
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltrasensitive small molecule fluorogenic probe for human heparanase†
Jun
Liu
ab,
Kelton A.
Schleyer
ab,
Tyrel L.
Bryan
a,
Changjian
Xie
a,
Gustavo
Seabra
b,
Yongmei
Xu
c,
Arjun
Kafle
a,
Chao
Cui
ab,
Ying
Wang
a,
Kunlun
Yin
a,
Benjamin
Fetrow
a,
Paul K. P.
Henderson
a,
Peter Z.
Fatland
a,
Jian
Liu
c,
Chenglong
Li
b,
Hua
Guo
 a and
Lina
Cui
*ab
a and
Lina
Cui
*ab
aDepartment of Chemistry and Chemical Biology, University of New Mexico, Albuquerque, NM 87131, USA
bDepartment of Medicinal Chemistry, College of Pharmacy, University of Florida, Gainesville, FL 32610, USA. E-mail: linacui@cop.ufl.edu
cDivision of Chemical Biology and Medicinal Chemistry, Eshelman School of Pharmacy, University of North Carolina, Chapel Hill, NC 27599, USA
First published on 20th October 2020
Abstract
Heparanase (HPA) is a critical enzyme involved in the remodeling of the extracellular matrix (ECM), and its elevated expression has been linked with diseases such as various types of cancer and inflammation. The detection of heparanase enzymatic activity holds tremendous value in the study of the cellular microenvironment, and search of molecular therapeutics targeting heparanase, however, no structurally defined probes are available for the detection of heparanase activity. Here we present the development of the first ultrasensitive fluorogenic small-molecule probe for heparanase enzymatic activity via tuning the electronic effect of the substrate. The probe exhibits a 756-fold fluorescence turn-on response in the presence of human heparanase, allowing one-step detection of heparanase activity in real-time with a picomolar detection limit. The high sensitivity and robustness of the probe are exemplified in a high-throughput screening assay for heparanase inhibitors.
Introduction
Heparanase (HPA), an endo-β-glucuronidase of the glycoside hydrolase 79 (GH79) family,1,2 is responsible for the cleavage of heparan sulfate (HS) chains of heparan sulfate proteoglycans (HSPG).3 These protein-polysaccharide conjugated macromolecules, abundantly expressed in the extracellular matrix (ECM), play an essential structural role in maintaining the ECM integrity. Moreover, the HS side chains bind to an array of biological effector molecules, such as growth factors, chemokines, and cytokines, thereby serving as their reservoir that can liberate the desired signaling molecules when needed. Therefore the HS-degrading activity of heparanase can lead to a variety of fundamental regulations to impact cellular behavior, such as remodeling and degradation of the ECM, generation of biologically active carbohydrate fragments, and liberation of biological mediators that are bound to HS side chains, thus controlling cellular signaling processes.4,5 Under normal physiological conditions, heparanase regulates cellular homeostasis, and its high-level expression is mainly observed in the placenta and blood-borne cells such as platelets, neutrophils, mast cells, and lymphocytes.1 Strikingly, the expression of heparanase is significantly elevated in most types of cancer tissues,6–9 and increased heparanase activity is regularly linked with increased angiogenesis, metastasis, and shortened post-surgical survival.7,9,10 The elevated enzymatic activity of heparanase has also been reported in various inflammatory diseases and autoimmune disorders that involve degradation of HS side chains of HSPGs and extensive ECM remodeling.11Despite the existence of other isoforms, heparanase-1 is the only known enzyme for HS cleavage.1 Heparanase enzymatic activity herein is referred to as the HS-degrading activity of heparanase-1. Furthermore, heparanase is initially expressed as a latent enzyme (proheparanase), which is processed in the lysosome and secreted to the ECM where it meets the substrate HS; therefore, its enzymatic activity cannot be represented by the mRNA level.12 Methods that can directly measure the enzymatic activity of heparanase are highly desired to facilitate the basic biological study of heparanase in the context of ECM and cellular microenvironment. Such methods are also indispensable in the medical examination of various physiopathological conditions associated with heparanase activity, and in search of molecular therapeutics targeting heparanase or its regulatory molecules.
However, the progress on the development of heparanase probes is disproportionate to the biological importance of the enzyme. Several heparanase assays have emerged over the past two decades, but these assays have encountered disadvantages that limit their broad application.1,13 Radioisotope-based assays are the most commonly used method for the detection of heparanase activity; these assays measure the radioactivity using radiolabeled heparan sulfate polysaccharides, and require a chromatographic separation of the reaction products, restricting their use to very small sample sizes.14,15 A more recent homogeneous time-resolved fluorescence assay, currently considered the most convenient approach available to detect heparanase activity,16,17 requires tedious multi-step reagent addition and the output signal and substrate degradation lacks linear correlation.18 Another common drawback of these assays developed so far is the use of heterogeneous heparan sulfate polysaccharides, suffering from difficulties in the standardization of the assays due to the structural complexity and diversity of the polymer substrates.13,19,20 To avoid the disadvantages resulting from heterogeneity, a colorimetric assay21 uses the pentasaccharide fondaparinux, a commonly anticoagulant, instead of heparan sulfate as the substrate,22 but this assay has not gained broad application due to its low sensitivity and requirement of high substrate concentration, high temperature, and long incubation time.
Herein, we describe a fluorogenic small molecule with a defined chemical structure that offers 756-fold fluorescence turn-on response in the presence of human heparanase, allowing one-step detection of heparanase activity in real-time with a picomolar detection limit. Using a standard fluorescence microplate reader, we demonstrate the high sensitivity, simplicity, and robustness of the probe through its application in a high-throughput screening assay for heparanase inhibitors.
Results and discussion
The challenge in the development of structurally defined heparanase probes with fluorescence readout largely stems from its elusive substrate recognition mode, despite extensive investigations being made.23–25 One known fact is that heparanase is an endoglycosidase that can cleave the internal glycosidic bond between a glucuronic acid (GlcUA) residue and an N-sulfoglucosamine (GlcN(NS)) residue bearing either a 3-O-sulfo or a 6-O-sulfo group.26 Regarding the substrate size, the minimum heparanase recognition sequence discovered was a trisaccharide [GlcN(NS,6S)-GlcUA-GlcN(NS,6S)];27 indeed, molecules containing the disaccharide substrate [GlcN(NS)-GlcUA] have shown mixed results.16,28 These known features pose a significant hurdle to the development of structurally defined heparanase probes with fluorescence readout. A dye-quencher system may be too bulky for the catalytic site of heparanase, in addition to the synthetic challenge, while a fluorogenic system requires the enzyme to have exoglycosidase properties to cleave the fluorophore off the reducing end (Fig. 1). To obtain a fluorogenic probe, can we shift heparanase's endoglycosidic mode of action to exoglycosidic? | ||
| Fig. 1 Heparanase's endoglycosidic cleavage mode. Blue circle: monosaccharide residue. Red arrow: enzyme cleavage site. | ||
To examine this hypothesis, we chemically synthesized compounds 1 to 3 (Scheme 1). In order to achieve the 1,2-cis-α-linkage of the disaccharide moiety, we prepared donor 529,30 bearing an azido group at its C-2 position, to take advantage of the anomeric effect during glycosylation with acceptor 6.29,30 The glycosylation of 6 with trichloroacetimidate 5 was carried out in the presence of trimethylsilyl triflate (TMSOTf) promoter in dry solvents. It is worth mentioning that only very little desired disaccharide was observed while the reaction was carried out in dichloromethane (DCM). The unexpected intermolecular aglycone transfer31–33 was predominant mainly due to the low reactivity of the C4-hydroxyl group. Switching the solvent to toluene increased formation of the desired disaccharide, and further screening of solvents achieved disaccharide 7 in 79% yield with complete 1,2-cis-α-selectivity, when the reaction was carried out in a 2/1 mixture of toluene and 1,4-dioxane.34 Transformation of 7 into the corresponding brominated product 8 and further coupling with the fluorophore afforded the expected product in satisfying yield (55%) using silver oxide (Ag2O) as the activating agent in acetonitrile. Global deacetylation of 9a, reduction of the azido group to the amine, and subsequent saponification and sulfation reactions led to the desired compound 1. Molecules 2 and 3 were obtained in a similar manner (ESI†).
 | ||
| Scheme 1 Synthetic route to compounds 1–4. Reagents and conditions: (a) TMSOTf, 4 Å MS, toluene/1,4-dioxane = 2/1, 0 °C; (b) IBr, DCM, rt; (c) 4-methylcoumarin derivatives, Ag2O, MeCN; (d) NaOMe, MeOH; (e) TrCl, Py; (f) 4-MeOC6H4COCl, DMAP, Py; (g) TFA, DCM; (h) Et3N·SO3, DMF; (i) DAST, DCM; (j) H2 balloon, Pd/C, MeOH, rt, 4 h; (k) H2O/THF = 2/1, NaOH (pH = 11), rt; (l) Py·SO3, rt. | ||
We tested the response of compounds 1 to 3 towards heparanase (Fig. 2a and b). These compounds are innately non-fluorescent and are stable towards solvolysis, but if the enzyme-catalyzed hydrolysis reaction occurs, the aglycon can be released and fluoresce due to restored intramolecular charge transfer (ICT).35,36 Our initial results indicated that probe 228 could not be activated by human heparanase, while compound 3 produced a slight fluorescence signal after a long incubation time. Compared with the structure of 2, the marginally activatable compound 3 bears an electron-withdrawing difluoromethyl group ortho to the glycosidic oxygen, presumably leading to the increased leaving group ability37 of the aglycon based on the enzymatic mechanism.38,39 We envisioned that the activation energy barrier of the 6,8-difluoro-7-hydroxy-4-methylcoumarin (DiFMU), carrying two electron-withdrawing fluorine atoms ortho to its phenolic hydroxyl group, would be further lowered. Consistent with our hypothesis, incubation of the resulting probe 1 with human recombinant heparanase produced dramatic fluorescence enhancement higher than the background signal (Fig. 2b), and we termed compound 1 the heparanase-activatable, difluorocoumarin-based probe (HADP). Additionally, the fluorine substituents of DiFMU increase the quantum yield and improve the photostability,40 further enhancing the sensitivity of HADP. A recent docking study41 suggested that 4 could have greater affinity for heparanase than 2 owing to an additional sulfate group on the 6-O position of the glucosamine moiety. Starting with the intermediate 9a, the 6-OH group on the glucosamine residue was selectively protected by a bulky trityl group that could be selectively removed later for sulfation. Upon completion of tritylation, the 4-methoxybenzoyl group was employed to block the rest of the hydroxyl groups. Then the trityl group was selectively removed under acid conditions and sulfation was conducted on the newly exposed 6-OH of the glucosamine motif to achieve the desired compound 9b. It is noteworthy that the 4-methoxybenzoyl group was used instead of an acetyl protecting group to avoid acyl migration42 from the O-4 to O-6 position during sulfation. The remaining steps are similar to that of compounds 1–3 described above. With compound 4 in hand, we tested its activation by human heparanase; the extra 6-O-sulfate at the GlcN residue was not advantageous as no fluorescent response was observed (Fig. 2b).
 | ||
| Fig. 2 Enzymatic response of synthetic compounds. (a) Chemical structures of 1 (HADP) and its analogues 2–4. (b) Fluorescence responses of probe HADP and control compounds 2–4 (each 5 μM) towards human heparanase (HPA) (1 μg) in 40 mM NaOAc buffer (pH 5.0) at 37 °C, fluorescence intensity was recorded on a Spectramax M5 Multimode plate reader (Molecular Devices, USA) at each timepoint. (c) UV-vis absorption and (d) fluorescence (λex = 365 nm) spectra of probe HADP (5 μM) before (blue line) and after (red line) incubation with human heparanase (HPA) (2 μg) in 40 mM NaOAc buffer (pH 5.0) for 2 h at 37 °C. Fluorescence spectra were recorded on a Fluorolog TAU-3 spectrofluorometer with a xenon lamp (Jobin Yvon-Spex, Instruments S. A., Inc.) (e) Fluorescence quantification of (d). Turn-on ratio is defined as the ratio of fluorescence intensity after activation to that before activation. (f) Time dependence of fluorescence spectra of HADP (5 μM) with human heparanase (HPA) (1 μg) from 0 min to 3 h in 40 mM NaOAc buffer (pH 5.0). Inset of (f) quantification of fluorescence intensity at 455 nm over time. λex = 365 nm. Vertical bars indicate mean ± SD. ****p < 0.0001. | ||
To confirm that the fluorescent response of HADP was due to liberation of the fluorophore by heparanase, the UV-vis absorbance and fluorescence emission profiles of HADP with or without human heparanase were examined. HADP alone displayed maximal absorption at 277 nm and 313 nm (Fig. 2c); upon incubation with heparanase, a large bathochromic shift was observed with an absorption bump ranging from 320 nm to 365 nm, consistent with the absorption of uncaged DiFMU (pH 5.0, NaOAc buffer). HADP should be nonfluorescent (Fig. 2d) because the hydroxyl group of DiFMU is caged with a disaccharide, resulting in suppressed intramolecular charge transfer (ICT). Upon addition of heparanase, enzyme-triggered cleavage of the glycosidic bond exposes the free hydroxyl group of DiFMU, which serves as a strong electron donor in the coumarin D–π–A system, thereby recovering the ICT process and restoring the fluorescence.36 Consistent with this mechanism, HADP incubated with heparanase (HPA) (2 μg) displayed a remarkable fluorescence enhancement of up to 756-fold (may vary due to different instrumentation settings) at 450 nm within 2 hours (Fig. 2e). Then the time dependence of the fluorescence spectra of HADP (5 μM) with heparanase (HPA) (1 μg) were investigated (Fig. 2f). The fluorescence increased dramatically in the initial stage and reached a plateau at 2.5 h. More importantly, the fluorescence intensity exhibited a good linearity to time in the initial 15 minutes (Fig. 2f), facilitating the determination of enzyme kinetics. To further verify this mechanism, we used HPLC equipped with a diode array detector to monitor the reactions (Fig. S1a and b†). After HADP (5 μM) was incubated with heparanase (1 μg) for 4 hours (red line), we observed a clean conversion of the probe to a new peak at 13.1 min, clearly different from the retention time of HADP (black line); the product also shared the same absorption as that of DiFMU (Fig. S1a inset†), suggesting the complete activation of HADP by heparanase. Analysis of the new peak by high-resolution electrospray ionization (HR-ESI) mass spectrometry reported two signals at m/z 213.0359 and 235.0181 for the protonated and sodiated adducts of DiFMU, respectively (Fig. S2†). Collectively, heparanase enzymatic activity cleaves the glycosidic linkage of HADP to liberate the fluorescent DiFMU. No such conversion was observed in the analysis of compounds 2, 3 and 4 treated under the same condition (Fig. S1b†). Recently, Davies and coworkers reported that compound 4 could be cleaved by heparanase at a high concentration.43
We further studied the varied reactivity of compounds 1–3 with heparanase via density functional theory (DFT) and found that the length of the glycosidic bond of compound 1 (HADP) was slightly longer than that of compound 2 or 3, and the energy barrier for compound 1 (HADP) (18.67 kcal mol−1) in the transition state is lower than that of compound 2 or 3 (Fig. 3, S3–S6†). Docking studies of the probes with the human heparanase (PDB id: 5E8M) revealed little differences in the binding mode – all three molecules bind to the enzyme in nearly identical mode, and the substituent groups on the coumarin residue in compounds 1 and 3 did not introduce additional binding energy (Fig. 3, S7–S9, Table S1 and Movies S1–S3†). Collectively, the much higher reactivity of 1 (HADP) than 2 or 3 results from inherent ground state destabilization44 and transition state stabilization.
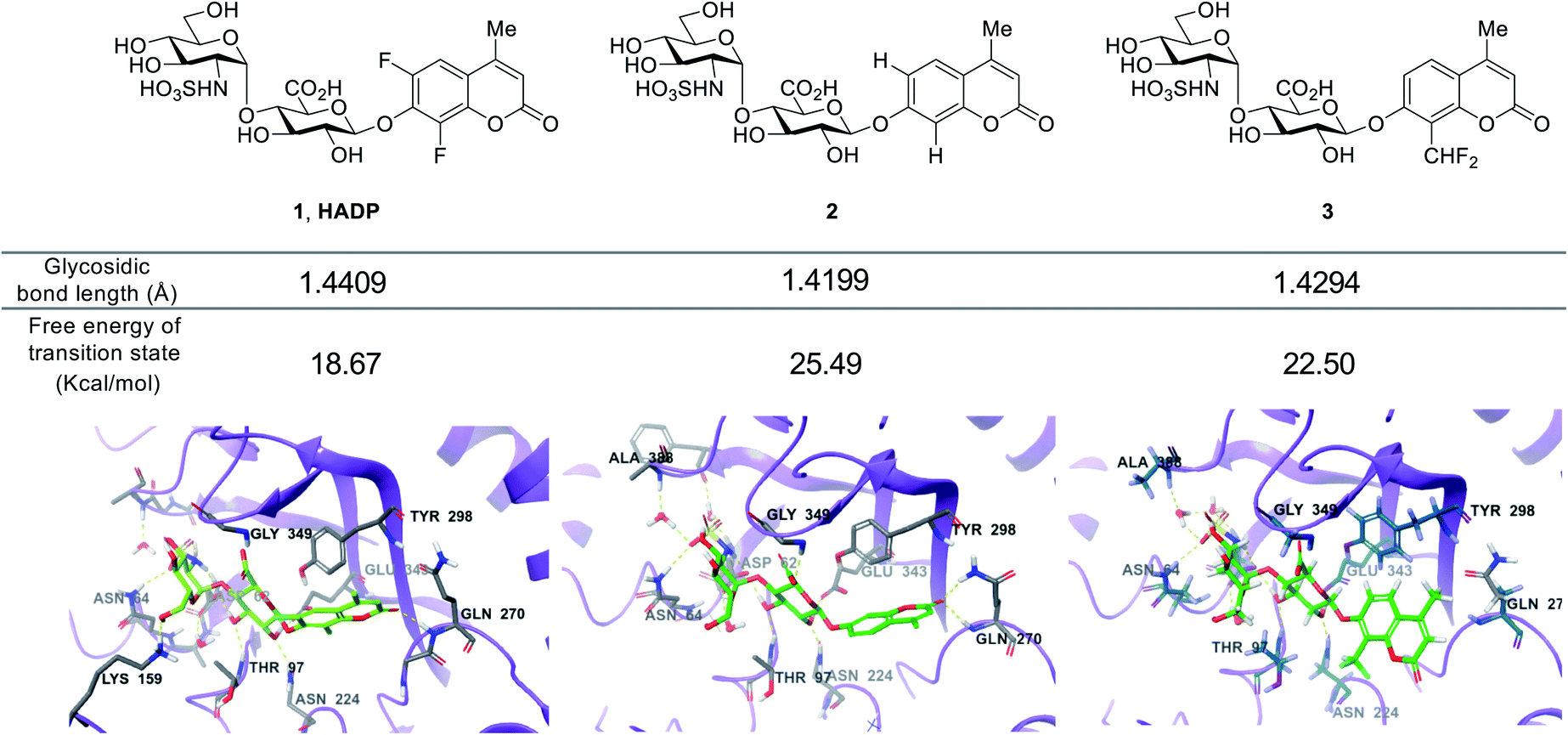 | ||
| Fig. 3 Calculated glycosidic bond length of compounds 1 (HADP), 2 and 3, free energy of transition state and binding modes in heparanase (HPA) (docking of the probes with the human heparanase, PDB 5E8M). | ||
Human heparanase has the highest enzymatic activity under acidic conditions (pH ∼5.4–6.8).45 We examined the effects of reaction buffer and pH on the activation of HADP by heparanase (Fig. S10†). The reported working buffers for heparanase include 50 mM 2-(N-morpholino)ethanesulfonic acid (MES) buffer (pH 6.0)26 and 40 mM sodium acetate (NaOAc) buffer (pH 5.0).2,28 To address the pH difference, we also tested 40 mM NaOAc (pH 6.0) buffer. At pH 6.0, greater probe activation was detected in the NaOAc buffer than what was observed in the MES buffer, and the reactivity increased further in NaOAc buffer when the pH was reduced to 5.0 (consistent with previous findings14).
Aware that pH also tremendously affects fluorescence intensity,46 we constructed pH profiles of HADP and the fluorophore DiFMU to determine the sensitivity of the probe at the optimal pH for heparanase activity (Fig. S11†). There was no remarkable fluorescence change at 455 nm for HADP in the pH ranging from 1 to 11 upon excitation, but the free fluorophore DiFMU demonstrated significantly greater fluorescence intensity at pH values above its reported pKa of 4.7.40 This indicates that HADP is inherently well-suited for heparanase activity assays, as the liberated fluorophore will generate strong signal at the ideal assay pH of 5.0. In contrast, the 4-methylumbelliferone fluorophore of compounds 2 and 4, with a pKa of 7.8, would generate markedly low fluorescence enhancement at this pH and would require a buffering step to optimize the assay sensitivity – if it were activated by heparanase at all. The pKa of DiFMU being lower than the pH of assay buffer contributes conveniently to achieving a simple one-step ‘mix-and-go’ assay without an additional basification step. Also, the cleavage rate of HADP can be qualitatively measured using the standard curve of DiFMU at pH of 5, facilitating the determination of kinetic parameters.
To confirm that HADP is selective for heparanase, the probe was evaluated against a series of possible interfering biological molecules and enzymes at excessively high levels (Fig. 4a). Heparanase exhibited the largest “off–on” response, while no fluorescence increase was observed upon addition of any other molecules or enzymes even at superphysiological levels. For instance, HADP showed no response to 100 μM H2O2, 5 mM glutathione (GSH), or 5 mM cysteine (Cys), common biological oxidizing and reducing species in cells. Hyaluronidase and chondroitinase, which are polysaccharide lyases catalyzing the cleavage of glyosidic bonds between N-acetyl-D-glucosamine and D-glucuronic acid in hyaluronic acid, and between D-hexosaminyl and D-glucuronic acid in chondroitin, respectively, did not afford obvious fluorescence change. Likewise, β-glucuronidase did not trigger a fluorescence response from HADP, which possesses a glycosyl substituent on the O-4 position of the glucuronic acid residue. Overall, HADP demonstrates remarkable selectivity for heparanase over other glycosidases and common biomolecules. To determine the detection limit of HADP for heparanase, concentration-dependent studies over time were performed with 5 μM HADP (Fig. S12a†). A good linear relationship between concentration and fluorescence intensity was obtained in the heparanase (HPA) concentration range of 0–0.01 μg μL−1, with an equation of F455 nm = 650443[HPA] + 12.03 (R2 = 0.9968) (Fig. 4b). Based on the 3σ/s method, the limit of detection (LOD) of HADP was calculated to be 0.35 ng mL−1 (67 pM), indicating the ultra-sensitivity of HADP for heparanase detection. To test if HADP was suitable for rapid and quantifiable heparanase detection, we evaluated the kinetics of the probe activation by heparanase. The kinetic parameters of the enzymatic activation of HADP by human heparanase were determined via time-dependent fluorescence intensity measurements in the presence of heparanase and HADP at different concentrations (Fig. S12c†). The Michaelis constant (KM), catalytic efficiency (kcat/KM) and turnover number (kcat) were calculated to be 8.3 μM, 0.29 μM−1 min−1 and 2.4 min−1, respectively. Compared with the KM of fondaparinux (46 μM),21 a pentasaccharide substrate for heparanase, HADP exhibited a significantly high affinity for heparanase.
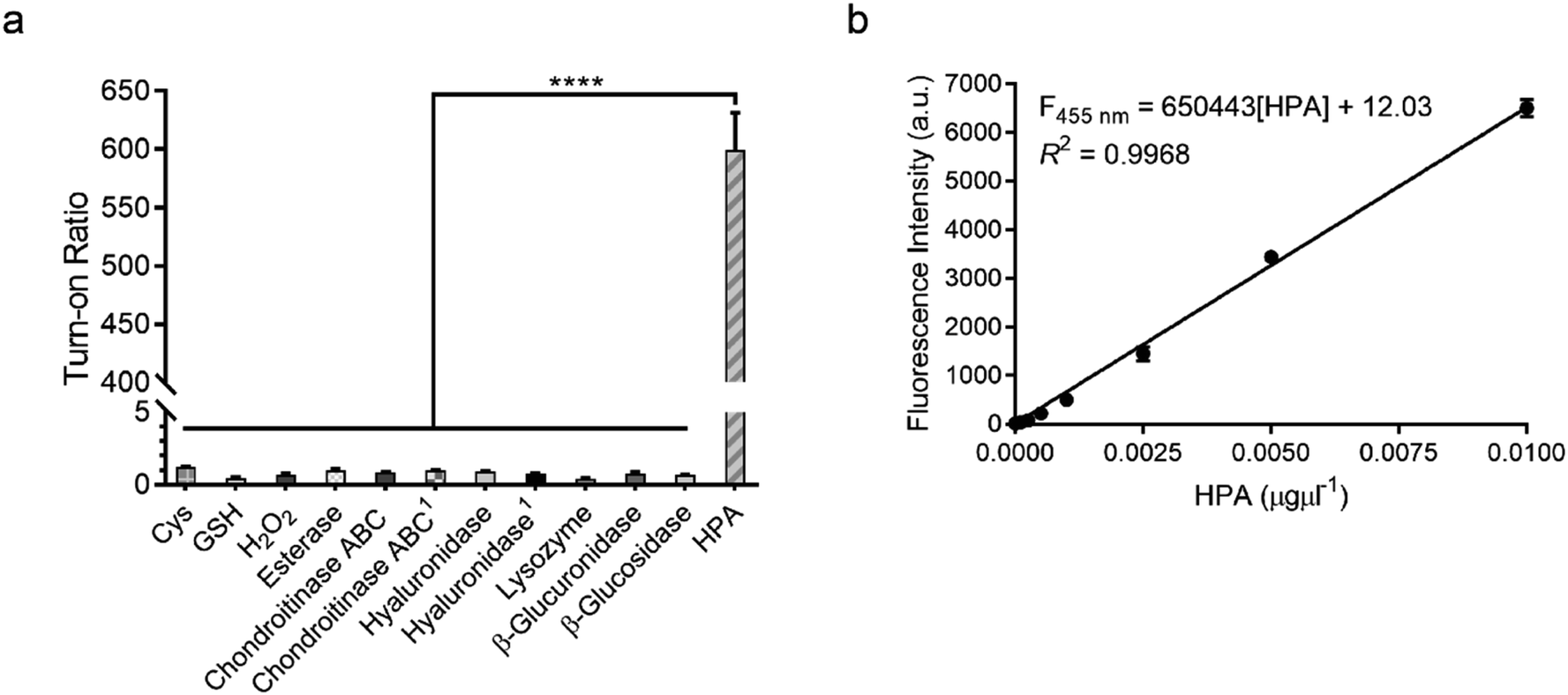 | ||
| Fig. 4 Sensitivity and selectivity of HADP. (a) Fluorescence responses of probe HADP to cysteine (Cys, 5 mM), glutathione (GSH, 5 mM), hydrogen peroxide (H2O2, 100 μM), esterase, chondroitinase ABC, hyaluronidase, lysozyme, β-glucuronidase, β-glucosidase (5 μg each) and human heparanase (HPA, 2 μg) for 4 h. 150 μg enzyme was used. (b) Linear plot of fluorescence intensity against various concentrations of human heparanase (HPA) at 28 min. λex/em = 365 nm/455 nm. Vertical bars indicate mean ± SD. ****p < 0.0001. | ||
Heparanase has been considered a drug target for cancer and inflammation.47–52 Nonetheless, only four saccharide-based inhibitors have been assessed in clinical trials.1,53 The major challenge in screening inhibitors for heparanase is the lack of robust assays.13 To evaluate the ability of HADP for inhibitor screening, we first tested whether it could accurately measure the potency of a known experimental heparanase inhibitor, suramin. Based on our assay, the calculated IC50 value of suramin was 1.0 μM (Fig. 5a), in good agreement with that determined by the previously mentioned HTRF assay.54
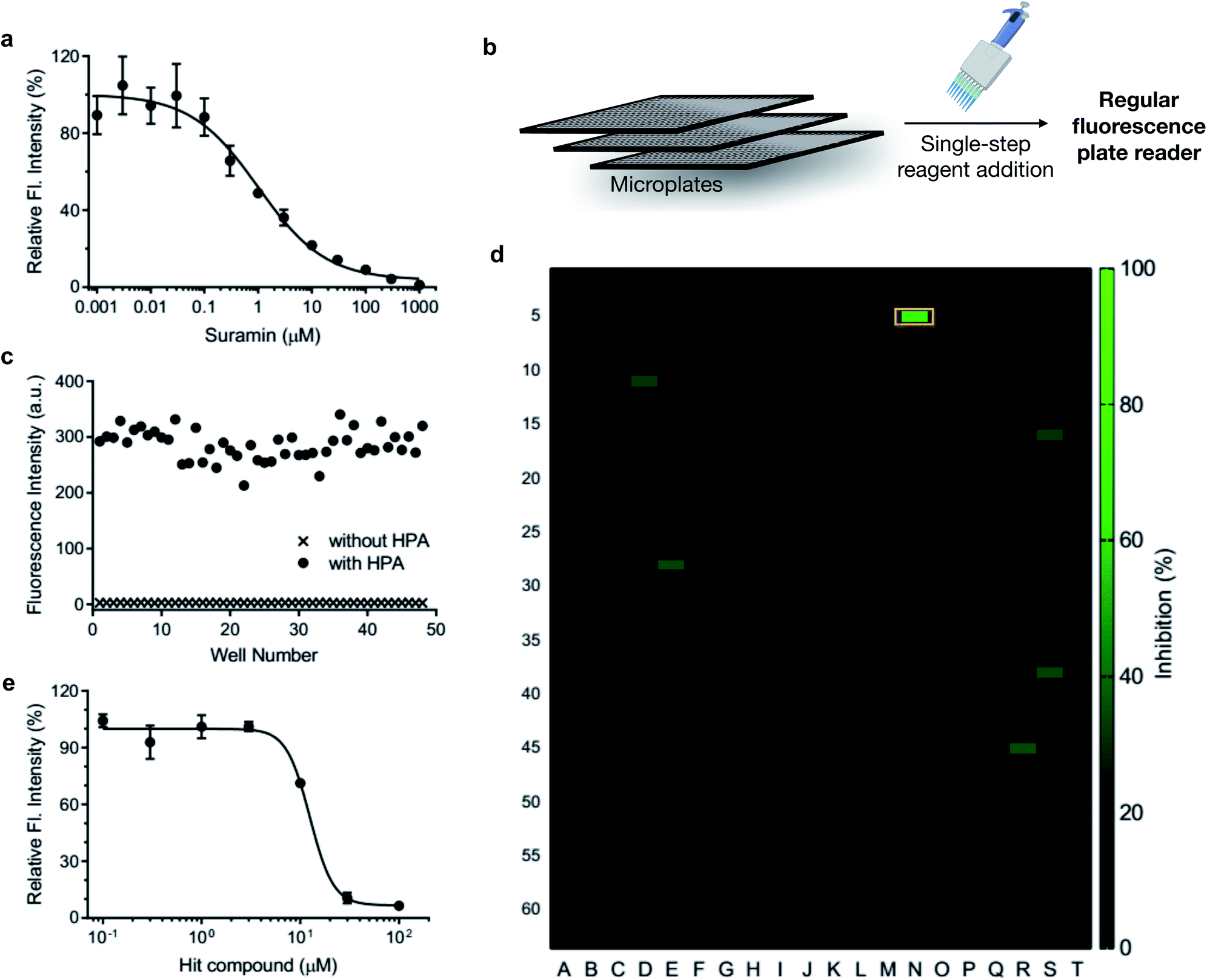 | ||
| Fig. 5 Application of HADP in high-throughput drug screening via a single-step fluorescence assay. (a) Inhibitory activity of suramin. (b) Schematic of single-step mix-and-go assay for high-throughput heparanase (HPA) inhibitor screening. (c) Quality assessment of the mix-and-go assay via manual pipetting. (d) Screening results of a 1280-compound library. Orange box: hit compound. (e) Validation of the inhibitory activity of the hit compound. λex/em = 365 nm/455 nm. | ||
The accuracy of this assay was further evaluated via manual pipette operation to give a Z′-factor of approximately 0.7 (Fig. 5c), indicating that the HADP-based assay can be an excellent platform for high-throughput drug screening.55 Next, we assessed the performance of HADP in a semi-high-throughput format, screening through a library of 1280 compounds purchased from Tocris at 10 μM in a 384-well plate assay against heparanase (Fig. 5b and d, detailed experiments can be found in ESI†). Several hits were identified, and further validation via an inhibition assay in triplicate identified a new drug scaffold with micromolar inhibitory activity (IC50 = 12.5 μM) toward heparanase (Fig. 5e). These results suggest that HADP is a sensitive and robust probe for high-throughput screening in seeking potential heparanase inhibitors.
Conclusions
In summary, via twisting heparanase's endoglycosidic mode of action through modulating the electronic properties of the algycon, we have developed the first structurally defined ultrasensitive fluorogenic probe HADP (1) for detecting heparanase activity; it offers up to 756-fold fluorescence turn-on response in the presence of human heparanase, allowing one-step detection of heparanase activity in real-time with a picomolar detection limit. We demonstrated the high sensitivity, simplicity, and robustness of HADP through high-throughput screening of novel heparanase inhibitors. The probe described will find broad applications in basic cell biology studies, the development of diagnostics, and drug discovery.Conflicts of interest
The University of Florida has filed a patent application. L. C., J. L., and K. A. S. are inventors for the patent on probes for heparanase.Acknowledgements
This work is supported by research grants to Prof. L. Cui from the National Institute of General Medical Sciences of National Institutes of Health (Maximizing Investigators' Research Award for Early Stage Investigators, R35GM124963), the Department of Defense (Congressionally Directed Medical Research Programs Career Development Award, W81XWH-17-1-0529), University of New Mexico (UNM Startup Award), and the University of Florida (UF Startup Fund). P. K. P. H. was supported by UNM-NIH Initiative for Maximizing Student Development (IMSD) Scholarship. P. Z. F. was supported by Dr Thomas Whaley Endowed Memorial Scholarship from the Department of Chemistry and Chemical Biology, UNM. Part of the high-resolution mass spectroscopy analysis was performed at the Mass Spectrometry Research and Education Center at the University of Florida supported by grant NIH S10 OD021758-01A1. We thank Profs. Wei Wang (UNM) and Weihong Tan (UF) for the use of their fluorescence spectrometers, and Robert Huigens III (UF) for the use of melting point apparatus.Notes and references
- S. Rivara, F. M. Milazzo and G. Giannini, Future Med. Chem., 2016, 8, 647–680 CrossRef CAS.
- L. Wu, C. M. Viola, A. M. Brzozowski and G. J. Davies, Nat. Struct. Mol. Biol., 2015, 22, 1016–1022 CrossRef CAS.
- J. P. Li and I. Vlodavsky, Thromb. Haemostasis, 2009, 102, 823–828 CrossRef CAS.
- I. Vlodavsky, Y. Friedmann, M. Elkin, H. Aingorn, R. Atzmon, R. Ishai-Michaeli, M. Bitan, O. Pappo, T. Peretz, I. Michal, L. Spector and I. Pecker, Nat. Med., 1999, 5, 793–802 CrossRef CAS.
- M. Gotte and G. W. Yip, Cancer Res., 2006, 66, 10233–10237 CrossRef.
- L. Fux, N. Ilan, R. D. Sanderson and I. Vlodavsky, Trends Biochem. Sci., 2009, 34, 511–519 CrossRef CAS.
- N. Ilan, M. Elkin and I. Vlodavsky, Int. J. Biochem. Cell Biol., 2006, 38, 2018–2039 CrossRef CAS.
- I. Shafat, M. W. Ben-Arush, J. Issakov, I. Meller, I. Naroditsky, M. Tortoreto, G. Cassinelli, C. Lanzi, C. Pisano, N. Ilan, I. Vlodavsky and F. Zunino, J. Cell Mol. Med., 2011, 15, 1857–1864 CrossRef CAS.
- V. Vreys and G. David, J. Cell Mol. Med., 2007, 11, 427–452 CrossRef CAS.
- E. Cohen, L. Doweck, I. Naroditsky, O. Ben-Lzhak, R. Kremer, L. A. Best, I. Viodavsky and N. Ilan, Cancer, 2008, 113, 1004–1011 CrossRef.
- R. Goldberg, A. Meirovitz, N. Hirshoren, R. Bulvik, A. Binder, A. M. Rubinstein and M. Elkin, Matrix Biol., 2013, 32, 234–240 CrossRef CAS.
- I. Shafat, E. Zcharia, B. Nisman, Y. Nadir, F. Nakhoul, I. Vlodavsky and N. Ilan, Biochem. Biophys. Res. Commun., 2006, 341, 958–963 CrossRef CAS.
- M. Chhabra and V. Ferro, Molecules, 2018, 23, 2971–2981 CrossRef.
- C. Freeman and C. R. Parish, Biochem. J., 1997, 325, 229–237 CrossRef CAS.
- C. Nardella and C. Steinkuhler, Anal. Biochem., 2004, 332, 368–375 CrossRef CAS.
- R. S. Loka, F. Yu, E. T. Sletten and H. M. Nguyen, Chem. Commun., 2017, 53, 9163–9166 RSC.
- R. S. Loka, E. T. Sletten, U. Barash, I. Vlodaysky and H. M. Nguyen, ACS Appl. Mater. Interfaces, 2019, 11, 244–254 CrossRef CAS.
- K. Enomoto, H. Okamoto, Y. Numata and H. Takemoto, J. Pharm. Biomed. Anal., 2006, 41, 912–917 CrossRef CAS.
- J. C. Sistla, S. Morla, A. B. Alabbas, R. C. Kalathur, C. Sharon, B. B. Patel and U. R. Desai, Carbohydr. Polym., 2019, 205, 385–391 CrossRef CAS.
- J. C. Sistla and U. Desai, Bio Protoc., 2019, 9, e3356–e3367 CAS.
- E. Hammond, C. P. Li and V. Ferro, Anal. Biochem., 2010, 396, 112–116 CrossRef CAS.
- H. Ben-Artzi, M. Ayal-Hershkovitz, I. Vlodavsky, I. Pecker, Y. Peleg and D. Miron, Method of Screening for Potential Anti-Metastatic and Anti-Inflammatory Agents Using Mammalian Heparanase as a Probe, US Pat. 6,190,875 B1, 20 Feb 2001.
- S. B. Peterson and J. Liu, Matrix Biol., 2013, 32, 223–227 CrossRef CAS.
- S. B. Peterson and J. Liu, J. Biol. Chem., 2012, 287, 34836–34843 CrossRef CAS.
- Y. Mao, Y. Huang, J. A. Buczek-Thomas, C. M. Ethen, M. A. Nugent, Z. L. L. Wu and J. Zaia, J. Biol. Chem., 2014, 289, 34141–34151 CrossRef CAS.
- S. B. Peterson and J. Liu, J. Biol. Chem., 2010, 285, 14504–14513 CrossRef CAS.
- Y. Okada, S. Yamada, M. Toyoshima, J. Dong, M. Nakajima and K. Sugahara, J. Biol. Chem., 2002, 277, 42488–42495 CrossRef CAS.
- A. G. Pearson, M. J. Kiefel, V. Ferro and M. von Itzstein, Org. Biomol. Chem., 2011, 9, 4614–4625 RSC.
- S. M. Rele, S. S. Iyer, S. Baskaran and E. L. Chaikof, J. Org. Chem., 2004, 69, 9159–9170 CrossRef CAS.
- M. Johannes, M. Reindl, B. Gerlitzki, E. Schmitt and A. Hoffmann-Roder, Beilstein J. Org. Chem., 2015, 11, 155–161 CrossRef.
- F. Belot and J. C. Jacquinet, Carbohydr. Res., 1996, 290, 79–86 CrossRef CAS.
- T. Zhu and G. J. Boons, Carbohydr. Res., 2000, 329, 709–715 CrossRef CAS.
- D. A. Leigh, J. P. Smart and A. M. Truscello, Carbohydr. Res., 1995, 276, 417–424 CrossRef CAS.
- A. Kafle, J. Liu and L. N. Cui, Can. J. Chem., 2016, 94, 894–901 CrossRef CAS.
- K. Setsukinai, Y. Urano, K. Kikuchi, T. Higuchi and T. Nagano, J. Chem. Soc., Perkin Trans. 2, 2000, 2453–2457, 10.1039/B006449l.
- H. M. Burke, T. Gunnlaugsson and E. M. Scanlan, Chem. Commun., 2015, 51, 10576–10588 RSC.
- C. Dubiella, H. Cui and M. Groll, Angew. Chem. Int. Ed., 2016, 55, 13330–13334 CrossRef CAS.
- D. E. Koshland, Biol. Rev. Biol. Proc. Camb. Phil. Soc., 1953, 28, 416–436 CrossRef CAS.
- L. Wu, J. Jiang, Y. Jin, W. W. Kallemeijn, C. L. Kuo, M. Artola, W. Dai, C. van Elk, M. van Eijk, G. A. van der Marel, J. D. C. Codee, B. I. Florea, J. Aerts, H. S. Overkleeft and G. J. Davies, Nat. Chem. Biol., 2017, 13, 867–873 CrossRef CAS.
- W. C. Sun, K. R. Gee and R. P. Haugland, Bioorg. Med. Chem. Lett., 1998, 8, 3107–3110 CrossRef CAS.
- A. Pennacchio, A. Capo, S. Caira, A. Tramice, A. Varriale, M. Staiano and S. D'Auria, Biotechnol. Appl. Biochem., 2018, 65, 89–98 CrossRef CAS.
- E. J. Corey, A. Guzmanperez and M. C. Noe, J. Am. Chem. Soc., 1995, 117, 10805–10816 CrossRef CAS.
- L. Wu, N. Wimmer, V. Ferro and G. J. Davies, ChemRxiv, 2020, DOI:10.26434/chemrxiv.12555272.v1.
- T. M. Duo, K. Robinson, I. R. Greig, H. M. Chen, B. O. Patrick and S. G. Withers, J. Am. Chem. Soc., 2017, 139, 15994–15999 CrossRef CAS.
- D. Gilat, R. Hershkoviz, I. Goldkorn, L. Cahalon, G. Korner, I. Vlodavsky and O. Lider, J. Exp. Med., 1995, 181, 1929–1934 CrossRef CAS.
- D. W. Fink and W. R. Koehler, Anal. Chem., 1970, 42, 990–993 CrossRef CAS.
- E. A. McKenzie, Br. J. Pharmacol., 2007, 151, 1–14 CrossRef CAS.
- V. Masola, M. F. Secchi, G. Gambaro and M. Onisto, Curr. Cancer Drug Targets, 2014, 14, 286–293 CrossRef CAS.
- R. D. Sanderson, M. Elkin, A. C. Rapraeger, N. Ilan and I. Vlodavsky, FEBS J., 2017, 284, 42–55 CrossRef CAS.
- A. Messore, V. N. Madia, L. Pescatori, F. Saccoliti, V. Tudino, A. De Leo, M. Bortolami, D. De Vita, L. Scipione, F. Pepi, R. Costi, S. Rivara, L. Scalvini, M. Mor, F. F. Ferrara, E. Pavoni, G. Roscilli, G. Cassinelli, F. M. Milazzo, G. Battistuzzi, R. Di Santo and G. Giannini, J. Med. Chem., 2018, 61, 10834–10859 CrossRef CAS.
- V. N. Madia, A. Messore, L. Pescatori, F. Saccoliti, V. Tudino, A. De Leo, M. Bortolami, L. Scipione, R. Costi, S. Rivara, L. Scalvini, M. Mor, F. F. Ferrara, E. Pavoni, G. Roscilli, G. Cassinelli, F. M. Milazzo, G. Battistuzzi, R. Di Santo and G. Giannini, J. Med. Chem., 2018, 61, 6918–6936 CrossRef CAS.
- C. D. Mohan, S. Hari, H. D. Preetham, S. Rangappa, U. Barash, N. Ilan, S. C. Nayak, V. K. Gupta, Basappa, I. Vlodavsky and K. S. Rangappa, Iscience, 2019, 15, 360–390 CrossRef CAS.
- E. Hammond, A. Khurana, V. Shridhar and K. Dredge, Front. Oncol., 2014, 4, 195 Search PubMed.
- Cisbio Heparanase Assay Toolbox. Data can be found at https://www.cisbio.com/usa/drug-discovery/heparanase-assay-toolbox, accessed: Jan 12, 2020.
- J. H. Zhang, T. D. Y. Chung and K. R. Oldenburg, J. Biomol. Screen, 1999, 4, 67–73 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc04872k |
| This journal is © The Royal Society of Chemistry 2021 |