
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReappraising Schmidpeter's bis(iminophosphoranyl)phosphides: coordination to transition metals and bonding analysis†
Adrien T.
Normand
 *,
E. Daiann
Sosa Carrizo
*,
Corentin
Magnoux‡
,
Esteban
Lobato
,
Hélène
Cattey
,
Philippe
Richard
,
Stéphane
Brandès
,
Charles H.
Devillers
,
Anthony
Romieu
,
Pierre
Le Gendre
and
Paul
Fleurat-Lessard
*
*,
E. Daiann
Sosa Carrizo
*,
Corentin
Magnoux‡
,
Esteban
Lobato
,
Hélène
Cattey
,
Philippe
Richard
,
Stéphane
Brandès
,
Charles H.
Devillers
,
Anthony
Romieu
,
Pierre
Le Gendre
and
Paul
Fleurat-Lessard
*
Institut de Chimie Moléculaire de l'Université de Bourgogne (ICMUB), UMR 6302, CNRS, Université de Bourgogne, 9, Avenue Alain Savary, 21000 Dijon, France. E-mail: adrien.normand@u-bourgogne.fr; eric-daiann.sosa-carrizo@u-bourgogne.fr; paul.fleurat-lessard@u-bourgogne.fr
First published on 26th October 2020
Abstract
The synthesis and characterization of a range of bis(iminophosphoranyl)phosphide (BIPP) group 4 and coinage metals complexes is reported. BIPP ligands bind group 4 metals in a pseudo fac-fashion, and the central phosphorus atom enables the formation of d0–d10 heterobimetallic complexes. Various DFT computational tools (including AIM, ELF and NCI) show that the phosphorus–metal interaction is either electrostatic (Ti) or dative (Au, Cu). A bridged homobimetallic Cu–Cu complex was also prepared and its spectroscopic properties were investigated. The theoretical analysis of the P–P bond in BIPP complexes reveals that (i) BIPP are closely related to ambiphilic triphosphenium (TP) cations; (ii) the P–P bonds are normal covalent (i.e. not dative) in both BIPP and TP.
Introduction
Geminal dianions stabilized by main group elements have been a topic of interest in main group and coordination chemistry since the 1970s.1–5 Our understanding of chemical bonding has evolved significantly as a result of numerous insightful studies focused on these species. For instance, double ylides (also known as carbodiphosphoranes, CDP) have been instrumental in validating the notion of dative bonding between non-metallic main group elements.6–11 Thus, CDP may be viewed as phosphine complexes of C(0), i.e. the C–P bonds are dative covalent.5,6 The arrow formalism (Scheme 1) is often used to highlight the “metal-like” character of the central carbon atom. | ||
| Scheme 1 Selected examples of geminal dianions (CDP, BIPM) and their phosphorus analogues (TP, BIPP), all depicted using the arrow formalism. | ||
The nature of the C–P bond in these compounds has been rather controversial,12–16 partly because evidence of C–P bond cleavage in CDP and related compounds is scarce:17 indeed, dative covalent bonds are generally perceived as weak, compared to normal covalent bonds.14,18 However, the defining feature of dative covalent bonds is their preference for heterolytic bond cleavage in the gas phase (or inert solvents),18–24 which does not equate with bond weakness.
Isolobal replacement of the central carbon atom in CDP by phosphorus yields triphosphenium cations (TP, Scheme 1).25–28 In this case, the dative nature of the P–P bonds seems to be corroborated by the reactivity of TP, which readily undergo heterolytic P–P bond cleavage in solution (“ligand exchange”, Scheme 2).29–39 In other words, the arrow formalism acts as a compelling heuristic device to explain the reactivity of TP. Whether it also relates to the bonding situation depends on one's understanding of the arrow formalism. Some authors, who follow Haaland's analysis and use the arrow formalism to depict metal–ligand bonds or main group donor–acceptor adducts (e.g. H3N → BH3),18 might conclude that P–P bonds in TP and related compounds are dative covalent (according to IUPAC).40 This would seem natural, given the reactivity of these compounds in solution. Other authors, who stick to Sidgwick's definition of the “coordinate link”19 and use arrows in a broader context (e.g. to “fix the octet”24 in compounds such as amine oxides, H2SO4, or XeO), will not conclude anything at all.§ Nevertheless, it remains that the bonding situation in TP has yet to be addressed with ad hoc theoretical tools.41
 | ||
| Scheme 2 “Ligand exchange” in triphosphenium cations (TP). | ||
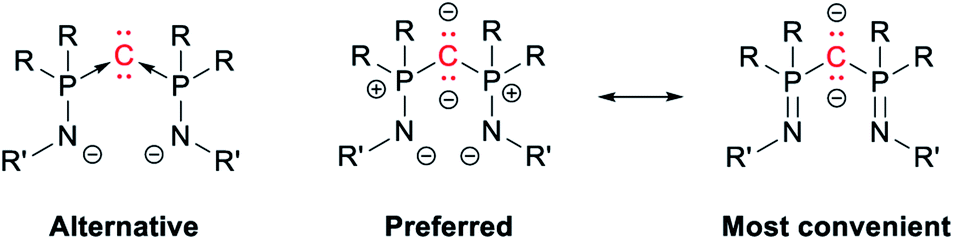
Replacing the phosphine “ligands” in CDP by phosphinoamides yields bis(iminophosphoranyl)methanediides (BIPM), which act as versatile ligands for a staggeringly broad range of metals.1–5,42–44 Along with related compounds, BIPM have helped challenge the widespread assumption that bonding in lanthanide and actinide complexes is essentially ionic, and that they are not conducive to metal–carbon double bonds.45–57 It was suggested that BIPM may also be viewed as C(0) complexes (Scheme 3, left),3 but the general agreement is that the P–C bonds are normal covalent.5,58 Thus most authors in the field feel more comfortable with the alternating dipolar representation (Scheme 3, center)—although those who perpetuate Sidgwick's intellectual heritage might argue that both representations are strictly equivalent. Note that for graphical convenience, the side arms are generally represented with a P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond (Scheme 3, right). This is of course chemically “wrong”, but much more legible than the charge-laden (and octet rule-abiding) resonance form.
N double bond (Scheme 3, right). This is of course chemically “wrong”, but much more legible than the charge-laden (and octet rule-abiding) resonance form.
 | ||
| Scheme 3 | ||
Very little is known about the phosphorus analogues of BIPM, i.e. bis(iminophosphoranyl)phosphides (BIPP): a single derivative was reported in 1985 by Schmidpeter, and characterized by 31P NMR spectroscopy only.59,60 We surmised that these compounds could offer a unique opportunity to introduce a phosphide ligand in the coordination sphere of group 4 metals (Scheme 4), an area which we have been investigating for some time.61–64 We also became interested in comparing their properties to those of TP, with a particular focus on the nature—normal or dative covalent—of P–P bonds, and the possibility to prepare early-late heterobimetallic complexes (by coordination of a late transition metal to the central phosphorus atom).32,34–37,65–68
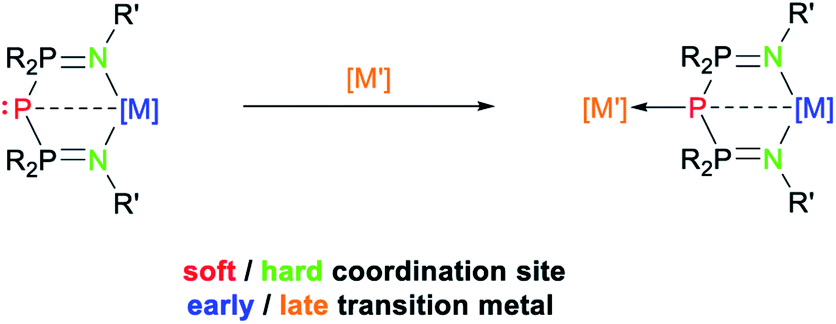 | ||
| Scheme 4 Targeted early (left) and early/late (right) metal complexes of BIPP. | ||
Results and discussion
Ligands synthesis
Schmidpeter described the synthesis of BIPP anions starting either from white phosphorus (P4) or from [K(18-crown-6)][P(CN)2].59,60 We found it more convenient and cost-effective to use P4 since the synthesis of the P(CN)2− anion also starts from P4.69 The downside of using P4—aside from its well-documented toxicity and pyrophoric character—is that it is not commercially available in Europe anymore, and therefore has to be prepared following a procedure developed by Baker in the XIXth century. This consists in the thermal allotropic conversion of red phosphorus into white phosphorus.70 More recently, Arnold, Hsu and Tsai adapted this procedure to modern day Schlenk-line techniques.71 We independently developed a safer variation of Baker's allotropic conversion, using a custom-made silica glass Schlenk flask for improved heat resistance (see the ESI† for details).Thus, heating lithium phosphinoamides 1 with an excess of P4 (2 to 2.4 eq.) at 100 °C in 1,4-dioxane afforded BIPP–Li proligands 2 in moderate to good yields (Scheme 5). Replacing 1,4-dioxane by THF in the case of phosphinoamide 1b afforded compound 2b′ in lower yield, but enabled to grow single crystals for X-ray diffraction analysis (vide infra). Whilst not completely optimal, this procedure easily affords multigram quantities of 2. Proligands 2 were characterized by multinuclear NMR spectroscopy, IR and UV-Vis spectroscopies, elemental analysis and, in the case of 2b′, XRD analysis.
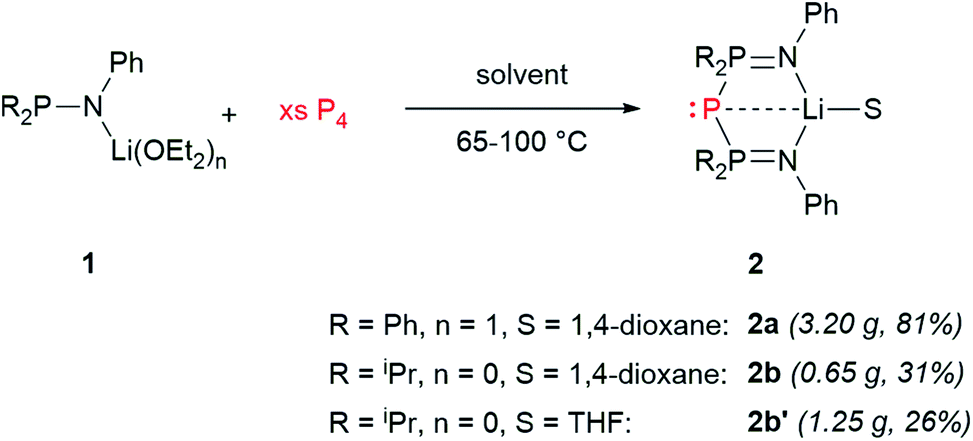 | ||
| Scheme 5 Synthesis of BIPP–Li proligands. | ||
All other compounds in this study were characterized similarly. Relevant 31P{1H} NMR parameters, such as the chemical shifts of lateral (δPL) and central (δPC) P atoms, along with the 1JPP coupling constants values, are gathered in Table 1.
| Compound | Metal | δPL (ppm) | δPC (ppm) | 1 J PP (Hz) |
|---|---|---|---|---|
| a Spectra recorded in CD2Cl2 at 298 K except for Li complexes (THF-d8), 2b (293 K), 4a (260 K), 5a (291 K), 6a/6a′ (292 K), 8b (290 K). b See ref. 34. | ||||
| 2a | Li | 31.8 | −118.7 | 411 |
| 2b | Li | 62.2 | −201.6 | 462 |
| 2b′ | Li | 61.0 | −190.8 | 450 |
| 4a | Ti | 32.5 | −212.9 | 381 |
| 5a | Zr | 27.2 | −210.2 | 351 |
| 5b | Zr | 56.9 | −223.0 | 384 |
| 6a | Ti | 37.4 | −179.2 | 416 |
| 6a-Py | Ti | 34.5 | −189.3 | 390 |
| 6b | Ti | 67.7 | −241.0 | 474 |
| 7b | Ti/Au | 73.7 | −160.4 | 347 |
| 8b | Ti/Cu | 70.2 | −212.8 | 414 |
| 9b | Cu | 59.2 | −146.2 | 349 |
| A | — | 34.1 | −220.9 | 414 |
| B | —/Au | 31.3 | −108.7 | 314 |
The 31P{1H} NMR spectrum of BIPP complexes corresponds to an A2X spin system: the lateral P atoms resonate as a doublet in the positive region of the spectrum, whilst the central P atom resonates as a strongly shielded triplet. The δPC value is quite sensitive, even to remote changes in the environment around the central P atom. Thus, whilst the δPL value of 2b and 2b′ are quite close (ΔδPL = 1.2 ppm), the difference between δPC values is much larger (ΔδPC = 10.8 ppm), especially considering that both compounds only differ by the solvent molecule coordinated to Li (1,4-dioxane vs. THF). The values reported in Table 1 for BIPP compounds are comparable to those reported for compounds A and B (Chart 1). The dicoordinate¶ central phosphorus of A shows typical “ligand exchange” reactivity in the presence of strongly donating phosphines.34
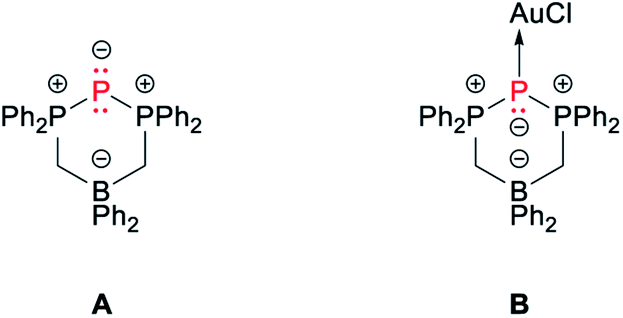 | ||
| Chart 1 Selected compounds from the literature. | ||
The XRD structure of 2b′ is shown in Fig. 1. The unit cell was found to contain one molecule of 2b′ with disordered iPr groups (borne by P1) and one disordered THF molecule. Relevant metric parameters for BIPP complexes and related compounds A and B are given in Table 2.
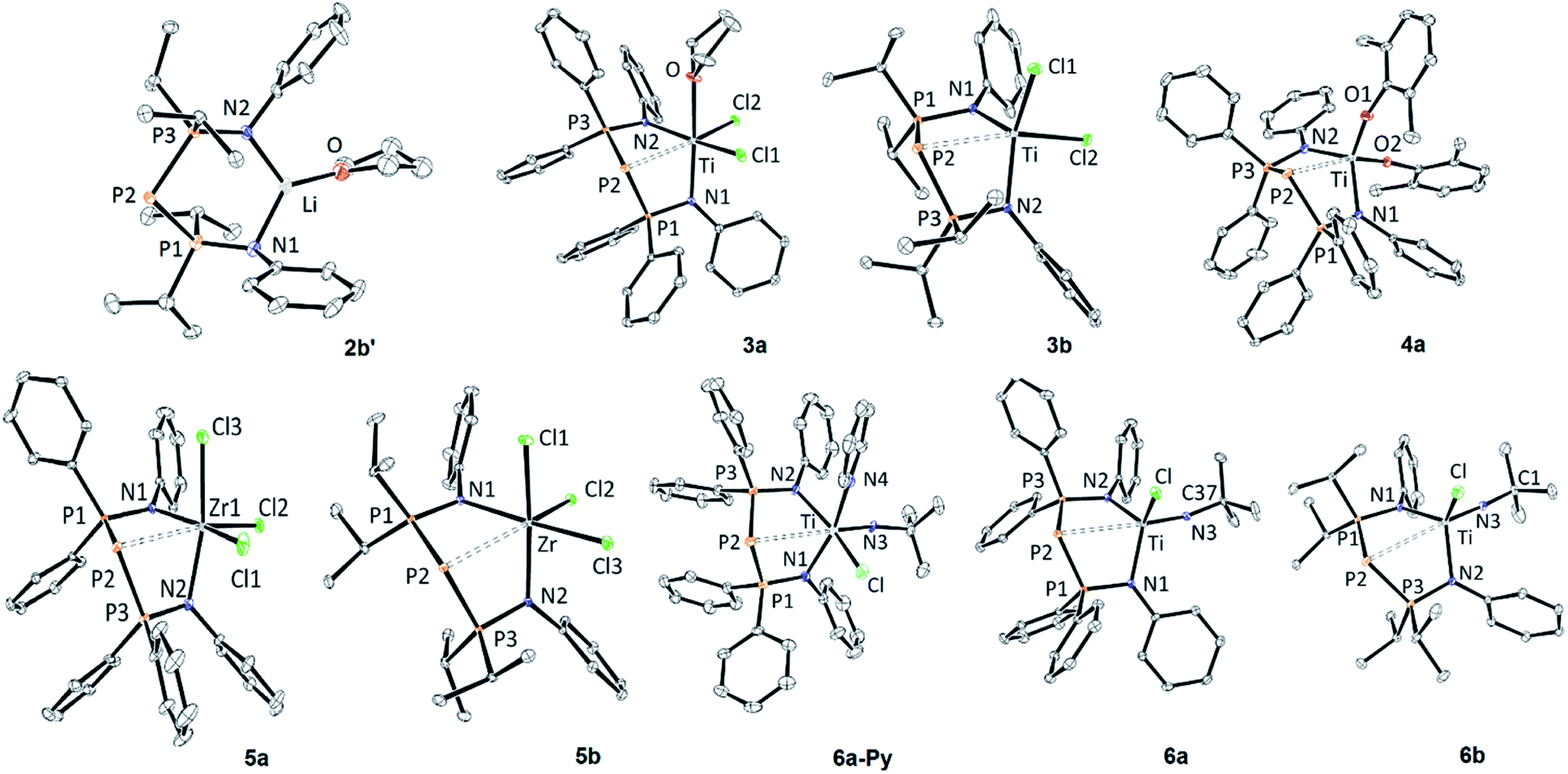 | ||
| Fig. 1 ORTEP depiction of the X-ray structures of monometallic BIPP complexes (thermal ellipsoids drawn at the 30% probability level, solvent molecules, BPh4− anion (for 4a), hydrogen and disordered atoms are not shown for clarity). For 5a and 6b only one independent molecule is shown (5a-1 and 6b-1). See Table 2 for metric parameters. See the ESI† for additional bond distance and angles. | ||
| Compound | Metal | P2–Ma | P1–P2 | P2–P3 | N1–P1 | N2–P3 | N1–M | N2–M | P1–P2–P3 | N1–M–N2 |
|---|---|---|---|---|---|---|---|---|---|---|
| a Italicized values indicate bond distance < sum of covalent radii (Alvarez, ref. 72). b Two independent molecules in the unit cell. c See ref. 34. d See ref. 78. e C–M distance. | ||||||||||
| 2b′ | Li | 3.909(6) | 2.1359(11) | 2.1509(10) | 1.619(2) | 1.614(2) | 1.962(6) | 1.936(6) | 102.64(4) | 111.5(2) |
| 3a | Ti | 2.7712(6) | 2.1558(7) | 2.1333(6) | 1.6345(14) | 1.6278(14) | 2.1024(14) | 2.1611(14) | 98.72(2) | 97.77(5) |
| 3b | Ti | 2.7832(6) | 2.1626(7) | 2.1424(6) | 1.6400(14) | 1.6374(14) | 2.0116(13) | 2.0501(14) | 108.45(2) | 110.02(6) |
| 4a | Ti | 2.8125(13) | 2.1415(14) | 2.1460(15) | 1.640(3) | 1.646(3) | 2.023(3) | 2.014(3) | 102.25(6) | 114.19(13) |
| 5a-1 | Zr | 2.8896(5) | 2.1566(6) | 2.1621(6) | 1.6236(14) | 1.6337(15) | 2.1960(15) | 2.1868(15) | 99.19(2) | 90.37(5) |
| 5a-2 | Zr | 2.8961(6) | 2.1649(7) | 2.1499(7) | 1.6336(15) | 1.6299(15) | 2.1960(15) | 2.1893(15) | 100.81(3) | 88.63(6) |
| 5b | Zr | 2.8327(5) | 2.1502(6) | 2.1553(6) | 1.6381(13) | 1.6371(13 | 2.2168(13) | 2.1713(13) | 106.11(2) | 99.22(5) |
| 6a-Py | Ti | 3.078(2) | 2.138(2) | 2.144(2) | 1.615(5) | 1.621(5) | 2.112(5) | 2.131(5) | 99.50(9) | 93.12(19) |
| 6a | Ti | 3.0308(7) | 2.1434(8) | 2.1388(8) | 1.6464(19) | 1.6379(19) | 2.0758(19) | 2.0256(19) | 103.57(3) | 107.72(7) |
| 6b-1 | Ti | 3.1049(6) | 2.1404(5) | 2.1531(5) | 1.6422(13) | 1.6409(13) | 2.0372(13) | 2.0257(13) | 108.98(2) | 108.19(5) |
| 6b-2 | Ti | 3.0706(5) | 2.1293(6) | 2.1494(6) | 1.6453(13) | 1.6496(13) | 2.0470(13) | 2.0391(13) | 106.74(2) | 108.87(5) |
| 7b | Ti/Au | 2.9823(12)/2.2240(10) | 2.1884(9) | 2.1884(9) | 1.637(2) | 1.637(2) | 2.037(2) | 2.037(2) | 106.31(5) | 103.38(10) |
| 8b | Ti/Cu | 3.1159(10)/2.1948(9) | 2.1682(11) | 2.1676(12) | 1.632(3) | 1.637(3) | 2.029(3) | 2.038(3) | 109.39(5) | 104.90(11) |
| 9b | Cu | 2.1798(5) | 2.1631(6) | 2.2332(6) | 1.6296(15) | 1.5814(16) | 1.8989(15) | — | 109.84(3) | — |
| A | — | — | 2.1371(9) | 2.1328(9) | — | — | — | — | 95.70(3) | — |
| B | —/Au | 2.2512(17) | 2.200(2) | 2.174(2) | — | — | — | — | 94.59(9) | — |
| C , | Ti | 2.599(2)e | — | — | 1.636(2) | 1.630(2) | 2.052(2) | 2.040(2) | — | 108.53(7) |
The main features of the solid-state structure of 2b′ are (i) its low symmetry, noticeable in the orientation of the Ph rings away and above the LiNPPPN ring; (ii) the large P2–Li distance (3.909(6) Å), far superior to the sum of covalent radii for Li and P (2.35 ± 0.10 Å),72 and close to the sum of van der Waals radii (4.02 Å);73 (iii) the slightly obtuse P1–P2–P3 angle (102.64(4)°). The latter feature is shared between all compounds reported in Table 2. Indeed, the angle between the central P atom and its substituents ranges from 94.59(9)° in B to 109.84(3)° in 9b. Likewise, the P–P bond distances in 2b′ (2.143 Å on average) fall within the rather narrow range of values reported in Table 2 (from 2.1293(6) Å for 6b-2 to 2.2332(6) for 9b).
BIPP coordination to group 4 metals
With ligands 2 in our hands, we turned to the synthesis of Ti and Zr complexes. Reaction of 2a and 2b with [TiCl3(THF)3] in THF gave 3a and 3b, respectively, after workup (Scheme 6).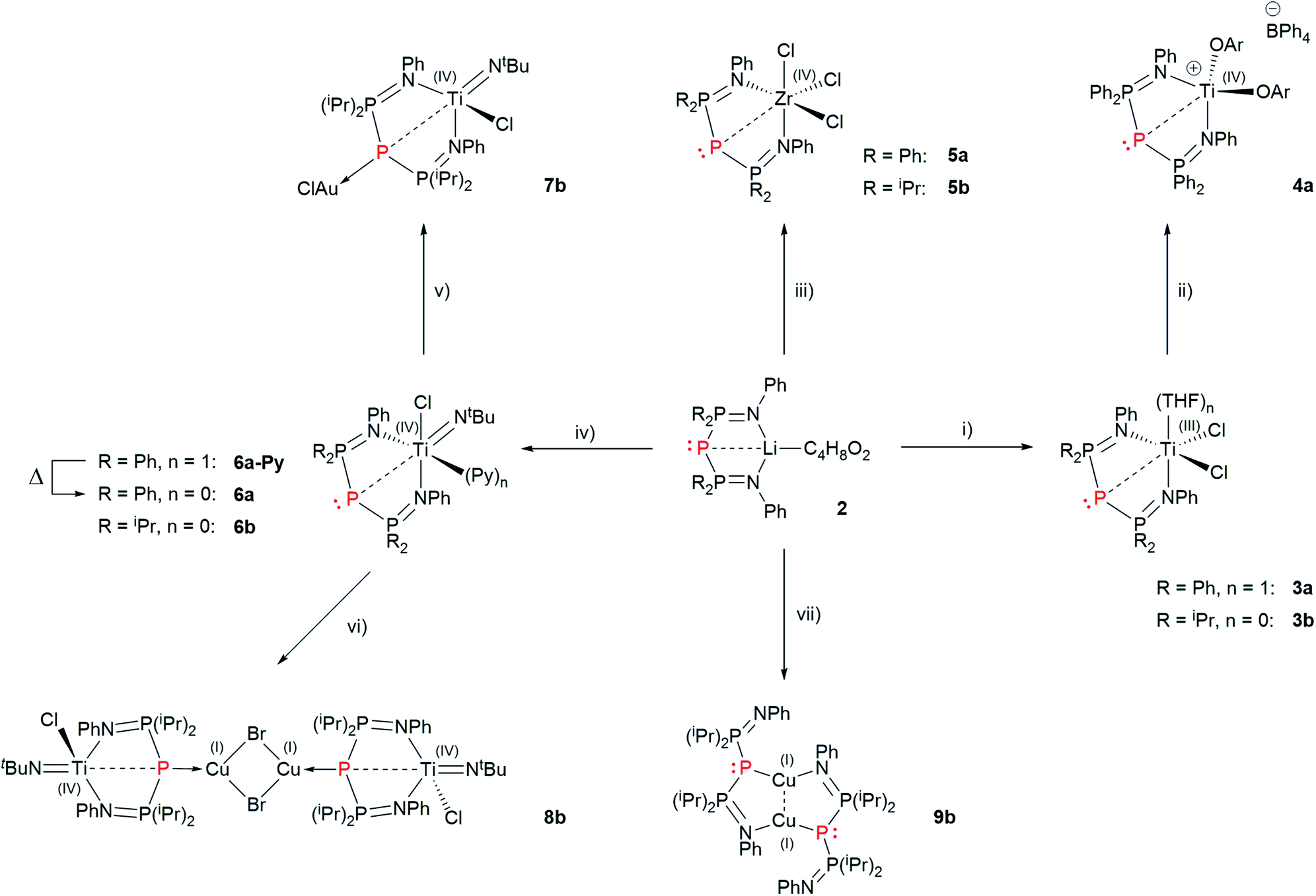 | ||
| Scheme 6 Synthesis of group 4 and coinage metal complexes of BIPP. Reagents and conditions: (i) 1 eq. [TiCl3(THF)3], THF; (ii) 1 eq. [Cp2Fe][BPh4], 2 eq. ArONa (OAr = 2,6-dimethylphenolato); (iii) 1 eq. ZrCl4, toluene; (iv) 1 eq. [TiCl2(NtBu)(Py)3], toluene; (v) 1 eq. [Au(THT)Cl], CH2Cl2; (vi) 1 eq. CuBr, CH2Cl2; (vii) 1 eq. CuBr, toluene. All reactions were conducted at room temperature (THT = tetrahydrothiophene). | ||
The room temperature EPR spectra of 3a and 3b in THF are very similar and characterized by a single absorption line (3a: g = 1.9524; 3b: g = 1.9520) with hyperfine coupling to Ti (3a, 3b: A47/79 Ti = 15.7 10−4 cm−1). Interestingly, no super-hyperfine coupling to P or N was observed.74
The X-ray structures of 3a and 3b are shown in Fig. 1. In contrast with 2b′, these compounds display much shorter P–M distances, with values reaching the upper limit of the sum of covalent radii (Table 2). Remarkably, complex 3a contains one coordinated molecule of THF, whilst 3b is solvent-free (both compounds crystallized from THF solutions). Consequently, 3a displays a distorted octahedral geometry along three axes (Cl1–Ti–N2; Cl2–Ti–P2; N1–Ti–O), whilst 3b is best described as a distorted trigonal bipyramid built along the Cl2–Ti–P2 unique axis. The extra ligand in 3a leads to elongated average Ti–N distances compared to 3b, as well as a narrower N1–Ti–N2 angle. On the other hand, intra-ligand distances, e.g. average P–P and P–N bond distances, are quite similar in both complexes (3a: P–P = 2.145 Å, P–N = 1.631 Å; 3b: P–P = 2.155 Å, P–N = 1.639 Å).
Together with Erker, we have shown that cationic d0 group 4 metallocene complexes containing a phosphorus ligand (phosphide or phosphine) display frustrated Lewis pair (FLP) behaviour;61–64,75 therefore we were keen to explore the possibility of generating post-metallocene Ti(IV) cations from complexes 3. Oxidation of 3a by [Cp2Fe][BPh4] in THF, followed by salt metathesis with sodium 2,6-dimethylphenolate etherate yielded complex 4a in 66% yield after workup (Scheme 6). In the case of 3b, the same procedure yielded an intractable mixture of compounds.
The 1H NMR spectrum of 4a at 300 K reveals the fluxionality of the PPh and OAr groups (Fig. S30†). Cooling to 260 K enabled to partly freeze the structure, although free rotation of each OAr group around O–CAr bonds was still observed. Compared to ligand 2a, the central P atom in 4a resonates at much higher field in the 31P{1H} spectrum (−212.9 vs. −118.7 ppm, see Table 1); however, in light of the δPC value for other BIPPPh compounds, it appears that 2a is the outlier.
Considering the sensitivity of the P2–M bond distance to crystal packing forces (vide infra), the P2–Ti bond distance in 4a is also very similar to that in 3a and 3b (Δd < 0.05 Å). At 2.8125(13) Å it is distinctly superior to the sum of covalent radii for Ti and P, hinting at the possibility of FLP reactivity.76 However, the reaction mixture of 4a and ferrocene carboxaldehyde (FcCHO) in CD2Cl2 remained unchanged; we had previously observed that FcCHO is one of the most reactive substrates against Ti/P FLPs,64,75 therefore we did not investigate the FLP behaviour of 4a any further. Instead, we turned our attention to Zr complexes, which generally display enhanced FLP reactivity compared to Ti.61,64
We synthesized Zr complexes 5 by salt metathesis between ZrCl4 and ligands 2. After workup, complexes 5a and 5b were obtained in 67% and 71% yield respectively (Scheme 5). The 31P{1H} NMR spectra of 5a and 5b both display a strongly shielded signal for the central P atom (−210.2 and −223.0 ppm, Table 1). Interestingly, these values are quite close (ΔδPC = 12.8 ppm), whereas the central P atoms in ligands 2a and 2b resonate at vastly different frequencies (ΔδPC = 82.9 ppm).
The X-ray structures of 5a and 5b are shown in Fig. 1. Two independent molecules of 5a are present in the unit cell, but their metric parameters are very similar (Table 2). The P2–Zr bond distance is very similar in 5a-1, 5a-2 and 5-b, just within the sum of covalent radii for Zr and P. Both 5a and 5b display a distorted octahedral geometry along Cl1–Zr–N2, Cl2–Zr–P2 and Cl3–Zr–N1 axes. Due to the pseudo octahedral geometry, the N1–Zr–N2 angles in both compounds (e.g.5a-2: 88.63(6)°; 5b: 99.22(5)°) are rather small compared to other less crowded complexes (e.g.6a: 107.72(7)°, 3b: 110.02(6)°). As mentioned above, the larger variations observed for the BIPPPh ligand are a probable consequence of the flexibility conferred by the PPh2 groups.
Interestingly, the Cl ligands in octahedral BIPP complexes experience a clear structural trans effect (STE) from the iminophosphorane side arms (see Table S13†).77 This is in line with the non-covalent nature of the interaction between P2 and M (vide infra).
Complexes 5 gave intractable mixtures of compounds upon reaction with sodium 2,6-dimethylphenolate, therefore we were unable to synthesize the Zr analogues of 4a. In an ultimate attempt to investigate the FLP behaviour of Zr complexes, we reacted 5b with FcCHO in C6D5Br, but we only observed a slight broadening of the central phosphorus atom signal at room temperature, and product decomposition at 60 °C.
The “boat” conformation of complexes 3–5 is reminiscent of the coordination behavior of bis(iminophosphoranyl)methanide ligands (BIPMH), which are the conjugate acids of BIPM.2 We have previously reported the only known examples of Ti–BIPMH complexes.78 Such compounds may be synthesized by salt metathesis between BIPMH anions and [TiCl2(NtBu)(Py)3] (Mountford's complex). Applying this procedure to ligands 2 gave complexes 6a-Py and 6b in 50% and 81% yield, respectively, after workup (Scheme 6). The pyridine-free complex 6a was obtained in 74% yield by heating a toluene solution of 6a-Py to reflux.
The 1H NMR spectrum of 6a-Py displays characteristic signals corresponding to the pyridine ligand (multiplets at 9.17, 7.68 and 7.32–7.19 ppm). Pyridine loss induces noticeable changes in the 31P{1H} spectrum of 6a compared to 6a-Py: the lateral and central P atoms resonate at higher field (ΔδPL = −2.9 ppm; ΔδPL = −10.1 ppm), and the 1JPP coupling constant is slightly weaker (Δ1JPP = −26 Hz).
Fig. 1 shows the X-ray structure of 6a and 6a-Py. For 6a-Py, the analysis confirmed that the pyridine molecule is bound to Ti, and occupies one of the axial positions of the distorted octahedron. Interestingly, complex 6a crystallizes with one disordered molecule of toluene, despite the fact that the crystals were obtained by diffusion of pentane into a CH2Cl2 solution of 6a (1 eq. of toluene was also observed by NMR spectroscopy in microcrystalline samples of 6a). The P2–Ti distances in both complexes are considerably longer than the sum of covalent radii, but other distances and angles are unremarkable within the series of compounds reported in Table 2.
The distorted trigonal planar geometry of 6a is very similar to that of its BIPMH analogue (C), as shown by the comparison of metric parameters in Table 2 and the superimposed structures (shown in Fig. S127†). The main difference between 6a and C is the longer E–Ti (E = P, C) bond distance between the metal and the central, negatively charged atom, due to the larger size of phosphorus.72,73 These observations highlight the structural analogy between BIPP and BIPMH ligands.
Fig. 1 shows the crystal structure of one of the two independent molecules of 6b. Again, a distorted planar geometry is observed for 6b-1 and 6b-2 (not shown). The P2–Ti distance is clearly superior to the sum of covalent radii for P and Ti in both cases, but it is noticeably shorter in 6b-1 (Δd = −0.0343(8) Å). As suggested by the superimposed structures of 6b-1 and 6b-2 (shown in Fig. S128†), this difference could originate from conformational changes, i.e. different orientations of the iPr and Ph groups.
BIPP coordination to group 10 metals
The long P2–Ti distance in 6a and 6b suggests that the interaction between both atoms is non-covalent (vide infra). A logical consequence is that it should be possible to coordinate one or two late transition metals to the central P atom, similarly to TP cations.Reaction of 6a with [AuCl(THT)] (THT = tetrahydrothiophene) in CD2Cl2 yielded a complex mixture. However, clean formation of a new compound was observed in the case of 6b. When the reaction was repeated on preparative scale, complex 7b was isolated in 48% yield. Similarly, reaction of 6b with CuBr yielded complex 8b in 78% isolated yield (Scheme 6).
Coordination of AuCl and CuBr fragments to 6b has a dramatic impact on the 31P{1H} NMR spectrum (Table 1). The 1JPP coupling constant decreases considerably (7b: Δ1JPP = −127 Hz; 8b: Δ1JPP = −60 Hz), whilst the central phosphorus atom resonates at much lower field (7b: ΔδPC = 80.6 ppm; 8b: ΔδPC = 28.2 ppm). In the case of 8b, the corresponding signal is very broad, due to the coordination to Cu. Finally, the lateral phosphorus atoms resonate at slightly lower field (7b: ΔδPL = 6.0 ppm; 8b: ΔδPL = 2.5 ppm).
Complex 7b decomposes quite rapidly in CD2Cl2 solutions: decomposition products can already be observed in freshly prepared samples. A probable explanation for this behaviour is that the electron-donating properties of 6b are insufficient. This is also consistent with the impossibility to stabilize AuCl with 6a. By contrast, 8b does not show signs of decomposition in solution. Since the central phosphorus atom in complexes 6 possesses two free lone pairs (vide infra), we attempted to coordinate two CuBr fragments to 6b, however we could only observe the formation of 8b.
Fig. 2 shows the structure of 7b and 8b. These are rare examples of structurally characterized examples of d0/d10 heterobimetallic complexes bridged by a phosphorus atom.79–83 Both complexes show some disorder (the tBu group in the case of 7b, one of the Ph groups in the case of 8b).
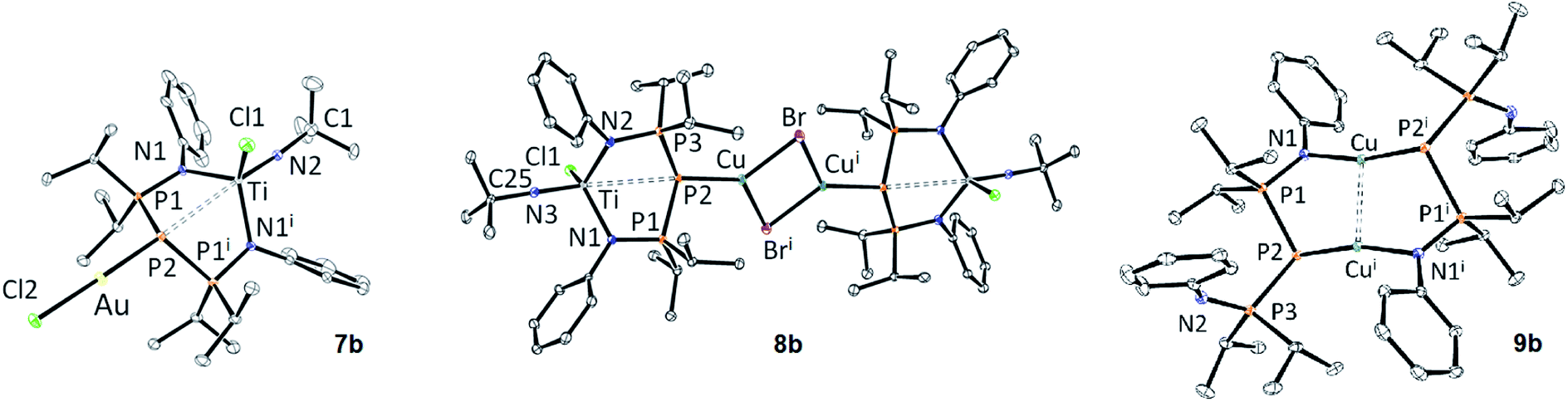 | ||
| Fig. 2 ORTEP depiction of the X-ray structures of 7b, 8b and 9b (thermal ellipsoids drawn at the 30% probability level, hydrogen atoms, solvent molecules and disorder are not shown for clarity). See Table 2 for metric parameters. See the ESI† for additional bond distance and angles. | ||
The structural changes observed upon coordination of the coinage metal fragment to 6b are quite subtle. In the case of gold (7b), the P2–Ti distance becomes slightly shorter, although still superior to the sum of covalent radii. The P–P distances become slightly longer, which could be due to back-bonding between the electron-rich d10 metal and the BIPP ligand (see the AIM results in the ESI†). In the case of copper (8b), the P2–Ti and P–P bond distances become slightly elongated compared to 6b. In both cases, the central P atom adopts an AX3E geometry (7b: ∑α(P2) = 327.51(8)°; 8b: ∑α(P2) = 333.31(8)°). The Au–P2 distance in 7b is close to those observed in Au–TP complex B (Table 2), as well as in [AuCl(PR3)] complexes reported by Schmidbaur. (R = Me: 2.233(3) Å; R = iPr: 2.239(2) Å).84
No Cu–TP complexes have been reported yet, but the Cu–P2 bond distance in 8b is very close to those observed for dimeric [Cu(μ-Br)(PR3)]2 complexes reported by Healy (∼2.20 Å).85
In light of the above, it appears that the net electron-donating properties of 6b are close to weakly basic TP cations such as A (which can only bind one AuCl fragment). We wondered whether the more reactive Li complexes 2 could entail stronger electron donation and/or different coordination modes to coinage metals. Reaction of 2a with [Au(THT)Cl] and CuBr in C6D6 gave a complex mixture and an insoluble solid that could not be analysed by NMR spectroscopy, respectively. Reaction of 2b′ with [Au(THT)Cl] gave a complex mixture.|| However, reaction of 2b with CuBr gave a single new product. When the reaction was conducted in toluene instead of C6D6, complex 9b was isolated in 67% yield (Scheme 6).
The 1H and 31P{1H} NMR spectra of 9b in CD2Cl2 at 298 K belie the solid-state structure depicted in Fig. 2: indeed, only one set of signals is observed for the Ph and iPr groups in the 1H NMR spectrum. The 31P{1H} NMR spectrum offers an explanation, since the signal of the lateral phosphorus atoms is visibly broad. Upon cooling to 250 K, this signal broadens to the extent that the 1JPP coupling constant is not distinguishable anymore (Fig. S104†). A possible explanation for the observed fluxionality of 9b would be iminophosphorane arm exchange following a dissociative mechanism, or central phosphorus exchange following an associative mechanism (via a P-bridged intermediate).
Compared to other BIPP complexes reported in this study, 9b displays remarkable optical spectroscopic features. For instance, the IR spectrum shows a strong absorption at 1335 cm−1 in an otherwise empty region of the spectrum (Fig. 3). Analysis of the normal modes for 9b by DFT revealed that the strong absorption at 1335 cm−1 is due to N–CPh stretching of the free iminophosphorane arm (see multimedia file), which explains why it is not visible in κ3-N,P,N-coordinated compounds.
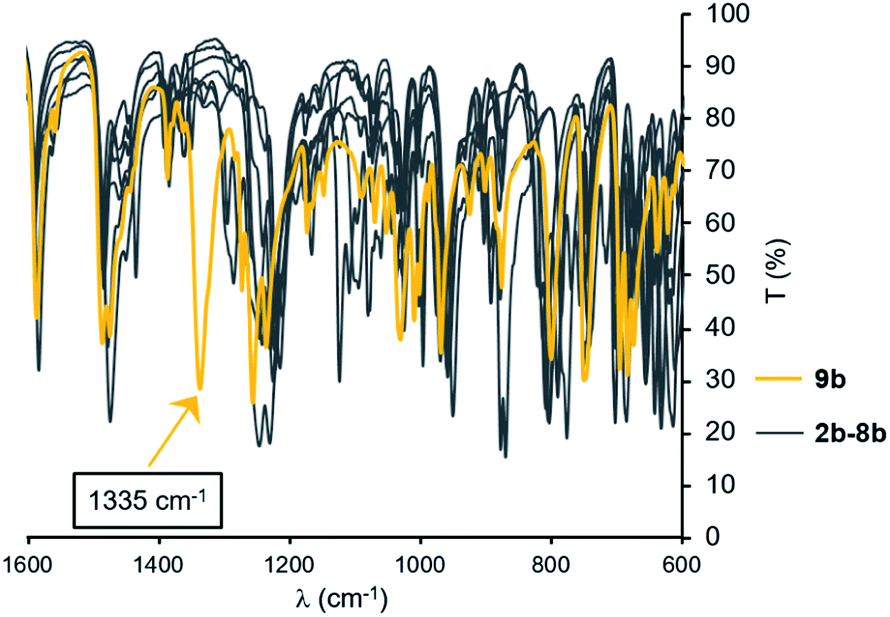 | ||
| Fig. 3 IR spectra of BIPP complexes 2b–9b (ATR, 1600–600 cm−1 range). | ||
Additionally, the UV-Vis spectrum in CH2Cl2 shows an intense absorption at 336 nm (ε = 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1). The latter is reminiscent of the behaviour of cationic dimeric complexes [Cu(dcpm)2]2[X2] reported by Che (D, Chart 2; dcpm = bis(dicyclohexylphosphine)methane). These complexes also display a strong absorption centred at 307–311 nm (ε ≈ 14000–17000 M−1 cm−1), which has been ascribed to a metal–metal 3d → 4p transition.86,87
000 M−1 cm−1). The latter is reminiscent of the behaviour of cationic dimeric complexes [Cu(dcpm)2]2[X2] reported by Che (D, Chart 2; dcpm = bis(dicyclohexylphosphine)methane). These complexes also display a strong absorption centred at 307–311 nm (ε ≈ 14000–17000 M−1 cm−1), which has been ascribed to a metal–metal 3d → 4p transition.86,87
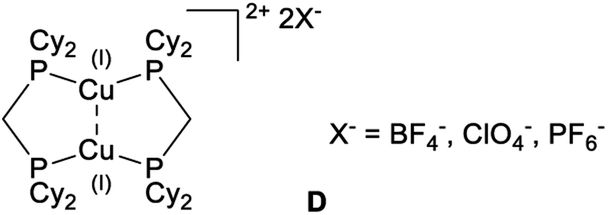 | ||
| Chart 2 Luminescent Cu(I) complexes resported by Che. | ||
Interestingly, the bicyclic structure of 9b can also be related to those of D. Focussing on DBF4, the Cu–Cu distance is close, although slightly shorter in 9b (2.5759(4) vs. 2.691(2) Å). The P2i–Cu distance in 9b (2.1798(5) Å) is also slightly shorter than the average P–Cu distance in DBF4 (2.22 Å). These spectroscopic and structural similarities prompted us to investigate the luminescence properties of 9b, since complexes D were found to be weakly emissive in solution and in the solid state; however, in our case no fluorescence could be observed in solution (CH2Cl2, THF) or in the solid state upon excitation at 336 nm.
In order to understand the lacklustre photophysical properties of 9b compared to D, we computed their absorption spectra with TD–DFT. The computed absorption wavelengths were found in fair agreement with the experimental one: 359 nm instead of 336 nm for 9b and 289 nm instead of 310 nm (average) for D+. In both computed complexes, this absorption corresponds to a HOMO to LUMO transition. As postulated by Che,86,87 this transition has 3d → 4p character. In the case of 9b, both the HOMO and the LUMO are actually quite developed on the phenyl groups of the phosphorus atoms (Fig. S119†), and hence the contribution of the Cu orbitals in this S0–S1 transition is smaller than in the case of D+. This suggests that the phenyl groups in 9b are probably responsible for quenching the fluorescence, as noted elsewhere.88 As a corollary, replacing the phenyl groups in 9b by alkyl groups should restore the fluorescence.
Finally, we conducted voltammetric analyses on 9b (THF, 0.1 M NaBPh4). Several non-reversible events took place both in oxidation and reduction, which could not be interpreted (see Fig. S105†).
Bonding analysis
The above experimental results raise a number of questions pertaining to the bonding situation in BIPP complexes. Bearing in mind the broader context of the nature of bonding in main group compounds, one may thus ask:(i) How do BIPP complexes compare to TP cations and related dicoordinate phosphorus compounds?
(ii) Is the arrow formalism relevant to represent the P–P bonding situation in BIPP complexes?
(iii) What is the nature of the P–Ti interaction?
In order to address these questions, density functional theory (DFT) calculations were conducted on complexes 6a/6b, 7a/7b and 9b, along with ambiphilic dicoordinate phosphorus compounds A and P(CN)2−. All compounds were computed in toluene using the Polarizable Continuum Model (PCM). The bonding situation was analysed and/or represented using the following computational tools: molecular orbitals, Electron Localization Function (ELF) analysis,89–93 Electrostatic Potential Maps (EPM),94 Bader's Atoms in Molecules (AIM),95–97 and Non-Covalent Interactions (NCI) plots.98,99
The computed bond lengths and angles are in good agreement with the experimental solid state structures. In general, the observed deviations are modest (<3%). Notable exceptions include the P2–M distances in 6a and 6b, and the P1–P2–P3 angle in 6a and A, which deviate by up to ±8%.
 | ||
| Fig. 4 HOMO and LUMO for complexes 6a, 6b, A, P(CN)2− and 9b (cut-off 0.05). | ||
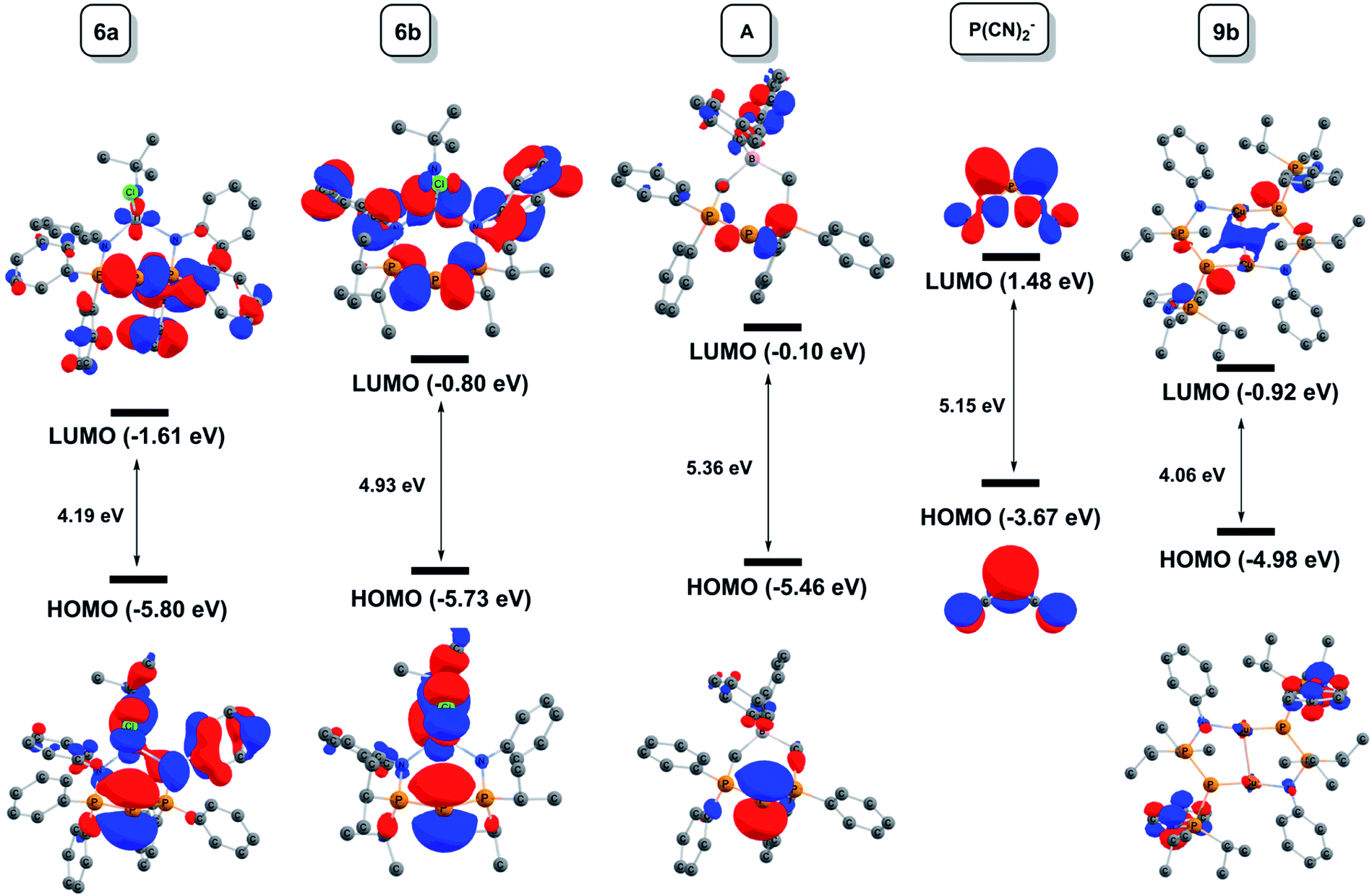
 | ||
| Fig. 5 Electrostatic potential maps for 6a, 6b, 7b, A and 9b plotted over the range −0.05 a.u. (red) to 0.05 a.u. (blue). The isosurfaces are drawn at 0.01 e a.u.−3. | ||
Coordination of AuCl (7b) visibly reduces the maximum charge on the central phosphorus atom, although AIM calculations show that the net (negative) charge actually increases upon going from 6b (Q(PC) = −0.15) to 7b (Q(PC) = −0.29).
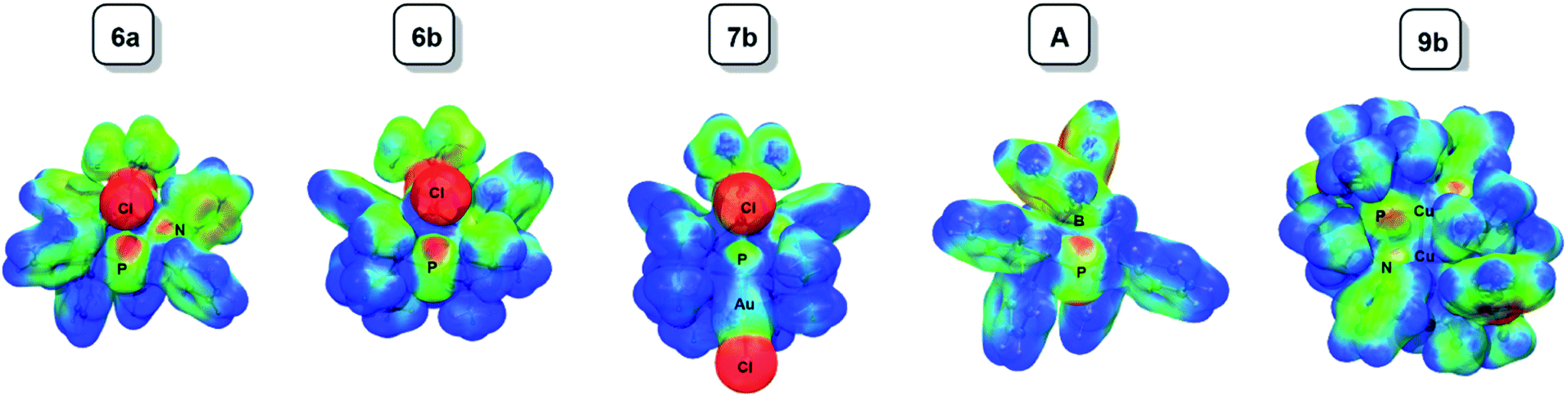
The ELF is a mathematical function that relates to the probability of finding an electron pair in the vicinity of a particular point in space. It is scaled so that 0 ≤ ELF ≤ 1 and increases with the increasing probability of finding a localized electron pair. The topological analysis of the ELF leads to a partition into several “basins” that can be mapped to chemical concepts: core electrons, bonds and lone pairs. In this paper, we have chosen to plot so-called f-localization domains, which are regions of space where ELF > f. Fig. 6 displays the f-localization domains (f = 0.8) of 6a, 6b, 7b, 9b, A and P(CN)2− separated into lone pair and valence basins.
 | ||
| Fig. 6 Graphical representation of the ELF localization domains (f = 0.8) for 6a, 6b, 7b, A and P(CN)2−. Top: lone pair basins; bottom: valence basins. | ||
Looking at the lone pair basins depicted in Fig. 6, the similarity between neutral BIPP complexes 6a–6b, zwitterionic TP compound A and anionic P(CN)2− is evident: indeed, these compounds all possess two lone pairs of electrons at the central phosphorus atom. Coordination of AuCl to this phosphorus atom (7b) logically leaves one available lone pair, as noted elsewhere.34–36,67
Looking at the cylindrically shaped valence basins related to the P–P bonds, the similarity is again evident in the case of 6a–6b and A, which lends further credence to the view of BIPP as TP compounds with anionic substituents. The situation is different in the case of P(CN)2−: the valence basins associated with the P–C bonds are visibly deformed and point towards the central phosphorus atom, suggesting the presence of polarized (or dative) bonds. The presence of AuCl in 7b also modifies the electron distribution associated with the P–P bonds, i.e. the valence basins point towards the AuCl moiety.
The QTAIM is a topological analysis of the properties of the electron density (ρ), its gradient (∇ρ) and its Laplacian (∇2ρ) in the three-dimensional Euclidean space. Within the framework of the QTAIM, atoms emerge as non-overlapping regions of space (atomic basins) defined by their kinetic energies, and separated by interatomic surfaces.96,100 Generally, bonded atoms are connected by a bond path (BP) crossing a bond critical point (BCP), i.e. a saddle point of the electron density.101
The numerical values of a series of topological indexes at BCPs enable to characterize the type of bonds between atoms: these are the delocalization index (DI, the number of delocalized electron pairs between two atoms), H/ρ and G/ρ ratios (where H is the total energy density, and G the kinetic energy density), in addition to ρ and ∇2ρ.†† Taken together, these parameters can help differentiate open-shell (normal covalent), intermediate (polar and/or dative covalent) and closed-shell (ionic, van der Waals) interactions. Of particular interest for our purposes is the DI, which has been shown to be a reliable measure of bond order;102 thus, normal covalent bonds typically display DI values close to the formal bond order (∼1 in the case of a single bond), whilst dative covalent bonds yield lower values (<1), and closed-shell interactions give DI values close to 0.22,103
We performed AIM calculations for complexes 6a, 6b, 7b and 9b, along with A and P(CN)2−. For the sake of conciseness, only the P–P bonds (P–C for P(CN)2−) and the P–Ti interaction are presented in Table 3.104
| BCP | ρ (e Å−3) | ∇2ρ (e Å−5) | G/ρ (Eh e−1) | H/ρ (Eh e−1) | DI | |
|---|---|---|---|---|---|---|
| 6a | P–P | 0.857 | −4.531 | 0.287 | −0.658 | 1.022 |
| P–P | 0.844 | −4.386 | 0.278 | −0.642 | 1.011 | |
| P–Ti | 0.115 | 0.940 | 0.647 | −0.071 | 0.085 | |
| 6b | P–P | 0.844 | −4.362 | 0.263 | −0.626 | 1.042 |
| P–P | 0.837 | −4.241 | 0.262 | −0.617 | 1.033 | |
| A | P–P | 0.837 | −4.265 | 0.286 | −0.644 | 1.027 |
| P–P | 0.857 | −4.579 | 0.298 | −0.670 | 1.044 | |
| P(CN)2− | P–C | 0.992 | 6.266 | 1.259 | −0.816 | 1.023 |
| 7b | P–P | 0.776 | −3.061 | 0.290 | −0.564 | 0.869 |
| P–P | 0.776 | −3.012 | 0.290 | −0.562 | 0.876 | |
| P–Ti | 0.135 | 1.060 | 0.627 | −0.070 | 0.099 | |
| 9b | P–P | 0.817 | −3.928 | 0.238 | −0.574 | 0.986 |
| P–P | 0.769 | −3.350 | 0.215 | −0.519 | 0.910 | |
P–P bonds. The principal outcome of these calculations is that P–P bonds in BIPP complexes and the model TP compound A are best described as normal covalent bonds. Indeed, in every computed molecule the P–P BCPs display low (i.e. <1) G/ρ and negative H/ρ ratios, along with DI values close to unity; these results unambiguously indicate a normal covalent bond between light atoms, rather than a donor–acceptor interaction.103 On the other hand, the moderate ρ (∼0.84 e Å−3) and │∇2ρ│ (∼4.0 e Å−5) suggest relatively low bond dissociation energies (BDEs), as observed experimentally in the case of A.34
In order to investigate the nature of the P–P interaction in 6a and 6b in more details, we plotted the contour plot of ∇2ρ in the P–P–P plane and the graph of ∇2ρ along the P–P BPs (Fig. 7). The charge concentration due to the normal covalent interaction between the lateral and central phosphorus atoms is clearly visible in the contour plot (top).
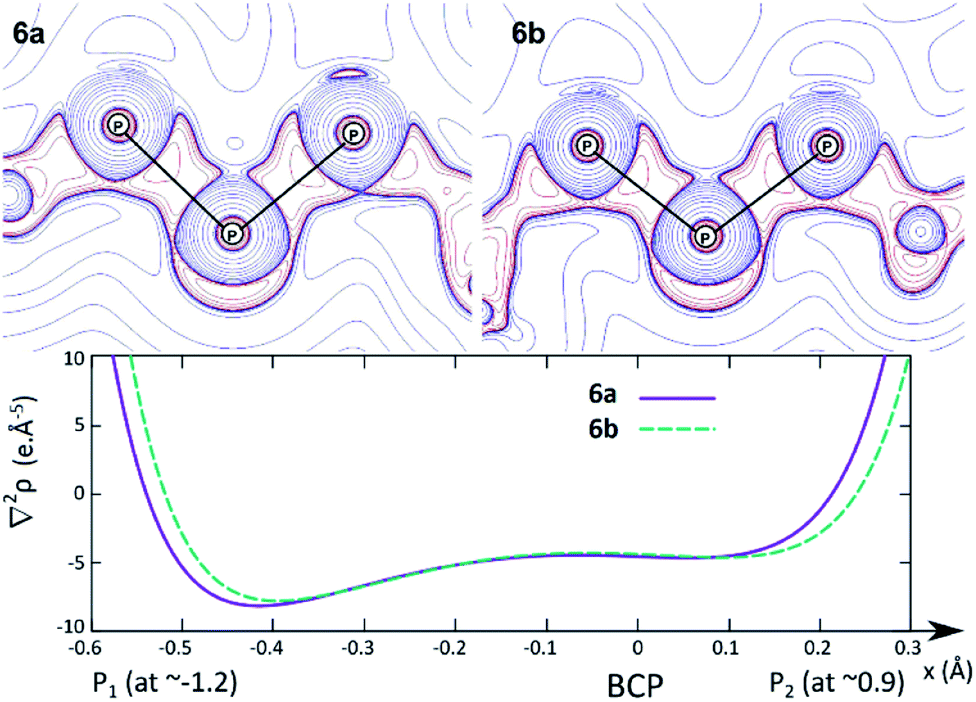 | ||
| Fig. 7 Top: contour map of ∇2ρ in the P–P–P plane for 6a and 6b (red lines indicate charge concentration and blue lines indicate charge depletion). Bottom: graph of ∇2ρ along the left-hand P–P BPs for 6a and 6b. | ||
Upon closer inspection of the Laplacian along the P–P bond path (bottom), additional features become apparent. Firstly, the BCP lies close to a local minimum (a characteristic of normal covalent bonds), and it is located in a rather flat region of the Laplacian; therefore, the P–P interaction cannot be described as a donor–acceptor bond, since this type of bonds entails much higher charge concentration in the atomic basin of the donor (i.e. the Laplacian profile should be much steeper). However, the BCP also lies ∼0.3 Å closer to the more electropositive central phosphorus. The slight polarisation of the P–P bond towards the lateral phosphorus atom is actually quite remarkable, given the large charge difference between both atoms (e.g. for 6a: Q(PL) = 2.11 and 2.13; Q(PC) = −0.06; see the ESI† for the full table). Overall, the best Lewis representation of BIPP complexes is the alternating dipolar form (see BIPM, Scheme 3, center), and our results suggest that this is also the case for TP compounds.
Different results were obtained for the P(CN)2− anion; in this case, the P–C bond is characterized by a positive value of ∇2ρ at the BCP, a high kinetic energy (G/ρ > 1), a negative total energy (H/ρ ≪ 0) and a DI close to unity. Moreover, the BCP lies closer to the more electropositive phosphorus atom. This picture is consistent either with a heteropolar covalent bond, or to a donor–acceptor interaction with considerable back-bonding, e.g. as in ([Co(CO)4]2).103 Therefore, although dicoordinate phosphorus compounds such as A and P(CN)2− readily undergo heterolytic P–P and P–C bond cleavage in solution, the indiscriminate use of the term “ligand exchange” to qualify these reactions could be deceptive in view of the contrasting bonding situations.
P–Ti interaction. The AIM calculations also gave insights into the nature of the PC–Ti interaction in BIPP complexes. According to the computed topological indexes at the BCP, the P–Ti interaction in 6a can be described as weakly electrostatic, as witnessed by the very low electron density (ρ = 0.115 e Å−3), the positive Laplacian (∇2ρ = 0.940 e Å−5), and the low value of the delocalization index (DI = 0.085). Also, the contour plot of the Laplacian in the PC–Ti–Cl plane shows that the valence shell charge concentration (VSCC) corresponding to the lone pair of P pointing towards Ti is not aligned with the P–Ti BP (Fig. 8). This misalignment—evidenced by the angle α—is strongly reminiscent of the situation observed by Mitzel in a family of aminosilanes exhibiting weak intramolecular N–Si interactions.105–108
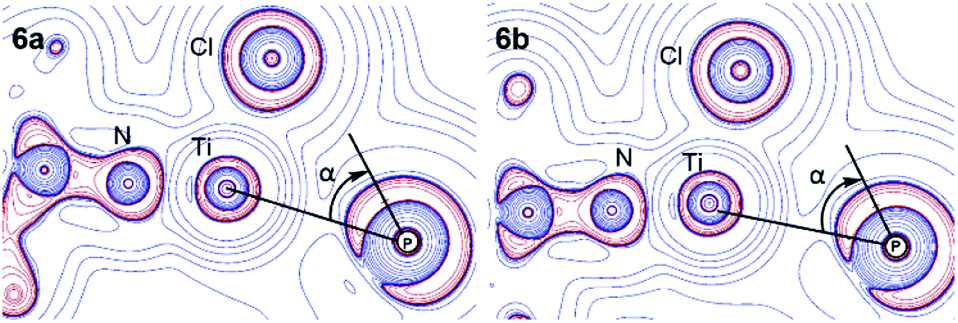 | ||
| Fig. 8 Contour map of ∇2ρ in the PC–Ti–Cl plane for 6a and 6b (red lines indicate charge concentration and blue lines indicate charge depletion). | ||
Interestingly, the values of G/ρ and H/ρ for the P–Ti interaction are at variance with the correspondence rules established by Macchi and Sironi,103,109 in particular the negative total energy density (H) is surprising given the closed-shell nature of the interaction. This is probably due to a small admixture of covalent character, and highlights the fact that correspondence rules between topological indexes and chemical concepts should be taken with the proverbial pinch of salt, especially in the case of unusual bonding situations.
It should also be noted that a BCP was not found between PC and Ti in the case of 6b; however, the similar aspects of the contour plots shown in Fig. 8 suggest that the absence of a BCP is probably due to a “catastrophe point” in the molecular graph of 6b (i.e. the BCP disappears after a threshold P–Ti distance).110 The fact that a BCP “reappears” upon coordination of AuCl (complex 7b) is consistent with this hypothesis. Essentially, the P–Ti interaction is similar in 6a and 6b.
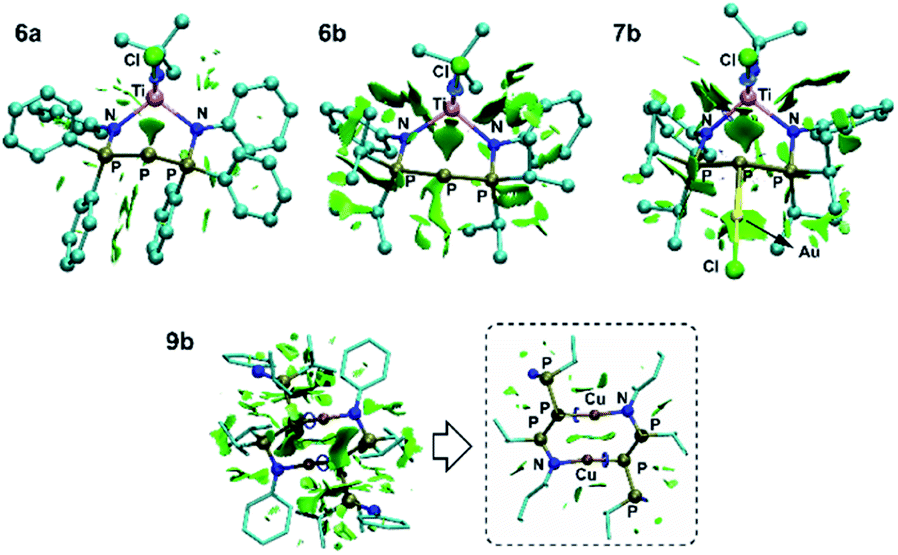 | ||
| Fig. 9 NCI plot for 6a, 6b, 7b and 9b. Gradient isosurface (s = 0.3 a.u.) are colored according to a blue-green-red scheme over the range of −8.00 < sign (λ2) (ρ < 8.00 a.u.). Green indicates very weak interaction. | ||
Conclusions
Although the first BIPP derivative was reported some 35 years ago by Schmidpeter, and despite the obvious analogy with P(IV)-stabilized methanediides (e.g.BIPM), the coordination chemistry of BIPP was an unexplored area before we undertook the present study. This apparent anomaly is undoubtedly related to their challenging synthesis, which requires the use of white phosphorus in order to install the central P atom. In the presence of early transition metals, BIPP act as peculiar fac-coordinating ligands: the soft central phosphorus atom mainly interacts electrostatically with the early metal, leaving enough electron density to bind late transition metals (such as Cu or Au). In the absence of an early metal, they switch to a κ2-P,N coordination mode in order to bind Cu more strongly. Interestingly they do not seem to share BIPM's propensity to engage in multiple bonding interactions with transition metals. These intriguing coordination properties certainly warrant further studies, e.g. with f-group elements and late transition metals.The key feature of BIPP is their similarity to triphosphenium cations (TP): indeed, as evidenced computationally, BIPPs may be regarded as TP bearing anionic substituents, and hence as ambiphilic phosphides. An important practical consequence is that they should undergo heterolytic P–P bond cleavage under relatively mild conditions. Crucially, the topological analysis of the computed electron density and its Laplacian demonstrate that the P–P bonds in these compounds are normal covalent: we therefore suggest reconsidering the terminology currently employed to describe their reactivity (i.e. nucleophilic substitution vs. ligand exchange). Our results also indicate that homolytic bond cleavage might be observed under suitable conditions (e.g. in nonpolar solvents).
Finally, a word of caution is in order: experimentally, TP behave like labile main group donor–acceptor adducts. Hence, the use of the arrow formalism will inevitably mislead some authors into thinking that the P–P bonds are dative covalent. To be sure, the arrow formalism is merely a heuristic device, but the key point is that it is not used consistently by chemists: it can take different meanings depending on one's perspective on chemical bonds and their representations. In the end, it does not really matter which representation is used, as long as chemists take precautions in order to avoid misinterpretations of their drawings.
Experimental section
Synthesis
Carefully read the risk assessment and cautionary notes in the ESI† before carrying out any procedure involving white phosphorus.Attempted syntheses and failed experiments
See the ESI.†Computational details
All calculations were performed using the Gaussian 16 package,111 and the B3PW91 hybrid functional on the real experimental systems.112 Gold and copper atoms were described with the LANL2DZ pseudopotential.113 The 6-31+G(d,p) basis set were employed for all other atoms. This basis set will be denoted as BS1. Optimizations were carried out without any symmetry restrictions taking into solvent effect (toluene) by means of the continuum standard solvation PCM model.114 All stationary points computed were fully optimized. All total energies and Gibbs free energies have been zero-point energy (ZPE) and temperature corrected using unscaled density functional frequencies. Molecular orbitals were plotted using ChemCraft.115In order to gain insight into the nature of the chemical bonding, we performed atoms in molecules analysis (QTAIM),95,96 using AIMALL software,116 and Electronic Localized Function analysis using TopChem package.117 The Electrostatic Potential Surface (EPS) map and NCIPLOT98,99 have been plotted with VMD software.118 For AIM, ELF and NCI analyses, wavefunction was recomputed using RIFIT basis sets for gold and Cu atoms (NCI only for Cu) and 6-31+G(d,p) for all other atoms using geometries obtained at the PCM (Toluene)-B3PW91-D3/BS1 level.
The B3PW91/BS1 level was also used for the time-dependant DFT computations. We tested that using the larger def2-TZVP basis set did not change the computed wavelengths.119
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are indebted to Dr Nicolas Mézailles for sharing his vast knowledge of white phosphorus with us. We thank T. A. Keith for his help in plotting the Laplacian along a bond critical path. The authors also thank the “Plateforme d'Analyse Chimique et de Synthèse Moléculaire de l'Université de Bourgogne” (PACSMUB, http://www.wpcm.fr) for access to spectroscopy and instrumentation, Mr Marcel Soustelle (University of Burgundy, ICMUB, PACSMUB) for elemental analyses and Prof Ewen Bodio (University of Burgundy, ICMUB, OCS team) for access to SAFAS Flx-Xenius XC spectrofluorimeter. Financial support from Centre National de la Recherche Scientifique (CNRS), Université de Bourgogne (UB), Agence Nationale de la Recherche (ALCATRAS, ANR-16-CE07-0001-01), Conseil Regional de Bourgogne (PARI CDEA program), Conseil Régional de Bourgogne Franche-Comté (PhosFerTiMn project), Fonds Européen de Développement Régional (FEDER) and the PIA-excellence ISITE-BFC program (COMICS project) is gratefully acknowledged. Calculations were performed using HPC resources from DSI-CCUB (Université de Bourgogne).Notes and references
- T. Cantat, N. Mezailles, A. Auffrant and P. Le Floch, Dalton Trans., 2008, 1957–1972 RSC.
- T. K. Panda and P. W. Roesky, Chem. Soc. Rev., 2009, 38, 2782–2804 RSC.
- S. T. Liddle, D. P. Mills and A. J. Wooles, Chem. Soc. Rev., 2011, 40, 2164–2176 RSC.
- S. Harder, Coord. Chem. Rev., 2011, 255, 1252–1267 CrossRef CAS.
- M. Fustier-Boutignon, N. Nebra and N. Mézailles, Chem. Rev., 2019, 119, 8555–8700 CrossRef CAS.
- R. Tonner, F. Öxler, B. Neumüller, W. Petz and G. Frenking, Angew. Chem., Int. Ed., 2006, 45, 8038–8042 CrossRef CAS.
- R. Tonner and G. Frenking, Chem.–Eur. J., 2008, 14, 3260–3272 CrossRef CAS.
- L. Zhao, M. Hermann, N. Holzmann and G. Frenking, Coord. Chem. Rev., 2017, 344, 163–204 CrossRef CAS.
- C. A. Dyker and G. Bertrand, Nat. Chem., 2009, 1, 265–266 CrossRef CAS.
- M. Alcarazo, C. W. Lehmann, A. Anoop, W. Thiel and A. Fürstner, Nat. Chem., 2009, 1, 295–301 CrossRef CAS.
- L. Zhao, S. Pan, N. Holzmann, P. Schwerdtfeger and G. Frenking, Chem. Rev., 2019, 119, 8781–8845 CrossRef CAS.
- H. Schmidbaur, Angew. Chem., Int. Ed., 2007, 46, 2984–2985 CAS.
- G. Frenking, B. Neumüller, W. Petz, R. Tonner and F. Öxler, Angew. Chem., Int. Ed., 2007, 46, 2986–2987 Search PubMed.
- H. Daniel, K. Ingo and S. Andreas, Angew. Chem., Int. Ed., 2014, 53, 6047–6048 Search PubMed.
- G. Frenking, Angew. Chem., Int. Ed., 2014, 53, 6040–6046 CrossRef CAS.
- D. Himmel, I. Krossing and A. Schnepf, Angew. Chem., Int. Ed., 2014, 53, 370–374 CrossRef CAS.
- Ligand exchange reactions have been observed in a P,S-double ylides and in a Si(0) silylene, see (a) N. Dellus, T. Kato, N. Saffon-Merceron, V. Branchadell and A. Baceiredo, Inorg. Chem., 2011, 50, 7949–7951 CrossRef CAS; (b) K. C. Mondal, H. W. Roesky, M. C. Schwarzer, G. Frenking, I. Tkach, H. Wolf, D. Kratzert, R. Herbst-Irmer, B. Niepötter and D. Stalke, Angew. Chem., Int. Ed., 2013, 52, 1801–1805 CrossRef CAS.
- A. Haaland, Angew. Chem., Int. Ed., 1989, 28, 992–1007 CrossRef.
- N. V. Sidgwick, The Theory of Electronic Valency, Oxford University Press, London, 1929 Search PubMed.
- V. I. Minkin, Pure Appl. Chem., 1999, 71, 1919 CAS.
- I. Abdellah, C. Lepetit, Y. Canac, C. Duhayon and R. Chauvin, Chem.–Eur. J., 2010, 16, 13095–13108 CrossRef CAS.
- N. A. Pushkarevsky, P. A. Petrov, D. S. Grigoriev, A. I. Smolentsev, L. M. Lee, F. Kleemiss, G. E. Salnikov, S. N. Konchenko, I. Vargas-Baca, S. Grabowsky, J. Beckmann and A. V. Zibarev, Chem.–Eur. J., 2017, 23, 10987–10991 CrossRef CAS.
- S. Mebs and J. Beckmann, J. Phys. Chem. A, 2017, 121, 7717–7725 CrossRef CAS.
- A. Nandi and S. Kozuch, Chem.–Eur. J., 2020, 26, 759–772 CrossRef CAS.
- A. Schmidpeter, Heteroat. Chem., 1999, 10, 529–537 CrossRef CAS.
- P. K. Coffer and K. B. Dillon, Coord. Chem. Rev., 2013, 257, 910–923 CrossRef CAS.
- B. D. Ellis and C. L. B. Macdonald, Coord. Chem. Rev., 2007, 251, 936–973 CrossRef CAS.
- E. M. Dionisi, J. F. Binder, J. H. W. LaFortune and C. L. B. Macdonald, Dalton Trans., 2020, 49, 12115–12127 RSC.
- S. Lochschmidt and A. Schmidpeter, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1985, 40, 765–773 Search PubMed.
- A. Schmidpeter, S. Lochschmidt and W. S. Sheldrick, Angew. Chem., Int. Ed., 1985, 24, 226–227 CrossRef.
- S. C. Kosnik, G. J. Farrar, E. L. Norton, B. F. T. Cooper, B. D. Ellis and C. L. B. Macdonald, Inorg. Chem., 2014, 53, 13061–13069 CrossRef CAS.
- J. F. Binder, A. a. Swidan, M. Tang, J. H. Nguyen and C. L. B. Macdonald, Chem. Commun., 2015, 51, 7741–7744 RSC.
- S. C. Kosnik and C. L. B. Macdonald, Dalton Trans., 2016, 45, 6251–6258 RSC.
- J. W. Dube, C. L. B. Macdonald and P. J. Ragogna, Angew. Chem., Int. Ed., 2012, 51, 13026–13030 CrossRef CAS.
- S. C. Kosnik, M. C. Nascimento, J. F. Binder and C. L. B. Macdonald, Dalton Trans., 2017, 46, 17080–17092 RSC.
- J. F. Binder, S. C. Kosnik and C. L. B. Macdonald, Chem.–Eur. J., 2018, 24, 3556–3565 CrossRef CAS.
- J. F. Binder, A. a. Swidan and C. L. B. Macdonald, Inorg. Chem., 2018, 57, 11717–11725 CrossRef CAS.
- J. Zhou, L. L. Liu, L. L. Cao and D. W. Stephan, Angew. Chem., Int. Ed., 2019, 58, 18276–18280 CrossRef CAS.
- For other “ligand exchange” reactions at phosphorus, see (a) N. Burford, T. S. Cameron, P. J. Ragogna, E. Ocando-Mavarez, M. Gee, R. McDonald and R. E. Wasylishen, J. Am. Chem. Soc., 2001, 123, 7947–7948 CrossRef CAS; (b) N. Burford, P. J. Ragogna, R. McDonald and M. J. Ferguson, J. Am. Chem. Soc., 2003, 125, 14404–14410 CrossRef CAS; (c) M. M. Hansmann, R. Jazzar and G. Bertrand, J. Am. Chem. Soc., 2016, 138, 8356–8359 CrossRef CAS; (d) M. M. Hansmann and G. Bertrand, J. Am. Chem. Soc., 2016, 138, 15885–15888 CrossRef CAS; (e) M. Donath, E. Conrad, P. Jerabek, G. Frenking, R. Fröhlich, N. Burford and J. J. Weigand, Angew. Chem., Int. Ed., 2012, 51, 2964–2967 CrossRef CAS.
- IUPAC, Compendium of Chemical Terminology (the “Gold Book”), compiled by A. D. McNaught and A. Wilkinson, Blackwell Scientific Publications, Oxford, 2nd edn, 1997, online version (2019) created by S. J. Chalk. ISBN 0-9678550-9-8, DOI:10.1351/goldbook.
- B. D. Ellis and C. L. B. Macdonald, Inorg. Chem., 2006, 45, 6864–6874 CrossRef CAS.
- A. Kasani, R. P. Kamalesh Babu, R. McDonald and R. G. Cavell, Angew. Chem., Int. Ed., 1999, 38, 1483–1484 CrossRef CAS.
- R. G. Cavell, R. P. Kamalesh Babu, A. Kasani and R. McDonald, J. Am. Chem. Soc., 1999, 121, 5805–5806 CrossRef CAS.
- C. M. Ong and D. W. Stephan, J. Am. Chem. Soc., 1999, 121, 2939–2940 CrossRef CAS.
- T. Cantat, T. Arliguie, A. Noël, P. Thuéry, M. Ephritikhine, P. L. Floch and N. Mézailles, J. Am. Chem. Soc., 2009, 131, 963–972 CrossRef CAS.
- J.-C. Tourneux, J.-C. Berthet, P. Thuery, N. Mezailles, P. Le Floch and M. Ephritikhine, Dalton Trans., 2010, 39, 2494–2496 RSC.
- J.-C. Tourneux, J.-C. Berthet, T. Cantat, P. Thuéry, N. Mézailles and M. Ephritikhine, J. Am. Chem. Soc., 2011, 133, 6162–6165 CrossRef CAS.
- J.-C. Tourneux, J.-C. Berthet, T. Cantat, P. Thuéry, N. Mézailles, P. Le Floch and M. Ephritikhine, Organometallics, 2011, 30, 2957–2971 CrossRef CAS.
- G. Ma, M. J. Ferguson, R. McDonald and R. G. Cavell, Inorg. Chem., 2011, 50, 6500–6508 CrossRef CAS.
- W. Ren, X. Deng, G. Zi and D.-C. Fang, Dalton Trans., 2011, 40, 9662–9664 RSC.
- O. J. Cooper, D. P. Mills, J. McMaster, F. Moro, E. S. Davies, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2011, 50, 2383–2386 CrossRef CAS.
- D. P. Mills, O. J. Cooper, F. Tuna, E. J. L. McInnes, E. S. Davies, J. McMaster, F. Moro, W. Lewis, A. J. Blake and S. T. Liddle, J. Am. Chem. Soc., 2012, 134, 10047–10054 CrossRef CAS.
- V. H. Gessner, F. Meier, D. Uhrich and M. Kaupp, Chem.–Eur. J., 2013, 19, 16729–16739 CrossRef CAS.
- O. J. Cooper, D. P. Mills, J. McMaster, F. Tuna, E. J. L. McInnes, W. Lewis, A. J. Blake and S. T. Liddle, Chem.–Eur. J., 2013, 19, 7071–7083 CrossRef CAS.
- M. Gregson, E. Lu, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2013, 52, 13016–13019 CrossRef CAS.
- M. Gregson, E. Lu, F. Tuna, E. J. L. McInnes, C. Hennig, A. C. Scheinost, J. McMaster, W. Lewis, A. J. Blake, A. Kerridge and S. T. Liddle, Chem. Sci., 2016, 7, 3286–3297 RSC.
- E. Lu, S. Sajjad, V. E. J. Berryman, A. J. Wooles, N. Kaltsoyannis and S. T. Liddle, Nat. Commun., 2019, 10, 634 CrossRef CAS.
- A recent study by Gessner and Frenking suggests that this is due to the increased electron-sharing character of the P–C bonds conferred by the phosphinoamide substituents: L. T. Scharf, D. M. Andrada, G. Frenking and V. H. Gessner, Chem.–Eur. J., 2017, 23, 4422–4434 CrossRef CAS.
- A. Schmidpeter and G. Burget, Angew. Chem., Int. Ed., 1985, 24, 580–581 CrossRef.
- A. Schmidpeter and G. Bürget, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1985, 40, 1306 Search PubMed.
- A. T. Normand, C. G. Daniliuc, B. Wibbeling, G. Kehr, P. Le Gendre and G. Erker, J. Am. Chem. Soc., 2015, 137, 10796–10808 CrossRef CAS.
- A. T. Normand, C. G. Daniliuc, G. Kehr, P. Le Gendre and G. Erker, Dalton Trans., 2016, 45, 3711–3714 RSC.
- A. T. Normand, C. G. Daniliuc, B. Wibbeling, G. Kehr, P. Le Gendre and G. Erker, Chem.–Eur. J., 2016, 22, 4285–4293 CAS.
- A. T. Normand, Q. Bonnin, S. Brandès, P. Richard, P. Fleurat-Lessard, C. H. Devillers, C. Balan, P. Le Gendre, G. Kehr and G. Erker, Chem.–Eur. J., 2019, 25, 2803–2815 CrossRef CAS.
- J. W. Dube, C. L. B. Macdonald, B. D. Ellis and P. J. Ragogna, Inorg. Chem., 2013, 52, 11438–11449 CrossRef CAS.
- P. K. Coffer, R. M. K. Deng, K. B. Dillon, M. A. Fox and R. J. Olivey, Inorg. Chem., 2012, 51, 9799–9808 CrossRef CAS.
- K. Schwedtmann, M. H. Holthausen, K.-O. Feldmann and J. J. Weigand, Angew. Chem., Int. Ed., 2013, 52, 14204–14208 CrossRef CAS.
- J. W. Dube, V. A. Béland, P. D. Boyle and P. J. Ragogna, Can. J. Chem., 2015, 93, 253–264 CrossRef CAS.
- Macdonald reported an alternative method for the synthesis of [PPh4][P(CN)2] during the course of our study: J. F. Binder, S. C. Kosnik, P. B. J. St Onge and C. L. B. Macdonald, Chem.–Eur. J., 2018, 24, 14644–14648 CrossRef CAS.
- H. B. Baker, Philos. Trans. R. Soc., A, 1888, 179, 571–591 Search PubMed.
- C. W. Hsu, Y. C. Tsai, B. M. Cossairt, C. Camp, J. Arnold, B. M. Cossairt, B. M. Cossairt, C. C. Cummins and Y. C. Tsai, Inorg. Synth., 2019, 123–134 Search PubMed.
- B. Cordero, V. Gomez, A. E. Platero-Prats, M. Reves, J. Echeverria, E. Cremades, F. Barragan and S. Alvarez, Dalton Trans., 2008, 2832–2838 RSC.
- S. Alvarez, Dalton Trans., 2013, 42, 8617–8636 RSC.
- This is in contrast with phosphidotitanocene compounds in which superhyperfine coupling to P was observed, see ref. 64.
- A. T. Normand, P. Richard, C. Balan, C. G. Daniliuc, G. Kehr, G. Erker and P. Le Gendre, Organometallics, 2015, 34, 2000–2011 CrossRef CAS.
- Methanediide complexes have been shown to display metal–ligand cooperativity (FLP-type behavior), see (a) R. P. Kamalesh Babu, R. McDonald and R. G. Cavell, Organometallics, 2000, 19, 3462–3465 CrossRef; (b) K.-S. Feichtner and V. H. Gessner, Chem. Commun., 2018, 54, 6540–6553 RSC.
- B. J. Coe and S. J. Glenwright, Coord. Chem. Rev., 2000, 203, 5–80 CrossRef CAS.
- A. T. Normand, A. Massard, P. Richard, C. Canovas, C. Balan, M. Picquet, A. Auffrant and P. Le Gendre, Dalton Trans., 2014, 43, 15098–15110 RSC.
- A. Glöckner, T. Bannenberg, S. Büschel, C. G. Daniliuc, P. G. Jones and M. Tamm, Chem.–Eur. J., 2011, 17, 6118–6128 CrossRef.
- F. Lindenberg, T. Shribman, J. Sieler, E. Hey-Hawkins and M. S. Eisen, J. Organomet. Chem., 1996, 515, 19–25 CrossRef CAS.
- R. T. Baker, W. C. Fultz, T. B. Marder and I. D. Williams, Organometallics, 1990, 9, 2357–2367 CrossRef CAS.
- F. Dielmann, E. V. Peresypkina, B. Krämer, F. Hastreiter, B. P. Johnson, M. Zabel, C. Heindl and M. Scheer, Angew. Chem., Int. Ed., 2016, 55, 14833–14837 CrossRef CAS.
- B. P. Johnson, F. Dielmann, G. Balázs, M. Sierka and M. Scheer, Angew. Chem., Int. Ed., 2006, 45, 2473–2475 CrossRef CAS.
- K. Angermaier, E. Zeller and H. Schmidbaur, J. Organomet. Chem., 1994, 472, 371–376 CrossRef CAS.
- G. A. Bowmaker, S. E. Boyd, J. V. Hanna, R. D. Hart, P. C. Healy, B. W. Skelton and A. H. White, J. Chem. Soc., Dalton Trans., 2002, 2722–2730 RSC.
- C.-M. Che, Z. Mao, V. M. Miskowski, M.-C. Tse, C.-K. Chan, K.-K. Cheung, D. L. Phillips and K.-H. Leung, Angew. Chem., Int. Ed., 2000, 39, 4084–4088 CrossRef CAS.
- W.-F. Fu, X. Gan, C.-M. Che, Q.-Y. Cao, Z.-Y. Zhou and N. N.-Y. Zhu, Chem.–Eur. J., 2004, 10, 2228–2236 CrossRef CAS.
- M. Nishikawa, T. Sano, M. Washimi, K. Takao and T. Tsubomura, Dalton Trans., 2016, 45, 12127–12136 RSC.
- A. D. Becke and K. E. Edgecombe, J. Chem. Phys., 1990, 92, 5397–5403 CrossRef CAS.
- B. Silvi and A. Savin, Nature, 1994, 371, 683–686 CrossRef CAS.
- P. Fuentealba, E. Chamorro and J. C. Santos, in Theoretical and Computational Chemistry, ed. A. Toro-Labbé, Elsevier, 2007, vol. 19, pp. 57–85 Search PubMed.
- M. Causá, A. Savin and B. Silvi, Found. Chem., 2014, 16, 3–26 CrossRef.
- Y. Grin, A. Shavin and B. Silvi, in The Chemical Bond, ed. G. Frenking and S. Shaik, 2014, vol. 1, pp. 345–382 Search PubMed.
- T. Clark, in The Chemical Bond, ed. G. Frenking and S. Shaik, 2014, vol. 2, pp. 523–536 Search PubMed.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- R. Bader, Atoms in molecules: a quantum theory, Oxford University Press, 1994 Search PubMed.
- P. Lode and A. Popelier, in The Chemical Bond, ed. G. Frenking and S. Shaik, 2014, vol. 1, pp. 271–308 Search PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CAS.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 Search PubMed.
- R. F. W. Bader, J. Phys. Chem. A, 2010, 114, 7431–7444 CrossRef CAS.
- However it should be emphasized that there is no strict equivalence between BCPs and chemical bonds, see (a) R. F. W. Bader, J. Phys. Chem. A, 2009, 113, 10391–10396 CrossRef CAS; (b) C. Foroutan-Nejad, S. Shahbazian and R. Marek, Chem.–Eur. J., 2014, 20, 10140–10152 CrossRef CAS; (c) Z. A. Keyvani, S. Shahbazian and M. Zahedi, Chem.–Eur. J., 2016, 22, 5003–5009 CrossRef CAS; (d) S. Shahbazian, Chem.–Eur. J., 2018, 24, 5401–5405 CrossRef CAS.
- C. Outeiral, M. A. Vincent, Á. Martín Pendás and P. L. A. Popelier, Chem. Sci., 2018, 9, 5517–5529 RSC.
- P. Macchi and A. Sironi, Coord. Chem. Rev., 2003, 238–239, 383–412 CrossRef CAS.
- For a detailed analysis of the P–Au, P–Cu and Cu–Cu interactions in 7b and 9b, see the ESI.† Our conclusions regarding the polar single bond nature of the P/N interaction in BIPP compounds (which should therefore be written P+–N−, not PN) are similar to those formulated by Stalke: N. Kocher, D. Leusser, A. Murso and D. Stalke, Chem.–Eur. J., 2004, 10, 3622–3631 CrossRef CAS.
- N. W. Mitzel, U. Losehand, A. Wu, D. Cremer and D. W. H. Rankin, J. Am. Chem. Soc., 2000, 122, 4471–4482 CrossRef CAS.
- N. W. Mitzel, K. Vojinović, T. Foerster, H. E. Robertson, K. B. Borisenko and D. W. H. Rankin, Chem.–Eur. J., 2005, 11, 5114–5125 CrossRef CAS.
- N. W. Mitzel, K. Vojinović, R. Fröhlich, T. Foerster, H. E. Robertson, K. B. Borisenko and D. W. H. Rankin, J. Am. Chem. Soc., 2005, 127, 13705–13713 CrossRef CAS.
- M. Hagemann, R. J. F. Berger, S. A. Hayes, H.-G. Stammler and N. W. Mitzel, Chem.–Eur. J., 2008, 14, 11027–11038 CrossRef CAS.
- P. Macchi, D. M. Proserpio and A. Sironi, J. Am. Chem. Soc., 1998, 120, 13429–13435 CrossRef CAS.
- Obviously, the computed distance for 6b is superior to that threshold, but we note that the experimental solid state value is lower by 6–7 % (Table 2). We do not think that this is due to a failure of the computations to reproduce the P–Ti bond distance accurately. Rather, this difference probably originates from the different permittivities (ε) of toluene and the bulk crystal. For enlightening studies of medium effects on chemical bonds, see ref. 105–108 and: (a) H. Jiao and P. v. R. Schleyer, J. Am. Chem. Soc., 1994, 116, 7429–7430 CrossRef CAS; (b) K. R. Leopold, M. Canagaratna and J. A. Phillips, Acc. Chem. Res., 1997, 30, 57–64 CrossRef CAS . For detailed discussions on the “chemical meaning” of molecular graphs and BCPs, see ref. 101.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (b) J. P. Perdew, in Electronic Structure of Solids, ed. P. Ziesche and H. Eschrig, Akademie-Verlag, Berlin, 1991, p. 11 Search PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS.
- ChemCraft – graphical software for visualization of quantum chemistry computations, https://www.chemcraftprog.com, last visited June, 3rd 2020 Search PubMed.
- T.-A. Keith, AIMAll (version 17.11.14B), TK Gristmill Software, Overland Park KS, USA, 2014 Search PubMed.
- D. Kozlowski and J. Pilmé, J. Comput. Chem., 2011, 32, 3207–3217 CrossRef CAS.
- (a) W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS; (b) http://www.ks.uiuc.edu/Research/vmd/ .
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- A. Kalemos and A. Mavridis, J. Phys. Chem. A, 2009, 113, 13972–13975 CrossRef CAS.
- B. A. Jackson, J. Harshman and E. Miliordos, J. Chem. Educ., 2020, 97, 3638–3646 CrossRef CAS.
- G. Parkin, J. Chem. Educ., 2006, 83, 791 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Compounds 2b′, 3a, 3b, 4a, 5a, 5b, 6a, 6a-Py, 6b, 7b, 8b, 9b; detailed procedure for the preparation of P4; spectra of new compounds. CCDC 1985134–1985146. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc04736h |
| ‡ Current address: School of Chemistry, The University of Manchester, Oxford Road, Manchester M13 9PL, UK. |
| § Incidentally, the usage of the arrow to “fix the octet” has recently been appraised theoretically, and it turns out that for some molecules (e.g. H2SO4, HNO3 or HClO4),120,121 the dissociation of oxygen is actually heterolytic; thus, for these molecules, the arrow formalism also corresponds to Haaland's definition. We thank an anonymous reviewer for opening our eyes to the different usages of the arrow formalism, and to the fact that according to Sidgwick, the D → A and D+–A− representations are strictly equivalent. |
| ¶ The term “divalent” instead of “dicoordinate” is often encountered in the literature. However, as pointed by a reviewer, the latter is less ambiguous since it does not presuppose the number of electrons involved in bonding. For a rigorous discussion on valence and coordination number, see ref. 122. |
| || We did not react 2b with [AuCl(THT)], however 2b and 2b′ can be used indifferently, for example to synthesize 3b or 6b. |
| ** Due to the negative charge, the HOMO and LUMO energies of P(CN)2− are considerably higher than those of BIPP and TP compounds; we computed the molecular orbitals of [P(CN)2][K] and found that the energies were lowered by 2.48 eV (LUMO) and 1.66 eV (HOMO). Hence the HOMO–LUMO gap of [P(CN)2][K] is comparable to those of 6a and 6b. |
| †† The ellipticity (ε) is also commonly used to assess bond multiplicity, however we have found that the obtained values are sometimes meaningless (e.g. for the P–Ti interaction). |
| This journal is © The Royal Society of Chemistry 2021 |