DOI:
10.1039/D0SC05777K
(Edge Article)
Chem. Sci., 2021,
12, 3929-3936
Ligand-enforced intimacy between a gold cation and a carbenium ion: impact on stability and reactivity†
Received
19th October 2020
, Accepted 8th January 2021
First published on 8th January 2021
Abstract
Controlling the reactivity of transition metal complexes by positioning non-innocent functionalities around the catalytic pocket is a concept that has led to significant advances in catalysis. Here we describe our efforts toward the synthesis of dicationic phosphine gold complexes of general formula [(o-Ph2P(C6H4)Carb)Au(tht)]2+ decorated by a carbenium moiety (Carb) positioned in the immediate vicinity of the gold center. While the most acidic examples of such compounds have limited stability, the dicationic complexes with Carb+ = 9-N-methylacridinium and Carb+ = [C(ArN)2]+ (ArN = p-(C6H4)NMe2) are active as catalysts for the cycloisomerization of N-propargyl-4-fluorobenzamide, a substrate chosen to benchmark reactivity. The dicationic complex [(o-Ph2P(C6H4)C(ArN)2)Au(tht)]2+, which also promotes hydroarylation and enyne cyclization reactions, displays a higher catalytic activity than its acridinium analog, indicating that the electrophilic reactivity of these complexes scales with the Lewis acidity of the carbenium moiety. These results support the role of the carbenium unit as a non-innocent functionality which can readily enhance the activity of the adjacent metal center. Finally, we also describe our efforts toward the generation and isolation of free γ-cationic phosphines of general formula [(o-Ph2P(C6H4)Carb)]+. While cyclization into phosphonium species is observed for Carb+ = [C(ArN)2]+, [C(Ph)(ArN)]+, and 9-xanthylium, [(o-Ph2P(C6H4)-9-N-methylacridinium)]+ can be isolated as an air stable, biphilic derivative with uncompromised Lewis acidic and basic properties.
Introduction
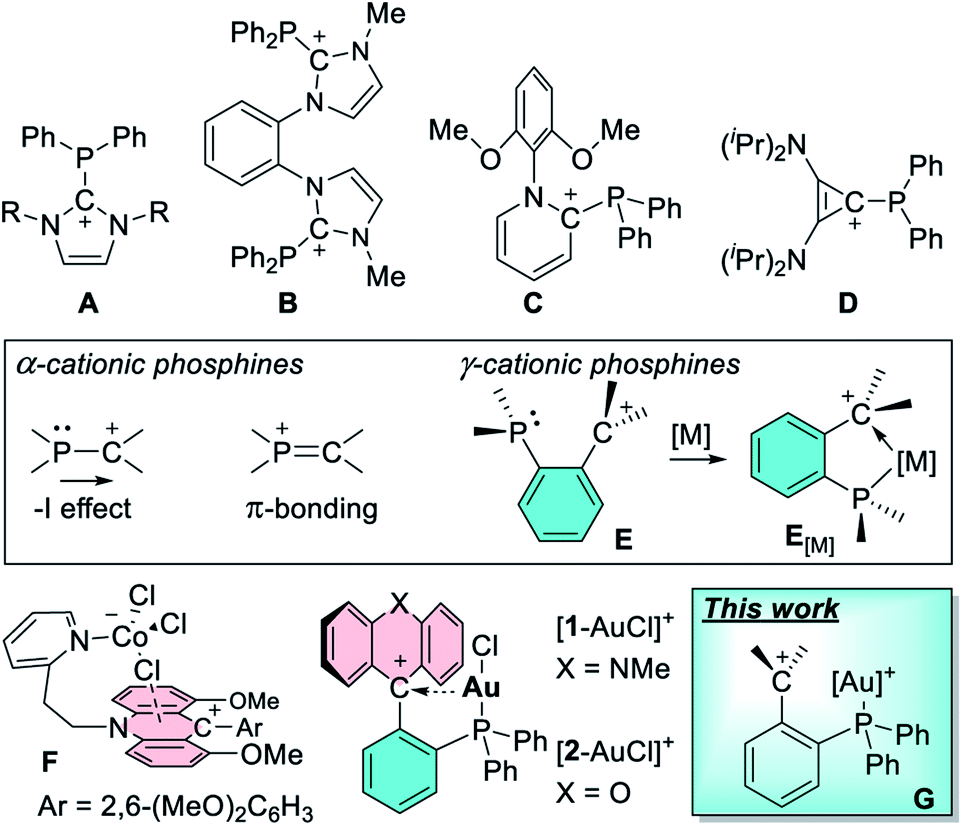
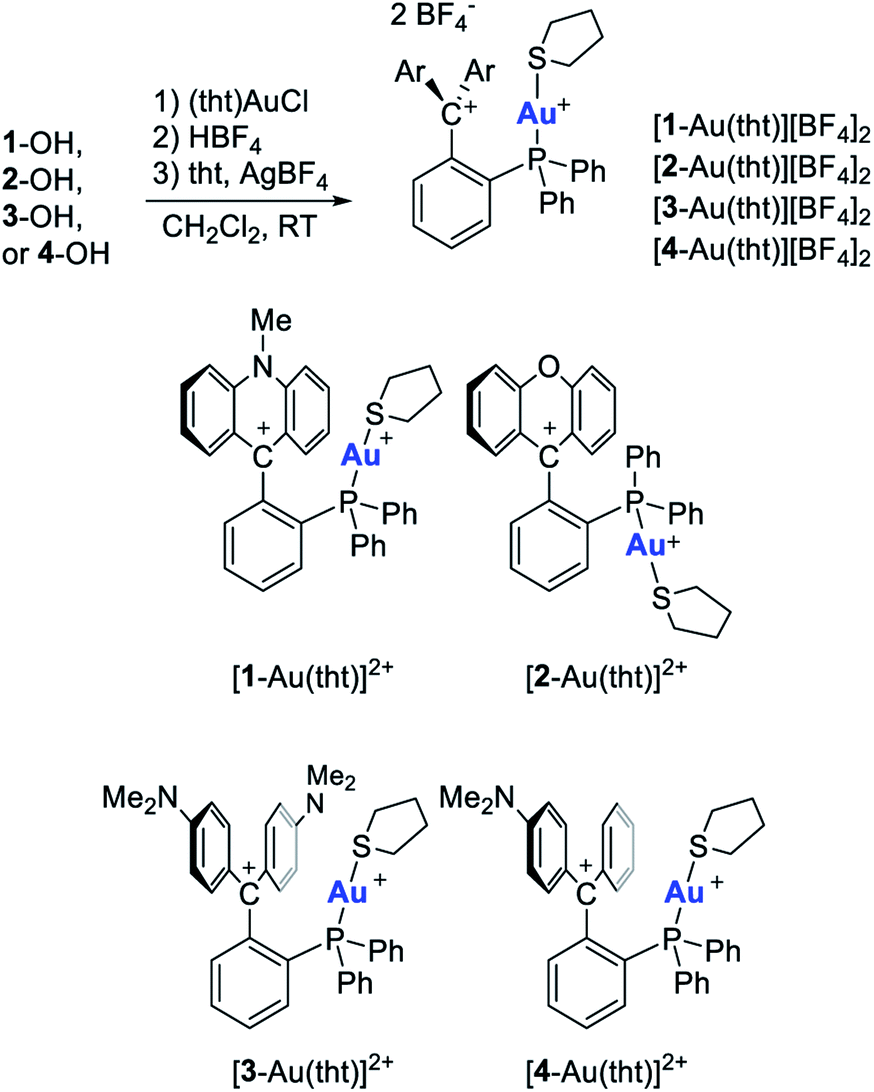
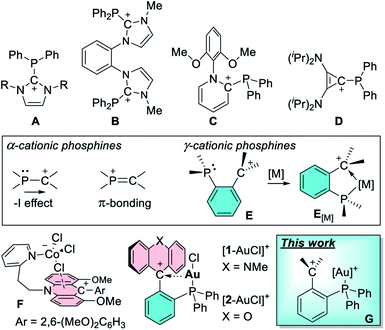
With the control of catalytic reactivity as an ultimate goal, efforts towards the design of new ligand scaffolds have remained a central thrust of modern coordination chemistry and one that has recently fueled numerous advances in the chemistry of phosphine-based ligands.1,2 While the coordination chemistry of phosphine ligands has traditionally emphasized their donor properties, recent efforts have explored the induction of π-acidity by incorporation of electron withdrawing substituents.3–8 This strategy, which rests on an energy lowering of phosphorus-centered σ*-orbitals,3 has been revitalized by the advent of cationic phosphines9–12 such as A,13,14B,15C,16 and D17 (Fig. 1). These phosphines bear a carbenium ion directly connected to the phosphorus center, setting the stage for an inductive depletion of electron density at the phosphorus center.18,19 At the same time the carbenium ion can also engage the phosphorus lone pair in a π-interaction, further accentuating electron-density depletion at the pnictogen atom.20 These effects, the significance of which depends on the nature of the phosphorus and carbenium ion substituents, elevate the π-acidity of the phosphorus atom, leading to notable benefits in late transition metal catalysis.9–11,17,21–27 Stimulated by these advances, we have recently become interested in a variant of the above-mentioned α-phosphine–carbenium motif in which an ortho-phenylene linker is inserted between the two moieties (E, Fig. 2).21 In these phosphines, which we refer to as γ-cationic phosphines, the carbenium ion and the phosphorus atom lack a direct connection and are thus partly insulated from one another. However, the carbenium ion is positioned to act as a Z-type ligand,28–32 capable of interacting directly with the metal center through a M → C+ interaction33–35 as in complexes of type E[M].36 Interactions are also possible between the carbenium unit and metal-bound anionic ligands, as evoked by Gianetti for complexes of type F.37 Because such γ-cationic phosphines are prone to phosphonium ion formation via “coordination” of the phosphorus atom to the carbenium center,38 we have developed a strategy that provides direct access to the gold complexes [1-AuCl]+ and [2-AuCl]+ (Fig. 1).39 We found that [1-AuCl]+ efficiently catalyze reactions such as the cyclo-isomerization of propargyl amide without the addition of any activators. We proposed that the emergence of catalytic activity for these gold chloride derivatives results from the formation of an Au → C+ interaction that hardens the gold center and enhances its electrophilic reactivity. Because these complexes possess an Au–Cl bond, we have now decided to target dicationic derivatives of type G by chloride anion abstraction. We speculated that the convergence of two cationic moieties at the core of these complexes may further enhance their electrophilic reactivity. In this article, we investigate this possibility by describing a series of such dicationic complexes as well as their evaluation as carbophilic catalysts. We also describe the isolation of an uncomplexed γ-cationic phosphine of type E.
 |
| | Fig. 1 Top: the carbenium resonance form of arbitrarily selected examples of α-cationic phosphines. Inset: possible electronic effects at play in α- and γ-cationic phosphines. Bottom: recently isolated examples of complexes featuring pendent carbenium units and dicationic complexes targeted in this study. | |
 |
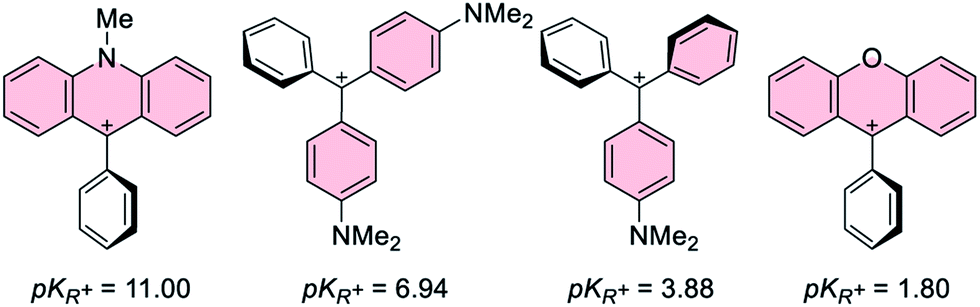
| | Fig. 2 pKR+ values of selected carbenium ions. The resonance structures shown are those corresponding to the carbenium ion.41,42 | |
Results and discussion
Synthesis and reactivity of cationic phosphine ligands
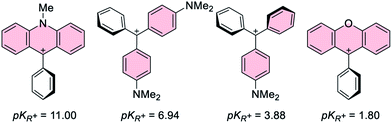
Given that the pKR+ values40 of the carbenium ions present in [1-AuCl]+ and [2-AuCl]+ differ by 9 orders of magnitude (Fig. 2), we reasoned that our understanding of these systems would benefit from the synthesis of additional derivatives with carbenium moieties of intermediate pKR+ values. With this in mind and guided by the known pKR+ values of [Ph2CArN]+ and [PhC(ArN)2]+ (ArN = p-(C6H4)NMe2) (Fig. 2),41 we decided to target complexes featuring carbenium ions stabilized by the p-dimethylaminophenyl substituent.
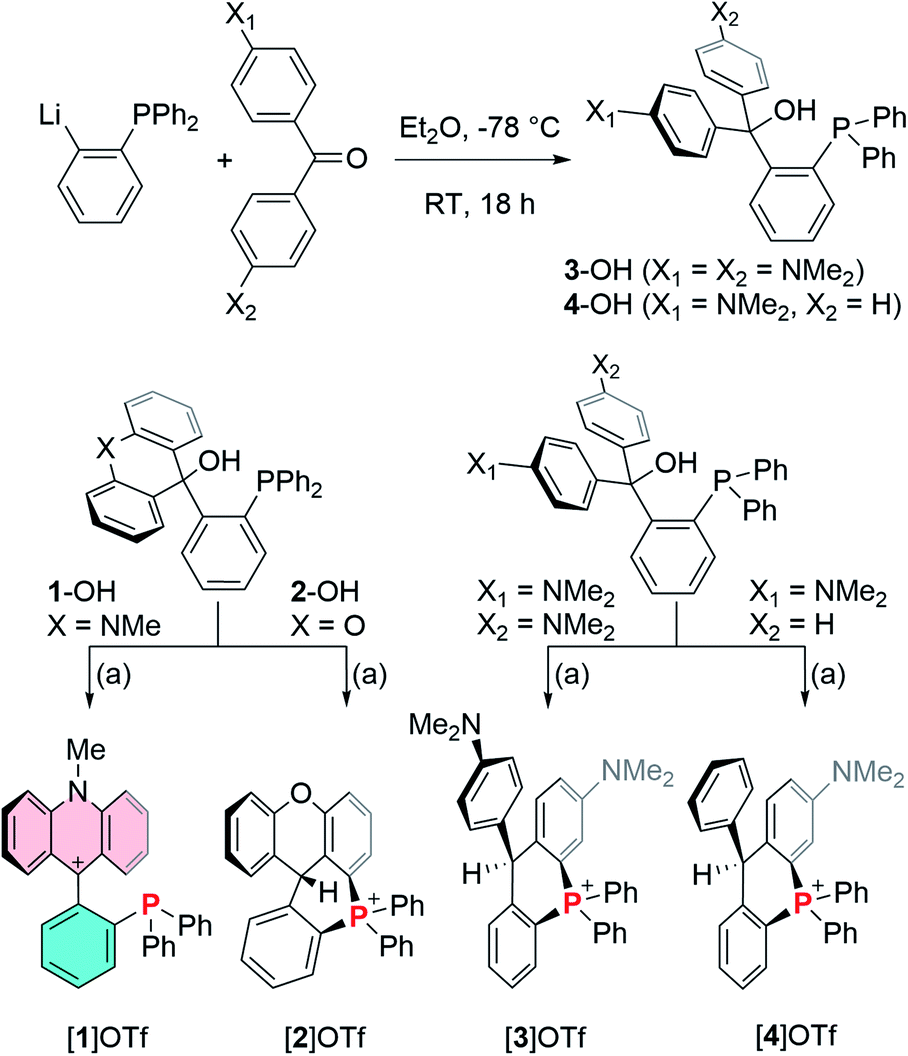
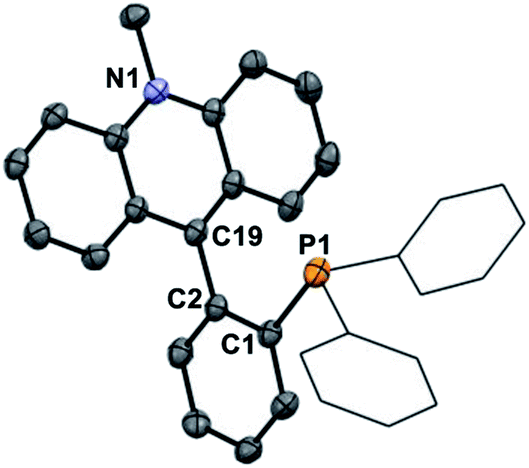
To this end, 1-lithio-2-diphenylphosphino-benzene was allowed to react with 4,4′-bis(dimethylamino)benzophenone or 4-(dimethylamino)benzophenone leading to the formation of the phosphinocarbinols 3-OH and 4-OH, respectively (Scheme 1). After characterization of 3-OH and 4-OH including by X-ray diffraction (see ESI†),43 we investigated their dehydroxylation reactions as a means to access the phosphine carbenium cations. Carbinols 1-OH and 2-OH,39 were also included in these studies. After investigating a few dehydrating agents, we found that treatment of 1-OH with trimethylsilyl triflate (TMSOTf) in CH2Cl2 afforded the phosphine acridinium [1]+ (referred to as EliPhos) as a triflate salt (Scheme 1). Conversion of 1-OH into [1]+ is accompanied with the appearance of a 31P NMR resonance at −14.3 ppm, significantly downfield from that of 1-OH (−19.9 ppm) (Scheme 2). The 1H and 13C NMR spectra of [1]+ is consistent with the presence of an acridinium unit.44–47 In particular, the nitrogen-bound methyl resonance at 4.81 ppm is shifted downfield with respect to that in the carbinol precursor 1-OH (3.64 ppm). The carbenium centre gives rise to a 13C NMR signal at 162.6 ppm which appears as a doublet (J = 6.3 Hz), which we presume arises from coupling to the neighboring phosphorus atom. Salt [1]OTf is air stable but should be stored in the dark to prevent oxidation. The crystal structure of this cationic phosphine has been determined (Fig. 3).43 The structure of [1]+ is reminiscent of that determined for boron-based ambiphilic derivatives, most of which exhibit significant P → C bonding.48–51 Despite a phosphorus–carbenium separation of 3.11 Å in [1]+, NBO analysis at the optimized geometry (see ESI†) does not show any notable P → C bonding thus indicating that the donor properties of the phosphorus atom are uncompromised. This conclusion is supported experimentally by the observation that [1]OTf reacts with (tht)AuCl to afford [1-AuCl]+.
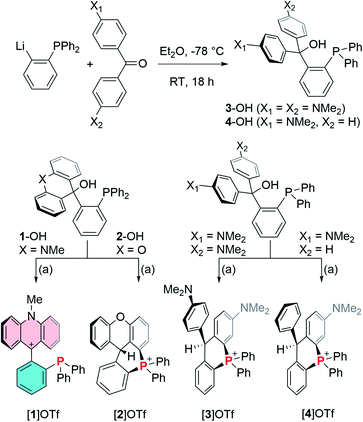 |
| | Scheme 1 Top: synthesis of carbinols 3-OH and 4-OH. Bottom: synthesis of [1]OTf, [2]OTf, [3]OTf and [4]OTf by dehydroxylation of the corresponding carbinols. (a) TMSOTf, CH2Cl2, RT. | |
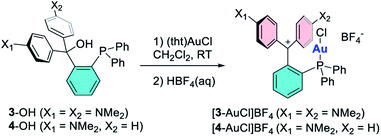 |
| | Scheme 2 Top: synthesis of the gold complexes [3-AuCl]BF4 and [4-AuCl]BF4. | |
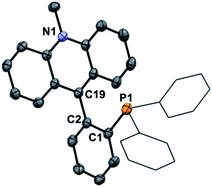 |
| | Fig. 3 ORTEP representation of the structure of [1]OTf. Hydrogen atoms and OTf− counterion omitted for clarity. Thermal ellipsoids drawn at 50% probability and phenyl groups drawn as thin lines. | |
Encouraged by the successful synthesis of [1]OTf, carbinols 2-OH, 3-OH and 4-OH were also treated with TMSOTf in CH2Cl2. A rapid screening of the product of these reactions by 31P NMR spectroscopy were inconsistent with the formation of the targeted cationic phosphines. Indeed, the products of these reactions, referred to as [2]OTf, [3]OTf and [4]OTf give rise to 31P NMR resonances at 8.6 ppm for [2]OTf, 3.4 ppm for [3]OTf, and 3.9 ppm for [4]OTf, consistent with the presence of phosphonium centers and indicative of cyclization reactions as depicted in Scheme 1. The formation of these cyclized phosphonium species has been confirmed by X-ray analysis (see ESI†).43 Formation of a six membered phosphonium ring is accompanied by a hydrogen atom migration from the aromatic CH group attacked by the phopshine to the methylium center. These rearrangements are reminiscent of those recently described by Takaya for related ortho-phenylene phosphinoboranes and their cyclization into phosphonium borates.52 A parallel also exists with the work of Romero-Nieto on the synthesis phosphonium-containing heterocycles by cyclization.53 Hydrogen atom migration can be confirmed by the appearance of an aliphatic signal in the 1H NMR spectrum of the compounds. A similar reaction was observed upon dehydroxylation and heating of o-(Ph2P)C6H4(CPh2(OH)) which also affords a cyclic phosphonium cations upon dehydroxylation.38 Bimolecular analogs of such reactions involving phosphines and tritylium cations are also known.54
Cationic gold complexes
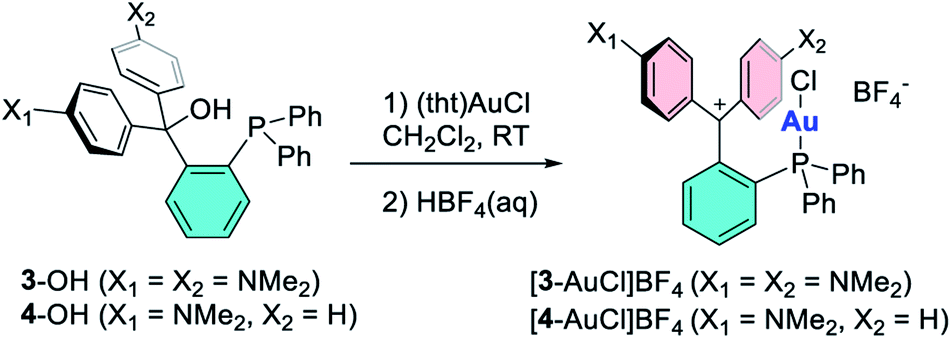
Since we were unable to isolate [3]+ and [4]+ in their uncyclized phosphine carbenium form, we decided to access their corresponding gold complexes by reaction of the carbinols with (tht)AuCl, followed by treatment with 1 equiv. of HBF4.39 This method afforded the target monocationic complexes [3-AuCl]+ and [4-AuCl]+ as air stable tetrafluoroborate salts that are blue/green and red, respectively (Scheme 2). The formation of these complexes is easily followed by NMR spectroscopy which shows a shift of the 31P NMR resonance from −15.9 to 25.9 ppm upon conversion of 3-OH into [3-AuCl]+ and from −16.0 to 25.8 ppm upon conversion of 4-OH into [4-AuCl]+. The presence of the dimethyl amine protons gives rise to a 1H NMR resonance at 2.88 ppm for both 3-OH and 4-OH. These resonances shift downfield upon dehydroxylation to 3.21 ppm for [3-AuCl]+ and 3.42 and 3.51 ppm in the case of [4-AuCl]+.
The main distinguishing feature among the four gold chloride complexes considered in this study is the nature of the carbenium unit. The pKR+ of the four parent carbenium derivatives shown in Fig. 2 suggests that the carbenium unit of these complexes falls in the following Lewis acidity order: [1-AuCl]+ < [3-AuCl]+ < [4-AuCl]+ < [2-AuCl]+.41,42 Given the metallobasic character of gold(I),55 one could anticipate that a more Lewis acidic carbenium center would result in a stronger Au → C+ interaction. However, the crystal structure of [3-AuCl][BF4] and [4-AuCl][BF4],43 and those previously determined for [1-AuCl][BF4] and [2-AuCl][BF4] do not indicate the existence of such a trend.39 Indeed, the Au–C+ distances observed for [3-AuCl][BF4] (3.209(3) Å) and [4-AuCl][BF4] (3.250(6) Å) are both longer than that in [1-AuCl][BF4] (3.168(9) Å) which possesses a less acidic carbenium unit. A discrepancy also exists between [3-AuCl][BF4] and [4-AuCl][BF4] since the less Lewis acidic carbenium unit of [3-AuCl][BF4] forms a Au–Ccarbenium contact that is shorter than that in [3-AuCl][BF4]. The lack of correlation between the Au–Ccarbenium distance and the Lewis acidity of the carbenium unit indicates that Au → Ccarbenium interactions, if present, are too weak or too sterically congested to impact the structures of these derivatives. Elaborating on the influence of steric congestion, we assign the larger Au–Ccarbenium distances in [3-AuCl][BF4] and [4-AuCl][BF4] to the greater steric demand of their carbenium moieties which are not planarized by incorporation in a six-membered ring as in the case of [1-AuCl][BF4] and [2-AuCl][BF4]. The greater encumbrance of the carbenium moieties in [3-AuCl][BF4] and [4-AuCl][BF4] also manifests in the large value of αd, defined as the dihedral angle existing between the Au1–P1–C1 plane and the C1–C2–C19 plane (Fig. 4). Indeed the αd values, which are listed in Table 1, are notably larger for [3-AuCl][BF4] (43.1(3)°) and [4-AuCl][BF4] (34.5(6)°) than for [1-AuCl][BF4] (10.0(9)°) and [2-AuCl][BF4] (1.36(18)°).
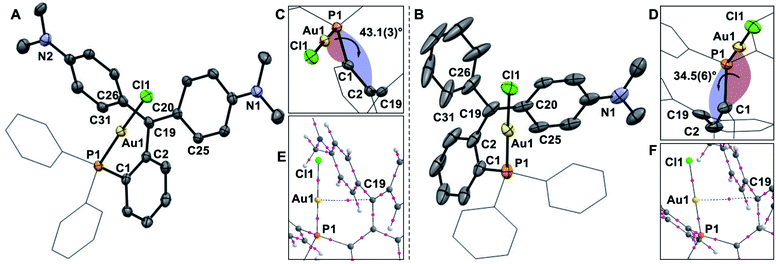 |
| | Fig. 4 ORTEP representations of the structures of [3-AuCl][BF4] (A) and [4-AuCl][BF4] (B), with a close up of the torsion angle (C and D). Hydrogen atoms and BF4− counterions omitted for clarity. Thermal ellipsoids drawn at 50% probability and phenyl groups drawn as thin lines. Selected bond lengths (Å) and angles (°) for [3-AuCl][BF4]: C(19)–C(2) 1.500(4), C(19)–C(20) 1.413(5), C(19)–C(26) 1.427(5); C(2)–C(19)–C(20) 117.2(3), C(20)–C(19)–C(26) 125.2(3), C(26)–C(19)–C(2) 117.6(3). Selected bond lengths (Å) and angles (°) for [4-AuCl][BF4]: C(19)–C(2) 1.494(11), C(19)–C(20) 1.365(10), C(19)–C(26) 1.473(7); C(2)–C(19)–C(20) 119.0(5), C(20)–C(19)–C(26) 124.8(6), C(26)–C(19)–C(2) 116.1(6). Graphical output of the AIM analysis (E and F) showing the bond critical point between Au1 and C19 at the optimized geometry of [3-AuCl]+ and [4-AuCl]+. | |
Table 1 Compilation of key spectroscopic and structural features of the gold complexes discussed in this study. [1-AuCl][BF4] and [1-AuCl][BF4] were described in an earlier publication39
|
|
δ(31P) |
Au–C+ (Å) |
α
d (°) |
| [1-AuCl][BF4] |
22.4 |
3.168(9) |
10.0(9)° |
| [1-Au(tht)][BF4]2 |
27.5 |
3.320(4) |
47.4(4)° |
| [2-AuCl][BF4] |
24.5 |
3.131(3) |
1.36(18)° |
| [2-Au(tht)][BF4]2 |
28.9 |
4.381(3) |
91.8(3)° |
| [3-AuCl][BF4] |
25.9 |
3.209(3) |
43.1(3)° |
| [3-Au(tht)][BF4]2 |
29.2 |
3.290(13) |
28.4(9)° |
| [4-AuCl][BF4] |
25.8 |
3.250(6) |
34.5(6)° |
| [4-Au(tht)][BF4]2 |
29.5 |
3.376(10) |
37.2(9)° |
A computational investigation of [3-AuCl][BF4] and [4-AuCl][BF4] leads to results that are consistent with the weakness of the Au → Ccarbenium interactions present in these structures. Indeed, an atoms-in-molecules (AIM) analysis shows that the electron density (ρ(r)) at the critical point of the path connecting the gold atom to the carbenium center are equal to 1.31 × 10−2 e per bohr3 for [3-AuCl]+ and 1.32 × 10−2 e per bohr3 for [4-AuCl]+ (Fig. 4). These values are about an order of magnitude lower than those of covalent Au–C bonds, indicating that the Au → Ccarbenium interactions in [3-AuCl]+ and [4-AuCl]+ are weak. This conclusion is supported by the results of natural bond orbital calculations that show only weak donor–acceptor bonding (E2 < 1 kcal) between the gold atom and the adjacent carbenium units (see ESI†).
Dicationic complexes
Cationic gold complexes of type H have been previously characterized,56–58 and also implicated as carbophilic catalysts.59–61 The flanking aryl ring in these structures provides stability to the cationic gold center, facilitating their isolation and deployment in catalysis. It occurred to us that chloride anion abstraction from [1-AuCl][BF4], [2-AuCl][BF4], [3-AuCl][BF4] and [4-AuCl][BF4] could provide access to the dicationic analogues of complexes of type H. To test this idea, a solution of [1-AuCl][BF4] in CH2Cl2 was treated with AgBF4 in the presence of tetrahydrothiophene (tht) (Scheme 3). This reaction proceeded smoothly to afford [1-Au(tht)][BF4]2 which could be isolated as an orange crystalline solid that slowly decomposes under ambient conditions, presumably because of liberation of the tht ligand as observed for other thioether–gold complexes.62 While the 1H NMR spectrum indicates an intact ligand backbone and the presence of a tht ligand, the 31P NMR resonance of [1-Au(tht)][BF4]2 appears at 27.5 ppm, downfield from the chemical shift of 22.4 ppm measured for [1-AuCl][BF4]. Going back to the pKR+ trend presented in Fig. 2, we became eager to establish whether dicationic complexes could also be obtained starting from the more electron-deficient complexes [2-AuCl][BF4], [3-AuCl][BF4], and [4-AuCl][BF4]. Gratifyingly, we observed formation of the corresponding dicationic complexes [2-Au(tht)][BF4]2, [3-Au(tht)][BF4]2, and [4-Au(tht)][BF4]2 (Scheme 3). As in the case of [1-Au(tht)][BF4]2, these new complexes show diagnostic NMR resonances that confirm the presence of a tht ligand bound to the gold atom. The 31P NMR chemical shift of the dicationic complexes (28.9 ppm for [2-Au(tht)][BF4]2, 29.2 ppm for [3-Au(tht)][BF4]2, and 29.5 ppm for [4-Au(tht)][BF4]2) are also slightly shifted downfield when compared to those of their gold chloride precursors (24.5 ppm for [2-AuCl][BF4], 25.9 ppm for [3-AuCl][BF4], and 25.8 ppm for [4-AuCl][BF4]) (Table 1). Attempts to isolate the acetonitrile (MeCN) complexes were also investigated. However, we only obtained evidence for the formation of [1-Au(MeCN)][BF4]2 but its isolation was irreproducible.
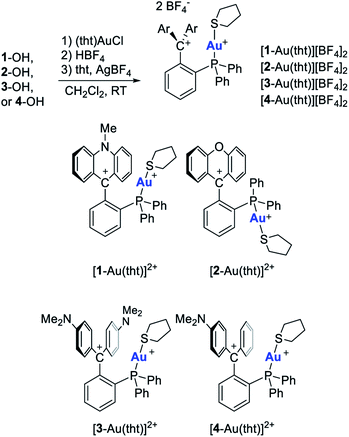 |
| | Scheme 3 Synthesis of the dicationic complexes. | |
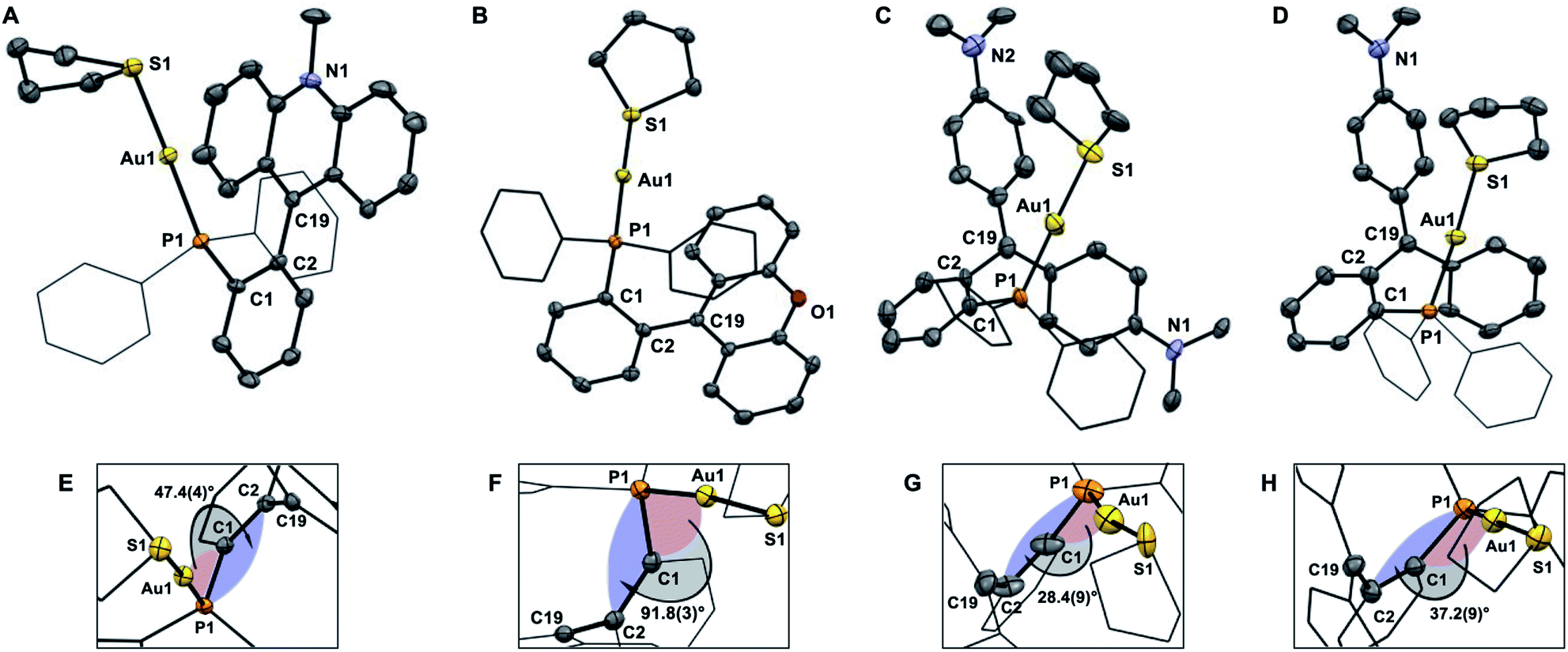
The crystal structures of the tetrafluoroborate salts of these dications have been determined crystallographically (Fig. 5).43 In all cases, we observe an increase in the separation between the gold center and the carbenium ion (see Table 1). This increase, which may reflect increased electrostatic repulsions between the cationic gold moiety and the carbenium ion, only amounts to ∼0.1–0.2 Å in the case of [1-Au(tht)][BF4]2, [3-Au(tht)][BF4]2, and [4-Au(tht)][BF4]2 (Table 1). By contrast, conversion of [2-AuCl]+ into [2-Au(tht)]2+ is accompanied by a 1.2 Å increase of the Au–C+ distance. At the same time, the dihedral angle of αd, increases from 1.36(18)° in [2-AuCl]+ to 91.8(3)° [2-Au(tht)]2+. We assign the unique structure change seen upon formation of [2-Au(tht)]2+ to the lower pKR+ of the xanthylium unit and the resulting increased electrostatic destabilization occurring at the core of these complexes.
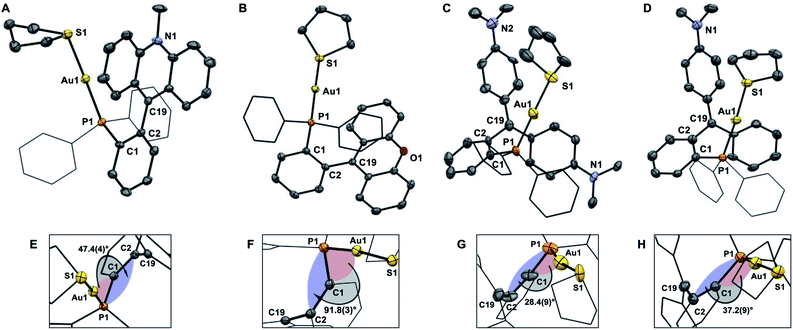 |
| | Fig. 5 ORTEP representations of the structures of [1-Au(tht)][BF4]2 (A), [2-Au(tht)][BF4]2 (B), [3-Au(tht)][BF4]2 (C), and [4-Au(tht)][BF4]2 (D), with a close up showing the torsion angle αd (E–H). Hydrogen atoms and BF4− counterions omitted for clarity. Thermal ellipsoids drawn at 50% probability and phenyl groups drawn as thin lines. Selected bond lengths (Å) and angles (°) for [1-Au(tht)][BF4]2: Au(1)–(P1) 2.2699(11), Au(1)–S(1) 2.3352(12), C(19)–C(2) 1.500(6), C(19)–C(20) 1.395(6), C(19)–C(26) 1.406(6); C(2)–C(19)–C(20) 122.1(4), C(20)–C(19)–C(26) 119.6(4), C(26)–C(19)–C(2) 118.3(4). Selected bond lengths (Å) and angles (°) for [2-Au(tht)][BF4]2: Au(1)–(P1) 2.2710(9), Au(1)–S(1) 2.3315(9), C(19)–C(2) 1.479(5), C(19)–C(20) 1.414(5), C(19)–C(26) 1.415(4); C(2)–C(19)–C(20) 121.5(3), C(20)–C(19)–C(26) 118.2(3), C(26)–C(19)–C(2) 120.2(3). Selected bond lengths (Å) and angles (°) for [3-Au(tht)][BF4]2: Au(1)–(P1) 2.251(5), Au(1)–S(1) 2.286(5), C(19)–C(2) 1.507(19), C(19)–C(20) 1.43(2), C(19)–C(26) 1.43(2); C(2)–C(19)–C(20) 116.3(15), C(20)–C(19)–C(26) 127.5(16), C(26)–C(19)–C(2) 116.2(15). Selected bond lengths (Å) and angles (°) for [4-Au(tht)][BF4]2: Au(1)–(P1) 2.281(2), Au(1)–S(1) 2.334(2), C(19)–C(2) 1.477(14), C(19)–C(20) 1.378(14), C(19)–C(26) 1.469(13); C(2)–C(19)–C(20) 120.3(9), C(20)–C(19)–C(26) 121.6(9), C(26)–C(19)–C(2) 117.2(9). | |
Reactivity studies
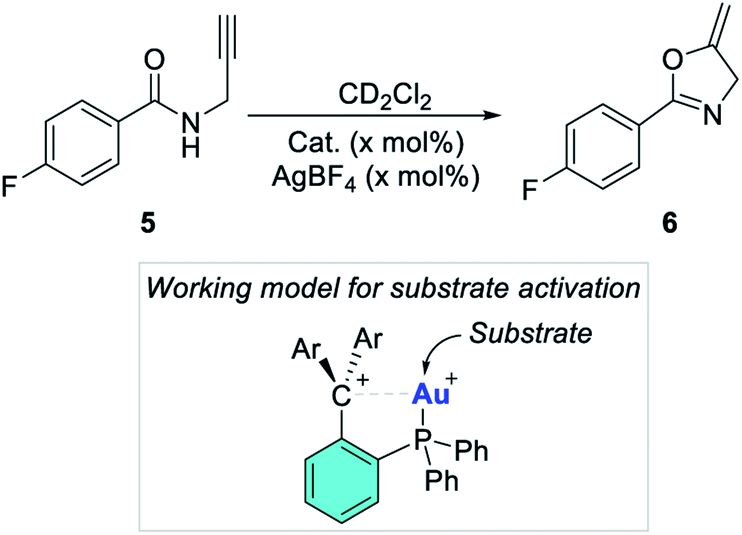
Having verified that the dicationic complexes are indeed accessible, we became eager to assess their performance in carbophilic catalysis. To this end, we selected the cycloisomerization of propargyl amide 5 as a benchmark reaction.63–65 We decided to investigate this reaction at room temperature with a 2% loading of catalyst in CD2Cl2 which was dried to avoid a possible interference of water with the carbenium ion. To establish a baseline, we first tested the activity of the four gold chloride complexes [1-AuCl]+, [2-AuCl]+, [3-AuCl]+, and [4-AuCl]+ in the absence of an activator (entries 1–4, Table 2). While [1-AuCl]+ and [3-AuCl]+ gave hardly any conversion (entries 1 and 3), [2-AuCl]+ and [4-AuCl]+ displayed moderate activity with conversion after 20 minutes of 10% and 7%, respectively (entries 2 and 4). The higher activity of [2-AuCl]+ and [4-AuCl]+ can be explained based on the greater acidity of the carbenium centres present in these structure and their ability to enhance the electrophilic character of the gold centre. This observation is aligned with the conclusion of our previous report on the catalytic properties of [1-AuCl]+ and [2-AuCl]+ in the same reaction, but at a higher temperature.39
Table 2 Catalytic activity of the complexes in the reaction shown in the Scheme 4. The conversion was measured by 1H NMR spectroscopy, in situ, using CD2Cl2 as a solvent
| Entry |
Catalyst (as BF4− salts) |
Mol% |
AgX, mol% |
Time (min) |
Conv. (%) |
| 1 |
[1-AuCl]+ |
2 |
0 |
20 |
0 |
| 2 |
[2-AuCl]+ |
2 |
0 |
20 |
10 |
| 3 |
[3-AuCl]+ |
2 |
0 |
20 |
2 |
| 4 |
[4-AuCl]+ |
2 |
0 |
20 |
7 |
| 5 |
[1-AuCl]+ |
2 |
AgBF4, 4 |
20 |
83 |
| 6 |
[3-AuCl]+ |
2 |
AgBF4, 4 |
20 |
>98 |
| 7 |
[3-AuCl]+ |
2 |
AgNTf2, 4 |
20 |
>98 |
| 8 |
[1-Au(tht)]2+ |
2 |
0 |
20 |
85 |
| 9 |
[3-Au(tht)]2+ |
2 |
0 |
20 |
>98 |
| 10 |
[1-Au(tht)]2+ |
0.5 |
0 |
180 |
39 |
| 11 |
[3-Au(tht)]2+ |
0.5 |
0 |
180 |
69 |
| 12 |
Ph3AuCl |
0.5 |
AgBF4, 1 |
180 |
14 |
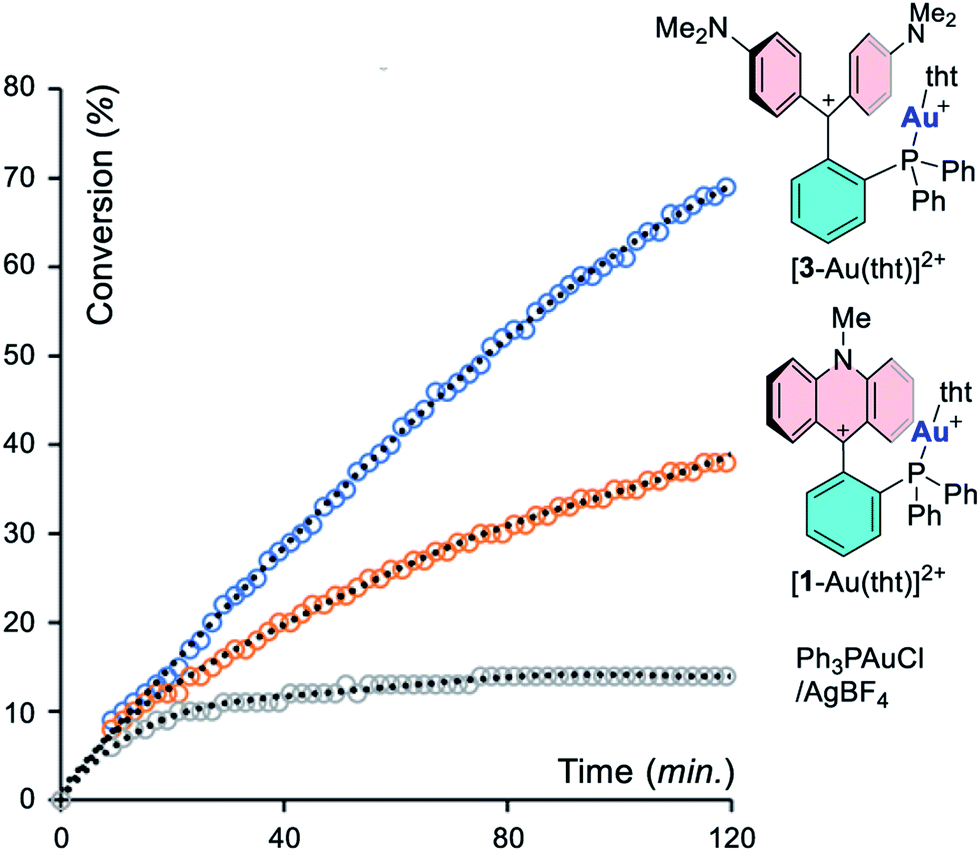
Next, we became eager to test the effect induced by the addition of an activator capable of engaging the gold-bound chloride ligand. To this end, the experiments in entries 1–4 were repeated in the presence of 2 mol% AgBF4. Treatment of [2-AuCl]+ and [4-AuCl]+ with AgBF4 resulted in the immediate production of a black precipitate indicating decomposition of the catalyst. We propose that the elevated acidity of the carbenium units present in these complexes jeopardizes the stability of the dications. In fact, addition of the reaction substrate to [2-Au(tht)]2+ and [4-Au(tht)]2+ also leads to decomposition as evinced by the formation of a dark residue. This decomposition suggests that the acidity of the 9-xanthylium and [C(Ph)(ArN)]+ carbenium units present in these complexes might be excessive. A different observation was made in the case of complexes [1-AuCl]+ and [3-AuCl]+ which underwent smooth activation with AgBF4. 1H NMR monitoring showed that the reaction reached 83% conversion after 20 minutes with [1-AuCl]+/AgBF4 (entry 5). The reaction was almost complete at the same time point when [3-AuCl]+/AgBF4 or [3-AuCl]+/AgNTf2 was employed (entry 6 and 7). We also observed that AgBF4 (10 mol%) alone did not promote this reaction. These results lead us to propose that the active catalysts are dicationic species resulting from abstraction of the gold-bound chloride ligand and resembling the tht adducts [1-Au(tht)]2+ and [3-Au(tht)]2+. In fact, these dicationic tht adducts can also be used as catalysts for these reactions (entries 8 and 9). Moreover, the activity of these two catalysts very closely matches those obtained with [1-AuCl]+/AgBF4 and [3-AuCl]+/AgBF4 suggesting that the active species are indeed dications as in the working model depicted in Scheme 4. We also note that the greater activity of [3-Au(L)]2+ correlates with the greater electron deficiency of the [CAr2N]+ moiety when compared to the 9-N-methyl-acridinium moiety present in [1-Au(L)]2+. This difference in activity allows us to introduce the notion that the reactivity of the metal center in these dicationic species is influenced by the acidity of the adjacent carbenium unit.
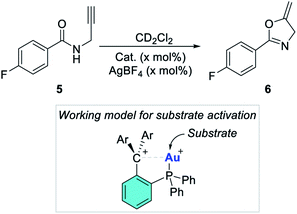 |
| | Scheme 4 Propargylamide cyclization used as a benchmark reaction. The inset shows a possible working model for substrate activation. | |
To better illustrate this effect, we repeated the experiment with a catalyst loading of 0.5% and monitored its progress by 1H NMR spectroscopy, using [1-Au(tht)][BF4]2 and [3-Au(tht)][BF4]2 as pre-catalysts (entries 10 and 11). This lower loading was selected to slow down the reaction such that its progress could be more easily monitored. The results of these experiments, presented in Fig. 6, unambiguously illustrates the superiority of [3-Au(L)]2+. We will also note that the reaction is initially catalyzed by Ph3PAuCl/AgBF4 (entry 12). However, after a few minutes, the reaction rate slows down dramatically, suggesting catalyst decomposition.
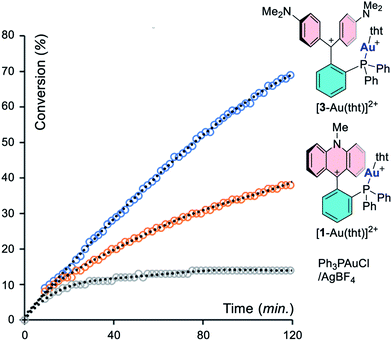 |
| | Fig. 6 Conversion of propargyl amide 5 into 6 as a function of time with different catalysts. | |
Given that [3-Au(tht)][BF4]2 emerged as the best catalyst, we decided to also test its activity in the hydroarylative cyclization of the propargyl ether 7 (ref. 66) as well as the cyclization of enyne 9 (Scheme 5).671H NMR monitoring showed that these reactions proceeded smoothly in 1–2 hours when carried out in CD2Cl2 with a 1 mol% catalyst loading. In the case of 9, the six membered-isomer 10a was formed predominantly which is not unusual when such reactions are mediated by phosphine–gold catalysts.68,69 Finally, we also tested these reactions using in situ-generated [(Ph3P)Au(tht)][BF4] as a catalyst.70 While this simple catalyst afforded the cycloisomerized isomer of 7 with a 32% conversion after 1 hour, a conversion of less than 1% was observed at the same time point for the enyne cyclization of 9.
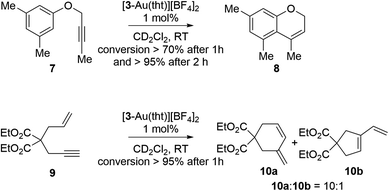 |
| | Scheme 5 Intramolecular hydroarylation and enyne cyclization reactions catalyzed by [3-Au(tht)][BF4]2. | |
Conclusions
This study demonstrates that ambiphilic ortho-phenylene-bridged γ-cationic phosphines can be isolated as in the case of the acridinium derivative [1]+ which resists cyclization via phosphonium ion formation. The stability of this derivative can be ascribed to the aromaticity of the acridinium core which tames its carbenium-like reactivity, leaving the phosphine exposed and available for subsequent reactions. Another important outcome of this study is the discovery that the ortho-phenylene γ-cationic phosphines considered in this work can also support the formation of dicationic gold(I) complexes of general formula [(o-Ph2P(C6H4)Carb)Au(tht)]2+, the stability of which appears to be influenced by the Lewis acidity of the carbenium unit (Carb+). Indeed when Carb+ = [C(Ph)(ArN)]+ or 9-xanthylium, the complexes are prone to facile decomposition. However in the presence of less electron deficient carbenium units (Carb+ = [C(ArN)2]+, and 9-N-methylacridinium), the complexes are sufficiently stable to be used as catalysts and readily promote the cycloisomerization of the propargyl amide used to benchmark these catalysts. Out of these two complexes, the variant possessing a more electron deficient bis-(para-dimethylamino-phenyl)carbenium unit is more active, thus suggesting that the carbophilic reactivity of the gold centre scales with the acidity of the carbenium unit. It follows that, if not excessive to the point of inducing decomposition of the catalysts, the carbenium ion of these complexes can be used to tune and enhance the reactivity of the adjacent cationic metal centre. Extension of this concept to other catalytic systems is ongoing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Acknowledgement is made to the Donors of the American Chemical Society Petroleum Research Fund for partial support of this research (61541-ND3). We also acknowledge support from the National Science Foundation (CHE-1856453), the Welch Foundation (A-1423), and Texas A&M University (Arthur E. Martell Chair of Chemistry).
Notes and references
-
A. Börner, Phosphorus Ligands in Asymmetric Catalysis: Synthesis and Applications, Wiley-VCH Weinheim, 2008 Search PubMed.
-
D. W. Allen, in Organophosphorus Chemistry: Volume 43, The Royal Society of Chemistry, 2014, vol. 43, pp. 1–51 Search PubMed.
- S. Hussein, D. Priester, P. Beet, J. Cottom, S. J. Hart, T. James, R. J. Thatcher, A. C. Whitwood and J. M. Slattery, Chem.–Eur. J., 2019, 25, 2262–2271 CrossRef CAS.
- K. D. Cooney, T. R. Cundari, N. W. Hoffman, K. A. Pittard, M. D. Temple and Y. Zhao, J. Am. Chem. Soc., 2003, 125, 4318–4324 CrossRef CAS.
- J. P. Genet, T. Ayad and V. Ratovelomanana-Vidal, Chem. Rev., 2014, 114, 2824–2880 CrossRef CAS.
- R. J. Harris, K. Nakafuku and R. A. Widenhoefer, Chem.–Eur. J., 2014, 20, 12245–12254 CrossRef CAS.
- K. K. Banger, A. K. Brisdon, C. J. Herbert, H. A. Ghaba and I. S. Tidmarsh, J. Fluorine Chem., 2009, 130, 1117–1129 CrossRef CAS.
- S. A. Blaszczyk, D. A. Glazier and W. Tang, Acc. Chem. Res., 2020, 53, 231–243 CrossRef CAS.
- Y. Canac, C. Maaliki, I. Abdellah and R. Chauvin, New J. Chem., 2012, 36, 17–27 RSC.
- M. Alcarazo, Chem.–Eur. J., 2014, 20, 7868–7877 CrossRef CAS.
- M. Alcarazo, Acc. Chem. Res., 2016, 49, 1797–1805 CrossRef CAS.
- K. Schwedtmann, G. Zanoni and J. J. Weigand, Chem.–Asian J., 2018, 13, 1388–1405 CrossRef CAS.
- N. Kuhn, G. Henkel and M. Göhner, Z. Anorg. Allg. Chem., 1999, 625, 1415–1416 CrossRef CAS.
- M. Azouri, J. Andrieu, M. Picquet, P. Richard, B. Hanquet and I. Tkatchenko, Eur. J. Inorg. Chem., 2007, 2007, 4877–4883 CrossRef.
- Y. Canac, N. Debono, L. Vendier and R. Chauvin, Inorg. Chem., 2009, 48, 5562–5568 CrossRef CAS.
- H. Tinnermann, L. D. M. Nicholls, T. Johannsen, C. Wille, C. Golz, R. Goddard and M. Alcarazo, ACS Catal., 2018, 8, 10457–10463 CrossRef CAS.
- J. Petuškova, H. Bruns and M. Alcarazo, Angew. Chem., Int. Ed., 2011, 50, 3799–3802 CrossRef.
- I. Abdellah, C. Lepetit, Y. Canac, C. Duhayon and R. Chauvin, Chem.–Eur. J., 2010, 16, 13095–13108 CrossRef CAS.
- C. Maaliki, C. Lepetit, Y. Canac, C. Bijani, C. Duhayon and R. Chauvin, Chem.–Eur. J., 2012, 18, 7705–7714 CrossRef CAS.
- A. Igau, A. Baceiredo, H. Gruetzmacher, H. Pritzkow and G. Bertrand, J. Am. Chem. Soc., 1989, 111, 6853–6854 CrossRef CAS.
- D. J. Brauer, K. W. Kottsieper, C. Liek, O. Stelzer, H. Waffenschmidt and P. Wasserscheid, J. Organomet. Chem., 2001, 630, 177–184 CrossRef CAS.
- N. Debono, Y. Canac, C. Duhayon and R. Chauvin, Eur. J. Inorg. Chem., 2008, 2008, 2991–2999 CrossRef.
- S. Saleh, E. Fayad, M. Azouri, J.-C. Hierso, J. Andrieu and M. Picquet, Adv. Synth. Catal., 2009, 351, 1621–1628 CrossRef CAS.
- J. Petuškova, M. Patil, S. Holle, C. W. Lehmann, W. Thiel and M. Alcarazo, J. Am. Chem. Soc., 2011, 133, 20758–20760 CrossRef.
- J. Carreras, M. Patil, W. Thiel and M. Alcarazo, J. Am. Chem. Soc., 2012, 134, 16753–16758 CrossRef CAS.
- J. Carreras, G. Gopakumar, L. Gu, A. Gimeno, P. Linowski, J. Petuškova, W. Thiel and M. Alcarazo, J. Am. Chem. Soc., 2013, 135, 18815–18823 CrossRef CAS.
- B. Vabre, Y. Canac, C. Lepetit, C. Duhayon, R. Chauvin and D. Zargarian, Chem.–Eur. J., 2015, 21, 17403–17414 CrossRef CAS.
- G. Parkin, Organometallics, 2006, 25, 4744–4747 CrossRef CAS.
- A. Amgoune and D. Bourissou, Chem. Commun., 2011, 47, 859–871 RSC.
- J. S. Jones and F. P. Gabbaï, Acc. Chem. Res., 2016, 49, 857–867 CrossRef CAS.
- H. Kameo and H. Nakazawa, Chem.–Asian J., 2013, 8, 1720–1734 CrossRef CAS.
- G. Bouhadir and D. Bourissou, Chem. Soc. Rev., 2016, 45, 1065–1079 RSC.
- H. Goodman, L. Mei and T. L. Gianetti, Front. Chem., 2019, 7, 365 CrossRef CAS.
- A. I. Yanovsky, Y. T. Struchkov, A. Z. Kreindlin and M. I. Rybinskaya, J. Organomet. Chem., 1989, 369, 125–130 CrossRef.
- S. Lupan, M. Kapon, M. Cais and F. H. Herbstein, Angew. Chem., Int. Ed., 1972, 11, 1025–1027 CrossRef CAS.
- T. Iwai, R. Tanaka and M. Sawamura, Organometallics, 2016, 35, 3959–3969 CrossRef CAS.
- L. Mei, J. M. Veleta, J. Bloch, H. J. Goodman, D. Pierce-Navarro, A. Villalobos and T. L. Gianetti, Dalton Trans., 2020, 49, 16095–16105 RSC.
- K. Chansaenpak, M. Yang and F. P. Gabbaï, Philos. Trans. R. Soc., A, 2017, 375, 20170003 CrossRef.
- L. C. Wilkins, Y. Kim, E. D. Litle and F. P. Gabbaï, Angew. Chem., Int. Ed., 2019, 58, 18266–18270 CrossRef CAS.
- N. C. Deno, J. J. Jaruzelski and A. Schriesheim, J. Am. Chem. Soc., 1955, 77, 3044–3051 CrossRef CAS.
- C. D. Ritchie, Can. J. Chem., 1986, 64, 2239–2250 CrossRef CAS.
- J. W. Bunting, V. S. F. Chew, S. B. Abhyankar and Y. Goda, Can. J. Chem., 1984, 62, 351–354 CrossRef CAS.
- CCDC 2039023, 2039025, 2039028, 2039030–2039034, 2039036 and 2039040–2039043 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via.
- E. R. Clark and M. J. Ingleson, Angew. Chem., Int. Ed., 2014, 53, 11306–11309 CrossRef CAS.
- Y.-K. Yang and J. Tae, Org. Lett., 2006, 8, 5721–5723 CrossRef CAS.
- S. Fukuzumi, H. Kotani, K. Ohkubo, S. Ogo, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2004, 126, 1600–1601 CrossRef CAS.
- T. W. Hudnall, C. L. Dorsey, J. S. Jones and F. P. Gabbaï, Chem.–Eur. J., 2016, 22, 2882–2886 CrossRef CAS.
- K. Sorochkina, K. Chernichenko, M. Nieger, M. Leskelä and T. Repo, Z. Naturforsch., B: J. Chem. Sci., 2017, 72, 903 CAS.
- R. Chuong, K. A. Luck, R. L. Luck, L. P. Nguyen, D. Phan, L. R. Pignotti, E. Urnezius and E. J. Valente, J. Organomet. Chem., 2013, 724, 45–50 CrossRef CAS.
- G.-Q. Chen, G. Kehr, C. G. Daniliuc, C. Mück-Lichtenfeld and G. Erker, Angew. Chem., Int. Ed., 2016, 55, 5526–5530 CrossRef CAS.
- M. Boudjelel, E. D. Sosa Carrizo, S. Mallet-Ladeira, S. Massou, K. Miqueu, G. Bouhadir and D. Bourissou, ACS Catal., 2018, 8, 4459–4464 CrossRef CAS.
- T. Ito, N. Iwasawa and J. Takaya, Angew. Chem., Int. Ed., 2020, 59, 11913–11917 CrossRef CAS.
- A. Belyaev, Y.-T. Chen, Z.-Y. Liu, P. Hindenberg, C.-H. Wu, P.-T. Chou, C. Romero-Nieto and I. O. Koshevoy, Chem.–Eur. J., 2019, 25, 6332–6341 CrossRef CAS.
- L. Cabrera, G. C. Welch, J. D. Masuda, P. Wei and D. W. Stephan, Inorg. Chim. Acta, 2006, 359, 3066–3071 CrossRef CAS.
- S. Bontemps, G. Bouhadir, K. Miqueu and D. Bourissou, J. Am. Chem. Soc., 2006, 128, 12056–12057 CrossRef CAS.
- E. Herrero-Gómez, C. Nieto-Oberhuber, S. López, J. Benet-Buchholz and A. M. Echavarren, Angew. Chem., Int. Ed., 2006, 45, 5455–5459 CrossRef.
- T. J. Brown, M. G. Dickens and R. A. Widenhoefer, Chem. Commun., 2009, 6451–6453 RSC.
- A. Zhdanko, M. Ströbele and M. E. Maier, Chem.–Eur. J., 2012, 18, 14732–14744 CrossRef CAS.
- A. Homs, C. Obradors, D. Lebœuf and A. M. Echavarren, Adv. Synth. Catal., 2014, 356, 221–228 CrossRef CAS.
- D. J. Gorin, B. D. Sherry and F. D. Toste, Chem. Rev., 2008, 108, 3351–3378 CrossRef CAS.
- R. A. Widenhoefer, Chem.–Eur. J., 2008, 14, 5382–5391 CrossRef CAS.
- S. P. Fisher, A. El-Hellani, F. S. Tham and V. Lavallo, Dalton Trans., 2016, 45, 9762–9765 RSC.
- A. S. K. Hashmi, J. P. Weyrauch, W. Frey and J. W. Bats, Org. Lett., 2004, 6, 4391–4394 CrossRef CAS.
- S. Sen and F. P. Gabbaï, Chem. Commun., 2017, 53, 13356–13358 RSC.
- J. P. Weyrauch, A. S. K. Hashmi, A. Schuster, T. Hengst, S. Schetter, A. Littmann, M. Rudolph, M. Hamzic, J. Visus, F. Rominger, W. Frey and J. W. Bats, Chem.–Eur. J., 2010, 16, 956–963 CrossRef CAS.
- H. Tinnermann, C. Wille and M. Alcarazo, Angew. Chem., Int. Ed., 2014, 53, 8732–8736 CrossRef CAS.
- A. Fürstner, F. Stelzer and H. Szillat, J. Am. Chem. Soc., 2001, 123, 11863–11869 CrossRef.
- K. Fourmy, S. Mallet-Ladeira, O. Dechy-Cabaret and M. Gouygou, Organometallics, 2013, 32, 1571–1574 CrossRef CAS.
- F. Inagaki, C. Matsumoto, Y. Okada, N. Maruyama and C. Mukai, Angew. Chem., Int. Ed., 2015, 54, 818–822 CrossRef CAS.
- R. Usón, A. Laguna, M. Laguna, J. Jiménez, M. P. Gómez, A. Sainz and P. G. Jones, J. Chem. Soc., Dalton Trans., 1990, 3457–3463 RSC.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*