
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegiodivergent synthesis of pyrazino-indolines vs. triazocines via α-imino carbenes addition to imidazolidines†‡
Alejandro
Guarnieri-Ibáñez
a,
Adiran
de Aguirre
 a,
Céline
Besnard
b,
Amalia I.
Poblador-Bahamonde
*a and
Jérôme
Lacour
*a
a,
Céline
Besnard
b,
Amalia I.
Poblador-Bahamonde
*a and
Jérôme
Lacour
*a
aDepartment of Organic Chemistry, University of Geneva, Quai Ernest Ansermet 30, 1211 Geneva 4, Switzerland. E-mail: Amalia.PobladorBahamonde@unige.ch; Jerome.Lacour@unige.ch
bLaboratory of Crystallography, University of Geneva, Quai Ernest Ansermet 24, 1211 Geneva 4, Switzerland
First published on 23rd November 2020
Abstract
Hexahydropyrazinoindoles were prepared in a single step from N-sulfonyl triazoles and imidazolidines. Under dirhodium catalysis, α-imino carbenes were generated and formed nitrogen ylide intermediates that, after subsequent aminal opening, afforded the pyrazinoindoles predominantly via formal [1,2]-Stevens and tandem Friedel–Crafts cyclizations. Of mechanistic importance, a regiodivergent reactivity was engineered through the use of a specific unsymmetrically substituted imidazolidine that promoted the exclusive formation of 8-membered ring 1,3,6-triazocines. Based on DFT calculations, an original Curtin–Hammett-like situation was demonstrated for the mechanism. Further derivatizations led to functionalized tetrahydropyrazinoindoles in high yields.
Introduction
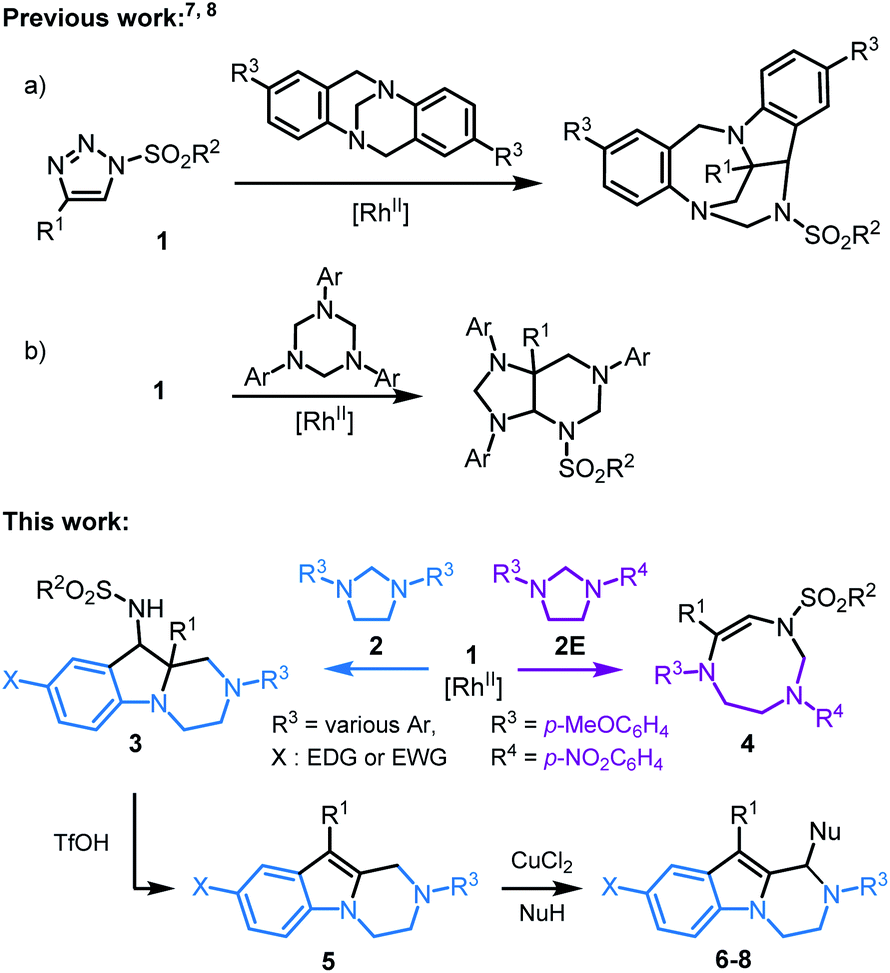
N-Sulfonyl-1,2,3-triazoles 1, readily accessible through Cu(I)-catalyzed azide alkyne cycloadditions (CuAACs),1 are key building blocks in synthetic, biological and medicinal chemistry.2 For the purpose of this study, they also decompose under metal-catalyzed conditions to afford α-imino carbenes.3 These electrophilic unsaturated intermediates have received much attention in recent years, as many synthetically useful and original processes can be afforded, from migrations to ylide-forming reactions and subsequent transformations.4,5 Only few studies have been reported on the reactivity of α-imino carbenes with tertiary amines or aminals, and on the subsequent ammonium ylide chemistry.6 Of special interest to the current study, Tröger bases were shown to react and yield polycyclic indoline-benzodiazepines through a cascade of [1,2]-Stevens, Friedel–Crafts, Grob and aminal formation reactions (Scheme 1, top, a).7 Also, 1,3,5-triazinanes form octahydro-1H-purine derivatives via formal [6 + 3] cycloaddition, ring-closure and rearrangements (Scheme 1, top, b).8 Herein, in a new development, the intermolecular reactivity of N-sulfonyl-1,2,3-triazoles 1 with imidazolidines 2 is reported (Scheme 1, bottom). Under dirhodium catalysis (3 mol%), hexahydropyrazino[1,2-a]indoles 3 are obtained in good yields (up to 90%, diastereomeric ratio (dr) up to 6.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Mechanistically, after the initial addition of α-imino carbenes to 2 and subsequent ylide formation, the transformation involves [1,2]-Stevens and Friedel–Crafts reactions. The process is general and yields systematically the polycyclic pyrazino-indolines 3. However, and importantly, with unsymmetrically substituted imidazolidine 2E (R3 = p-MeOC6H4 and R4 = p-NO2C6H4), a regiodivergent pathway is obtained favoring the selective formation of 8-membered ring hexahydro-1,3,6-triazocines 4.
:1). Mechanistically, after the initial addition of α-imino carbenes to 2 and subsequent ylide formation, the transformation involves [1,2]-Stevens and Friedel–Crafts reactions. The process is general and yields systematically the polycyclic pyrazino-indolines 3. However, and importantly, with unsymmetrically substituted imidazolidine 2E (R3 = p-MeOC6H4 and R4 = p-NO2C6H4), a regiodivergent pathway is obtained favoring the selective formation of 8-membered ring hexahydro-1,3,6-triazocines 4.
 | ||
| Scheme 1 Reactivity of N-sulfonyl triazoles with aminals. | ||
Based on first principles, detailed mechanistic analysis will show that, after regioselective ylide formation and aminal ring opening, N-cyclization rather than C-cyclization occurs in this case to form the medium sized heterocycle 4. The influence of kinetic and thermodynamic preferences, and the occurrence of a Curtin–Hammett type situation in most cases, will be demonstrated and discussed. Furthermore, compounds 3 can be transformed into tetrahydropyrazino[1,2-a]indoles 5 and further derivatives 6–8 by oxidative C–C bond forming reactions. Such compounds 3, 5–8 ought to interest medicinal chemists as pyrazino[1,2-a]indoles are commonly found in biologically active compounds with 5-HT2C receptor agonist,9 antifungal,10 antibacterial,11 anticancer,12 and antiviral activities,13 among others (Fig. 1).14
 | ||
| Fig. 1 Selected biologically active pyrazino[1,2-a]indoles.9b,10,11,12b | ||
Results and discussion
Initial experiments were performed by mixing N-tosyl-4-phenyl-1,2,3-triazole 1a (1.5 equiv.), 1,3-di-p-tolylimidazolidine 2A (1 equiv.) and Rh2(Piv)4 (1 mol%) in CHCl3 (0.25 M), and heating the mixture at 80 °C during 63 h. 1H-NMR spectroscopic analysis of the crude reaction mixture revealed the presence of unreacted starting materials and two new diastereomeric products 3aA′ and 3aA′′ (24% and 20% yields respectively). Their structures were determined with certainty, and the relative configuration relationship in particular, only after X-ray diffraction analysis (Scheme 1 and ESI‡).With this result in hand, optimization studies were conducted to improve the synthesis of 3aA and results are reported in the ESI.‡ Best conditions involve the use of CH2Cl2 as solvent and 3 mol% of Rh2(Piv)4 as catalyst.15 Complete consumption of 2A is then achieved after 14 h at 80 °C with only a slight excess of 1a (1.5 equiv., 0.25 M). Diastereomeric products 3aA′ and 3aA′′ are then formed in 61% and 17% yields respectively. Treatment of 3aA′ with sodium (Na) and naphthalene in THF led to a complete conversion and afforded free amino 3A in 83% yield (Scheme 2, top). A 1 mmol scale reaction was also performed affording products 3aA with comparable yield and diastereoselectivity (77%, dr 3.6:1).16
 | ||
| Scheme 2 Hexahydropyrazino[1,2-a]indoles 3. aFrom 3aA′: Na, naphthalene, THF, 17 h, −78 °C to 25 °C. bStick view of the crystal structure of 3aA′ (major diastereoisomer). cInseparable mixture of diastereoisomers. d3 equiv. of N-sulfonyl-1,2,3-triazole were used. | ||
With the optimized conditions in hand, a series of N-sulfonyl-1,2,3-triazoles 1a–1h was prepared by CuAAC17 and tested with imidazolidines 2A–2D (Scheme 2). Mesyl 1b and nosyl 1c triazoles afforded products 3bA and 3cA in moderate yields and diastereoselectivity (dr up to 6.8:1). Triazole reagents 1d to 1f with electron donating (EDG) or electron withdrawing (EWG) groups at regioisomeric positions on aromatic group R1 led to the corresponding heterocycles 3dA–3fA in average to excellent yields (58–88%), but poor to moderate diastereoselectivity. Thiophene derived 1g and 6-methoxynaphthalene 1h were also compatible leading to the expected products 3gA and 3hA in 82% and 71% yields, respectively.
A series of imidazolidines were prepared following reported procedures18 and tested with N-tosyl-triazole 1a (Scheme 2). These symmetrical substrates containing EDGs or EWGs at R3 positions led to 3aB–3aD with very good yields (74–90%) and moderate levels of diastereoselectivity.
To get some insight on the mechanism of this transformation, unsymmetrically substituted 2E was synthesized, with electron-withdrawing p-NO2 and -donating p-OMe substituents on the aromatic rings of the imidazolidine. Compound 2E was then treated with N-tosyl triazole 1a and Rh2(Piv)4 under the optimized conditions. Careful analysis by NMR spectroscopy revealed the presence of a major product as a single regioisomer (57%). This compound was however not the expected hexahydropyrazino[1,2-a]indole 3aE but 8-membered triazocine 4aE. Interestingly and importantly for further mechanistic discussions, compounds 4aE contains an aminal –NCH2N– bridge that is formed with the nitrogen atom carrying the p-NO2C6H4 ring. The structure of 4aE was confirmed later by X-ray diffraction analysis (Scheme 3, top). Treatment of 2E with triazoles 1e and 1i also led to hexahydro-1,3,6-triazocines 4eE and 4iE in 56% and 36% yield, respectively. As mentioned above, these results are important to understand the reactivity pathways involved in this Rh(II)-catalyzed α-imino diazo decomposition and subsequent nitrogen ylide/cyclization processes.
 | ||
| Scheme 3 Access to substituted hexahydro-1,3,6-triazocines 4. aStick view of the crystal structure of 4aE. Hydrogen atoms are omitted for clarity and the MeO group presents a local disorder. | ||
Theoretical study
To rationalize the above substrate-dependent synthetic divergence toward either pyrazino-indole or triazocine products, DFT calculations were performed using the α-imino carbene complex A as starting point. Geometrically, in relation with the imidazolidine skeleton, the Rh-carbene faces either the aminal bridge or the –CH2CH2– backbone of the 5-membered ring. Both approximation modes were studied and only the most favored path is detailed here (Fig. 2 and S8‡). From complex A, the computed transition state TS A-B involves the C–N bond formation between the carbene complex and the imidazolidine substrate (ΔG‡ = 7.7 kcal mol−1), achieving the nitrogen ylide intermediate B. The system rapidly evolves to B′ by switching the Rh catalyst from the C to the N atom of the former α-imino carbene, in an exergonic step of 2.7 kcal mol−1. In this disposition, the aminal opening occurs through TS B′-C with a low activation barrier (ΔG‡ = 2.2 kcal mol−1) leading to the iminium intermediate C with a relative energy of 13.1 kcal mol−1 below the initial reactants (full details in Fig. S1‡). The computed results for the donor OMe – acceptor NO2 system (2E) show the same two-step process although in this case the aminal opening requires higher activation energy (ΔG‡ = 7.2 kcal mol−1) and the final iminium intermediate C(OMe–NO2) is stabilized by only 3.7 kcal mol−1 (see further details in Fig. S4‡).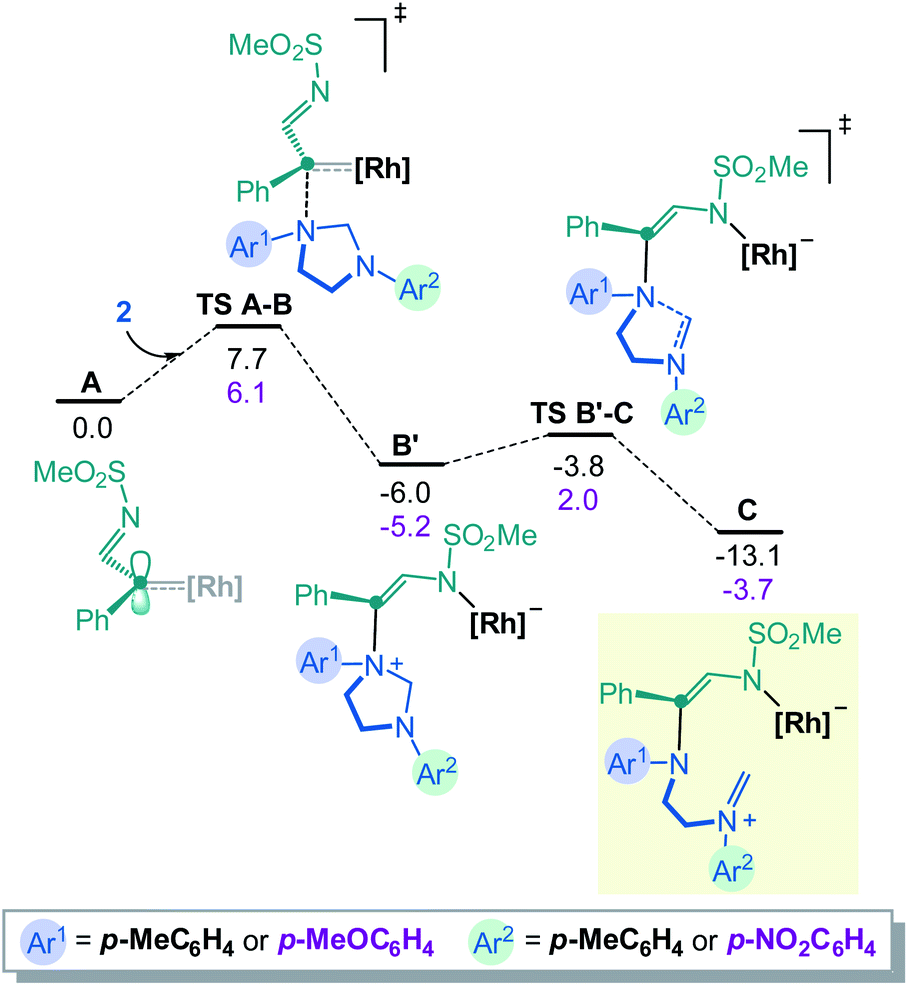 | ||
| Fig. 2 Computed Gibbs energy profile for the formation of the iminium intermediate C. Donor–donor energies (Ar1, Ar2 EDGs) in black and donor–acceptor (Ar1 EDG, Ar2 EWG) in magenta, all in kcal mol−1. | ||
At this stage, intermediate C can evolve either towards N- or C-cyclizations. Pathway A (Fig. 3, left) depicts the progression of intermediate C through an N-cyclization process, TS C-4, in a quasi-barrier less step (ΔG‡ = 1.3 kcal mol−1) affording the formation of the 8-membered-[Rh] complex. The Rh catalyst is then released and hexahydro-1,3,6-triazocine product 4 is formed in an exergonic reversible manner (ΔG = 11.7 kcal mol−1 below C). The C-cyclization reaction is shown in Pathway B (Fig. 3, right). First, viaTS C-D′, the 6-membered-[Rh] complex D′ is formed with an activation barrier of 8.1 kcal mol−1. This transition state involves the bond formation between the C atom of the iminium ion and the C atom of the former carbene. It is worth to mention that the process described from B to D′ corresponds to a formal [1,2]-Stevens rearrangement.19 Finally, to proceed towards the formation of hexahydropyrazino[1,2-a]indole 3, intermediate D evolves through a Friedel–Crafts cyclization with the assistance of the Rh2(Piv)4 catalyst (see details in Fig. S3‡). These final steps require an overall activation energy close to 20 kcal mol−1 and lead to the formation of the most stable product 3 with a relative energy of 37.4 kcal mol−1 below the initial reactants. The analysis of the full reaction path follows a clear Curtin–Hammett behavior. Compound 4, the kinetically-preferred product formed viaTS C-4, can reopen in the reaction conditions again and, viaTS C-D′ (ΔG‡ = 19.8 kcal mol−1), the formation of the thermodynamic product 3 is then favored. This last step is irreversible as 32.4 kcal mol−1 are required to overcome TS C-D′ from 3.
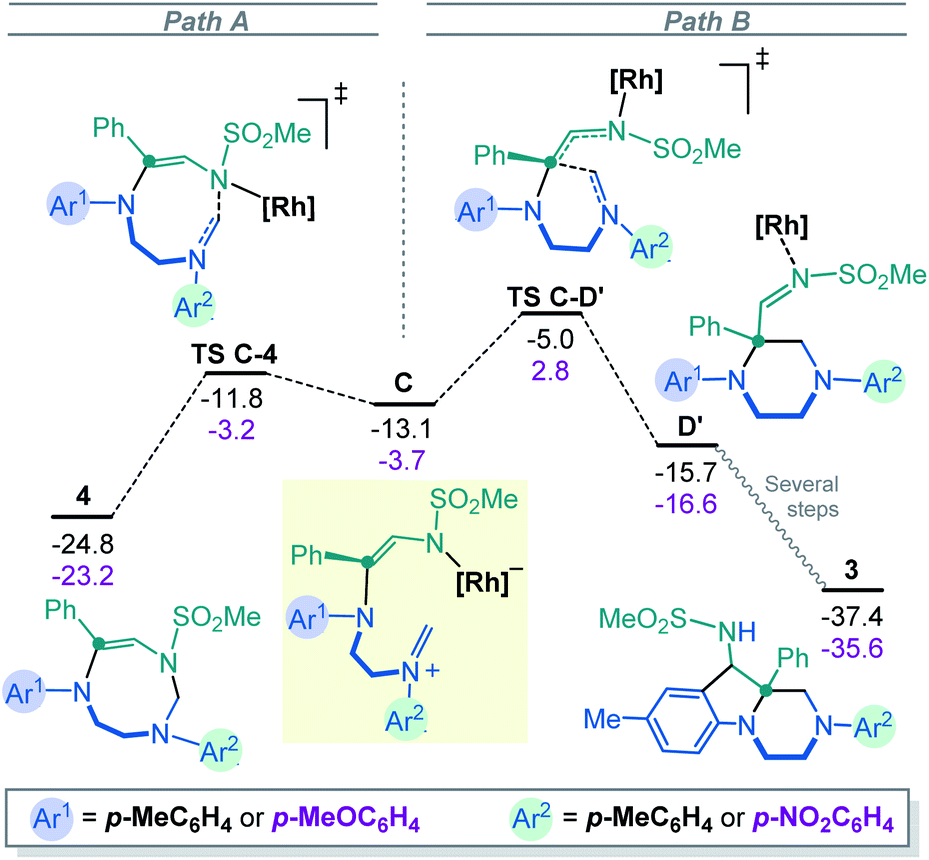 | ||
| Fig. 3 Computed Gibbs energy profile for the 8-membered (left, N-cyclization) or 6-membered (right, C-cyclization) ring formation from the iminium intermediate C. Donor–donor (Ar1, Ar2 EDGs) energies in black and donor–acceptor (Ar1 EDG, Ar2 EWG) in magenta, all in kcal mol−1. | ||
Fig. 3 also depicts the energies for the donor–acceptor system (Ar1 EDG, Ar2 EWG, numerical values in magenta). Interestingly, in this case, TS C-D′(OMe–NO2)20 has an energy of 2.8 kcal mol−1 above the initial reactants which is sufficient to suppress the Curtin–Hammett behavior. In fact, once product 4 is formed, the reversibility of the opening reaction is no longer feasible. The formation of product 3 would require ca. 26 kcal mol−1, which is not permitted in the reaction conditions. The selectivity of the carbene towards the different N atoms was also calculated when unsymmetrically substituted imidazolidine 2E is used as substrate. The results show that the attack on the N-atom bearing the p-MeOC6H4 group is favored, in agreement with the experimental observations (see details in Fig. S12‡).
Overall, thanks to the DFT calculations that are summarized in Scheme 4, an elaborate Curtin–Hammett behavior is evidenced with derivatives 3 and 4 as the thermodynamically- and kinetically-preferred products, respectively. With symmetrical imidazolidines as substrates (Ar1 = Ar2 EDGs or weakly EWGs), the corresponding 8-membered ring derivatives of type 4 are unstable in the reaction conditions and evolve towards hexahydropyrazino[1,2-a]indoles 3, after a reversible ring opening. Such a behavior does not occur anymore when donor–acceptor 2E is used as reactant (Ar1 strong EDG, Ar2 strong EWG). This substrate-dependent reactivity is controlled by TS C-D′, the transition state at the origin of the synthetic divergence.
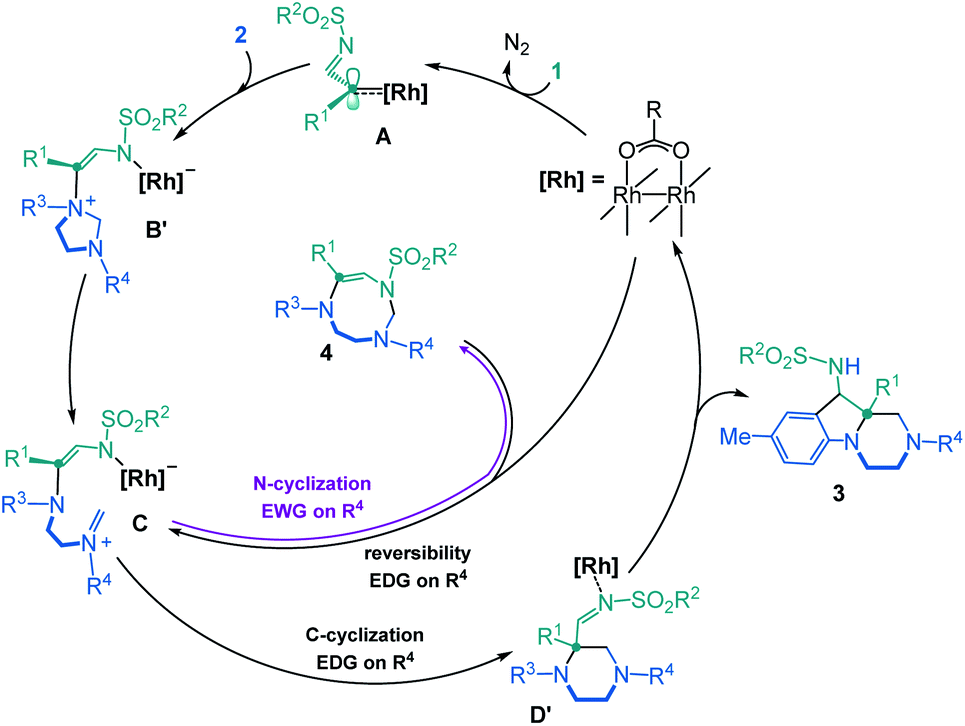 | ||
| Scheme 4 Global mechanistic rationale. | ||
Extension
Finally, with hexahydropyrazino[1,2-a]indoles 3 in hand, care was taken to extend the chemistry to further heterocycles and addition derivatives. First, treatment of 3aA (dr 3.6:1) with concentrated triflic acid (25 °C, 14 h) led to further derivatization. In fact, 1H-NMR analysis of the crude reaction mixture revealed a full conversion and the formation of single product 5aA, which was easily isolated in good yield (82%). Its exact structure was determined by X-ray diffraction analysis (Scheme 5, bottom). Such 1,2,3,4-tetrahydropyrazino[1,2-a]indole of type 5 is probably formed through protonation and elimination of TsNH2 followed by a 1,2-shift of the aryl group (R1) and a final proton loss (see Scheme S2‡). Isolated single diastereoisomers 3aA′ and 3aA′′ were treated independently with TfOH under the reaction conditions. Product 5aA was formed in both instances and isolated with the same yield. A series of derivatives 3A was treated under these reaction conditions. Both electron donating and electron withdrawing substituents on the migrating phenyl group (R1) afforded the corresponding products 5dA and 5fA in very good yields (87% and 88%, respectively). Other substituents R1 such as thiophene 3gA and 6-methoxynaphthalene 3hA were also compatible leading to the expected products 5gA and 5hA in 77% and 86% yields, respectively. p-MeO and p-Br substituents at R3 position led to 5aB and 5aC in 78% and 77% yields.
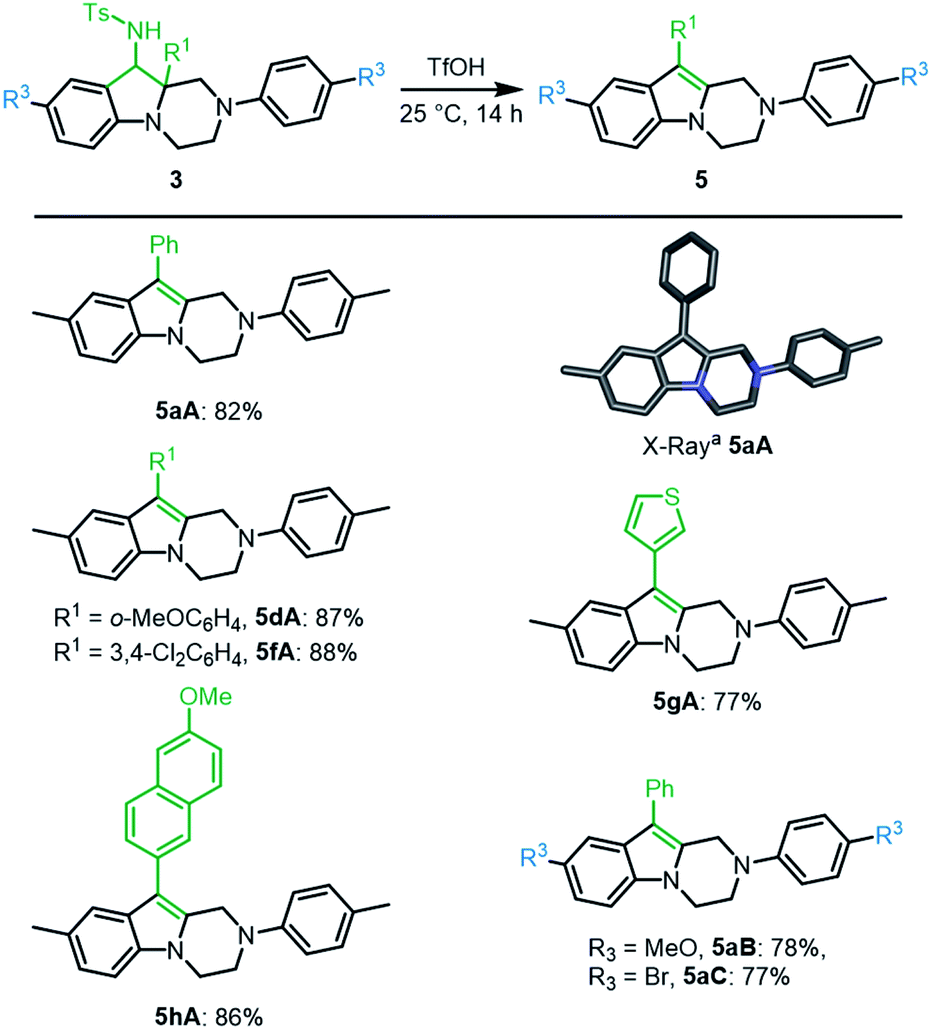 | ||
| Scheme 5 One-step access to tetrahydropyrazino[1,2-a]indoles 5. aStick view of the crystal structure of 5aA. Hydrogen atoms are omitted for clarity. | ||
Then, compounds 5 were further functionalized via oxidative C–C bond forming reactions using conditions similar to that reported for tetrahydroisoquinolines.21 In fact, treatment of 5aA with one equivalent of CuCl2 in THF/MeNO2 (1:1) led to the corresponding iminium intermediate,22,23 which upon addition of Hünig's base (iPr2EtN, 1.1 equiv.) led to the clean formation of 6 (92% yield) by an in situ aza-Henry reaction (Scheme 6). Further oxidative derivatizations of 5aA were possible with silyl ketene acetals and cyanide anion as nucleophiles to afford the corresponding ester 7 and nitrile 8 in 73% and 93% yields, respectively.
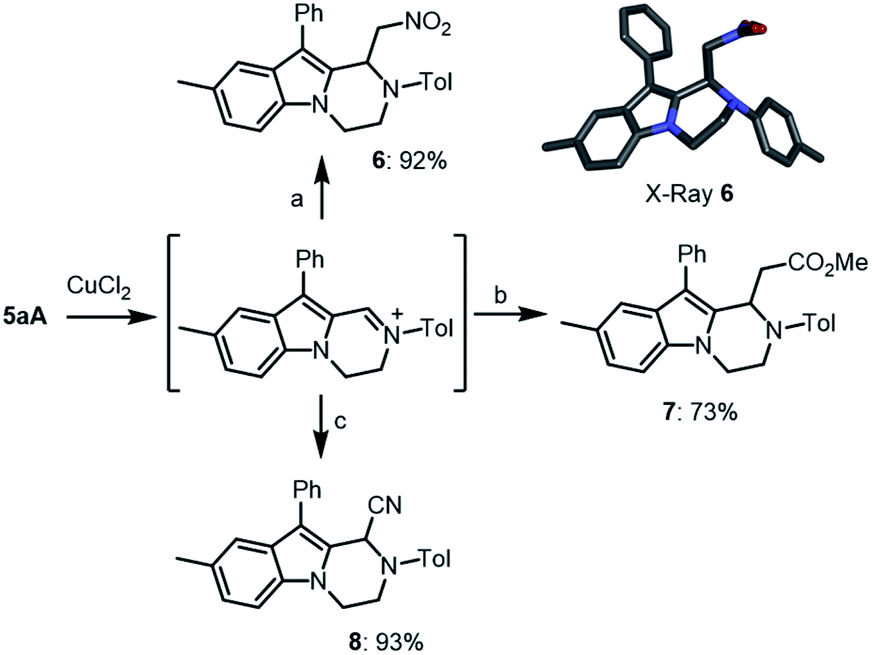 | ||
| Scheme 6 Late stage functionalization of 5aAvia oxidative C–C bond formation: (a) (i) CuCl2 (1 equiv.), THF/MeNO2 (1:1), 25 °C, 3 h; (ii) iPr2EtN (1.1 equiv.), 25 °C, 2 h. (b) (i) CuCl2 (1 equiv.), THF, 25 °C, 16 h; (ii) 1-(tert-butyldimethylsilyloxy)-1-methoxyethene (2 + 1 equiv.), 25 °C, 6 h. (c) CuCl2 (1 equiv.), THF, 25 °C, 16 h; (ii) NaCN (1.5 + 1.5 equiv.), MeOH, 25 °C, 6 h. Insert: stick view of the crystal structure of 6. Hydrogen atoms are omitted for clarity and the CH2NO2 chain presents a local disorder. | ||
Conclusions
In summary, novel hexahydropyrazinoindoles 3 were prepared in a single step from N-sulfonyl triazoles 1 and imidazolidines 2. Under dirhodium catalysis, α-imino carbenes were generated and formed nitrogen ylide intermediates that, after subsequent aminal opening, afforded derivatives 3 predominantly via a formal C-reactivity/[1,2]-Stevens pathway and tandem Friedel–Crafts cyclization. Of mechanistic importance, a regiodivergent reactivity could be engineered through the use of unsymmetrically substituted imidazolidine substrate 2E that promoted the exclusive formation of 8-membered ring derivative 4. Importantly, DFT computed results demonstrated a mechanistic Curtin–Hammett-like situation. First, compounds of type 4 are formed as kinetic products and reopened reversibly with the aid of the Rh2(Piv)4 catalyst. Given time and energy, the system evolves towards the formation of the thermodynamically preferred product 3, after an irreversible Friedel–Crafts alkylation that seals the multi-step pathway. With imidazolidine 2E as reagent, the formation of 4 becomes irreversible and the 8-membered heterocycle is formed preferentially as a unique regioisomer. Finally, with derivatives 3 in hand, tetrahydropyrazinoindoles 5 were prepared efficiently upon treatment with triflic acid; compounds 5 being themselves suitable for late-stage functionalization via oxidative C–C bond forming reactions giving products 6 to 8.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Geneva and the Swiss National Science Foundation for financial support (JL: 200020-172497 and 200020-184843). We also acknowledge the contributions of the Sciences Mass Spectrometry (SMS) platform at the Faculty of Sciences, University of Geneva. We also thank Carmine Chiancone for technical support.Notes and references
- (a) M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS; (b) J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC; (c) B. Schulze and U. S. Schubert, Chem. Soc. Rev., 2014, 43, 2522–2571 RSC; (d) V. K. Tiwari, B. B. Mishra, K. B. Mishra, N. Mishra, A. S. Singh and X. Chen, Chem. Rev., 2016, 116, 3086–3240 CrossRef CAS; (e) M. M. Haugland, S. Borsley, D. F. Cairns-Gibson, A. Elmi and S. L. Cockroft, ACS Nano, 2019, 13, 4101–4110 CrossRef CAS.
- (a) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS; (b) W. G. Lewis, L. G. Green, F. Grynszpan, Z. Radić, P. R. Carlier, P. Taylor, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 1053–1057 CrossRef CAS; (c) F. Amblard, J. H. Cho and R. F. Schinazi, Chem. Rev., 2009, 109, 4207–4220 CrossRef CAS; (d) C. Le Droumaguet, C. Wang and Q. Wang, Chem. Soc. Rev., 2010, 39, 1233–1239 RSC; (e) P. Thirumurugan, D. Matosiuk and K. Jozwiak, Chem. Rev., 2013, 113, 4905–4979 CrossRef CAS.
- (a) P. Grünanger, P. V. Finzi and C. Scotti, Chem. Ber., 1965, 98, 623–628 CrossRef; (b) M. E. Hermes and F. D. Marsh, J. Am. Chem. Soc., 1967, 89, 4760–4764 CrossRef CAS; (c) R. E. Harmon, F. Stanley, S. K. Gupta and J. Johnson, J. Org. Chem., 1970, 35, 3444–3448 CrossRef CAS; (d) R. E. Harmon, R. A. Earl and S. K. Gupta, J. Chem. Soc. D, 1971, 296–297 RSC.
- (a) B. Chattopadhyay and V. Gevorgyan, Angew. Chem., Int. Ed., 2012, 51, 862–872 CrossRef CAS; (b) A. V. Gulevich and V. Gevorgyan, Angew. Chem., Int. Ed., 2013, 52, 1371–1373 CrossRef CAS; (c) H. M. Davies and J. S. Alford, Chem. Soc. Rev., 2014, 43, 5151–5162 RSC; (d) P. Anbarasan, D. Yadagiri and S. Rajasekar, Synthesis, 2014, 46, 3004–3023 CrossRef CAS; (e) Y. Wang, X. Lei and Y. Tang, Synlett, 2015, 26, 2051–2059 CrossRef; (f) Y. Jiang, R. Sun, X.-Y. Tang and M. Shi, Chem.–Eur. J., 2016, 22, 17910–17924 CrossRef CAS.
- (a) S. Chuprakov, F. W. Hwang and V. Gevorgyan, Angew. Chem., Int. Ed., 2007, 46, 4757–4759 CrossRef CAS; (b) S. Chuprakov, S. W. Kwok, L. Zhang, L. Lercher and V. V. Fokin, J. Am. Chem. Soc., 2009, 131, 18034–18035 CrossRef CAS; (c) S. Chuprakov, J. A. Malik, M. Zibinsky and V. V. Fokin, J. Am. Chem. Soc., 2011, 133, 10352–10355 CrossRef CAS; (d) M. Zibinsky and V. V. Fokin, Org. Lett., 2011, 13, 4870–4872 CrossRef CAS; (e) D. Yadagiri and P. Anbarasan, Chem.–Eur. J., 2013, 19, 15115–15119 CrossRef CAS; (f) E. E. Schultz and R. Sarpong, J. Am. Chem. Soc., 2013, 135, 4696–4699 CrossRef CAS; (g) T. Miura, T. Tanaka, K. Matsumoto and M. Murakami, Chem.–Eur. J., 2014, 20, 16078–16082 CrossRef CAS; (h) T. Miura, T. Nakamuro, C.-J. Liang and M. Murakami, J. Am. Chem. Soc., 2014, 136, 15905–15908 CrossRef CAS; (i) D. Yadagiri and P. Anbarasan, Org. Lett., 2014, 16, 2510–2513 CrossRef CAS; (j) F. Medina, C. Besnard and J. Lacour, Org. Lett., 2014, 16, 3232–3235 CrossRef CAS; (k) V. N. G. Lindsay, H. M. F. Viart and R. Sarpong, J. Am. Chem. Soc., 2015, 137, 8368–8371 CrossRef CAS; (l) R. W. Kubiak, J. D. Mighion, S. M. Wilkerson-Hill, J. S. Alford, T. Yoshidomi and H. M. L. Davies, Org. Lett., 2016, 18, 3118–3121 CrossRef CAS; (m) A. Guarnieri-Ibáñez, F. Medina, C. Besnard, S. L. Kidd, D. R. Spring and J. Lacour, Chem. Sci., 2017, 8, 5713–5720 RSC; (n) T. Miura, Q. Zhao and M. Murakami, Angew. Chem., Int. Ed., 2017, 56, 16645–16649 CrossRef CAS; (o) X. Ma, X. Xie, L. Liu, R. Xia, T. Li and H. Wang, Chem. Commun., 2018, 54, 1595–1598 RSC; (p) Z. Liu, Q. Du, H. Zhai and Y. Li, Org. Lett., 2018, 20, 7514–7517 CrossRef CAS; (q) Z. J. Garlets and H. M. L. Davies, Org. Lett., 2018, 20, 2168–2171 CrossRef CAS; (r) R. Jia, J. Meng, J. Leng, X. Yu and W. P. Deng, Chem.–Asian J., 2018, 13, 2360–2364 CrossRef CAS; (s) D. Yadagiri, M. Chaitanya, A. C. S. Reddy and P. Anbarasan, Org. Lett., 2018, 20, 3762–3765 CrossRef CAS; (t) Z.-F. Xu, L. Shan, W. Zhang, M. Cen and C.-Y. Li, Org. Chem. Front., 2019, 6, 1391–1396 RSC; (u) P. B. De, S. Atta, S. Pradhan, S. Banerjee, T. A. Shah and T. Punniyamurthy, J. Org. Chem., 2020, 85, 4785–4794 CrossRef CAS; (v) A. C. S. Reddy, K. Ramachandran, P. M. Reddy and P. Anbarasan, Chem. Commun., 2020, 56, 5649–5652 RSC; (w) H. J. Dequina, J. Eshon, W. T. Raskopf, I. Fernández and J. M. Schomaker, Org. Lett., 2020, 22, 3637–3641 CrossRef CAS; (x) T. Miura, T. Nakamuro, Y. Ishihara, Y. Nagata and M. Murakami, Angew. Chem., Int. Ed., 2020, 59, 20475–20479 CrossRef CAS.
- (a) S. Chuprakov, S. W. Kwok and V. V. Fokin, J. Am. Chem. Soc., 2013, 135, 4652–4655 CrossRef CAS; (b) H. J. Jeon, D. J. Jung, J. H. Kim, Y. Kim, J. Bouffard and S.-g. Lee, J. Org. Chem., 2014, 79, 9865–9871 CrossRef CAS; (c) D. J. Lee, H. S. Han, J. Shin and E. J. Yoo, J. Am. Chem. Soc., 2014, 136, 11606–11609 CrossRef CAS; (d) D. J. Lee, D. Ko and E. J. Yoo, Angew. Chem., Int. Ed., 2015, 54, 13715–13718 CrossRef CAS; (e) X. Lei, L. Li, Y.-P. He and Y. Tang, Org. Lett., 2015, 17, 5224–5227 CrossRef CAS; (f) H.-D. Xu, Z.-H. Jia, K. Xu, H. Zhou and M.-H. Shen, Org. Lett., 2015, 17, 66–69 CrossRef CAS; (g) T. Ryu, Y. Baek and P. H. Lee, J. Org. Chem., 2015, 80, 2376–2383 CrossRef CAS; (h) Y.-Z. Zhao, H.-B. Yang, X.-Y. Tang and M. Shi, Chem.–Eur. J., 2015, 21, 3562–3566 CrossRef CAS; (i) Y. Wang, X. Lei and Y. Tang, Chem. Commun., 2015, 51, 4507–4510 RSC; (j) N. V. Rostovskii, J. O. Ruvinskaya, M. S. Novikov, A. F. Khlebnikov, I. A. Smetanin and A. V. Agafonova, J. Org. Chem., 2017, 82, 256–268 CrossRef CAS.
- (a) A. Bosmani, A. Guarnieri-Ibáñez, S. Goudedranche, C. Besnard and J. Lacour, Angew. Chem., Int. Ed., 2018, 57, 7151–7155 CrossRef CAS; (b) A. Bosmani, A. Guarnieri-Ibáñez and J. Lacour, Helv. Chim. Acta, 2019, 102, e1900021 CrossRef.
- J. Ge, X. Wu and X. Bao, Chem. Commun., 2019, 55, 6090–6093 RSC.
- (a) M. Bös, F. Jenck, J. R. Martin, J. L. Moreau, V. Mutel, A. J. Sleight and U. Widmer, Eur. J. Med. Chem., 1997, 32, 253–261 CrossRef; (b) S. Röver, D. R. Adams, A. Bénardeau, J. M. Bentley, M. J. Bickerdike, A. Bourson, I. A. Cliffe, P. Coassolo, J. E. P. Davidson, C. T. Dourish, P. Hebeisen, G. A. Kennett, A. R. Knight, C. S. Malcolm, P. Mattei, A. Misra, J. Mizrahi, M. Muller, R. H. P. Porter, H. Richter, S. Taylor and S. P. Vickers, Bioorg. Med. Chem. Lett., 2005, 15, 3604–3608 CrossRef; (c) H. G. F. Richter, D. R. Adams, A. Benardeau, M. J. Bickerdike, J. M. Bentley, T. J. Blench, I. A. Cliffe, C. Dourish, P. Hebeisen, G. A. Kennett, A. R. Knight, C. S. Malcolm, P. Mattei, A. Misra, J. Mizrahi, N. J. T. Monck, J. M. Plancher, S. Roever, J. R. A. Roffey, S. Taylor and S. P. Vickers, Bioorg. Med. Chem. Lett., 2006, 16, 1207–1211 CrossRef CAS; (d) N. Krogsgaard-Larsen, A. A. Jensen and J. Kehler, Bioorg. Med. Chem. Lett., 2010, 20, 5431–5433 CrossRef CAS.
- R. K. Tiwari, A. K. Verma, A. K. Chhillar, D. Singh, J. Singh, V. Kasi Sankar, V. Yadav, G. L. Sharma and R. Chandra, Bioorg. Med. Chem., 2006, 14, 2747–2752 CrossRef CAS.
- R. K. Tiwari, D. Singh, J. Singh, V. Yadav, A. K. Pathak, R. Dabur, A. K. Chhillar, R. Singh, G. L. Sharma, R. Chandra and A. K. Verma, Bioorg. Med. Chem. Lett., 2006, 16, 413–416 CrossRef CAS.
- (a) Z. Shiokawa, K. Hashimoto, B. Saito, Y. Oguro, H. Sumi, M. Yabuki, M. Yoshimatsu, Y. Kosugi, Y. Debori, N. Morishita, D. R. Dougan, G. P. Snell, S. Yoshida and T. Ishikawa, Bioorg. Med. Chem., 2013, 21, 7938–7954 CrossRef CAS; (b) Y. J. Kim, J. S. Pyo, Y.-S. Jung and J.-H. Kwak, Bioorg. Med. Chem. Lett., 2017, 27, 607–611 CrossRef CAS.
- G. Zoidis, E. Giannakopoulou, A. Stevaert, E. Frakolaki, V. Myrianthopoulos, G. Fytas, P. Mavromara, E. Mikros, R. Bartenschlager, N. Vassilaki and L. Naesens, RSC Med. Chem., 2016, 7, 447–456 RSC.
- (a) R. A. Bit, P. D. Davis, L. H. Elliott, W. Harris, C. H. Hill, E. Keech, H. Kumar, G. Lawton, A. Maw, J. S. Nixon, D. R. Vesey, J. Wadsworth and S. E. Wilkinson, J. Med. Chem., 1993, 36, 21–29 CrossRef CAS; (b) D. M. Vigushin, G. Brooke, D. Willows, R. C. Coombes and C. J. Moody, Bioorg. Med. Chem. Lett., 2003, 13, 3661–3663 CrossRef CAS; (c) D. R. Goldberg, Y. Choi, D. Cogan, M. Corson, R. DeLeon, A. Gao, L. Gruenbaum, M. H. Hao, D. Joseph, M. A. Kashem, C. Miller, N. Moss, M. R. Netherton, C. P. Pargellis, J. Pelletier, R. Sellati, D. Skow, C. Torcellini, Y. C. Tseng, J. Wang, R. Wasti, B. Werneburg, J. P. Wu and Z. Xiong, Bioorg. Med. Chem. Lett., 2008, 18, 938–941 CrossRef CAS; (d) Z. Xiong, D. A. Gao, D. A. Cogan, D. R. Goldberg, M.-H. Hao, N. Moss, E. Pack, C. Pargellis, D. Skow, T. Trieselmann, B. Werneburg and A. White, Bioorg. Med. Chem. Lett., 2008, 18, 1994–1999 CrossRef CAS; (e) H. G. F. Richter, C. Freichel, J. Huwyler, T. Nakagawa, M. Nettekoven, J. M. Plancher, S. Raab, O. Roche, F. Schuler, S. Taylor, C. Ullmer and R. Wiegand, Bioorg. Med. Chem. Lett., 2010, 20, 5713–5717 CrossRef CAS; (f) D. J. Buzard, T. O. Schrader, X. Zhu, J. Lehmann, B. Johnson, M. Kasem, S. H. Kim, A. Kawasaki, L. Lopez, J. Moody, S. Han, Y. Gao, J. Edwards, J. Barden, J. Thatte, J. Gatlin and R. M. Jones, Bioorg. Med. Chem. Lett., 2015, 25, 659–663 CrossRef CAS.
- With chiral catalysts such Rh2(R-DOSP)4 or Rh2(S-TCPTTL)4, only racemic product 3aA is obtained. This could be an indication that a racemization of intermediate D′ occurs in situ. See ref. 7b for such a possibility involving a sequence of Mannich and retro-Mannich reactions. Calculations further indicate that the barrier for the retro-Mannich is affordable in the experimental conditions (see Fig. 3).
- Somewhat surprisingly, the Friedel–Crafts reaction leads to the preferred formation of the cis-adduct over the trans-diastereoisomer. This predominance can be rationalized in silico by a difference (ΔΔG‡ 2.3 kcal mol−1) between the corresponding transition states. See Fig. S13‡ for details.
- J. Raushel and V. V. Fokin, Org. Lett., 2010, 12, 4952–4955 CrossRef CAS.
- (a) C. H. Yoder and J. J. Zuckerman, Inorg. Chem., 1967, 6, 103–107 CrossRef CAS; (b) A. R. Katritzky, W. Fan and C. Fu, J. Org. Chem., 1990, 55, 3209–3213 CrossRef CAS; (c) A. J. Arduengo, R. Krafczyk, R. Schmutzler, H. A. Craig, J. R. Goerlich, W. J. Marshall and M. Unverzagt, Tetrahedron, 1999, 55, 14523–14534 CrossRef CAS; (d) M. K. Denk, S. Gupta, J. Brownie, S. Tajammul and A. J. Lough, Chem.–Eur. J., 2001, 7, 4477–4486 CrossRef CAS.
- R. Bach, S. Harthong and J. Lacour, in Comprehensive Organic Synthesis II, ed. P. Knochel, Elsevier, Amsterdam, 2nd edn, 2014, pp. 992–1037 Search PubMed.
- This transition state was computed as single point since full optimization led to problems with the convergence due to the intrusion of imaginary frequencies related to the methyl groups of the Rh2(Piv)4 catalyst.
- (a) D. Sureshkumar, A. Sud and M. Klussmann, Synlett, 2009, 10, 1558–1561 Search PubMed; (b) E. Boess, D. Sureshkumar, A. Sud, C. Wirtz, C. Farès and M. Klussmann, J. Am. Chem. Soc., 2011, 133, 8106–8109 CrossRef CAS; (c) E. Boess, C. Schmitz and M. Klussmann, J. Am. Chem. Soc., 2012, 134, 5317–5325 CrossRef CAS; (d) J. Dhineshkumar, M. Lamani, K. Alagiri and K. R. Prabhu, Org. Lett., 2013, 15, 1092–1095 CrossRef CAS; (e) G. Bergonzini, C. S. Schindler, C.-J. Wallentin, E. N. Jacobsen and C. R. J. Stephenson, Chem. Sci., 2014, 5, 112–116 RSC; (f) Y. Liu, C. Wang, D. Xue, M. Xiao, C. Li and J. Xiao, Chem.–Eur. J., 2017, 23, 3051–3061 CrossRef CAS; (g) G. Oss, S. D. de Vos, K. N. H. Luc, J. B. Harper and T. V. Nguyen, J. Org. Chem., 2018, 83, 1000–1010 CrossRef CAS.
- N. Saleh, A. Bosmani, C. Besnard, T. Bürgi, D. Jacquemin and J. Lacour, Org. Lett., 2020, 22, 7599–7603 CrossRef CAS.
- Synthetic attempts to isolate and characterize the in situ generated iminium salt were unfortunately unsuccessful.
Footnotes |
| † The dataset for this article can be found at the following DOI: 10.26037/yareta:u5rrhkxqyrgbhpijt7uhyqi6eq. It will be preserved for 10 years. |
| ‡ Electronic supplementary information (ESI) available: Synthetic protocols, 1H/13C NMR and HR mass spectra. CCDC 2015200–2015204. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc05725h |
| This journal is © The Royal Society of Chemistry 2021 |