
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRitter-enabled catalytic asymmetric chloroamidation of olefins†
Daniel C.
Steigerwald
,
Bardia
Soltanzadeh‡
,
Aritra
Sarkar
,
Cecilia C.
Morgenstern
,
Richard J.
Staples
 and
Babak
Borhan
*
and
Babak
Borhan
*
Michigan State University, Department of Chemistry, East Lansing, MI 48824, USA. E-mail: babak@chemistry.msu.edu
First published on 7th December 2020
Abstract
Intermolecular asymmetric haloamination reactions are challenging due to the inherently high halenium affinity (HalA) of the nitrogen atom, which often leads to N-halogenated products as a kinetic trap. To circumvent this issue, acetonitrile, possessing a low HalA, was used as the nucleophile in the catalytic asymmetric Ritter-type chloroamidation of allyl-amides. This method is compatible with Z and E alkenes with both alkyl and aromatic substitution. Mild acidic workup reveals the 1,2-chloroamide products with enantiomeric excess greater than 95% for many examples. We also report the successful use of the sulfonamide chlorenium reagent dichloramine-T in this chlorenium-initiated catalytic asymmetric Ritter-type reaction. Facile modifications lead to chiral imidazoline, guanidine, and orthogonally protected 1,2,3 chiral tri-amines.
Introduction
The last decade has witnessed dramatic progress in the asymmetric halofunctionalization of olefins as a relatively new addition to the field of asymmetric catalysis.1 To reach this point, catalytic events have had to outcompete a number of intrinsic difficulties associated with halonium ions, including olefin-to-olefin halenium transfer and equilibrium of the putative cyclic haliranium to the open β-halocarbenium ion.2 Overcoming these challenges has enabled the construction of carbon–halogen bonds, as well as the vicinal formation of carbon–halogen,3 carbon–carbon,4 carbon–oxygen5 and carbon–nitrogen6 bonds greatly increasing the molecular and stereochemical complexity in a single step transformation.The importance of the catalytic asymmetric halofunctionalization chemistry is reflected by the large number of reports on intramolecular halocyclizations,4,5h–aa,6k–aa along with a growing list of intermolecular halofunctionalizations.3,5a–g,6a–j Early investigations focused on the more accessible intramolecular cases, although recent reports demonstrate that the entropically challenged intermolecular events have also succumbed to excellent strategies in delivering products in high yield and enantioexcess. Nonetheless, intermolecular asymmetric haloamination and haloamidation reactions have not seen the same level of progress, especially with organocatalysts with unactivated olefins.6a–j An important challenge in the development of such reactions can be attributed to the high halenium affinity (HalA) of the nitrogen atom as compared to other nucleophiles, which leads to the direct halogenation of the nitrogen atom as opposed to the target functionality, such as the olefin (see Fig. 1b, HalA (Cl) values).2a This is especially detrimental in asymmetric halogenations, as the transfer of the halenium to the nitrogen atom would shunt the path of the halogen through the catalyst, which is necessary to achieve enantiofacial selectivity. As a result, haloamination reactions differ from the successful catalytic asymmetric haloetherifications/esterifications since alcohols and other oxygen nucleophiles have a lower HalA than the corresponding nitrogen nucleophiles. There are a few elegant examples of intermolecular asymmetric catalytic solutions to circumvent the high halonium affinity of nitrogen in the literature (Fig. 1a). These solutions have required either, (1) the use of a “pro-nucleophile”, i.e., the halenium addition to the olefin revealing a more nucleophilic nitrogen atom (see the work of Masson, Zhou, and Burns),6c,e,f,h or (2) the addition of a nitrogen nucleophile to an α,β-unsaturated system that subsequently captures a halenium ion (Feng and coworkers).6a,b,d,g,i,j
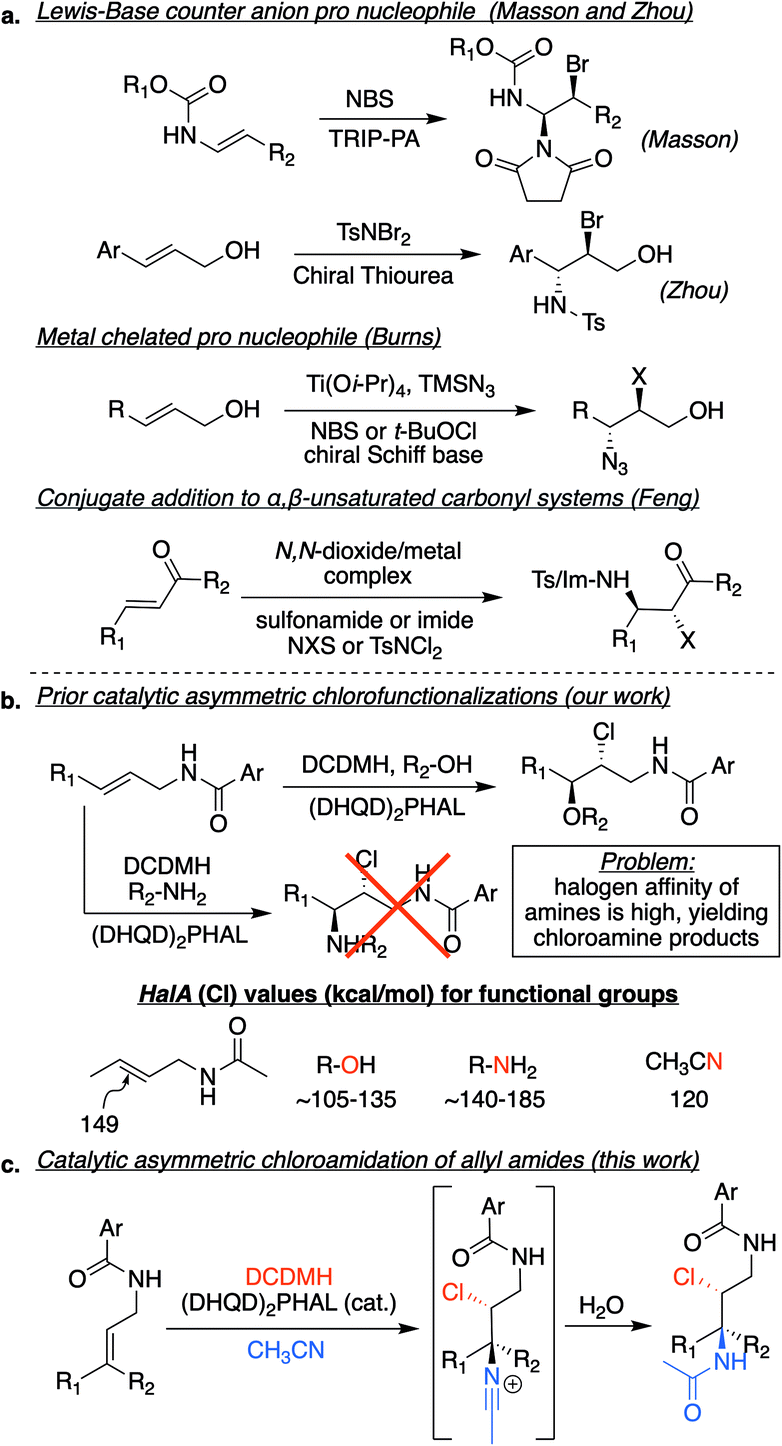 | ||
| Fig. 1 (a) Prior art in catalytic asymmetric haloamination (or equivalent functionality) of olefins; (b) generalized example of catalytic asymmetric chlorofunctionalizations. Initial attempts using this strategy failed to deliver the chloroamination product. HalA (Cl) values suggest the need for a nucleophilically tempered nitrogen source. The HalA (Cl) of nitrile is close to the HalA (Cl) of alcohols, which have successfully succumbed to haloetherifications; (c) putative strategy for the Ritter-mediated chloroamidation reaction. | ||
In our approach, we envisioned the use of a nitrogen atom with attenuated HalA in the same range calculated for oxygen nucleophiles. Presumably, this would circumvent the ability for the nitrogen atom to participate prematurely in abstraction of the halenium from its donor. A cursory look at HalA (Cl) values led to the nitrile functionality as a potential candidate (see Fig. 1b).2a We imagined the use of an alkyl nitrile would lead to a halenium-induced Ritter reaction, effectively introducing a nitrogen atom. Previous reports have shown nitriles as nucleophilic participants in different halenium induced Ritter-type reactions, although not in an asymmetric fashion.7 The closest related example is from Pasquato and coworkers that employed acetonitrile to open a pre-formed enantiopure thiiranium ion, resulting in the corresponding acetamide.8 In fact, we had also observed what we presumed to be a Ritter side product previously, while developing an effective strategy towards catalytic asymmetric dihalogenation. During our optimizations, we had observed that although acetonitrile was capable of delivering the desired dihalogenated product, albeit not as the optimum solvent system, the Ritter side product was evident.3e This observation laid the foundation for the development of this reaction as a route for haloamidation of olefins in an asymmetric fashion.
Utilizing nitriles as the nucleophilic partners in chloroamidations, we demonstrate the use of cinchona alkaloid dimer catalysts, along with a variety of chlorenium sources, in an efficient methodology to deliver products in high yields and enantioselectivity. Furthermore, we show that the immediate product of the Ritter-type reaction is the trapping of the putative nitrilium cation with the donor of the chlorenium, and in fact, when dichloramine-T is used, the sulfonylamidine product is stable for isolation. Vicinal chloramines9 and in particular enantioenriched vicinal chloroamides are useful in downstream synthesis of chiral aziridine, oxazoline, and amino alcohol moieties.10 To expand on this chemistry, we employ vicinal chloroamidines to synthesize enantioenriched imidazoline, guanidine, and orthogonally protected 1,2,3 chiral tri-amines.
Results and discussion
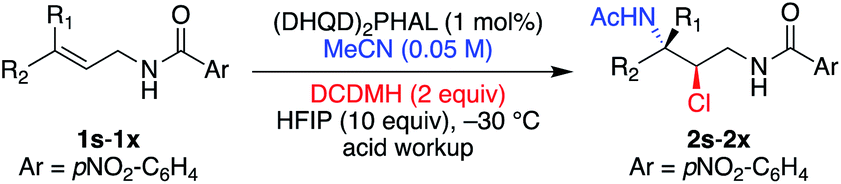
Our prior success in using the cinchona alkaloid dimers in the catalytic asymmetric intra- and intermolecular halofunctionalization of allyl amides prompted the following investigation for developing a process for haloamidations.3e,5e,11 Cognizant of the issues with using amine or amide nucleophiles, namely, their elevated reactivity towards abstracting haleniums from their respective sources, we opted to use nitriles, possessing an attenuated affinity for halenium ions.Ritter-type exploratory investigations
The study was initiated with substrate 1a, which had previously shown excellent results in delivering enantioenriched 1,2-chloroethers.5e Early exploration of the reaction with acetonitrile, catalytic (DHQD)2PHAL, and 1,3-dichloro-5,5-dimethylhydantoin (DCDMH) revealed the presence of Ritter-type products. Nonetheless, unlike the product of a classical Ritter-reaction that yields the corresponding amide by trapping of the nitrilium ion intermediate with water,7g,j,12 the observed product was the result of the hydantoin anion trap of the nitrilium ion intermediate as indicated by the mass spectrum of the crude product (see 2a′, Table 1, as well as Tables S2 and S3† for NMR evidence). Mild acid workup hydrolyzed the amidine product 2a′ to provide the α-chloroamide 2a. Interestingly, without the presence of (DHQD)2PHAL, the nitrilium ion is trapped by water, as indicated by direct amide formation that yields 2a. The control over product formation suggests that the catalyst is not innocent in the addition of the hydantoin ion to the nitrilium ion. This divergent pathway hints towards an associative complex between (DHQD)2PHAL and DCDMH.5j We also discovered that polarization of the olefin, resulting from the inductive electron withdrawing nature of the amide, albeit not large, is sufficient in dictating the regiochemistry in the Ritter type mechanism for 1a in both catalyzed and noncatalyzed reactions (see Table S4,† entries 1 and 2 for uncatalyzed vs. catalyzed regiochemical outcomes).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Additive (equiv.) | Cl+ source | Cat. (mol%) | Time (h) | Yielda (%) | eeb (%) |
| a NMR yield on a 0.05 mmol scale. b Enantiomeric excess determined by chiral HPLC. c Reaction completed at room temperature. d Major product was the incorporation of the p-tolyl sulfonamide from DiCh-T (see 3a for structure). e 0.6 equiv. of DCDMH was used. f Reaction completed in dichloromethane (0.10 M) with 10 equiv. of acetonitrile. | ||||||
| 1 | None | DCDMH | 10 | 72 | 68 | 96 |
| 2 | HFIP (2) | DCDMH | 10 | 0.5 | 71 | 99 |
| 3 | HFIP (10) | DCDMH | 10 | 0.5 | 78 | 99 |
| 4c | HFIP (10) | DCDMH | 10 | 0.5 | 78 | 98 |
| 5c | HFIP (10) | NCS | 10 | 96 | 70 | 98 |
| 6 | HFIP (10) | TCCA | 10 | 0.5 | 42 | 98 |
| 7d | HFIP (10) | DiCh-T | 10 | 0.5 | 12 | 99 |
| 8 | HFIP (10) | DCDMH | 1 | 0.5 | 76 | 99 |
| 9e | HFIP (10) | DCDMH | 1 | 9 | 67 | 99 |
| 10 | TFE (10) | DCDMH | 1 | 5 | 67 | 96 |
| 11 | PhCO2H (10) | DCDMH | 1 | 2 | 29 | 97 |
| 12f | HFIP (10) | DCDMH | 1 | 4 | 53 | 99 |
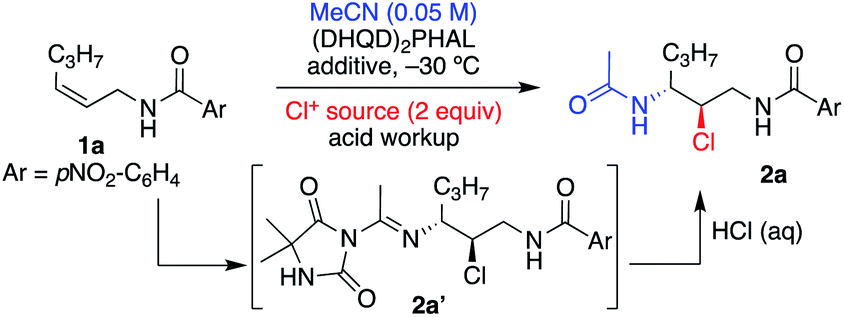
Table 1 illustrates the optimization of the reaction under various conditions with the Z aliphatic substrate 1a. The reaction provides 2a in 68% yield (96% ee), however, requiring 72 h to reach completion (entry 1, Table 1). In our previously reported studies on asymmetric halofunctionalization reactions,3e,11a,b,13 we had observed an increased performance, both in terms of rate of reaction and yield of products, when a fluorinated alcohol additive was employed. Presumably, the acidic nature of the alcohol, and its attenuated nucleophilicity, are good combinations that lead to rate acceleration without nucleophilic participation in the reaction.14 There is also evidence that protonation of cinchona alkaloid dimeric catalysts could lead to altered conformations.15 An early screening of solvents showed that the addition of 1,1,1,3,3,3-hexafluoroisopropanol (HFIP, entries 2 and 3, Table 1) improved the enantiomeric excess of 2a, while tremendously increasing the rate of the reaction.
DCDMH proved to be the optimal chlorenium source as the less active NCS (entry 5) was sluggish and gave slightly lower ee, while the more active chlorenium TCCA (entry 6) gave a lower yield. Use of dichloramine-T returned the product in high ee, although in low yields. Interestingly, the mass balance was identified as the p-tolyl sulfonylamidine product 3a (addition of the p-tolyl sulfonamide to the Ritter intermediate, yielding a stable product, vide infra). Lowering the catalyst loading (entry 8) led to a negligible change in reaction proficiency, and thus 1 mol% (DHQD)2PHAL was chosen as standard for ensuing reactions. Less reactive substrates required increased catalyst loading to achieve optimal proficiency (see Table S1† for experiments and discussion). Varying the equivalents of DCDMH had no effect on the enantiopurity of the final product, although the yield suffered slightly with lower amounts (entry 9). A quick screen of acidic additives (entries 10 and 11) proved HFIP's superiority and was thus maintained as part of the standard reaction condition. Decreasing nucleophile equivalents (entry 12) provided slightly lower yield and longer reaction times but retained high enantioselectivity for 1a.
Next, we examined the nature of the amide on the performance of the reaction (Table 2). Comparing to the standard substrate 1a, electronic perturbations to the aryl of the amide group did not alter the course or results of the reactions, delivering products 2b–2f in good yields and high enantioselectivity (entries 1–6, Table 2). The acetamide substrate 1g, though sluggish, provided the chloroamidation product 2g with good enantiocontrol (94% ee). Nonetheless, the results were inferior in terms of yield, enantiopurity of product, and time to completion of the reaction in comparison to arylamide substrates 1a–1f. Interestingly, the E aliphatic substrate 1h was nonreactive without HFIP, but reacted under the standard condition to yield product 2h in good yield and high enantioselectivity (entry 8, Table 2).
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Pdt | Time (h) | R1 | R2 | R3 | Yielda (%) | dr | eeb (%) |
| a Isolated yield on a 0.1 mmol scale. b Enantiomeric excess determined by chiral HPLC. c Absolute stereochemical determination was verified by X-ray crystal analysis (see ESI). | ||||||||
| 1 | 2a | 0.5 | pNO2-C6H4 | C3H7 | H | 90 | >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
99 |
| 2 | 2b | 2 | Ph | C3H7 | H | 81 | >20:1 |
98 |
| 3c | 2c | 0.5 | pMeO-C6H4 | C3H7 | H | 89 | >20:1 |
99 |
| 4 | 2d | 0.5 | pF-C6H4 | C3H7 | H | 85 | >20:1 |
99 |
| 5 | 2e | 0.5 | pt-Bu-C6H4 | C3H7 | H | 79 | >20:1 |
99 |
| 6 | 2f | 0.5 | pBr-C6H4 | C3H7 | H | 91 | >20:1 |
99 |
| 7 | 2g | 18 | Me | C3H7 | H | 58 | >20:1 |
94 |
| 8 | 2h | 5 | pNO2-C6H4 | H | C3H7 | 81 | >20:1 |
97 |
| 9c | 2i | 5 | pBr-C6H4 | H | C3H7 | 59 | >20:1 |
95 |

A note regarding the absolute stereochemistry of the molecules reported in this manuscript: products 2c and 2i were crystalline, and their 3-dimensional structures were solved, revealing the stereochemistry of the (DHQD)2PHAL catalyzed reaction. The absolute stereochemistry of other molecules in this report are by analogy to these two structures.16
The requirement for a secondary amide substrate was briefly examined with the analogous imide 1j, ester 1k, and N-methylated tertiary amide 1l (Scheme 1). Substrates 1j and 1k yielded their respective chloroamide products 2j and 2k, respectively, albeit with less enantiocontrol than the aryl amide substrates, while requiring a higher catalyst loading (10 mol%). The anticipated chloroamide product was not observed upon treatment of 1l under slightly modified conditions (10 mol% catalyst instead of 1 mol%, and 0 °C instead of −30 °C), but instead chloroester 2l′′ was isolated in good yield. As depicted in Scheme 1, 2l′′ is presumably obtained from the hydrolysis of the presumed intermediate 2l′. Taken together, these results not only indicate the need for a hydrogen bonding element supplied by the 2° amide, but also the amide confirmation presumably plays a significant role in the success of these asymmetric catalytic reactions.
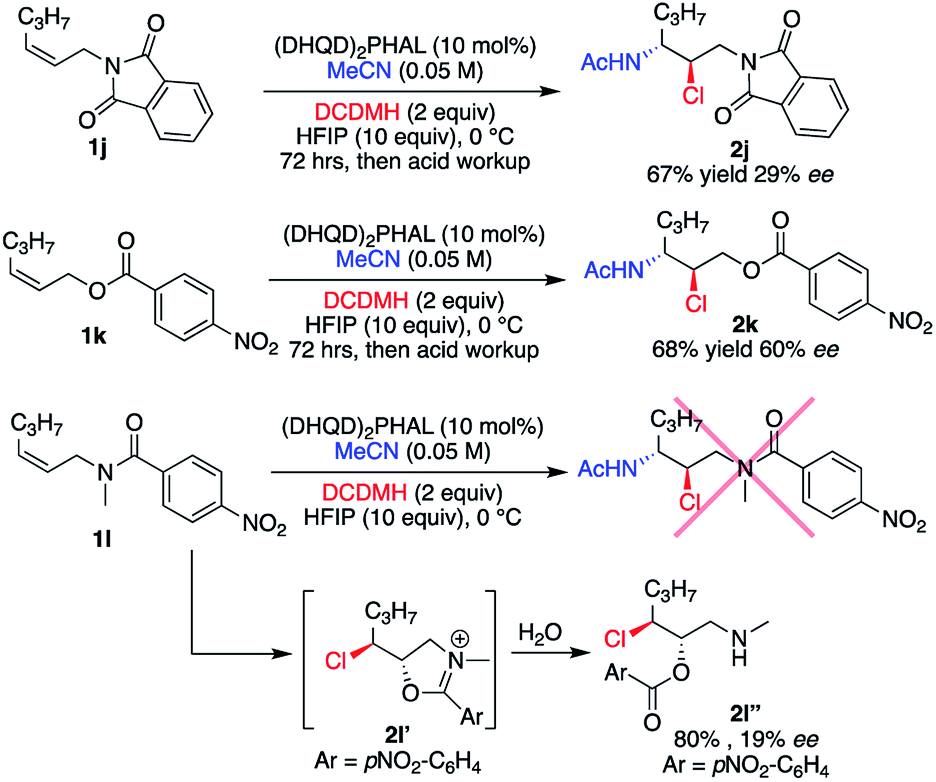 | ||
| Scheme 1 Alternative functional handles. | ||
Exploring the substrate scope of the Ritter-type asymmetric chloroamidation reaction
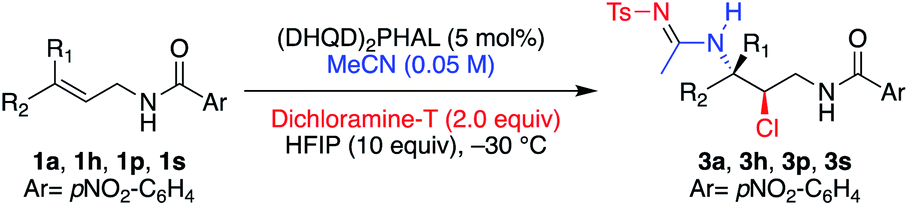
Table 3 illustrates the results of the substrate scope for E and Z aliphatic allyl-amides. In all cases, the minor diastereomer was not observed. Z-Olefins (entries 1–5) reacted smoothly to yield the corresponding chloroamide products in high yields, enantioselectivities (99% ee for all examples), and regioselectivities. This was true of the less electronically biased examples 1o and 1p, which often result in lower performance due to inductive changes in polarity, leading to regioisomeric products.3a,e The extended reaction time required for 1p led to the over-chlorinated product 2p′ (resulting from the α-chlorination of the acetamide moiety) in ∼2:1 ratio (2p′:2p).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Pdt | Time (h) | R1 | R2 | Yielda (%) | dr | eeb (%) |
| a Isolated yields on a 0.1 mmol scale. b Enantioselectivity determined by chiral HPLC. c Isolated yields on a 1.0 mmol scale. d 15 mol% (DHQD)2PHAL was added over the course of the reaction (3 days), maintaining the temperature at 0 °C. e Combined yield of the acetamide product and the α-chlorinated acetamide product (see ESI). f Both acetamide and α-chlorinated acetamide were obtained with 99% ee (see ESI). g Reaction performed with quasi-enantiomeric (DHQ)2PHAL. | |||||||
| 1 | 2a | 0.5 | C3H7 | H | 90 (83)c | >20:1 |
99 |
| 2 | 2m | 0.5 | C6H13 | H | 79 | >20:1 |
99 |
| 3 | 2n | 0.5 | C2H5 | H | 73 | >20:1 |
99 |
| 4 | 2o | 5 | TBDPSOC2H4 | H | 62 | >20:1 |
99 |
| 5d | 2p | 72 | BnOCH2 | H | 23 (69)e | >20:1 |
99f |
| 6 | 2h | 5 | H | C3H7 | 81 | >20:1 |
96 |
| 7 | 2q | 6 | H | C6H13 | 83 | >20:1 |
94 |
| 8 | 2r | 6 | Me | Me | 79 | na | 99 |
| 9g | ent-2a | 0.5 | C3H7 | H | 87 | >20:1 |
99 |
| 10g | ent-2h | 5 | H | C3H7 | 87 | >20:1 |
97 |
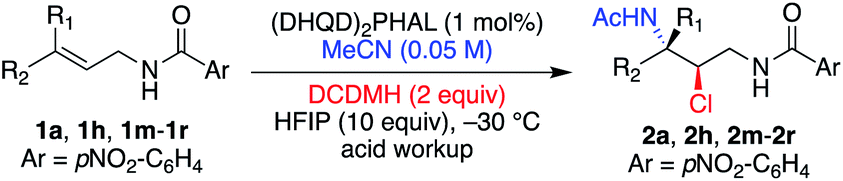
The same success was observed for the corresponding E-isomeric substrates, providing the chloroamide products with slightly less enantiocontrol (≥94% ee) and excellent yields (entries 6 and 7). The tri-substituted allyl amide 1r was also not problematic, providing the product 2r in high yield as well as high ee (entry 8). The quasi-enantiomeric (DHQ)2PHAL catalyst gave comparable results for the Z and E isomeric substrates 1a and 1h, yielding ent-2a and ent-2h, in 99% ee and 97% ee, respectively (entries 9 and 10).
Aryl substituted allyl amide substrates proved more problematic, leading to diastereomeric products, presumably as a result of carbocationic stabilization afforded by the aromatic group (Table 4).3e,5e,11b As expected, the more electron rich systems, having the ability to stabilize the benzylic carbocation, resulted in lower selectivity (entries 1–3), while the electron deficient pCF3-Ph substituent restored the high diastereomeric selectivity observed with the alkyl systems (entry 4).11b,17 Similar to 1p, the extended reaction time required for full conversion of 1v to the product led to α-chlorination of the acetamide functionality as the major product (∼5:1 2v′:2v). Nonetheless, while the chloroamidation of electron rich aryls led to low drs, each diastereomer was isolated in high enantiomeric excess, suggesting the olefinic face selectivity during the chlorination is preserved.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Pdta | R1 | R2 | Yieldb (%) | drc | ee majord (%) | ee minord (%) |
| a Time of completion for products 2s, 2u, 2x, and ent-2s was 1 h, while 2w and ent-2w required 10 h, with 2v the most sluggish, necessitating 120 h. b Isolated yield on a 0.1 mmol scale. c Diastereomeric ratio determined by NMR. d Enantiomeric excess determined by chiral HPLC. e 15 mol% (DHQD)2PHAL was added over the course of the reaction (3 days), maintaining the temperature at 23 °C. f Combined yield of acetamide product and α-chlorinated acetamide product (see ESI). g The α-chlorinated acetamide product had enantiomeric excess of 87% (see ESI). h Reaction performed with quasi-enantiomeric (DHQ)2PHAL. | |||||||
| 1 | 2s | Ph | H | 95 | 65:35 |
99 | 99 |
| 2 | 2t | pCl-C6H4 | H | 92 | 66:34 |
97 | 98 |
| 3 | 2u | pMe-C6H4 | H | 78 | 50:50 |
99/99 | 99/99 |
| 4e | 2v | pCF3-C6H4 | H | 12 (76)f | >20:1 |
89g | na |
| 5 | 2w | H | Ph | 53 | 74:26 |
99 | 93 |
| 6 | 2x | Me | Ph | 57 | 61:39 |
99 | 97 |
| 7h | ent-2s | Ph | H | 84 | 63:37 |
99 | 99 |
| 8h | ent-2w | H | Ph | 66 | 70:30 |
92 | 99 |
Efforts to improve diastereoselectivity, such as employing a less polar co-solvent, decreasing equivalents of HFIP, and increasing catalyst loading were unfruitful. Neither the E-substituted alkene 1w, nor the trisubstituted alkene 1x were immune to the observed diminished diastereoselectivity, although in both cases high enantioselectivity of their products were maintained (entries 5 and 6). The reduced yield for product 2w was attributed to competing intramolecular halocyclization, not observed with Z alkenes. The quasi-enantiomeric (DHQ)2PHAL provided ent-2s and ent-2w with similar efficiencies in all categories.
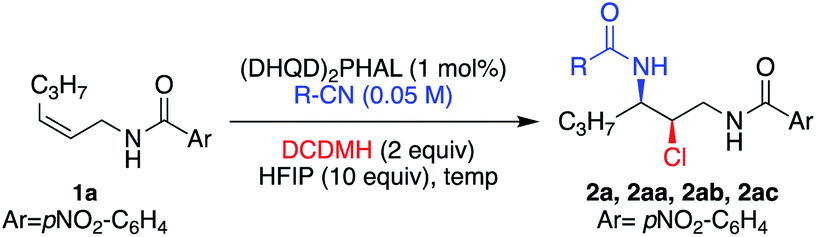
The next variable examined was the nitrile nucleophile, yielding different amide products (Table 5). Reactions of 1a proceeded smoothly with propionitrile (entry 2), benzonitrile (entry 3), and the bulky pivalonitrile (entry 4). Although the latter two reactions required slightly higher temperatures to accommodate the higher melting points of their respective nitrile solvents, there were no significant observed erosion in enantioselectivities. The versatility in choosing different nitrile nucleophiles enables the assembly of more complex amide structures.
Asymmetric sulfonylamidine products via trapping of the Ritter intermediate
As described above, the use of dichloramine-T as the chlorenium source led to low yields of the chloroamide product (see Table 1, entry 7), although reaction conversion was high. Analysis of the reaction products led to the identification of the corresponding chlorosulfonylamidine (see 3a, Table 7), which results from the capture of the nitrilium ion intermediate with the sulfonylamide generated upon transfer of the halogen. While the amidine products from addition of the hydantoin to the Ritter intermediate were observed before (see structure 2a′ in Table 1), they were not stable to isolation. Interestingly, the sulfonyl functionality lends stability to the structure, so much so that it does not hydrolyze with aqueous acid treatment.A quick screen led to a slight modification from conditions used in the Ritter-type reactions with DCDMH (Table 6). Standard conditions used with DCDMH led to a 5.4:1 3h:4h ratio (entry 1). Not surprisingly, increasing equivalents of HFIP worsened the selectivity (entry 2). As illustrated in entry 3, however, omission of HFIP to eliminate the side product 4h reduces the enantioselectivity of 3h, similar to reactions that employed DCDMH as the chlorenium source. Interestingly, increased equivalents of dichloramine-T greatly enhanced the product ratio (16:1, 3h:4h), while maintaining high ee (entry 4). Further verification of the latter was the observed diminution of the same ratio (2.6:1) when 1.25 equivalents of dichloramine-T was employed (entry 5). Alternatively, increase in the amount of catalyst (from 1 mol% to 5 mol%), without increasing dichloramine-T (2 equivalents), led to the same high product ratio (entry 6). It is likely that 4h originates from the trap of the nitrilium intermediate, as incubation of 3h in neat HFIP over a prolonged period did not return any 4h.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | DiCh-T (equiv.) | HFIP (equiv.) | Yield 3ha (%) |
3h:4hb |
eec (%) 3h |
| a NMR yield on a 0.05 mmol scale. b Ratios are obtained from NMR of crude reaction mixture. c Enantiomeric excess determined by chiral HPLC. | ||||||
| 1 | 1 | 2.00 | 10 | 43 | 5.4:1 |
94 |
| 2 | 1 | 2.00 | 20 | 39 | 2.6:1 |
94 |
| 3 | 1 | 2.00 | 0 | 45 | na | 62 |
| 4 | 1 | 3.00 | 10 | 49 | 16:1 |
92 |
| 5 | 1 | 1.25 | 10 | 50 | 2.6:1 |
95 |
| 6 | 5 | 2.00 | 10 | 56 | >20:1 |
96 |
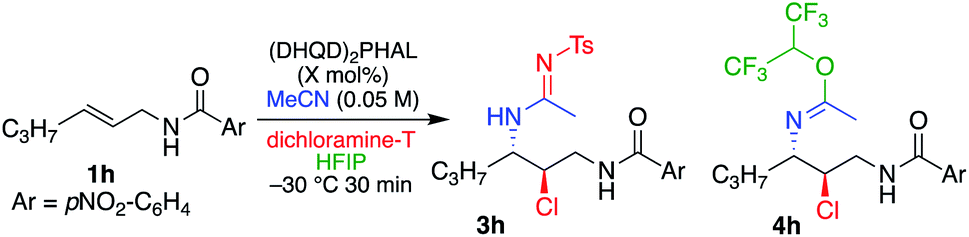
Table 7 lists a short survey of substrates that highlights a similar level of efficiency for the dichloramine-T mediated reaction that yield the chlorosulfonylamidines as compared to the chloroamides obtained with DCDMH. Z and E aliphatic allyl amides 1a and 1h are converted to their corresponding products 3a and 3h in good yields and high enantiomeric excess (99% and 95%, respectively). The benzyl protected allylic alcohol 1p also returned product 3p with no observable evidence for regio-isomeric products in high enantiomeric excess (entry 3). As detailed above, the aryl substituted olefin 1s was more problematic, leading to diastereomeric products, although with high ee for each isomer.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Prda | R1 | R2 | Yieldb (%) | drc | ee majord (%) | ee minord (%) |
| a Time for completion for products 3a and 3h was 30 min, while 3s required 2 h, with 3p the most sluggish, necessitating 24 h. b Isolated yield on a 0.1 mmol scale. c Diastereomeric ratio determined by NMR. d Enantiomeric excess determined by chiral HPLC. e NMR yield on a 0.05 mmol scale. | |||||||
| 1 | 3a | C3H7 | H | 71 | >20:1 |
99 | na |
| 2 | 3h | H | C3H7 | 65 | >20:1 |
95 | na |
| 3 | 3p | BnOCH2 | H | 65 | >20:1 |
96 | na |
| 4 | 3s | Ph | H | 54e | 61:39 |
99 | 97 |
Reactions of chlorosulfonylamidines
The utility of the sulfonylamide product 3a was demonstrated via its cyclization to form the imidazoline 5a (Scheme 2).7d,h This product was then easily hydrolyzed to the chiral tri-amine 6a upon treatment with dilute HCl, yielding the orthogonally protected triamine product with two contiguous chiral centers. This could be of synthetic value, as there are few known methods to deliver chiral triamines,18 in addition, this allows for orthogonal protection. Also illustrated in Scheme 2, is the conversion of 1a to 7a, using dimethylcyanamide as the nucleophile, en route to the cyclic guanidine 8a.7e The enantioselectivity obtained in the asymmetric transformation is maintained in subsequent reaction for both sequences described above.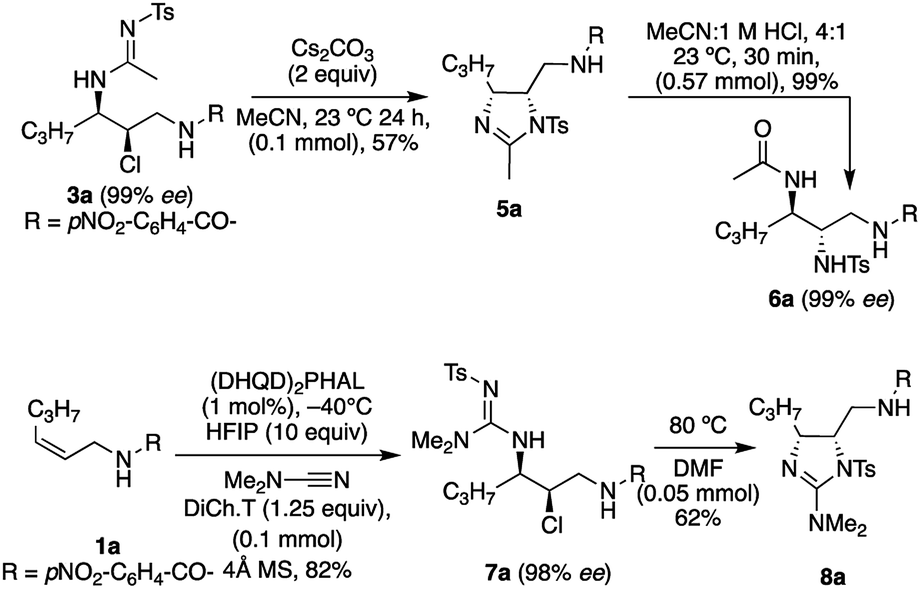 | ||
| Scheme 2 Chemical elaborations of chlorosulfonylamidines. | ||
Conclusions
In summary, we describe the catalytic asymmetric chloroamidation of allylic-amides via a chlorenium induced Ritter-type reaction. This is achieved through the use of a nucleophilically tempered source, a nitrile, that circumvents the high halonium affinity of most sp3 nitrogen atoms. The methodology was successful in producing a broad range of chloroamides in good yields and high enantioselectivity. Interestingly, the use of two different chlorenium sources, although both deliver products with high ee, proceed through intermediates that exhibit different chemical stabilities. Presumably, the intermediates are produced by the trapping of the nitrilium with either the residual hydantoin or chloramine-T, after having the chlorenium transferred. The hydantoin-trapped molecule is not as stable to isolation, and is readily hydrolyzed to the corresponding amide. The chloramine-T trapped product, on the other hand, is stable, isolable, and can be manipulated to yield new categories of products.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Generous support was provided by the NIH (GM110525).Notes and references
- (a) M. Khalid and S. Mohammed, Indian J. Heterocycl. Chem., 2018, 28, 507 CAS; (b) S. E. Denmark, W. E. Kuester and M. T. Burk, Angew. Chem., Int. Ed., 2012, 51, 10938 CrossRef CAS; (c) W. J. Chung and C. D. Vanderwal, Angew. Chem., Int. Ed., 2016, 55, 4396 CrossRef CAS PubMed; (d) Y. A. Cheng, W. Z. Yu and Y. Y. Yeung, Org. Biomol. Chem., 2014, 12, 2333 RSC; (e) J. Chen and L. Zhou, Synthesis, 2014, 46, 586 CrossRef.
- (a) K. D. Ashtekar, N. S. Marzijarani, A. Jaganathan, D. Holmes, J. E. Jackson and B. Borhan, J. Am. Chem. Soc., 2014, 136, 13355 CrossRef CAS PubMed; (b) S. E. Denmark, M. T. Burk and A. J. Hoover, J. Am. Chem. Soc., 2010, 132, 1232 CrossRef CAS PubMed.
- (a) K. C. Nicolaou, N. L. Simmons, Y. C. Ying, P. M. Heretsch and J. S. Chen, J. Am. Chem. Soc., 2011, 133, 8134 CrossRef CAS; (b) D. X. Hu, G. M. Shibuya and N. Z. Burns, J. Am. Chem. Soc., 2013, 135, 12960 CrossRef CAS PubMed; (c) D. X. Hu, F. J. Seidl, C. Bucher and N. Z. Burns, J. Am. Chem. Soc., 2015, 137, 3795 CrossRef CAS; (d) M. L. Landry, D. X. Hu, G. M. McKenna and N. Z. Burns, J. Am. Chem. Soc., 2016, 138, 5150 CrossRef CAS; (e) B. Soltanzadeh, A. Jaganathan, Y. Yi, H. Yi, R. J. Staples and B. Borhan, J. Am. Chem. Soc., 2017, 139, 2132 CrossRef CAS PubMed; (f) M. L. Landry and N. Z. Burns, Acc. Chem. Res., 2018, 51, 1260 CrossRef CAS PubMed; (g) F. Scheidt, M. Schafer, J. C. Sarie, C. G. Daniliuc, J. J. Molloy and R. Gilmour, Angew. Chem., Int. Ed., 2018, 57, 16431 CrossRef CAS PubMed; (h) B. B. Gilbert, S. T. C. Eey, P. Ryabchuk, O. Garry and S. E. Denmark, Tetrahedron, 2019, 75, 4086 CrossRef CAS; (i) M. K. Haj, S. M. Banik and E. N. Jacobsen, Org. Lett., 2019, 21, 4919 CrossRef CAS PubMed; (j) V. Wedek, R. Van Lommel, C. G. Daniliuc, F. De Proft and U. Hennecke, Angew. Chem., Int. Ed., 2019, 58, 9239 CrossRef CAS PubMed.
- (a) Q. X. Cao, J. Luo and X. D. Zhao, Angew. Chem., Int. Ed., 2019, 58, 1315 CrossRef CAS PubMed; (b) Z. M. Chen, Q. W. Zhang, Z. H. Chen, H. Li, Y. Q. Tu, F. M. Zhang and J. M. Tian, J. Am. Chem. Soc., 2011, 133, 8818 CrossRef CAS; (c) H. Li, F. M. Zhang, Y. Q. Tu, Q. W. Zhang, Z. M. Chen, Z. H. Chen and J. Li, Chem. Sci., 2011, 2, 1839 RSC; (d) Y. Liu, Y. L. S. Tse, F. Y. Kwong and Y. Y. Yeung, ACS Catal., 2017, 7, 4435 CrossRef CAS; (e) C. H. Muller, M. Wilking, A. Ruhlmann, B. Wibbeling and U. Hennecke, Synlett, 2011, 2043, DOI:10.1055/s-0030-1260983; (f) F. Romanov-Michailidis, L. Guenee and A. Alexakis, Angew. Chem., Int. Ed., 2013, 52, 9266 CrossRef CAS PubMed; (g) R. C. Samanta and H. Yamamoto, J. Am. Chem. Soc., 2017, 139, 1460 CrossRef CAS PubMed; (h) Y. Sawamura, Y. Ogura, H. Nakatsuji, A. Sakakura and K. Ishihara, Chem. Commun., 2016, 52, 6068 RSC.
- (a) G. X. Li, Q. Q. Fu, X. M. Zhang, J. Jiang and Z. Tang, Tetrahedron: Asymmetry, 2012, 23, 245 CrossRef CAS; (b) W. Zhang, N. Liu, C. M. Schienebeck, X. Zhou, I. I. Izhar, I. A. Guzei and W. P. Tang, Chem. Sci., 2013, 4, 2652 RSC; (c) Y. Zhang, H. Xing, W. Q. Xie, X. L. Wan, Y. S. Lai and D. W. Ma, Adv. Synth. Catal., 2013, 355, 68 CrossRef CAS; (d) L. J. Li, C. X. Su, X. Q. Liu, H. Tian and Y. A. Shi, Org. Lett., 2014, 16, 3728 CrossRef CAS PubMed; (e) B. Soltanzadeh, A. Jaganathan, R. J. Staples and B. Borhan, Angew. Chem., Int. Ed., 2015, 54, 9517 CrossRef CAS PubMed; (f) P. F. Zhou, Y. F. Cai, X. Zhong, W. W. Luo, T. F. Kang, J. Li, X. H. Liu, L. L. Lin and X. M. Feng, ACS Catal., 2016, 6, 7778 CrossRef CAS; (g) J. Li, Z. Q. Li, X. Zhang, B. Xu and Y. Shi, Org. Chem. Front., 2017, 4, 1084 RSC; (h) K. Murai, T. Matsushita, A. Nakamura, S. Fukushima, M. Shimura and H. Fujioka, Angew. Chem., Int. Ed., 2010, 49, 9174 CrossRef CAS PubMed; (i) G. E. Veitch and E. N. Jacobsen, Angew. Chem., Int. Ed., 2010, 49, 7332 CrossRef CAS PubMed; (j) D. C. Whitehead, R. Yousefi, A. Jaganathan and B. Borhan, J. Am. Chem. Soc., 2010, 132, 3298 CrossRef CAS PubMed; (k) W. Zhang, S. Q. Zheng, N. Liu, J. B. Werness, I. A. Guzei and W. P. Tang, J. Am. Chem. Soc., 2010, 132, 3664 CrossRef CAS PubMed; (l) L. Zhou, C. K. Tan, X. J. Jiang, F. Chen and Y. Y. Yeung, J. Am. Chem. Soc., 2010, 132, 15474 CrossRef CAS PubMed; (m) U. Hennecke, C. H. Muller and R. Frohlich, Org. Lett., 2011, 13, 860 CrossRef CAS PubMed; (n) V. Rauniyar, A. D. Lackner, G. L. Hamilton and F. D. Toste, Science, 2011, 334, 1681 CrossRef CAS PubMed; (o) M. C. Dobish and J. N. Johnston, J. Am. Chem. Soc., 2012, 134, 6068 CrossRef CAS PubMed; (p) C. Fang, D. H. Paull, J. C. Hethcox, C. R. Shugrue and S. F. Martin, Org. Lett., 2012, 14, 6290 CrossRef CAS PubMed; (q) K. Ikeuchi, S. Ido, S. Yoshimura, T. Asakawa, M. Inai, Y. Hamashima and T. Kan, Org. Lett., 2012, 14, 6016 CrossRef CAS; (r) D. H. Paull, C. Fang, J. R. Donald, A. D. Pansick and S. F. Martin, J. Am. Chem. Soc., 2012, 134, 11128 CrossRef CAS PubMed; (s) A. Armstrong, D. C. Braddock, A. X. Jones and S. Clark, Tetrahedron Lett., 2013, 54, 7004 CrossRef CAS; (t) C. H. Muller, C. Rosner and U. Hennecke, Chem.–Asian J., 2014, 9, 2162 CrossRef; (u) H. Nakatsuji, Y. Sawamura, A. Sakakura and K. Ishihara, Angew. Chem., Int. Ed., 2014, 53, 6974 CrossRef CAS PubMed; (v) T. Arai, O. Watanabe, S. Yabe and M. Yamanaka, Angew. Chem., Int. Ed., 2015, 54, 12767 CrossRef CAS PubMed; (w) Y. Kawato, A. Kubota, H. Ono, H. Egami and Y. Hamashima, Org. Lett., 2015, 17, 1244 CrossRef CAS PubMed; (x) M. Aursnes, J. E. Tungen and T. V. Hansen, J. Org. Chem., 2016, 81, 8287 CrossRef CAS PubMed; (y) S. E. Denmark, P. Ryabchuk, M. T. Burk and B. B. Gilbert, J. Org. Chem., 2016, 81, 10411 CrossRef CAS PubMed; (z) F. Gelat, M. Coffinet, S. Lebrun, F. Agbossou-Niedercorn, C. Michon and E. Deniau, Tetrahedron: Asymmetry, 2016, 27, 980 CrossRef CAS; (a a) Y. M. Cao, D. Lentz and M. Christmann, J. Am. Chem. Soc., 2018, 140, 10677 CrossRef CAS PubMed.
- (a) Y. Cai, X. Liu, Y. Hui, J. Jiang, W. Wang, W. Chen, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2010, 49, 6160 CrossRef CAS PubMed; (b) Y. Cai, X. Liu, J. Jiang, W. Chen, L. Lin and X. Feng, J. Am. Chem. Soc., 2011, 133, 5636 CrossRef CAS PubMed; (c) A. Alix, C. Lalli, P. Retailleau and G. Masson, J. Am. Chem. Soc., 2012, 134, 10389 CrossRef CAS PubMed; (d) Y. F. Cai, X. H. Liu, P. F. Zhou, Y. L. Kuang, L. L. Lin and X. M. Feng, Chem. Commun., 2013, 49, 8054 RSC; (e) J. Qi, G. T. Fan, J. Chen, M. H. Sun, Y. T. Dong and L. Zhou, Chem. Commun., 2014, 50, 13841 RSC; (f) C. Lebee, F. Blanchard and G. Masson, Synlett, 2016, 27, 559 CAS; (g) P. F. Zhou, L. L. Lin, L. Chen, X. Zhong, X. H. Liu and X. M. Feng, J. Am. Chem. Soc., 2017, 139, 13414 CrossRef CAS; (h) F. J. Seidl, C. Min, J. A. Lopez and N. Z. Burns, J. Am. Chem. Soc., 2018, 140, 15646 CrossRef CAS PubMed; (i) Y. F. Cai, X. H. Liu, P. F. Zhou and X. M. Feng, J. Org. Chem., 2019, 84, 1 CrossRef CAS PubMed; (j) P. F. Zhou, X. H. Liu, W. B. Wu, C. R. Xu and X. M. Feng, Org. Lett., 2019, 21, 1170 CrossRef CAS PubMed; (k) D. Huang, H. Wang, F. Xue, H. Guan, L. Li, X. Peng and Y. Shi, Org. Lett., 2011, 13, 6350 CrossRef CAS PubMed; (l) D. Huang, X. Liu, L. Li, Y. Cai, W. Liu and Y. Shi, J. Am. Chem. Soc., 2013, 135, 8101 CrossRef CAS PubMed; (m) L. Zhou, D. W. Tay, J. Chen, G. Y. C. Leung and Y. Y. Yeung, Chem. Commun., 2013, 49, 4412 RSC; (n) B. Egart and C. Czekelius, Chem.–Asian J., 2014, 9, 2088 CrossRef CAS PubMed; (o) P. Mizar, A. Burrelli, E. Gunther, M. Softje, U. Farooq and T. Wirth, Chem.–Eur. J., 2014, 20, 13113 CrossRef CAS PubMed; (p) C. B. Tripathi and S. Mukherjee, Org. Lett., 2014, 16, 3368 CrossRef CAS PubMed; (q) Y. A. Cheng, W. Z. Yu and Y. Y. Yeung, Angew. Chem., Int. Ed., 2015, 54, 12102 CrossRef CAS PubMed; (r) H. Huang, H. Pan, Y. Cai, M. Liu, H. Tian and Y. Shi, Org. Biomol. Chem., 2015, 13, 3566 RSC; (s) Z. Li and Y. Shi, Org. Lett., 2015, 17, 5752 CrossRef CAS PubMed; (t) W. Liu, H. Pan, H. Tian and Y. Shi, Org. Lett., 2015, 17, 3956 CrossRef CAS PubMed; (u) H. Pan, H. Huang, W. Liu, H. Tian and Y. Shi, Org. Lett., 2016, 18, 896 CrossRef CAS PubMed; (v) H. J. Jiang, K. Liu, J. Yu, L. Zhang and L. Z. Gong, Angew. Chem., Int. Ed., 2017, 56, 11931 CrossRef CAS PubMed; (w) Y. H. Lu, H. Nakatsuji, Y. Okumura, L. Yao and K. Ishihara, J. Am. Chem. Soc., 2018, 140, 6039 CrossRef CAS; (x) K. M. Mennie, S. M. Banik, E. C. Reichert and E. N. Jacobsen, J. Am. Chem. Soc., 2018, 140, 4797 CrossRef CAS; (y) P. D. Parker, B. C. Lemercier and J. G. Pierce, J. Org. Chem., 2018, 83, 12 CrossRef CAS PubMed; (z) T. J. Struble, H. M. Lankswert, M. Pink and J. N. Johnston, ACS Catal., 2018, 8, 11926 CrossRef CAS PubMed; (a a) X. Y. Tan, H. J. Pan, H. Tian and Y. Shi, Sci. China: Chem., 2018, 61, 656 CrossRef CAS.
- (a) K. K. Rajbongshi, I. Saikia, L. D. Chanu, S. Roy and P. Phukan, J. Org. Chem., 2016, 81, 5423 CrossRef CAS; (b) D. W. Tay, I. T. Tsoi, J. C. Er, G. Y. C. Leung and Y. Y. Yeung, Org. Lett., 2013, 15, 1310 CrossRef CAS PubMed; (c) J. M. Huang, Z. J. Ye, D. S. Chen and H. Zhu, Org. Biomol. Chem., 2012, 10, 3610 RSC; (d) L. Zhou, J. Zhou, C. K. Tan, J. Chen and Y. Y. Yeung, Org. Lett., 2011, 13, 2448 CrossRef CAS; (e) L. Zhou, J. Chen, J. Zhou and Y. Y. Yeung, Org. Lett., 2011, 13, 5804 CrossRef CAS; (f) Y. Y. Yeung, S. Hong and E. J. Corey, J. Am. Chem. Soc., 2006, 128, 6310 CrossRef CAS; (g) Y. Y. Yeung, X. Gao and E. J. Corey, J. Am. Chem. Soc., 2006, 128, 9644 CrossRef CAS; (h) K. I. Booker-Milburn, D. J. Guly, B. Cox and P. A. Procopiou, Org. Lett., 2003, 5, 3313 CrossRef CAS PubMed; (i) A. Hassner, L. A. Levy and R. Gault, Tetrahedron Lett., 1966, 3119 CrossRef CAS; (j) D. H. Jiang, T. He, L. Ma and Z. Y. Wang, RSC Adv., 2014, 4, 64936 RSC.
- V. Lucchini, G. Modena and L. Pasquato, J. Chem. Soc., Chem. Commun., 1994, 1565, 10.1039/c39940001565.
- L. Legnani, G. Prina-Cerai, T. Delcaillau, S. Willems and B. Morandi, Science, 2018, 362, 434 CrossRef CAS PubMed.
- S. Inuki, Y. Yoshimitsu, S. Oishi, N. Fujii and H. Ohno, J. Org. Chem., 2010, 75, 3831 CrossRef CAS PubMed.
- (a) A. Jaganathan, R. J. Staples and B. Borhan, J. Am. Chem. Soc., 2013, 135, 14806 CrossRef CAS PubMed; (b) A. Jaganathan, A. Garzan, D. C. Whitehead, R. J. Staples and B. Borhan, Angew. Chem., Int. Ed., 2011, 50, 2593 CrossRef CAS PubMed; (c) A. Garzan, A. Jaganathan, N. S. Marzijarani, R. Yousefi, D. C. Whitehead, J. E. Jackson and B. Borhan, Chem.–Eur. J., 2013, 19, 9015 CrossRef CAS PubMed.
- A. Guerinot, S. Reymond and J. Cossy, Eur. J. Org. Chem., 2012, 2012, 19 CrossRef CAS.
- A. Jaganathan and B. Borhan, Org. Lett., 2014, 16, 3616 CrossRef CAS PubMed.
- (a) A. M. Arnold, A. Pothig, M. Drees and T. Gulder, J. Am. Chem. Soc., 2018, 140, 4344 CrossRef CAS PubMed; (b) A. Roth and S. E. Denmark, J. Am. Chem. Soc., 2019, 141, 13767 CrossRef CAS PubMed.
- R. Yousefi, A. Sarkar, K. D. Ashtekar, D. C. Whitehead, T. Kakeshpour, D. Holmes, P. Reed, J. E. Jackson and B. Borhan, J. Am. Chem. Soc., 2020, 142, 7179 CrossRef CAS PubMed.
- The absolute stereochemistry of 2c and 2i were established by single crystal X-ray diffraction (their corresponding Cambridge Structural Database deposition numbers are provided following each compound name). These are: 2c (1864170) and 2i (1909607). A note regarding the absolute stereochemistries reported here as compared to our previous work for the catalytic asymmetric dihalogenation of olefins (see ref. 3e); we had observed the Ritter product as a side-product of the dihalogenation reaction when acetonitrile was used as a solvent. Although the relative stereochemistry reported in this manuscript is the same as the one reproted previously, our crystallographic analysis of products obtained here show the opposite olefin facial selectivity. The stereochemistry shown for the Ritter product in the dihalogenation study (ref. 3e) was based on the absolute stereochemistry of the dihalogenation products, which we now realize is different as compared to the Ritter medicated halofunctionalization. The source of this change in olefin facial selectivity is currently under scrutiny through mechanistic investigations.
- K. D. Ashtekar, M. Vetticatt, R. Yousefi, J. E. Jackson and B. Borhan, J. Am. Chem. Soc., 2016, 138, 8114 CrossRef CAS.
- (a) L. Lykke, B. D. Carlsen, R. S. Rambo and K. A. Jorgensen, J. Am. Chem. Soc., 2014, 136, 11296 CrossRef CAS PubMed; (b) B. M. Trost, S. Malhotra, D. E. Olson, A. Maruniak and J. Du Bois, J. Am. Chem. Soc., 2009, 131, 4190 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1864170 and 1909607. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc05224h |
| ‡ Present Address: Apeel Sciences, 71 S Los Carneros Rd., Goleta, CA 93117. |
| This journal is © The Royal Society of Chemistry 2021 |