
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCu-catalyzed hydroxycyclopropanol ring-opening cyclization to tetrahydrofurans and tetrahydropyrans: short total syntheses of hyperiones†
Weida
Liang
,
Xinpei
Cai
and
Mingji
Dai
 *
*
Department of Chemistry and Center for Cancer Research, Purdue University, West Lafayette, IN 47907, USA. E-mail: mjdai@purdue.edu
First published on 20th November 2020
Abstract
Tetrahydrofurans (THFs) and tetrahydropyrans (THPs) are important core scaffolds frequently found in many molecules of medicinal importance. Herein, we report a novel copper-catalyzed hydroxycyclopropanol ring-opening cyclization methodology to synthesize di- or tri-substituted THFs and THPs. In this reaction, a strained C–C bond was cleaved and a new Csp3–O bond was formed to produce the aforementioned O-heterocycles. The new THF synthesis features a broad substrate scope, scalability, and good functional-group tolerability. It enabled us to complete the shortest enantioselective syntheses of hyperiones A and B (3 and 4 steps, respectively), which is significantly shorter than the previously reported two total syntheses (≥10 steps).
Introduction
O-Heterocycles, particularly tetrahydrofurans (THFs) and tetrahydropyrans (THPs) are privileged scaffolds in bioactive natural products1 and lifesaving drug molecules.2 For example, THF-containing macrolide amphidinolide C1 (1, Fig. 1) has demonstrated potent inhibition activity against cancer cell proliferation.3 Eribulin features three THFs and three THPs in its backbone and is an FDA-approved drug for treating various cancers.4 Sofosbuvir, a nucleoside analog, is a pricy, but effective medication to treat hepatitis C.5 Due to the importance of THFs and THPs in natural products, human medicines, and other fields, many synthetic strategies and methodologies have been developed for their syntheses.6 Regardless of the recent advances, achieving high stereoselectivity via a ring forming process in preparing disubstituted or polysubstituted THFs and THPs still remains a synthetic challenge.7 One commonly used ring closing strategy for THF and THP synthesis is via an intramolecular C–O bond formation. Intramolecular nucleophilic substitution via a 5 or 6-exo-tet cyclization process (cf.5 → 4) falls into this category.6d–g Related transition metal, electrophilic reagent (such as I2 or PhSeCl) or radical promoted additions to the activated π-systems including olefins, alkynes, and allenes have been developed as well.6h–o A transition metal-catalyzed direct Csp3–O ring closure is deemed an attractive strategy (cf.6 → 4), but poses significant challenges. When metalloalkyl species are involved, β-H elimination is always a potential problem, not to mention the difficulties involved in generating such metalloalkyl species. Furthermore, both the metalloalkyl species and the tethered alcohols are nucleophilic, therefore, an external oxidant is needed.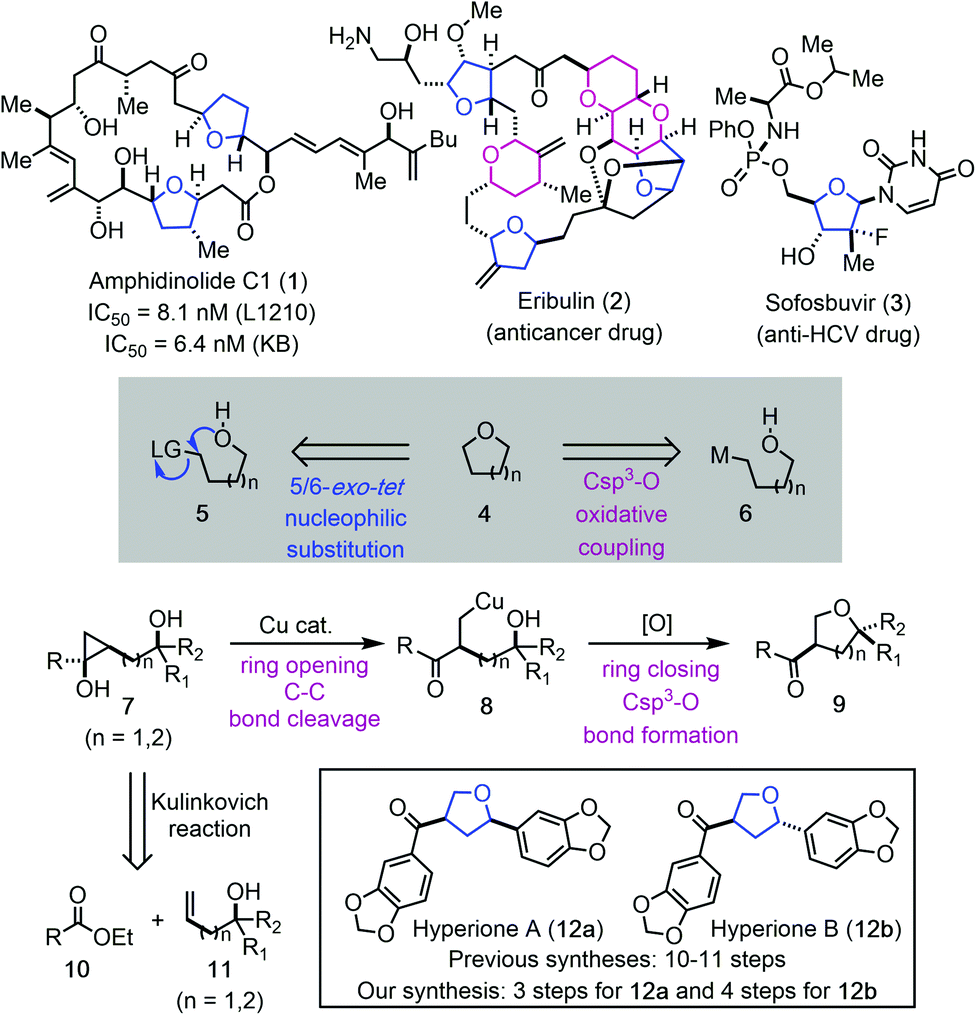 | ||
| Fig. 1 THFs and THPs in natural products and drugs and our synthetic strategy. | ||
Our continued interest in cyclopropanol ring-opening cross coupling reactions made us wonder if metalloalkyl species such as 6 could be generated from a cyclopropanol precursor (cf.7 → 8, Fig. 1). Cyclopropanols are highly strained molecules and can be readily prepared via the Kulinkovich reaction, the Simmons–Smith reaction, and other protocols.8 Despite of their intrinsic ring strain, cyclopropanols are bench stable and can be stored for months or years. Meanwhile, their strain offers unusual reactivity. Synthetic chemists have been harnessing this reactivity and developed a variety of cyclopropanol ring-opening reactions to install different functional groups such as aryl, alkynyl, alkenyl, acyl, activated alkyl, halogen, nitrile, azide, amine, and others at the β-position of carbonyl compounds.9 We have developed palladium-catalyzed hydroxycyclopropanol ring-opening carbonylative lactonization to facilitate total syntheses of complex natural products.10 Meanwhile, we developed a series of copper-catalyzed cyclopropanol ring-opening trifluoromethylation, trifluoromethylthioation, amination, and alkylation with activated alkyl electrophiles.11 However, cyclopropanol ring-opening β-oxygenation via a transition metal-catalyzed Csp3–O cross coupling reaction has not been realized. Our experience in this area taught us that copper-homoenolates derived from cyclopropanol ring opening are less prone to β-H elimination in comparison to the corresponding palladium intermediates. We wondered the possibility of trapping the copper-homoenolate derived from a cyclopropanol with an alcohol via an oxidative cross coupling process. If such process happens in an intramolecular fashion, it would lead to valuable O-heterocycles such as THFs and THPs (cf.7 → 8 → 9). It would also represent one of the very few methods to rearrange strained molecules to O-heterocycles.12 This method would offer several advantages. First, the starting material can be convergently assembled from readily available ester 10 and (bis)homoallylic alcohol 11 by using the diastereoselective cyclopropanol synthesis method developed by Cha and co-workers.13 Second, the stereochemistry of the THF or THP products would rely on the stereochemistry of 7 not the ring forming process. Third, the intramolecular process may enable the Csp3–O bond formation of sterically hindered tertiary alcohols. Fourth, the product would be equipped with an aryl or alkyl ketone for further elaboration. Meanwhile, several challenges lie in the proposed cyclopropanol ring-opening cyclization process. First, for unsymmetrical cyclopropanol such as 7, competitive ring-opening processes exist. Metal-mediated ring opening is required to ensure selective cleavage of the less substituted C–C bond and radical-mediated ring opening needs to be suppressed to avoid the cleavage of the more substituted C–C bond and formation of a more stable radical. Second, once intermediate 8 is formed, reductive elimination to form the Csp3–O bond should be facilitated before the copper-homoenolate decomposes to an enone or other byproducts. Third, a proper oxidant needs to be identified which could promote the catalytic cycle without oxidizing the tethered alcohol or promoting undesired radical cyclopropanol ring-opening processes. Herein, we report the details of our efforts in realizing such a copper-catalyzed cyclopropanol ring-opening cyclization to synthesize substituted THFs or THPs and its application in the shortest total syntheses of natural products hyperiones A (12a, 3 steps) and B (12b, 4 steps).
Results and discussion
Our investigation started with hydroxycyclopropanol 13a, which was prepared from ethyl acetate and 1-phenylbut-3-en-1-ol using the method developed by Cha and co-workers.13 To prove the concept, we first explored the use of stoichiometric Cu(II) salt and were delightful to discover that desired cis 3-methylketone-5-phenyl substituted THF product 14a was obtained in 42% or 35% yield when 2.0 equiv. of Cu(OTf)2 or Cu(OAc)2 were used, respectively (entry 1 and 2, Table 1). Encouraged by these results, we started to evaluate different oxidants to render the Cu(II) salt catalytic. Both oxygen (entry 3) and 1,4-benzoquinone (BQ, entry 5) were effective oxidants when 0.1 equiv. of Cu(OTf)2 was used, but tBuOOH failed the task (entry 4). Cu(I) salt such as CuBr was less efficient than Cu(OTf)2 (entry 6). Solvent evaluation revealed that toluene was better than 1,2-dichloroethane (DCE), dichloromethane (DCM), benzene, and others. Molecule sieves were not necessary. Interestingly, commonly used phenanthroline (entry 10) and bathophenanthroline (entry 11) ligands for copper-catalyzed reactions completely inhibited the transformation. Further reducing the amount of BQ led to significantly decreased yield. We then explored several commercially available BQ derivatives including p-xyloquinone (entry 13), 2,6-dimethyl BQ (entry 15), methyl BQ (entry 16), and 1,4-naphthoquinone (entry 17) and identified p-xyloquinone as the superior one. Notably, a combination of BQ (0.5 equiv., entry 12) or p-xyloquinone (0.5 equiv., entry 14) with oxygen (balloon) gave similar results to the use of 2.0 equiv. of BQ or p-xyloquinone. For the convenience of operations, we decided to use a single oxidant, p-xyloquinone, for the substrate scope investigations. Additionally, when the reaction was conducted at gram-scale, 73% yield of 14a was obtained. Compound 14a could also be produced in enantioenriched form (92% ee) from the corresponding enantioenriched homoallylic alcohol (92% ee).|
|
||
|---|---|---|
| Entry | Reaction conditions | Yielda |
| a Isolated yield. b 73% yield for gram scale. c 92% ee when 14a was prepared from the corresponding enantioenriched homoallylic alcohol with 92% ee; L1: phenanthroline; L2: bathophenanthroline; DCE: 1,2-dichloroethane; BQ: 1,4-benzoquinone; M.S.: molecular sieve. | ||
| 1 | Cu(OTf)2 (2.0), DCE, M.S., 24 h | 42% |
| 2 | Cu(OAc)2 (2.0), DCE, M.S., 24 h | 35% |
| 3 | Cu(OTf)2 (0.1), O2, DCE, M.S., 12 h | 41% |
| 4 | Cu(OTf)2 (0.1), tBuOOH (2.0), DCE, M.S., 6 h | 0% |
| 5 | Cu(OTf)2 (0.1), BQ (2.0), DCE, M.S., 12 h | 51% |
| 6 | CuBr (0.1), BQ (2.0), DCE, M.S., 12 h | 27% |
| 7 | Cu(OTf)2 (0.1), BQ (2.0), DCM, 4 h | 67% |
| 8 | Cu(OTf)2 (0.1), BQ (2.0), benzene, 4 h | 57% |
| 9 | Cu(OTf)2 (0.1), BQ (2.0), tol., 4 h | 68% |
| 10 | Cu(OTf)2 (0.1), BQ (2.0), L1 (0.2 equiv.), tol., 4 h | 0% |
| 11 | Cu(OTf)2 (0.1), BQ (2.0), L2 (0.2 equiv.), tol., 4 h | 0% |
| 12 | Cu(OTf)2 (0.1), BQ (0.5), O2 (balloon), tol., 4 h | 65% |
| 13 | Cu(OTf) 2 (0.1), p-xyloquinone (2.0), tol., 4 h | 76 , % |
| 14 | Cu(OTf) 2 (0.1), p-xyloquinone (0.5), O 2 (balloon), tol., 4 h | 75% |
| 15 | Cu(OTf)2 (0.1), 2,6-dimethyl BQ (2.0), tol., 4 h | 70% |
| 16 | Cu(OTf)2 (0.1), methyl BQ (2.0), tol., 4 h | 74% |
| 17 | Cu(OTf)2 (0.1), 1,4-naphthoquinone (2.0), tol., 4 h | 62% |
| 18 | Cu(OTf)2 (0.1), tol., 16 h | 6% |
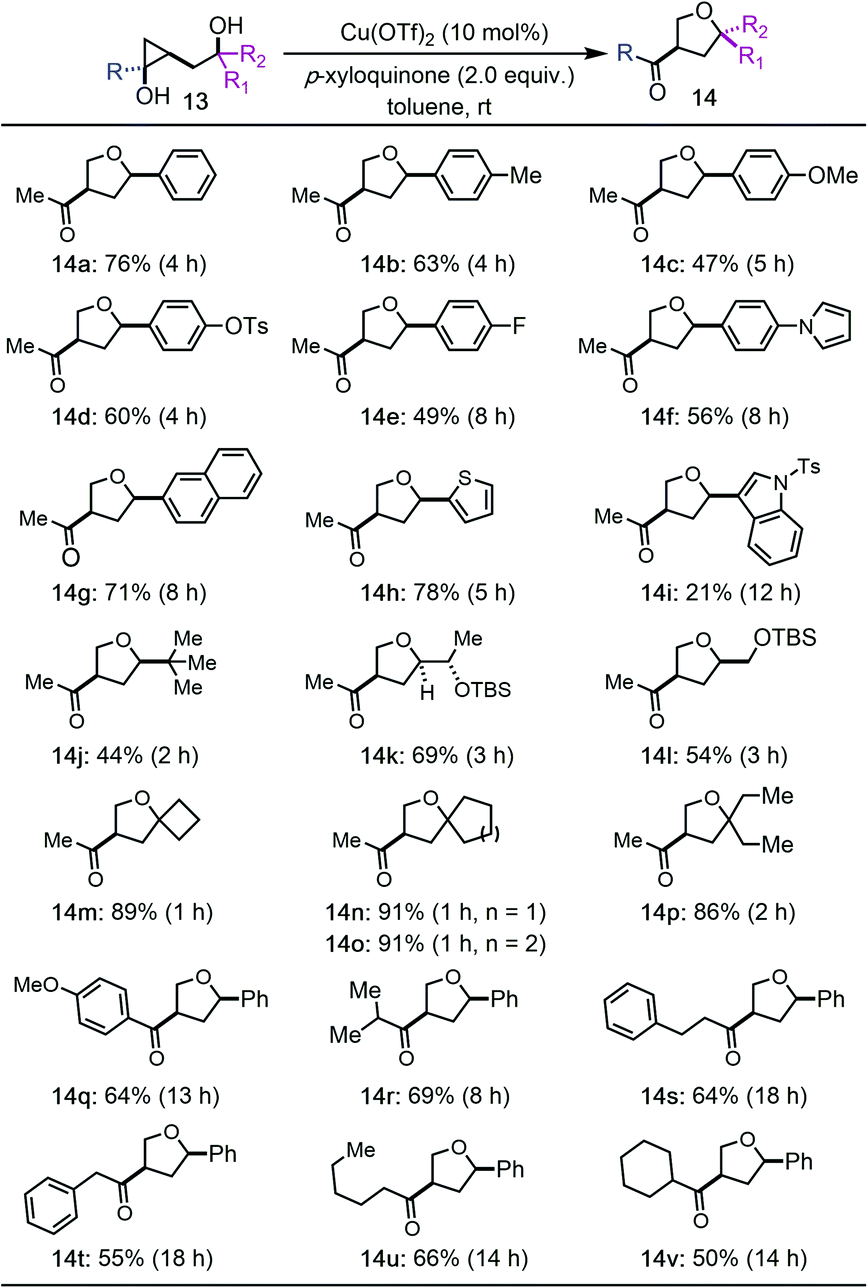
We then prepared a collection of hydroxycyclopropanols and evaluated them under the optimized reaction conditions (Tables 2 and 3). In general, the reaction has a broad substrate scope for the synthesis of substituted THFs. A good functional group tolerability was observed as well. For example, 3-methylketone-5-aryl substituted THFs can be prepared (14a–i). Both electron rich and electron deficient aryl groups worked effectively (14a–g). Heteroaryl groups such as pyrrole (14f), thiophene (14h), and indole (14i) were tolerated, but not furan. The aryl groups can also be switched to alkyl groups such as bulky tert-butyl group (14j) and TBS-ether containing alkyl groups (14k and 14l). All of these cases led to the formation of 3-methylketone-5-aryl/alkyl substituted THFs with cis stereochemistry, which are otherwise difficult to synthesize. Notably, tertiary alcohols are suitable substrates as well (14m–p). Higher reaction yields were obtained for these tertiary alcohols, which indicates a Thorpe–Ingold effect to facilitate the Csp3–O ring closing process despite the steric hindrance of the tertiary alcohols. Additionally, hydroxycyclopropanols derived from different esters can be used to deliver products with different substituents on the resulting ketones (14q–v). Again, both aryl and alkyl groups are tolerated.
|
a A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereomers was used as starting material. :1 mixture of diastereomers was used as starting material.
|
|---|
|
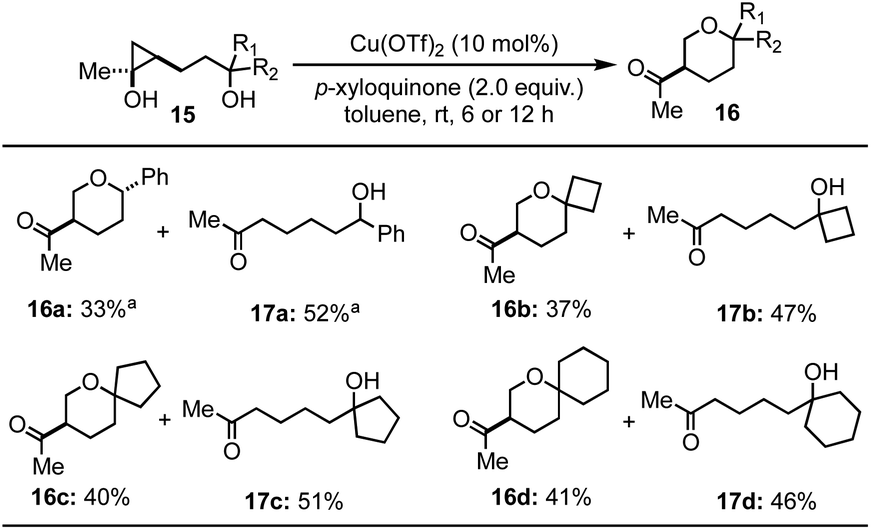
We then tried to expand the established reaction conditions for the syntheses of THFs to THPs, but were less successful in this direction (Table 3). For the cases of cyclopropanols of type 15, which are derived from the Kulinkovich reaction between ethyl acetate and the corresponding bishomoallylic alcohols, while we were able to obtain the desired THP products 16a–16d, the yields were significantly lower than the corresponding THF cases. In addition to the desired THP products, for each case, we observed the formation of cyclopropanol ring-opening protonation product (cf.17a–d). The isolation of 17a–d as the major products suggests a radical cyclopropane C–C bond cleavage mode to break the more substituted C–C bond. The resulting secondary radical intermediate was then quenched or underwent hydrogen atom transfer (HAT) process to give side products 17a–d in the end. Notably, when an inseparable epimeric mixture of 15a (R1, R2 = H, Ph or Ph, H) was used, the formation of 16a with a 3,6-trans stereochemistry was observed, but not the one with a 3,6-cis stereochemistry, which indicates that the ring-opening cyclization process is sensitive to the stereochemistry of the tether secondary alcohol.
To provide insights about the reaction mechanism of the copper-catalyzed cyclopropanol ring-opening intramolecular Csp3–O cross coupling reaction, a series of experiments were conducted. First, Cu(OTf)2 catalyst is essential for the cyclopropanol ring opening as well as the THF or THP ring closure. Without Cu(OTf)2, 95% of the starting material 13a was recovered without any 14a formation. Interestingly, when 13a′, the epimer of 13a, was used, desired THF product 14a′ with trans stereochemistry was produced in 30% yield together with 9% of 18 and 44% of enone 19a (Fig. 2). The formation of enone 19a as the major product from 13a′ indicates again that the stereochemistry of the secondary alcohol of 13a or 13a′ plays an important role in determining the cyclopropanol ring-opening pathways, which will be discussed in the proposed catalytic cycle. Additionally, when compound 18 was subjected to the optimized reaction conditions, we didn't observe any 14a or 14a′, which rules out the formation of the THF ring via a Baldwin's rule disfavored 5-endo-trig oxa-Michael addition. Radical inhibitors such as TEMPO and BHT were explored as well. The formation of 14a or 16c was dramatically inhibited by the addition of 1.2 equiv. of TEMPO to the reaction mixture. Cyclopropanol 13a or 15c were recovered in 84% or 50% yield, respectively. For the case of 15c, a new byproduct 19b was isolated in 20% yield, which presumably came from a 5-exo-trig cyclization of the corresponding enone byproduct or direct trapping of a β alkyl carbocation with the tertiary alcohol. However, when BHT was used, desired THF product 14a was still produced in 67% yield from 13a. For the case of 15c, the yield of THP product 16c dropped from 40% to 12% and the yield of 17c increased slight from 51% to 60%. The latter is likely due to the fact that BHT is a superb hydrogen atom donor, which could quickly quench the β alkyl radical derived from a radical cyclopropane ring opening process.
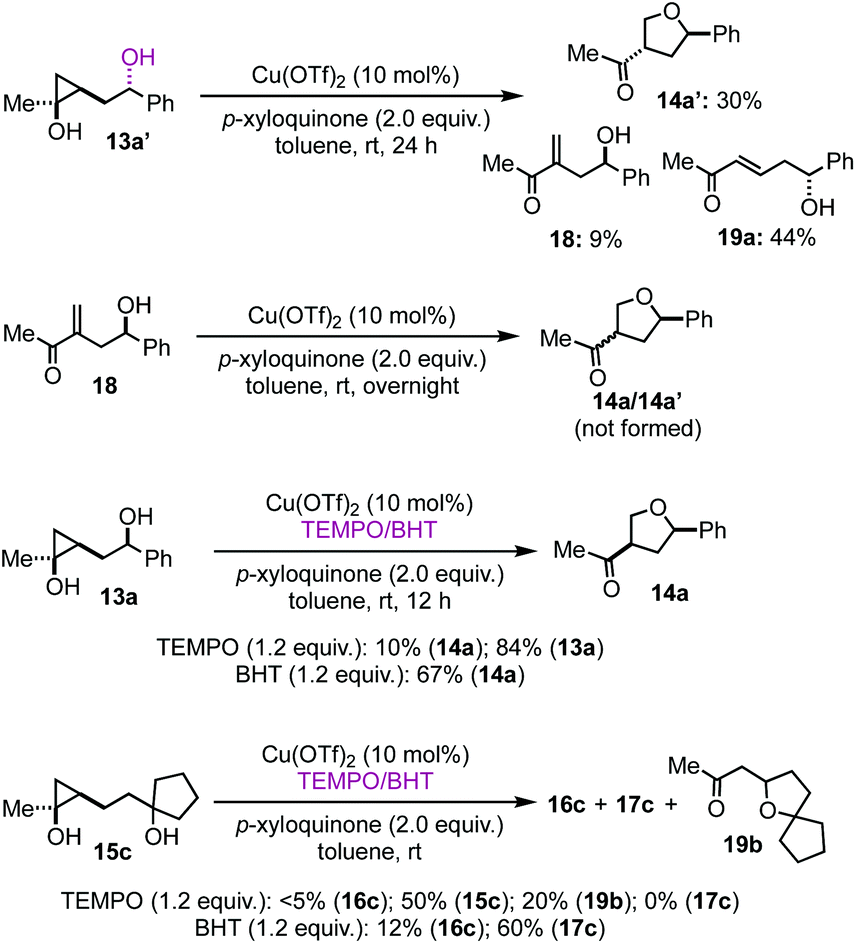 | ||
| Fig. 2 Mechanistic studies. | ||
Based on the above experimental results, a plausible catalytic cycle was proposed in Fig. 3. Cu(OTf)2 would enter the catalytic process by reacting with 13a to form a 7-membered metallocycle intermediate (A). Once intermediate A is formed, selective cyclopropane ring opening would lead to copper-homoenolate B, which would be further oxidized by p-xyloquinone to the corresponding Cu(III) intermediate. Reductive elimination similar to the Chan–Lam process14 would give product 14a and generate a Cu(I) species, which would be the starting point for the rest of the catalytic cycles. This Cu(I) species would coordinate with substrate 13avia a ligand exchange process to form alkoxy–Cu(I) intermediate C. In our previous studies,11 we showed that Cu(I) salts were ineffective to promote cyclopropanol ring opening. Thus, intermediate C would be oxidized to a Cu(II) intermediate by p-xyloquinone. The secondary alcohol on the appended side chain would coordinate with the copper center to form the same 7-membered metallocycle A to continue the rest of the cycle (A → B → 14a) and regenerate the Cu(I) species. When compound 13a was used, intermediate A with the large phenyl group in the pseudo equatorial position would be formed. With compound 13a′, intermediate D would be formed. The phenyl group of D would take the pseudo axial position and experience strong steric repulsion with the methylene group of the cyclopropane ring. Therefore, intermediate D would not be favored. When 15a was used, the coordination of the secondary alcohol on the copper center would require an 8-membered ring formation, which is energetically undesirable. Overall, the stereochemistry of the secondary alcohol as well as its distance from the cyclopropanol are significant for which reaction pathway will occur. It enforces a strong directing effect on the reaction.
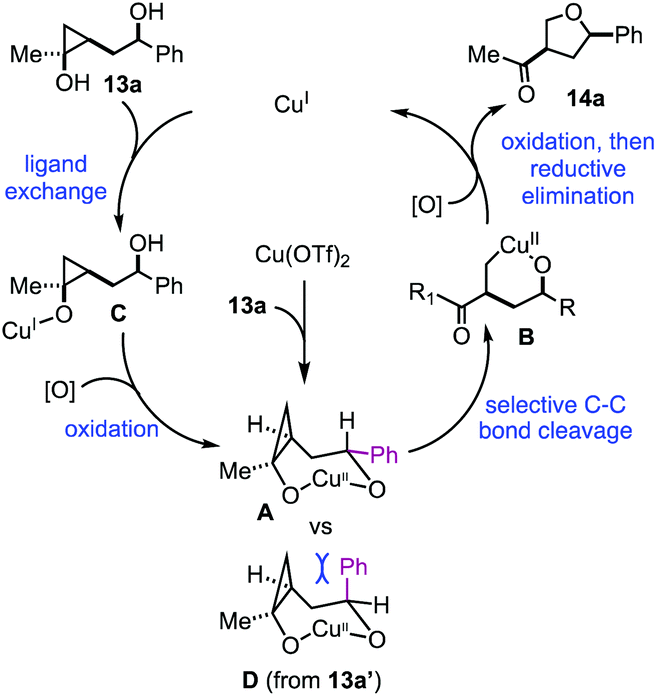 | ||
| Fig. 3 Proposed reaction mechanism. | ||
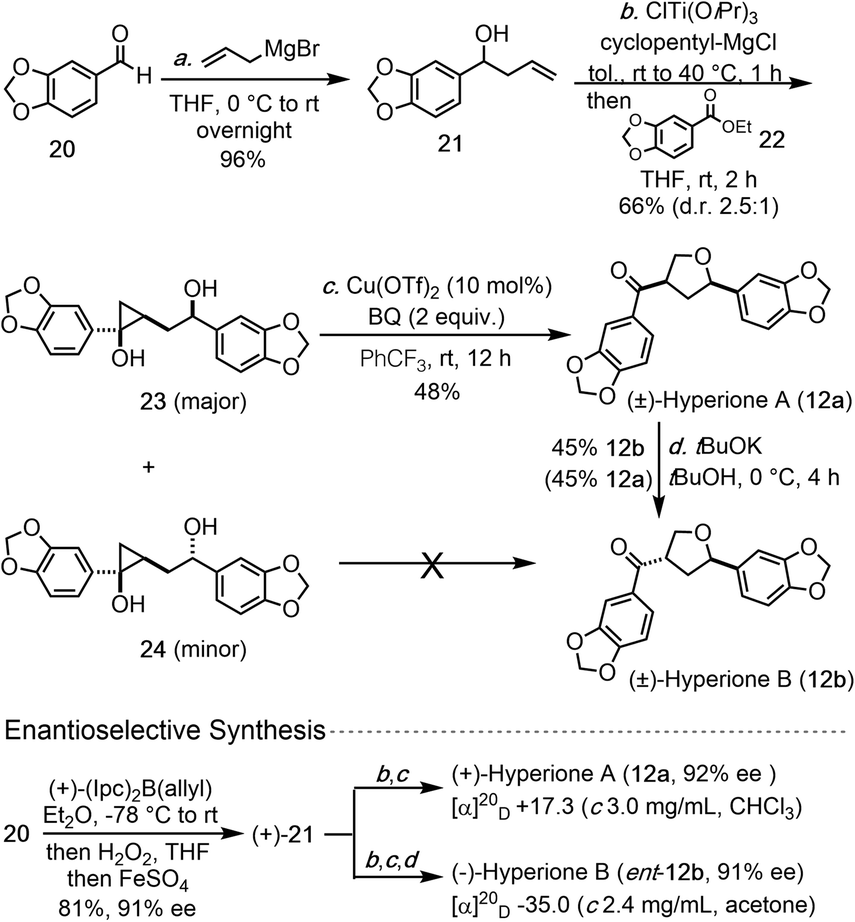 | ||
| Scheme 1 Total syntheses of hyperiones. | ||
We then began to apply the copper-catalyzed cyclopropanol ring-opening cyclization to prepare two THF-containing natural products, hyperiones A and B. Both of them were isolated from the leaves of Hypericum chinense,15 an important herb medicine to treat fever, hepatitis, and sepsis. Hyperiones A and B are two norlignan natural products derived from the well-known dietary lignan sesamin or its epimer asarinin. In 2013, the groups of Barker16 and Deska17 independently reported their total syntheses of these two natural products. Barker's syntheses of hyperiones A and B involve 10 steps started from a known oxazolidinone, which can be prepared in one step from commercially available starting material. Their key THF ring formation is a low yielding acid-catalyzed cyclization (23%). The Deska syntheses require 10 steps and involves two enzymatic reactions and one Ag-catalyzed cycloisomerization to form a dihydrofuran ring which was reduced later.
The newly developed copper-catalyzed cyclopropanol ring-opening cyclization enabled us to complete the total synthesis of hyperione A in 3 steps and hyperione B in 4 steps (Scheme 1), which are significantly shorter than the reported ones. Our synthesis started with commercially available aldehyde 20, which underwent Grignard 1,2-addition with allylMgBr to give known allylic alcohol 21 in 96% yield. Kulinkovich reaction between 21 and ester 22 gave a 2.5:1 separable mixture of cyclopropanols 23 and 24 in 66% total yield. Cyclopropanol 23 underwent the copper-catalyzed ring-opening cyclization smoothly to complete the total synthesis of hyperione A (12a) in only 3 steps, but cyclopropanol 24 failed to deliver hyperione B (12b) directly. Alternatively, tBuOK-promoted epimerization converted hyperione A to B in 45% yield with 45% of hyperione A recovered. Notably, for the conversion of 23 to 12a, BQ is a better oxidant than p-xyloquinone and trifluorotoluene is superior to toluene as the solvent. Additionally, homoallylic alcohol 21 could be prepared in enantioenriched form (91% ee) by using an asymmetric 1,2-addition,18 which eventually led to asymmetric total syntheses of (+)-hyperione A (12a, 92% ee) and (−)-hyperione B (ent-12b, 91% ee). The optical rotation of synthetic (+)-hyperione A is +17.3 (c 3.0 mg mL−1, CHCl3), same in sign and about an order magnitude smaller than the reported value for natural hyperione A (+195.6; c 0.045, CHCl3).15 Similar observation was reported by Barker et al. (−18.5; c 0.016, CHCl3)16 and Deska et al. (+19.2; c 0.045, CHCl3).17 The optical rotation of synthetic (−)-hyperione B is −35.0 (c 2.4 mg mL−1, acetone), opposite in sign and similar in magnitude to the reported value for natural hyperione B.
As mentioned earlier, one advantage of the new THF/THP synthesis is that the product will be equipped with an aryl/alkyl ketone at the C3 position, which could be further functionalized to produce a variety of products. For example, the carbonyl group of 14a could be easily converted to a gem difluoro group (cf.25, Scheme 2) upon the treatment with DAST.19 Both 14a and 16a could undergo spirocyclization with aryl hydrazine 26 to afford 27 and 28 in 82% and 62% yield, respectively.20 Additionally, methyl ketone 14a could be converted to hydrazone 29, whose structure was confirmed by X-ray analysis.21 Hydrazone 29 could undergo a Pd-catalyzed cross coupling reaction with aryl bromide 30 to afford 31 in good yield.22
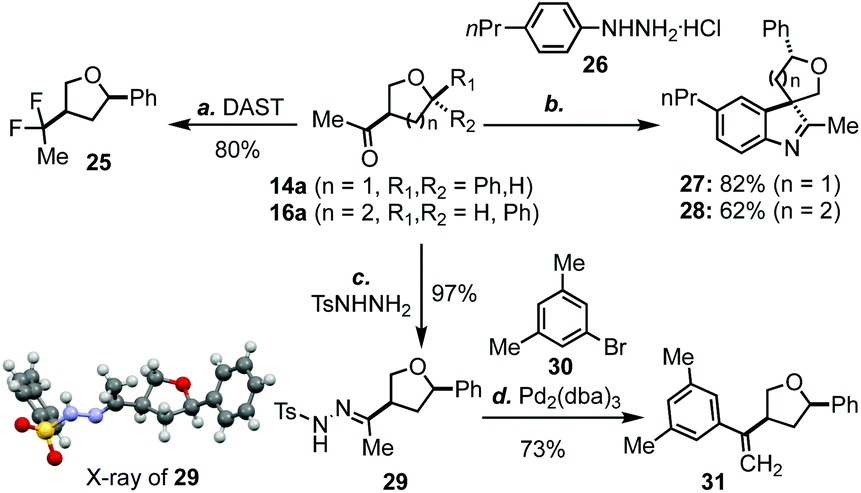 | ||
| Scheme 2 Synthetic applications. (a). DAST, CH2Cl2, reflux, 24 h, 80%; (b) 26, AcOH, reflux, 6 h, 82% (27), 62% (28); (c) TsNHNH2, MeOH, rt, overnight, 97%; (d) 30, Pd2(dba)3 (1 mol%), Xphos (2 mol%), tBuOLi (2.2 equiv.), dioxane, reflux, 12 h, 73%. | ||
Conclusions
In summary, we developed a novel copper-catalyzed cyclopropanol ring-opening cyclization to synthesize O-heterocycles including THFs and THPs. The reaction has a broad substrate scope for synthesizing THFs. Its synthetic potential was demonstrated by a 3 or 4-step total synthesis of hyperione A or B, respectively, as well as the diversifications of the ketone containing THFs/THPs to more sophisticated products. Mechanistically, a selective C–C bond cleavage followed by an oxidative Csp3–O bond formation occurred in the reaction process. The alcohol on the appended side chain functions not only as a nucleophile for the Csp3–O bond formation, but also a directing group to guide the cyclopropane ring opening. This new method is conceptually different from all the other THF/THP synthesis and is expected to have broad application in facilitating the synthesis of natural products and other bioactive molecules.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by NIH R35 GM128570 and NSF CAREER 1553820. We thank the NIH P30CA023168 for supporting shared NMR resources to Purdue Center for Cancer Research. The XRD data was collected on a new single crystal X-ray diffractometer supported by the NSF through the Major Research Instrumentation Program under Grant No. CHE 1625543.Notes and references
- (a) A. Lorente, J. Lamariano-Merketegi, F. Albericio and M. Alvarez, Chem. Rev., 2013, 113, 4567 CrossRef CAS; (b) M. M. Faul and B. E. Huff, Chem. Rev., 2000, 100, 2407 CrossRef CAS.
- (a) M. D. Delost, D. T. Smith, B. J. Anderson and J. T. Njardarson, J. Med. Chem., 2018, 61, 10996 CrossRef CAS; (b) A. K. Ghosh and D. D. Anderson, Future Med. Chem., 2011, 3, 1181 CrossRef CAS.
- J. Kobayashi, M. Ishibashi, M. R. Walchli, H. Nakamura, Y. Hirata, T. Sasaki and Y. Ohizumi, J. Am. Chem. Soc., 1988, 110, 490 CrossRef CAS.
- U. Swami, I. Chaudhary, M. H. Ghalib and S. Goel, Crit. Rev. Oncol. Hematol., 2012, 81, 163 CrossRef.
- G. M. Keating, Drugs, 2014, 74, 1127 CrossRef CAS.
- (a) J. P. Wolfe and M. B. Hay, Tetrahedron, 2007, 63, 261 CrossRef CAS; (b) M. C. Elliott and E. Williams, J. Chem. Soc., Perkin Trans., 2001, 1, 2303 RSC; (c) N. M. Nasir, K. Ermanis and P. A. Clarke, Org. Biomol. Chem., 2014, 12, 3323 RSC; (d) T. Fukuyama, B. Vranesic, D. P. Negri and Y. Kishi, Tetrahedron Lett., 1978, 31, 2741 CrossRef; (e) T. R. Hoye and Z. Ye, J. Am. Chem. Soc., 1996, 118, 1801 CrossRef CAS; (f) B. Kang, J. Mowat, T. Pinter and R. Britton, Org. Lett., 2009, 11, 1717 CrossRef CAS; (g) P. Li, J. Yang and K. Zhao, J. Org. Chem., 1999, 64, 2259 CrossRef CAS; (h) T. Hosokawa, M. Hirata and S.-I. Murahachi, Tetrahedron Lett., 1976, 17, 1821 CrossRef; (i) S. Inoki and T. Mukaiyama, Chem. Lett., 1990, 19, 67 CrossRef; (j) M. F. Semmelhack and N. Zhang, J. Org. Chem., 1989, 54, 4483 CrossRef CAS; (k) R. M. Trend, Y. K. Ramtohul, E. M. Ferreira and B. M. Stoltz, Angew. Chem., Int. Ed., 2003, 42, 2892 CrossRef CAS; (l) M. B. Hay, A. R. Hardin and J. P. Wolfe, J. Org. Chem., 2005, 70, 3099 CrossRef CAS; (m) K. Fujita, K. Murata, M. Iwaoka and S. Tomoda, Tetrahedron, 1997, 53, 2029 CrossRef CAS; (n) S. H. Kang, S. B. Lee and C. M. Park, J. Am. Chem. Soc., 2003, 125, 15748 CrossRef CAS; (o) M. Zlotorzynska, H. Zhai and G. M. Sammis, Org. Lett., 2008, 10, 5083 CrossRef CAS.
- S. M. Nicolle, W. Lewis, C. J. Hayes and C. J. Moody, Angew. Chem., Int. Ed., 2015, 54, 8485 CrossRef CAS.
- O. G. Kulinkovich, Chem. Rev., 2003, 103, 2597 CrossRef CAS.
- (a) A. Nikolaev and A. Orellana, Synthesis, 2016, 48, 1741 CrossRef CAS; (b) X. Wu and C. Zhu, Chem. Rec., 2018, 18, 587 CrossRef CAS; (c) L. R. Mills and S. A. L. Rousseaux, Eur. J. Org. Chem., 2019, 8–26 CrossRef CAS; (d) X. Cai, W. Liang and M. Dai, Tetrahedron, 2019, 75, 193 CrossRef CAS; (e) Q. Tan, Z. Yang, D. Jiang, Y. Cheng, J. Yang, S. Xi and M. Zhang, Angew. Chem., Int. Ed., 2019, 58, 6420 CrossRef CAS; (f) P. P. Das, K. Belmore and J. K. Cha, Angew. Chem., Int. Ed., 2012, 51, 9517 CrossRef CAS; (g) N. N. Rao and J. K. Cha, J. Am. Chem. Soc., 2015, 137, 2243 CrossRef CAS; (h) X. Zhou, S. Yu, L. Kong and X. Li, ACS Catal., 2016, 6, 647 CrossRef CAS; (i) H. Zhao, X. Fan, J. Yu and C. Zhu, J. Am. Chem. Soc., 2015, 137, 3490 CrossRef CAS; (j) Y. F. Wang, K. K. Toh, E. P. Ng and S. Chiba, J. Am. Chem. Soc., 2011, 133, 6411 CrossRef CAS.
- (a) D. C. Davis, K. L. Walker, C. Hu, R. N. Zare, R. M. Waymouth and M. Dai, J. Am. Chem. Soc., 2016, 138, 10693 CrossRef CAS; (b) K. Ma, X. Yin and M. Dai, Angew. Chem., Int. Ed., 2018, 57, 15209 CrossRef CAS; (c) X. Yin, K. Ma, Y. Dong and M. Dai, Org. Lett., 2020, 22, 5001 CrossRef CAS; (d) X. Cai, W. Liang, M. Liu, X. Li and M. Dai, J. Am. Chem. Soc., 2020, 142, 13677 CrossRef CAS.
- (a) Y. Li, Z. Ye, T. M. Bellman, T. Chi and M. Dai, Org. Lett., 2015, 17, 2186 CrossRef CAS; (b) Z. Ye and M. Dai, Org. Lett., 2015, 17, 2190 CrossRef CAS; (c) Z. Ye, K. E. Gettys, X. Shen and M. Dai, Org. Lett., 2015, 17, 6074 CrossRef CAS; (d) Z. Ye, X. Cai, J. Li and M. Dai, ACS Catal., 2018, 8, 5907 CrossRef CAS.
- (a) M. Brichacek, L. A. Batory and J. T. Njardarson, Angew. Chem., Int. Ed., 2010, 49, 1648 CrossRef CAS; (b) L. A. Batory, C. E. Mclnnis and J. T. Njardarson, J. Am. Chem. Soc., 2006, 128, 16054 CrossRef CAS; (c) T. F. Schneider, J. Kaschel, B. Dittrich and D. B. Werz, Org. Lett., 2009, 11, 2317 CrossRef CAS; (d) C. Brand, G. Rauch, M. Zanoni, B. Dittrich and D. B. Werz, J. Org. Chem., 2009, 74, 8779 CrossRef CAS.
- (a) L. G. Quan, S.-H. Kim, J. C. Lee and J. K. Cha, Angew. Chem., Int. Ed., 2002, 41, 2160 CrossRef CAS; (b) J. Lee, H. J. Kim and J. K. Cha, J. Am. Chem. Soc., 1996, 118, 4198 CrossRef CAS.
- (a) I. Munir, A. F. Zahoor, N. Rasool, S. A. R. Naqvi, K. M. Zia and R. Ahmad, Mol. Diversity, 2019, 23, 215 CrossRef CAS; (b) M. J. West, J. W. B. Fyfe, J. C. Vantourout and A. J. B. Watson, Chem. Rev., 2019, 119, 12491 CrossRef CAS.
- W. Wang, Y. H. Zeng, K. Osman, K. Shinde, M. Rahman, S. Gibbons and Q. Mu, J. Nat. Prod., 2010, 73, 1815 CrossRef CAS.
- N. Duhamel, C. E. Rye and D. Barker, Asian J. Org. Chem., 2013, 2, 491 CrossRef CAS.
- C. Manzuna Sapu and J. Deska, Org. Biomol. Chem., 2013, 11, 1376 RSC.
- H. Sun and W. R. Roush, Org. Synth., 2011, 88, 87 CrossRef CAS.
- W. J. Middleton, J. Org. Chem., 1975, 40, 574 CrossRef CAS.
- L. Yin, H. Li, W. Liu, Z. Yao, Z. Cheng, H. Zhang and H. Zou, Eur. J. Med. Chem., 2018, 144, 1 CrossRef CAS.
- CCDC 1996070 contains the supplementary crystallographic data for compound 29.
- (a) J. Barluenga, P. Moriel, C. Valdes and F. Aznar, Angew. Chem., Int. Ed., 2007, 46, 5587 CrossRef CAS; (b) Q. Xiao, Y. Zhang and J. Wang, Acc. Chem. Res., 2013, 46, 236 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1996070. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc05556e |
| This journal is © The Royal Society of Chemistry 2021 |