
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSmall molecule microarray identifies inhibitors of tyrosyl-DNA phosphodiesterase 1 that simultaneously access the catalytic pocket and two substrate binding sites†
Xue Zhi
Zhao
 a,
Evgeny
Kiselev
b,
George T.
Lountos
c,
Wenjie
Wang
b,
Joseph E.
Tropea
d,
Danielle
Needle
d,
Thomas A.
Hilimire
a,
John S.
Schneekloth
Jr
a,
David S.
Waugh
d,
Yves
Pommier
b and
Terrence R.
Burke
Jr
*a
a,
Evgeny
Kiselev
b,
George T.
Lountos
c,
Wenjie
Wang
b,
Joseph E.
Tropea
d,
Danielle
Needle
d,
Thomas A.
Hilimire
a,
John S.
Schneekloth
Jr
a,
David S.
Waugh
d,
Yves
Pommier
b and
Terrence R.
Burke
Jr
*a
aChemical Biology Laboratory, Center for Cancer Research, National Cancer Institute, Frederick, MD, USA. E-mail: burkete@mail.nih.gov
bDevelopmental Therapeutics Branch, Laboratory of Molecular Pharmacology, Center for Cancer Research, National Cancer Institute, Bethesda, MD, USA
cBasic Science Program, Frederick National Laboratory for Cancer Research, Frederick, MD, USA
dCenter for Structural Biology, Center for Cancer Research, National Cancer Institute, Frederick, MD, USA
First published on 28th January 2021
Abstract
Tyrosyl-DNA phosphodiesterase 1 (TDP1) is a member of the phospholipase D family of enzymes, which catalyzes the removal of both 3′- and 5′-DNA phosphodiester adducts. Importantly, it is capable of reducing the anticancer effects of type I topoisomerase (TOP1) inhibitors by repairing the stalled covalent complexes of TOP1 with DNA. It achieves this by promoting the hydrolysis of the phosphodiester bond between the Y723 residue of human TOP1 and the 3′-phosphate of its DNA substrate. Blocking TDP1 function is an attractive means of enhancing the efficacy of TOP1 inhibitors and overcoming drug resistance. Previously, we reported the use of an X-ray crystallographic screen of more than 600 fragments to identify small molecule variations on phthalic acid and hydroxyquinoline motifs that bind within the TDP1 catalytic pocket. Yet, the majority of these compounds showed limited (millimolar) TDP1 inhibitory potencies. We now report examining a 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-member library of drug-like Small Molecules in Microarray (SMM) format for their ability to bind Alexa Fluor 647 (AF647)-labeled TDP1. The screen identified structurally similar N,2-diphenylimidazo[1,2-a]pyrazin-3-amines as TDP1 binders and catalytic inhibitors. We then explored the core heterocycle skeleton using one-pot Groebke–Blackburn–Bienayme multicomponent reactions and arrived at analogs having higher inhibitory potencies. Solving TDP1 co-crystal structures of a subset of compounds showed their binding at the TDP1 catalytic site, while mimicking substrate interactions. Although our original fragment screen differed significantly from the current microarray protocol, both methods identified ligand–protein interactions containing highly similar elements. Importantly inhibitors identified through the SMM approach show competitive inhibition against TDP1 and access the catalytic phosphate-binding pocket, while simultaneously providing extensions into both the substrate DNA and peptide-binding channels. As such, they represent a platform for further elaboration of trivalent ligands, that could serve as a new genre of potent TDP1 inhibitors.
000-member library of drug-like Small Molecules in Microarray (SMM) format for their ability to bind Alexa Fluor 647 (AF647)-labeled TDP1. The screen identified structurally similar N,2-diphenylimidazo[1,2-a]pyrazin-3-amines as TDP1 binders and catalytic inhibitors. We then explored the core heterocycle skeleton using one-pot Groebke–Blackburn–Bienayme multicomponent reactions and arrived at analogs having higher inhibitory potencies. Solving TDP1 co-crystal structures of a subset of compounds showed their binding at the TDP1 catalytic site, while mimicking substrate interactions. Although our original fragment screen differed significantly from the current microarray protocol, both methods identified ligand–protein interactions containing highly similar elements. Importantly inhibitors identified through the SMM approach show competitive inhibition against TDP1 and access the catalytic phosphate-binding pocket, while simultaneously providing extensions into both the substrate DNA and peptide-binding channels. As such, they represent a platform for further elaboration of trivalent ligands, that could serve as a new genre of potent TDP1 inhibitors.
Introduction
Tyrosyl-DNA phosphodiesterase 1 (TDP1) is a member of the phospholipase D family of enzymes that catalyses the removal of both 3′- and 5′-DNA phosphodiester adducts.1 It serves a variety of physiological roles, including the repair of a broad spectrum of lesions induced by anticancer drugs. Among its important substrates are the stalled topoisomerase type I (TOP1) and DNA covalent complexes produced when a TOP1 tyrosyl residue cleaves one strand of DNA.2–4 Functioning in this capacity, TDP1 inhibitors could potentially increase the anticancer activity of TOP1 inhibitors by reducing the repair of TOP1-DNA lesions.5,6 Accordingly, such agents may represent a promising new class of therapeutics with potential use for the treatment of cancer in synergistic combination with current TOP1 inhibitors.4,7–19While significant effort has been devoted to developing TDP1 inhibitors and a variety of inhibitors have been reported, the structural basis for how these inhibitors interact with TDP1 is generally unknown, which may limit the derivation of new analogues through structure-based approaches.6,20 In addition, many TDP1 inhibitors exhibit physiochemical properties, which could potentially endow them with promiscuous mechanisms of inhibition that would make them of questionable value for further development.21–23 Recently, we reported the structures of several small molecules bound to TDP1 that resulted from the crystallographic screening of more than 600 fragments.24 Surprisingly, despite the relatively large size of the enzyme, all molecules were found to bind to the same site of TDP1, specifically within the catalytic phosphate-binding pocket. The uniform topological distribution of bound fragments could be related to the fact that they resulted from soaking preformed crystals, where the conformational mobility of the solid crystalline protein may be more limited than would be found with protein in solution.
Progress in the development of TDP1 inhibitors could be significantly assisted by expanding the diversity of molecular structures that are capable of accessing and binding to the catalytic machinery. Following up on the reasoning that contacts between neighbouring proteins in the crystal lattice during our initial fragment screens may have limited access to some potential binding sites, we performed a second screen using a small molecule microarray (SMM) approach, where protein–ligand interactions would occur in an aqueous milieu. Toward this end, we screened fluorescently labeled TDP1 in a binding assay against a SMM of 21000 spatially arrayed drug-like molecules bound to a glass surface and achieved an approximate overall 0.5% hit rate. Based on structural similarities, we examined a subset of compounds for their ability to inhibit TDP1 catalysis in vitro. This led us to identify the N,2-diphenylimidazo[1,2-a]pyrazin-3-amine nucleus as a new TDP1-binding motif exhibiting low-micromolar inhibitory potencies. We further extended this class of compounds to the N,2-diphenylimidazo[1,2-a]pyridin-3-amine nucleus using one-pot Groebke–Blackburn–Bienayme multicomponent reactions that employ readily available building blocks of aldehyde, isocyanide and pyrazin-2-amine or pyridin-2-amine. By combining structural features of N,2-diphenylimidazo[1,2-a]pyrazin-3-amine with phthalic acid we were able to confirm by X-ray crystallography that these inhibitors bind within the catalytic site of TDP1, where they replicate aspects of the DNA-TOP1 substrate complex. Remarkably, ligands resulting from the SMM approach bound to the same phosphate-binding site as those obtained from crystallographic screening. Additionally, inhibitors identified through the SMM approach not only accessed the catalytic phosphate-binding pocket, but also simultaneously provided extensions into both the substrate DNA and peptide-binding channels. As such, they represent a potential platform for further elaboration into trivalent ligands, that could serve as a new genre of potent TDP1 inhibitors.
Results
Small molecule microarray (SMM) screening to identify new TDP1-binding motif
SMM can offer a highly efficient and compact platform to interrogate large numbers of compounds.25 Previously, we utilized the catalytic domain of TDP1 (residues 148-608) to screen for TDP1-binding fragments using X-ray crystallography.24 Within this catalytic domain there are three surface-situated Cys residues (C205, C251 and C413) that are potentially accessible for fluorescence labelling using maleimide–thiol coupling reactions. In our current work, we labeled TDP1 (148-608) with Alexa Fluor 647 (AF647) employing a commercially available AF647 C2 maleimide dye kit (Fig. 1A).26 We then compared the enzymatic activity of the AF647-tagged TDP1 (148-608) construct with unlabelled catalytic domain as well as with the full-length wild-type TDP1 using an in vitro gel-based TDP1 assay (Fig. 1B).24 The AF647-labeled TDP1 was found to have catalytic activity intermediate between the more active TDP1 (148-608) and the less active wild-type TDP1 (Fig. 1C), indicating that the AF647-labeled TDP1 (148-608) was suitable for SMM binding screen.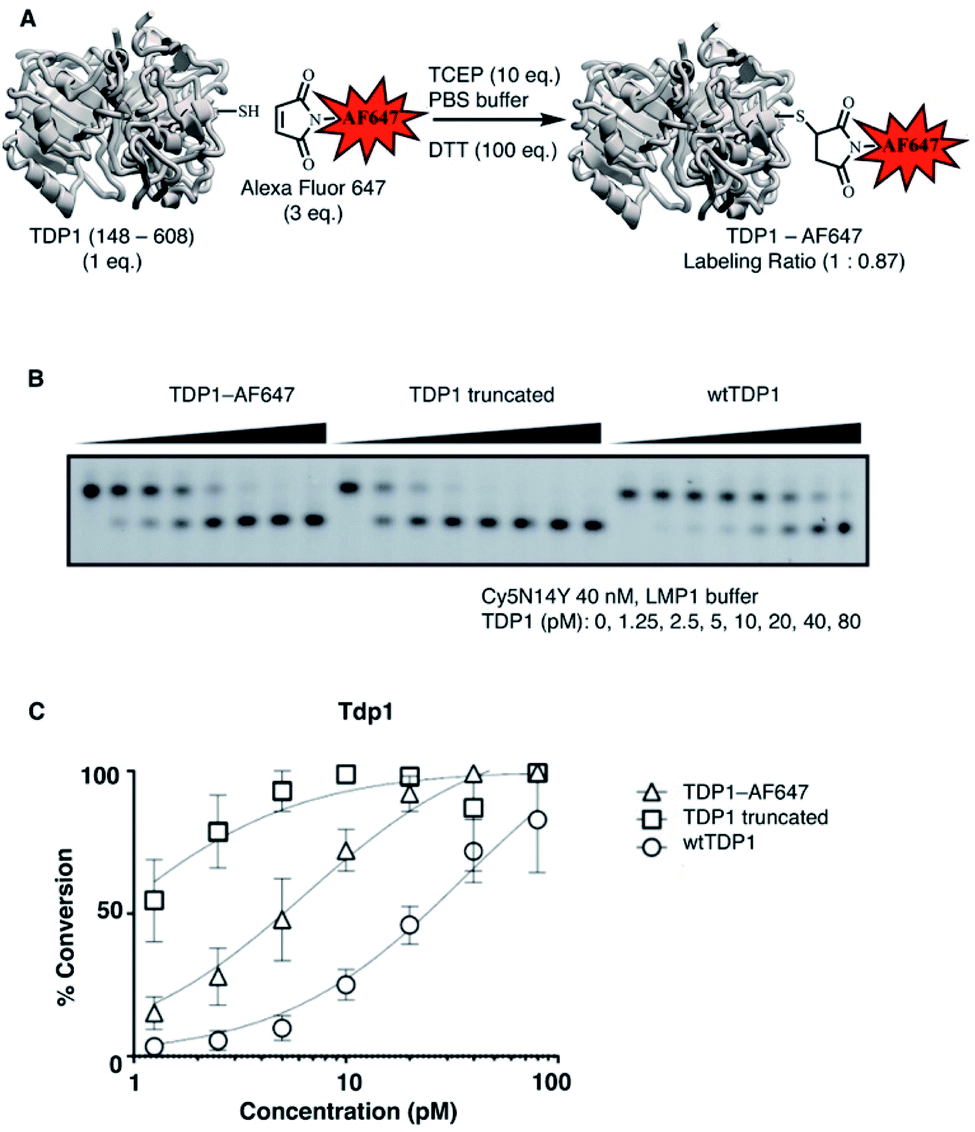 | ||
| Fig. 1 Labelling TDP1 (148-608) with Alexa Fluor 647. (A) Procedural schematic of reacting the catalytic domain of TDP1 (148-608) with Alexa Fluor 647 (AF647) maleimide; (B) gel-based assay showing the activity of AF647-labeled TDP1 (148-608) (TDP1-AF647) as compared with the catalytic domain of unlabelled TDP1 (148-608) (TDP1 truncated) and wild-type full-length TDP1 (wtTDP1). (C) Plot of enzyme activity data shown in panel (B). | ||
Next, to identify compounds that selectively bind to TDP1, we screened AF647-labeled TDP1 (148-608) against drug-like small molecules using a SMM (Fig. 2A).27,28 Briefly, a library of 21000 compounds was covalently immobilized on glass slides using isocyanate surface chemistry, as described.29 Incubating the SMM with the AF647-labeled TDP1 (148-608) identified 109 hits (0.5% hit rate), which were assigned visually into similar structural groups (Table S1†). Based on this grouping, 37 representative compounds were evaluated for their ability to inhibit TDP1 catalysis in gel-based TDP1 assays (Fig. S1 and S2†).30 Compounds M7 and M8 (Fig. 2B) showed micromolar inhibitory potencies (M7 IC50 = 3 μM; M8 IC50 = 233 μM). Both M7 and M8 share an imidazo[1,2-a]pyrazin-3-amine core, with M7 having a 4-aminobenzoic acid group and M8 having a 3-aminobenzoic acid moiety. By this process, we identified the 2-phenylimidazo[1,2-a]pyrazin-3-phenylamine nucleus as a preferred TDP1-binding motif.
 | ||
| Fig. 2 (A) Schematic representation of the SMM screen using AF647-labeled TDP1 (148-608) to identify hits that bind to TDP1. (B) Chemical structures of M7 and M8 and microscope images of their SMM spots. The shared N,2-diphenylimidazo[1,2-a]pyrazin-3-amine core is highlighted in blue. | ||
Inhibition of TDP1 by an imidazo[1,2-a]pyrazin-3-amine is competitive
We evaluated a series of commercially available analogues having an imidazo[1,2-a]pyrazin-3-amine core as well as a small number of synthetic analogues (Table S1†). We identified as being approximately two-fold more potent than M7, compound 7b, which lacks the 2-hydroxyl group on the 2-phenyl ring of the imidazo[1,2-a]pyrazine-3-amine nucleus (Fig. 3A and S2†). In addition, we confirmed that 7b exhibits a competitive mode of inhibition with a Ki value of 0.309 ± 0.122 μM (Fig. 3B). | ||
| Fig. 3 (A) Structures of TDP1 inhibitors discussed in the text. (B) Lineweaver–Burk plot (LB plot) of TDP1 inhibition by 7b showing a competitive mode of inhibition (constant of inhibition Ki = 0.309 ± 0.122 μM). (C) TDP1 inhibition curves of 7b (commercial solid), 7bp (HPLC-purified 7b), and 7′bp (prepared and HPLC-purified sample from our laboratory). (D) Gel-based TDP1 inhibition assay using 7b, 7bp, and 7′bp. Drug concentration for each band: 4.6, 13.7, 41.2, 123.5, 370, 1111, 3333, 10000 (μM). | ||
The imidazo[1,2-a]pyrazin-3-amines were initially all obtained commercially (Table S2†). However, these can be prepared by one-pot Groebke–Blackburn–Bienayme multicomponent reactions, in which freshly synthesized arylisocyanides (3) are coupled with aromatic aldehydes (4) in the presence of pyrazine-2-amines (5) (Scheme 1).31 Typically, the resulting products can be obtained in high purity by simple precipitation out of the reaction mixtures. We observed that product yields were generally at least 40%. However, yields were often substantially lower when electron withdrawing substituents were present on either the aldehyde or isocyanate components. Similar trends have been previously reported.32 Using this procedure and isolating product by precipitation, we obtained 7′b as a white amorphous solid spectroscopically identical to the original commercial sample (low resolution mass spectrum, 1H- and 13C-NMR, Fig. 3A). The synthetic 7′b obtained in this fashion behaved in similar fashion to the commercial material 7b in TDP1 assays.
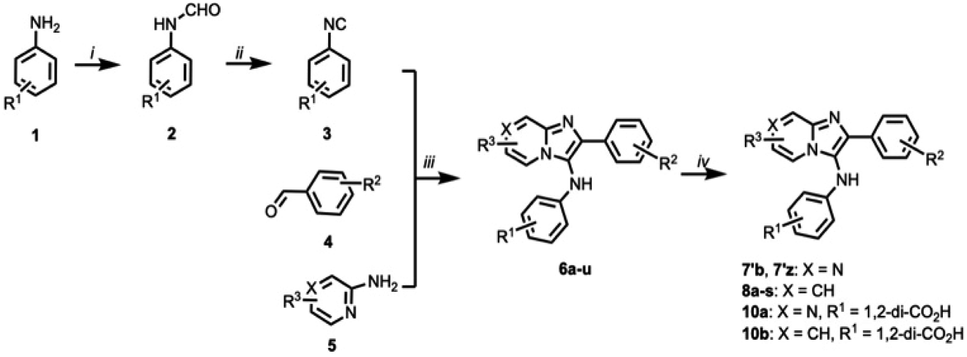 | ||
| Scheme 1 Preparation of TDP1 inhibitors by one-pot Groebke–Blackburn–Bienayme multicomponent reactions. Reagents and Conditions: (i) Ac2O, HCO2H, 50 °C; (ii) POCl3, Et3N, 0 °C; (iii) AcOH, MeOH, rt. (X = N: imidazo[1,2-a]pyrazin-2-amine or X = CH: imidazo[1,2-a]pyridin-2-amine); (iv) NaOH (2N, aq.), MeOH. | ||
Examination of structural variation of the imidazo[1,2-a]pyrazin-3-amine core
We next examined analogues by removing various nitrogen atoms from the parent nucleus. Deletion of the pyrazine 7-nitrogen of 7b yielded the corresponding 2-phenyl-3-phenylamine-[1,2-a]pyridine nucleus (8a, Fig. 3A). Shifting the bridgehead 4-nitrogen of 8a to the 3-position and conversion of the 3-phenylamine to 3-benzyl moiety provided the 1-benzyl-2-phenyl-1H-benzo[d]imidazole ring system (9a). Finally, removal of the 3-nitrogen from 9a gave the 1-benzyl-2-phenyl-1H-benzo[d]imidazole nucleus (9b) (Fig. 3A). For comparison purposes, we retained the 4-carboxy group found in the parent 7b. In the biochemical assays, analogues based on the [1,2-a]pyridine and 1H-benzo[d]imidazole nuclei (9a and 9b, respectively) gave IC50 values > 100 μM. Deletion of the pyrazine 7-nitrogen of 7b to yield 4-((2-phenylimidazo[1,2-a]pyridine-3-yl)amino)benzoic acid (8a) resulted in retention of inhibitory potency (IC50 = 9.3 μM) relative to the parent 7b (IC50 = 4.5 μM). We prepared a variety of analogues based the imidazo[1,2-a]pyrazin-3-amine core and subjected them to in vitro gel-based TDP1 inhibition assays (Table S2†).When we subjected either the commercial sample 7b or the synthetic sample 7′b to reverse-phase HPLC purification (acetonitrile and H2O with 0.1% TFA as eluents) and then lyophilized the collected eluent, we obtained the corresponding solids, 7bp and 7′bp, respectively (“p” indicating purification by HPLC). Both compounds gave identical 1H NMR spectra, which showed slight differences relative to 7bp and 7′bp that were readily attributed to their different salt forms (HCl prior to HPLC purification). We also observed that both 7bp and 7′bp showed reduced inhibitory potencies in the TDP1 gel-based assay (Fig. 3D). The reasons for the loss of inhibitory potency are not clear.
Structural variation of the N,2-diphenylimidazo[1,2-a]pyridine-3-amine core
Using the one-pot multicomponent Groebke–Blackburn–Bienayme protocol, we prepared a set of analogues based on the N,2-diphenylimidazo[1,2-a]pyridine-3-amine core (Scheme 1, wherein X = CH). We evaluated these analogues using the in vitro gel-based TDP1 assay (Table 1). Removal of the carboxylic acid group at the R1 position (8a) resulted in significant loss of inhibitory potency (6t, IC50 > 100 μM). While keeping a carboxylic acid at the R1 position, a variety of substituents were examined at the R3 position. Those substituents that were not proton donors retained inhibitory IC50 values of from 3 μM to 15 μM (i.e., R3 = CH3, OBn, CF3 and NO2; 8b, c, d, e, respectively). However, inhibitory potencies were severely diminished (IC50 > 100 μM) when proton donors were introduced (i.e., R3 = OH, SO3H, CO2H; 8f, g, h, respectively, Table 1). Good inhibitory potency was regained by moving the carboxyl group in 8h from the R3 position to the R5 position (8m, IC50 = 0.9 μM) or to the X position (8s, IC50 = 4.0 μM). Shifting the carboxyl group to the R2 position was also well tolerated (10b, IC50 = 11.0 μM). Removing the R1 carboxyl from 8m resulted in good retention of inhibitory potency (8k, IC50 = 2.4 μM), while shifting that carboxyl to the R3 position gave a modest loss in potency (8i, IC50 = 40.1 μM). Adding a carboxyl group to the R4 position of 8m was highly deleterious (8j, IC50 > 100 μM). Replacing the R5 carboxyl in 8m with a bromine (8o, IC50 = 1.6 μM) or methylsulfone (8q, IC50 = 1.8 μM) was well tolerated, causing little effect on the good inhibitory potency of 8m. Replacing the R1 carboxyl in 8m with a nitro group (8l) lowered potency approximately 20-fold (from IC50 = 0.9 μM to 22.0 μM). Doing a similar replacement in 8q caused an approximate 10-fold loss of potency (6u, IC50 = 16.1 μM). Finally, adding a phenyl ring to the R3 position of 8m was well tolerated (8n, IC50 = 1.5 μM), but when a similar change was done to 8q, a modest loss of potency was observed (8r, IC50 = 9.9 μM). In summary, a carboxyl group at the R1 position was generally important for inhibitory potency. While retaining a carboxyl group at the R1 position, substitution at the R3 position was well tolerated (8b, c, d, e), except when the R3 substituent was a proton donor and, in these cases, there was a significant loss of inhibitory potency (8f, g, h). Introduction of a carboxyl group at the R5 or X positions was well tolerated (8m, n, s), but was deleterious at the R4 position (8j). A carboxyl group at the R5 position gave good inhibitory potency both in the presence (8m) and absence (8k) of a carboxyl group at the R1 position. The presence of carboxyl groups simultaneously at the R1 and R2 positions was tolerated (10b). These compounds showed TDP1 selectivity over TDP2 in gel-based in vitro assay (Table S4†). Several of the compounds showed micromolar inhibition in assays employing whole cell extracts (WCE).30 While having little cytotoxicity, some of the compounds were shown to act synergistically with the TOP1 inhibitor camptothecin (CPT) in human colon cancer cell assays (see below). Taken together, the data are consistent with the compounds selectively targeting TDP1.|
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | 8a | 6t | 8b | 8c | 8d | 8e | 8f | 8g | 8h | 8i | 8j |
| a Half maximal inhibitory concentrations (IC50 values) obtained from in vitro TDP1 gel-based assays. | |||||||||||
| R1 | CO2H | H | CO2H | CO2H | CO2H | CO2H | CO2H | CO2H | CO2H | H | CO2H |
| R2 | H | H | H | H | H | H | H | H | H | H | H |
| R3 | H | H | CH3 | OBn | CF3 | NO2 | OH | SO3H | CO2H | CO2H | H |
| R4 | H | H | H | H | H | H | H | H | H | H | CO2H |
| R5 | H | H | H | H | H | H | H | H | H | H | CO2H |
| X | CH | CH | CH | CH | CH | CH | CH | CH | CH | CH | CH |
| IC50a (μM) | 9.3 | >100 | 14.8 | 12.7 | 4.2 | 3.2 | >100 | >100 | >100 | 40.1 | >100 |

| Compound | 8k | 8l | 8m | 8n | 8o | 8p | 8q | 8r | 6u | 8s | 10a | 10b |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| R1 | H | NO2 | CO2H | CO2H | CO2H | CO2H | CO2H | CO2H | NO2 | CO2H | CO2H | CO2H |
| R2 | H | H | H | H | H | H | H | H | H | H | CO2H | CO2H |
| R3 | H | H | H | Ph | H | H | H | Ph | H | H | H | H |
| R4 | H | H | H | H | H | H | H | H | H | H | H | H |
| R5 | CO2H | CO2H | CO2H | CO2H | Br | H | SO2Me | SO2Me | SO2Me | H | H | H |
| X | CH | CH | CH | CH | CH | CPh | CH | CH | CH | CCO2H | N | CH |
| IC50a (μM) | 2.4 | 22.0 | 0.9 | 1.5 | 1.6 | 1.8 | 1.8 | 9.95 | 16.1 | 4.0 | 45.5 | 11.0 |
Determination of Lineweaver–Burk plots for 8a, 10a and 10b
We had initially prepared a Lineweaver–Burk analysis of the mode of TDP1 inhibition by 7b and found that it was competitive in nature (Fig. 3B). Additionally, we now subjected 8a (the corresponding N,2-diphenylimidazo[1,2-a]pyridine-3-amine version of 7b), to Lineweaver–Burk analysis and found that it also inhibited TDP1 in a competitive fashion (Fig. S3†). Similarly, we subjected 10a (the 3,4-dicarboxy variant of 4-carboxy-containing 7b) and 10b (the 3,4-dicarboxy variant of 4-carboxy-containing 8a) to Lineweaver–Burk analysis and found that these also exhibited competitive modes of inhibition with micromolar Ki values (Fig. S3†).Demonstration that the TDP1 inhibitors act in synergistic fashion with the TOP1 inhibitor camptothecin (CPT)
To establish whether our TDP1 inhibitors could act in cells as TDP1 inhibitors, we performed combination experiments with the classical TOP1 inhibitor camptothecin (CPT)3 in assays using the human colon cancer cell line HCT116 (Table S4 and Fig. S4–S7†). Synergy scores were calculated based on SynergyFinder 2.0 (Table S4 and Fig. S6†).33 Synergy scores were larger than 10 for M7 (15.6), 6u (10.8), 8m (13.4), 8n (11.3), 8o (16.3), 8s (20.7) and 10b (17.9), indicating that the our TDP1 inhibitors act synergistically with CPT in human cancer cells.Determination of protein–ligand interactions by X-ray crystallography
Recently, we disclosed the first X-ray crystal structures of low molecular weight molecules bound within the TDP1 catalytic pocket.24 To determine the ligand–protein interactions for our current SMM-derived inhibitors, we solved the X-ray crystal structures of 10a and 10b bound to the TDP1 catalytic domain at 1.86 and 1.70 Å, respectively (Fig. 4 and Table S3†). Compound 10a is anchored in the active site pocket of TDP1 by direct hydrogen bonds via its carboxylate head group. Hydrogen bonds are formed between the carboxylate oxygen atoms and the side chain Oγ atom of S399 (2.8 Å hydrogen bond distance), K495 Nζ atom (2.7 Å), H493 Nε2 atom (2.9 Å), N283 Nδ2 atom (3.0 Å), K265 Nζ atom (2.6 Å), and H263 Nδ1 atom (2.8 Å). A water molecule (A823), forms a hydrogen bond bridge to the carboxylate O27 atom of 10a (2.6 Å) and the backbone carbonyl oxygen of S514 (2.7 Å). Stabilizing hydrophobic interactions with 10a are provided by the side chain atoms of Y204, P461, and W590 (Fig. 4A). Compound 10b binds to the active site in essentially the same manner with the carboxylate headgroups bound to the catalytic residues. Hydrogen bonds are formed between the carboxylate oxygen atoms of 10b and the side chain Oγ atom of S399 (2.8 Å), the Nζ atom of K495 (2.6 Å), Nε2 atom of H493 (2.4 Å), Nδ2 atom of N283 (2.7 Å), and Nζ atom of K265 (2.7 Å), and Nε2 atom of H263 (2.7 Å) (Fig. 4B).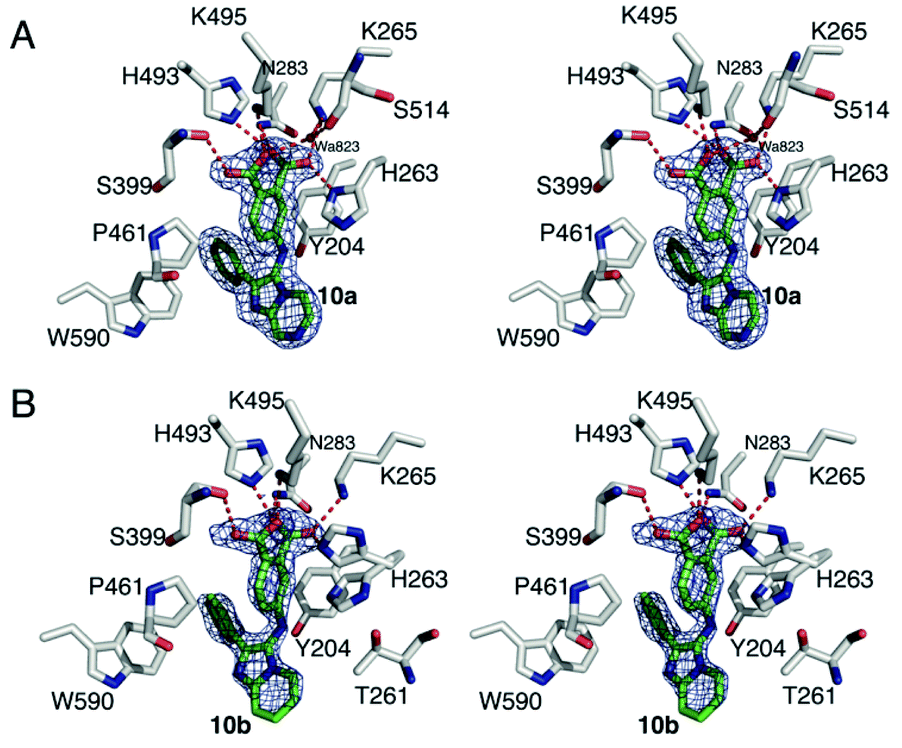 | ||
| Fig. 4 Crystal structures of TDP1 bound 10a and 10b. (A) Stereoview of the active site of TDP1 (carbon atoms in gray, oxygen atoms in red, nitrogen atoms in blue) from the crystal structure of TDP1 bound to compound 10a (PDB code: 6W7L, carbon atoms in green). The fit of compound 10a is shown to the final 2Fo − Fc electron density map (blue) at 1.86 Å resolution and contoured at 1.0σ level. (B) Stereoview of the active site of TDP1 (carbon atoms in gray, oxygen atoms in red, nitrogen atoms in blue) from the crystal structure of TDP1 bound to compound 10b (PDB code: 6W7K, carbon atoms in green). The fit of compound 10b is shown to the final 2Fo − Fc electron density map (blue) at 1.7 Å resolution and contoured at 1.0σ level (atoms in green, nitrogen atoms in blue, and oxygen atoms in red). The binding site of a TOP1 derived peptide (cyan sticks) and a DNA tetranucleotide AGTT (yellow sticks) and bound vanadate (VO4, magenta sphere) are shown. | ||
Discussion
As with many other phospholipase D members, the active site of TDP1 contains two conserved signature “HKN” motifs (H263, K265, N283 and H459, K495, N516) in close proximity to a substrate-binding channel.5,34–36 The enzymatic reaction occurs through two stages. In the first step, the Nε2 nitrogen of H263 executes an in-line SN2 attack onto the DNA 3′-phosphate ester group (Fig. 5A). This proceeds through a bipyramidal transition state, which is thought to be stabilized by the amine side chains of flanking K265 and K495 residues. A phosphoryl-mediated covalent bond is formed between the H263 imidazole ring and the 3′-phosphate group of the DNA substrate, while the incipient negative charge on the departing alcohol is neutralized by protonation from the H493 residue, which acts a general acid (Fig. 5A and B).35 When the substrate consists of a DNA-TOP1 cleavage complex, the alcohol component represents the sidechain hydroxyl of the Y723 residue of TOP1. However, because a complex between native TOP1 and DNA is poorly cleaved, it has been shown that TOP1 must be processed prior to serving as a TDP1 substrate.35,37,38 The reaction cycle is completed by hydrolysis of the DNA – H263 phosphonamide bond through nucleophilic attack of a water molecule under general base-catalysed activation by the H493 Nε2 nitrogen (Fig. 5C). | ||
| Fig. 5 Schematic representation of TDP1 catalysis. (A) Nucleophilic attacks on the phosphate group of the TOP1-DNA complex by the imidazole N atom of H263 (B) Formation of a phosphoryl-histidine covalent intermediate with displacement of the TOP1 tyrosyl residue. (C) Nucleophilic attack on the phosphoryl-histidine intermediate by a H493-activated water molecule. (D) Generation and release of a final 3′-phosphate DNA product. Residue numbers refer to human TDP1. TOP1-DNA complex are color-coded (phosphate group is tan; TOP1-derived peptide is cyan with tyrosyl residue in blue; DNA is magenta). Curved arrows denote the transfer of electron pairs. | ||
Insights into the mechanisms of substrate binding and phosphate ester cleavage have been informed by X-ray crystal structures of fully active N-terminally truncated TDP1 (149-608) without34 and with vanadate or tungstate bound within the catalytic site both without35 and with single-strand DNA and TOP1-derived peptide substrates (Fig. 6).36,39 Vanadate and tungstate serve as phosphoryl hydrolysis transition state intermediate mimetics that clarify the catalytic functions of residues within the TDP1 active site. More recently, crystal structures of TDP1 in complex with double-strand DNA have shed further light on binding interactions of DNA.40
 | ||
| Fig. 6 The crystal structure of TDP1 (wheat ribbons, PDB code: 1RFF) illustrating the overall 3D-structure of the enzyme and highlighting features of the active site region. The residues forming the HKN motifs are depicted in stick format (carbon atoms in green, nitrogen atoms in blue, and oxygen atoms in red). The binding site of a TOP1 derived peptide (cyan sticks) and a DNA tetranucleotide AGTT (yellow sticks) and bound vanadate (VO4, magenta sphere) are shown. | ||
X-ray crystal structures of 10a and 10b bound to TDP1
Although the HKN-containing phosphoryl-binding pocket is well defined, to date structure-based inhibitor development has been challenging. A potentially important advance was recently made by our report of several low molecular weight compounds bound within the TDP1 phosphate binding pocket identified by soaking TDP1 crystals with fragment cocktails. These ligands bound deeply within the phosphate-binding pocket where they recapitulated features of substrate interactions with key residues of the catalytic machinery.24 Many of the compounds were simple phthalic acids (Fig. 7). | ||
| Fig. 7 Structures of low molecular weight TDP1-binding compounds identified by soaking TDP1 crystals with fragment cocktails.24 | ||
The current crystal structures of 10a (PDB code: 6W7L) and 10b (PDB code: 6W7K) show that they include the same important interactions with the catalytic phosphate-binding pocket as our previously identified structurally more simple phthalic acid-containing inhibitors.24 An important distinction of our SMM-derived inhibitors, is that they contain additional 2-phenylimidazo[1,2-a]pyrazine (for 10a) or 2-phenylimidazo[1,2-a]pyridine (for 10b) nuclei that are held above the phthalic acid moiety, with the latter serving to anchor the molecules to the phosphate-binding pocket. This is exemplified by overlaying 10b with TDP1-bound 4-aminophthalic acid (Fig. 8A).24
 | ||
| Fig. 8 View of the TDP1 active site (carbon atoms in gray, nitrogen in blue, and oxygen in red). (A) Comparison of the binding modes of 4-aminophthalic acid (depicted as space-filling with carbons in magenta; PDB code: 6DJJ)24 and 10b (carbons in cyan; PDB code: 6W7K) with black circles showing sites of potential extension above and beyond the phosphate-binding pocket. (B) Overlay of 10b onto with previously reported semi-transparent structures of single-strand DNA (carbons in grey) and TOP1-derived substrate peptide (carbons in tan) with vanadate as a phosphate mimetic (bound space filling) (PDB code: 1NOP).35 Electrostatic surface colored with positive charge blue, negative charge red and neutral white. (C) Bound 10b with phosphate-binding catalytic pocket colored red; DNA-binding pocket colored magenta and peptide-binding pocket colored tan. Arrows indicate positions on 10b extending into substrate binding channels. | ||
Our new TDP1-binding platforms represent a potentially important advance beyond our previously identified phthalic acid inhibitors, due to the possibilities they afford for exploring substrate binding regions outside the catalytic pocket. A three-component picture emerges relating TDP1 structure to enzymatic function. First, there is a well-formed catalytic pocket (red surface, Fig. 8C), whose bottom is formed by the two HKN motifs. Second, extending from one side of the active site is a long narrow positively charged cleft, where the single-stranded DNA binds (magenta surface, Fig. 8C). Third, projecting outward from the catalytic center in the direction opposite to the DNA-binding cleft is a more open and less positively charged channel, which is occupied by the TOP1-derived peptide (tan surface, Fig. 8C) (Fig. 8B and C). While low molecular weight molecules, such as 4-aminophthalic acid efficiently access elements of the phosphate-binding pocket, they are largely confined within this region.24 The more structurally complex molecular architectures of agents discovered as a result of the current SMM screen, such as 10a and 10b, extend above the phosphate-binding pocket and overlap with both the peptide and DNA components of the bi-substrate complex (Fig. 8B). This provides projections directed toward both the DNA and peptide-binding channels that potentially afford points from which to elaborate extensions into these channels (Fig. 8C). Therefore, inhibitors identified in our current work represent a platform for rationale-based construction of trivalent ligands capable of simultaneously accessing the catalytic phosphate-binding pocket and the neighbouring DNA and peptide-binding channels.
Conclusions
A number of TDP1 inhibitors have previously been reported with some exerting synergy with TOP1 inhibitors.10,12,14,16,18,19 However, the precise manner in which these ligands bind to the TDP1 protein are unknown, which limits further development using structure-based approaches. Our current work represents a qualitative departure from previous efforts in the way that it first interrogates the ability of ligands to bind to TDP1 using a microarray including more than 20000 ligands. It then secondarily examines the ability of binders to inhibit the catalytic activity of TDP1 using highly specific biochemical assays. The SMM screen identified structurally similar N,2-diphenylimidazo[1,2-a]pyrazin-3-amines as TDP1 binders and catalytic inhibitors, which we then explored using one-pot Groebke–Blackburn–Bienayme multicomponent reactions. In so doing, we have achieved analogues having high inhibitory potencies. We have also shown that members of the series exhibit competitive modes of inhibition and that certain compounds can function in synergistic fashion with a TOP1 inhibitor. Finally, we solved the crystal structures of a subset of compounds bound within the TDP1 catalytic site. Importantly, inhibitors identified through the SMM approach are anchored within the catalytic phosphate-binding pocket, while simultaneously providing extensions into both the substrate DNA and peptide-binding channels.
In summary, using a SMM approach, our current work has permitted the elaboration of our previously discovered mono-valent fragments into platforms that may form a foundation upon which a new genre of potent multivalent TDP1 inhibitors can be built. Multivalency affords a well-known means of achieving significant enhancement in binding affinity, where it is not uncommon for increases in affinity to be orders-of-magnitude relative to mono-valent ligands.41,42 Our compounds present a new class of ligands that have been established by X-ray crystallography to bind within the catalytic pocket and extend out of the pocket with projections toward the DNA and peptide substrate binding channels. Given the current state of TDP1 inhibitor development, this achievement is significant in its implications for future development of trivalent ligands with potentially high TDP1 affinity and selectivity. Our findings make a particularly important contribution to the field, given the historic challenges faced by developing TDP1 inhibitors.
Conflicts of interest
Aspects of the work presented are contained within one or more patent applications.Acknowledgements
This project has been funded in whole or in part with Federal Funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health under contract HHSN26120080001E and the Staff Scientist/Staff Clinician Research Award (SS/SC-RA) and Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research (Z01-BC 006150 and Z01-BC 006198). X-ray diffraction data were collected on the Southeast Regional Collaborative Access Team (SER-CAT) beamline 22-ID at the Advanced Photon Source, Argonne National Laboratory. Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under contract No. W-31-109-Eng-38. Supporting institutions of SER-CAT may be found at http://www.ser-cat.org/members.html. We thank the Biophysics Resource in the Structural Biophysics Laboratory, NCI at Frederick for use of the LC/ESMS instrument. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does the mention of trade names, commercial products or organizations imply endorsement by the US Government.References
- A. S. Kawale and L. F. Povirk, Nucleic Acids Res., 2018, 46, 520–537 CrossRef CAS
.
- H. U. Barthelmes, M. Habermeyer, M. O. Christensen, C. Mielke, H. Interthal, J. J. Pouliot, F. Boege and D. Marko, J. Biol. Chem., 2004, 279, 55618–55625 CrossRef CAS
- Y. Pommier, Chem. Rev., 2009, 109, 2894–2902 CrossRef CAS
- G. L. Beretta, G. Cossa, L. Gatti, F. Zunino and P. Perego, Curr. Med. Chem., 2010, 17, 1500–1508 CrossRef CAS
- Y. Pommier, S.-y. N. Huang, R. Gao, B. B. Das, J. Murai and C. Marchand, DNA Repair, 2014, 19, 114–129 CrossRef CAS
- S. S. Laev, N. F. Salakhutdinov and O. I. Lavrik, Bioorg. Med. Chem., 2016, 24, 5017–5027 CrossRef CAS
- H. Interthal, H. J. Chen, T. E. Kehl-Fie, J. Zotzmann, J. B. Leppard and J. J. Champoux, EMBO J., 2005, 24, 2224–2233 CrossRef CAS
- E. Q. Comeaux and R. C. A. M. van Waardenburg, Drug Metab. Rev., 2014, 46, 494–507 CrossRef CAS
- R. Gao, B. B. Das, R. Chatterjee, O. D. Abaan, K. Agama, R. Matuo, C. Vinson, P. S. Meltzer and Y. Pommier, DNA Repair, 2014, 13, 1–9 CrossRef CAS
- A. Zakharenko, N. Dyrkheeva and O. Lavrik, Med. Res. Rev., 2019, 39, 1427–1441 CrossRef CAS
- E. J. Brettrager and C. A. M. van Waardenburg, Cancer Drug Resist., 2019, 2, 1153–1163 Search PubMed
- E. V. Koldysheva, A. P. Men'shchikova, E. L. Lushnikova, N. A. Popova, V. I. Kaledin, V. P. Nikolin, A. L. Zakharenko, O. A. Luzina, N. F. Salakhutdinov and O. I. Lavrik, Bull. Exp. Biol. Med., 2019, 166, 661–666 CrossRef CAS
- A. Thomas and Y. Pommier, Clin. Cancer Res., 2019, 25, 6581 CrossRef CAS
- A. L. Zakharenko, O. A. Luzina, D. N. Sokolov, V. I. Kaledin, V. P. Nikolin, N. A. Popova, J. Patel, O. D. Zakharova, A. A. Chepanova, A. Zafar, J. Reynisson, E. Leung, I. K. H. Leung, K. P. Volcho, N. F. Salakhutdinov and O. I. Lavrik, Eur. J. Med. Chem., 2019, 161, 581–593 CrossRef CAS
- O. Luzina, A. Filimonov, A. Zakharenko, A. Chepanova, O. Zakharova, E. Ilina, N. Dyrkheeva, G. Likhatskaya, N. Salakhutdinov and O. Lavrik, J. Nat. Prod., 2020, 83, 2320–2329 CrossRef CAS
- A. L. Zakharenko, M. S. Drenichev, N. S. Dyrkheeva, G. A. Ivanov, V. E. Oslovsky, E. S. Ilina, I. A. Chernyshova, O. I. Lavrik and S. N. Mikhailov, Molecules, 2020, 25, 3694 CrossRef CAS
- E. V. Koldysheva, A. P. Men’shchikova, E. L. Lushnikova, N. A. Popova, V. I. Kaledin, V. P. Nikolin, A. L. Zakharenko, O. A. Luzina, N. F. Salakhutdinov and O. I. Lavrik, Bull. Exp. Biol. Med., 2019, 166, 661–666 CrossRef CAS
- T. M. Khomenko, A. L. Zakharenko, A. A. Chepanova, E. S. Ilina, O. D. Zakharova, V. I. Kaledin, V. P. Nikolin, N. A. Popova, D. V. Korchagina, J. Reynisson, R. Chand, D. M. Ayine-Tora, J. Patel, I. K. H. Leung, K. P. Volcho, N. F. Salakhutdinov and O. I. Lavrik, Int. J. Mol. Sci., 2020, 21, 126 CrossRef CAS
- E. D. Gladkova, I. V. Nechepurenko, R. A. Bredikhin, A. A. Chepanova, A. L. Zakharenko, O. A. Luzina, E. S. Ilina, N. S. Dyrkheeva, E. M. Mamontova, R. O. Anarbaev, J. Reynisson, K. P. Volcho, N. F. Salakhutdinov and O. I. Lavrik, Int. J. Mol. Sci., 2020, 21, 7162 CrossRef CAS
- T. S. Dexheimer, S.-y. N. Huang, S. Antony, C. Marchand and Y. Pommier, Adv. Anticancer Agents Med. Chem., 2013, 2, 444–471 CAS
- J. B. Baell, L. Ferrins, H. Falk and G. Nikolakopoulos, Aust. J. Chem., 2013, 66, 1483–1494 CrossRef CAS
- D. A. Erlanson, J. Med. Chem., 2015, 58, 2088–2090 CrossRef CAS
- J. B. Baell and J. W. M. Nissink, ACS Chem. Biol., 2018, 13, 36–44 CrossRef CAS
- G. T. Lountos, X. Z. Zhao, T. R. Burke, E. Kiselev, Y. Pommier, J. E. Tropea, D. Needle and D. S. Waugh, Nucleic Acids Res., 2019, 47, 10134–10150 CrossRef CAS
- M. Uttamchandani and S. Q. Yao, Methods Mol. Biol., 2010, 669, 1–15 CrossRef CAS
- K. Zlotkowski, W. M. Hewitt, R. S. Sinniah, J. E. Tropea, D. Needle, G. T. Lountos, J. J. Barchi, D. S. Waugh and J. S. Schneekloth, SLAS Discovery, 2017, 22, 760–766 CAS
- J. E. Bradner, O. M. McPherson and A. N. Koehler, Nat. Protoc., 2006, 1, 2344–2352 CrossRef CAS
- J. L. Duffner, P. A. Clemons and A. N. Koehler, Curr. Opin. Chem. Biol., 2007, 11, 74–82 CrossRef CAS
- J. Sztuba-Solinska, S. R. Shenoy, P. Gareiss, L. R. H. Krumpe, S. F. J. Le Grice, B. R. O'Keefe and J. S. Schneekloth, J. Am. Chem. Soc., 2014, 136, 8402–8410 CrossRef CAS
- C. Marchand, S.-y. N. Huang, T. S. Dexheimer, W. A. Lea, B. T. Mott, A. Chergui, A. Naumova, A. G. Stephen, A. S. Rosenthal, G. Rai, J. Murai, R. Gao, D. J. Maloney, A. Jadhav, W. L. Jorgensen, A. Simeonov and Y. Pommier, Mol. Cancer Ther., 2014, 13, 2116–2126 CrossRef CAS
- S. Shaaban and B. F. Abdel-Wahab, Mol. Diversity, 2016, 20, 233–254 CrossRef CAS
- A. Boltjes and A. Doemling, Eur. J. Org. Chem., 2019, 2019, 7007–7049 CrossRef CAS
- A. Ianevski, A. K. Giri and T. Aittokallio, Nucleic Acids Res., 2020, 48, W488–W493 CrossRef CAS
- D. R. Davies, H. Interthal, J. J. Champoux and W. G. J. Hol, Structure, 2002, 10, 237–248 CrossRef CAS
- D. R. Davies, H. Interthal, J. J. Champoux and W. G. J. Hol, J. Mol. Biol., 2002, 324, 917–932 CrossRef CAS
- D. R. Davies, H. Interthal, J. J. Champoux and W. G. J. Hol, Chem. Biol., 2003, 10, 139–147 CrossRef CAS
- Y. Sun, L. K. Saha, S. Saha, U. Jo and Y. Pommier, DNA Repair, 2020, 94, 102926 CrossRef CAS
- Y. Sun, L. M. Miller Jenkins, Y. P. Su, K. C. Nitiss, J. L. Nitiss and Y. Pommier, Sci. Adv., 2020, 6, eaba6290 CrossRef CAS
- D. R. Davies, H. Interthal, J. J. Champoux and W. G. J. Hol, J. Med. Chem., 2004, 47, 829–837 CrossRef CAS
- F. J. Flett, E. Ruksenaite, L. A. Armstrong, S. Bharati, R. Carloni, E. R. Morris, C. L. Mackay, H. Interthal and J. M. Richardson, Nat. Commun., 2018, 9, 24 CrossRef
-
V. M. Krishnamurthy, L. A. Estrofi and G. M. Whitesides, in Fragment-based Approaches in Drug Discovery, ed. W. J. a. D. A. Erlanson, Wiley-VCH GmbH & Co., Weinheim, DE, 2006, pp. 11–53 Search PubMed
- C. Chittasupho, Ther. Delivery, 2012, 3, 1171–1187 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc05411a |
| This journal is © The Royal Society of Chemistry 2021 |