
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrocatalytic redox neutral [3 + 2] annulation of N-cyclopropylanilines and alkenes†
Qi
Wang
a,
Qile
Wang
b,
Yuexiang
Zhang
c,
Yasmine M.
Mohamed
a,
Carlos
Pacheco
 a,
Nan
Zheng
*b,
Richard N.
Zare
*d and
Hao
Chen
*a
a,
Nan
Zheng
*b,
Richard N.
Zare
*d and
Hao
Chen
*a
aDepartment of Chemistry & Environmental Science, New Jersey Institute of Technology, Newark, New Jersey 07102, USA. E-mail: hao.chen.2@njit.edu
bDepartment of Chemistry and Biochemistry, University of Arkansas, Fayetteville, Arkansas 72701, USA. E-mail: nzheng@uark.edu
cDepartment of Chemistry and Biochemistry, Ohio University, Athens, Ohio 45701, USA
dDepartment of Chemistry, Stanford University, Stanford, California 94305-5080, USA. E-mail: zare@stanford.edu
First published on 9th November 2020
Abstract
Although synthetic organic electrochemistry (EC) has advanced significantly, net redox neutral electrosynthesis is quite rare. Two approaches have been employed to achieve this type of electrosynthesis. One relies on turnover of the product by the reactant in a chain mechanism. The other involves both oxidation on the anode and reduction on the cathode in which the radical cation or the radical anion of the product has to migrate between two electrodes. Herein, a home-built electrochemistry/mass spectrometry (EC/MS) platform was used to generate an N-cyclopropylaniline radical cation electrochemically and to monitor its reactivity toward alkenes by mass spectrometry (MS), which led to the discovery of a new redox neutral reaction of intermolecular [3 + 2] annulation of N-cyclopropylanilines and alkenes to provide an aniline-substituted 5-membered carbocycle via direct electrolysis (yield up to 81%). A chain mechanism, involving the regeneration of the substrate radical cation and the formation of the neutral product, is shown to be responsible for promoting such a redox neutral annulation reaction, as supported by experimental evidence of EC/MS.
Introduction
Mass spectrometry (MS) has become a powerful technique for studying reaction mechanisms since the advent of soft ionization methods such as electrospray ionization (ESI).1–12 The combination of electrochemistry (EC) with MS, EC/MS, can be applied to produce drugs in vivo metabolites, or cleave proteins/peptides followed by MS analysis.13–17 It can also be used to reduce disulfide bonds to facilitate MS sequencing of proteins/peptides,18–20 oxidize tyrosine to perform absolute MS quantitation,21 and oxidize lipids to determine double bond locations of unsaturated lipids.22,23 It has also been used to capture elusive reaction intermediates24–33 and to screen electrosynthetic reactions.34 The advantages of EC/MS for reaction screening are multiple. It is very sensitive and uses a tiny amount of reactants (nmoles to pmoles). It allows the monitoring of the reactivity of electrochemically generated short-lived reactive species by online MS detection. In spite of these advantages, the integrated online EC/MS platform has not been extensively used to screen electrosynthetic reactions.Recently, synthetic organic electrochemistry has achieved a dramatic uptick in popularity.35–51 Electrosynthesis uses a pair of electrodes to add or subtract electrons to or from the substrate, which triggers the formation of the target product.37,52–63 Compared with non-electrochemical synthesis, electrosynthesis has the advantages of offering more selective, safer, and less energy consumption approaches.64–74 One of the more challenging and thus elusive electrosyntheses that attracts much attention75–79 is net redox neutral reactions, in which both oxidation and reduction steps are involved to achieve overall redox neutrality. Initial anode oxidation or cathode reduction of the substrate to form a product radical cation or anion is usually paired with an opposite single-electron transfer (SET) event to furnish a neutral product.
Two possible conditions can trigger the occurrence of redox neutral electrochemical reactions. One involves the product radical cation or anion being stable enough to migrate to the cathode or anode so that a SET reduction or oxidation occurs to yield the final neutral product.77–80 The other requires the product radical cation or anion to undergo a SET oxidation or reduction with the starting material in a chain mechanism. The former is more arduous to meet as a tandem oxidation/reduction has to occur on a macroscopically separated anode and cathode. Most of the reported redox-neutral electrochemical syntheses are centered on the latter condition. Notable examples include [2 + 2] cycloaddition81–84 and [4 + 2] Diels–Alder85–88 cycloaddition reactions mediated by anodically produced radical cations of dienes or dienophiles. Chiba40 raised the importance of a redox tag on the chain event of these reactions. Radical anions of activated alkenes generated by cathodic reduction are equally capable of promoting these two classes of cycloaddition reactions. Xu89 developed a redox-neutral method for intramolecular hydroamidation of alkenes without relying on a chain event. The hydroamidation was achieved by an intramolecular addition of an amidyl radical generated via ferrocene as the redox catalyst to an alkene. The resulting carbon radical then abstracted a hydrogen atom from 1,4-cyclohexadiene (1,4-CHD) to complete the redox neutral process. For a handful of examples invoking a chain mechanism, catalytic current efficiency was usually used as the primary evidence,81,90 and limited kinetic studies were sometimes reported to strengthen the argument.91 Ambiguity about the chain event often remains.
We previously developed a [3 + 2] annulation of N-cyclopropylanilines (CPA) with alkenes by photoredox catalysis92 and subsequently investigated its mechanism.93 As shown in Scheme 1 (top panel), upon irradiation, Ru(II)(bpz)32+ is promoted to the excited triplet state Ru(II)*(bpz)32+, which oxidizes CPA to the radical cation. The radical cation subsequently undergoes ring opening, and then adds to styrene to produce the [3 + 2] annulation product radical cation. Finally, the radical cation is reduced via two mechanisms: a photoredox reaction and a chain reaction. We questioned whether we could achieve the annulation reaction by direct electrolysis presumably via the chain process completely. However, converting the photoredox catalysis to electrochemistry for this annulation reaction was nontrivial, as net redox neutral reactions are known to be problematic for electrochemistry but facile by photoredox catalysis. Herein, we report our studies in developing the electrocatalytic redox neutral [3 + 2] annulation of N-cyclopropylanilines and alkenes by the chain mechanism (Scheme 1, bottom panel). We took full advantage of the capabilities of the integrated online EC/MS platform as a screening tool to expedite the discovery. As evidenced by net redox neutral reactions, photochemistry and electrochemistry complement each other. This study overcame the inherent limitation of net redox neutral reactions in electrochemistry and used it as a template to address other types of challenging electrosynthetic reactions.
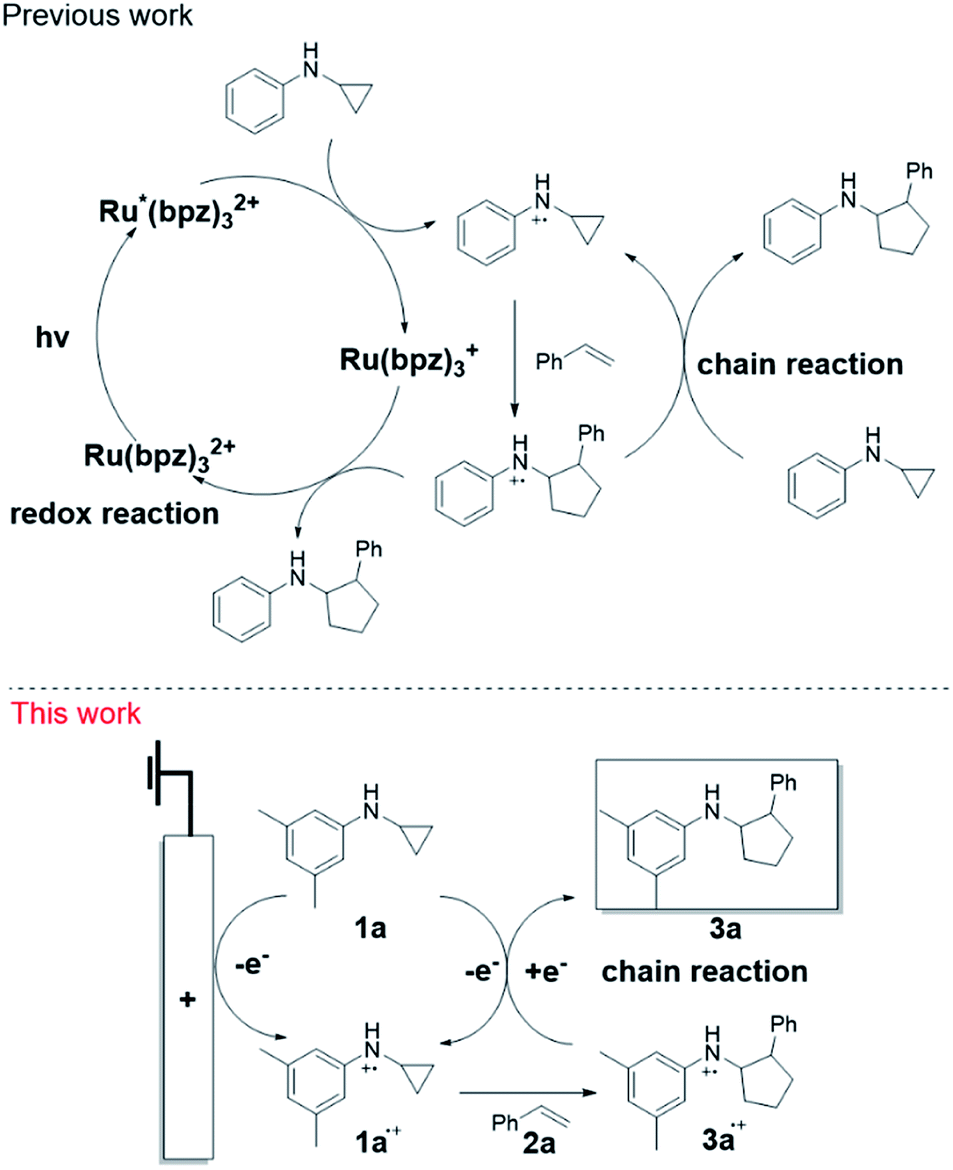 | ||
| Scheme 1 Proposed mechanism for the [3 + 2] annulation reaction of N-cyclopropylanilines and styrene (top panel illustrates the reaction catalyzed with a photocatalyst; bottom panel displays the reaction triggered electrochemically without using a catalyst). | ||
Results and discussion
We started our investigation using a home-built electrochemistry/mass spectrometry (EC/MS) platform (Scheme 2). It consisted of an electrochemical thin-layer flow cell, a short piece of a fused silica capillary as a microreactor and an online MS detector, with one reactant fused into the flow cell via channel 1 and another reactant introduced for reaction via channel 2. The flow cell was equipped with a glassy carbon disc (i.d., 6 mm) as the working electrode (WE), Ag/AgCl (3 M NaCl) as the reference electrode (RE), and the cell stainless steel body serving as a counter electrode (CE). The solution flowing out of the capillary microreactor was soft ionized by sonic spray ionization (SSI). To prove that the electrochemical method can be used to generate the [3 + 2] annulation reaction product, N-cyclopropyl-3,5-dimethylaniline (CPDA, 1a), and styrene (2a) were first chosen as reactants (Scheme 3). A small-scale test was performed using an electrochemical thin-layer flow cell along with online MS monitoring. A solution of 1a (1 mM) and lithium triflate (LiOTf, 1 mM) in MeCN was infused into the cell via channel 1 and MeCN was infused via channel 2 (flow rate: 50 μL min−1 for each channel). When a potential of +3.0 V was applied to the WE, as shown in the recorded SSI-MS spectrum (Fig. 1a), the 1a+˙ of m/z 161 (theoretical m/z 161.11990, measured m/z: 161.12017, mass error 1.68 ppm) was detected, indicating the occurrence of the electro-oxidation of 1a. When 2a was introduced to replace MeCN via channel 2, indeed, the protonated [3 + 2] annulation product [3a + H]+ (theoretical m/z 266.19033, measured m/z: 266.18972, mass error 2.29 ppm) was observed (Fig. 1b). Upon collision induced dissociation (CID), the ion of m/z 266 gave rise to fragment ions of m/z 145, 122, and 91 by losses of C8H11N, C11H12, and C12H17N, respectively, consistent with its assigned structure (Fig. 1c). At the same time, the intensity of the 1a+˙ decreased from 1.6 × 106 (Fig. 1a) to 1.9 × 104 (Fig. 1b), indicating that 1a+˙ did react with 2a to produce the [3 + 2] annulation product. The +1 ion of 3a instead of 3a+˙ was observed probably due to the charge transfer between 3a+˙ and 1a to form 3a which was ionized as the +1 ion by SSI.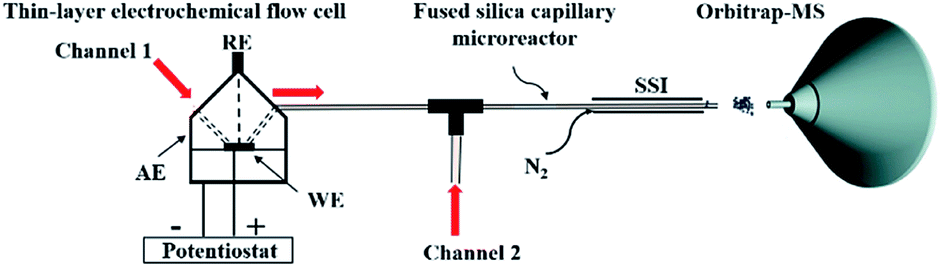 | ||
| Scheme 2 Home-built electrochemistry/mass spectrometry (EC/MS) setup. | ||
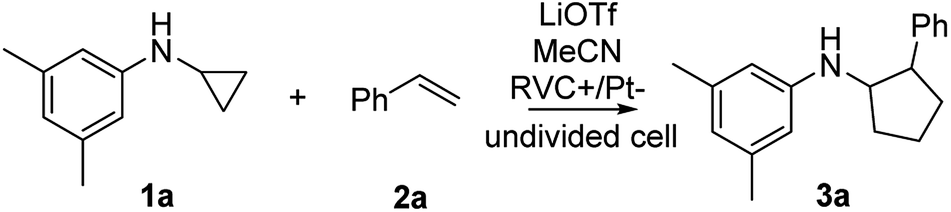 | ||
| Scheme 3 Intermolecular [3 + 2] annulation of N-cyclopropyl-3,5-dimethylaniline (CPDA) 1a and styrene 2a by electrolysis. | ||
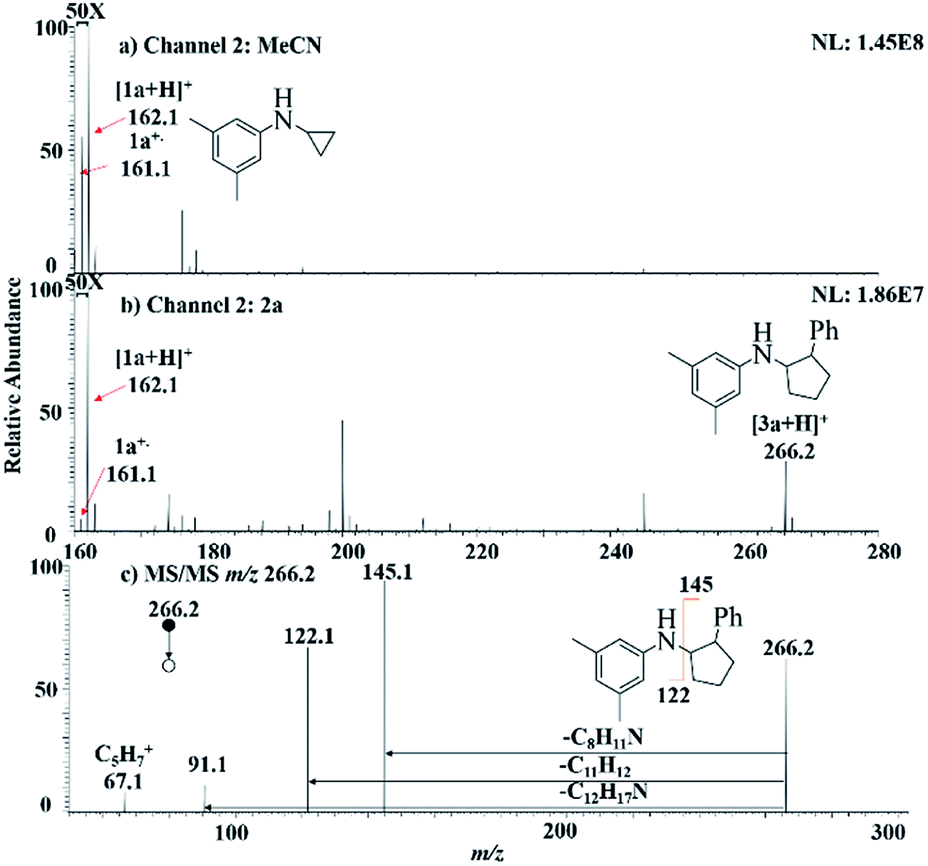 | ||
| Fig. 1 MS spectra showing (a) the formation of radical cation 1a+˙ when the cell was turned on with 1a being introduced into the flow cell via channel 1 and MeCN being introduced via channel 2; (b) the product ion [3a + H]+ was observed when the cell was turned on with 2a being introduced via channel 2. (c) MS/MS spectrum of m/z 266. | ||
The EC/MS setup (Scheme 2) also allowed us to verify whether or not the charge transfer between 3a+˙ and 1a could take place, a key step in the chain reaction mechanism (Fig. 2a), that would be needed to generate the neutral product 3a. Using the EC/MS setup, a solution of the annulation product 3a (1 mM) and LiOTf (1 mM) in MeCN was infused into the flow cell through channel 1, and MeCN was injected via channel 2. A potentiostat was used to supply the potential for electro-oxidation. The injection flow rate for both channels was 15 μL min−1. A voltage of +3.0 V was applied to the flow cell to trigger the oxidation of 3a. The expected radical cation 3a+˙ was detected (Fig. 2b, black line, theoretical 265.18250, measured 265.18234, mass error 0.60 ppm). When the solvent (MeCN) was replaced by 1a (0.1 mM in MeCN) in channel 2, the intensity of 3a+˙ decreased (Fig. 2b, red line), indicating the consumption of 3a+˙ by 1a. At the same time, radical cation 1a+˙ was detected (Fig. 2c, red line, theoretical 161.11990, measured 161.12002, mass error 0.74 ppm). To confirm that the observed 1a+˙ was not due to oxidation of 1a by other oxidative species from the cell, a control experiment was performed in which only LiOTf (1 mM) in MeCN (without 3a) was infused into the cell for oxidation under the same conditions and then mixed with 1a. In this control experiment, no 1a+˙ was generated (Fig. 2c, black line). This set of data confirms that the oxidation of 1a by the annulation product radical cation 3a+˙ (Fig. 2a) did occur, being the critical step responsible for completing the redox neutral reaction.
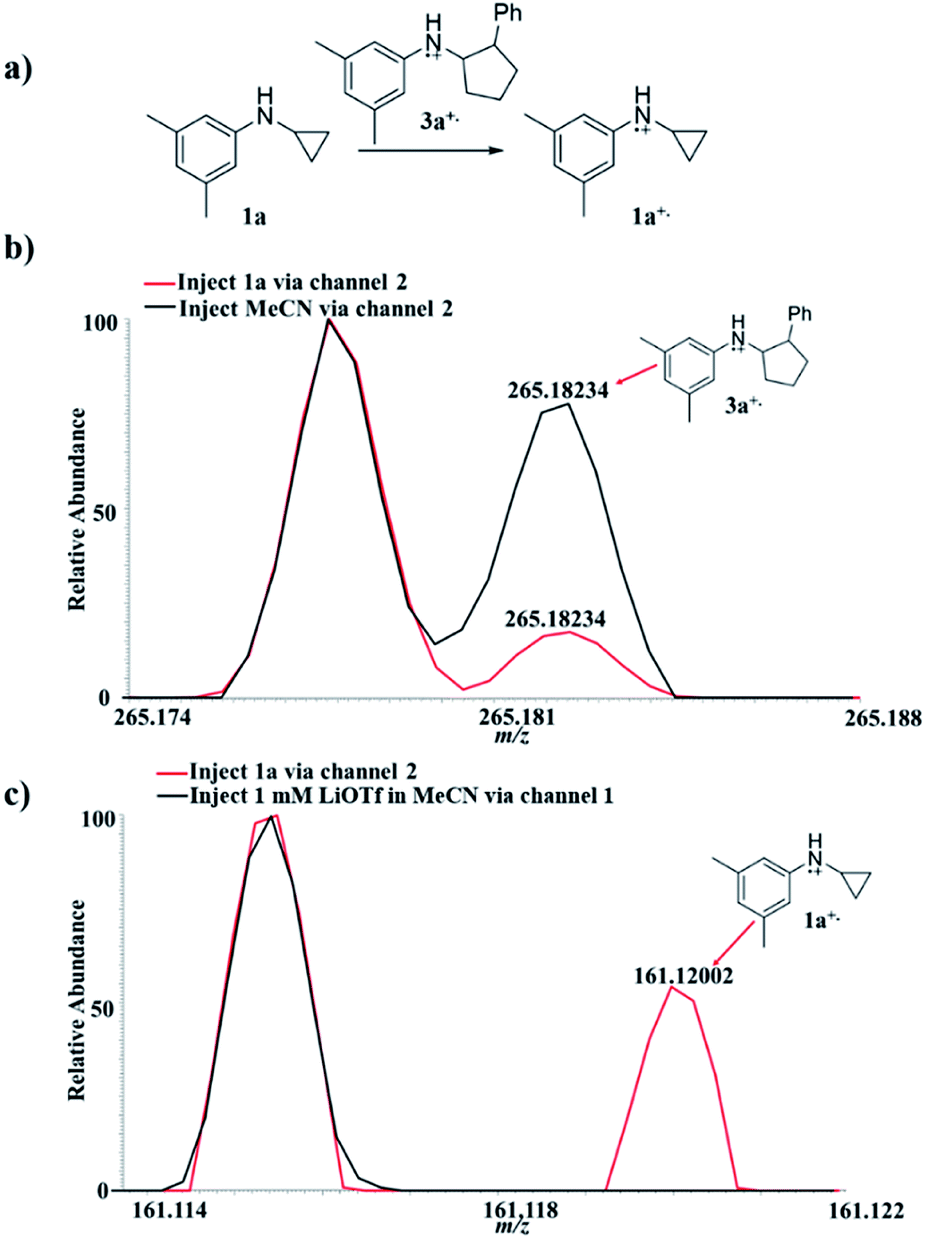 | ||
| Fig. 2 Online MS monitoring of the oxidation of 1a by 3a+˙: (a) chemical equation showing the reaction between 1a and 3a+; (b) MS spectra showing the formation of the radical cation 3a+˙ when the oxidation potential was applied to the cell. The signal of 3a+˙ was lower when the solution injected via channel 2 changed from MeCN (black line) to 1a (red line). (c) MS spectra showing the formation of radical cation 1a+˙ (red line) when the cell was turned on with 3a being introduced into the flow cell via channel 1 and 1a being introduced via channel 2. No formation of radical cation 1a+˙ was observed (black line) when the cell was turned on with 1 mM LiOTf in MeCN being introduced into the flow cell via channel 1 and 1a being introduced via channel 2. | ||
As encouraged by the success of observing individual key reaction steps that are needed for electrosynthesis of 3a from 1a and 2a, we attempted bulk solution electrolysis. A piece of Pt plate and reticulated vitreous carbon (RVC, a porous carbon electrode) were inserted into a 20 mL clear screw glass vial, serving as the cathode and anode, respectively. A solution of 1a (100 mM, 1 mmol), styrene 2a (1 M, 10 mmol), and LiOTf (1 M, 10 mmol) in 10 mL MeCN was added into the electrolysis cell (Fig. 3a, more details are shown in General procedure 2 of the ESI†). After 9 h at 1 mA direct current provided by a direct current (DC) power supply, the protonated [3 + 2] annulation product [3a + H]+ (theoretical m/z: 266.19033, measured m/z: 266.19003, mass error −1.13 ppm) was detected by MS and appeared as the dominant peak in the MS spectrum (Fig. 3b), indicating a good yield of the reaction. Upon CID, the ion m/z 266 gave rise to fragment ions of m/z 145, 122, and 91 upon CID by losses of C8H11N, C11H12, and C12H17N, respectively, consistent with its assigned structure. Using dibromomethane as the internal standard, the NMR yield was measured to be 68%. This result showed that the scale-up electrolysis for intermolecular [3 + 2] annulation reaction worked.
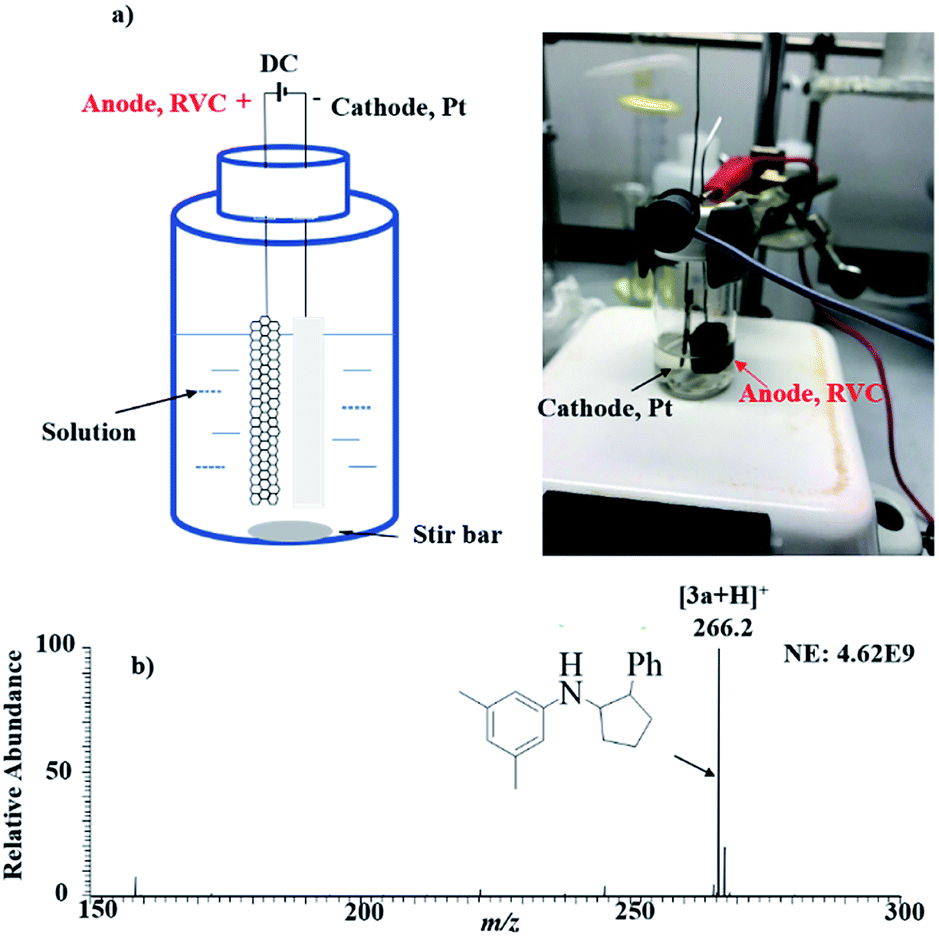 | ||
| Fig. 3 Large-scale electrolysis: (a) setup and actual apparatus picture, and (b) MS spectrum showing the formation of the product 3a by electrolysis. | ||
We thus went ahead to optimize the electrolysis conditions, and again, 1a and 2a were chosen as the model substrates (Table 1). A constant current (entries 1 and 2) or a constant voltage (entries 3 and 4) was used for electrolysis. No product was observed using a higher current (entry 2) while a higher voltage led to a lower yield (entry 4). Compared with the constant voltage mode, the constant current was preferred because it gave the same yield (NMR yield) in a shorter reaction time (75%, 9 h for entry 1 vs. 75%, 12 h for entry 3). Switching the anode from porous RVC to planar Pt furnished the product in a lower yield (entry 5), probably because the porous RVC electrode provided a large electrode surface area to react with reactants. Doubling the amount of LiOTf (entry 6) or replacing LiOTf with other electrolytes (entries 7 and 8) all resulted in lower yields. The yield was also reduced when the amount of styrene was doubled (entries 9), probably caused by the side effect from styrene polymerization on the electrode. Likewise, halving the amount of LiTOf or styrene resulted in lower yields with more unreacted N-cyclopropylaniline. Finally, the addition of water completely inhibited the product formation (entry 10) and replacing MeCN with MeOH led to a lower yield (entry 11). Thus entry 1 experimental conditions were adopted for electrolysis of different substrates.
|
|
||||
|---|---|---|---|---|
| Entry | Variation from standard condition | Reaction yielda | Reactant leftover (Y/N) | Reaction time |
| a NMR yield with dibromomethane used as the reference. | ||||
| 1 | None | 75% | N | 9 h |
| 2 | I cell = 4 mA | No product | N | 9 h |
| 3 | E cell = 3.5 V | 75% | N | 12 h |
| 4 | E cell = 4.5 V | 42% | N | 12 h |
| 5 | Pt+/Pt−, Ecell = 3.5 V | 40% | Y | 12 h |
| 6 | LiOTf (20 equiv.) | 65% | N | 9 h |
| 7 | Electrolyte LiClO4 | 15% | Y | 9 h |
| 8 | Electrolyte Me4NOAc | 42% | Y | 9 h |
| 9 | Styrene (20 equiv.) | 20% | Y | 9 h |
| 10 | Solvent MeCN/H2O 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
No product | Y | 9 h |
| 11 | Solvent MeOH | 35% | Y | 9 h |
After optimizing the reaction conditions, a variety of N-cyclopropylamines and alkenes were investigated, and good isolation yields were obtained (Scheme 4). An aryl group on N-cyclopropylamines could promote the initial oxidation to occur as it decreased the redox potential of N-cyclopropylamines.94 Substituents such as methyl and chlorine on the aromatic ring of cyclopropylamine were tolerated (3a and 4a). N-Cyclopropylamines substituted with other arenes such as biphenyl and naphthalene also worked (5a and 6a). Substituents on the phenyl group of styrenes such as ortho-bromine and para-methoxy groups had little effect on the yields (3c, 3d, 4c, 4e, 5b, and 5d). Substitution of the phenyl group of styrene by a naphthyl group lowered the yield (4d). Other types of pi bonds were explored. Acrylonitrile gave acceptable to good yields of the annulation products (3b, 4b, 5c, and 6b). The diastereoselectivity for the annulation reaction was generally poor, as most of them gave a 1 : 1 mixture except 4a and 5c (2 : 1 and 4 : 1 were observed for the two products, respectively, with the cis isomer as the major product). Phenylacetylene was shown to be a viable annulation partner (4f).
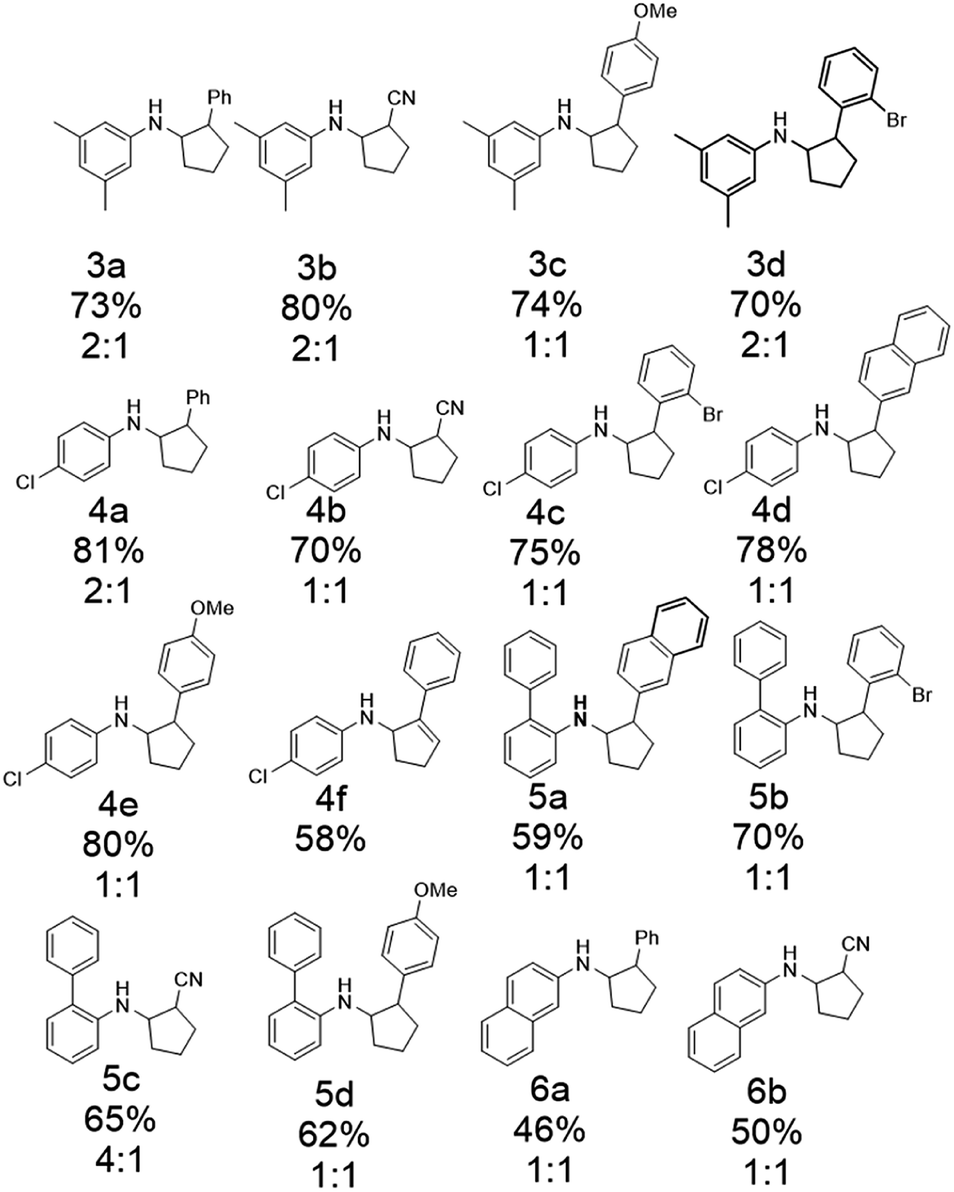 | ||
| Scheme 4 The substrate scope of intermolecular [3 + 2] annulation by the electrochemical approach (percentages show the isolation yields). The ratios for cis/trans product isomers are also listed. | ||
Mechanistically, after establishing the chain mechanism as a viable pathway to achieve the redox neutral reaction, we questioned the feasibility of the other conditions in which the product radical cations were reduced on the cathode. We carried out the reaction of 1a and 2a using an H-type divided cell with two compartments separated with a frit membrane. The two electrodes, RVC and Pt electrodes, were inserted into one chamber each separately (Fig. S1†). A solution of 1a (32 mM, 0.64 mmol), 2a (300 mM, 6 mmol), and LiOTf (300 mM) in 20 mL MeCN was added into one chamber of the electrolysis cell (the anode RVC side) and 20 mL of MeCN containing 300 mM LiOTf was added into the other chamber (the cathode Pt side). After 9 h of electrolysis with an applied 1 mA constant current across the two electrodes, interestingly, the protonated [3 + 2] annulation product [3a + H]+ (theoretical m/z: 266.19033, measured m/z: 266.18938, mass error −3.57 ppm) was detected from the anode side chamber by MS (Fig. S2,† the NMR yield was 23%). In contrast, no product or reactant was detected in the cathode side chamber, indicating that no product or reactant could pass through the frit membrane. The formation of 3a in the divided cell suggested that the reduction of the 3a radical cation was caused by the starting reactant 1a with simultaneous formation of the 1a radical cation via the chain mechanism rather than caused by the cathode reduction. The yield was lower than in the un-divided cell. Presumably because the membrane increased the resistance, a higher voltage was needed to apply to the cell to maintain a constant current (5.0–6.5 V for 1 mA electrolysis). As shown in Table 1 (entry 4), a higher voltage harmed the annulation reaction.
A comparison between the total charge required for the reaction and the electrical input could also help to clarify if the reaction involves the chain reaction mechanism.81 According to Faraday's law, the total electric charge (Q) responsible for oxidizing/reducing a reactant in a redox reaction, is directly proportional to the quantity of the oxidized/reduced substance: Q = nzF, where n is the moles of the reactant, z is the number of electrons transferred per molecule for the redox reaction (z = 1 for oxidation of 1a), and F is the Faraday constant (9.65 × 104 C mol−1). According to Q = nzF, 1 × 10−3 mol × 1 × 9.65 × 104 C mol−1 = 96.5 C electricity would be needed for the complete oxidation of 1 mmol 1a experimentally. As mentioned before, 1 mA was applied to the undivided electrolysis cell for 9 h to complete the electrolysis. The actual consumption of electricity was calculated to be 1 × 10−3 A × 9 × 3600 s = 32.4 C based on Q = it, where i was the applied current and t was the reaction time. Therefore, the reaction was driven to completion with 0.34 equiv. of the current. The catalytic current used for the reaction also supported the chain reaction mechanism.
Conclusions
A new type of challenging electrochemical redox neutral reaction was developed using our homemade online EC/MS platform. Without using any catalyst, the [3 + 2] annulation of N-cyclopropylamines and alkenes proceeded by direct electrolysis instead. Various mechanistic investigations including the use of a divided cell, measurements of the catalytic current, and online MS monitoring of the key electron transfer step between the product radical cation and the reactant supported that the reaction was completed via a chain reaction process. Considering that a chain reaction mechanism is often accompanied by a photoredox reaction in photocatalysis, we expect that electrolysis can be applied to many other visible-light-mediated reactions as an alternative catalyst-free approach.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NSF (CHE-1915878) and NIH (1R15GM137311-01).Notes and references
- A. A. Sabino, A. H. L. Machado, C. R. D. Correia and M. N. Eberlin, Angew. Chem., Int. Ed. Engl., 2004, 43, 2514–2518 CrossRef CAS.
- D. Feichtinger and D. A. Plattner, Angew. Chem., Int. Ed. Engl., 1997, 36, 1718–1719 CrossRef CAS.
- D. J. Wilson and L. Konermann, Anal. Chem., 2003, 75, 6408–6414 CrossRef CAS.
- S. Fürmeier, J. Griep-Raming, A. Hayen and J. O. Metzger, Chem.–Eur. J., 2005, 11, 5545–5554 CrossRef.
- M. M. Meyer, G. N. Khairallah, S. R. Kass and R. A. J. O'Hair, Angew. Chem., Int. Ed. Engl., 2009, 48, 2934–2936 CrossRef CAS.
- L. P. E. Yunker, R. L. Stoddard and J. S. McIndoe, J. Mass Spectrom., 2014, 49, 1–8 CrossRef CAS.
- S. Chen, Q. Wan and A. K. Badu-Tawiah, Angew. Chem., Int. Ed. Engl., 2016, 55, 9345–9349 CrossRef CAS.
- S. Fürmeier and J. O. Metzger, J. Am. Chem. Soc., 2004, 126, 14485–14492 CrossRef.
- W. Ai, Y. Gao, J. Xue, X. Liu, H. Liu, J. Wang and Y. Bai, Chem. Commun., 2020, 56, 2163–2166 RSC.
- T. Chen, Q. Yao, R. R. Nasaruddin and J. Xie, Angew. Chem., Int. Ed. Engl., 2019, 58, 11967–11977 CrossRef CAS.
- H. Zhang, X. Li, K. Yu, N. Li, J. He, H. You and J. Jiang, Anal. Chim. Acta, 2018, 1013, 36–42 CrossRef CAS.
- M. Lu, Y. Su, P. Zhao, X. Ye, Y. Cai, X. Shi, E. Masson, F. Li, J. L. Campbell and H. Chen, Chem.–Eur. J., 2018, 24, 2144–2150 CrossRef CAS.
- S. Bruckenstein and R. R. Gadde, J. Am. Chem. Soc., 1971, 93, 793–794 CrossRef CAS.
- F. Zhou and G. J. Van Berkel, Anal. Chem., 1995, 67, 3643–3649 CrossRef CAS.
- G. Diehl and U. Karst, Anal. Bioanal. Chem., 2002, 373, 390–398 CrossRef CAS.
- H. P. Permentier, A. P. Bruins and R. Bischoff, Mini-Rev. Med. Chem., 2008, 8, 46–56 CrossRef CAS.
- M. C. S. Regino and A. Brajter-Toth, Anal. Chem., 1997, 69, 5067–5072 CrossRef CAS.
- Y. Zhang, W. Cui, H. Zhang, H. D. Dewald and H. Chen, Anal. Chem., 2012, 84, 3838–3842 CrossRef CAS.
- Y. Zhang, H. D. Dewald and H. Chen, J. Proteome Res., 2011, 10, 1293–1304 CrossRef CAS.
- J. Li, H. D. Dewald and H. Chen, Anal. Chem., 2009, 81, 9716–9722 CrossRef CAS.
- P. Zhao, Q. Wang, M. Kaur, Y.-I. Kim, H. D. Dewald, O. Mozziconacci, Y. Liu and H. Chen, Anal. Chem., 2020, 92, 7877–7883 CrossRef CAS.
- S. Tang, H. Cheng and X. Yan, Angew. Chem., Int. Ed. Engl., 2020, 59, 209–214 CrossRef CAS.
- K. Chintalapudi and A. K. Badu-Tawiah, Chem. Sci., 2020, 11, 9891–9897 RSC.
- J. Hu, N. Zhang, P.-K. Zhang, Y. Chen, X.-H. Xia, H.-Y. Chen and J.-J. Xu, Angew. Chem., Int. Ed. Engl., 2020, 59, 209–214 CrossRef.
- T. A. Brown, H. Chen and R. N. Zare, J. Am. Chem. Soc., 2015, 137, 7274–7277 CrossRef CAS.
- T. A. Brown, H. Chen and R. N. Zare, Angew. Chem., Int. Ed. Engl., 2015, 54, 11183–11185 CrossRef CAS.
- R. Qiu, X. Zhang, H. Luo and Y. Shao, Chem. Sci., 2016, 7, 6684–6688 RSC.
- H. Cheng, X. Yan and R. N. Zare, Anal. Chem., 2017, 89, 3191–3198 CrossRef CAS.
- J. Yu, Y. Zhou, X. Hua, S. Liu, Z. Zhu and X.-Y. Yu, Chem. Commun., 2016, 52, 10952–10955 RSC.
- F. T. G. van den Brink, L. Büter, M. Odijk, W. Olthuis, U. Karst and A. van den Berg, Anal. Chem., 2015, 87, 1527–1535 CrossRef CAS.
- Z. Wang, Y. Zhang, B. Liu, K. Wu, S. Thevuthasan, D. R. Baer, Z. Zhu, X.-Y. Yu and F. Wang, Anal. Chem., 2017, 89, 960–965 CrossRef CAS.
- E. L. Clark and A. T. Bell, J. Am. Chem. Soc., 2018, 140, 7012–7020 CrossRef CAS.
- P. Khanipour, M. Löffler, A. M. Reichert, F. T. Haase, K. J. J. Mayrhofer and I. Katsounaros, Angew. Chem., Int. Ed. Engl., 2019, 58, 7145 CrossRef.
- Q. Wan, S. Chen and A. K. Badu-Tawiah, Chem. Sci., 2018, 9, 5724–5729 RSC.
- E. J. Horn, B. R. Rosen, Y. Chen, J. Tang, K. Chen, M. D. Eastgate and P. S. Baran, Nature, 2016, 533, 77–81 CrossRef CAS.
- A. Badalyan and S. S. Stahl, Nature, 2016, 535, 406–410 CrossRef CAS.
- A. Wiebe, D. Schollmeyer, K. M. Dyballa, R. Franke and S. R. Waldvogel, Angew. Chem., Int. Ed. Engl., 2016, 55, 11801–11805 CrossRef CAS.
- L. Schulz, M. Enders, B. Elsler, D. Schollmeyer, K. M. Dyballa, R. Franke and S. R. Waldvogel, Angew. Chem., Int. Ed. Engl., 2017, 56, 4877–4881 CrossRef CAS.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS.
- Y. Okada and K. Chiba, Chem. Rev., 2018, 118, 4592–4630 CrossRef CAS.
- W. Zhang, K. L. Carpenter and S. Lin, Angew. Chem., Int. Ed. Engl., 2020, 59, 409–417 CrossRef CAS.
- K. D. Moeller, Chem. Rev., 2018, 118, 4817–4833 CrossRef CAS.
- R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC.
- N. Fu, G. S. Sauer and S. Lin, Nat. Protoc., 2018, 13, 1725–1743 CrossRef CAS.
- W. R. Leow, Y. Lum, A. Ozden, Y. Wang, D.-H. Nam, B. Chen, J. Wicks, T.-T. Zhuang, F. Li, D. Sinton and E. H. Sargent, Science, 2020, 368, 1228 CrossRef CAS.
- X. Hu, L. Nie, G. Zhang and A. Lei, Angew. Chem., Int. Ed. Engl., 2020, 59, 15238–15243 CrossRef CAS.
- E. B. Corcoran, M. T. Pirnot, S. Lin, S. D. Dreher, D. A. DiRocco, I. W. Davies, S. L. Buchwald and D. W. C. MacMillan, Science, 2016, 353, 279–283 CrossRef CAS.
- J. J. Murphy and P. Melchiorre, Nature, 2015, 524, 297–298 CrossRef CAS.
- J. L. Jeffrey, J. A. Terrett and D. W. C. MacMillan, Science, 2015, 349, 1532–1536 CrossRef CAS.
- Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437–440 CrossRef CAS.
- L. Shi and W. Xia, Chem. Soc. Rev., 2012, 41, 7687–7697 RSC.
- T. Gieshoff, A. Kehl, D. Schollmeyer, K. D. Moeller and S. R. Waldvogel, J. Am. Chem. Soc., 2017, 139, 12317–12324 CrossRef CAS.
- R. Hayashi, A. Shimizu and J.-i. Yoshida, J. Am. Chem. Soc., 2016, 138, 8400–8403 CrossRef CAS.
- S. Lips, D. Schollmeyer, R. Franke and S. R. Waldvogel, Angew. Chem., Int. Ed. Engl., 2018, 57, 13325–13329 CrossRef CAS.
- S. Lips, B. A. Frontana-Uribe, M. Dörr, D. Schollmeyer, R. Franke and S. R. Waldvogel, Chem.–Eur. J., 2018, 24, 6057–6061 CrossRef CAS.
- A. Wiebe, S. Lips, D. Schollmeyer, R. Franke and S. R. Waldvogel, Angew. Chem., Int. Ed. Engl., 2017, 56, 14727–14731 CrossRef CAS.
- S. Lips, A. Wiebe, B. Elsler, D. Schollmeyer, K. M. Dyballa, R. Franke and S. R. Waldvogel, Angew. Chem., Int. Ed. Engl., 2016, 55, 10872–10876 CrossRef CAS.
- T. Wirtanen, E. Rodrigo and S. R. Waldvogel, Chem.–Eur. J., 2020, 44, 777–780 Search PubMed.
- K. Mahanty, D. Maiti and S. De Sarkar, J. Org. Chem., 2020, 85, 3699–3708 CrossRef CAS.
- X. Ye, P. Zhao, S. Zhang, Y. Zhang, Q. Wang, C. Shan, L. Wojtas, H. Guo, H. Chen and X. Shi, Angew. Chem., Int. Ed. Engl., 2019, 58, 17226–17230 CrossRef CAS.
- P. Xiong, H. Long, J. Song, Y. Wang, J.-F. Li and H.-C. Xu, J. Am. Chem. Soc., 2018, 140, 16387–16391 CrossRef CAS.
- A. J. J. Lennox, S. L. Goes, M. P. Webster, H. F. Koolman, S. W. Djuric and S. S. Stahl, J. Am. Chem. Soc., 2018, 140, 11227–11231 CrossRef CAS.
- Y. Liang, F. Lin, Y. Adeli, R. Jin and N. Jiao, Angew. Chem., Int. Ed. Engl., 2019, 58, 4566–4570 CrossRef CAS.
- Y. N. Ogibin, M. N. Elinson and G. I. Nikishin, Russ. Chem. Rev., 2009, 78, 89–140 CrossRef CAS.
- J. B. Sperry and D. L. Wright, Chem. Soc. Rev., 2006, 35, 605–621 RSC.
- J.-i. Yoshida, K. Kataoka, R. Horcajada and A. Nagaki, Chem. Rev., 2008, 108, 2265–2299 CrossRef CAS.
- B. A. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Green Chem., 2010, 12, 2099–2119 RSC.
- E. J. Horn, B. R. Rosen and P. S. Baran, ACS Cent. Sci., 2016, 2, 302–308 CrossRef CAS.
- Y. Jiang, K. Xu and C. Zeng, Chem. Rev., 2018, 118, 4485–4540 CrossRef CAS.
- Y. Kawamata, M. Yan, Z. Liu, D.-H. Bao, J. Chen, J. T. Starr and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 7448–7451 CrossRef CAS.
- S. Möhle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe and S. R. Waldvogel, Angew. Chem., Int. Ed. Engl., 2018, 57, 6018–6041 CrossRef.
- A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed. Engl., 2018, 57, 5594–5619 CrossRef CAS.
- A. S. S. Doobary and H. P. Caldora, Angew. Chem., Int. Ed., 2020, 59, 1155–1160 CrossRef.
- A. J. J. Lennox, J. E. Nutting and S. S. Stahl, Chem. Sci., 2018, 9, 356–361 RSC.
- H. Wang and H. Huang, Chem. Rec., 2016, 16, 1807–1818 CrossRef CAS.
- Q. Qin and S. Yu, Org. Lett., 2014, 16, 3504–3507 CrossRef CAS.
- K. D. Moeller, M. R. Marzabadi, D. G. New, M. Y. Chiang and S. Keith, J. Am. Chem. Soc., 1990, 112, 6123–6124 CrossRef CAS.
- K. D. Moeller and L. D. Rutledge, J. Org. Chem., 1992, 57, 6360–6363 CrossRef CAS.
- K. D. Moeller, P. W. Wang, S. Tarazi, M. R. Marzabadi and P. L. Wong, J. Org. Chem., 1991, 56, 1058–1067 CrossRef CAS.
- C. Zhu, H. Yue, P. Nikolaienko and M. Rueping, CCS Chem., 2020, 2, 179–190 CrossRef CAS.
- M. Arata, T. Miura and K. Chiba, Org. Lett., 2007, 9, 4347–4350 CrossRef CAS.
- K. Chiba, T. Miura, S. Kim, Y. Kitano and M. Tada, J. Am. Chem. Soc., 2001, 123, 11314–11315 CrossRef CAS.
- Y. Okada, A. Nishimoto, R. Akaba and K. Chiba, J. Org. Chem., 2011, 76, 3470–3476 CrossRef CAS.
- Y. Okada, R. Akaba and K. Chiba, Org. Lett., 2009, 11, 1033–1035 CrossRef CAS.
- Y. Imada, N. Shida, Y. Okada and K. Chiba, Chin. J. Chem., 2019, 37, 557–560 CrossRef CAS.
- K. Chiba and M. Tada, J. Chem. Soc., Chem. Commun., 1994, 2485–2486 RSC.
- Y. Okada, Y. Yamaguchi, A. Ozaki and K. Chiba, Chem. Sci., 2016, 7, 6387–6393 RSC.
- A. Ozaki, Y. Yamaguchi, Y. Okada and K. Chiba, ChemElectroChem, 2017, 4, 1852–1855 CrossRef CAS.
- L. Zhu, P. Xiong, Z.-Y. Mao, Y.-H. Wang, X. Yan, X. Lu and H.-C. Xu, Angew. Chem., Int. Ed. Engl., 2016, 55, 2226–2229 CrossRef CAS.
- Y. Imada, Y. Okada and K. Chiba, Beilstein J. Org. Chem., 2018, 14, 642–647 CrossRef CAS.
- K. T. Lorenz and N. L. Bauld, J. Am. Chem. Soc., 1987, 109, 1157–1160 CrossRef CAS.
- S. Maity, M. Zhu, R. S. Shinabery and N. Zheng, Angew. Chem., Int. Ed. Engl., 2012, 51, 222–226 CrossRef CAS.
- Y. Cai, J. Wang, Y. Zhang, Z. Li, D. Hu, N. Zheng and H. Chen, J. Am. Chem. Soc., 2017, 139, 12259–12266 CrossRef CAS.
- G. J. Kavarnos and N. J. Turro, Chem. Rev., 1986, 86, 401–449 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General considerations, general procedures, and compound characterization. See DOI: 10.1039/d0sc05665k |
| This journal is © The Royal Society of Chemistry 2021 |