
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProbing conformational hotspots for the recognition and intervention of protein complexes by lysine reactivity profiling†
Zheyi
Liu‡
a,
Wenxiang
Zhang‡
ab,
Binwen
Sun
ac,
Yaolu
Ma
ac,
Min
He
ac,
Yuanjiang
Pan
 b and
Fangjun
Wang
*ac
b and
Fangjun
Wang
*ac
aCAS Key Laboratory of Separation Sciences for Analytical Chemistry, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: wangfj@dicp.ac.cn
bDepartment of Chemistry, Zhejiang University, Hangzhou 310027, China
cUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 23rd November 2020
Abstract
Probing the conformational and functional hotspot sites within aqueous native protein complexes is still a challenging task. Herein, a mass spectrometry (MS)-based two-step isotope labeling-lysine reactivity profiling (TILLRP) strategy is developed to quantify the reactivities of lysine residues and probe the molecular details of protein–protein interactions as well as evaluate the conformational interventions by small-molecule active compounds. The hotspot lysine sites that are crucial to the SARS-CoV-2 S1–ACE2 combination could be successfully probed, such as S1 Lys417 and Lys444. Significant alteration of the reactivities of lysine residues at the interaction interface of S1-RBD Lys386–Lys462 was observed during the formation of complexes, which might be utilized as indicators for investigating the S1-ACE2 dynamic recognition and intervention at the molecular level in high throughput.
Introduction
The conformational hotspot sites within native proteins play essential roles in the protein interaction and function regulations.1 Characterization of these hotspot sites could facilitate the elucidation of protein function mechanisms and the development of targeted drugs.2,3 Conventional methods for protein hotspot site characterization include theoretical prediction via sequence alignment of a functional site among a given set of proteins4 as well as experimental mutation of a specific site to elucidate the influence on the protein structure and function.5,6 Recently, affinity probe based chemical proteomic strategies have been developed for profiling the conformation- or function-specific active sites in high throughput.7,8 However, it is still challenging to characterize intrinsic active sites within native proteins without the introduction of exogenous chemical probes.Lysine residue (K) bears a positively charged amino group on its side chain under the native physiological statuses of proteins. It could interact with a negatively charged amino acid such as aspartic acid (D) and glutamic acid (E) or anionic ligands to form salt bridges or hydrogen bonds, which is essential for stabilizing the protein structure and keeping the protein activity.9–12 Therefore, many lysine residues are protein function centers and involved in protein structure modulation and biological function regulation such as kinase activation.13–17 Besides, the protein conformation modulation levels are directly related to the alteration values of lysine reactivity in the modulated protein regions.18–22
Recently, a novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has spread over the world and causes atypical viral pneumonia COVID-19.23,24 Both structural modelling and experimental evidence have demonstrated the strong interaction of the spike (S) glycoprotein of SARS-CoV-2 with the human cellular receptor angiotensin-converting enzyme 2 (ACE2), which is crucial to the SARS-CoV-2 invasion of the host cells.25–27 The S glycoprotein contains two functional subunits S1 and S2 for binding the membrane receptor ACE2 and fusion of the viral and cellular membranes, respectively.25,28,29 The structures of the SARS-CoV-2 S trimer and ACE2–S1 receptor binding domain (RBD) complex have been successfully determined by cryo-EM.25,28,29 However, it is still challenging to probe the molecular details of the dynamic interactions and pharmaceutical interventions of S1 with ACE2 within aqueous solution in high throughput. Recently, much effort has been devoted to the molecular docking (MD) prediction of potential inhibitors for SARS-CoV-2 protein targets as well as human membrane receptor ACE2.30–34 It is urgent to develop a high-throughput and highly sensitive experimental strategy to monitor the dynamic interaction and intervention of SARS-CoV-2 S1 with ACE2, which might promote the discovery and progress of an effective therapeutic agent.
Herein, we developed a two-step isotope labeling-lysine reactivity profiling (TILLRP) strategy to probe the conformational hotspot sites within native protein–protein interactions. Briefly, the protein sample is labeled with heavy and light isotopic dimethyl tags under native and denatured conditions in sequence. After comprehensive proteomics quantification, the labeling reactivities of lysine residues in native states could be confidently quantified (Fig. S1†). We applied the TILLRP strategy to monitor the dynamic conformational hotspots of the recognition and intervention of SARS-CoV-2 S1 with the ACE2 receptor (Fig. 1). The labeling reactivities of lysine residues at the complex interaction interface of S1-RBD Lys386–Lys462 are directly related to the protein complex formation and might be utilized as indicators for investigating the S1–ACE2 dynamic recognition and intervention. This TILLRP strategy exhibits capability in probing and evaluating the dynamic conformational alterations of S1–ACE2 recognition and intervention by exogenous compounds at the molecular level in high throughput.
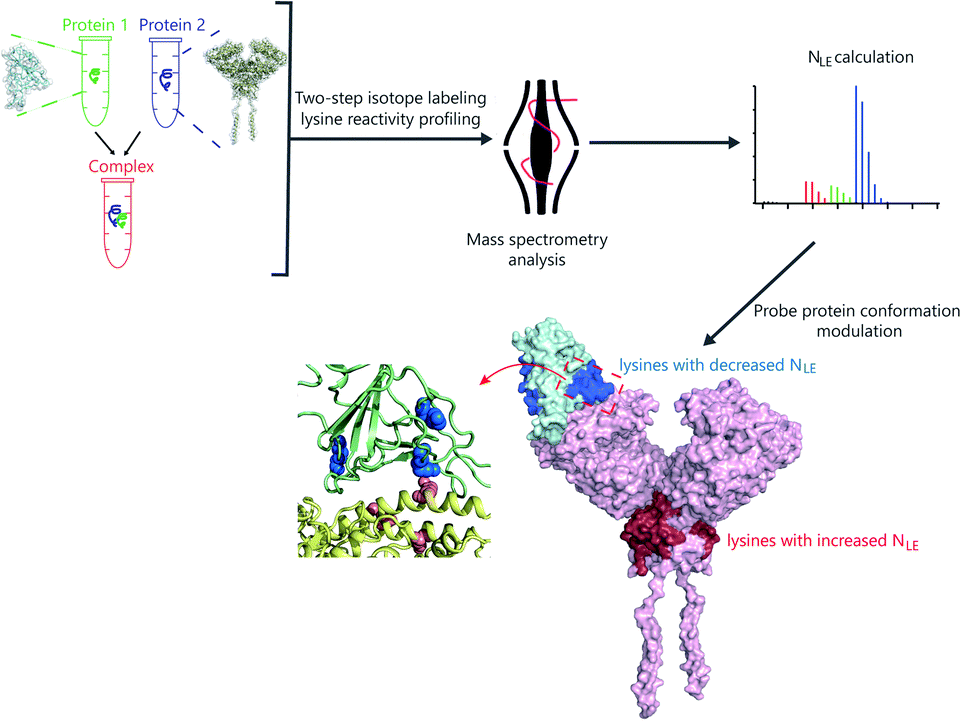 | ||
| Fig. 1 The workflow of the mass spectrometry-based two-step isotope labeling-lysine reactivity profiling (TILLRP) strategy for probing the conformational hotspots of the dynamic interaction and pharmaceutical intervention of SARS-CoV-2 S1 with ACE2. | ||
Results and discussion
Probing conformational hotspots of native proteins
The reactivities of lysine residues are greatly dependent on their proximal micro-environments in native protein conformations. Hotspot lysine sites usually engage in crucial salt–bridge interactions and exhibit relatively low side-chain reactivities. It is possible to characterize the functional and conformational hotspots by lysine reactivity profiling.21Bovine serum albumin (BSA) was utilized to benchmark the TILLRP strategy at first. The normalized labeling efficiency (NLE) values of 23 BSA lysine residues were obtained with high heterogeneity (Fig. 2A and B). The NLE values of sites Lys76, Lys322, and Lys499 are lower than 0.25, indicating the strong interactions within their proximal microenvironments (Fig. 2C–E). In contrast, the site Lys350 could be completely labeled (NLE = 0.98) and no interaction could be observed in its native microenvironments (Fig. S2A†). One-third of the charged residues in native proteins are involved in salt-bridges and a distance below 4 Å between the charged groups is a criterion to judge the formation of strong salt–bridge interactions.35,36 We summarized the distances between lysine residues and their proximal acidic residues in the BSA structure (Fig. S2B†). A good correlation between the NLE values and the distances could be observed, demonstrating that the relative reactivities (labeling efficiency) of lysine residues are dependent on their proximal salt–bridge interactions (Fig. S2B†). In contrast, the lysine NLE values exhibit a poor relationship with solvent-accessible surface areas (SASAs), which is different from previous protein foot-printing strategies (Fig. S2C†).37,38
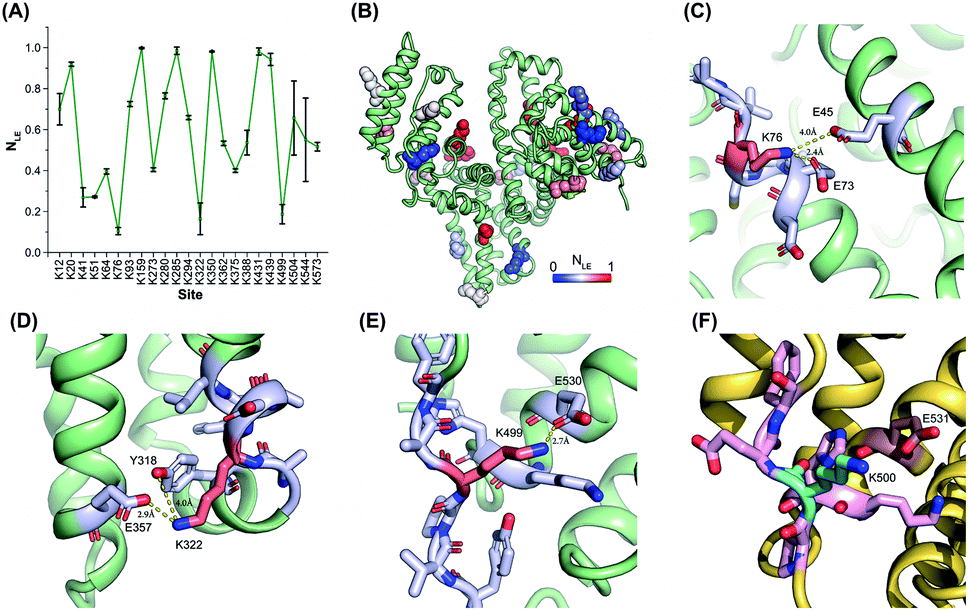 | ||
| Fig. 2 Lysine reactivity profiling of bovine and human serum albumins by using the TILLRP strategy. (A) The NLE values of 23 BSA lysine residues; (B) the conformational distribution of the quantified lysine sites; (C–F) the proximal microenvironments of Lys76, Lys322, and Lys499 in BSA (PDB: 4f5s), and Lys500 in HSA (PDB: 1ao6). | ||
The relative reactivities of lysine residues in the human serum albumin (HSA) were further investigated. Most of the lysine NLE values quantified in both purified and serum native HSA are consistent, indicating that the serum native structure is mainly maintained in its purified form (Fig. S3A†). Further, most of the conserved lysine residues between HSA and BSA exhibit similar NLE values except for Lys41, Lys51, Lys500, and Lys545 (Fig. S3B†). Compared to the BSA Lys499, HSA Lys500 is not engaged in any proximal interaction (Fig. 2F).
Then, the relative reactivities of lysine sites in catechol-O-methyl transferase (COMT) were further analyzed before and after the combination with a small-molecule inhibitor tolcapone. Finally, only the NLE of Lys144 was significantly decreased from 0.68 to 0.13, while little effect was observed for the other lysine residues (Fig. S4A†). COMT Lys144 is known as the crucial site that mediates the binding of COMT with a small-molecule inhibitor (Fig. S4B†),39,40 demonstrating that the lysine reactivity is sensitive to the conformation alteration modulated by exogenous small molecules. Overall, the relative reactivities (NLE) of lysine residues could be utilized to probe the conformational interactions and interventions of native proteins.
Conformational hotspots in the S1–ACE2 interaction
It is still challenging to monitor the dynamic molecular details of conformation alterations in aqueous native proteins, especially for membrane protein complexes with large molecular weights (MW). In this study, lysine residues were utilized as endogenous probes within native proteins to monitor the dynamic conformational hotspots in the S1–ACE2 interaction.The relative reactivities of lysine residues within SARS-CoV2 S1 and human ACE2 were systematically analyzed before and after the formation of the specific-recognition complex. The structure of S1 protein is relatively loose and flexible for membrane receptor adaptation and combination, while the structure of ACE2 is more compact with many helix-rich regions.28 Thus, the NLE values of ACE2 lysine residues were generally lower than the ones of S1 under identical labeling conditions (Fig. 3A). For example, Lys288 within the ACE2 peptidase domain (PD) with strong proximal interactions exhibits a relative low NLE value (0.01), while the NLE of Lys444 within the S1 receptor-binding domain (RBD) is as high as 0.90 due to little proximal interaction existing at its native state (Fig. S5†).
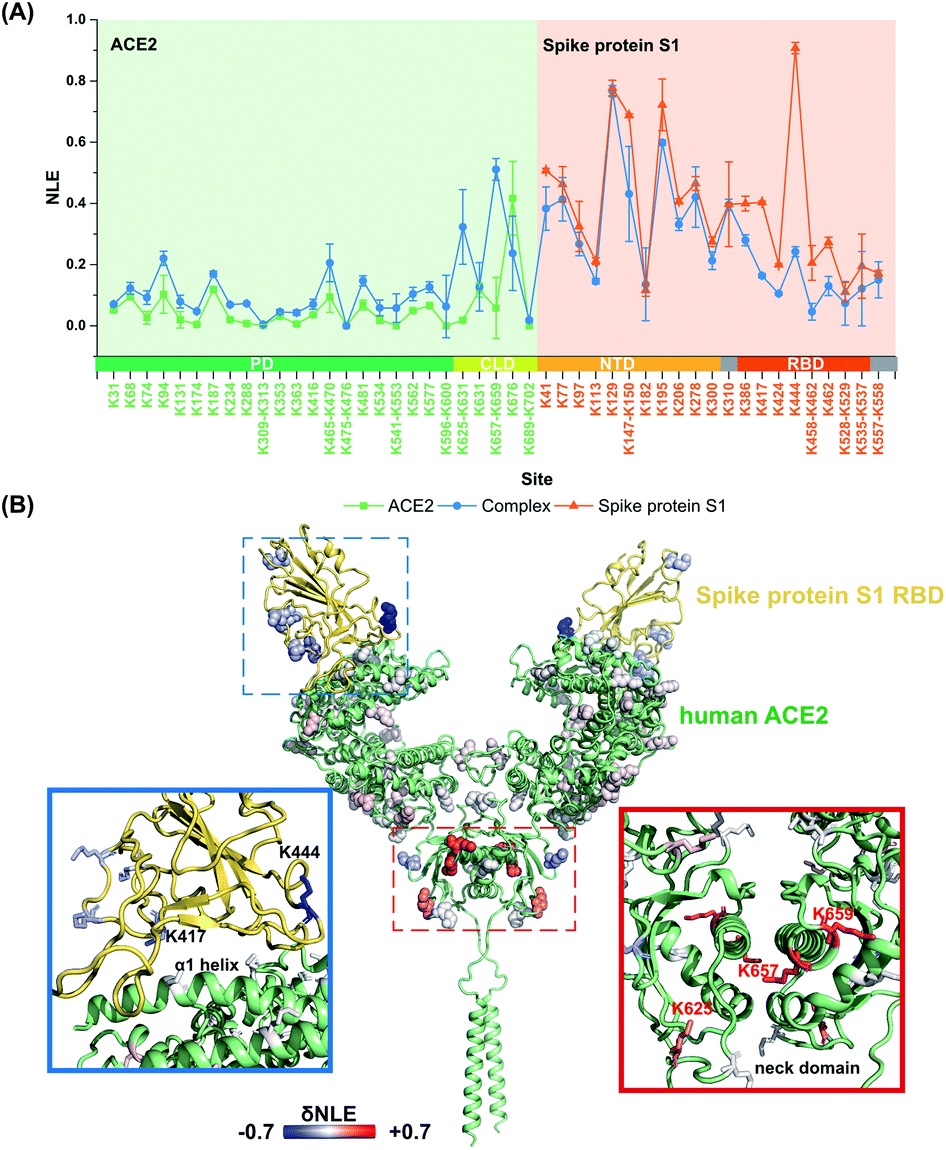 | ||
| Fig. 3 Quantitative lysine reactivity profiling of S1–ACE2 before and after the formation of the complex. (A) The profiles of the lysine NLE values of S1, ACE2 and the S1–ACE2 complex; (B) the δNLE values of the lysine residues in S1 and ACE2 between free and complex states. | ||
After the formation of the S1–ACE2 complex, the conformation alteration regions could be clearly observed as the lysine NLE values obtained in these regions are changed significantly (Fig. 3A). Obviously, the sequence region of Lys386–Lys462 belonging to the S1-RBD exhibits the most significant decreasing levels in the NLE values. This region is known as the flexible loop region of S1-RBD that spans over the α1 helix of ACE2-PD, and thus is the direct interaction interface of S1 in the formation of the complex.28 Interestingly, the NLE values of S1-RBD Lys417 and Lys444 decreased from 0.40 and 0.91 to 0.16 and 0.24, respectively, after binding to ACE2. In the native conformation of the trimer-S glycoprotein,26 Lys444 is distributed at the top of the S1 trimer, and no interaction is found in its side chain proximal microenvironment (Fig. S6†). During the combination with ACE2, the conformation of one of the RBD domains changes from the closed (all RBD down) or semi-closed state (one RBD up) to a relatively stable open state (RBD up) and combines with ACE2.29 Then, the side chain of Lys444 in this conformation forms a salt bridge with the Gly447 skeleton (Fig. S7†). The decrease of NLE from 0.91 to 0.24 can reflect the conformation difference of the Lys444 local microenvironment during the combination of S1 and ACE2. Similarly, Lys417 can only form a salt bridge at most with Asn370 on the adjacent chain in the native conformation of the S protein (Fig. S6†). After the combination with ACE2, the conformation of the S1-RBD region changes to a relatively stable open state,26,41 and finally the Lys417 site could form salt bridges with both ACE2 Asp30 and S1 Leu455 (Fig. S7†). This is highly consistent with the Lys417NLE value decreasing from 0.40 to 0.16 during the formation of the S1–ACE2 complex. Thus, the Lys417 site is a crucial hotspot in the interaction and combination of S1 and ACE2. The substitution of Val404 in the SARS-CoV-RBD with Lys417 in the SARS-CoV-2-RBD is also recognized as one of the most important substitutions that enhance the binding affinity for ACE2.28
For the other part of ACE2, we discovered that the overall conformation of ACE2 from the N-terminal PD to C-terminal collectrin-like domain (CLD) becomes more relaxed after the formation of the S1–ACE2 complex as many lysine NLE values in these domains exhibited consistent increasing trends (Fig. 3A). The most significant changes of labeling reactivity were observed for residues Lys625 and Lys657 in the ACE2 neck domain, which is located between the PD and transmembrane helix and mainly mediates the dimerization of ACE2. The ACE2 dimer has two conformations, open and closed, while only the closed state is retained after the combination with S1-RBD.28 Thus, the increase of the NLE values of most of the lysine residues might be induced by the ACE2 dimer conformation change. Interestingly, little reactivity change was observed for the lysine residues within the direct interaction interface regions of ACE2 Lys31–Lys68 and Lys353–Lys363 during the formation of the S1–ACE2 complex. This might be because the α1 and α2 helices of ACE2 are relatively compact and stable and the combination of flexible S1-RBD will not modulate their conformations significantly.
Above all, the conformational details of the S1–ACE2 dynamic interaction including the direct interaction interface and related conformational changes could be probed by the TILLRP strategy with high sensitivity and high residue resolution. As the interaction interface of S1-RBD is modulated much more significantly as described above, we speculate that the relative reactivity (NLE values) of lysine residues within S1-RBD Lys386–Lys462 could be utilized as indicators for evaluating the dynamic recognition and intervention of the S1–ACE2 complex.
Conformational interventions on S1–ACE2 by active compounds
Due to the outbreak of COVID-19, many potential small-molecule compounds have been proposed to target S1 and ACE2 via MD theoretical prediction, including glycyrrhizic acid, promethazine hydrochlorine, baicalin, chloroquine, scutellarin, hesperetin, and nicotinamide.42–44 However, evaluating the conformational interventions of these compounds on native S1–ACE2 quantitatively is still challenging.At first, glycyrrhizic acid-induced conformational effects were investigated by using the TILLRP strategy. Interestingly, the NLE value of Lys444 within the interaction interface of S1-RBD Lys386–Lys462 exhibited further a decreasing trend after the introduction of glycyrrhizic acid into the S1–ACE2 solution (Fig. 4). In contrast, little intervention was observed on the overall structure of ACE2, demonstrating that the target of glycyrrhizic acid might be S1. Although the glycyrrhizic acid exhibits significant intervention on the S1 conformation in the local regions of Lys77, Lys129, Lys182, Lys310, and Lys444 (t-test < 0.05, |δNLE| ≥ 0.1), we think the S1–ACE2 complex could be still retained as the reactivities of most of the lysine residues at the S1-RBD interaction interface rarely exhibit an increasing trend. The results obtained in MD simulations also demonstrate that the proximal interactions around Lys77, Lys182, Lys310, and Lys444 are enhanced after the combination with glycyrrhizic acid (Fig. S8†), consistent with their NLE value decrease.
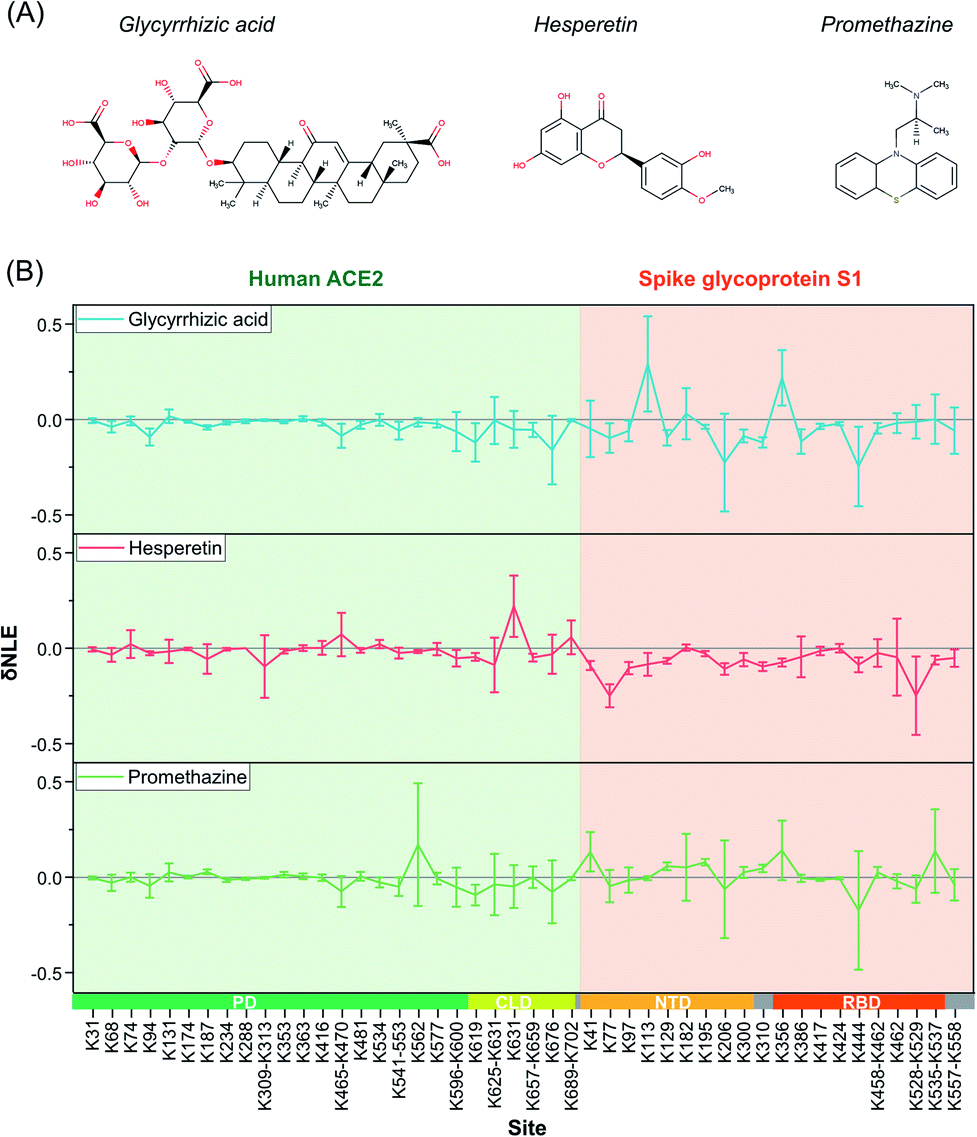 | ||
| Fig. 4 The δNLE of lysine residues in S1–ACE2 complexes induced by the treatment of exogenous small-molecule compounds. | ||
In the case of hesperetin, most of the conformational effects also focused on protein S1 (Fig. 4 and S9†). Briefly, the reactivities of lysine residues Lys77, Lys97, Lys206, Lys310, and Lys557 decreased significantly, implying that the local regions of these lysine residues are modulated by hesperetin (Fig. S9†). However, lysine residues at the direct interaction interface of S1-RBD Lys386–Lys462 have been rarely disturbed by hesperetin. Similar results were also observed when promethazine, one of the reported active drugs for SARS-CoV,45 was incubated with the S1–ACE2 solution (Fig. 4). Other active molecules including scutellarin, nicotinamide and sulfobutyl ether-β-cyclodextrin46 were also evaluated in a similar manner (Fig. S10†). Although some significant conformational effects on the S1–ACE2 complex could be successfully observed with high sensitivity, no obvious conformational intervention was discovered for disrupting the reactivities of lysine residues at the direct interaction interface of S1-RBD Lys386–Lys462. Therefore, the TILLRP strategy might be a useful experimental method for evaluating the pharmaceutical effects of active compounds on the S1–ACE2 complex at the molecular level.
Conclusions
Structural characterization of protein and protein complexes plays a fundamental important role in the elucidation of the related functions and drug developments. Aside from the great success of solid-phase based methods such as X-ray crystallography and cryogenic electron microscopy, direct analysis of native proteins in aqueous solution could also compliment the understanding of the structure and dynamics upon the formation of complexes and binding of ligands. The reactivities of lysine residues within native proteins are inherent characteristics of specific conformation and lysine residues can be utilized as endogenous probes for monitoring protein conformational fluctuations. In this study, a two-step isotope labeling-lysine reactivity profiling (TILLRP) strategy is developed to quantify the relative reactivity (NLE) of lysine sites within native proteins. We demonstrated that the NLE values are directly related to the local microenvironments of lysine sites and could be utilized to monitor the protein–protein interactions and interventions induced by exogenous small-molecule compounds.This novel TILLRP strategy was applied to investigate the dynamic conformational hotspots of S1–ACE2 complex recognition and intervention in native aqueous solution. The direct interaction interface of S1–ACE2 recognition and related conformational changes could be successfully monitored. We suggest that the NLE values of lysine residues at the interaction interface of S1-RBD Lys386–Lys462 might be utilized as indicators for investigating S1–ACE2 recognition and intervention. Although no efficient active compound was discovered to disrupt the formation of the S1–ACE2 complex directly, dynamic conformational interventions on S1–ACE2 induced by small molecules such as glycyrrhizic acid could be successfully monitored with high sensitivity and high throughput. Furthermore, we think that the TILLRP strategy could also be applied to investigate the pharmaceutical interventions of active compounds to other SARS-CoV2 protein targets due to its broad applicability with little limitation on protein types. Overall, we believe that the TILLRP strategy might provide a promising experimental alternative for potential anti-SARS-CoV2 drug evaluation at the molecular level.
Experimental
Materials
All the chemicals and proteins with no additional specification were all purchased from Sigma-Aldrich. The S1 and ACE2 recombinant proteins with Hig-tag at the C-terminus were purchased from Sino Biological (Beijing, China) with purities ≥95% (determined by SEC-HPLC).Two-step isotope labeling of native proteins in aqueous solution
The serum albumin samples were prepared at 1 mg mL−1 with 50 mM Hepes (pH 7.4). Then, the serum albumin samples were subjected to consecutive two-step isotope labeling. In the first-step labeling at the native state, the serum albumin samples were labeled with 10 mM 13CD2O and 20 mM NaCNBD3 for 10 min, and the labeling reaction was quenched by the adding of 5 fold sample volumes of precipitation buffer (acetone/ethanol/acetic acid, 50/50/0.1, v/v/v). In the second-step labeling at the denatured state, the participated protein samples were collected by centrifugation and re-dissolved in denaturing buffer with 6 M guanidine hydrochloride and 50 mM Hepes (pH 7.4). The partially labeled serum albumin samples were then labeled with 40 mM pyridine-borane and 20 mM CH2O for 2 h at 37 °C for completed dimethylation of the lysine residues.20,47 The labeling reaction was quenched by adding ammonium acetate. Finally, the excess labeling reagents were depleted by ultrafiltration.The recombinant SARS-Cov-2 spike glycoprotein S1, human ACE2 and the S1–ACE2 complex samples were diluted to 0.3 mg mL−1 with 50 mM phosphate buffer (pH 7.4). The protein samples were firstly labeled with 5 mM 13CD2O and 10 mM NaCNBD3 for 5 min in native aqueous solution, and the labeling reaction was quenched by adding 5 fold sample volumes of precipitation buffer. The second step of denature isotope labeling was identical to that of the serum samples as described above.
Protein sample digestion and preparation
The buffer systems of all labeled protein samples were exchanged to 8 M urea and 10 mM NH4HCO3 (pH 8.0) by ultrafiltration with centrifugal ultrafiltration units with a nominal molecular weight cut-off of 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (Ultracel YM-30, Merck Millipore Ltd). Then, the proteins were reduced with 5 mM TCEP and alkylated with 10 mM IAA in sequence. Finally, the protein samples were digested by endoproteinase Glu-C (Roche Life Science) or chymotrypsin with a substrate/enzyme ratio of 1:50 (w/w) at 25 °C overnight. The digested peptide samples were collected by centrifugation and acidified with 10% TFA. Finally, the peptide samples were lyophilized and stored at −80 °C until LC-MS analysis.
000 (Ultracel YM-30, Merck Millipore Ltd). Then, the proteins were reduced with 5 mM TCEP and alkylated with 10 mM IAA in sequence. Finally, the protein samples were digested by endoproteinase Glu-C (Roche Life Science) or chymotrypsin with a substrate/enzyme ratio of 1:50 (w/w) at 25 °C overnight. The digested peptide samples were collected by centrifugation and acidified with 10% TFA. Finally, the peptide samples were lyophilized and stored at −80 °C until LC-MS analysis.
LC-MS analyses
The labeled serum albumin samples were analyzed using an LTQ-Orbitrap XL coupled with an Accela 600 HPLC system (Thermo Scientific). Briefly, 0.02 μg BSA peptides were firstly loaded onto a 5 cm × 200 μm i.d. trap column (C18, 5 μm, 120 Å, SunChrom) with a flowrate of 5 μL min−1 and buffer A (0.1% FA in Milli-Q water) in 10 min. Then, the peptides were separated by a 15 cm × 75 μm i.d. capillary column (C18, 3 μm, 120 Å, SunChrom). The binary reversed phase (RP) separation gradient was set as: from 5% to 35% buffer B (0.1% FA in acetonitrile) in 30 min. MS was operated in data-dependent acquisition. The full MS spectra were collected using an orbitrap analyzer with a resolution of 60000 and the top 10 abundant ions in the full MS spectrum were subjected to CID with an isolation window of 2 Da and a normalized energy of 35%. The fragment ions (MS2) were collected in the LTQ. The AGC of full MS and MS2 was set at 1 × 106 and 1 × 104, respectively. The dynamic exclusion was enabled with an exclusion time of 120 s.
The protein samples of spike glycoprotein S1, ACE2 and the S1–ACE2 complex were analyzed using an Orbitrap Fusion Lumos Tribrid MS coupled to a Vanquish Flex HPLC system (Thermo Fisher Scientific). Briefly, 0.2 μg protein digests were firstly loaded onto a 5 cm × 150 μm C18 trap column (5 μm C18, SunChrom) and separated by a 15 cm × 150 μm C18 capillary column (1.9 μm C18, Dr Maisch) at a flow rate 0.3 μL min−1. Mass spectrometry was operated in positive mode in a data-dependent manner. The MS1 spectra were collected with the orbitrap with a resolution of 120000. The MS2 spectra were collected in a ‘top-speed’ manner (3 seconds) with a resolution of 15000 using the orbitrap. The precursor ions with a charge state of 2 to 5 were isolated with a m/z window of 1.4 m/z and subjected to HCD with a normalized energy of 28%. The dynamic exclusion was enabled with an exclusion time of 60 s.
Data analysis
The collected MS datasets were firstly searched against the database of corresponding protein sequences downloaded from Uniprot using Maxquant (version 1.6.7). Quantification multiplicity was set as 3 and DimethyLys0, DimethylLys4 (modified to +32.0535 Da) and DimethylLys8 were selected. Glu-C was selected with a maximum missed cleavage of 4. The function of match between runs was enabled. The other parameters were set as default values.The native labeling efficiency (NLE) values could be calculated via the formula: NLE = 1 − IL/(IH + IM + IL), where IH, IM and IL are the intensity of heavy, medium and light peptide labeling forms containing corresponding lysine residues, respectively.
Lysine microenvironment docking based on the changes of labeling efficiency
The minimized model of Spike-ACE2 is from targeting COVID-19: GHDDI Info Sharing Portal (https://www.ghddi-ailab.github.io/Targeting2019-nCoV/nCov_Structures/). The lysine sites with P value < 0.05 and |δNLE| ≥ 0.1 were chosen as significant sites for following K sites.Using AutoDockTools (version1.5.6),48 each K site with a significant NLE difference between S1–ACE2 free and complex states was transformed into flexible residues and preserved, the rest of corresponding protein structure was kept rigid respectively, and the small-molecule compounds (glycyrrhizic acid (ZINC96015174), hesperetin (ZINC39092), nicotinamide (ZINC5878), promethazine (ZINC20250), scutellarin (ZINC21992916) and sulfobutyl ether-β-cyclodextrin (Captisol)) were preserved after torsion. The above three parts of the file format are saved as *.pdbqt. Using AutoDock Vina version 1.1.2,49 each lysine site of each chain was docked three times. The docking parameters were as follows: the center_xyz set as the N atom position of the branched chain of each lysine, the grid box size is set to 30 (30 × 30 × 30 Å), num_modes was set to 20, and exhaustiveness was set to 30. In order to improve the simulated accuracy of the interaction region of small molecules, the docking conformations should be consistent with the changing trend of NLE as much as possible. The docking conformations at the top in the ranking of binding affinity energy were selected as representative results. Protein and ligands structures were rendered by PyMol.
Author contributions
Z. L., W. Z. and F. W. conceived and designed the study. Z. L., B. S. and M. H. performed the sample preparation. Z. L., Y. M. performed mass spectrometry analysis. W. Z. conducted MD analysis. W. Z., Z. L. and F. W. analyzed the experimental data and discussed the results. Z. L., W. Z., Y. P. and F. W. wrote the manuscripts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Key R&D Program of China (2016YFF0200504), the National Natural Science Foundation of China (91853101 and 21675152), the Original Innovation Project of CAS (ZDBS-LY-SLH032), and the grant from DICP (DICPI202007) to F.W is gratefully acknowledged.Notes and references
- P. C. Ng and S. Henikoff, Annu. Rev. Genomics Hum. Genet., 2006, 7, 61–80 CrossRef CAS.
- R. Krishnamurty, J. L. Brigham, S. E. Leonard, P. Ranjitkar, E. T. Larson, E. J. Dale, E. A. Merritt and D. J. Maly, Nat. Chem. Biol., 2013, 9, 43–50 CrossRef CAS.
- P. C. Ng and S. Henikoff, Genome Res., 2001, 11, 863–874 CrossRef CAS.
- J. U. Bowie, J. F. Reidhaar-Olson, W. A. Lim and R. T. Sauer, Science, 1990, 247, 1306–1310 CrossRef CAS.
- G. A. Weiss, C. K. Watanabe, A. Zhong, A. Goddard and S. S. Sidhu, Proc. Natl. Acad. Sci. U.S.A., 2000, 97, 8950–8954 CrossRef CAS.
- K. L. Morrison and G. A. Weiss, Curr. Opin. Chem. Biol., 2001, 5, 302–307 CrossRef CAS.
- Y. Liu, M. P. Patricelli and B. F. Cravatt, Proc. Natl. Acad. Sci. U.S.A., 1999, 96, 14694–14699 CrossRef CAS.
- B. F. Cravatt, A. T. Wright and J. W. Kozarich, Annu. Rev. Biochem., 2008, 77, 383–414 CrossRef CAS.
- C. D. Waldburger, J. F. Schildbach and R. T. Sauer, Nat. Struct. Mol. Biol., 1995, 2, 122–128 CrossRef CAS.
- D. E. Anderson, W. J. Becktel and F. W. Dahlquist, Biochemistry, 1990, 29, 2403–2408 CrossRef CAS.
- S. Kumar and R. Nussinov, J. Mol. Biol., 1999, 293, 1241–1255 CrossRef CAS.
- C.-H. Chan, T.-H. Yu and K.-B. Wong, PLoS One, 2011, 6, e21624 CrossRef CAS.
- W. N. Burnette, V. Mar, B. Platler, J. Schlotterbeck, M. McGinley, K. Stoney, M. Rohde and H. Kaslow, Infect. Immun., 1991, 59, 4266–4270 CrossRef CAS.
- M. C. O'Brien, K. M. Flaherty and D. B. McKay, J. Biol. Chem., 1996, 271, 15874–15878 CrossRef.
- M. L. Plater, D. Goode and M. J. C. Crabbe, J. Biol. Chem., 1996, 271, 28558–28566 CrossRef CAS.
- R. Lahti, K. Pohjanoksa, T. Pitkäranta, P. Heikinheimo, T. Salminen, P. Meyer and J. Heinonen, Biochemistry, 1990, 29, 5761–5766 CrossRef CAS.
- J. Reinstein, I. Schlichting and A. Wittinghofer, Biochemistry, 1990, 29, 7451–7459 CrossRef CAS.
- T. R. Chen, L. H. Wei, X. Q. Guan, C. Huang, Z. Y. Liu, F. J. Wang, J. Hou, Q. Jin, Y. F. Liu, P. H. Wen, S. J. Zhang, G. B. Ge and W. Z. Guo, Bioorg. Chem., 2019, 92, 103199 CrossRef CAS.
- J. Chen, A. Wang, B. Liu, Y. Zhou, P. Luo, Z. Zhang, G. Li, Q. Liu and F. Wang, Anal. Chem., 2019, 91, 13222–13229 CrossRef CAS.
- Z. Liu, Y. Zhou, J. Liu, J. Chen, A. J. R. Heck and F. Wang, TrAC, Trends Anal. Chem., 2019, 118, 771–778 CrossRef CAS.
- Y. Zhou, Z. Liu, J. Zhang, T. Dou, J. Chen, G. Ge, S. Zhu and F. Wang, Chem. Commun., 2019, 55, 4311–4314 RSC.
- Y. Zhou, Y. Wu, M. Yao, Z. Liu, J. Chen, J. Chen, L. Tian, G. Han, J. R. Shen and F. Wang, Anal. Chem., 2016, 88, 12060–12065 CrossRef CAS.
- R. Castagnoli, M. Votto, A. Licari, I. Brambilla, R. Bruno, S. Perlini, F. Rovida, F. Baldanti and G. L. Marseglia, JAMA Pediatrics, 2020, 174, 882–889 CrossRef.
- C.-C. Lai, T.-P. Shih, W.-C. Ko, H.-J. Tang and P.-R. Hsueh, Int. J. Antimicrob. Agents, 2020, 105924 CrossRef CAS.
- J. Lan, J. Ge, J. Yu, S. Shan, H. Zhou, S. Fan, Q. Zhang, X. Shi, Q. Wang, L. Zhang and X. Wang, Nature, 2020, 581, 215–220 CrossRef CAS.
- A. C. Walls, Y. J. Park, M. A. Tortorici, A. Wall, A. T. McGuire and D. Veesler, Cell, 2020, 181, 281–292 CrossRef CAS.
- N. J. Matheson and P. J. Lehner, Science, 2020, 369, 510–511 CrossRef CAS.
- R. Yan, Y. Zhang, Y. Li, L. Xia, Y. Guo and Q. Zhou, Science, 2020, 367, 1444–1448 CrossRef CAS.
- Q. Wang, Y. Zhang, L. Wu, S. Niu, C. Song, Z. Zhang, G. Lu, C. Qiao, Y. Hu, K.-Y. Yuen, Q. Wang, H. Zhou, J. Yan and J. Qi, Cell, 2020, 181, 894–904 CrossRef CAS.
- M. K. Gupta, S. Vemula, R. Donde, G. Gouda, L. Behera and R. Vadde, J. Biomol. Struct. Dyn., 2020, 1–11, DOI:10.1080/07391102.2020.1751300.
- D. C. Hall Jr and H. F. Ji, Trav. Med. Infect. Dis., 2020, 35, 101646 CrossRef.
- C. Wu, Y. Liu, Y. Yang, P. Zhang, W. Zhong, Y. Wang, Q. Wang, Y. Xu, M. Li, X. Li, M. Zheng, L. Chen and H. Li, Acta Pharm. Sin. B, 2020, 10, 766–788 CrossRef.
- A. Ubani, F. Agwom, O. RuthMorenikeji, S. Nathan, P. Luka, A. Umera, U. Umar, S. Omale, N. E. Nnadi and J. C. Aguiyi, bioRxiv, 2020 DOI:10.1101/2020.03.31.017657.
- B. T. P. Thuy, T. T. A. My, N. T. T. Hai, L. T. Hieu, T. T. Hoa, H. Thi Phuong Loan, N. T. Triet, T. T. V. Anh, P. T. Quy, P. V. Tat, N. V. Hue, D. T. Quang, N. T. Trung, V. T. Tung, L. K. Huynh and N. T. A. Nhung, ACS Omega, 2020, 5, 8312–8320 CrossRef CAS.
- D. J. Barlow and J. M. Thornton, J. Mol. Biol., 1983, 168, 867–885 CrossRef CAS.
- S. Kumar and R. Nussinov, Chembiochem, 2002, 3, 604–617 CrossRef CAS.
- M. S. Wanigasekara and S. M. Chowdhury, Anal. Chim. Acta, 2016, 935, 197–206 CrossRef CAS.
- D. Suckau, M. Mak and M. Przybylski, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 5630–5634 CrossRef CAS.
- J. Vidgren, L. A. Svensson and A. Liljas, Nature, 1994, 368, 354–358 CrossRef CAS.
- L. E. Kiss and P. Soares-da-Silva, J. Med. Chem., 2014, 57, 8692–8717 CrossRef CAS.
- D. Wrapp, N. Wang, K. S. Corbett, J. A. Goldsmith, C.-L. Hsieh, O. Abiona, B. S. Graham and J. S. McLellan, Science, 2020, 367, 1260–1263 CrossRef CAS.
- J. Zhou and J. Huang, Front. Cell Dev. Biol., 2020, 8, 589 CrossRef.
- H. Javelot, J. Petrignet, F. Addiego, J. Briet, M. Solis, W. El-Hage, C. Hingray and L. Weiner, Med. Hypotheses, 2020, 110025 CrossRef CAS.
- R. Yu, L. Chen, R. Lan, R. Shen and P. Li, Int. J. Antimicrob. Agents, 2020, 106012 CrossRef CAS.
- T. Y. Ho, S. L. Wu, J. C. Chen, C. C. Li and C. Y. Hsiang, Antiviral Res., 2007, 74, 92–101 CrossRef CAS.
- S. T. Jones, V. Cagno, M. Janeček, D. Ortiz, N. Gasilova, J. Piret, M. Gasbarri, D. A. Constant, Y. Han and L. Vuković, Sci. Adv., 2020, 6, eaax9318 CrossRef CAS.
- Z. Liu, R. Wang, J. Liu, R. Sun and F. Wang, J. Proteome Res., 2019, 18, 2185–2194 CrossRef CAS.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc05330a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |