DOI:
10.1039/D0SC05193D
(Edge Article)
Chem. Sci., 2021,
12, 3558-3567
Cerium(IV) complexes with guanidinate ligands: intense colors and anomalous electronic structures†
Received
18th September 2020
, Accepted 9th December 2020
First published on 9th December 2020
Abstract
A series of cerium(IV) mixed-ligand guanidinate–amide complexes, {[(Me3Si)2NC(NiPr)2]xCeIV[N(SiMe3)2]3−x}+ (x = 0–3), was prepared by chemical oxidation of the corresponding cerium(III) complexes, where x = 1 and 2 represent novel complexes. The Ce(IV) complexes exhibited a range of intense colors, including red, black, cyan, and green. Notably, increasing the number of the guanidinate ligands from zero to three resulted in significant redshift of the absorption bands from 503 nm (2.48 eV) to 785 nm (1.58 eV) in THF. X-ray absorption near edge structure (XANES) spectra indicated increasing f occupancy (nf) with more guanidinate ligands, and revealed the multiconfigurational ground states for all Ce(IV) complexes. Cyclic voltammetry experiments demonstrated less stabilization of the Ce(IV) oxidation state with more guanidinate ligands. Moreover, the Ce(IV) tris(guanidinate) complex exhibited temperature independent paramagnetism (TIP) arising from the small energy gap between the ground- and excited states with considerable magnetic moments. Computational analysis suggested that the origin of the low energy absorption bands was a charge transfer between guanidinate π orbitals that were close in energy to the unoccupied Ce 4f orbitals. However, the incorporation of sterically hindered guanidinate ligands inhibited optimal overlaps between Ce 5d and ligand N 2p orbitals. As a result, there was an overall decrease of ligand-to-metal donation and a less stabilized Ce(IV) oxidation state, while at the same time, more of the donated electron density ended up in the 4f shell. The results indicate that incorporating guanidinate ligands into Ce(IV) complexes gives rise to intense charge transfer bands and noteworthy electronic structures, providing insights into the stabilization of tetravalent lanthanide oxidation states.
Introduction
Open-shell lanthanide ions exhibit a range of electric or magnetic dipole transitions corresponding to the ultra-violet, visible, and near-infrared absorptions, giving rise to a variety of colors and electronic structures.1 A common transition for such lanthanide(III) ions is the 4f → 4f transition, which is parity-forbidden, sharp, and weakly intense (ε < 1 M−1 cm−1).2,3 The 4f orbitals are usually treated as “core-like” orbitals, meaning that the compact 4f shells are efficiently shielded by the filled 5s and 5p congeners. Consequently, the 4f → 4f transitions are, to a first approximation, unperturbed by crystal field splitting and the transition energies show only small differences between lanthanide(III) complexes and the idealized free ions.1,4
Unlike other lanthanides, the trivalent and tetravalent oxidation states are readily accessible in molecular cerium complexes.5 The 4f → 4f transitions for the Ce3+ ion fall in the infrared region, hence the colors (and the emission colors) of cerium(III) complexes are affected by 4f → 5d absorptions.1,5–12 The 4f electron, upon absorption of light, becomes chemically accessible through promotion to 5d orbital sets. As a result, unprecedented luminescent properties as well as photoredox catalysis involving cerium(III) complexes as photosensitizers can be achieved by judicious ligand selection.8,12–19
With respect to cerium(IV) complexes, their intense colors are primarily due to parity-allowed ligand-to-metal charge transfer (LMCT) transitions of the Ce4+ cation.20–24 The electronic structures of the cerium(IV) complexes are sensitive to the ligand environment.25 Since the electron is transferred into a metal-based orbital, the LMCT can result in the reduction of the metal cation. For example, in the presence of excess NEt4Cl, irradiation at the absorption region of [CeIVCl6]2− leads to the formation of the hexachlorocerate(III) anion ([CeIIICl6]3−).26 Taking advantage of the strong excited-state reduction power, fast electron-transfer kinetics of [CeIIICl6]2− and photoreduction of [CeIVCl6]2−, photoinduced dehalogenation and borylation of aryl chlorides by [CeIIICl6]3− have been accomplished.16,27
In addition to the reactivity of such LMCT excited states, we hypothesize that LMCT transitions can be used to achieve unprecedented electronic structures of cerium(IV) complexes. Most of the known cerium(IV) complexes display absorption bands in two major regions of the spectrum. The first one is ∼300–400 nm, where Ce[NP(pip)3]4 (pip = piperidinyl, 335 nm),23 Ce[η2-ON(tBu)(2-OMe-5-tBu-C6H3)]4 (359 nm),28 Ce(tmhd)4 (tmhd = 2,2,6,6-tetramethyl-3,5-heptanedionato, 372 nm),6,29 [NEt4]2[CeCl6] (375 nm),26,30,31 and Ce(IV) oxo complexes [Li(2,2,2-cryptand)]{O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ce[N(SiMe3)2]3} (306 nm),15 and [Rb(OCe(TriNOx))] (TriNOx3− = {(2-tBuNO)C6H4CH2}3N]3−, 380 nm),32 fall into this category. The second one is ∼500–650 nm, which has Ce{N[CH2CH2N(SiMe2tBu)]3}I (ca. 500 nm),33 CeX[N(SiMe3)2]3 (X = F, Cl, Br, I, ca. 500 nm),22 Ce(IV) imido complexes [M(solv)x][CeN(3,5-(CF3)2C6H3)(TriNOx)] (M = Li, K, Rb, Cs, solv = TMEDA, THF, Et2O, or DME, ca. 510 nm),32 Ce[2-(tBuNO)py]4 ([2-(tBuNO)py]− = N-tert-butyl-N-2-pyridylnitroxide, 524 nm),34 Ce(COT)2 (COT = cyclooctatetraenyl, 569 nm),35 and Ce(Odpp)4 (dpp = 2,6-(C6H5)2-C6H3, 624 nm)36 in this category. For the complexes in the second category, noteworthy electronic structures have been observed. For example, the LMCT band at 569 nm of Ce[2-(tBuNO)py]4 indicates mixing of vacant Ce 4f and 5d orbitals with filled ligand-based orbitals, that provides a strong stabilization of the CeIV oxidation state in this complex.34 Moreover, Ce(COT)2 has a multiconfigurational ground-state and displays temperature-independent paramagnetism.37,38 We stress that it has been shown that the degree of multiconfigurational character determined from a calculation depends on rotations of the natural orbitals in the active space without any effect on the calculated energy.39 This was used to indicate partial equivalence39 between multiconfigurational character and single configurational character with significant covalence in Ce(COT)2,40 although in that system a purely single configurational calculation was not obtainable. With that fact in mind, metal–ligand covalency in the cerium(IV) imido complexes is more significant than that of the thorium(IV) analogs.32,41,42 In terms of other lanthanide compounds with low energy LMCT bands, Cp*2Yb(bipy) (Cp* = pentamethylcyclopentadienyl, bipy = 2,2′-bipyridine), which has a low-lying LMCT band at ca. 2105 nm, shows significant metal–ligand orbital overlap and strong exchange coupling (2J = −0.11 eV).43 A molecular Tb(IV) compound, Tb(NP(1,2-bis-tBu-diamidoethane)(NEt2))4, with a LMCT band at 575 nm demonstrates a small HOMO–LUMO gap (1.28 eV), covalent metal–ligand bonds, and a multiconfigurational ground state.44 Remarkable lanthanide-ligand orbital mixing has also been spectroscopically determined in tetravalent lanthanide oxides, LnO2 (Ln = Ce, Pr, and Tb),45 and Pr(IV) and Tb(IV) siloxide compounds.46–48 Inspired by these findings, we are interested in developing and studying cerium(IV) complexes that show red-shifted absorption bands (>650 nm). We postulate that cerium(IV) complexes with such low energy LMCT bands would have strong metal–ligand interactions, leading to unusual electronic structures. Herein, we report that applying the guanidinate ligands to the Ce(IV) cation gives rise to intense colors and anomalous electronic structures, characteristic of 4f and 5d covalency. These results promote the understanding of bonding and electronic structures for lanthanide complexes and inform design strategies for manifesting and exploring multi-configurational electronic structures.
Ce[N(SiMe3)2]3} (306 nm),15 and [Rb(OCe(TriNOx))] (TriNOx3− = {(2-tBuNO)C6H4CH2}3N]3−, 380 nm),32 fall into this category. The second one is ∼500–650 nm, which has Ce{N[CH2CH2N(SiMe2tBu)]3}I (ca. 500 nm),33 CeX[N(SiMe3)2]3 (X = F, Cl, Br, I, ca. 500 nm),22 Ce(IV) imido complexes [M(solv)x][CeN(3,5-(CF3)2C6H3)(TriNOx)] (M = Li, K, Rb, Cs, solv = TMEDA, THF, Et2O, or DME, ca. 510 nm),32 Ce[2-(tBuNO)py]4 ([2-(tBuNO)py]− = N-tert-butyl-N-2-pyridylnitroxide, 524 nm),34 Ce(COT)2 (COT = cyclooctatetraenyl, 569 nm),35 and Ce(Odpp)4 (dpp = 2,6-(C6H5)2-C6H3, 624 nm)36 in this category. For the complexes in the second category, noteworthy electronic structures have been observed. For example, the LMCT band at 569 nm of Ce[2-(tBuNO)py]4 indicates mixing of vacant Ce 4f and 5d orbitals with filled ligand-based orbitals, that provides a strong stabilization of the CeIV oxidation state in this complex.34 Moreover, Ce(COT)2 has a multiconfigurational ground-state and displays temperature-independent paramagnetism.37,38 We stress that it has been shown that the degree of multiconfigurational character determined from a calculation depends on rotations of the natural orbitals in the active space without any effect on the calculated energy.39 This was used to indicate partial equivalence39 between multiconfigurational character and single configurational character with significant covalence in Ce(COT)2,40 although in that system a purely single configurational calculation was not obtainable. With that fact in mind, metal–ligand covalency in the cerium(IV) imido complexes is more significant than that of the thorium(IV) analogs.32,41,42 In terms of other lanthanide compounds with low energy LMCT bands, Cp*2Yb(bipy) (Cp* = pentamethylcyclopentadienyl, bipy = 2,2′-bipyridine), which has a low-lying LMCT band at ca. 2105 nm, shows significant metal–ligand orbital overlap and strong exchange coupling (2J = −0.11 eV).43 A molecular Tb(IV) compound, Tb(NP(1,2-bis-tBu-diamidoethane)(NEt2))4, with a LMCT band at 575 nm demonstrates a small HOMO–LUMO gap (1.28 eV), covalent metal–ligand bonds, and a multiconfigurational ground state.44 Remarkable lanthanide-ligand orbital mixing has also been spectroscopically determined in tetravalent lanthanide oxides, LnO2 (Ln = Ce, Pr, and Tb),45 and Pr(IV) and Tb(IV) siloxide compounds.46–48 Inspired by these findings, we are interested in developing and studying cerium(IV) complexes that show red-shifted absorption bands (>650 nm). We postulate that cerium(IV) complexes with such low energy LMCT bands would have strong metal–ligand interactions, leading to unusual electronic structures. Herein, we report that applying the guanidinate ligands to the Ce(IV) cation gives rise to intense colors and anomalous electronic structures, characteristic of 4f and 5d covalency. These results promote the understanding of bonding and electronic structures for lanthanide complexes and inform design strategies for manifesting and exploring multi-configurational electronic structures.
Results and discussion
Syntheses and structures
In previous studies from our group, a complete series of mixed-ligand guanidinate amide Ce(III) complexes (1–4, Scheme 1) were prepared and their photochemical properties were demonstrated.12–14 These Ce(III) complexes were all light yellow in color arising from f → d transitions. We observed that the tris(guanidinate) Ce(IV) complex, {[(Me3Si)2NC(NiPr)2]3CeIV}[BArF4] ([4+][BArF4], ArF = 3,5-bis(trifluoromethyl)phenyl) exhibited an intense and unusual dark green color.14 In order to expand the scope of cerium(IV) guanidinate complexes and compare their electronic structures, we set out to prepare the corresponding bis- and mono(guanidinate) cerium(IV) complexes. Note that 2-Cl− and [3+][BArF4] are new complexes with full characterization available in the ESI.†
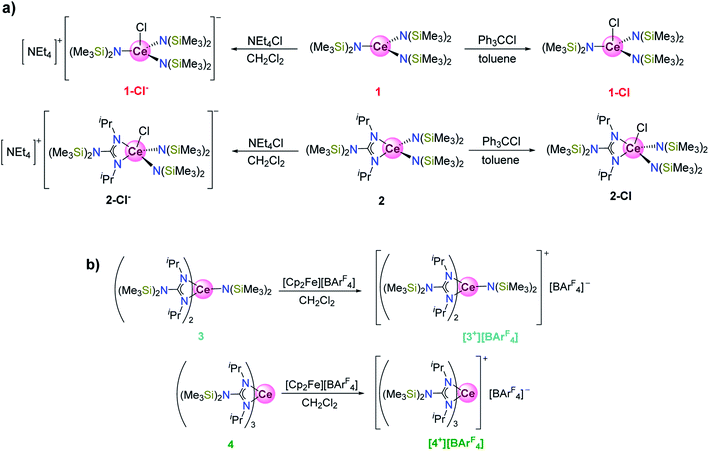 |
| | Scheme 1 (a) Syntheses of 1-Cl, 1-Cl−, 2-Cl, and 2-Cl−. (b) Syntheses of [3+][BArF4] and [4+][BArF4]. The colors for each of the compound labels were selected accurately represent their actual colors (see Fig. 2 inset, vide infra). | |
Treatment of the bis(guanidinate) cerium(III) complex [(Me3Si)2NC(NiPr)2]2CeIII[N(SiMe3)2] (3) with [Cp2Fe][BArF4] in CH2Cl2 followed by recrystallization from CH2Cl2/toluene layering, afforded dark cyan crystals of complex {[(Me3Si)2NC(NiPr)2]2Ce[N(SiMe3)2]}[BArF4] ([3+][BArF4]) in 51% yield. Single crystal X-ray diffraction confirmed the bis(guanidinate) structural motif for [3+][BArF4] (Fig. 1). The closed shell nature of complex [3+][BArF4] was evident from 1H NMR, 13C NMR, and 19F NMR spectra collected in CD2Cl2 (Fig. S1–S3†). In contrast, our attempts to prepare the corresponding cationic cerium(IV) complex of 1 and 2 using oxidants with non-coordinating anions (e.g., [Cp2Fe][BArF4]) all resulted in intractable mixtures. Therefore, the previously reported CeIV–Cl complexes, CeIVCl[N(SiMe3)2]3 (1-Cl)22,49 and [(Me3Si)2NC(NiPr)2]CeIVCl[N(SiMe3)2]2 (2-Cl)13 were used for comparison in this study. Complexes 1-Cl and 2-Cl were prepared by oxidation of 1 and 2 with Ph3CCl in 80% and 72% yields, respectively. The corresponding one electron reduced products of 1-Cl and 2-Cl, 1-Cl− (ref. 14) and 2-Cl−, respectively, were also prepared by reacting 1 or 2 with NEt4Cl in dichloromethane. Attmepts to synthesize 3-Cl− and 4-Cl− by reacting 3 and 4 with NEt4Cl in CH2Cl2, respectively were unsuccessful as evidenced by 1H NMR spectroscopy in CD2Cl2. The average Ce–Cl bond length of 2-Cl− (2.808(3) Å) was longer than that of 1-Cl− (2.7611(9) Å), suggesting more steric bulk around the Ce3+ cation in 2-Cl− than that in 1-Cl− due to only one guanidinate ligand in the coordination sphere. However, when we attempted to synthesize the corresponding CeIV–Cl complexes for 3 and 4, no reaction was observed when 3 and 4 were treated with Ph3CCl. This result can be attributed to the large steric bulk of ligands around the Ce(III) cation in the bis- and tris(guanidinate) complexes (3 and 4), that prevent oxidation of the Ce(III) cation by an inner-sphere electron transfer pathway.14 Even using a less sterically demanding inner-sphere oxidant, C2Cl6, resulted in no color change when added to 3 and 4 where only starting material was observed by 1H NMR spectroscopy in C6D6.
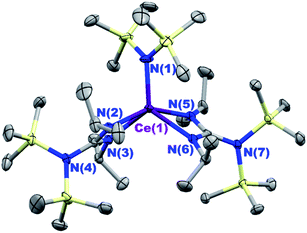 |
| | Fig. 1 Thermal ellipsoid plot of {[(Me3Si)2NC(NiPr)2]2CeIV[N(SiMe3)2]}[BArF4] ([3+][BArF4]) at the 30% probability level. The [BArF4]− anion was omitted for clarity. Selected bond length (Å) and angles (deg): Ce(1)–N(1) 2.175(5), Ce(1)–N(2) 2.377(4), Ce(1)–N(3) 2.361(3), Ce(1)–N(5) 2.391(4), Ce(1)–N(6) 2.367(3); N(2)–Ce(1)–N(3) 56.5(1), N(5)–Ce(1)–N(6) 56.3(1). | |
Electronic absorption spectra
We noted that complexes 2-Cl, [3+][BArF4], and [4+][BArF4] that had one, two, and three guanidinate ligands, respectively, demonstrated distinctive colors compared to 1-Cl. Whereas 1-Cl was red in a THF solution, black, cyan and green colors were observed for complexes 2-Cl, [3+][BArF4], and [4+][BArF4], respectively. Such differences in colors are reflected in the electronic absorption spectra of these complexes in THF solutions (Fig. 2). All absorption spectra were fit to a minimum number of overlapping Gaussian bands to reveal the lowest energy absorption features (Fig. S9†). The lowest energy absorption bands with extinction coefficients of 103 M−1 cm−1 were found at 667 nm (1.86 eV), 685 nm (1.81 eV) and 785 nm (1.58 eV) for complexes 2-Cl, [3+][BArF4] and [4+][BArF4], respectively. These absorption bands were significantly red-shifted compared to those of 1-Cl (503 nm, 2.48 eV) and other reported cerium(IV) complexes (Table 1), such as Ce(COT)2 (569 nm, 2.18 eV),35 Ce(NiPr2)4 (ca. 570 nm, 2.20 eV),50 CeI[N(SiMe3)2]3 (529 nm, 2.34 eV),22 Ce[2-(tBuNO)py]4 (524 nm, 2.37 eV),34 [Li(TMEDA)][CeN(3,5-(CF3)2C6H3)(TriNOx)] (519 nm, 2.38 eV),32 Ce{N[CH2CH2N(SiMe2tBu)]3}I (ca. 500 nm, 2.50 eV),33 Ce[N(SiHMe2)2]4 (ca. 440 nm, 2.80 eV),36 and Ce[NP(pip)3]4 (335 nm, 3.70 eV).23
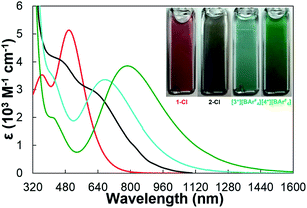 |
| | Fig. 2 Electronic absorption spectra of 1-Cl (red), 2-Cl (black), [3+][BArF4] (cyan), and [4+][BArF4] (green). Inset: pictures of 0.10 M THF solutions of 1-Cl, 2-Cl, [3+][BArF4], and [4+][BArF4] in 1.0 mm pathlength quartz cuvettes. | |
Table 1 Comparison of energies of the lowest energy absorption bands for reported cerium(IV) complexes and 1-Cl, 2-Cl, [3+][BArF4], and [4+][BArF4]
| Complex |
E (eV) |
| Ce[NP(pip)3]4 |
3.70 |
| Ce[N(SiHMe2)2]4 |
2.80 |
| Ce{N[CH2CH2N(SiMe2tBu)]3}I |
2.50 |
|
1-Cl
|
2.48 |
| [Li(TMEDA)][CeN(3,5-(CF3)2C6H3)(TriNOx)] |
2.38 |
| Ce[2-(tBuNO)py]4 |
2.37 |
| CeI[N(SiMe3)2]3 |
2.34 |
| Ce(NiPr2)4 |
2.20 |
| Ce(COT)2 |
2.18 |
|
2-Cl
|
1.86 |
|
[3+][BArF4]
|
1.81 |
|
[4+][BArF4]
|
1.58 |
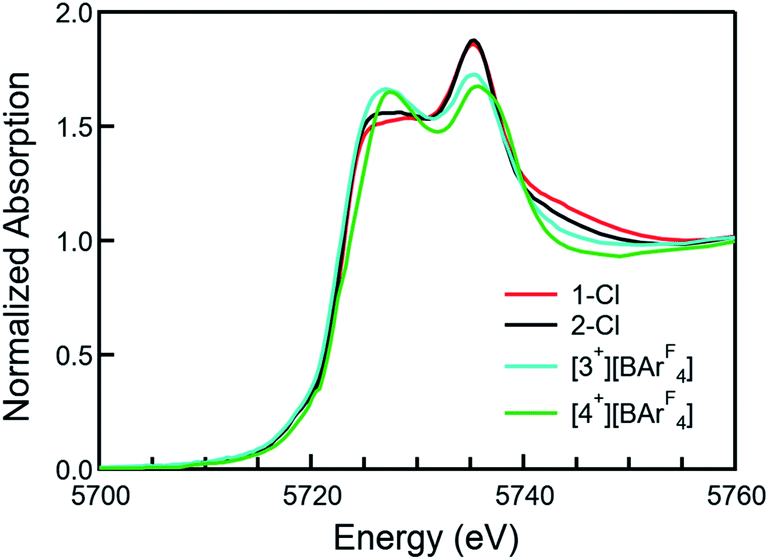
XANES spectra
XANES (X-ray absorption near edge structure) spectra were collected to confirm the Ce(IV) oxidation states of 1-Cl, 2-Cl, [3+][BArF4], and [4+][BArF4] (Fig. 3, see the ESI† for details). All the Ce L3-edge spectra exhibited two peaks, one at ca. 5726 eV and another one at ca. 5736 eV, corresponding to the core-hole excitations from a Ce(IV) cation to the final states, 2![[p with combining low line]](https://www.rsc.org/images/entities/char_0070_0332.gif) 4f1
4f1![[L with combining low line]](https://www.rsc.org/images/entities/char_004c_0332.gif) 5d1 and 24f05d1, respectively, in which indicates a ligand hole. No temperature dependence was observed in the XANES spectra for any of the complexes. Given that Ce is in a very similar crystallographic site for each compound, these data indicate that all complexes are formal Ce(IV) compounds with significant multiconfigurational character, possibly due to some form of covalence.51–53 The overall f occupancy (nf) was determined from the relative area of the lower energy XANES peak to the total weight of the doublet peak. Fitting details are included in the SI. The nf values from the 1-Cl, 2-Cl, [3+][BArF4], and [4+][BArF4] data are 0.51(3), 0.52(3), 0.57(3), and 0.57(3), respectively, which are comparable to the values of Ce(trop)4 (0.50), Ce(acac)4 (0.51), CeO2 (0.58), and Ce(tmtaa)2 (0.59), but larger than that of Ce[NP(pip)3]4 (0.21) and smaller than that of Ce(COT)2 (0.82).23,44,52,53 Moreover, compounds with lower energy absorption bands tend to show larger nf values (Fig. S18†).
5d1 and 24f05d1, respectively, in which indicates a ligand hole. No temperature dependence was observed in the XANES spectra for any of the complexes. Given that Ce is in a very similar crystallographic site for each compound, these data indicate that all complexes are formal Ce(IV) compounds with significant multiconfigurational character, possibly due to some form of covalence.51–53 The overall f occupancy (nf) was determined from the relative area of the lower energy XANES peak to the total weight of the doublet peak. Fitting details are included in the SI. The nf values from the 1-Cl, 2-Cl, [3+][BArF4], and [4+][BArF4] data are 0.51(3), 0.52(3), 0.57(3), and 0.57(3), respectively, which are comparable to the values of Ce(trop)4 (0.50), Ce(acac)4 (0.51), CeO2 (0.58), and Ce(tmtaa)2 (0.59), but larger than that of Ce[NP(pip)3]4 (0.21) and smaller than that of Ce(COT)2 (0.82).23,44,52,53 Moreover, compounds with lower energy absorption bands tend to show larger nf values (Fig. S18†).
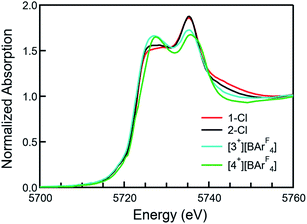 |
| | Fig. 3 Normalized X-ray absorption spectra at the Ce L3 absorption edge of 1-Cl (red), 2-Cl (black), [3+][BArF4] (cyan), and [4+][BArF4] (green). | |
Electrochemistry
Cyclic voltammetry was performed on 1-Cl, 2-Cl, [3+][BArF4], and [4+][BArF4] at room temperature in THF to evaluate the relative stability of the formal Ce(IV) oxidation states. Complexes 1-Cl, 2-Cl, [3+][BArF4], and [4+][BArF4] exhibited quasi-reversible Ce(IV/III) redox waves at −0.30 V,54 −0.46 V,13 −0.08 V, and +0.01 V versus Cp2Fe0/+, respectively (see the SI). The data indicate that 2-Cl has the most stabilized Ce(IV) oxidation state across the series, whereas the tris(guanidinate) ligand framework of [4+][BArF4] provides the least stabilization of the Ce(IV) oxidation state. Previously our group conducted density functional theory (DFT) calculations to predict the reduction potentials of organic molecules55,56 and cerium(III/IV) complexes.57,58 Here we applied a computational-time-saving DFT method, the LUMO energy correlation method of molecular Ce(IV) complexes,58 to predict the reduction potentials of 1-Cl, 2-Cl, [3+][BArF4], and [4+][BArF4] (Fig. 4). The LUMO energy correlation method was previously applied to distinguish multiconfigurational Ce(IV) porphyrinate and phthalocyanate complexes (Fig. 4, shown as green circles).58,59 The geometries of 1-Cl, 2-Cl, [3+][BArF4], and [4+][BArF4] were optimized by DFT calculations in dichloromethane solvent continuum field, and the experimental data were normalized using a method described previously (see the ESI†).58 When including these complexes into the previous correlation, an accurate prediction of their redox potentials was obtained (Fig. 4). Therefore, unlike the Ce(IV) porphyrinate and phthalocyanate complexes, the Ce(IV) guanidinate-amide complexes were successfully fit into the LUMO energy correlation. The LUMO energy successfully captured the stabilizing effects from the multiconfigurational ground states of the Ce(IV) guanidinate–amide complexes, and the stabilizing effects from any multiconfigurational ground states were less significant than the apparent stabilization of the Ce(IV) porphyrinate and phthalocyanate complexes, which also exhibited two peaks in their Ce L3-edge XANES spectra.
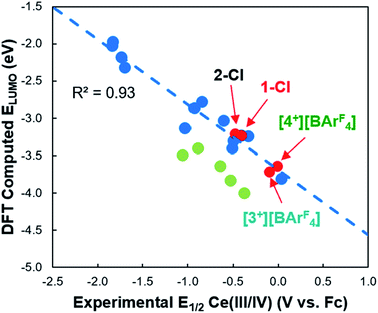 |
| | Fig. 4 Correlation of calculated LUMO energy versus experimental E1/2 of 1-Cl, 2-Cl, [3+][BArF4], and [4+][BArF4] (red circles). The previous LUMO energy correlation (blue dashed line) was performed on a series of Ce(IV) complexes with a range of ligand fields and coordination geometries (blue circles).58 Data for multiconfigurational Ce(IV) porphyrinate and phthalocyanate complexes are shown as light green circles. | |
Magnetism
Van Vleck temperature-independent paramagnetism (TIP) has been observed for molecular Ce(IV) complexes, where the physical basis for the observed TIP has been the small energy difference between the open-shell singlet ground state and the low-lying triplet first excited state.37,38,52,53,60 Magnetometry studies were carried out on [4+][BArF4] as an example. The corresponding Ce(III) tris(guanidinate) complex 4 was measured for comparison.
The χT products were measured from 300 to 2 K at an applied field of 0.5 T for 4 and [4+][BArF4] (Fig. 5). At room temperature, the χT value of 4 was 0.68 emu K mol−1, consistent with a Ce(III) cation in a 2F5/2 ground state (Fig. 5, left).38,61–64 Upon cooling, the χT product decreased steadily to 0.33 emu K mol−1 at 10 K. This decrease in moments at lower temperature was attributed to the depopulation of crystal-field energy levels created by perturbations of the J = 5/2 manifold. Complex [4+][BArF4] showed a different magnetic susceptibility response, where a linear decrease from high (300 K) to low temperature (∼50 K) in the χT product was observed. After removing diamagnetic contribution using Pascal's constants,65 the paramagnetic contribution from a residual impurity of 4 (1.2%, J = 5/2) was removed through least squares fitting over a temperature range of 50–300 K (see the ESI† for details). The resulting susceptibility plots for [4+][BArF4] are shown in Fig. 5, right, affording χTIP = 3.2(3) × 10−4 emu mol−1. The resulting χTIP value of [4+][BArF4] was similar in magnitude to those of Ce(COT)2 (χTIP = 1.4(2) × 10−4 emu mol−1),53 Ce(acac)4 (acac = acetylacetonate, χTIP = 2.1(2) × 10−4 emu mol−1),52 Ce(tmtaa)2 (tmtaaH2 = tetramethyldibenzotetraaza[14]annulene, χTIP = 2.33(6) × 10−4 emu mol−1),52 and Ce[(1,4-SiiPr3)2C8H4]2 (χTIP = 4.5(3) × 10−4 emu mol−1).60
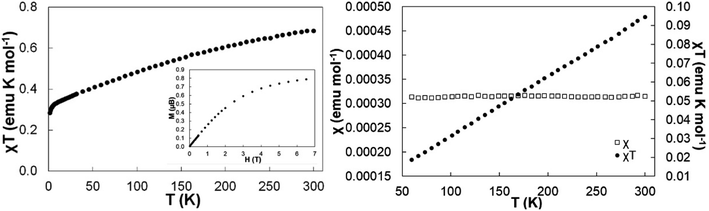 |
| | Fig. 5 (Left) Temperature-dependent magnetic data with a 0.5 T applied magnetic field for 4 and (inset) field-dependent magnetization at 2 K. (Right) Temperature-independent magnetic data with 0.5 T applied magnetic field for [4+][BArF4]. A fit is provided for χ versus T plot, giving χTIP = 3.2(3) × 10−4 emu mol−1. | |
Overall, our experimental data confirm the Ce(IV) oxidation states and elucidate structure–property relationships for the Ce(IV) guanidinate-amide complexes. Compounds with more guanidinate ligands tend to show larger steric encumbrance about the Ce(IV) cations, lower energy absorption bands, larger nf values, and more positive Ce(IV/III) reduction potentials. We next carried out quantum mechanical calculations to understand the role of guanidinate ligands further, especially the impact on LMCT transitions, 4f and 5d covalency, and stabilization of the Ce(IV) oxidation states.
Computational analysis
To understand the origin of the low energy absorption bands in these Ce(IV) guanidinate complexes, time-dependent density functional theory (TD-DFT) calculations were performed for 1-Cl, 2-Cl, [3+], and [4+] (see the ESI† for details). There was good agreement between the geometrical parameters in the DFT optimized and X-ray structures (<0.05 Å). Moreover, the absorption spectra predicted by TD-DFT calculations were in good agreement with experimental data (Fig. 6). The predicted vertical excitations corresponding to the lowest energy absorption bands for 2-Cl, [3+], and [4+], with reasonably large oscillator strengths, were ligand-to-metal charge transfer (LMCT) transitions6,20–22,30,34–36,54,66–69 from guanidinate π orbitals70,71 to cerium 4f orbitals in each case. For example, the major orbital contributions of the vertical excitation at 1.63 eV for [4+] were illustrated by TD-DFT calculations as HOMO−2 → LUMO+4 (13%), HOMO → LUMO (19%), and HOMO → LUMO+6 (48%). The computed HOMO−2 to HOMO for [4+] were primarily guanidinate π orbitals (Fig. 7 and S62–S64†). The major orbital contributions (i.e., reasonably large oscillator strengths) of the lowest energy vertical excitation for 2-Cl, [3+], and [4+] are listed in Tables S15–S17.† On the other hand, the lowest energy transition at 2.21 eV for 1-Cl was found to be mainly from amide p orbitals to cerium 4f orbitals (Table S14†).
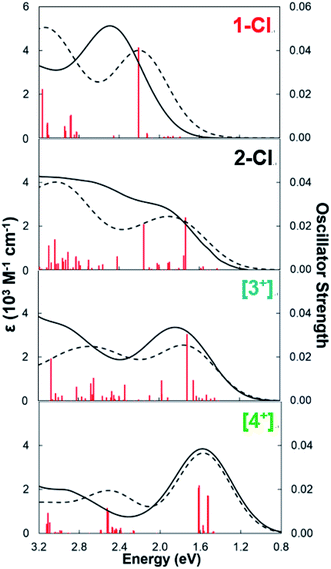 |
| | Fig. 6 Experimental (black solid line) and TD-DFT predicted (black dashed line) electronic absorption spectra of 1-Cl, 2-Cl, [3+] and [4+]. The predicted spectra are rendered with a fwhm of 5000 cm−1. Oscillator strengths for the electronic transitions are shown as red vertical lines. | |
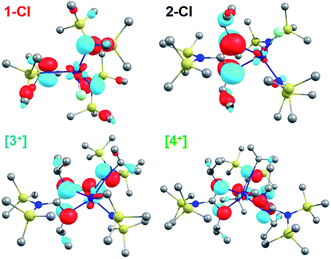 |
| | Fig. 7 Calculated HOMOs for 1-Cl, 2-Cl, [3+], and [4+]. Significant contributions from π orbitals of guanidinate ligands were evident for 2-Cl, [3+], and [4+]. | |
The presence of low energy absorption bands in CeIV guanidinate complexes was also evident from the computed HOMO–LUMO gaps. Computed energy gaps of 2.42, 2.36 and 2.20 eV were found between the HOMO and LUMO orbitals for 2-Cl, [3+], and [4+]. These HOMO–LUMO gaps were smaller than that of 1-Cl at 3.07 eV. While LUMO orbitals had primarily cerium 4f character (>89%) and only minor 5d character (<2%) in these CeIV complexes (Table 2), the atomic orbital contributions to the HOMOs were distinctly different between 1-Cl and other CeIV complexes with guanidinate ligands (Fig. 7). The computed HOMO of 1-Cl was found to be mainly composed of amide p character (Table S18†). In contrast, significant AO contributions from guanidinate p character were identified for the computed HOMOs of 2-Cl, [3+], and [4+] (Table 2). An increase in guanidinate AO character of HOMOs through 2-Cl, [3+], to [4+] was observed. Only minor Ce 4f (<6%) and 5d (<4%) AO character was found in HOMO (Table S18†). Hence, the unusual colors and low-lying absorption bands of CeIV guanidinate complexes are attributed to the presence of guanidinate π orbitals which are ∼2 eV below the empty cerium 4f orbitals.
Table 2 % contribution of guanidinate p AO character in HOMO and Ce 4f AO character in LUMO of 1-Cl, 2-Cl, [3+], and [4+]
| Complex |
1-Cl
|
2-Cl
|
[3+]
|
[4+]
|
| Guanidinate p in HOMO |
— |
82.61 |
85.26 |
89.28 |
| Ce 4f in LUMO |
92.84 |
93.57 |
90.76 |
89.83 |
The computed HOMO of [4+] also indicated the correct symmetry for π-bonding interactions between the linear combination of guanidinate π orbitals and fy(3x2−y2) atomic orbital of Ce (Fig. 8). To illustrate possible bonding interactions between the ligands and cerium, we depicted a schematic molecular orbital diagram of [4+] with an ideal C3v local symmetry. Besides the HOMO with a2 symmetry resulting from interaction between the ligand π orbitals and the cerium 4f orbital, the computed degenerate HOMO−1 and HOMO−2 with e symmetry correspond to π interactions between ligand orbitals and cerium fxyz and fz(x2−y2) atomic orbital, respectively. As a result, we postulated that despite the small overlap of the core-like 4f orbitals with guanidinate π orbitals, these orbital arrangements with matched symmetries provided stabilization to the Ce(IV) oxidation state in complex [4+][BArF4].
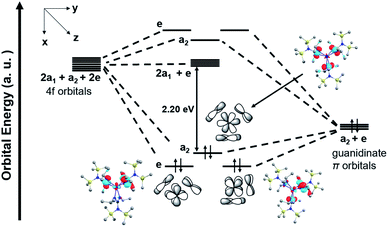 |
| | Fig. 8 Schematic molecular orbital diagram of [4+] with ideal C3v local symmetry and computed a2 and e MOs. The HOMO–LUMO energy gap is 2.20 eV. | |
An assessment of the covalency of cerium–ligand bonds can be made based on the electron populations of the natural atomic orbitals (NAOs), which are listed in Table 3, along with the charges of the Ce centers determined by the natural bond orbital (NBO) analyses. The extent of covalency is another indicator of the degree of LMCT in the ground state wavefunctions. The data in Table 3 showed that the Ce charge increased from 1-Cl to [4+] no matter which computational method was used. [4+] with the most positive reduction potential had the largest Ce charge. The 5d population, calculated by all methods, consistently decreased from 1-Cl to [4+]. However, the 4f population, calculated by DFT methods, increased from 1-Cl (0.91 based on PBE-DFT) to [4+] (0.94 based on PBE-DFT). The trend was opposite to that calculated by complete active space self-consistent field (CASSCF) method. The CASSCF calculations are affected by limitations of the active spaces, due to the sheer sizes of the systems, in particular [3+] and [4+]. The 4f populations obtained from the hybrid B3LYP-DFT and, especially, the non-hybrid Kohn–Sham (KS)-DFT (PBE-DFT) were significantly higher. The 4f populations obtained with KS-DFT and non-hybrid functionals such as PBE were shown previously to be somewhat inflated by the KS delocalization error.72 However, the static correlation error73 (i.e., the error that arises from treating multi-configurational states) was numerically probed to be the smallest with non-hybrid functionals.72,74 Therefore, the PBE calculations would seem to be a good compromise for describing the complexes. Moreover, the PBE calculations reproduce the trend in the spectroscopic nf values (Fig. S73†). Since the 4f population increased to a lesser extent compared to the decrease of 5d population, there is an overall decrease of the ligand-to-metal electron donation and increase of the Ce charge from 1-Cl to [4+].
Table 3 4f and 5d electron populations and charges of the Ce metal center, from NBO analyses based on wavefunction (CASSCF) and density functional calculations (B3LYP and PBE). Small populations of more diffuse NBOs centered on Ce are not listed
| Complex |
1-Cl
|
2-Cl
|
[3+]
|
[4+]
|
| CASSCF |
Ce charge |
2.34 |
2.37 |
2.65 |
2.78 |
| 4f |
0.41 |
0.40 |
0.36 |
0.34 |
| 5d |
1.03 |
1.00 |
0.83 |
0.73 |
| B3LYP |
Ce charge |
2.18 |
2.26 |
2.44 |
2.51 |
| 4f |
0.71 |
0.69 |
0.72 |
0.74 |
| 5d |
0.84 |
0.80 |
0.61 |
0.53 |
| PBE |
Ce charge |
1.98 |
2.05 |
2.25 |
2.31 |
| 4f |
0.91 |
0.90 |
0.93 |
0.94 |
| 5d |
0.84 |
0.79 |
0.60 |
0.53 |
To further understand the differences in 4f and 5d population and metal–ligand covalency in the Ce(IV) guanidinate complexes, 2-Cl, [3+], and [4+], natural localized molecular orbitals (NLMOs)75 were obtained from the NBO analyses. Fig. 9 shows the NLMOs for different types of Ce–N bonds in 2-Cl. 2-Cl (Fig. 9a) was used as an example to establish the bonding model, as it demonstrated the most stabilized Ce(IV) oxidation state. Only the PBE-DFT method was used here because it afforded results consistent with the experimental data. Notably, different bonding models for Ce–amide bonds and Ce–guanidinate bonds were illustrated (Fig. 9b–f). Each amide ligand interacted with the Ce atom through one Ce–N σ bond (Fig. 9b) and one Ce–N π bond (Fig. 9c). However, the two N atoms, N(3) and N(4), of the guanidinate ligand interacted with the Ce atom unequally, resulted from the steric congestion of the guanidinate ligand. The bond distance of Ce–N(3) was slightly shorter than that of Ce–N(4) by 0.03 Å. The shorter Ce–N(3) bond had one Ce–N σ bond (Fig. 9d) and one Ce–N–C π bond (Fig. 9e), while the Ce–N(4) bond had only σ bond weight (Fig. 9f). Similar bonding situations were observed for [3+] and [4+]. Moreover, the NLMOs indicated that the Ce–N–C π bond for the guanidinate ligand had larger Ce 4f weight (84%) than that of the Ce–N π bond formed for the amide ligand (66%). The larger Ce 4f weight was consistent with a better energy match between Ce 4f orbitals and guanidinate π orbitals than that between Ce 4f orbitals and amide nitrogen p orbitals. However, the Ce–N–C π bond had the least Ce 5d character (16%) among all bonding types. The Ce–N bond distances for the guanidinate ligand were about 0.2 Å longer than that for the amide ligand. Therefore, increasing the number of sterically hindered guanidinate ligands from 2-Cl to [4+] resulted in less spatial overlap between Ce 5d orbitals and ligand π orbitals. Overall, the consecutive replacements of the amide ligand by the rigid guanidinate ligand decrease the ligand donation into the metal 5d shell, due to less optimal overlap when going from 2-Cl to [4+], while simultaneously increasing the 4f participation in the metal–ligand bonding orbitals. Related scenarios have been reported for Ce(IV)/Th(IV) imido complexes,42 lanthanide dioxides and sesquioxides,45,76 [LnCl6]x− (x = 2, 3; Ln = CeIV, CeIII, NdIII, SmIII, EuIII, GdIII),30 and [AnCl6]2− (An(IV) = Th, U, Np, Pu).77 The NLMOs show one Ce–Cl σ bond and two Ce–Cl π bonds, which have similar bonding characteristics to the computed Ce-amide bonds. This is also reflected on the positive charges of 1-Cl and 2-Cl compared to [3+] and [4+] for the Ce atom (Table 3).
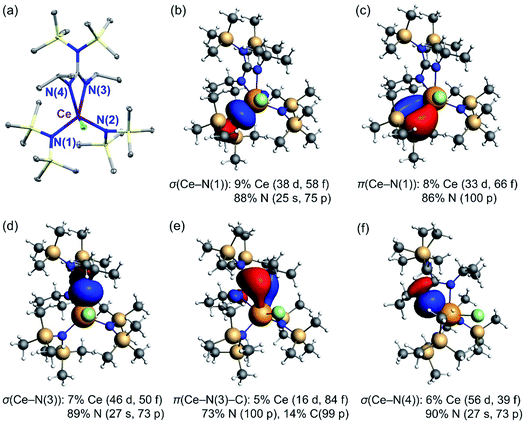 |
| | Fig. 9 Crystal structure and Ce–N natural localized molecular orbitals (NLMOs) (±0.03 a.u. isosurfaces) and weight-% of atomic shell contributions to the NLMOs of 2-Cl. (a) Thermal ellipsoid plot of 2-Cl at the 30% probability level.13 Selected bond lengths (Å): Ce(1)–N(1) 2.384(4), Ce(1)–N(2) 2.422(4), Ce(1)–N(3) 2.529(3), Ce(1)–N(4) 2.545(4). (b) σ(Ce–N(1)) for the amide ligand. (c) π(Ce–N(1)) for the amide ligand. (d) σ(Ce–N(3)) for the guanidinate ligand. (e) π(Ce–N(3)–C) for the guanidinate ligand. (f) σ(Ce–N(4)) for the guanidinate ligand. | |
Overall, our computational analyses elucidate the impacts of guanidinate ligand frameworks on the experimental observables. First, the nature of unusual colors and low-lying absorption bands for Ce(IV) guanidinate complexes, 2-Cl, [3+][BArF4], and [4+][BArF4], is ligand-to-metal charge transfer (LMCT) transitions from guanidinate π orbitals to the cerium 4f shell. Moreover, a better energy match between the guanidinate π orbitals and the cerium 4f orbitals increases the participation of Ce 4f orbitals in the metal–ligand bonds, consistent with the increase of nf values extracted from the Ce L3-edge XANES spectra. Incorporation of sterically bulky guanidinate ligands decreases the overlap between cerium 5d orbitals and ligand π orbitals, resulting in a reduction of overall ligand-to-metal electron donation and thereby larger Ce charge, more positive Ce(IV/III) reduction potential, and less stabilized Ce(IV) oxidation state. Similarly, tuning the Ce(IV/III) reduction potentials for Ce(III) bis(guanidinate) mono(amide) photocatalysts by a modification of ligand steric profiles has been achieved by some of us.17
Conclusion
In summary, the electronic structures of a series of Ce(IV) guanidinate–amide mixed-ligand complexes incorporating zero, one, two, and three guanidinate ligands were investigated by comprehensive spectroscopic and computational studies. Unusually intense colors arising from low energy LMCT bands were observed for these complexes. The origin of the low energy absorption bands was attributed to the presence of guanidinate π orbitals, which were energetically close to the cerium 4f orbitals. Moreover, XANES spectra showed an increase of nf values with more guanidinate ligands, consistent with the increase of computed Ce 4f population. Although all Ce(IV) guanidinate-amide complexes were multiconfigurational in the sense that the sizable 4f populations can be thought of as a mixture of Ce(III) LMCT configurations with pure ‘crystal field’ Ce(IV) configurations in the ground state wavefunctions, the LUMO-energy-reduction-potential correlation revealed that the influence from the multiconfigurational ground states on the stabilization of the Ce(IV) oxidation state was not significant. Among the Ce(IV) guanidinate complexes, steric effects on the 5d covalency were observed. Compounds with fewer guanidinate ligands demonstrated greater Ce 5d population, less Ce charge, and a more stabilized Ce(IV) oxidation state. And temperature independent paramagnetism (TIP) observed in the Ce(IV) tris(guanidinate) complex was attributed to the small energy gap between the ground and excited states with considerable magnetic moments. Overall, leveraging the 4f and 5d covalency by the number of guanidinate ligands results in anomalous electronic structures and unusually intense colors for these compounds.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
E. J. S. and J. A. gratefully acknowledge Office of Basic Energy Sciences, Chemical Sciences, Geosciences and Biosciences Division, Materials and Chemical Sciences Research for Quantum Information Science Program, of the U.S. Department of Energy under Award DE-SC0020169. E. J. S. also thanks the National Science Foundation for partial financial support of Y.Q. and H.Y. (CHE-1362854/1955724.) E. J. S also acknowledges the University of Pennsylvania for support. Work at Lawrence Berkeley National Laboratory was supported by the Director, Office of Energy Research, Office of Basic Energy Sciences, Chemical Sciences, Geosciences and Biosciences Division, Separation Science Program, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by U.S. National Science Foundation Grant No. OCI-1053575. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. Robert F. Higgins thanks the Nature Conservancy for support through the NatureNet Science Fellowship. Dr Huayi Fang and Dr Walter Dorfner were acknowledged for helpful discussion. Dr Kimberly Mullane was acknowledged for assistance of SQUID measurements. We thank Dr Stefan Minasian for assistance in loading the X-ray absorption holder for the synchrotron measurements.
Notes and references
-
S. Cotton, Lanthanide and Actinide Chemistry, John Wiley and Sons, West Sussex, U. K., 2006 Search PubMed.
- K. B. Yatsimirskii and N. K. Davidenko, Coord. Chem. Rev., 1979, 27, 223–273 CrossRef CAS.
- S. N. Misra and S. O. Sommerer, Appl. Spectrosc. Rev., 1991, 26, 151–202 CrossRef.
-
P. A. Tanner, in Transition Metal and Rare Earth Compounds: Excited States, Transitions, Interactions III, Springer Berlin Heidelberg, Berlin, Heidelberg, 2004, pp. 167–278, DOI:10.1007/b96863.
- L. J. Nugent, R. D. Baybarz, J. L. Burnett and J. L. Ryan, J. Inorg. Nucl. Chem., 1971, 33, 2503–2530 CrossRef CAS.
- A. Vogler and H. Kunkely, Inorg. Chim. Acta, 2006, 359, 4130–4138 CrossRef CAS.
- X. Qin, X. Liu, W. Huang, M. Bettinelli and X. Liu, Chem. Rev., 2017, 117, 4488–4527 CrossRef CAS.
- Y. Qiao and E. J. Schelter, Acc. Chem. Res., 2018, 51, 2926–2936 CrossRef CAS.
- P. Lindqvist-Reis, F. Réal, R. Janicki and V. Vallet, Inorg. Chem., 2018, 57, 10111–10121 CrossRef CAS.
- X.-L. Zheng, Y. Liu, M. Pan, X.-Q. Lü, J.-Y. Zhang, C.-Y. Zhao, Y.-X. Tong and C.-Y. Su, Angew. Chem., Int. Ed., 2007, 46, 7399–7403 CrossRef CAS.
- M. Suta, N. Harmgarth, M. Kühling, P. Liebing, F. T. Edelmann and C. Wickleder, Chem. Asian J., 2018, 13, 1038–1044 CrossRef CAS.
- Y. Qiao, D.-C. Sergentu, H. Yin, A. V. Zabula, T. Cheisson, A. McSkimming, B. C. Manor, P. J. Carroll, J. M. Anna, J. Autschbach and E. J. Schelter, J. Am. Chem. Soc., 2018, 140, 4588–4595 CrossRef CAS.
- H. Yin, P. J. Carroll, J. M. Anna and E. J. Schelter, J. Am. Chem. Soc., 2015, 137, 9234–9237 CrossRef CAS.
- H. Yin, P. J. Carroll, B. C. Manor, J. M. Anna and E. J. Schelter, J. Am. Chem. Soc., 2016, 138, 5984–5993 CrossRef CAS.
- M. K. Assefa, G. Wu and T. W. Hayton, Chem. Sci., 2017, 8, 7873–7878 RSC.
- Y. Qiao, Q. Yang and E. J. Schelter, Angew. Chem., Int. Ed., 2018, 57, 10999–11003 CrossRef CAS.
- Y. Qiao, T. Cheisson, B. C. Manor, P. J. Carroll and E. J. Schelter, Chem. Commun., 2019, 55, 4067–4070 RSC.
- V. R. Yatham, P. Bellotti and B. König, Chem. Commun., 2019, 55, 3489–3492 RSC.
- Y. Kuramochi, S. Sayama and A. Satake, Chem. -Eur. J., 2019, 25, 12042–12045 CrossRef CAS.
- J. A. Bogart, A. J. Lewis, M. A. Boreen, H. B. Lee, S. A. Medling, P. J. Carroll, C. H. Booth and E. J. Schelter, Inorg. Chem., 2015, 54, 2830–2837 CrossRef CAS.
-
O. Horváth and K. L. Stevenson, Charge transfer photochemistry of coordination compounds, VCH Publishers, Inc, New York, 1993 Search PubMed.
- U. J. Williams, P. J. Carroll and E. J. Schelter, Inorg. Chem., 2014, 53, 6338–6345 CrossRef CAS.
- N. T. Rice, J. Su, T. P. Gompa, D.
R. Russo, J. Telser, L. Palatinus, J. Bacsa, P. Yang, E. R. Batista and H. S. La Pierre, Inorg. Chem., 2019, 58, 5289–5304 CrossRef CAS.
- J. Friedrich, Y. Qiao, C. Maichle-Mössmer, E. J. Schelter and R. Anwander, Dalton Trans., 2018, 47, 10113–10123 RSC.
-
A. K. Brisdon, Inorganic Spectroscopic Methods, Oxford University Press, Oxford, UK, 1998 Search PubMed.
- L. L. Costanzo, S. Pistarà and G. Condorelli, J. Photochem., 1983, 21, 45–51 CrossRef CAS.
- H. Yin, Y. Jin, J. E. Hertzog, K. C. Mullane, P. J. Carroll, B. C. Manor, J. M. Anna and E. J. Schelter, J. Am. Chem. Soc., 2016, 138, 16266–16273 CrossRef CAS.
- W. L. Dorfner, P. J. Carroll and E. J. Schelter, Dalton Trans., 2014, 43, 6300–6303 RSC.
- H. Kunkely and A. Vogler, J. Photochem. Photobiol. A: Chem., 2001, 146, 63–66 CrossRef CAS.
- M. W. Löble, J. M. Keith, A. B. Altman, S. C. E. Stieber, E. R. Batista, K. S. Boland, S. D. Conradson, D. L. Clark, J. Lezama Pacheco, S. A. Kozimor, R. L. Martin, S. G. Minasian, A. C. Olson, B. L. Scott, D. K. Shuh, T. Tyliszczak, M. P. Wilkerson and R. A. Zehnder, J. Am. Chem. Soc., 2015, 137, 2506–2523 CrossRef.
- J. L. Ryan and C. K. Jørgensen, J. Phys. Chem., 1966, 70, 2845–2857 CrossRef CAS.
- L. A. Solola, A. V. Zabula, W. L. Dorfner, B. C. Manor, P. J. Carroll and E. J. Schelter, J. Am. Chem. Soc., 2017, 139, 2435–2442 CrossRef CAS.
- C. Morton, N. W. Alcock, M. R. Lees, I. J. Munslow, C. J. Sanders and P. Scott, J. Am. Chem. Soc., 1999, 121, 11255–11256 CrossRef CAS.
- J. A. Bogart, A. J. Lewis, S. A. Medling, N. A. Piro, P. J. Carroll, C. H. Booth and E. J. Schelter, Inorg. Chem., 2013, 52, 11600–11607 CrossRef CAS.
- A. Streitwieser, S. A. Kinsley, J. T. Rigsbee, I. L. Fragala and E. Ciliberto, J. Am. Chem. Soc., 1985, 107, 7786–7788 CrossRef CAS.
- U. J. Williams, D. Schneider, W. L. Dorfner, C. Maichle-Mossmer, P. J. Carroll, R. Anwander and E. J. Schelter, Dalton Trans., 2014, 43, 16197–16206 RSC.
- C. H. Booth, M. D. Walter, M. Daniel, W. W. Lukens and R. A. Andersen, Phys. Rev. Lett., 2005, 95, 267202 CrossRef CAS.
- M. D. Walter, R. Fandos and R. A. Andersen, New J. Chem., 2006, 30, 1065–1070 RSC.
- O. Moossen and M. Dolg, Chem. Phys. Lett., 2014, 594, 47–50 CrossRef CAS.
- A. Kerridge, R. Coates and N. Kaltsoyannis, J. Phys. Chem. A, 2009, 113, 2896–2905 CrossRef CAS.
- L. A. Solola, A. V. Zabula, W. L. Dorfner, B. C. Manor, P. J. Carroll and E. J. Schelter, J. Am. Chem. Soc., 2016, 138, 6928–6931 CrossRef CAS.
- T. Cheisson, K. D. Kersey, N. Mahieu, A. McSkimming, M. R. Gau, P. J. Carroll and E. J. Schelter, J. Am. Chem. Soc., 2019, 141, 9185–9190 CrossRef.
- W. W. Lukens, N. Magnani and C. H. Booth, Inorg. Chem., 2012, 51, 10105–10110 CrossRef CAS.
- N. T. Rice, I. A. Popov, D. R. Russo, J. Bacsa, E. R. Batista, P. Yang, J. Telser and H. S. La Pierre, J. Am. Chem. Soc., 2019, 141, 13222–13233 CrossRef CAS.
- S. G. Minasian, E. R. Batista, C. H. Booth, D. L. Clark, J. M. Keith, S. A. Kozimor, W. W. Lukens, R. L. Martin, D. K. Shuh, S. C. E. Stieber, T. Tylisczcak and X.-d. Wen, J. Am. Chem. Soc., 2017, 139, 18052–18064 CrossRef CAS.
- C. T. Palumbo, I. Zivkovic, R. Scopelliti and M. Mazzanti, J. Am. Chem. Soc., 2019, 141, 9827–9831 CrossRef CAS.
- A. R. Willauer, C. T. Palumbo, R. Scopelliti, I. Zivkovic, I. Douair, L. Maron and M. Mazzanti, Angew. Chem., Int. Ed., 2020, 59, 3549–3553 CrossRef CAS.
- A. R. Willauer, C. T. Palumbo, F. Fadaei-Tirani, I. Zivkovic, I. Douair, L. Maron and M. Mazzanti, J. Am. Chem. Soc., 2020, 142, 5538–5542 CrossRef CAS.
- P. L. Arnold, Z. R. Turner, N. Kaltsoyannis, P. Pelekanaki, R. M. Bellabarba and R. P. Tooze, Chem.–Eur. J., 2010, 16, 9623–9629 CrossRef CAS.
- D. Schneider, T. Spallek, C. Maichle-Mossmer, K. W. Tornroos and R. Anwander, Chem. Commun., 2014, 50, 14763–14766 RSC.
- R. L. Halbach, G. Nocton, J. I. Amaro-Estrada, L. Maron, C. H. Booth and R. A. Andersen, Inorg. Chem., 2019, 58, 12083–12098 CrossRef CAS.
- R. L. Halbach, G. Nocton, C. H. Booth, L. Maron and R. A. Andersen, Inorg. Chem., 2018, 57, 7290–7298 CrossRef CAS.
- M. D. Walter, C. H. Booth, W. W. Lukens and R. A. Andersen, Organometallics, 2009, 28, 698–707 CrossRef CAS.
- J. R. Robinson, P. J. Carroll, P. J. Walsh and E. J. Schelter, Angew. Chem., Int. Ed., 2012, 51, 10159–10163 CrossRef CAS.
- J. A. Bogart, H. B. Lee, M. A. Boreen, M. Jun and E. J. Schelter, J. Org. Chem., 2013, 78, 6344–6349 CrossRef CAS.
- W. L. Dorfner, P. J. Carroll and E. J. Schelter, Org. Lett., 2015, 17, 1850–1853 CrossRef CAS.
- J. A. Bogart, A. J. Lewis and E. J. Schelter, Chem.–Eur. J., 2015, 21, 1743–1748 CrossRef CAS.
- J. R. Levin, W. L. Dorfner, A. X. Dai, P. J. Carroll and E. J. Schelter, Inorg. Chem., 2016, 55, 12651–12659 CrossRef CAS.
- Y. Bian, J. Jiang, Y. Tao, M. T. M. Choi, R. Li, A. C. H. Ng, P. Zhu, N. Pan, X. Sun, D. P. Arnold, Z.-Y. Zhou, H.-W. Li, T. C. W. Mak and D. K. P. Ng, J. Am. Chem. Soc., 2003, 125, 12257–12267 CrossRef CAS.
- G. Balazs, F. G. N. Cloke, J. C. Green, R. M. Harker, A. Harrison, P. B. Hitchcock, C. N. Jardine and R. Walton, Organometallics, 2007, 26, 3111–3119 CrossRef CAS.
- A. C. Behrle, J. R. Levin, J. E. Kim, J. M. Drewett, C. L. Barnes, E. J. Schelter and J. R. Walensky, Dalton Trans., 2015, 44, 2693–2702 RSC.
- S. D. Stults, R. A. Andersen and A. Zalkin, Organometallics, 1990, 9, 115–122 CrossRef CAS.
- W. J. Evans and M. A. Hozbor, J. Organomet. Chem., 1987, 326, 299–306 CrossRef CAS.
- S. S. Galley, S. A. Pattenaude, C. A. Gaggioli, Y. Qiao, J. M. Sperling, M. Zeller, S. Pakhira, J. L. Mendoza-Cortes, E. J. Schelter, T. E. Albrecht-Schmitt, L. Gagliardi and S. C. Bart, J. Am. Chem. Soc., 2019, 141, 2356–2366 CrossRef CAS.
- G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532 CrossRef CAS.
- B. D. Mahoney, N. A. Piro, P. J. Carroll and E. J. Schelter, Inorg. Chem., 2013, 52, 5970–5977 CrossRef CAS.
- M. Ciampolini, F. Mani and N. Nardi, J. Chem. Soc., Dalton Trans., 1977, 1325–1328, 10.1039/DT9770001325.
- J. R. Robinson, Y. Qiao, J. Gu, P. J. Carroll, P. J. Walsh and E. J. Schelter, Chem. Sci., 2016, 7, 4537–4547 RSC.
- Y.-M. So, G.-C. Wang, Y. Li, H. H.-Y. Sung, I. D. Williams, Z. Lin and W.-H. Leung, Angew. Chem., Int. Ed., 2014, 53, 1626–1629 CrossRef CAS.
- N. E. Mansfield, M. P. Coles and P. B. Hitchcock, Dalton Trans., 2005, 2833–2841, 10.1039/B506332A.
- A. M. Willcocks, T. Pugh, J. A. Hamilton, A. L. Johnson, S. P. Richards and A. J. Kingsley, Dalton Trans., 2013, 42, 5554–5565 RSC.
- T. J. Duignan and J. Autschbach, J. Chem. Theory Comput., 2016, 12, 3109–3121 CrossRef CAS.
- A. J. Cohen, P. Mori-Sánchez and W. Yang, Chem. Rev., 2012, 112, 289–320 CrossRef CAS.
- J. Autschbach and M. Srebro, Acc. Chem. Res., 2014, 47, 2592–2602 CrossRef CAS.
- M. K. Assefa, D.-C. Sergentu, L. A. Seaman, G. Wu, J. Autschbach and T. W. Hayton, Inorg. Chem., 2019, 58, 12654–12661 CrossRef CAS.
- A. B. Altman, J. I. Pacold, J. Wang, W. W. Lukens and S. G. Minasian, Dalton Trans., 2016, 45, 9948–9961 RSC.
- J. Su, E. R. Batista, K. S. Boland, S. E. Bone, J. A. Bradley, S. K. Cary, D. L. Clark, S. D. Conradson, A. S. Ditter, N. Kaltsoyannis, J. M. Keith, A. Kerridge, S. A. Kozimor, M. W. Löble, R. L. Martin, S. G. Minasian, V. Mocko, H. S. La Pierre, G. T. Seidler, D. K. Shuh, M. P. Wilkerson, L. E. Wolfsberg and P. Yang, J. Am. Chem. Soc., 2018, 140, 17977–17984 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1965035–1965036. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc05193d |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Haolin
Yin
ab,
Haolin
Yin