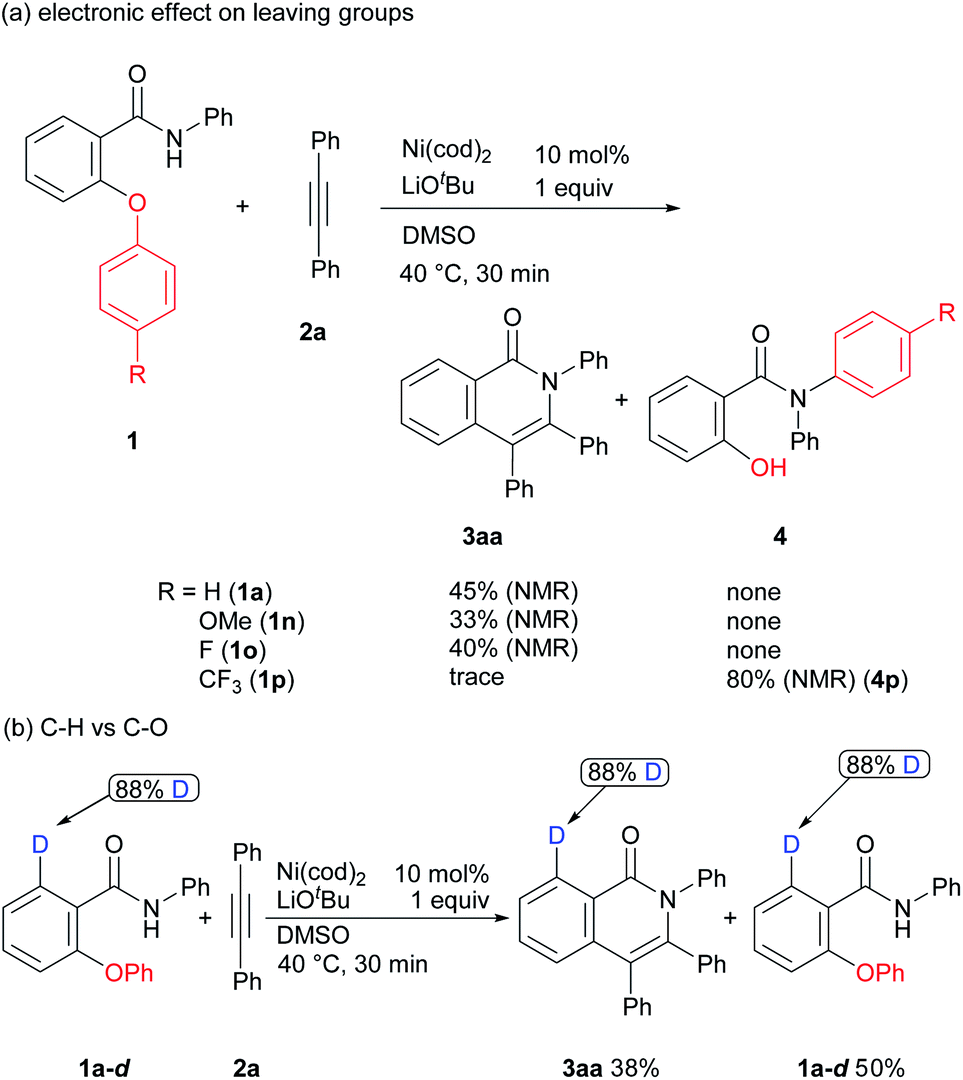 | ||
| Scheme 5 Mechanistic studies. | ||
A plausible reaction mechanism is shown in Scheme 6. The amide 1a reacts with a base to produce the lithium amidate A. The amidate A reacts with a Ni catalyst to give the anionic Ni amide complex B or C. The oxidative addition of a C–O bond produces a five-membered nickellacycle D with the generation of LiOPh. The insertion of an alkyne followed by reductive elimination gives the product 3aa with the regeneration of the Ni(0) species. A possible pathway for the formation of 4a involves an intramolecular SNAr type reaction viaF, which is consistent with the experimental results showing that a trifluoromethyl group facilitates the formation of 4a, as shown in Scheme 5a.
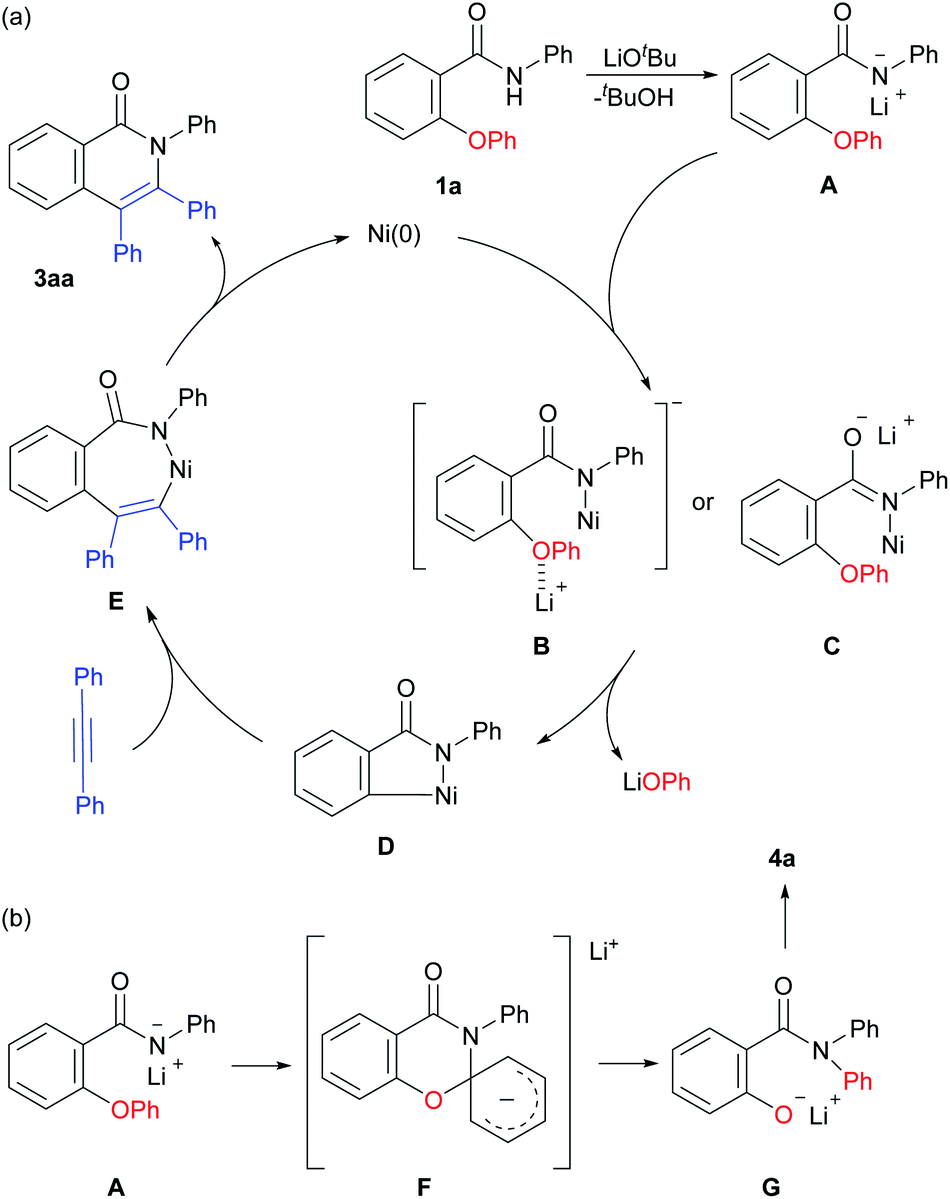 | ||
| Scheme 6 A plausible mechanism. | ||
In the above mechanism, LiOPh is generated in the reaction. We hypothesized that a catalytic amount of LiOPh could be used instead of a stoichiometric amount of LiOtBu. As expected, the reaction proceeded even in the presence of a catalytic amount of LiOPh (Scheme 7). However, a higher reaction temperature was necessary for the reaction to proceed efficiently, so we concluded that LiOtBu was the optimal base.
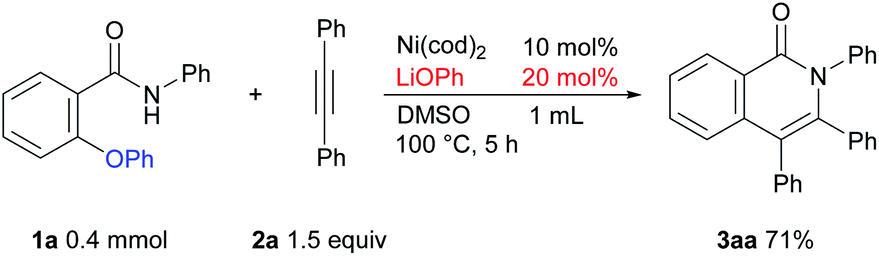 | ||
| Scheme 7 The reaction using a catalytic amount of LiOPh. | ||
We also examined the issue of whether this methodology might also be applicable to other strong bonds (Scheme 8). Gratifyingly, it was found that C–S bond activation also occurs to give the desired products.12 We also examined the possibility of C–CN bond activation, which afforded the desired products.13
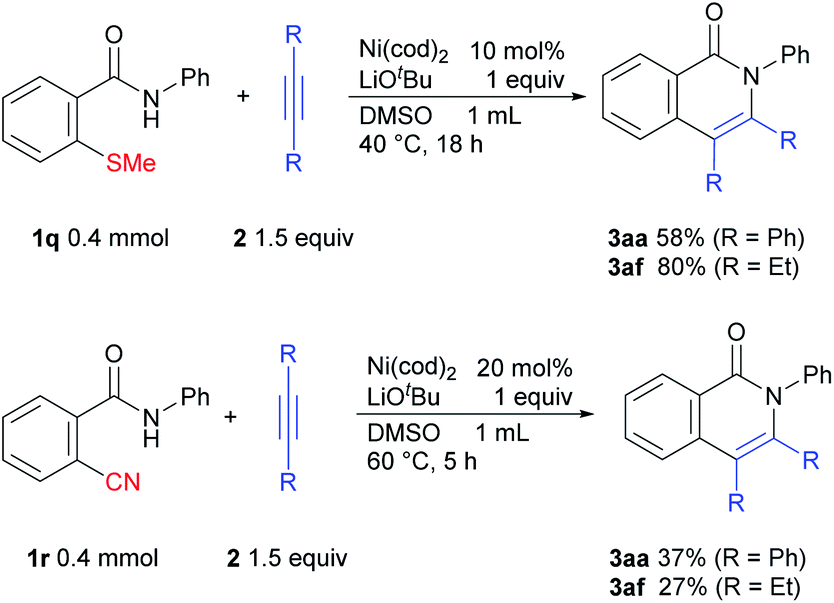 | ||
| Scheme 8 C–S and C–CN bond activation reactions. | ||
Conclusions
In summary, we report herein on the Ni-catalyzed C–O/N–H, C–S/N–H and C–CN/N–H annulation of amides with alkynes, leading to the production of 1(2H)-isoquinolinones. The reaction proceeded in the absence of ligands under low temperature and a wide variety of important functional groups was tolerated. The new methodology reported herein, such as the amidate-promoted activation of C–O bonds is applicable to the activation of other unreactive bonds, such as C–S and C–CN. Studies of the use of this methodology are currently underway and will be reported in due course.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a Grant in Aid for Specially Promoted Research by MEXT (No. 17H06091). We wish to thank the Instrumental Analysis Center, Faculty of Engineering, Osaka University, for assistance with the elemental analyses.Notes and references
- For selected recent reviews on C–O bond activation, see:
(a) B.-J. Li, D.-G. Yu, C.-L. Sun and Z.-J. Shi, Chem.–Eur. J., 2011, 17, 1728–1759 CrossRef CAS PubMed
; (b) B. M. Rosen, K. W. Quasdorf, D. A. Wilson, N. Zhang, A.-M. Resmerita, N. K. Garg and V. Percec, Chem. Rev., 2011, 111, 1346–1416 CrossRef CAS PubMed
- For transformation of C–O bonds with unsaturated bonds, see:
(a) A. R. Ehle, Q. Zhou and M. P. Watson, Org. Lett., 2012, 14, 1202–1205 CrossRef CAS PubMed
- I. Nohira, S. Liu, R. Bai, Y. Lan and N. Chatani, J. Am. Chem. Soc., 2020, 142, 17306–17311 CrossRef CAS PubMed
- For selected recent reviews on the synthesis of isoquinolinones via the cleavage of C–H bonds, see:
(a) V. P. Boyarskiy, D. S. Ryabukhin, N. A. Bokach and A. V. Vasilyev, Chem. Rev., 2016, 116, 5894–5986 CrossRef CAS PubMed
- For selected examples on the synthesis of isoquinolinones via the cleavage of C–X bonds, see:
(a) Y. Kajita, S. Matsubara and T. Kurahashi, J. Am. Chem. Soc., 2008, 130, 6058–6059 CrossRef CAS PubMed
- Hartwig reported on the hydrogenolysis of diaryl ether. A. G. Sergeev and J. F. Hartwig, Science, 2011, 332, 439–443 CrossRef CAS PubMed
- Wang and Uchiyama reported on the Ni-catalyzed reaction of aromatic esters with organoaluminum reagents. H. Ogawa, Z.-K. Yang, H. Minami, K. Kojima, T. Saito, C. Wang and M. Uchiyama, ACS Catal., 2017, 7, 3988–3994 CrossRef CAS
- For recent reviews on C–F activation, see:
(a)
S.-D. Yan, in Homogenous Catalysts for Unreactive Bond Activation, ed. Z.-J. Shi, John Wiley & Sons, Hoboken, N. J., 2015, pp. 203–268 Search PubMed
- For recent reviews on C–CN activation, see:
(a)
Y. Nakao, in C–C Bond Activation, ed. G. Dong, Springer Berlin Heidelberg, Berlin, 2014, pp. 33–58 Search PubMed
- See ESI† for details..
-
(a) A. Obata, Y. Ano and N. Chatani, Chem. Sci., 2017, 8, 6650–6655 RSC
- For recent reviews on C–S activation, see:
(a) F. Pang and Z.-J. Shi, ACS Catal., 2014, 4, 280–288 CrossRef
- The reaction was not optimized yet. 2-Cyanobenzamides are known to eaisly undergo cyclization to 3-imino-1-oxoisoindolines in the presence of a base.
(a) H. Li, W. Li, A. Spannenberg, W. Baumann, H. Neumann, M. Beller and X.-F. Wu, Chem.–Eur. J., 2014, 20, 8541–8544 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc06056a |
| This journal is © The Royal Society of Chemistry 2021 |