
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmyloid binding and beyond: a new approach for Alzheimer's disease drug discovery targeting Aβo–PrPC binding and downstream pathways†
James D.
Grayson
a,
Matthew P.
Baumgartner
b,
Cleide Dos
Santos Souza
a,
Samuel J.
Dawes
ac,
Imane Ghafir
El Idrissi
d,
Jennifer C.
Louth
a,
Sasha
Stimpson
a,
Emma
Mead
e,
Charlotte
Dunbar
e,
Joanna
Wolak
e,
Gary
Sharman
e,
David
Evans
 e,
Anastasia
Zhuravleva
c,
Margarita Segovia
Roldan
g,
Nicola Antonio
Colabufo
df,
Ke
Ning
g,
Claire
Garwood
g,
James A.
Thomas
a,
Benjamin M.
Partridge
a,
Antonio
de la Vega de Leon
h,
Valerie J.
Gillet
h,
Amélia P.
Rauter
i and
Beining
Chen
*a
e,
Anastasia
Zhuravleva
c,
Margarita Segovia
Roldan
g,
Nicola Antonio
Colabufo
df,
Ke
Ning
g,
Claire
Garwood
g,
James A.
Thomas
a,
Benjamin M.
Partridge
a,
Antonio
de la Vega de Leon
h,
Valerie J.
Gillet
h,
Amélia P.
Rauter
i and
Beining
Chen
*a
aDepartment of Chemistry, University of Sheffield, Brookhill, Sheffield S3 7HF, UK. E-mail: b.chen@sheffield.ac.uk
bComputational Chemistry and Cheminformatics, Eli Lilly and Company, Lilly Biotechnology Center, San Diego, CA 92121, USA
cFaculty of Biological Sciences, University of Leeds, Leeds LS2 9JT, UK
dUniv Bari, Biofordrug, Via Edoardo Orabona 4, I-70125 Bari, Italy
eComputational Chemistry and Chemoinformatics, Eli Lilly and Company, Erl Wood, Windlesham, GU20 6PH, UK
fUniv Bari, Dipartimento Farm Sci Farmaco, Via Edoardo Orabona 4, I-70125 Bari, Italy
gSheffield Institute of Translational Neuroscience, University of Sheffield, Sheffield S10 2HQ, UK
hInformation School, University of Sheffield, Sheffield S1 4DP, UK
iCentro de Química Estrutural, Faculdade de Ciências, Universidade de Lisboa, ED C8, 5 piso, 1749-016 Lisboa, Portugal
First published on 1st February 2021
Abstract
Amyloid β oligomers (Aβo) are the main toxic species in Alzheimer's disease, which have been targeted for single drug treatment with very little success. In this work we report a new approach for identifying functional Aβo binding compounds. A tailored library of 971 fluorine containing compounds was selected by a computational method, developed to generate molecular diversity. These compounds were screened for Aβo binding by a combined 19F and STD NMR technique. Six hits were evaluated in three parallel biochemical and functional assays. Two compounds disrupted Aβo binding to its receptor PrPC in HEK293 cells. They reduced the pFyn levels triggered by Aβo treatment in neuroprogenitor cells derived from human induced pluripotent stem cells (hiPSC). Inhibitory effects on pTau production in cortical neurons derived from hiPSC were also observed. These drug-like compounds connect three of the pillars in Alzheimer's disease pathology, i.e. prion, Aβ and Tau, affecting three different pathways through specific binding to Aβo and are, indeed, promising candidates for further development.
Introduction
Dementia is a family of age-related, incurable, and debilitating conditions which are characterised by a serious loss of cognitive ability beyond normal ageing that affects both men and women.1 Currently, 50 million people are suffering from dementia globally and this number is expected to increase to over 152 million by 2050.2 The global healthcare cost of dementia in 2018 escalated to $1 trillion dollars and is expected to double to $2 trillion by 2030. This hampers social and economic development and overwhelms health and social services, including the long-term care service.2Alzheimer's disease (AD) accounts for about 80% of all dementia cases. The onset of AD normally occurs in the later stages of human life (60–70 years old) and is triggered by many different pathological and environmental factors. The accumulations of the protein fragment β-amyloid (called β-amyloid plaques) deposited outside neurons, and an abnormal form of the protein tau (called tau tangles) accumulated inside neurons are two of several pathological changes associated with AD.3 β-amyloid plaques are believed to contribute to cell death by interfering with neuron-to-neuron communication at synapses,4–9 while tau tangles block the transport of nutrients and other essential molecules inside neurons.10,11 Most AD cases are of sporadic origin and the onset of cognitive behavioural impairment occurs well before clinical symptoms are seen.12
Over the past three decades, no single curative treatment for AD has been developed although some reached advanced clinical trial stages. For example, solanezumab, a monoclonal antibody targeting the central epitope of monomeric amyloid-β, KLVFFAED, with picomolar affinity, was developed by Eli Lilly in 2002.13–15 Solanezumab entered Phase II clinical trials in 2006, and was withdrawn from development in 2018 after over 15 years of studies and a billion dollar investment.16 The latest casualty is aducanumab from Biogen which is another monoclonal antibody targeting the aggregated forms of Aβ amyloid. Biogen halted development of the drug in March 2019 after preliminary data from two Phase III trials suggested it would not meet the primary endpoint. However, in October 2019 the company announced their intention to seek regulatory approval, following a reanalysis of the data.17
There are many reasons for such a high failure rate in AD drug discovery and development.18 The most pressing one is that the vast majority of current therapeutic approaches as demonstrated above only focus on a single target (mainly around amyloid beta) which alone is insufficient to cure AD, a complex disease with multiple causes.19 While amyloid beta is still a viable and clinically validated anti-oligomeropathy drug target, we believe that looking beyond just amyloid binding, i.e. the downstream effects of Aβ rather than on its accumulation and aggregation alone, especially how the binding affects its binding partners and other related signalling pathways, may be advantageous in improving the success rate of drug discovery for AD and produce urgently needed therapies.
It is commonly accepted that pathogenic amyloid beta (Aβ or Abeta), Aβ1-42, is the main component of the amyloid plaques found in the brains of Alzheimer patients.4,16 This peptide is the product of proteolytic cleavage of the amyloid precursor protein (APP) by β-secretase and γ-secretase.20,21 Monomeric Aβ1-42 can aggregate to form flexible soluble oligomers which may exist in several forms from small oligomers to fibrils.8 The formation of oligomeric species precedes the formation of amyloid plaques and the presence of protofibrils and oligomers correlates well with the neurotoxicity showing that oligomers have played a critical role in the pathogenesis and progression of AD (oligomeropathy).22–24 In addition, the misfolded oligomers (known as “seeds”) can induce Aβ molecules to also take the misfolded oligomeric form, leading to a chain reaction akin to a ‘prion’ infection.25–27 The other key protein, tau, which is also involved in AD forms ‘prion-like’ misfolded oligomers (Tauopathy).28 It is also believed that there is a connection between oligomeropathy and tauopathy, i.e. misfolded Aβ oligomers can induce tau to misfold.10,23
In addition, several receptors for Aβ1-42 oligomers (Aβo will be used throughout this paper unless otherwise stated) at synapses were discovered. Notably, the cellular prion protein (PrPC) has high affinity for Aβo.29 PrPC, a glycosylphosphatidylinositol (GPI)-anchored protein of 231 amino acids encoded by the PrnP gene located on chromosome 20 in humans, is widely expressed in the central nervous system during early development, and in adult neurons and glial cells. In the adult brain, maximal PrnP mRNA expression is observed in the neocortex and cerebellum. Although its involvement in transmissible spongiform encephalopathies is well known, PrPC is thought to be related to several normal and abnormal physiological processes.11 In AD, conditional deletion of the PrnP gene by anticancer drug, tamoxifen,30,31 rescued synapse loss in APP/PS1 mice models. The interaction between PrPC and Aβo appears to be involved in maintaining cognitive impairment in later stages of AD and endogenous or, synthetic ligands of PrPC interrupt Aβo mediated signalling and prevent neurotoxicity in neurons.12,29,32 Evidence also shows that PrPC deletion influences tau hyperphosphorylation because Fyn has been linked to somatodendritic accumulation of Tau. Therefore, cellular prion protein, PrPC, plays a vital role in the central dogma of AD aetiology connecting oligomeropathy with Tauopathy. Therefore, Aβ oligomers, PrPC, Fyn and Tau could all become potential polypharmacological drug discovery targets for AD.33
Here, we present a combined computational, biophysical, biochemical, and cellular effort in developing novel drug discovery approaches against AD. We developed a virtual screening strategy for identifying putative Aβo binders. We identified compounds reported as inhibitors for Aβ in the public domain and in the literature and used these compounds to search a library of fluorine-containing compounds using a fragment-based approach. Sub-libraries of compounds suggested from virtual screening were examined in a 19F NMR assay to identify compounds that bind to Aβo. The hit compounds were further tested in a number orthogonal NMR based assays to confirm their specific binding to Aβo.
Furthermore, we have identified several promising chemical scaffolds and tested them in cellular assays to validate their bindings and biological activities. We have developed a cellular-based Aβo–PrPC binding assay using HEK293 cell line. The selected compounds were also tested in more disease relevant models using induced human pluripotent stem cells (ihPSC) for their abilities in inhibiting hyperphosphorylations of Fyn and Tau. Together with an enzymatic BACE assay, these compounds were shown to specifically bind to Aβo, disrupt the Aβo–PrPC interaction, inhibit hyperphosphorylation of Fyn and Tau.
Results and discussion
Computational library design
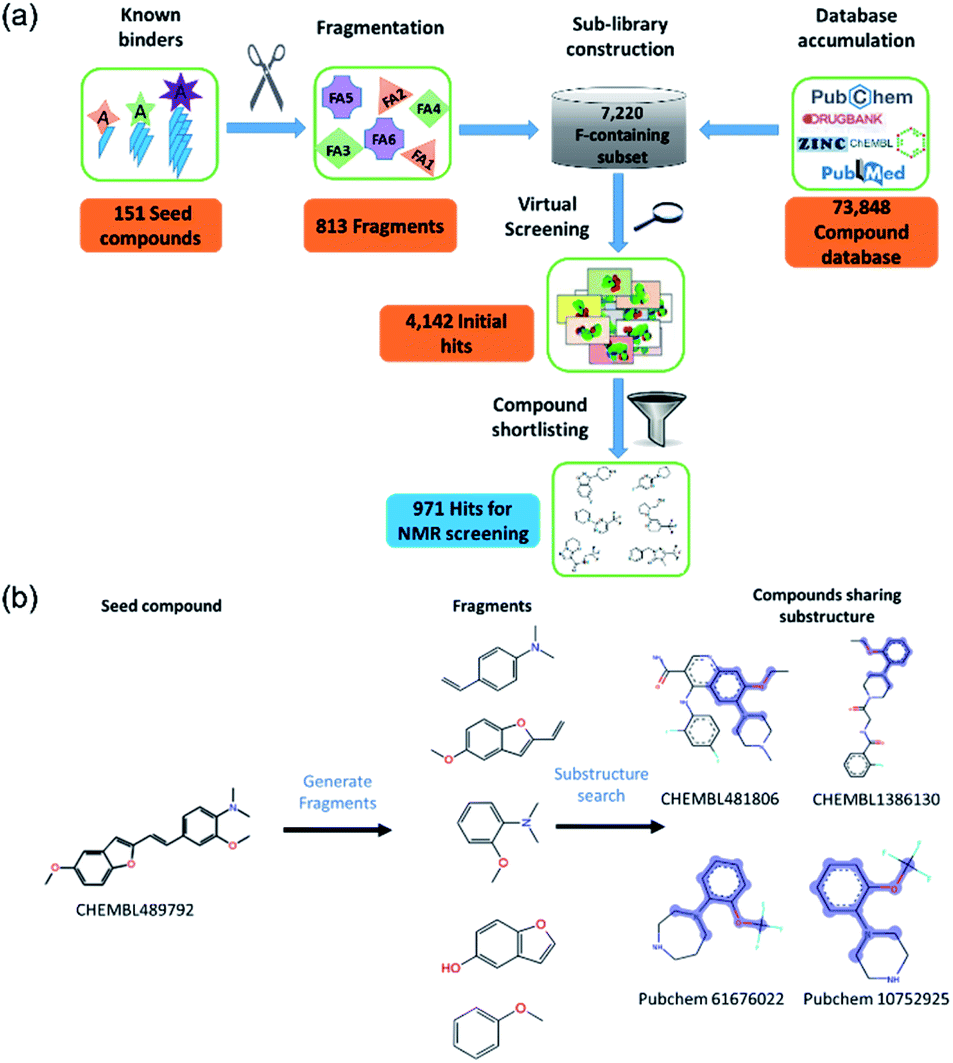 | ||
| Fig. 1 Computational design of fluorine containing antioligomeropathy compound library. (a) Flowchart for database construction and screening cascade. (b) A schematic illustration of computational fragment-based approach from a known Aβ binder CHEMBL489792. | ||
We identified 151 known Aβo inhibitors from the literature and public databases (ESI Table 1†). These seed compounds were fragmented and then these fragments (after filtering very simple fragments like phenyl rings) were used to perform a substructure search on a tailored chemical library consisting of 7220 fluorine-containing compounds. This library was a subset of a larger library of 73![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 848 compounds constructed from public databases such as ChEMBL, PubChem, Drugbank, and ZINC. The sub-library consists of compounds containing at least one fluorine atom and the compounds were available in Lilly's internal inventory (to allow for rapid procurement of the compounds). This fluorine subset was specifically selected to allow 19F NMR screening to be employed.35
848 compounds constructed from public databases such as ChEMBL, PubChem, Drugbank, and ZINC. The sub-library consists of compounds containing at least one fluorine atom and the compounds were available in Lilly's internal inventory (to allow for rapid procurement of the compounds). This fluorine subset was specifically selected to allow 19F NMR screening to be employed.35
The initial substructure search against the F-containing chemical library yielded 4142 initial hits from which 2000 diverse compounds were selected. Further assessment on their predicted solubility resulted in 971 compounds which were taken forward into the 19F NMR screening (ESI Table 2†). The design cascade can be illustrated using a known Aβ binder CHEMBL489792 as an example (Fig. 1b). CHEMBL489792 is an aminostyrybenzofuran derivative which was reported to be a potent inhibitor for Aβ fibril formation with IC50 of 0.07 mM in thioflavin T (ThT) assay.36 Fragments were generated from this compound and these fragments were screened against the complied fluorine-containing database subset resulting in compounds that share the identified fragment substructures.
848 compounds was applied to the 8 hits from the initial 19F NMR screening in order to produce more analogues in the hit expansion exercise. 36 fluorine and non-fluorine containing structural analogues were selected computationally (ESI Table 4†). These compounds were screened in a competition assay using the 19F NMR technique employed in the primary screening. This round of hit expansion exercise yielded 6 more hit compounds (Table 2). All 14 hits were then further validated and characterised by STD NMR to give 9 confirmed hits of which 6 were progressed to biological validation.
|
| Entry | Library location | Seed compound public ID | Analogue public ID | Chemical structure | Height reduction | Noise | Reduction/noise | Height decrease (%) |
|---|---|---|---|---|---|---|---|---|
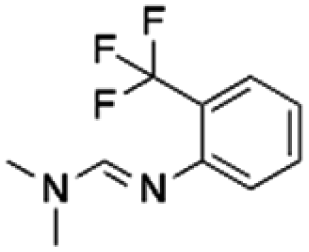
| 1 | 1009 | CHEMBL1673279 | PBCHM45210798 |
|
153.86 | 37.44 | 4.11 | 8.74 |
| 2 | 1048 | PBCHM3049683 | PBCHM57223647 |
|
167.81 | 37.10 | 4.52 | 2.96 |
| 3 | ZINC00159801 |
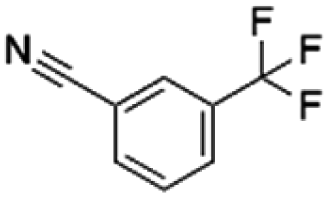
|
130.88 | 37.44 | 3.50 | 2.32 | ||
| 4 | ZINC00057047 |
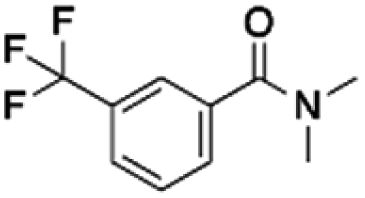
|
75.89 | 37.48 | 2.02 | 1.32 | ||
| 5 | 1055 | PBCHM120765 | CHEMBL448523 |
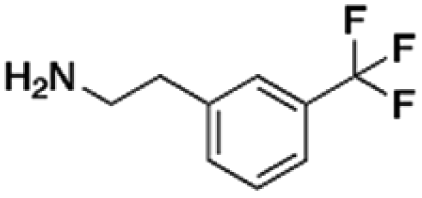
|
298.75 | 36.90 | 8.10 | 5.91 |
| 6 | ZINC00120199 |
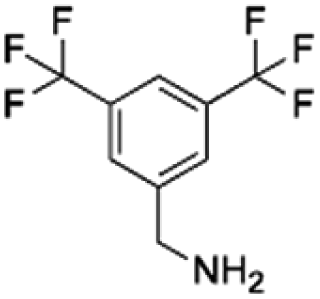
|
159.16 | 36.94 | 4.31 | 1.63 |
NMR screening and characterisation of binding
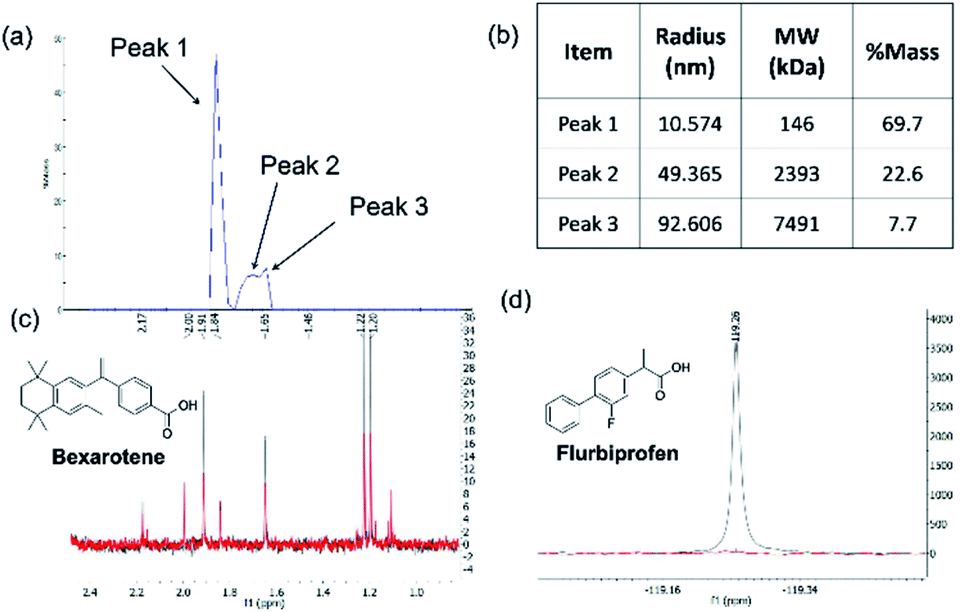 | ||
| Fig. 2 Aβ1-42 oligomer preparation and validation of NMR protocols. (a) size exclusion profile of the preparation over 1 hour (b) size of the oligomers analysed by dynamic light scattering (DLS) technique; (c) Section of 1H NMR spectra of 50 μM bexarotene in the presence (red) and absence (blue) of 1 μM of Aβo. The reduction of peak height upon the addition of the Aβ1-42 oligomer preparation indicates an interaction between bexarotene and the oligomer preparation; (d) section of 19F NMR spectra of 50 μM flurbiprofen in the presence (blue) and absence (red) of 1 μM of bovine serum albumin (BSA). | ||
The same protocol was applied to a model system to test if the experimental conditions for bexarotene and Aβo were applicable for 19F NMR screening of virtual hits from computer modelling. A model system consisting of bovine serum albumin (BSA) and its known fluorine-containing binding ligand, flurbiprofen, was constructed. The flurbiprofen is a nonsteroidal anti-inflammatory agent (NSAIA) with antipyretic and analgesic activity. It is an analogue of ibuprofen and >99% of it was bound to albumin after administration.49,50 Suppression of the 19F signals (Fig. 2d) of flurbiprofen upon BSA binding clearly demonstrated that the 19F NMR protocol can be used as a primary screening tool for virtual hits against Aβo prepared.
19F CPMG NMR is becoming an increasingly popular tool in the field of drug screening51 because each 19F atom, which is a 100% naturally abundant fluorine NMR-visible isotope, is absent in biomolecules such as proteins, F-containing compounds generates a unique chemical shift in the spectrum which is often simple with no erroneous background noise and interference from other signals, and hence can be easily identified.52 Moreover the fluorine nucleus is very sensitive to changes in the chemical environment, and can be a very sensitive probe even for the weakest binders. In addition, the large chemical shift range and the strong chemical shift anisotropy make it very simple to measure mixtures of up to 30 fragments in a single sample. When a small molecule is bound to a large protein, the signal intensity of the binding ligand is significantly attenuated by Carr–Purcell–Meiboom–Gill (CPMG) or spin-lock pulse due to interaction with protein. In this experiment, the 19F NMR methodology provides a sensitive tool not only for probing compound binding to Aβo, but also for rapidly deconvoluting library members from the mixture due to the unique shift of each library member in 19F NMR spectra.53 Therefore, the throughput of the NMR screening can be increased and the amount of Aβo required for the screening reduced. The only disadvantage of this technique is that compounds tested (in direct binding measurement at least) need to have at least one fluorine atom.
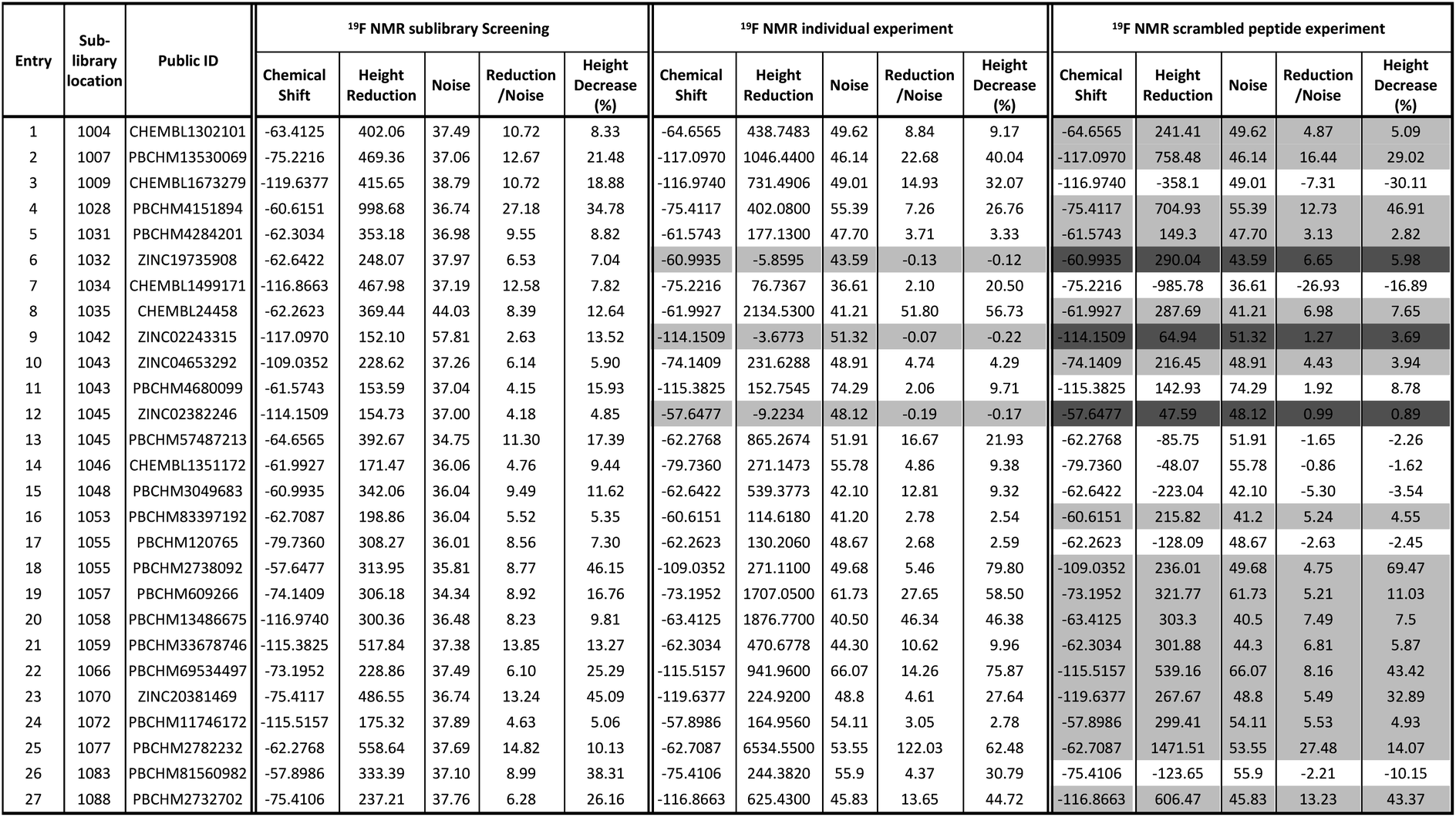
The 19F CPMG NMR experimental protocol described above was then used to screen compounds in 91 fluorine-containing sub-libraries in the presence and absence of Aβo. An example of sub-library screening is illustrated in Fig. 3. A section of spectra of three compounds from the 19F CPMG NMR experiment of sub-library mixture 1045 containing six compounds in the absence (blue) and the presence (red) of Aβo is shown in Fig. 3a. It was seen that compound PBCHM57487213 showed a noticeable reduction in the peak height.
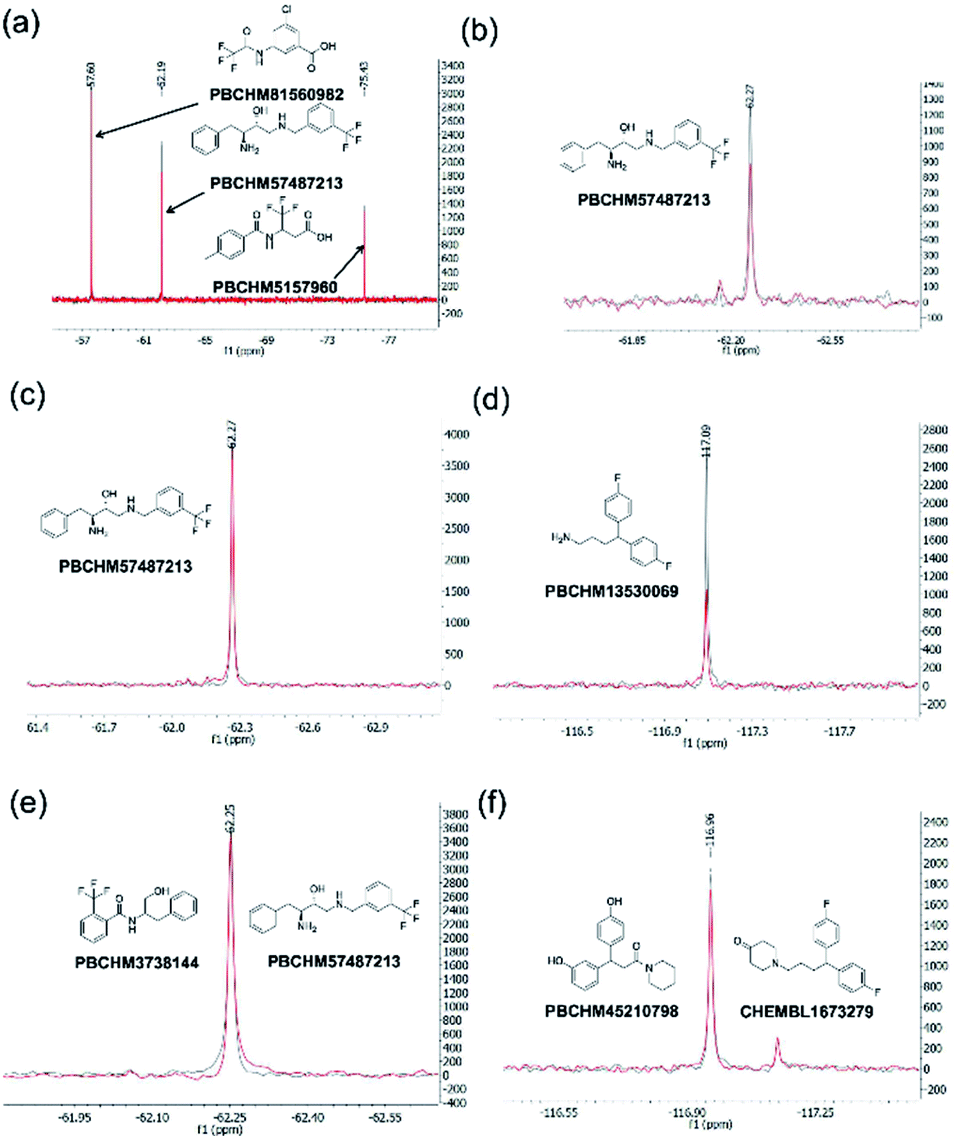 | ||
| Fig. 3 19F CPMG NMR screening. (a) Section 19F CPMG NMR spectra of sub-library 1045 containing 50 μM of compound ZINC02382246 (−57.6 ppm), PBCHM57487213 (−62.3 ppm) and PBCHM5157960 (−75.5 ppm) in the presence (red) and absence (blue) of 1 μM (concentration as monomer) Aβo; (b) section 19F CPMG NMR spectra of PBCHM57487213 (−62.3 ppm) in the presence (red) and absence (blue) of 1 μM (concentration as monomer) Aβo in the individual 19F NMR confirmation run; (c) section 19F CPMG NMR spectra of PBCHM57487213 (−62.3 ppm) in the presence (red) and absence (blue) of 1 μM (concentration as monomer) scrambled peptide; (d) section 19F CPMG NMR spectra of PBCHM13530069 (−62.3 ppm) in the presence (red) and absence (blue) of 1 μM (concentration as monomer) scrambled peptide; (e) 19F CPMG NMR spectra of 50 μM of seed compound PBCHM57487213 binding to 1 μM (concentration as monomer) Aβo in competition with 50 μM of analogue PBCHM3738144 in the presence (red) and absence (blue) of the analogue compound in the hit expansion experiment; (f) 19F CPMG NMR spectra of 50 μM of seed compound CHEMBL1673279 binding to 1 μM (concentration as monomer) Aβo in competition with 50 μM of analogue PBCHM45210798 in the presence (red) and absence the analogue compound in the hit expansion experiment. | ||
In order to assess if any changes in peak intensity are statistically significant, the ratio of peak reduction/noise and % of peak height reduction are calculated for each compound. The ratio of peak reduction/noise represents the significance of the peak reduction in reference to the base-line noise. A ratio of >2 has been assigned as the cut-off for a positive hit. The larger the ratio, the more reliable the data is. The percent peak reduction gives an indication of the relative binding abilities of compounds in each sub-library, therefore was used as a key parameter for measuring binding strength.
For each compound, hit compound entry, its sub-library number where it was tested, public ID, chemical shift, reduction in the peak height upon binding to Aβo, baseline noise, signal reduction/noise ratio as well as percent of peak height reduction compared to the peak height in the absence of Aβo are collected, presented and analysed. The 27 compounds that showed positive responses in the presence of Aβ are shown under 19F NMR sublibrary screening column in Table 1.
It can be seen that 27 hit compounds came from 24 sub-libraries and displayed a spectrum of abilities in peak height reduction. Judged by % of peak height reduction, four compounds (Table 1, entries 4, 18, 23 and 26) produced over 30% reduction in peak height, hence are classified as strong binders. 3 compounds (Table 1, entries 2, 22, and 27) reduced peak height by over 20%, hence are classified as medium binders. The rest of the compounds gave between 5 and 16% reduction in peak height in the presence of Aβo making them weak binders. Although the binding ability is arbitrarily assigned, it provides a tool for ranking the binding significance of the compounds. It is tempting to conclude stronger binders generally display a bigger ratio of peak reduction/noise than weaker binders: fraction bound will indeed be one component of the response, but other factors may also be important, so too much significance should not be placed on this. The chemical structures for all 27 hit compounds with their public IDs are shown in Fig. 4.
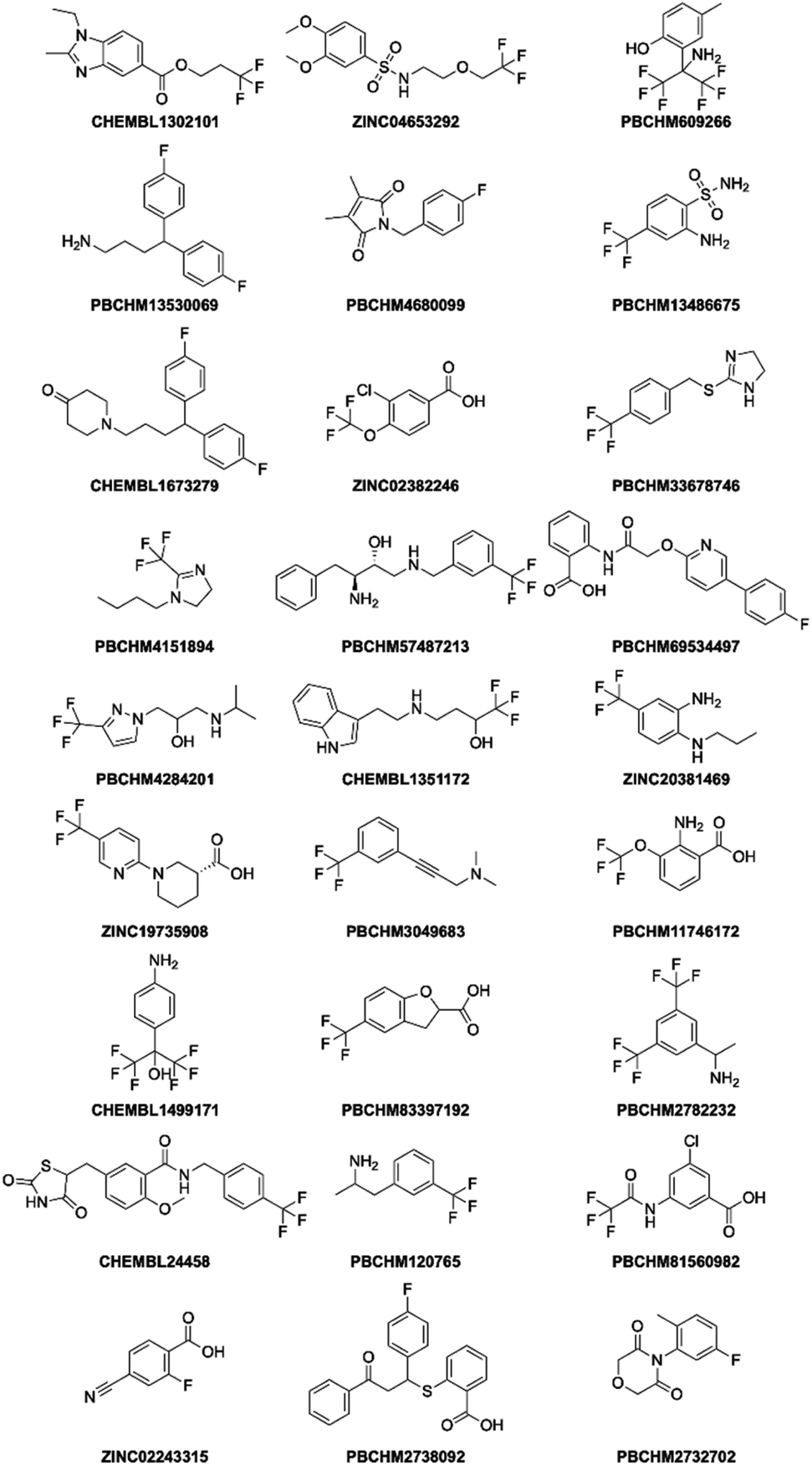 | ||
| Fig. 4 Structure of 27 hits from sub-library screening. | ||
To remove false positives caused by compound–compound interactions, compounds which were active within the mixture screen were re-tested via the 19F CPMG NMR experiment individually under the same conditions as they were screened in the library mixtures and a sample is shown in Fig. 3b. Using the same criteria and cut-offs applied in the primary sub-library screening, 24 out of 27 compounds showed reductions in the peak height in the presence of Aβo in the individual binding experiments, hence were deemed to be confirmed hit binders (under 19F NMR individual experiment column Table 1). It is interesting to observe that the binding of most compounds (Table 1, entries 1, 4, 10, 13, 14, 15, 21, 24 and 26) to Aβo in the individual confirmation experiment remained at a similar level as displayed in the primary sub-library screening. This demonstrates that the binding of these compounds was not affected by other compounds present in the library. Compound entries 2, 3, 7, 8, 13, 18, 19, 20 and 27 showed noticeable increases (>50%) in the % peak height reduction, when compared with the reduction seen in the sublibrary screening, showing that presence of other compounds in the sub-library inhibits their binding to Aβo. Compound entries 5, 11, 16, 17, 21, and 23 displayed significant reduction in the % peak height reduction when compared with the reduction seen in the sublibrary screening, showing that the presence of other compounds in the sub-library enhances their binding to Aβo.
It is worth mentioning that compound entries 10 and 11, belong to the same sub-library (Library 1043), but behave differently in the individual experiments. The presence of compound entry 10 seems to enhance the binding of compound entry 11 to Aβo while the presence of compound entry 11 does not seem to affect the binding of compound entry 10. However, the interference of other compounds in the sub-library cannot be completely ruled out. Compound entries 12 and 13 (Library 1045) displayed the opposite influence of one compound to the other when compared with compound entries 10 and 11. Compound entries 17 and 18 (Library 1055) affected each other significantly when present in the sub-library mixture. The presence of compound entry 17 significantly inhibits the binding of compound entry 18 to Aβo in the sub-library while the presence of compound entry 18 significantly enhances the binding of compound entry 17. This shows that they not only bind to Aβo individually, but also together they exhibit the strongest effects of compound–compound interaction upon the binding compared with the other two pairs. Three compounds (Table 1, entries 6, 9 and 12) displayed a negligible reduction in peak height when re-tested individually. This shows their bindings to Aβo rely on their interaction with other compounds in the corresponding sub-libraries. It may also mean that they are interacting with each other. Ligand–ligand interactions cannot be ruled out either.
The number of compounds which belong to strong binders increased from 4 to 11 and the % of peak reduction of some compounds has even doubled (Table 1, entries 2, 8, 19, 20, 22 and 25). The number of compounds in the medium-binder category increased by 1 and the number of weak binders reduced by 8. 3 compounds (Table 1, entries 6, 9 and 12) became non-binders and were removed from the hit list.
To avoid false positives arising from non-specific binding, these 27 initial hits were also subjected to a counter screen against a scrambled (non-amyloidal) peptide which has the same amino acid composition and peptide chain length, but a different primary sequence to Aβ1-42. The scramble was prepared using the same protocol as that of Aβo from Aβ1-42.
A counter screen of each compound against a scrambled peptide was also carried out in individual experiments using the same 19F CPMG NMR technique. The scrambled peptide possesses the same composition of amino acids, but different primary sequence as that of disease-causing Aβ1-42 sequence. It does not aggregate to form amyloid fibrils, therefore is often used as a control peptide to examine the specificity for Aβ binders. The scrambled peptide was subjected to the same preparation protocol as that of Aβo prepared from synthetic Aβ1-42 monomers. The binding between the hit compounds and the scrambled peptide was performed using the same protocol as the one used for Aβo binding.
All 27 initial 19F NMR sub-library hits (including the 3 negative hits in the individual confirmation experiment) were tested against the scrambled peptide and data is displayed in the “19F NMR scrambled peptide experiment” column in Table 1. An example of spectra for compound PBCHM57487213 (Table 1, entry 13) in the absence (blue) and presence (red) of scramble peptides are shown (Fig. 3c). No change in the peak height reduction was observed demonstrating that it does not bind to the scrambled peptide, hence is a specific binder for Aβo. An example of non-specific Aβo binder can be seen with compound PBCHM13530069 (Table 1, entry 2). This compound a positive hit in the initial sub-library screening as well as the individual confirmation 19F-CPMG NMR screening (Fig. 3d). However, it showed significant binding to the scramble peptide, hence is removed from the list of compounds for further investigation.
Out of 27 compounds, 11 were identified as specific Aβo binders that have a ratio of peak height reduction/noise less than 2 in the presence of scrambled peptide with negative or small % of peak height reduction. Compound entries 9 and 12, having already shown to be non-binders in the individual NMR experiment, also displayed no binding to the scrambled peptide. Notably, compound entry 6, also a non-binder in the individual experiment, displayed significant binding to the scrambled peptide. A compound can only be classified as a hit if they showed peak height reduction in both sub-library screening, individual experiment, and no peak reduction in the scrambled peptide experiment. Hence, compound entry 6 was also removed from the hit list. This leaves 8 confirmed hits in total (Table 1, entries 3, 7, 11, 13, 14, 15, 17, 26).
From the initial 614 compound library to 8 confirmed hits, a hit rate of 1.3% was obtained which is higher than average random high throughput campaign hit rate (0.01% and 0.14%).54 These hits were taken forward to the hit expansion experiment.
848 compounds. These compounds are non-proprietary, commercially available and were in stock in the Lilly inventory.
The 36 analogues were screened against their respective “seed/parent compounds” in a competition experiment using 19F NMR technique employed in the primary screening to rapidly acquire data and compound deconvolution.55 A solution was prepared containing the same concentration of the seed compound and its corresponding analogue, and the change in peak height of the original F-containing seed compound in the presence of its analogue and Aβo was analysed. If the original seed compound still displayed a reduction of peak height twice that of the noise in the presence of the structural analogue, it was deemed that the analogue displayed specific binding to the same location as the seed compound. In the competition experiment between seed compound PBCHM57487213 and its near neighbour analogue PBCHM3738144, it is clear that there is no competition between the compounds as no peak height reduction is observed (Fig. 3e). This indicated that the analogue PBCHM3738144 is either a weaker binder than its seed compound PBCHM57487213 or it binds to a different site from where its seed compound binds and does not interrupt the binding of the seed compound. Conversely, analogue PBCHM45210798 clearly competes with its seed compound CHEMBL1673279 (Fig. 3f) suggesting that this analogue binds at the same site on Aβo as its seed compound. Its binding strength is strong enough to compete its seed compound partially off. From the expansion competition experiment, 6 out of 36 compounds were identified as potential binders. Their original location, seed compound public ID, analogue public ID, chemical structure, NMR data on competition assay (peak height reduction, noise, reduction/noise ratio, % peak height reduction) are shown in Table 2.
Although this competition experiment would suggest competitive binding between the analogue and the seed for the same site on Aβo it does not eliminate the potential for the analogues to allosterically inhibit the seed compound. From this expansion experiment, 6 further hit compounds were identified as potential binders (Table 2) giving a total of 14 hit compounds from the 19F CPMG NMR experiments described above. The hit rate has increased by 13-folder, from 1.3% in the primary screening to 17% in the hit expansion exercise. This hit rate enhancement demonstrates the viabilities of both the computational and NMR approaches.
Characterisation of the binding using saturation transfer difference (STD) NMR
To further validate the hit compounds obtained from the initial 19F NMR screening, a STD screen was used as an orthogonal binding study to characterise binding affinity and identify hot spots on the molecule that are involved in the binding.56Saturation Transfer Difference (STD)-NMR is often employed during the drug discovery process as a method of binding validation which is complementary to 19F NMR screening. STD NMR is increasingly used as a semi-quantitative method for epitope mapping on ligand moieties interacting with the protein.57 An STD NMR experiment starts with the selective irradiation of the protons of the large biomolecule, such as a protein, using Gaussian Rf pulses. The resulting Rf saturation is then rapidly propagated across the entire protein through a spin diffusion effect via non-scalar magnetization transfer. If a smaller molecule ligand binds the receptor, saturation will also spread onto the ligand. As a result, intensity of the proton signals on the ligand will be attenuated. Subtraction of resulting spectrum from a reference spectrum without saturation yields the STD spectrum containing only signals of the binding ligand.56 STD NMR can be used to characterise weak ligand binding (Kd ∼ mM to μM) and map the hot spots on the ligand that are involved in the binding to large protein molecules (MW > 20 kDa).57 It does not require expensive stable isotopes or radioisotope labelling and only requires small amount of protein (nM to pM), hence is an economical method to analyse protein–ligand interactions especially when a large quantity of the proteinunder study is not achievable. During the experiment, the small molecules are usually used in large excess (20–1000 times excess) of the protein concentration.
The STD-NMR protocol used in the current study was verified by using two model systems: BSA and its known small ligand binder, Iburpofen (Fig. 5a), and Aβo and bexarotene (Fig. 5b). Examples of STD NMR spectra of two hit compounds are shown in Fig. 5c and d.
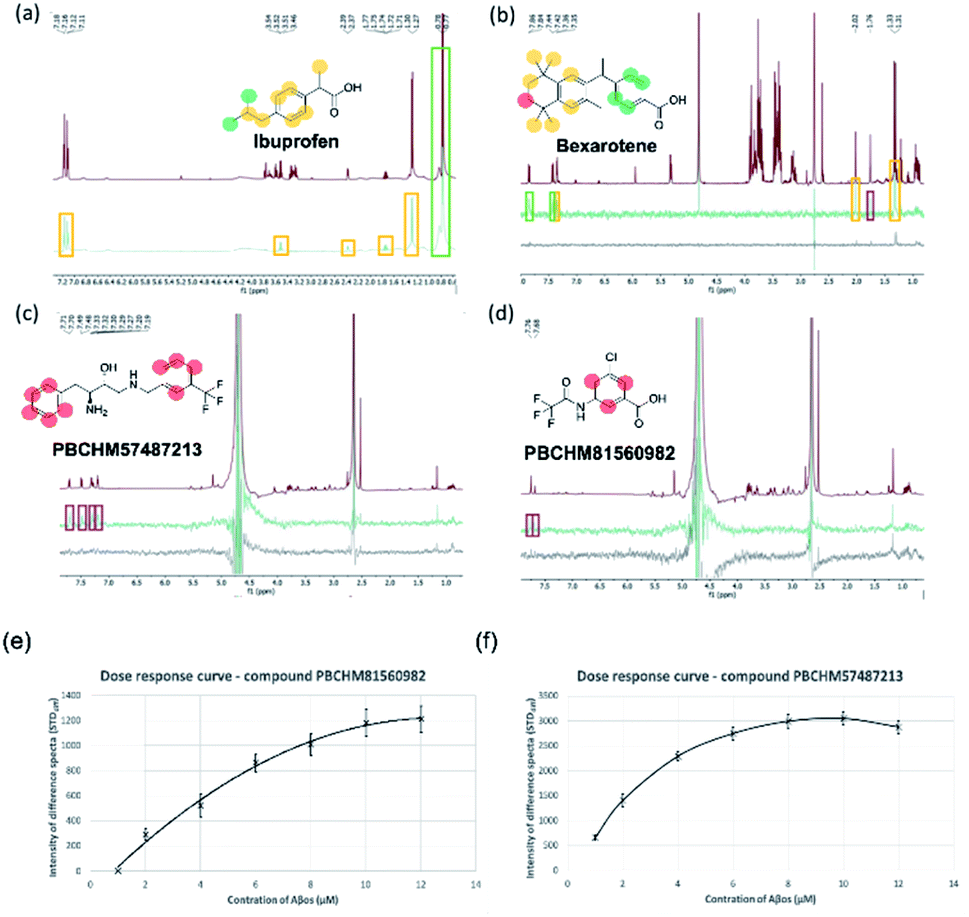 | ||
| Fig. 5 STD NMR characteristics (a) section of an STD spectra of 200 μM ibuprofen and 2 μM of bovine serum albumin (BSA) containing a glucose (200 μM) control. Reference spectra (grey) and on-resonance spectra (black); (b) section of an STD spectra of 200 μM bexarotene and 2 μM (concentration as monomer) Aβo. Reference spectra (red), on-resonance spectra (green), control (black). (c) STD NMR of 200 μM of PBCHM57487213 and 2 μM (concentration as monomer) Aβo. (d) STD NMR of 200 μM of compounds PBCHM81560982 and 2 μM (concentration as monomer) Aβo. Reference (red) spectra, on-resonance spectra (green), control (black). Color scheme on the structure: ranges of values for %STD moiety analysis: red = ≤5%, yellow = ≥10%, green = >10%. (e) Dose response curve – compound PBCHM57487213; (f) dose response curve – compound PBCHM81560982. | ||
In each of the model systems, the STD NMR experiment produces a “difference” spectrum by subtracting two contrasting “saturation” spectra. The first NMR spectra recorded is an off-resonance saturation (no protein saturation ‘STDoff’) spectra where the excitation pulse is away from the protein 1H signal. This STDoff spectra is referred to as the “reference” spectra as it is comparable with a standard 1H-NMR spectrum of the ligand (Fig. 5a (grey), Fig. 5b–d (red)). The second NMR spectra is an on-resonance saturation (selective protein proton saturation ‘STDon’) spectra where the excitation pulse is directed at a known protein 1H signal and does not interact directly with the added ligand. This STDon spectra displays decreases in peak intensity for the bound ligands. This is due to the transfer of energy from the excited protein to the ligands in closest proximity via the proton network. The difference spectra (STDdiff) is the subtraction of the STDoff spectra from the STDon (STDdiff = STDoff − STDon) (Fig. 5a (black), Fig. 5b–d (green)). The STDdiff highlights the small changes in peak height from the STDoff to the STDon that are not identifiable.
Finally, to eliminate potential ligand–ligand interactions interfering with the experiment the same excitation pulses and STDdiff were recorded without the presence of a protein and referred to as a “control” (Fig. 5b–d (black)). If a response was seen in the “control” it was believed to be due to ligand–ligand interactions and this false positive discarded.
From the STD-NMR experiment, additional information on the closest proximity proton functionalities ‘hot spots’ on each of the compound moieties can be measured semi-quantitatively from the magnitude of the STD effect. The STD effect can be represented numerically as a percentage ((STDdiff integral/STDoff integral) × 100). For each of the binders, a maximum STD effect of each moiety was calculated and mapped onto the atoms in each compound. The level of the STD effect was then mapped onto the atoms of each compound and colour-coded to reflect the semi quantitative nature of the technique.
The binding between BSA and ibuprofen, with non-binding spy compound glucose present, is shown in Fig. 5a. In the STDoff spectrum, chemical shifts for glucose can be seen between 3 and 4 ppm which are not presented in the STDdiff as glucose is not involved in the binding (Fig. 5a). The atoms on the ibuprofen that are involved in the binding to BSA and their level of commitments (% difference) in the binding are labelled and colour-coded in the STDdiff. It is clear that the carboxylic acid group should have the strongest interactions with BSA as BSA is the most abundant hydrophilic globular protein in blood serum acting primarily as a carrier protein for hormones, fatty acids, trace minerals, vitamins and iron.58 Strong interactions between BSA and ibuprofen involve the aromatic region and connected sidechains. This is expected because there are hydrophobic side chains and peptide backbones in BSA which favour hydrophobic interactions. This study complements to the molecular interactions identified through the X-ray studies of co-crystallisation of BSA and ibuprofen where hydrophobic part of the molecular interacts strongly with the binding pocket around Val349A and carboxylic acid group forms hydrogen bonds with Tyr355A and Arg120A.59,60
In the more relevant model system, atoms involved in the binding between Aβo and bexarotene are labelled and colour-coded in the STDdiff spectrum (STDoffFig. 5b (red)), (STDdiffFig. 5b (green)) and (STDcontrolFig. 5b (black)). As expected, Aβo are hydrophobic in nature, it is therefore not surprising to see the interactions heavily rely on the hydrophobic part of the molecule. The involvement of the carboxylic acid group cannot be defined from the STD experiment although some ionic interactions between the group and amine side chains should play roles in driving the initial interaction or enhancing/stabilising the interaction.
The protocols developed were applied to study the binding between Aβo and our hit compounds from the 19F NMR screen (Fig. 5c and d). STDoff, (red), STDdiff (green) and control (black) spectra were recorded for PBCHM57487213 and PBCHM81560982. Atoms involved in the binding between Aβo and the ligands are labelled and color-coded in the STDdiff spectra. Both compounds were shown to be medium to strong binders in the sub-library screening and individual confirmation 19F NMR experiments. Additionally, they were confirmed specific binders from the scrambled peptide screening. Although the percent peak differences are small and the involvement of non-hydrogen atoms cannot be confirmed, the aromatic protons are all shown to be affected upon Aβo binding just like bexarotene in this STD NMR experiment. This again demonstrates that the π–π and hydrophobic interactions are important in this type of molecular interactions.
The STD-NMR experiment was carried out with each of the 14 hit compounds with the addition of Aβo. Of the 8 initial hit compounds from the first round of screening, 7 of them were confirmed binders by STD NMR experiments (Table 3). Compound CHEMBL1499171 showed no change of signal in the STD NMR experiment, and was thus deemed inactive. Compared to other hit binders, it was noted that this molecule is smaller than the others (Fig. 4). We might speculate that this smaller fragment is either of lower affinity or makes less contacts with the protein, both of these would result in a weaker STD signal. There is no direct correlation between 19F NMR signal strength and STD-NMR signal change for any of the compounds.
| Entry | Public ID | Analogue public ID | Structure | Highest% STD |
|---|---|---|---|---|
| 1 | CHEMBL1673279 |
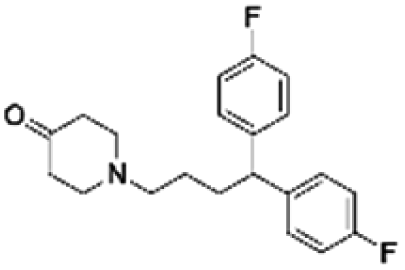
|
4.31 | |
| 2 | PBCHM45210798 |
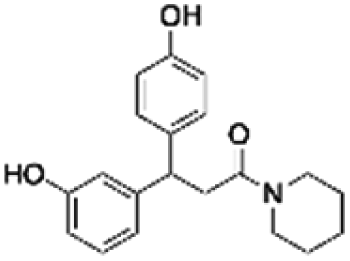
|
15.51 | |
| 3 | CHEMBL1499171 |
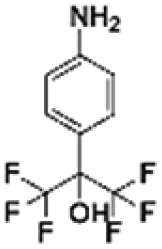
|
0.00 | |
| 4 | PBCHM4680099 |
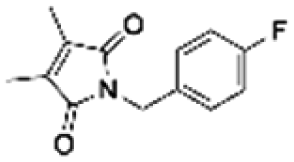
|
7.40 | |
| 5 | PBCHM57487213 |
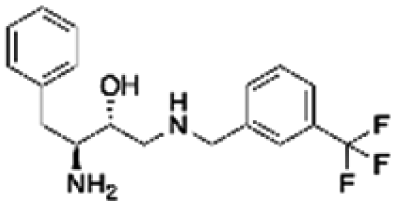
|
0.89 | |
| 6 | CHEMBL1351172 |
|
2.77 | |
| 7 | PBCHM3049683 |
|
6.02 | |
| 8 | PBCHM57223647 |
|
0.00 | |
| 9 | ZINC00159801 |
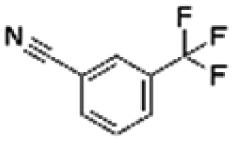
|
0.00 | |
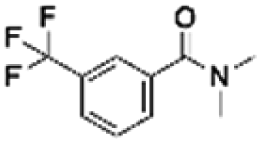
| 10 | ZINC00057047 |
|
0.00 | |
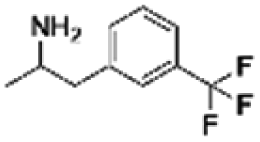
| 11 | PBCHM120765 |
|
8.07 | |
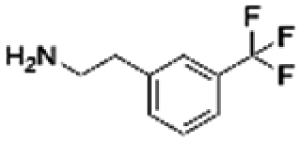
| 12 | CHEMBL448523 |
|
0.00 | |
| 13 | ZINC00120199 |
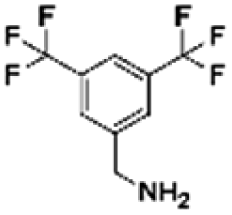
|
8.13 | |
| 14 | PBCHM81560982 |
|
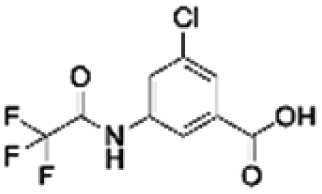
0.75 | |
| 15 | Bexarotene |
|
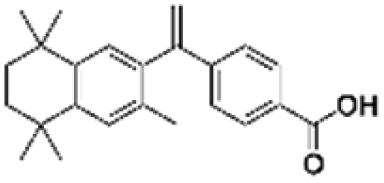
14.60 | |
Only 2 out of the 6 compounds from the hit expansion experiment showed noticeable changes in STD-NMR signals. It is interesting to note that none of the 3 analogues from the same seed-compound, PBCHM3049683, showed any changes in STD signals while the parent compound itself showed a good response. As above, it is noted that the smaller more fragment-like molecules give weaker STD signals, which may be due to weaker binding or less efficient contracts with the protein. This could indicate that competition assay alone may not be the best method used for hit identification. Other validation assays such as STD NMR should be used together to confirm the hits. Of course, it can also indicate that STD NMR is not the best technique to pick up small molecular weight binders. It is interesting to note that hit compound CHEMBL1673279 and its analogue PBCHM45210798 consistently showed good binding abilities in all 19F NMR and STD NMR experiments. The same trend is seen with hit compound PBCHM120765 and its analogue ZINC00120199. 14 hit compounds from 19F CPMG NMR techniques were subject to the STD filter and 9 compounds were confirmed as binders from STD NMR experiments (Table 3).
Compounds PBCHM57487213 and PBCHM81560982 were chosen for dose response studies (Fig. 5e and f). As STD NMR experiments are performed in an excess of ligand, in order to measure a dose–response curve, the concentration of the ligand was kept consistent while the concentration of Aβo was incrementally increased. The spectra were acquired over the same period of time and the integral of the STDdiff was measured. Both compounds adhere to a standard response of increased protein concentration. Initially, there is an increase in response as the protein concentration is increased and the curve then begins to plateau indicating the active sites of the proteins are completely occupied and this is the maximum response for the STD experiment. Finally, the curve began to decrease as the ligand and protein will no longer be in fast exchange. This decreases the response of the STD experiment. The estimated Kd for compound PBCHM57487213 is ∼2 μM and compound PBCHM81560982 is 4 μM. The dose response for other STD NMR confirmed hits were not carried out due to the availability of those compounds.
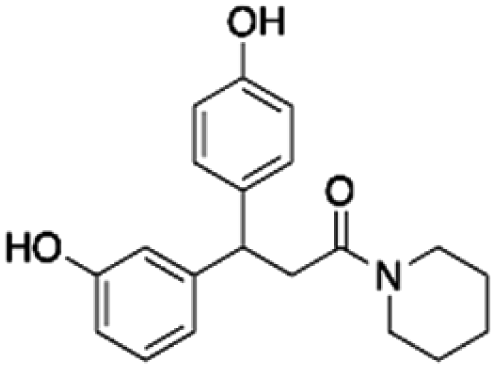
From the 9 potential binders identified from the 19F CPMG and STD-NMR experiments, 6 compounds (CHEMBL1673279, PBCHM4680099, PBCHM57487213, ZINC00120199, PBCHM120765, and PBCHM81560982) were chosen to be taken forward and tested in a biological assay to assess the effect of their binding upon the interfere the binding between Aβo and PrPC, and their effects on Fyn and Tau. These compounds were chosen due to their structural diversity, biophysical results and availabilities.
Biological and functional evaluation of selected hit compounds
To further evaluate the compounds that we identified in the biochemical assay, we sought to answer the following questions: (1) if Aβo prepared interacts with PrPC as previously reported;11,61 (2) if the Aβo binders identified from NMR screening interfere with Aβo–PrPC binding in a biologically relevant manner; (3) if so, do they have polypharmacological properties, i.e. any effects on downstream signalling (pFYN and pTau activities). In order to address these issues, different cellular screening models were developed.We examined the level of PRNP and other Alzheimer's disease related genes in each cell line, including FYN, NMDA receptor genes (GRIN1, GRIN2A, GRIN2B), mGluR5(GRM5), NACE1 and Tau producing gene (MAPT) (Fig. 6a). We found that iCells showed the greatest expression of all examined genes while SH-SY5Y cells contain the least relevant genes, PRNP gene in particular. However, the wild type HEK293 cell line was selected for studying Aβo and PrPC binding because it had moderate expression of the studied genes and it is also a robust cell line, easy to maintain at low cost, and amenable for high throughput screening. The expression of PrPC protein in the HEK293 cell line is visualised by staining against anti-PrPC antibody 8H4 (Fig. 6b) confirmed and quantified by flow cytometry (Fig. 6c).
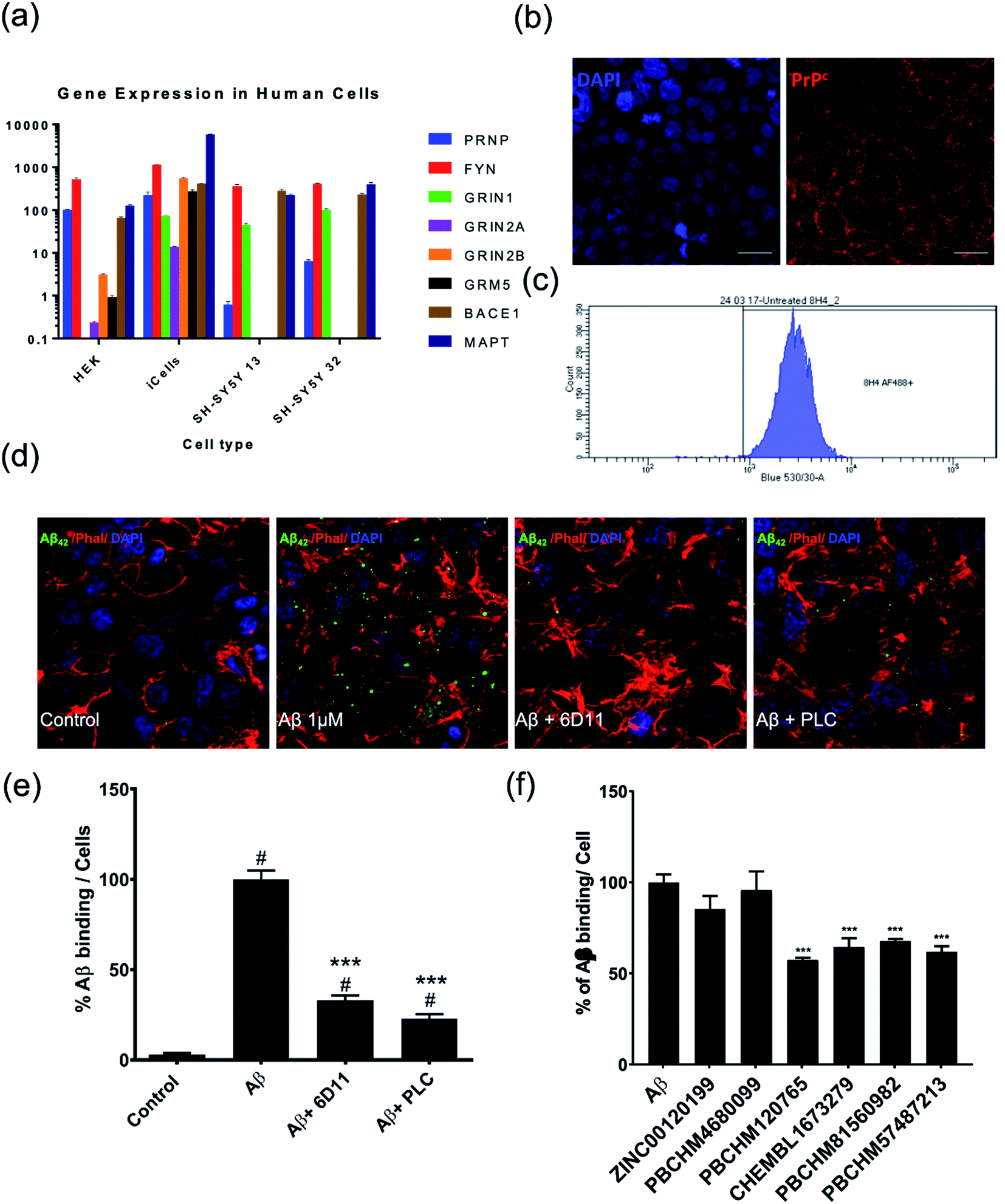 | ||
| Fig. 6 Assessment of PrPC expression level and Aβo binding to PrPC and inhibition of hit compounds. (a) Expression of a selection of neural genes in relevant neural cell lines; (b) HEK293 staining with DAPI (left), PrPC antibody 8H4 (right); (c) quantification of PrPC in HEK293 by flow cytometry; (d) from left to right: control HEK293 cells, HEK293 cells treated with Aβo at 1000 pg mL−1 for 2 hours, HEK293 cells treated with Aβo and anti-PrPC antibody 6D11, HEK293 cells treated with Aβo and PLC. Cells are stained with anti Aβ antibody (green), Phalloidin (red) and DAPI (blue); (e) quantification of Aβo binding to PrPC and inhibition of 6D11 and PLC; (f) percentage of binding of Aβo to PrPC on HEK293 in presence and absence of each compounds by immunofluorescence. Values expressed as mean ± SD (n ≥ 3), statistical significance indicated by #p < 0.05 in comparison with control groups and *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 in comparison with Aβo treated group, following one-way ANOVA and Tukey's post-hoc test. | ||
With the HEK293 cell line being selected as a cellular model for direct binding studies, the Aβo preparation used in the NMR screening was tested for their binding to PrPC on the surface of HEK293 cells. The cells were stained by anti-Aβ antibodies, Phallodin (cytoskeleton stain) and DAPI (nuclei). Both synthetic Aβ1-42 oligomers as described above and recombinant Aβo produced by CHO 7PA2 cells were examined in the binding studies to PrPC in the HEK293 cell model and both showed similar level bindings (data not shown). However, Aβo prepared from synthetic Aβ1-42 monomers displayed noticeable interference in immunostaining in confocal and ICC assays due to some large particles present which did not pose much problems in NMR experiments. The recombinant Aβo were obtained from the supernatant after harvest of the cells and debris removed. They contain a heterogeneous population of monomers, dimers, trimers, tetramers, higher state soluble oligomers and other cellular proteins as previously reported by western blotting. The recombinant Aβo were therefore used in all cellular assays (HEK cell binding assay and subsequent hiPSC functional assays). The recombinant Aβo were produced by CHO 7PA2 cells transfected with cDNA encoding APP751 containing Val 717Phe familial AD mutation and quantified by ELISA.
When compared with the control (Fig. 6d, first panel), Aβo were shown to be able to bind to PrPC on the cell surface of HEK293 cells (Fig. 6d, second panel). The binding of Aβo–PrPC can be significantly blocked by anti-prion antibody 6D11, raised against the epitope containing amino acids 93–109 of PrPC. This antibody was reported to block the interaction between Aβo and PrPC.61,63 Its inhibitory effect was confirmed in our HEK293 direct binding assay (Fig. 6d, third panel). The inhibition of 6D11 on the binding of Aβo to PrPC on the cell surface was further reduced by PLC treatment as previously reported64 (Fig. 6d, forth panel). The binding of Aβo on endogenous PrPC on wild type HEK293 cells and inhibitory effect of known inhibitors, 6D11 and PLC are quantified (Fig. 6e).
The ability of our hit compounds to disrupt the Aβo–PrPC interaction in HEK293 cells was assessed using a live-cell binding assay. The binding was visualised and quantified using immunocytochemistry (ICC). Prior to the binding assay, the cytotoxicity of selected compounds was assessed using MTT assay on HEK293 cells. The viabilities of all compounds are excellent with LD50 > 50 μM except compound PBCHM57487213 having LD50 at around 27 μM (ESI Fig. 1†).
In the binding assay, HEK293 cells were incubated with Aβo at a concentration of 1000 pg mL−1 for 2 hours. The culture medium was then removed and the cells were washed. This was followed by adding the tested compound in fresh medium at a final concentration of 10 μM. The cells were then left for 1 hour prior to ICC analysis. The percentage of the binding of Aβo in presence and absence of the tested compound is presented in (Fig. 6f). Compounds ZINC00120199 and PBCHM4680099 both showed very little inhibitory effects upon the Aβo–PrPC binding while the other 4 compounds all showed clear and statistically significant inhibition effects. These two compounds seem to have relatively small molecular weight and fewer rotatable bonds.
To assess the effects of hit compounds on pFyn activity, iPSC-derived NPCs from a health control individual (Cell line MIFF1 https://web.expasy.org/cellosaurus/CVCL_1E69) in under 3 weeks (much shorter than the time it takes to produce natural neurons) and express all essential biomarkers as mature neurons. The expression of the NPC marker, Nestin, and Aβo binding partner, PrPC, and PRNP and FYN genes were confirmed using immunostaining (Fig. 7a), flow cytometry (Fig. 7b) and qPCR (Fig. 7c).
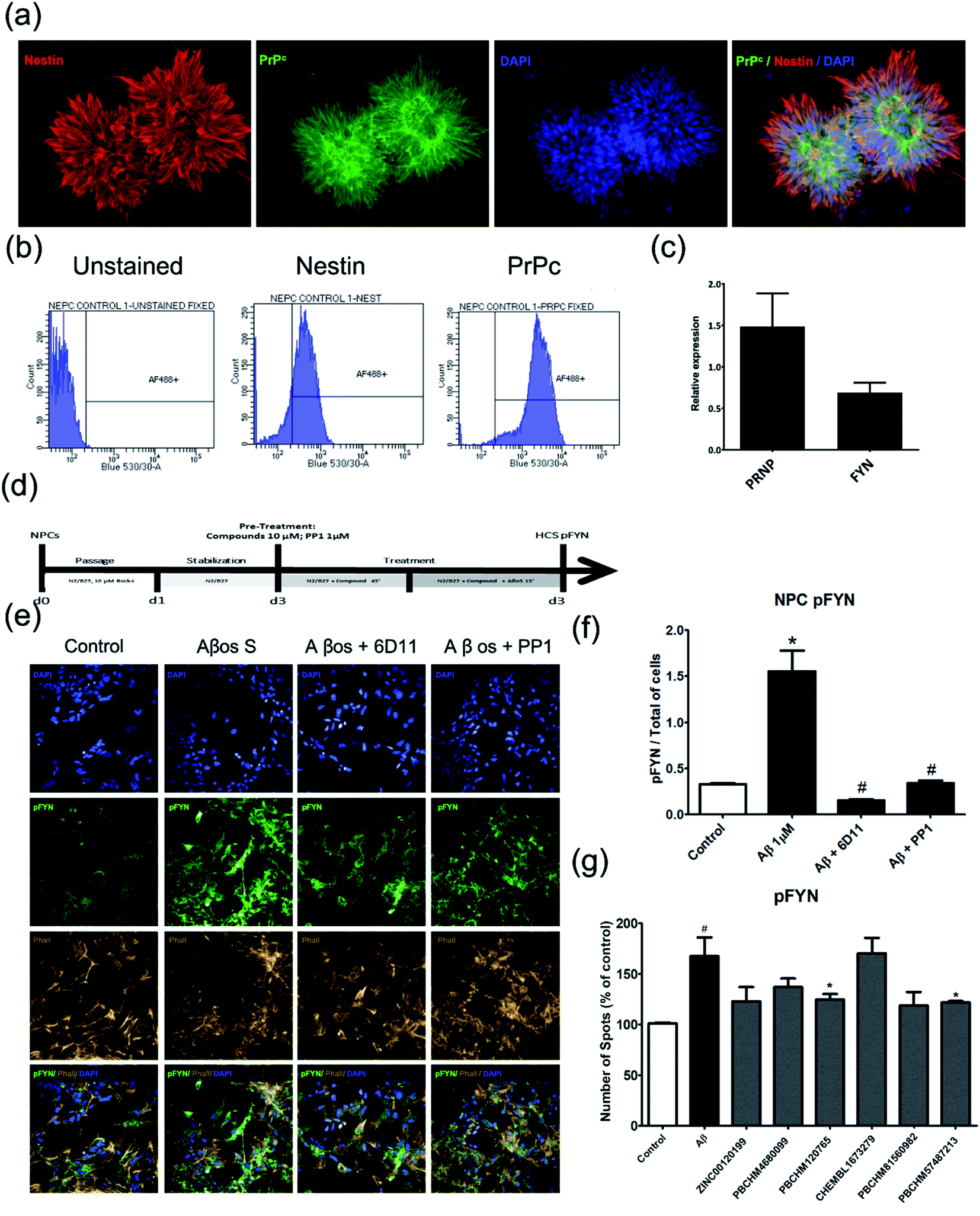 | ||
| Fig. 7 pFyn deactivation and kinase activity assay using NPC. (a) Immunofluorescence double staining of NPC for nestin a classical neural progenitor cell marker (red) and anti-prion (green), the nucleus was stained by DAPI (blue) and overlays; (b) confirmation of nestin and PrPC expression in NPC cells by flow cytometry. (c) Quantification of PRNP and FYN gene expression in NPC cells by qPCR; (d) compound treatment plan; (e) confirmation of Aβo induced pFYN activation using natural Aβo and inhibition of the pFYN activation by anti-prion antibody (6D11), and PP1 a well-known pFyn inhibitor using immunostaining; (f) quantification of the level of inhibition of pFYN activation with 6D11 and PP1; (g) inhibition of Aβo binders in pFYN activation caused by Aβo; values expressed as mean ± SD (n ≥ 3), statistical significance indicated by #p < 0.05, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, following one-way ANOVA and Tukey's post-hoc test. | ||
The Aβo binders were tested using the same protocol and results are shown (Fig. 7d). The screening protocol was developed using commercial anti-prion antibody, 6D11 and a well-known pFYN inhibitor PP1. 6D11 is a monoclonal anti-mouse IgG against epitope 93–109 on PrPC sequence. Recently, this antibody has been found to improve the cognitive deficits in an Alzheimer's disease mice model63,66,67 and prevent the binding of Aβo to PrPC causing Fyn alteration and Tau hyperphosphorylation.68 We used 6D11 as a positive control for developing the Fyn functional assay. PP1 is a cell-permeable pyrazolopyrimidine compound that is shown to inhibit Src family tyrosine kinases Lck, Fyn, Hck, and Src (IC50 = 5, 6, 20, and 170 nM, respectively) in in vitro kinase assays with an application as an anticancer agent.69–71
Both 6D11 and PP1 were used to treat 3 day old NPC in culture for 1 hour prior to Aβo treatment. The cells were harvested for immunofluorescence analysis 15 minutes after the treatment. When compared to the untreated control cells, Aβo caused hyperphosphorylation of Fyn, hence the activation of the Fyn kinase. Both 6D11 and PP1 significantly inhibit the activation of pFyn (Fig. 7e & f). PP1 was chosen as a positive control Aβo binder screening in our pFyn assay as it is a small molecule inhibitor (Fig. 7g). While 5 out of the 6 compounds reduced the Fyn hyperphosphorylation triggered by Aβo, compound PBCHM9815618 and PBCHM57487213 produced the most profound effects.
We were specifically interested in pFyn because it is widely expressed in the brain, it is abundant in neurons and it plays an important role in regulating cell proliferation and differentiation during the development of the CNS.72 It is also involved in signal transduction pathways that regulate survival metabolism and neuronal migration.73 We focused our studies on the effects of the compounds upon the reduction of the elevated level of pFyn (hyperphosphorylation) because any compound that reduces the pFyn level below the basal levels could be deleterious for the homeostasis of the cells and their mode-of-actions are unrelated to the activation of Fyn caused by the Aβo.
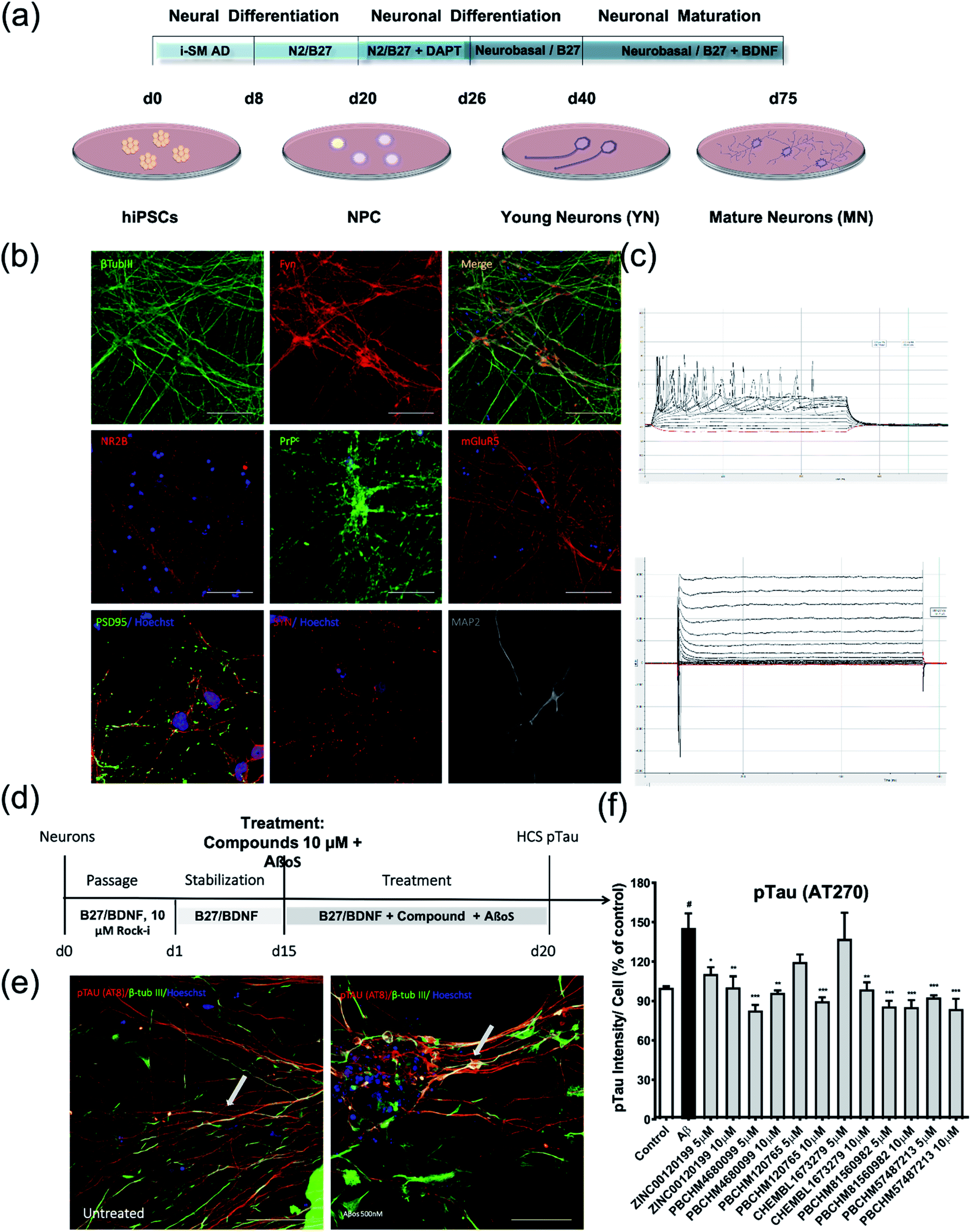 | ||
| Fig. 8 pTau assay using mature cortical neurons derived from iPSC. (a) Protocol for differentiating iPSC into neuroprogenitor cells and then mature cortical neurons; (b) characterisation of iPSC derived nature cortical neurons by immunostaining of neuronal markers (β-tubulin III, Fyn, NR2B, PrPC, mGluR5, PSD95, SYN and MAP2); (c) electrophysiological characterization of iPSC-derived mature cortical neurons. Action potential (top) K+–Na+ ion channel current (bottom) for mature cortical neurons; (d) treatment protocol for Aβo binders for their inhibitory effects on pTau production using mature cortical neurons; (e) pTau production induced by Aβo; (f) inhibition of pTau by 6 Aβo binders. Values expressed as mean ± SD (n ≥ 3), statistical significance indicated by #p < 0.05, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, following one-way ANOVA and Tukey's post-hoc test. | ||
Cells were stained for major protein biomarkers in signalling pathways related to AD development in neurons including general neuronal markers such as β-tubulin 3 (βtub-III) and MAP2, specific glutamatergic markers for cortical neurons such as N-methyl D-aspartate receptor subtype 2B (NMDAR2B or NR2B) and mGluR5, PrPC, synaptic markers such as synaptophysin (SYP) and PSD-95 (postsynaptic density protein 95) (Fig. 8b). Both NPC and neurons showed positive staining for those markers. The physiological functions of these mature neurons were characterized using electrophysiological functional parameters, i.e. action potential (Fig. 8c top) and voltage-gated potassium and sodium ion channel current (Fig. 8c bottom). The data clearly show that iPSCs have been successfully differentiated into mature cortical neurons over an extended period of time and function as normal neurons physiologically. The treatment regime for compounds and Aβo is illustrated in Fig. 8d. Upon the treatment of Aβo binders, significant pTau deposits were clearly seen while the amount of pTau in the untreated control is negligible (Fig. 8e).
Briefly, the iPSC derived nature neurons were passaged and grown for 15 days in B27/BDNF medium, then treated by a mixture of recombinant Aβo at 500 nM and tested compounds at 5 and 10 μM. The inhibitory effects of the 6 hit Aβo binders upon the pTau production induced by Aβo can be clearly seen in Fig. 8f. Amongst all Aβo binders, ZINC0011291995 is the weakest inhibitor, PBCHM4680099 somewhat displays an inverse dose–response trend. Clearer dose-responses effects can be seen with compounds PBCHM120765 and CHEMBL1673279 although PBCHM120765 seems to be a slightly stronger inhibitor. Inhibitory effects of compounds PBCHN81560982 and PBCHM57487213 stayed the same which is similar to that PBCHM120765 possesses at 10 mM concentration.
Positive outcomes from each assay can be clearly seen from individual cell-based assay. In HEK293 binding assay, 4 out of 6 compounds (CHEMBL1673279, PBCHM57487213, PBCHM120765, and PBCHM81560982) showed clear effects in disrupting Aβo–PrPC binding through specific binding to Aβo although their direct binding to PrPC can not be excluded. Compounds, PBCHM57487213 and PBCHM120765 showed noticeable effect in reducing the elevated pFyn level in NPC triggered by Aβo treatment while inhibitory effects of other compounds are observed but not significant. Inhibitory effects on pTau production in mature cortical neurons can be seen with most of the compounds, but statically significant inhibitory effects were observed with compounds CHEMBL1673279, PBCHM57487213, PBCHM120765, and PBCHM81560982.
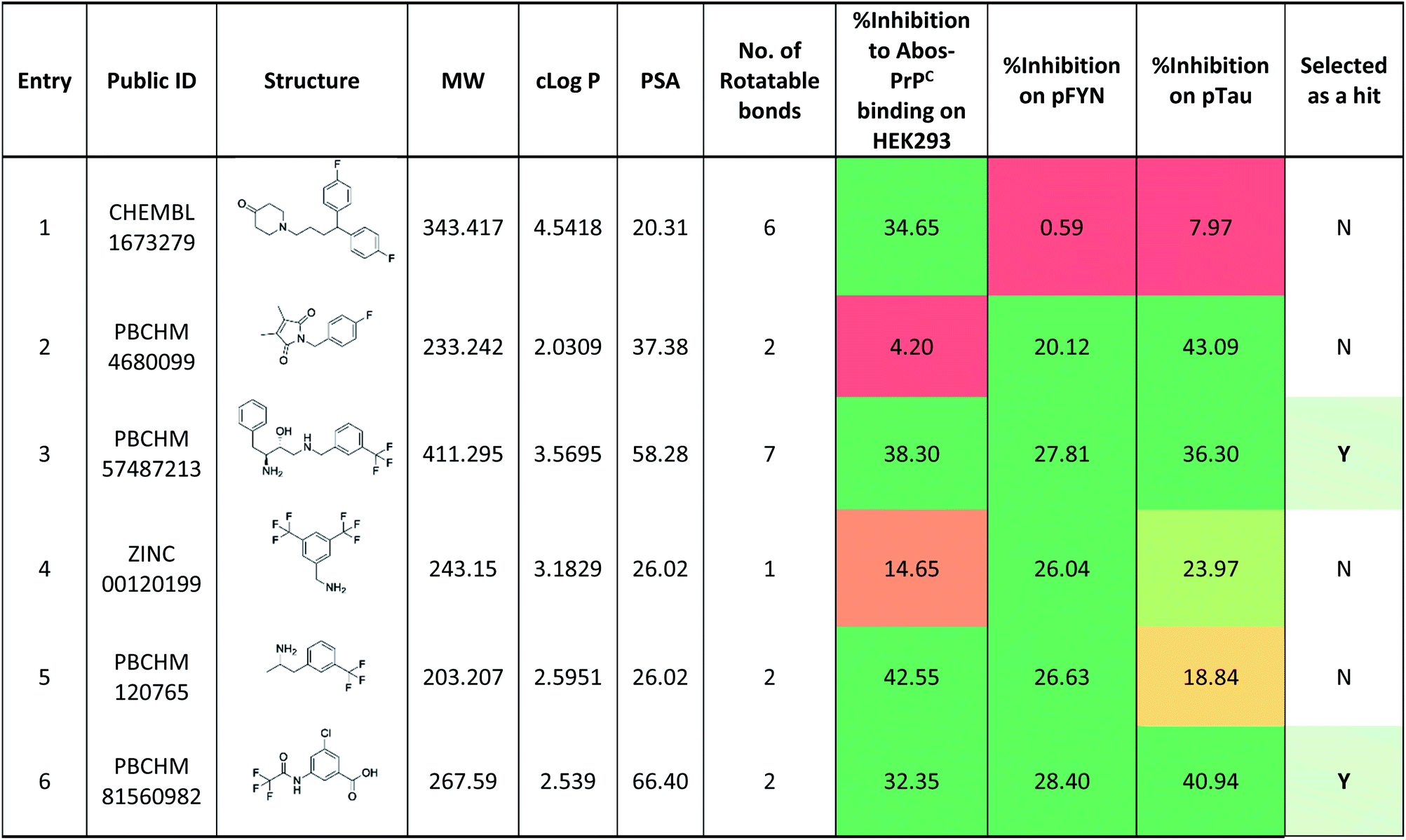
When the data from NMR and biological assays looks a little dispersed which make cross-board comparison a little difficult. In order to give a holistic assessment of the data, activities of all 6 compound under each of the 3 biological experiments were categorised as such 0–10 (red), 10–20 (orange/yellow) and >20 (green). Compounds with cross-board high activities (all >20, in green) were selected as lead compounds (Table 4). The other approach for lead compounds shortlisting was to use results from each one of the 5 assays (19F NMR, STD NMR plus 3 biological assays) which was arbitrarily ranked with highest activity assigned 6 and lowest one assigned as 1 in a descend order. The combined rank was produced. The one with the highest value was selected as lead compounds (data not shown). Both approaches yielded the same shortlist of lead compounds two final leads (PBCHM57487213 and PBCHM81560982) were selected after assessment of overall data although compound PBCHM120765 sits on the board line.
|
Both lead compounds came from the PUBCHEM collection. Both are fluorine-containing small molecules. Compound PBCHM57487213 is a chiral amino alcohol with 2 stereogenic centres. It can be synthesised by the ring opening of appropriately protected epoxide with correct stereo configurations using a trifluoromethyl benzylamine. This compound was patented (US20120053200A1) as a BACE-2 inhibitor which is potential drug target associated with both Alzheimer's diseases and diabetes. Compound PBCHM81560982 is a simple acylated amino benzoic acid and its biological activity of has not been reported in any literature so far. Both lead compounds are drug-like, obeying Lipinski's and Veber's rules. They can be further optimised and developed into anti-AD drugs. As both compounds have relatively low molecular weight and have some fragment features, an obvious strategy for optimisations could simply be merge these two compounds into one molecule to see if a synergistic activity can be achieved.
These lead structures connect three pillars of Alzheimer's diseases, i.e. prion, Aβ and Tau pathways. Aβo binding is a key feature in Aβ pathway, Aβo–PrPC inhibition connects the Aβ pathway with prion pathway while pTau are key components in Tau pathways and can be triggered by Aβo and linked with PrPC and the activation of Fyn.68,74 The experiment was designed to test the effects of the hit compounds on the downstream pathways.
Conclusions
In this work, we sought to identify compounds which bind to soluble Aβ1-42 oligomers (Aβo) using a suite of computational, biophysical and biological methods. Soluble Aβo are the toxic subunits of Alzheimer's disease (AD). A growing body of literature has indicated that the cellular prion protein (PrPC) acts as a receptor for Aβo in AD and connects the Aβ pathway and the Tau pathway. It is expected that Aβo binders that disrupt the Aβo and PrPC interaction may have effects on downstream signalling such as Fyn and Tau activities, therefore, possess polypharmacological activities.Solving the crystal structure of the major disease-causing amyloid peptide, Aβ1-42 and its oligomers, has proven to be extremely challenging. Most structural information came from solution or solid-state NMR studies. There is neither X-ray crystal structure of Aβ1-42 in the oligomeric form nor ligand co-crystallised structures being solved so far, although there are several structures of Aβ fibrils (PDB codes: 2MXU, 2LNQ, 6OC9, 6Y1A). This has made structure-based ligand design or high throughput screening unattainable. Therefore, a ligand-based computational approach was developed to rationalize the selection of potential candidate molecules for biophysical and biochemical screening.
With 19F NMR as a primary screening tool in mind, a computational method was employed that uses fragments of known binders to amyloidal proteins to search for compounds containing similar substructures and at least one F-atom in our compiled database of compounds which are in the public domain as well as available in Lilly's internal inventory. 614 soluble compounds out of 971 virtual hits were screened in 91 sub-libraries in 19F NMR primary screening and 27 compounds were identified as the Aβo binders. Further validation in individual experiments by 19F NMR against Aβo and against a scrambled Aβ-peptide to eliminate false positives and non-specific binding generated 8 confirmed hits. Further hit expansion on these 8 initial hits produced 36 analogues from which 6 more hits were obtained. This gave a total of 14 hit binders which were taken into the STD NMR experiment.
The STD experiment on the 14 initial hit compounds resulted in 9 confirmed hits. The hot spots which are involved in the binding of the hit compounds to Aβo were mapped out during the STD experiments. Semi-quantitative dose–response curve gave KD value at low micromolar levels. The combinations of various experiments using computer-aided design tools, 19F and STD NMR provided the most stringent assessment of potential binders to Aβo. 6 out 9 hit binders (CHEMBL1673279, PBCHM4680099, PBCHM57487213, ZINC00120199, PBCHM120765, and PBCHM81560982) were taken forward in the biological evaluation studies.
The compounds were tested in three cellular assays, one measuring Aβo–PrPC binding using a HEK293 cell line and the other two assessing changes in the levels of other key AD protein biomarkers including Fyn and Tau using NPC and neurons derived from iPSC. 4 out of 6 compounds (CHEMBL1673279, PBCHM57487213, PBCHM120765, and PBCHM81560982) showed good anti-oligmeropathy effects in disrupting Aβo–PrPC binding although these compounds did not show direct binding to PrPC. The polypharmacological effects of the hit compounds on pFyn and pTau were evaluated in hiPSC derived NPC and cortical neuron models. Compounds PBCHM57487213 and PBCHM120765 showed noticeable effect in reducing the elevated pFyn level in NPC triggered by Aβo treatment while inhibitory effects of other compounds were observed but not statistically significantly. Inhibitory effects on pTau production in mature cortical neurons can be seen with most of the compounds, but statistically significant inhibitory effects were observed with compounds CHEMBL1673279, PBCHM57487213, PBCHM120765, and PBCHM81560982.
The two final leads (PBCHM57487213 and PBCHM81560982) were selected after assessment of overall data. The work successfully demonstrated a combined computational, biophysical and biochemical effort in AD drug discovery. The computational method provides a plausible hit rationale for suggesting compounds for NMR screening; 19F and STD NMR have been shown to be effective tools for validating the compounds suggested by the computational design. Cellular models provided sound biochemical and functional validation of Aβo binders in the anti-oligmeropathy and polypharmacology context. The rate has been improved from 1.3% in initial 19F NMR screening to 17% in hit expansion, to 62% in STD NMR. A hit rate of 50% was achieved after the biochemical and functional assays. The lead structures that were discovered, connect 3 pillars in Alzheimer's disease pathology, i.e. prion, Aβ and Tau pathways. They showed polypharmacological effects on 3 different pathways through specific binding to Aβo, i.e. antioligomeropathy mechanism. These compounds are drug-like and can be further optimised to produce useful AD therapeutic drugs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Studies using human induced pluripotent stem cells were performed in strict accordance with the European Framework 7 Marie Curies guidelines. They are either commercially certified or approved by local institutions. The research leading to these results has received funding from the European Union's Seventh Framework Programme (FP7/2007–2013) under grant agreement no. 612347. The authors are grateful for funding from White Rose University Consortium (WRUC) BBSRC Doctoral Training Programme for Samual J Dawes; from EPSRC Molecular Scale Engineering DTC for Sasha Stimpson.References
- M. Goedert, M. G. Spillantini, B. Ghetti, R. A. Crowther and A. Klug, in Alzheimer: 100 Years and Beyond, ed. M. Jucker, K. Beyreuther, C. Haass, R. M. Nitsch and Y. Christen, 2006, pp. 297–304, DOI: 10.1007/978-3-540-37652-1_38 Search PubMed.
- https://www.alz.co.uk/research/statistics .
- A. I. Placido, C. M. F. Pereira, A. I. Duarte, E. Candeias, S. C. Correia, R. X. Santos, C. Carvalho, S. Cardoso, C. R. Oliveira and P. I. Moreira, Biochim. Biophys. Acta, Mol. Basis Dis., 2014, 1842, 1444–1453 CrossRef CAS.
- J. A. Hardy and G. A. Higgins, Science, 1992, 256, 184–185 CrossRef CAS.
- K. Herrup, Nat. Neurosci., 2015, 18, 794–799 CrossRef CAS.
- D. J. Selkoe and J. Hardy, EMBO Mol. Med., 2016, 8, 595–608 CrossRef CAS.
- S. Campioni, B. Mannini, M. Zampagni, A. Pensalfini, C. Parrini, E. Evangelisti, A. Relini, M. Stefani, C. M. Dobson, C. Cecchi and F. Chiti, Nat. Chem. Biol., 2010, 6, 140–147 CrossRef CAS.
- B. Mannini, E. Mulvihill, C. Sgromo, R. Cascella, R. Khodarahmi, M. Ramazzotti, C. M. Dobson, C. Cecchi and F. Chiti, ACS Chem. Biol., 2014, 9, 2309–2317 CrossRef CAS.
- P. Arosio, R. Cukalevski, B. Frohm, T. P. J. Knowles and S. Linse, J. Am. Chem. Soc., 2014, 136, 219–225 CrossRef CAS.
- G. S. Bloom, JAMA Neurol., 2014, 71, 505–508 CrossRef.
- H. H. Jarosz-Griffiths, E. Noble, J. V. Rushworth and N. M. Hooper, J. Biol. Chem., 2016, 291, 3174–3183 CrossRef CAS.
- A. E. Barry, I. Klyubin, J. M. Mc Donald, A. J. Mably, M. A. Farrell, M. Scott, D. M. Walsh and M. J. Rowan, J. Neurosci., 2011, 31, 7259–7263 CrossRef CAS.
- R. B. DeMattos, K. R. Bales, D. J. Cummins, J. C. Dodart, S. M. Paul and D. M. Holtzman, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8850–8855 CrossRef CAS.
- G. A. N. Crespi, S. J. Hermans, M. W. Parker and L. A. Miles, Sci. Rep., 2015, 5, 9649 CrossRef CAS.
- A. D. Watt, G. A. N. Crespi, R. A. Down, D. B. Ascher, A. Gunn, K. A. Perez, C. A. McLean, V. L. Villemagne, M. W. Parker, K. J. Barnham and L. A. Miles, Acta Neuropathol., 2014, 127, 803–810 CrossRef CAS.
- V. L. Villemagne, S. Burnham, P. Bourgeat, B. Brown, K. A. Ellis, O. Salvado, C. Szoeke, S. L. Macaulay, R. Martins, P. Maruff, D. Ames, C. C. Rowe, C. L. Masters and B. Australian Imaging, Lancet Neurol., 2013, 12, 357–367 CrossRef CAS.
- J. Sevigny, P. Chiao, T. Bussiere, P. H. Weinreb, L. Williams, M. Maier, R. Dunstan, S. Salloway, T. Chen, Y. Ling, J. O'Gorman, F. Qian, M. Arastu, M. Li, S. Chollate, M. S. Brennan, O. Quintero-Monzon, R. H. Scannevin, H. M. Arnold, T. Engber, K. Rhodes, J. Ferrero, Y. Hang, A. Mikulskis, J. Grimm, C. Hock, R. M. Nitsch and A. Sandrock, Nature, 2016, 537, 50–56 CrossRef CAS.
- J. Toyn, Expet Rev. Clin. Pharmacol., 2015, 8, 267–269 CrossRef CAS.
- M. S. Parihar and T. Hemnani, J. Clin. Neurosci., 2004, 11, 456–467 CrossRef CAS.
- B. De Strooper, R. Vassar and T. Golde, Nat. Rev. Neurol., 2010, 6, 99–107 CrossRef CAS.
- R. J. O'Brien and P. C. Wong, in Annual Review of Neuroscience, Vol 34, ed. S. E. Hyman, T. M. Jessell, C. J. Shatz, C. F. Stevens and H. Y. Zoghbi, 2011, vol. 34, pp. 185–204 Search PubMed.
- S. T. Ferreira, M. V. Lourenco, M. M. Oliveira and F. G. De Felice, Front. Cell. Neurosci., 2015, 9, 191 Search PubMed.
- T. Bilousova, C. A. Miller, W. W. Poon, H. V. Vinters, M. Corrada, C. Kawas, E. Y. Hayden, D. B. Tepow, C. Glabe, R. Albay III, G. M. Cole, E. Teng and K. H. Gylys, Am. J. Pathol., 2016, 186, 185–198 CrossRef CAS.
- T. Yang, S. Li, H. Xu, D. M. Walsh and D. J. Selkoe, J. Neurosci., 2017, 37, 152–163 CrossRef CAS.
- M. Costanzo and C. Zurzolo, Biochem. J., 2013, 452, 1–17 CrossRef CAS.
- M. Jucker and L. C. Walker, Nat. Neurosci., 2018, 21, 1341–1349 CrossRef CAS.
- S.-J. Lee, P. Desplats, C. Sigurdson, I. Tsigelny and E. Masliah, Nat. Rev. Neurol., 2010, 6, 702–706 CrossRef.
- B. B. Holmes and M. I. Diamond, J. Biol. Chem., 2014, 289, 19855–19861 CrossRef CAS.
- J. Lauren, D. A. Gimbel, H. B. Nygaard, J. W. Gilbert and S. M. Strittmatter, Nature, 2009, 457, 1128–U1184 CrossRef CAS.
- K. O'Neill, S. H. Chen and R. D. Brinton, Exp. Neurol., 2004, 188, 268–278 CrossRef.
- J. Luis Herrera, C. Fernandez, M. Diaz, D. Cury and R. Marin, Steroids, 2011, 76, 840–844 Search PubMed.
- S. V. Salazar, C. Gallardo, A. C. Kaufman, C. S. Herber, L. T. Haas, S. Robinson, J. C. Manson, M. K. Lee and S. M. Strittmatter, J. Neurosci., 2017, 37, 9207–9221 CrossRef CAS.
- C. Li and J. Gotz, EMBO J., 2017, 36, 3120–3138 CrossRef CAS.
- P. Joshi, S. Chia, J. Habchi, T. P. J. Knowles, C. M. Dobson and M. Vendruscolo, ACS Comb. Sci., 2016, 18, 144–153 CrossRef CAS.
- C. Dalvit, Prog. Nucl. Magn. Reson. Spectrosc., 2007, 51, 243–I CrossRef CAS.
- J. H. Byun, H. Kim, Y. Kim, I. Mook-Jung, D. J. Kim, W. K. Lee and K. H. Yoo, Bioorg. Med. Chem. Lett., 2008, 18, 5591–5593 CrossRef CAS.
- K. N. Dahlgren, A. M. Manelli, W. B. Stine, L. K. Baker, G. A. Krafft and M. J. LaDu, J. Biol. Chem., 2002, 277, 32046–32053 CrossRef CAS.
- I. Kuperstein, K. Broersen, I. Benilova, J. Rozenski, W. Jonekheere, M. Debulpaep, A. Vandersteen, I. Segers-Nolten, K. Van der Werf, V. Subramaniam, D. Braeken, G. Callewaert, C. Bartic, R. D'Hooge, I. C. Martins, F. Rousseau, J. Schymkowitz and B. De Strooper, EMBO J., 2010, 29, 3408–3420 CrossRef CAS.
- T. J. Esparza, N. C. Wildburger, H. Jiang, M. Gangolli, N. J. Cairns, R. J. Bateman and D. L. Brody, Sci. Rep., 2016, 6, 38187 CrossRef CAS.
- E. Y. Hayden and D. B. Teplow, Alzheimers Res. Ther., 2013, 5, 60 CrossRef.
- G.-f. Chen, T.-h. Xu, Y. Yan, Y.-r. Zhou, Y. Jiang, K. Melcher and H. E. Xu, Acta Pharmacol. Sin., 2017, 38, 1205–1235 CrossRef CAS.
- R. Gniadecki, C. Assaf, M. Bagot, R. Dummer, M. Duvic, R. Knobler, A. Ranki, P. Schwandt and S. Whittaker, Br. J. Dermatol., 2007, 157, 433–440 CrossRef CAS.
- K. H. Dragnev, W. J. Petty, S. J. Shah, L. D. Lewis, C. C. Black, V. Memoli, W. C. Nugent, T. Hermann, A. Negro-Vilar, J. R. Rigas and E. Dmitrovsky, Clin. Cancer Res., 2007, 13, 1794–1800 CrossRef CAS.
- F. J. Esteva, J. Glaspy, S. Baidas, L. Laufman, L. Hutchins, M. Dickler, D. Tripathy, R. Cohen, A. DeMichele, R. C. Yocum, C. K. Osborne, D. F. Hayes, G. N. Hortobagyi, E. Winer and G. D. Demetri, J. Clin. Oncol., 2003, 21, 999–1006 CrossRef CAS.
- C. Di Scala, H. Chahinian, N. Yahi, N. Garmy and J. Fantini, Biochemistry, 2014, 53, 4489–4502 CrossRef CAS.
- Z. Mirza and M. A. Beg, Curr. Alzheimer Res., 2017, 14, 327–334 CAS.
- H. Pham Dinh Quoc, T. Nguyen Quoc, Z. Bednarikova, P. Le Huu, L. Huynh Quang, Z. Gazova and M. S. Li, ACS Chem. Neurosci., 2017, 8, 1960–1969 CrossRef.
- N. Bibi, S. M. Danish Rizvi, A. Batool and M. A. Kamal, Curr. Pharmaceut. Des., 2019, 25(27), 2989–2995 CrossRef CAS.
- B. G. Jenkins, Life Sci., 1991, 48, 1227–1240 CrossRef CAS.
- J. Oravcova, V. Mlynarik, S. Bystricky, L. Soltes, P. Szalay, L. Bohacik and T. Trnovec, Chirality, 1991, 3, 412–417 CrossRef CAS.
- H. Chen, S. Viel, F. Ziarelli and L. Peng, Chem. Soc. Rev., 2013, 42, 7971–7982 RSC.
- C. Dalvit, P. E. Fagerness, D. T. A. Hadden, R. W. Sarver and B. J. Stockman, J. Am. Chem. Soc., 2003, 125, 7696–7703 CrossRef CAS.
- C. Dalvit and A. Vulpetti, Magn. Reson. Chem., 2012, 50, 834 CrossRef.
- T. Zhu, S. Y. Cao, P. C. Su, R. Patel, D. Shah, H. B. Chokshi, R. Szukala, M. E. Johnson and K. E. Hevener, J. Med. Chem., 2013, 56, 6560–6572 CrossRef CAS.
- C. Dalvit, A. D. Gossert, J. Coutant and M. Piotto, Magn. Reson. Chem., 2011, 49, 199–202 CrossRef CAS.
- M. Mayer and B. Meyer, Angewandte Chemie-International Edition, 1999, 38, 1784–1788 CrossRef CAS.
- M. Mayer and B. Meyer, J. Am. Chem. Soc., 2001, 123, 6108–6117 CrossRef CAS.
- A. Farrugia, Transfus. Med. Rev., 2010, 24, 53–63 CrossRef.
- P. J. Loll, B. S. Selinsky, K. Gupta and C. T. Sharkey, Worldwide Protein Data Bank, 2001, DOI: 10.2210/pdb1eqg/pdb Search PubMed.
- B. S. Selinsky, K. Gupta, C. T. Sharkey and P. J. Loll, Biochemistry, 2001, 40, 5172–5180 CrossRef CAS.
- C. Peters, M. P. Espinoza, S. Gallegos, C. Opazo and L. G. Aguayo, Neurobiol. Aging, 2015, 36, 1369–1377 CrossRef CAS.
- J. W. Um, H. B. Nygaard, J. K. Heiss, M. A. Kostylev, M. Stagi, A. Vortmeyer, T. Wisniewski, E. C. Gunther and S. M. Strittmatter, Nat. Neurosci., 2012, 15, 1227–U1285 CrossRef CAS.
- E. Chung, Y. Ji, Y. J. Sun, R. J. Kascsak, R. B. Kascsak, P. D. Mehta, S. M. Strittmatter and T. Wisniewski, BMC Neurosci., 2010, 11, 130 CrossRef.
- A. Muller, C. Kloppel, M. Smith-Valentine, J. Van Houten and M. Simon, Biochim. Biophys. Acta Biomembr., 2012, 1818, 117–124 CrossRef.
- R. Dolmetsch and D. H. Geschwind, Cell, 2011, 145, 831–834 CrossRef CAS.
- J. E. Pankiewicz, S. Sanchez, K. Kirshenbaum, R. B. Kascsak, R. J. Kascsak and M. J. Sadowski, Mol. Neurobiol., 2019, 56, 2073–2091 CrossRef CAS.
- J. F. McEwan, M. L. Windsor and S. D. Cullis-Hill, Tumor Biol., 2009, 30, 141–147 CrossRef CAS.
- M. Larson, M. A. Sherman, F. Amar, M. Nuvolone, J. A. Schneider, D. A. Bennett, A. Aguzzi and S. E. Lesne, J. Neurosci., 2012, 32, 16857 CrossRef CAS.
- A. H. Sikkema, S. H. Diks, W. F. A. den Dunnen, A. ter Elst, F. J. G. Scherpen, E. W. Hoving, R. Ruijtenbeek, P. J. Boender, R. de Wijn, W. A. Kamps, M. P. Peppelenbosch and E. de Bont, Cancer Res., 2009, 69, 5987–5995 CrossRef CAS.
- R. Karni, S. Mizrachi, E. Reiss-Sklan, A. Gazit, O. Livnah and A. Levitzki, FEBS Lett., 2003, 537, 47–52 CrossRef CAS.
- J. H. Hanke, J. P. Gardner, R. L. Dow, P. S. Changelian, W. H. Brissette, E. J. Weringer, K. Pollok and P. A. Connelly, J. Biol. Chem., 1996, 271, 695–701 CrossRef CAS.
- S. Schenone, C. Brullo, F. Musumeci, M. Biava, F. Falchi and M. Botta, Curr. Med. Chem., 2011, 18, 2921–2942 CrossRef CAS.
- S. J. Parsons and J. T. Parsons, Oncogene, 2004, 23, 7906–7909 CrossRef CAS.
- L. A. Gomes, S. A. Hipp, A. R. Upadhaya, K. Balakrishnan, S. Ospitalieri, M. J. Koper, P. Largo-Barrientos, V. Uytterhoeven, J. Reichwald, S. Rabe, R. Vandenberghe, C. A. F. von Arnim, T. Tousseyn, R. Feederle, C. Giudici, M. Willem, M. Staufenbiel and D. R. Thal, Acta Neuropathol., 2019, 138, 913–941 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc04769d |
| This journal is © The Royal Society of Chemistry 2021 |