
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
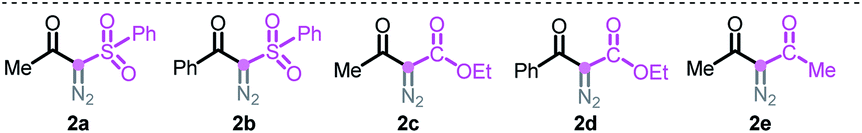
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal-free tandem carbene N–H insertions and C–C bond cleavages†
Pu
Chen
a,
Jiang
Nan
 *a,
Yan
Hu
a,
Yifan
Kang
a,
Bo
Wang
a,
Yangmin
Ma
*a and
Michal
Szostak
*ab
*a,
Yan
Hu
a,
Yifan
Kang
a,
Bo
Wang
a,
Yangmin
Ma
*a and
Michal
Szostak
*ab
aShaanxi Key Laboratory of Chemical Additives for Industry, College of Chemistry and Chemical Engineering, Shaanxi University of Science and Technology, Xi'an 710021, China. E-mail: nanjiang@sust.edu.cn
bDepartment of Chemistry, Rutgers University, 73 Warren Street, Newark, New Jersey 07102, USA
First published on 10th November 2020
Abstract
A metal-free C–H [5 + 1] annulation reaction of 2-arylanilines with diazo compounds has been achieved, giving rise to two types of prevalent phenanthridines via highly selective C–C cleavage. Compared to the simple N–H insertion manipulation of diazo, this method elegantly accomplishes a tandem N–H insertion/SEAr/C–C cleavage/aromatization reaction, and the synthetic utility of this new transformation is exemplified by the succinct syntheses of trisphaeridine and bicolorine alkaloids.
Introduction
Carbene conversion represents one of the most fundamental and synthetically useful reactions in modern organic syntheses.1 Of particular interest are diazo compounds, a class of “smart” carbene precursors that are being extensively utilized as versatile synthetic feedstock in the chemical as well as biological arena.2 A myriad of synthetically significant applications of diazo compounds have been realized in Wolff rearrangements,3 cyclopropanations,4 ylide formations,5 X–H insertions,6 C–H functionalizations7 and so forth (Scheme 1a). Regarding these examples, it is well known that metal-carbenes play a pivotal role; namely, diazo chemistry mainly relies on metal catalysts (e.g. Rh, Ru, Pd, Ir, Ag, Cu and Co) for arriving at the carbenoid reactivity. On account of strict constraints imposed by trace levels of metals in pharmaceuticals and also reaction economy,8 however, this “metal-engaged” mode of reactivity dramatically overshadows the prospective applications of diazo chemistry. In striking contrast, the evolvement of metal-free technologies heavily lags behind, albeit several elegant yet sporadic reactions have emerged in recent years.9 Moreover, these studies commonly refer to a “one-fold” operation forming only a single chemical bond within the entire process.10 Against this background, the exploration of more synthetically innovative streamlined approaches and strategies through multiple bond breaking/forming events in the absence of transition metals, such as domino and cascade reactions of diazo compounds, would be a desirable goal. In this paper, we report a new platform for upgrading a transform of model diazo compounds through a Brønsted acid-promoted N–H insertion/SEAr/C–C cleavage/oxidative aromatization four-fold domino reaction in a single chemical operation.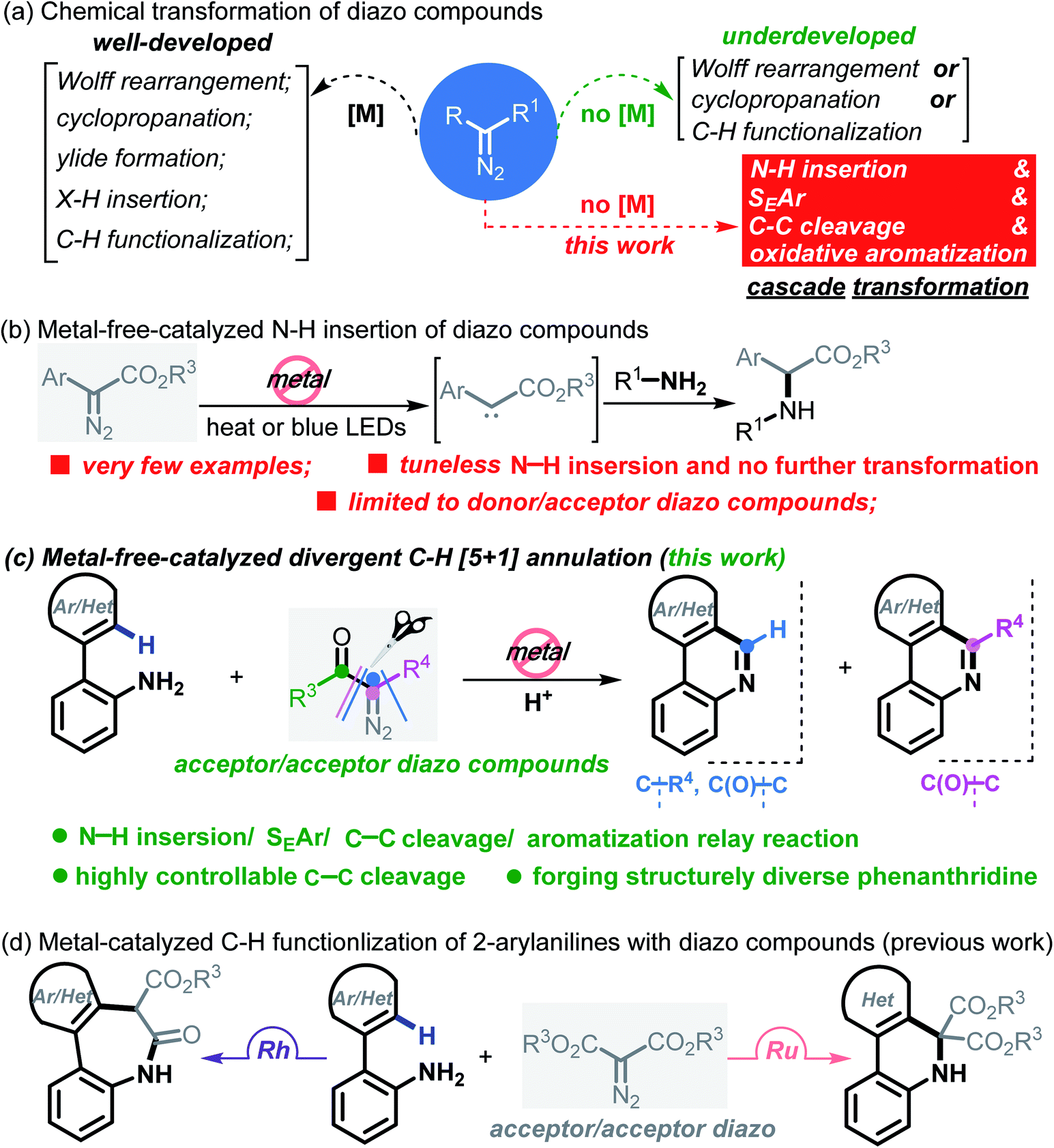 | ||
| Scheme 1 Chemical reactivity of diazo and this work. | ||
It is worth noting that N–H insertion/conversion is among the most promising platforms for constructing the ubiquitous C–N bonds widely encountered in an enormous number of bioactive molecules and countless top-selling marketed drugs, and this mode of reactivity has attracted significant endeavors in recent years and has been used to build numerous natural products.11 To date, while impressive progress has been made in the systems capitalizing on metal catalysts,12 there are only a handful of metal-free transformations between amines and diazo compounds13 (Scheme 1b). Pertinent work was done by Davies14 in 2012, wherein a thermolysis strategy enabled donor/acceptor carbenes to couple with amines. More recently, the groups of Davies15 and Jurberg16 demonstrated a blue LED-triggered N–H insertion reaction with amines and carbazoles, respectively. Notwithstanding the fundamental challenges in metal-free carbene insertions, these cases simply operated by N–H insertion and no further derivatization took place, which sharply hampered the formation of highly functionalized, structurally complex nitrogen-containing molecules, such as N-heterocycles. Indeed, these examples could be merely applied to more active, more nucleophilic, donor/acceptor carbenes. Within our recent strong interest in aniline chemistry,17 herein, we report an unprecedented Brønsted acid-promoted divergent [5 + 1] annulation between 2-arylanilines and acceptor/acceptor diazo carbenes in the absence of transition-metal catalysts (Scheme 1c), which fully differs from the precedent reaction versions between 2-arylanilines and diazo compounds mediated by metal catalysts (Scheme 1d). This newly established methodology performs a highly selective C–C bond cleavage with diazo compounds in a divergent, substrate-controlled mode, thus rapidly building two types of prevalent phenanthridine cores that are persistent in natural products and functional materials,18 but their de novo synthesis often requires intricate metal-catalyzed or light-promoted systems19 (Scheme 2).
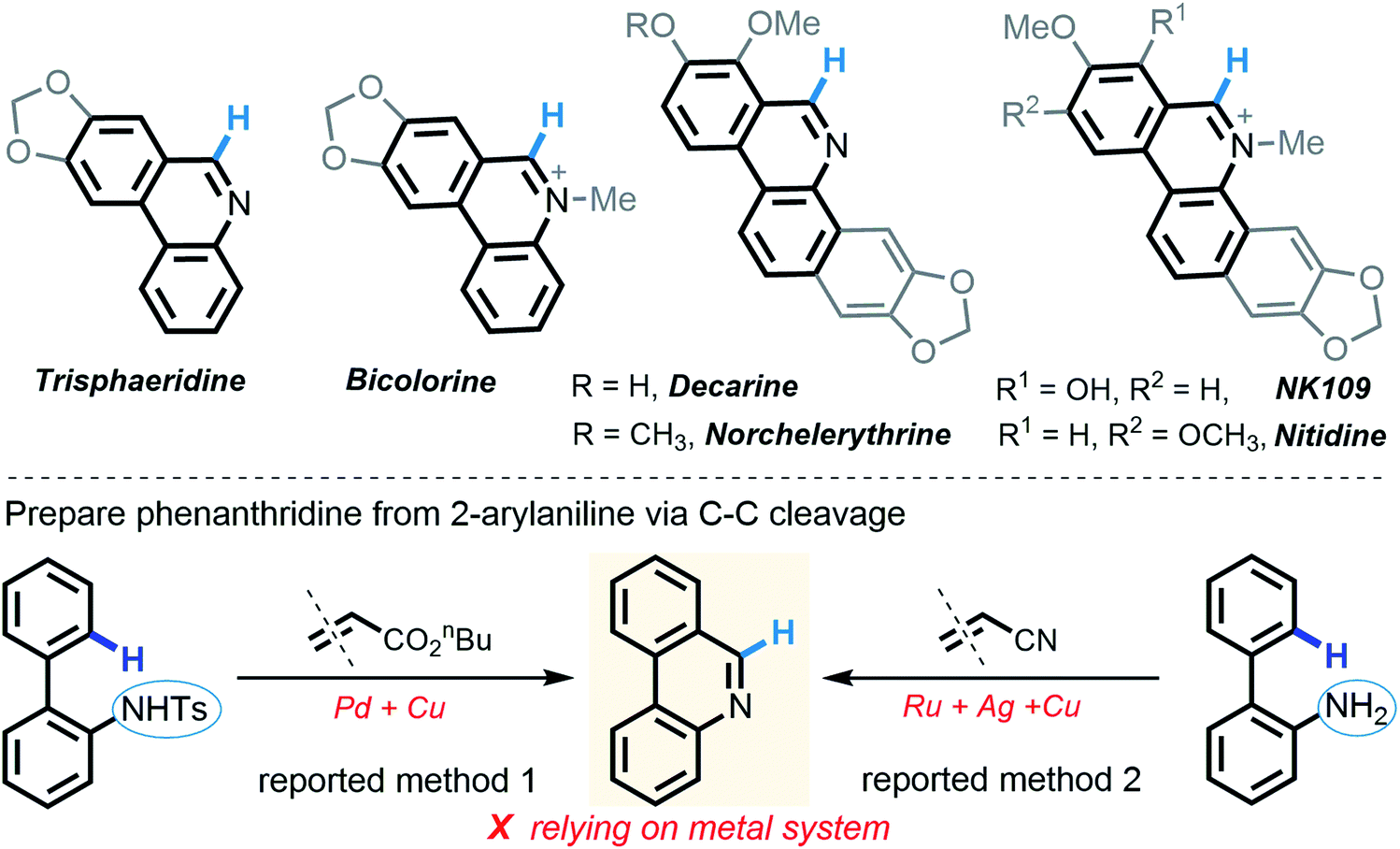 | ||
| Scheme 2 Biologically active phenanthridine derivatives and existing synthetic methods. | ||
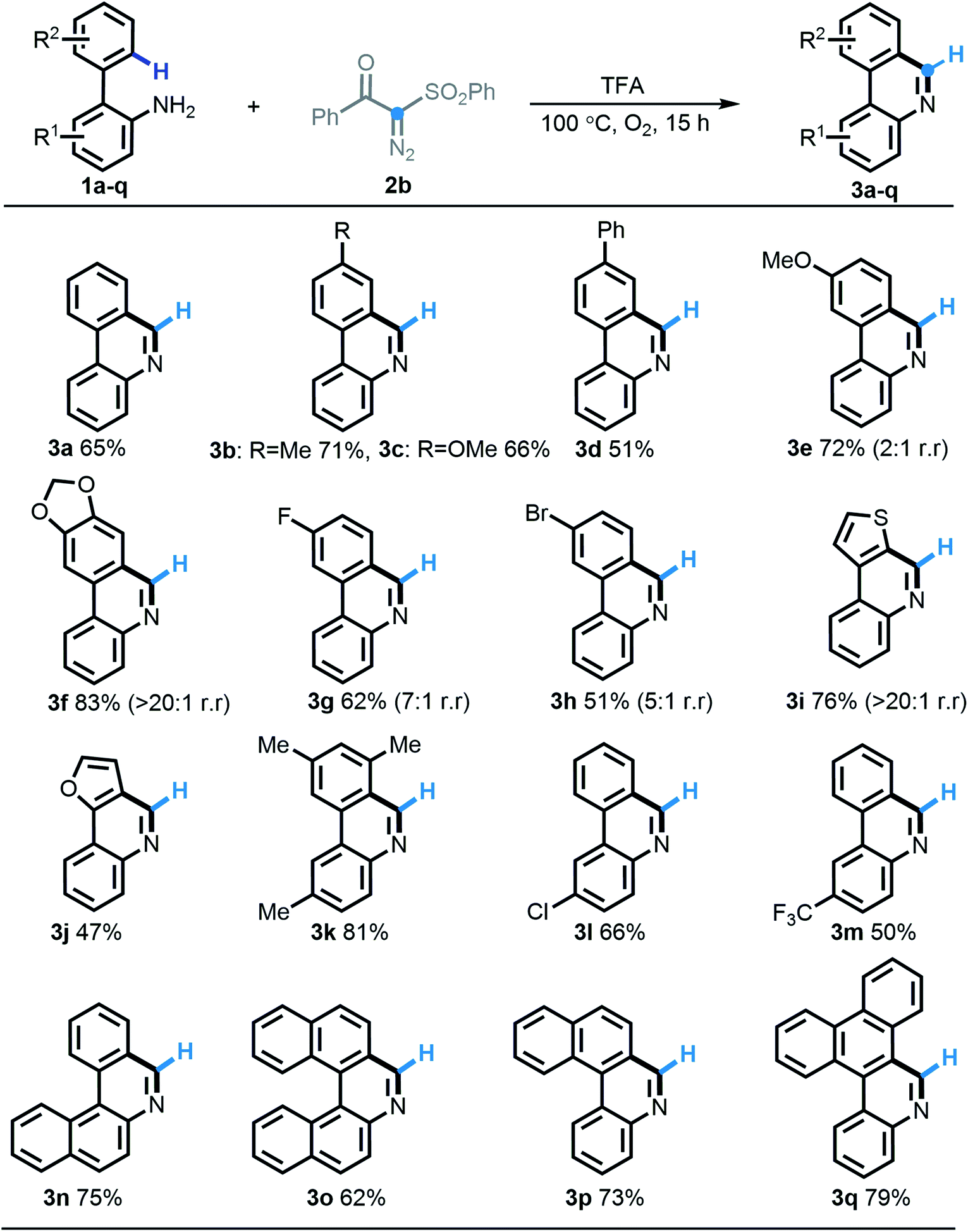 | ||
| Scheme 3 Substrate scope of [5 + 1] annulation. | ||
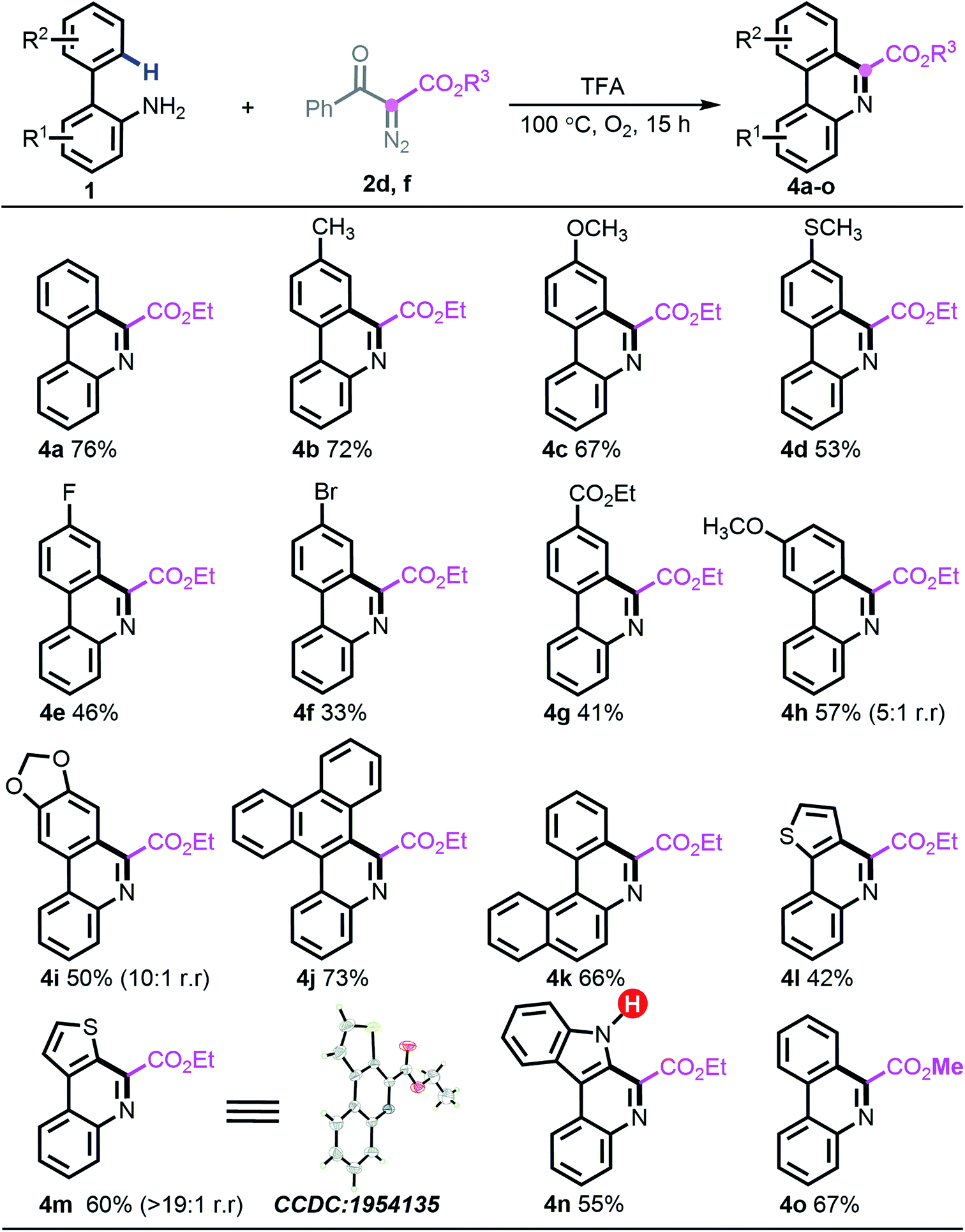 | ||
| Scheme 4 Substrate scope of deacylative [5 + 1] annulation. | ||
Results and discussion
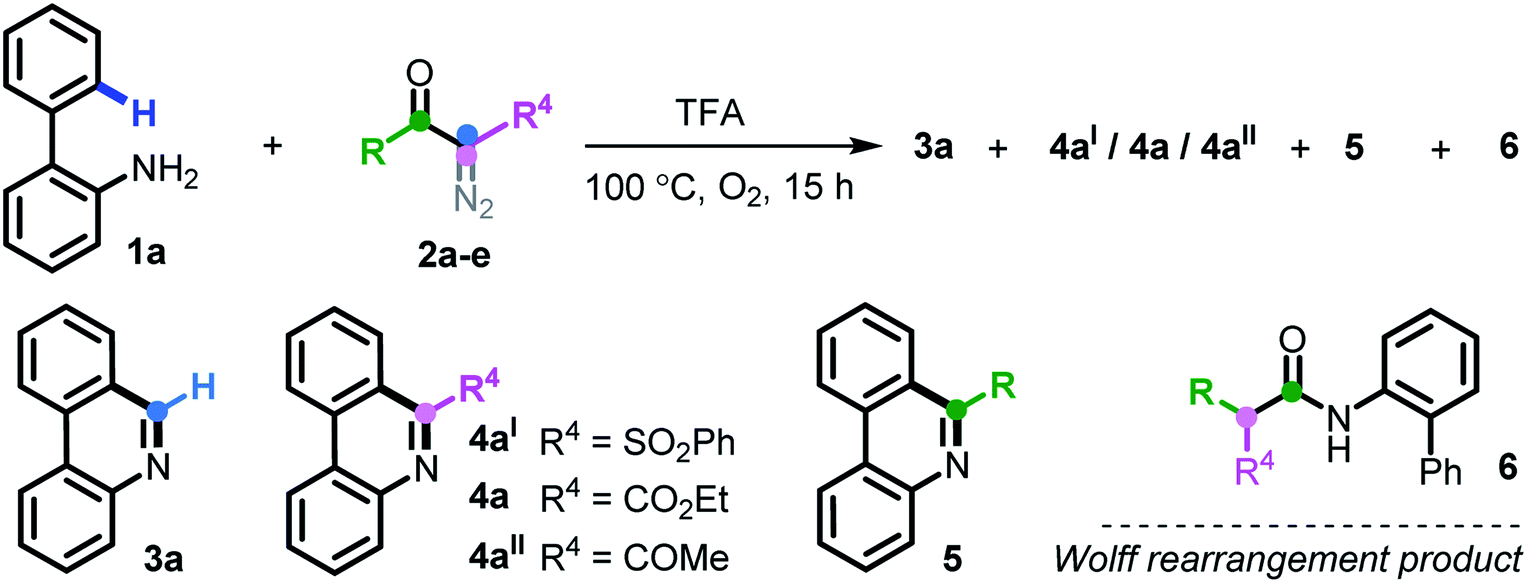
At the outset, we executed this study by reacting 2-phenylaniline 1a with diazo compound 2a (Table 1). After investigating several reaction parameters, the desired phenanthridine 3a was obtained in 50% isolated yield in trifluoroacetic acid (TFA) at 100 °C under O2 for 15 h (entry 1). Experimental results showed that employing TFA as the reaction solvent was vital, and other common solvents such as DMF, THF, HFIP, toluene, dioxane, and acetic acid were all absolutely futile, wherein only a certain amount of Wolff rearrangement product 6 was formed (entries 2–7). A decreased yield occurred when the reaction proceeded under air atmosphere; especially a nonaerobic system just resulted in 13% yield of 3a (entries 8–9), which undoubtedly clarified the significance of an aerobic environment. Changing the reaction temperature to either lower or higher levels diminished the efficiency (entries 10–11). When we turned our attention to phenyl-substituted substrate 2b, gratifyingly, the efficacy of the title transformation improved and yielded the target 3a in 65% yield (entry 12). Motivated by the above structure–activity relationship, we investigated the ester-modified diazo compound 2c in an attempt to further the optimization; unexpectedly, this procedure delivered exclusively another type of phenanthridine 4a in 37% yield (entry 13). In this method, phenyl-ester diazo compound 2d also behaved more efficiently and led to 76% yield (entry 14). Interestingly, persistency with diketo-derived diazo compound 2e failed to generate the corresponding 3a and 4aII; however, the third chemoselective product 5 was furnished in 18% yield (entry 15). Apparently, this divergent outcome hinges upon the exact structure of the diazo precursor to construct structurally distinct phenanthridines.|
|
|||||
|---|---|---|---|---|---|
| Entry | Variation from the assigned conditions | Yieldb (%) | |||
| 3a | 4 | 5 | 6 | ||
| a Assigned conditions: 1a (0.20 mmol), 2a (0.30 mmol), TFA (1.6 mL), 100 °C, 15 h, and O2 atmosphere. b Isolated yields (n.d: not detected). | |||||
| 1a | None | 50 | 4aI/n.d | n.d | n.d |
| 2 | DMF + TFA(10.0 equiv.) | n.d | 4aI/n.d | n.d | 14 |
| 3 | THF + TFA(10.0 equiv.) | n.d | 4aI/n.d | n.d | 33 |
| 4 | HFIP + TFA(10.0 equiv.) | 5 | 4aI/n.d | n.d | 6 |
| 5 | Toluene + TFA(10.0 equiv.) | 5 | 4aI/n.d | n.d | 35 |
| 6 | Dioxane + TFA(10.0 equiv.) | n.d | 4aI/n.d | n.d | 46 |
| 7 | AcOH + TFA(10.0 equiv.) | n.d | 4aI/n.d | n.d | 20 |
| 8 | Air instead of O2 | 43 | 4aI/n.d | n.d | n.d |
| 9 | N2 instead of O2 | 13 | 4aI/n.d | n.d | n.d |
| 10 | 80 °C instead of 100 °C | 33 | 4aI/n.d | n.d | n.d |
| 11 | 120 °C instead of 100 °C | 26 | 4aI/n.d | n.d | n.d |
| 12 | 2b instead of 2a | 65 | 4aI/n.d | n.d | n.d |
| 13 | 2c instead of 2a | n.d | 4a/37 | n.d | n.d |
| 14 | 2d instead of 2a | 4 | 4a/76 | n.d | n.d |
| 15 | 2e instead of 2a | n.d | 4aII/n.d | 18 | n.d |
|
|||||
Having identified the optimal reaction conditions, we firstly strived to test the substrate scope of this acid-induced [5 + 1] annulation reaction by choosing benzoyl-phenylsulfonyl diazo compound 2b as a synthetic partner. Overall, an extensive range of 2-arylanilines (1a–1q) participated in this predicted transformation quite well, furnishing the homologous phenanthridine products 3a–3q in 51–83% yields. With respect to either an ortho-arene system or an aniline segment, the substituted functionalities with different electronic groups including methyl (3b, 3k), methoxy (3c, 3e), phenyl (3d), methylenedioxy (3f), fluoro (3g), bromo (3h), chloro (3l), and trifluoromethyl (3m) were all compatible to undergo this protocol efficiently. As expected, the electron-deficient substrates behaved slightly less expediently. Gratifyingly, heterocyclic substrates such as thienyl (2i) and furyl (2j) were also efficient in this [5 + 1] annulation procedure. Notably, 2-arylanilines 2f–2i, possessing two potential C–H cyclization sites, preferentially operated at the least sterically congested position with excellent regioselectivity, except for the less selective performance with substrate 2e. More strikingly, the very useful π-extended phenanthridines widely found in functional materials, such as tetracycles (3n, 3p) and pentacycles (3o, 3q), were also efficiently obtained according to this highly selective [5 + 1] annulation in good yields.
Considering the distinct chemoselective behaviour with ester-derived diazo compound 2d (entry 14, Table 1), building highly functionalized 6-phenanthridines, we attempted to assess the generality of the deacylative [5 + 1] annulation process. Under the identified reaction conditions, reacting diazo 2d or 2f with a series of 2-arylanilines generated 6-carboalkoxy-phenanthridines (4a–o) in generally good yields. Notably, all the vacant carbons around the ortho-phenyl ring were applicable to introduce either electron-donating groups (4b–d and 4h–i), electron-withdrawing groups (4e–4g), or sterically encumbered phenanthrene scaffold (4j). The naphthylamine substrate 1n was amenable to this programed synthesis smoothly as well. Similar to the preceding desulfonylative/deacylative [5 + 1] annulation, this deacylative protocol generally showed slightly inferior results with electron-deficient substrates, although it is worth nothing that the appendage of bromo (4f) and ester (4g) groups could be valuable in downstream chemical transformations. Heterocyclic substrates 1v and 1i also allowed forming the corresponding products with excellent regioselectivity (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Meanwhile, the structure of 4m was unambiguously assigned by X-ray crystallographic analysis. It is noteworthy that the unprotected indole substrate 1w remained intact to produce the anticipated phenanthridine 4n in 55% yield. Further experimental results also showed that introducing other ester-carrying diazo compounds, such as 2f, was well supported, giving rise to the expected product 4o in 67% yield (Scheme 4).
:1). Meanwhile, the structure of 4m was unambiguously assigned by X-ray crystallographic analysis. It is noteworthy that the unprotected indole substrate 1w remained intact to produce the anticipated phenanthridine 4n in 55% yield. Further experimental results also showed that introducing other ester-carrying diazo compounds, such as 2f, was well supported, giving rise to the expected product 4o in 67% yield (Scheme 4).
In consideration of the capacity of this newly established method to rapidly assemble phenanthridines, we embarked on building trisphaeridine and other value-added derivatives constituting the core components of Amaryllidaceae alkaloids.20 Much to our delight, 2-arylaniline 1f, which was derived directly from commercially available reagents via Suzuki coupling, strongly performed in the scaled-up transformation, yielding trisphaeridine molecule 3f in 75% isolated yield on a gram scale. More importantly, N-methylation of 3f produced phenanthridine salt 3f1 and the subsequent anion exchange gave rise to bicolorine 3f2, a natural product which was isolated from Narcissus bicolor and shows promising anticancer activity. Additionally, after reduction and carbonyl derivatization, two other alkaloids, dihydrobicolorine 3f3 and N-methylcrisanidine 3f4, were also prepared in good yields (Scheme 5).
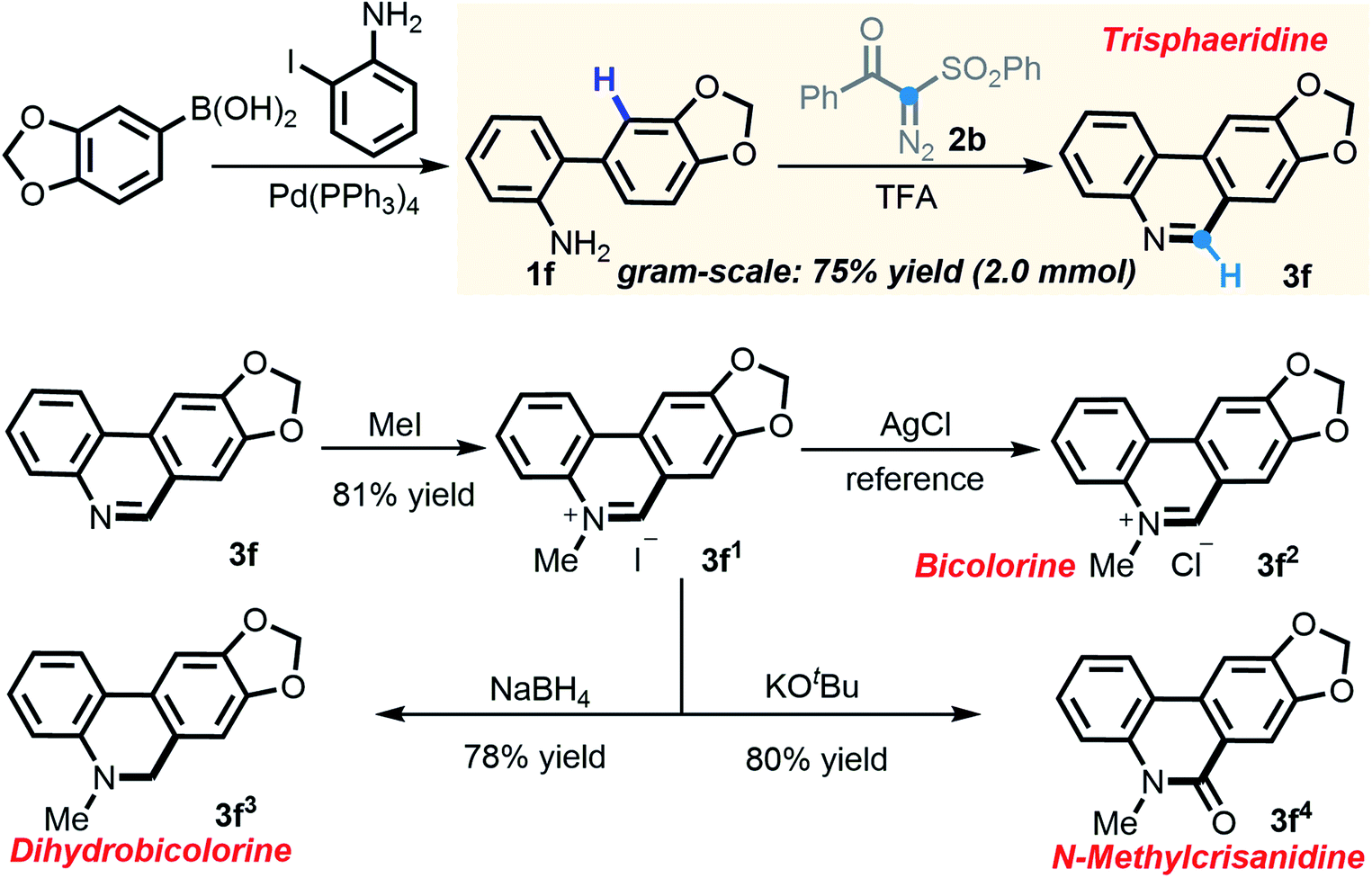 | ||
| Scheme 5 Synthetic application of established [5 + 1] annulation. | ||
To investigate the reaction pathway, a set of preliminary experiments was conducted. As shown in Scheme 6, the values (KH/KD = 1.3:1 and 1.1:1) of kinetic isotope effects implied that SEAr was not involved in the rate-determining step during this divergent [5 + 1] annulation process (Scheme 6a). Upon treatment with deuterated TFA, the experimental outcome suggested that the hydrogen from TFA participated in the product formation (Scheme 6b). With respect to the formation of phenanthridine 3a, the hypothetical electronically neutral secondary amine intermediate 7 was synthesized but failed to execute the envisioned cyclization. In contrast, the electron-deficient amine 8 delivered the product 3a, albeit in a lower yield of 25% (Scheme 6c), which revealed that an imine species might be engaged in the titular transformation and that the more electron-withdrawing substrate nature favors the reaction. Indeed, the prepared secondary ester-carrying amine 9 also led to the construction of phenanthridine 4a in 23% yield (Scheme 6d). Although our persistent attempts to synthesize other potential intermediates were frustrated, the above-outlined set of uniform experimental results indicated that an imine intermediate containing electron-deficient both R1 and R2 might enable this [5 + 1] protocol smoothly.
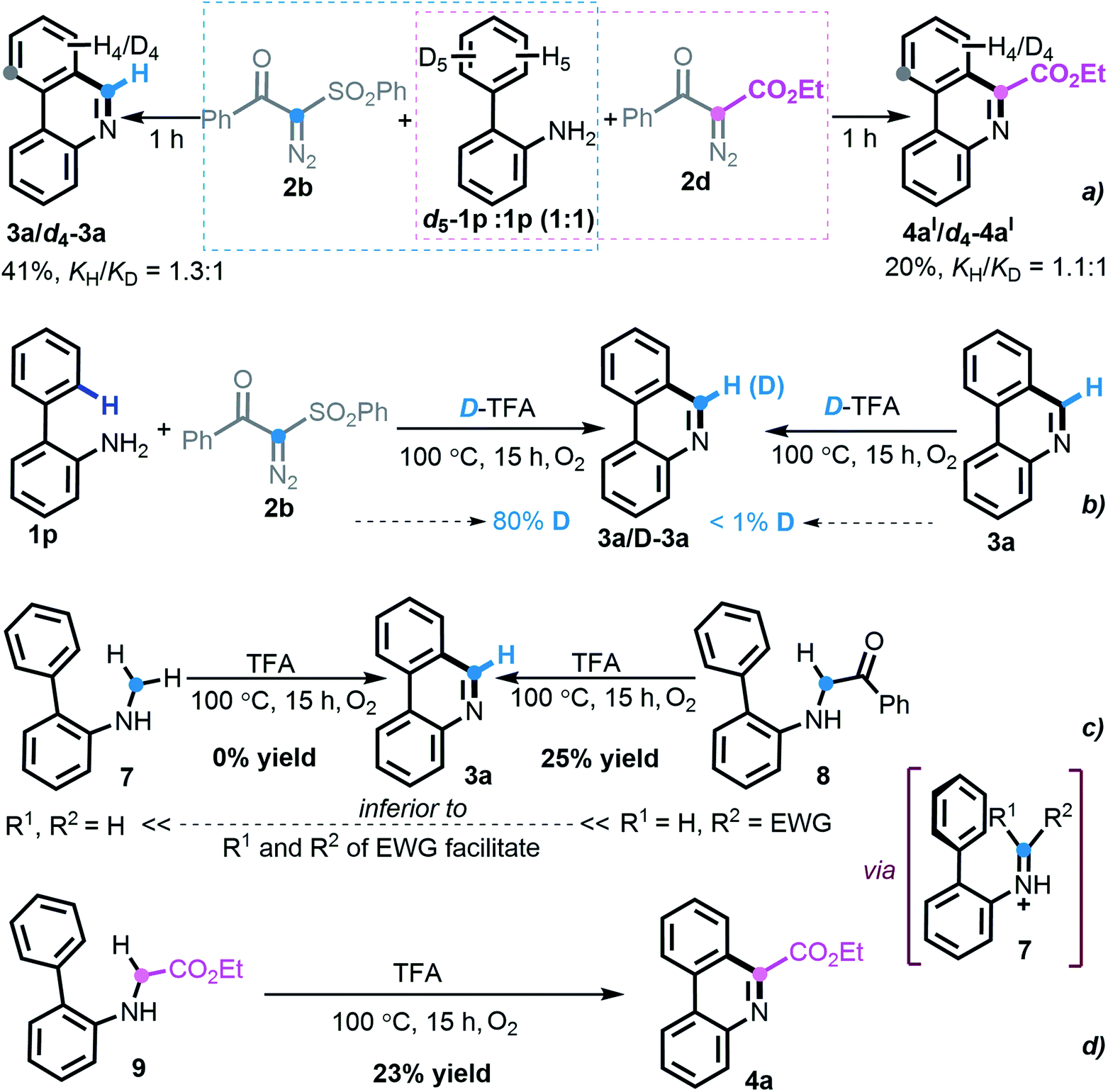 | ||
| Scheme 6 Mechanistic studies. (a) Kinetic isotope effect experiments. (b) Deuterium-labeling experiments. (c) Investigating intermediate toward phenanthridine 3a. (d) Investigating intermediate toward phenanthridine 3a. | ||
According to our mechanistic findings and literature precedents,21 the tentative reaction pathway for assembling the two classes of phenanthridines by divergent [5 + 1] annulation is proposed (Scheme 7). Under acidic conditions, the protonation of a diazo compound initially takes place to deliver species A, followed by N2 extrusion21a–c to produce secondary amine Bvia nucleophilic attack of NH2. This key intermediate as verified undergoes oxidation and subsequently performs the SEAr process, which is supported by the superior result of electron-rich 2-arylanilines in Schemes 3 and 4, to generate C–H functionalization product D. At this point, when the R unit consists of an ester group, selective C–C bond cleavage occurs via deacylation of the more electrophilic acyl group21d,e and concomitant release of benzoic acid that was identified by NMR spectroscopy, followed by oxidative aromatization21f,g to give product 4a. Comparatively, adjusting R to a more active SO2Ph group promotes a successive de-sulfonylation/de-acylation/oxidative aromatization to finally furnish phenanthridine 3a.
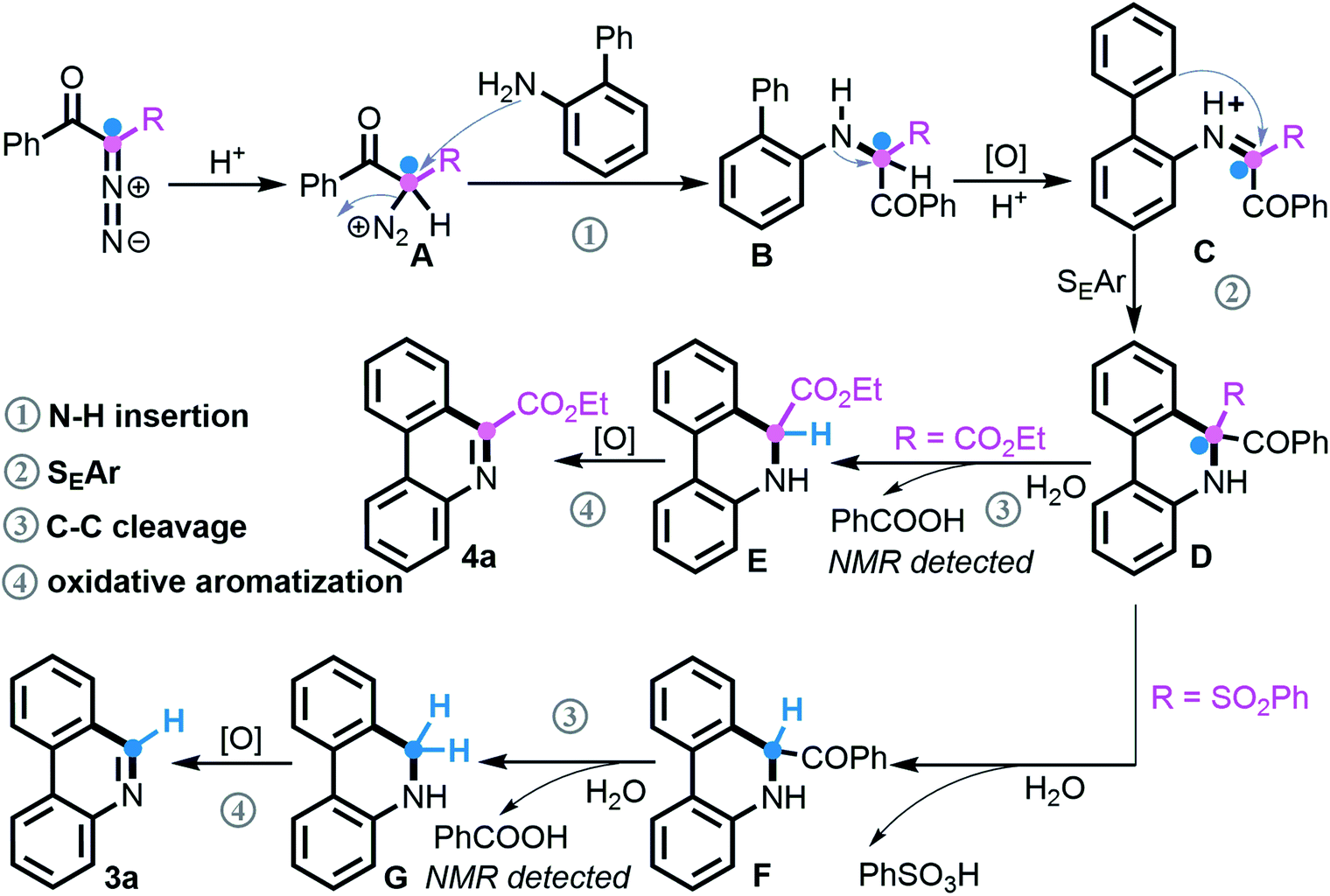 | ||
| Scheme 7 Proposed reaction pathway. | ||
Conclusions
In conclusion, we have demonstrated a substrate-controlled selective [5 + 1] annulation reaction of 2-arylanilines with diazo compounds by a tandem N–H insertion/SEAr/C–C cleavage/aromatization under metal-free conditions. Using sulfonyl-derived diazo compounds as the coupling partners, substituent-free phenanthridines are prepared by a dual desulfonylation and deacylation process. Alternatively, introducing ester-derived diazo compounds into the system divergently leads to the exclusive assembly of C6-carboalkoxy-substituted phenanthridines. Remarkably, the methodology has also been applied to a concise assembly of biologically active trisphaeridine and bicolorine Amaryllidaceae alkaloids as well as their derivatives. This study to the best of our knowledge represents the first example of Brønsted acid-promoted metal-free N–H insertion/multifold bond reorganization of diazo compounds. Metal-free domino transformations of diazo compounds will greatly expand the synthesis of omnipresent synthetic motifs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the NSFC (21801159), the China Post-doctoral Science Foundation (2018M640944), the Natural Science Basic Research Plan in Shaanxi Province of China (2020JQ-705), and the Education Department of Shaanxi Province (18JK0110).Notes and references
- (a) A. J. Arduengo and G. Bertrand, Chem. Rev., 2009, 109, 3209 CrossRef CAS; (b) Y. Xia, D. Qiu and J. Wang, Chem. Rev., 2017, 117, 13810 CrossRef CAS; (c) D. Zhu, L. Chen, H. Fan, Q. Yao and S. Zhu, Chem. Soc. Rev., 2020, 49, 908 RSC; (d) Z. Yang, M. L. Stivanin, I. D. Jurberg and R. M. Koenigs, Chem. Soc. Rev., 2020, 49, 6833 RSC.
- (a) A. Homberg and J. Lacour, Chem. Sci., 2020, 11, 6362 RSC; (b) H. Kato, I. Musha, M. Komatsuda, K. Muto and J. Yamaguchi, Chem. Sci., 2020, 11, 8779 RSC; (c) Q. Zhang, Q. Xiong, M. Li, W. Xiong, B. Shi, Y. Lan, Q. Lu and W. Xiao, Angew. Chem., Int. Ed., 2020, 59, 14096 CrossRef CAS; (d) A. Dasgupta, K. Stefkova, R. Babaahmdi, L. Gierlichs, A. Ariafard and R. L. Melen, Angew. Chem., Int. Ed., 2020, 59, 15492 CrossRef CAS; (e) Y. Su, G. Liu, J. Liu, L. Tram, H. Qiu and M. P. Doyle, J. Am. Chem. Soc., 2020, 142, 13846 CrossRef CAS.
- For selected examples, see: (a) R. Sarpong, J. T. Su and B. M. Stoltz, J. Am. Chem. Soc., 2003, 125, 13624 CrossRef CAS; (b) X. Hu, X. Chen, Y. Shao, H. Xie, Y. Deng, Z. Ke, H. Jiang and W. Zeng, ACS Catal., 2018, 8, 1308 CrossRef CAS; (c) X. Hu, F. Chen, Y. Deng, H. Jiang and W. Zeng, Org. Lett., 2018, 20, 6140 CrossRef CAS; (d) J. Liu, M. Li, B. Qu, L. Lu and W. Xiao, Chem. Commun., 2019, 55, 2031 RSC; (e) J. Meng, W. Ding and Z. Han, Org. Lett., 2019, 21, 9801 CrossRef CAS.
- For selected examples, see: (a) D. A. Vargas, R. L. Khade, Y. Zhang and R. Fasan, Angew. Chem., Int. Ed., 2019, 58, 10148 CrossRef CAS; (b) B. Wang, I. G. Howard, J. W. Pope, E. D. Conte and Y. Deng, Chem. Sci., 2019, 10, 7958 RSC; (c) Z. Yang, M. Möller and R. M. Koenigs, Angew. Chem., Int. Ed., 2020, 59, 5572 CrossRef CAS; (d) Z. Zhang, M. Zheng, X. Xue, I. Marek, F. Zhang and J. Ma, Angew. Chem., Int. Ed., 2019, 58, 18191 CrossRef CAS; (e) K. L. Smith, C. L. Padgett, W. D. Mackay and J. S. Johnson, J. Am. Chem. Soc., 2020, 142, 6449 CrossRef CAS.
- For selected examples, see: (a) D. M. Hodgson, F. Y. T. M. Pierard and P. A. Stupple, Chem. Soc. Rev., 2001, 30, 50 RSC; (b) A. DeAngelis, M. T. Taylor and J. M. Fox, J. Am. Chem. Soc., 2009, 131, 1101 CrossRef CAS.
- For selected examples, see: (a) W. Chan, S. Lo, Z. Zhou and W. Yu, J. Am. Chem. Soc., 2012, 134, 13565 CrossRef CAS; (b) Q. Zhou, S. Li, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2017, 56, 16013 CrossRef CAS; (c) Y. Jang, Ł. Woźniak, J. Pedroni and N. Cramer, Angew. Chem., Int. Ed., 2018, 57, 12901 CrossRef CAS; (d) A. Olmos, R. Gava, B. Noverges, D. Bellezza, K. Jacob, M. Besora, W. M. C. Sameera, M. Etienne, F. Maseras, G. Asensio, A. Caballero and P. J. Pérez, Angew. Chem., Int. Ed., 2018, 57, 13848 CrossRef CAS; (e) Z. Yu, Y. Li, P. Zhang, L. Liu and J. Zhang, Chem. Sci., 2019, 10, 6553 RSC.
- For selected examples, see: (a) X. Li and D. P. Curran, J. Am. Chem. Soc., 2013, 135, 12076 CrossRef CAS; (b) J. R. Jagannathan, J. C. Fettinger, J. T. Shaw and A. K. Franz, J. Am. Chem. Soc., 2020, 142, 11674 CrossRef CAS; (c) Y. Li, Y. Zhao, T. Zhou, M. Chen, Y. Li, M. Huang, Z. Xu, S. Zhu and Q. Zhou, J. Am. Chem. Soc., 2020, 142, 10557 CrossRef CAS.
- (a) D. Nair, J. Scarpello, L. White, L. Freista dos Santos, I. Vankelecom and A. Livingston, Tetrahedron Lett., 2001, 42, 8219 CrossRef CAS; (b) C. E. Garrett and K. Prasad, Adv. Synth. Catal., 2004, 346, 889 CrossRef CAS.
- (a) S. R. Ovalles, J. H. Hansen and H. M. L. Davies, Org. Lett., 2011, 13, 4284 CrossRef CAS; (b) H. Lam, Z. Qureshi, M. Wegmann and M. Lautens, Angew. Chem., Int. Ed., 2018, 57, 16185 CrossRef CAS; (c) A. Suneja and C. Schneider, Org. Lett., 2018, 20, 7576 CrossRef CAS; (d) J. Ghorai and P. Anbarasan, Org. Lett., 2019, 21, 3431 CrossRef CAS; (e) J. Ye, L. Quach, T. Paulisch and F. Glorius, J. Am. Chem. Soc., 2019, 141, 16227 CrossRef CAS; (f) X. Yi, J. Feng, F. Huang and J. B. Baell, Chem. Commun., 2020, 56, 1243 RSC; (g) H. Zheng, K. Dong, D. Wherritt, H. Arman and M. P. Doyle, Angew. Chem., Int. Ed., 2020, 59, 13613 CrossRef CAS; (h) N. Holmberg-Douglas, N. P. R. Onuska and D. A. Nicewicz, Angew. Chem., Int. Ed., 2020, 59, 7425 CrossRef CAS; (i) K. Wu, L. Wu, C. Zhou and C. Che, Angew. Chem., Int. Ed., 2020, 59, 16202 CAS.
- (a) J. Barluenga, M. Tomás-Gamasa, F. Aznar and C. Valdés, Angew. Chem., Int. Ed., 2010, 49, 4993 CrossRef CAS; (b) C. Zhai, D. Xing, C. Jing, J. Zhou, C. Wang, D. Wang and W. Hu, Org. Lett., 2014, 16, 2934 CrossRef CAS; (c) C. Tortoreto, D. Rackl and H. M. L. Davies, Org. Lett., 2017, 19, 770 CrossRef CAS; (d) R. P. Pandit, S. T. Kim and D. H. Ryu, Angew. Chem., Int. Ed., 2019, 58, 13427 CrossRef CAS; (e) R. D. C. Gallo, P. B. Momo, D. P. Day and A. C. B. Burtoloso, Org. Lett., 2020, 22, 2339 CrossRef CAS; (f) N. Holmberg-Douglas, N. P. R. Onuska and D. A. Nicewicz, Angew. Chem., Int. Ed., 2020, 59, 7425 CrossRef CAS; (g) Z. Li, J. Chen, L. Wu, A. Ren, P. Lu and Y. Wang, Org. Lett., 2020, 22, 26 CrossRef CAS; (h) Z. Zhang, Y. Yu, F. Huang, X. Yi, Y. Xu, Y. He, J. B. Baell and H. Huang, Green Chem., 2020, 22, 1594 RSC.
- (a) T. N. Salzmann, R. W. Ratcliffe, B. G. Christensen and F. A. Bouffard, J. Am. Chem. Soc., 1980, 102, 6161 CrossRef CAS; (b) C. F. Garcia, M. A. McKervey and T. Ye, Chem. Commun., 1996, 1465 RSC; (c) M. C. Bagley, K. E. Bashford, C. L. Hesketh and C. J. Moody, J. Am. Chem. Soc., 2000, 122, 3301 CrossRef CAS; (d) B. Xu, S. Zhu, X. Zuo, Z. Zhang and Q. Zhou, Angew. Chem., Int. Ed., 2014, 126, 3994 CrossRef; (e) K. Liu, C. Zhu, J. Min, S. Peng, G. Xu and J. Sun, Angew. Chem., Int. Ed., 2015, 54, 12962 CrossRef CAS.
- (a) A. C. B. Burtoloso, R. M. P. Dias and B. Bernardim, Acc. Chem. Res., 2015, 48, 921 CrossRef CAS; (b) J. Yoo, N. Park, J. H. Park, J. H. Park, S. Kang, S. M. Lee, H. J. Kim, H. Jo, J. Park and S. U. Son, ACS Catal., 2015, 5, 350 CrossRef CAS; (c) M. Li, J. Yu, Y. Li, S. Zhu and Q. Zhou, Science, 2019, 366, 990 CrossRef CAS; (d) S. M. Bierschenk, R. G. Bergman, K. N. Raymond and F. D. Toste, J. Am. Chem. Soc., 2020, 142, 733 CrossRef CAS.
- (a) Z. J. Wang, N. E. Peck, H. Renata and F. H. Arnold, Chem. Sci., 2014, 5, 598 RSC; (b) G. Sreenilayam and R. Fasan, Chem. Commun., 2015, 51, 1532 RSC; (c) X. Xu, C. Li, Z. Tao and Y. Pan, Adv. Synth. Catal., 2015, 357, 3341 CrossRef CAS.
- S. R. Hansen, J. E. Spangler, J. H. Hansen and H. M. L. Davies, Org. Lett., 2012, 14, 4626 CrossRef CAS.
- I. D. Jurberg and H. M. L. Davies, Chem. Sci., 2018, 9, 5112 RSC.
- M. L. Stivanin, A. A. G. Fernandes, A. F. daSilva, C. Y. Okada Jr and I. D. Jurberg, Adv. Synth. Catal., 2020, 362, 1106 CrossRef CAS.
- (a) J. Nan, Y. Hu, P. Chen and Y. Ma, Org. Lett., 2019, 21, 1984 CrossRef CAS; (b) P. Chen, J. Nan, Y. Hu, Q. Ma and Y. Ma, Org. Lett., 2019, 21, 4812 CrossRef CAS; (c) X. Wang, J. Zhang, Y. He, D. Chen, C. Wang, F. Yang, W. Wang, Y. Ma and M. Szostak, Org. Lett., 2020, 22, 5187 CrossRef CAS.
- (a) B. D. Krane, M. O. Fagbule, M. Shamma and B. Gozler, J. Nat. Prod., 1984, 47, 1 CrossRef CAS; (b) S. Prado, S. Michel, F. Tillequin, M. Koch, B. Pfeiffer, A. Pierré, S. Léonce, P. Colson, B. Baldeyrou, A. Lansiaux and C. Bailly, Bioorg. Med. Chem., 2004, 12, 3943 CrossRef CAS; (c) P. H. Bernardo, K. F. Wan, T. Sivaraman, J. Xu, F. K. Moore, A. W. Hung, H. Y. K. Mok, V. C. Yu and C. L. L. Chai, J. Med. Chem., 2008, 51, 6699 CrossRef CAS; (d) Z. Luo, F. Wang, J. Zhang, X. Li, M. Zhang, X. Hao, Y. Xue, Y. Li, F. D. Horgen, G. Yao and Y. Zhang, J. Nat. Prod., 2012, 75, 2113 CrossRef CAS.
- (a) T. Gerfaud, L. Neuville and J. Zhu, Angew. Chem., Int. Ed., 2009, 48, 572 CrossRef CAS; (b) D. A. Candito and M. Lautens, Angew. Chem., Int. Ed., 2009, 48, 6713 CrossRef CAS; (c) Z. Liang, L. Ju, Y. Xie, L. Huang and Y. Zhang, Chem. – Eur. J., 2012, 18, 15816 CrossRef CAS; (d) Y. Liu, R. Song, C. Wu, L. Gong, M. Hu, Z. Wang, Y. Xie and J. Li, Adv. Synth. Catal., 2012, 354, 347 CrossRef CAS; (e) J. Liu, C. Fan, H. Yin, C. Qin, G. Zhang, X. Zhang, H. Yia and A. Lei, Chem. Commun., 2014, 50, 2145 RSC; (f) W. Chen, C. Chen, Y. Chen and J. Hsieh, Org. Lett., 2015, 17, 1613 CrossRef CAS; (g) H. Jiang, X. An, K. Tong, T. Zheng, Y. Zhang and S. Yu, Angew. Chem., Int. Ed., 2015, 54, 4055 CrossRef CAS; (h) J. Rong, L. Deng, P. Tan, C. Ni, Y. Gu and J. Hu, Angew. Chem., Int. Ed., 2016, 55, 2743 CrossRef CAS; (i) Z. Wang, T. Li, J. Zhao, X. Shi, D. Jiao, H. Zheng and C. Chen, Org. Lett., 2018, 20, 6640 CrossRef CAS; (j) D. Chowdhury, S. Dana, A. Mandal and M. Baidya, Chem. Commun., 2019, 55, 11908 RSC; (k) J. Fan, L. Li, J. Zhang and M. Xie, Chem. Commun., 2020, 56, 2775 RSC; (l) S. Liu, W. Pan, S. Wu, X. Bu, S. Xin, J. Yu, H. Xu and X. Yang, Green Chem., 2019, 21, 2905 RSC; (m) T. Xiao, L. Li, G. Lin, Q. Wang, P. Zhang, Z. Mao and L. Zhou, Green Chem., 2014, 16, 2421 Search PubMed.
- Z. Jin, Nat. Prod. Rep., 2013, 30, 849 RSC.
- (a) N. Fei, B. Sauter and D. Gillingham, Chem. Commun., 2016, 52, 7501 RSC; (b) R. D. C. Gallo and A. C. B. Burtoloso, Green Chem., 2018, 20, 4547 RSC; (c) R. D. C. Gallo, P. B. Momo, D. P. Day and A. C. B. Burtoloso, Org. Lett., 2020, 22, 2339 CrossRef CAS; (d) B. Li, B. Zhang, X. Zhang and X. Fan, Chem. Commun., 2017, 53, 1297 RSC; (e) X. Guo, J. Han, Y. Liu, M. Qin, X. Zhang and B. Chen, J. Org. Chem., 2017, 82, 11505 CrossRef CAS; (f) Y. Huang, H. Song, Yu. Liu and Q. Wang, Chem. – Eur. J., 2018, 24, 2065 CrossRef CAS; (g) P. Natarajan, D. Chuskita and Priya, Green Chem., 2019, 21, 4406 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1954135. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc05763k |
| This journal is © The Royal Society of Chemistry 2021 |