
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cerium–quinone redox couples put under scrutiny†
Uwe
Bayer
 ,
Daniel
Werner
,
Andreas
Berkefeld
,
Cäcilia
Maichle-Mössmer
and
Reiner
Anwander
*
,
Daniel
Werner
,
Andreas
Berkefeld
,
Cäcilia
Maichle-Mössmer
and
Reiner
Anwander
*
Institut für Anorganische Chemie, Eberhard Karls Universität Tübingen (EKUT), Auf der Morgenstelle 18, 72076 Tübingen, Germany. E-mail: reiner.anwander@uni-tuebingen.de; Web: [http://uni-tuebingen.de/syncat-anwander]
First published on 23rd November 2020
Abstract
Homoleptic cerous complexes Ce[N(SiMe3)2]3, [Ce{OSi(OtBu)3}3]2 and [Ce{OSiiPr3}3]2 were employed as thermally robust, weakly nucleophilic precursors to assess their reactivity towards 1,4-quinones in non-aqueous solution. The strongly oxidizing quinones 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) or tetrachloro-1,4-benzoquinone (Cl4BQ) readily form hydroquinolato-bridged ceric complexes of the composition [(CeIVL3)2(μ2-O2C6R4)]. Less oxidising quinones like 2,5-di-tert-butyl-1,4-benzoquinone (tBu2BQ) tend to engage in redox equilibria with the ceric hydroquinolato-bridged form being stable only in the solid state. Even less oxidising quinones such as tetramethyl-1,4-benzoquinone (Me4BQ) afford cerous semiquinolates of the type [(CeIIIL2(thf)2)(μ2-O2C6Me4)]2. All complexes were characterised by X-ray diffraction, 1H, 13C{1H} and 29Si NMR spectroscopy, DRIFT spectroscopy, UV-Vis spectroscopy and CV measurements. The species putatively formed during the electrochemical reduction of [CeIV{N(SiMe3)2}3]2(μ2-O2C6H4) could be mimicked by chemical reduction with CoIICp2 yielding [(CeIII{N(SiMe3)2}3)2(μ2-O2C6H4)][CoIIICp2]2.
Introduction
Quinones are multifunctional organic molecules exhibiting intriguing redox behaviour.1,2 Of particular note is their importance in biological electron–transfer processes (photosynthesis, respiration)3 and in industrial catalysis (anthraquinone process for hydrogen peroxide production).4 Quinones can engage in one or two electron redox processes involving the formation of either semiquinolates or hydroquinolates.5 Strikingly, the reduction potential of 1,4-benzoquinones (para-benzoquinones) can easily be modified by introducing electron-withdrawing or donating substituents into the benzene ring.5,6 As a consequence, tetrachloro-1,4-benzoquinone (chloranil, Cl4BQ) and even more so 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) emerged as efficient oxidants in organic synthesis.7 DDQ has been further successfully applied in photoredox catalysis.8 Moreover, anionic η4-1,4-benzoquinone manganese tricarbonyl features a quinoid π-complex, broadly used for the fabrication of supramolecular metal–organometallic coordination networks.9 Relatedly, deprotonated variants of 2,5-dihydroxy-1,4-benzoquinone (DHBQ) were shown to act as rigid ditopic linkers,10e.g., to support the formation of pentagonal dodecahedral Ce2(H2O)18 cages or in permanently porous aluminium frameworks.11 DHBQ was also probed as a bridging redox-active ligand in bimetallic [LnCl2(thf)3]2(μ-bobq) (Ln = Y, Dy; bobq = 2,5-bisoxide-1,4-benzoquinolato) to build single-molecule magnets.12 More recently, the related semiquinolato radical-bridged dimeric complexes [LnCl2(thf)3(μ-Me4sq)2]2 (Ln = Y, Gd) were obtained by oxidation of the corresponding in situ formed hydroquinolate complexes with FeCl3.13 Semiquinolato-bridged scandium(III) species were reported to promote self-organised electron transfer from d-transition metals (Ir, Fe) to 1,4-quinones.14,15Targeted metal-redox chemistry with quinones has been a recurring issue for the rare-earth-metal couples Ln(II)/Ln(III)16 and Ce(III)/Ce(IV).17 Especially in the case of molecular cerium chemistry,17 its unique single-electron-transfer (SET) pathway has recently been extended beyond the traditional application of ceric ammonium nitrate (CAN; redox potential of 1.61 V vs. NHE) in organic synthesis18 to photoredox catalysis.19 On the other hand, redox protocols are known to provide efficient access to metalorganic CeIV complexes. Typically, such CeIII → CeIV transformations are promoted by halogenating oxidants (e.g. C2Cl6, Ph3CCl, PhICl2, TeCl4, FcPF6, FcBF4, Ph3CBF4, Ph3CPF6, I2),20 silver salts (AgX, X = F, I, BF4, OTf)21 or dioxygen.20b,22
Archetypical 1,4-benzoquinone (BQ) has been established as a versatile oxidant for the synthesis of homoleptic ceric complexes CeL4 from cerous ate complexes [CeL4M(do)x] via tandem oxidation-ligand redistribution protocols (L = monoanionic ligand, M = alkali metal and do = donor solvent; separation of an alkali-metal hydro-/semiquinolate).23 In the presence of sterically demanding ligands L, BQ was also shown to form hydroquinolato (hq)–bridged ceric complexes of the general composition [L3Ce–OC6H4O–CeL3].20g,24 This very CeIII → CeIV transformation was pioneered by Sen et al. in 1992, resulting in the isolation of [(tBu3CO)3Ce(OC6H4O)Ce(OCtBu3)3] (Chart 1, I).24a In the same paper, the oxidation of Ce(OCtBu3)3 with 2,6-di-tert-butyl-1,4-benzoquinone to the terminal CeIV-semiquinolate radical (tBu3CO)3Ce(O2C6H2tBu2) was described as evidenced by 1H NMR and EPR spectroscopic measurements.24a More recently, Schelter et al. reported on hq-bridged complex II resulting from the oxidation of cerous Ce(BINOlate)3(thf)Li3(thf)4 with 0.5 equivalents of BQ.24b Similarly, our group synthesized [Ce{N(SiMe3)2}3]2(μ2-O2C6H4)24c (III) and (CeCpR3)2(μ2-O2C6H4) (CpR = C5H4Me (IV) and C5H4(SiMe3) (V)).20g In contrast, the reaction of BQ with [Ce(Me2pz)3]x featuring the sterically less demanding and increasingly nucleophilic 3,5-dimethylpyrazolato ligand (Me2pz) led in fact to a transient CeIV hydroquinolate species (as indicated by the characteristic colour change), which, however, at ambient temperature was converted into the isolable trimetallic CeIII complex Ce3(pchd)2(Me2pz)5(thf)2 (pchd = 1,4-bis(3,5-dimethylpyrazol-1-yl)cyclohex-2,5-diene-1,4-diolato).23c Apparently, the new pchd ligand formed via 1,4-nucleophilic attack at bq by two adjacent Me2pz ligands. This nucleophilic reaction pathway could be prevented by using bulky tBu groups on the pz ligand, but homolpetic Ce(tBu2pz)4 was formed as the main ceric product via irreversible ligand rearrangement.23c
 | ||
| Chart 1 Structurally characterised dicerium(IV) hydroquinolate complexes (I–V),20g,24 obtained via oxidation of cerous precursor with BQ. | ||
As such, cerium–1,4-benzoquinone couples have revealed distinct redox chemistry, we became curious about as to what extent such redox transformations are affected by both the type of 1,4-benzoquinone oxidant and the molecular CeIII precursor. The present study uncovers some unexpected correlation between CeIV–hydroquinolato stabilisation and quinone oxidant strength, as well as a new path to p-semiquinolato–radical-bridged rare-earth-metal complexes.
Results and discussion
Molecular redox precursors
The quinones used in this study comprise 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ), tetrachloro-1,4-benzoquinone (Cl4BQ), 1,4-benzoquinone (BQ), tetramethyl-1,4-benzoquinone (Me4BQ), 2,5-di-tert-butyl-1,4-benzoquinone (tBu2BQ), 1,4-naphthoquinone (NQ), and 9,10-anthraquinone (AQ). All are commercially available and were selected according to their reduction potentials spanning a E0 range of 89 to 887 mV (2e−/2H+, vs. NHE, cf., Scheme 1).5,25 The cerous precursors were chosen according to the criteria solubility, weak nucleophilicity, proven access to the tetravalent state, and a stabilizing effect on the latter. Furthermore, the use of sterically bulky ligands was assumed to minimise the occurrence of ligand redistribution reactions. Accordingly, homoleptic Ce[N(SiMe3)2]3 (1) appeared to be an ideal benchmark system.24c After additional investigations into the respective pyrazolate chemistry, the abovementioned [Ce(R2pz)3] (R = Me, tBu) were discarded because of persisting alternative reaction pathways like 1,4-nucleophilic attack of BQ by Me2pz and ligand redistribution (formation of Ce(tBu2pz)4).23c The new pyrazolate studies clearly confirmed that steric hindrance of both the pyrazolato ligand and the 1,4-benzoquinone can minimise/counteract such undesired reactions, but the formation of product mixtures seems inevitable. Products crystallised from these reactions include minor amounts of ceric [Ce(tBu2pz)3(thf)]2(Me4hq) or a cerous product of partial pyrazolyl-promoted nucleophilic attack Ce3(bpad)(pasq)(Me2pz)6(thf) (bpad = 1,4-bis(3,5-dimethylpyrazol-1-yl)anthra-1,4-diolato; pasq = 1-(3,5-dimethylpyrazol-1-yl)anthra-1,4-semiquinolato) (84%) mixed with semiquinolate [Ce(Me2pz)2(thf)2(asq)]2 (asq = anthra-semiquinolato; cf. ESI† for structural details). The use of CeIII halides was discarded mainly for solubility issues. | ||
| Scheme 1 Oxidation of trivalent cerium complexes with 1,4-quinone derivatives under formation of hydroquinolato-bridged ceric complexes. To enable better assessment of the relative oxidation ability 2e−/2H+ reductions potentials are given in mV versus NHE according to ref. 5. (a) Calculated value. | ||
In addition to silylamide 1, the siloxide derivatives [Ce{OSi(OtBu)3}3]2 (2)21d,26 and [Ce(OSiiPr3)3]2 (3) were assessed as suitable cerous precursors. Complexes 2 and 3, with and without intramolecular donor site, respectively, were readily obtained in pure form via protonolysis of 1 with the corresponding silanol.26 The crystal structure of the new complex 3 revealed a dimeric arrangement with two μ2-bridging and four terminal siloxy groups (Fig. 1), similar to that found for tris(tert-butoxy)siloxy congener 2 or [Ce(OSiPh3)3]2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 27 or [Ce(OCHtBu2)3]2.28 The Ce–Oterminal (2.1659(14) and 2.1671(14) Å) and the Ce–Oμ2 distances (2.3951(12) and 2.4030(12) Å) of 3 are slightly shorter than those in 2 (Ce–Oterminal 2.202(3), 2.186(3) Å; Ce–Oμ2 2.532(2) Å) and [Ce(OSiPh3)3]2 (Ce–Oterminal 2.141(7), 2.185(6) Å; Ce–Oμ2 2.345(6), 2.583(5) Å) reflecting the lower coordination number (CN 4 vs. 5), but slightly longer than in [Ce(OCHtBu2)3]2 (Ce–Oterminal 2.142(2), 2.152(3) Å; Ce–Oμ2 2.363(3) Å).28 The 1H NMR spectrum of 3 in C6D6 shows two singlets at −28.82 and −17.23 ppm for the μ2-OSiiPr3 groups and two singlets at 6.46 and 9.09 ppm for the terminal siloxy ligands indicating a non-fluxional dimeric species in non-coordinating solvents. When recorded in THF-d8, only two signals for the OSiiPr3 groups appeared, in accordance with the formation of a monomeric adduct [Ce{OSiiPr3}3(thf-d8)x].
27 or [Ce(OCHtBu2)3]2.28 The Ce–Oterminal (2.1659(14) and 2.1671(14) Å) and the Ce–Oμ2 distances (2.3951(12) and 2.4030(12) Å) of 3 are slightly shorter than those in 2 (Ce–Oterminal 2.202(3), 2.186(3) Å; Ce–Oμ2 2.532(2) Å) and [Ce(OSiPh3)3]2 (Ce–Oterminal 2.141(7), 2.185(6) Å; Ce–Oμ2 2.345(6), 2.583(5) Å) reflecting the lower coordination number (CN 4 vs. 5), but slightly longer than in [Ce(OCHtBu2)3]2 (Ce–Oterminal 2.142(2), 2.152(3) Å; Ce–Oμ2 2.363(3) Å).28 The 1H NMR spectrum of 3 in C6D6 shows two singlets at −28.82 and −17.23 ppm for the μ2-OSiiPr3 groups and two singlets at 6.46 and 9.09 ppm for the terminal siloxy ligands indicating a non-fluxional dimeric species in non-coordinating solvents. When recorded in THF-d8, only two signals for the OSiiPr3 groups appeared, in accordance with the formation of a monomeric adduct [Ce{OSiiPr3}3(thf-d8)x].
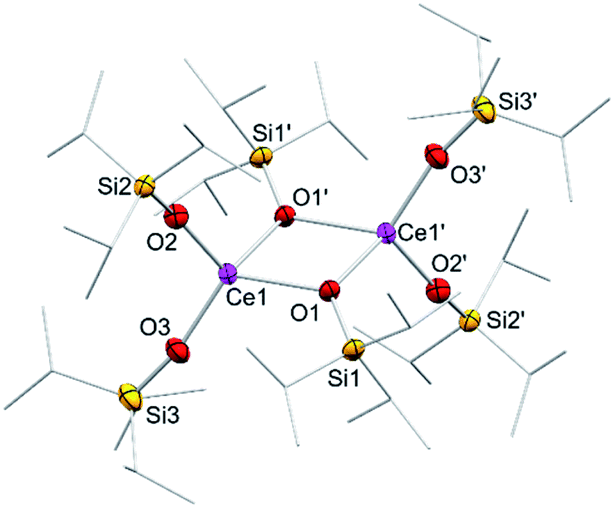 | ||
| Fig. 1 Crystal structure of [Ce{OSiiPr3}3]2 (3). Ellipsoids are shown at the 50% probability level. Hydrogen atoms are omitted for clarity. Selected interatomic distances [Å] and angles [°]: Ce1–O1 2.3951(12), Ce1–O2 2.1659(14), Ce1–O3 2.1671(14); Ce1–O2–Si2 163.03(9), Ce1–O3–Si3 167.40(10). | ||
Quinone oxidation of Ce[N(SiMe3)2]3 (1)
Treatment of Ce[N(SiMe3)2]3 (1) with each 0.5 equivalents of DDQ, Cl4BQ, Me4BQ, tBu2BQ and NQ, in a mixture of toluene and n-hexane, immediately led to a colour change from yellow to dark brown. Upon recrystallisation from toluene/n-hexane mixtures it was possible to isolate the hydroquinolato-bridged complexes [Ce{N(SiMe3)2}3]2(μ2-O2C6Cl4) (4Cl4hq), [Ce{N(SiMe3)2}3]2(μ2-O2C6Cl2(CN)2) (4ddhq), [Ce{N(SiMe3)2}3]2(μ2-O2C6Me4) (4Me4hq), [Ce{N(SiMe3)2}3]2(μ2-O2C6tBu2H2) (4tBu2hq) and [Ce{N(SiMe3)2}3]2(μ2-O2C10H6) (4nhq) in very good crystalline yields of 71 to 90% (Scheme 1). The crystal structures of the new complexes 4xhq are isostructural to the previously reported derivative 4hq24c and only differ in the bridging hq linker (Fig. 2). The Ce1–O1 distances of 2.084(6) to 2.173(2) Å (for a full list of interatomic distances see Table 1) are in the same range as found for other hq-bridged cerium complexes (2.086(10)–2.143(5) Å).20g,24 Likewise, the Ce1–N bond lengths compare well to other CeIV silylamides like [Ce{N(SiMe3)2}3]2(μ2-O2C6H4) (2.2388(14)–2.2487(14) Å),24c Ce[N(SiMe3)2]3Cl (2.217(3) Å),20a and Ce[N(SiHMe2)2]4 (2.2378(11)–2.2574(11) Å).20d Also, the C–C distances of the hq linker converge as the expected aromatic ring is formed and the C–O distances of 1.318(3) to 1.378(3) Å corroborate the formation of C–O single bonds. | ||
| Fig. 2 Crystal structures of [Ce{OSi(OtBu)3}3(Me-thf)2]2(μ2-O2C6H4) (5hq·2MeTHF), [Ce{OSiiPr3}3(thf)]2(μ2-O2C6Cl4) (6Cl4hq), [Ce{N(SiMe3)2}3]2(μ2-O2C6Cl2CN2) (4ddq), [Ce{N(SiMe3)2}3]2(μ2-O2C6Me4) (4Me4hq), and [Ce{OSiiPr3}3(thf)]2(μ2-O2C6tBu2H2) (6tBu2hq) (from top down). Ellipsoids are shown at the 50% probability level. Hydrogen atoms, disordered ligands and lattice solvents are omitted for clarity. Selected interatomic distances for 5hq, 6Cl4hq, 4ddq, and 4Me4hq are given in Tables 1 and 2. Selected bond lengths for 6tBu2hq [Å]: Ce1–O1 2.109(3), Ce1–O2 2.104(3), Ce1–O3 2.097(3), Ce1–O4 2.107(3), C1–C2 1.391(6), C2–C3 1.391(6), C1–C3′ 1.405(6), C1–O1 1.350(5). | ||
| Complex | 4hq 24c | 4Cl4hq | 4ddhq | 4Me4hq | 4tBu2hq | 4nhq |
|---|---|---|---|---|---|---|
| a NMR spectra recorded in C6D6. b UV-Vis spectra recorded in toluene. c Determined in THF using c(analyte) = 2 mM and c(electrolyte) = 0.1 M and a scan rate of 50 mV s−1. d Determined in toluene-d8 at 0 °C. | ||||||
| Ce1–O1 | 2.0895(13) | 2.149(4) | 2.1731(15) | 2.082(6) | 2.117(2) | — |
| Ce1–N1 | 2.2388(14) | 2.265(5) | 2.2206(18) | 2.280(7) | 2.235(2) | — |
| Ce1–N2 | 2.2398(15) | 2.230(5) | 2.2112(18) | 2.229(7) | 2.246(2) | — |
| Ce1–N3 | 2.2487(14) | 2.244(5) | 2.2467(19) | 2.238(7) | 2.259(2) | — |
| C–Carom | 1.387(3)–1.399(2) | 1.383(8)–1.392(8) | 1.390(4)–1.407(3) | 1.390(11)–1.408(12) | 1.388(4)–1.405(4) | — |
| C1–O1 | 1.356(2) | 1.324(7) | 1.318(3) | 1.366(10) | 1.378(3) | — |
| Ce1–O1–C1 | 173.13(11) | 156.1(4) | 161.02(15) | 162.4(6) | 146.79(18) | — |
| 1H NMRa | 0.43 | 0.45 | 0.45 | 0.43d | 0.49 | 0.44 |
| 13C{1H} NMRa | — | — | 5.6 | — | — | 5.6 |
| 29Si{1H} NMRa | — | −8.1 | −7.3 | −8.8 | — | −8.1 |
| μ eff | 0.67 | 0.68 | 0.59 | 0.89 | — | 1.19 |
| UV-Vis absorption bandsb | 485 | 319/518 | 384/511 | 362/411/681 | — | 339/425/474/677 |
| E pc | −0.699/−0.966 | −0.415/−0.624 | −0.546 | — | — | — |
| E pa | −0.558 | −0.290 | −0.1685 | — | — | — |
| E 1/2 | −0.76 | −0.46 | −0.36 | |||
| ΔE | 0.408 | 0.334 | 0.377 | — | — | — |
The 1H NMR spectra of compounds 4xhq in C6D6 show singlets for the trimethylsilyl (TMS) groups at 0.43 to 0.45 ppm along with signals for the bridging hydroquinolato moieties. Further, the 13C{1H} NMR spectra of 4ddhq and 4nq display a singlet for the TMS groups at 5.6 ppm and signals in the aromatic region for the different hydroquinolates, indicative of a successful reduction of the respective benzoquinone derivatives. The characterisation of 4tBu2hq in solution (C6D6) was not feasible, due to the prevailing equilibrium shown in Scheme 2, and ready back-formation of 1 and 2,5-di-tert-butyl-1,4-benzoquinone.
 | ||
| Scheme 2 Equilibrium between 4tBu2hq and reformation of reactants in solution and solid state. | ||
While the 1H NMR spectrum of 4tBu2hq primarily shows signals for the starting materials and only minor product signals, its DRIFT spectrum indicated the absence of any strong C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O absorption band, and therefore the stability of 4tBu2hq in the solid state (see Fig. S10 and S45 in ESI†). In contrast, complexes 4hq, 4Cl4hq and 4ddhq derived from the stronger oxidizing quinones are very stable in the solid state and in solution. This fits again well with the already pronounced instability of 4Me4hq and 4nhq which slowly decompose in n-hexane and toluene at ambient temperature and rapidly undergo decomposition in THF. Tracking of the progress of the decomposition by 1H NMR spectroscopy revealed the formation of 1 and other paramagnetic CeIII species which, however, could not be identified. The progressing decomposition can also be seen in the ligand-to-metal charge transfers observed in the UV-Vis spectra (Fig. S68, ESI†). As the spectra of 4hq, 4Cl4hq and 4ddhq show mainly one strong absorption band at around 500 nm (ε > 5060 L mol−1 cm−1), the spectra of 4Me4hq and 4nhq show several absorption bands with significantly lower intensities (ε < 4400 L mol−1 cm−1) indicative of CeIII species and therefore redox decomposition of the compounds.
O absorption band, and therefore the stability of 4tBu2hq in the solid state (see Fig. S10 and S45 in ESI†). In contrast, complexes 4hq, 4Cl4hq and 4ddhq derived from the stronger oxidizing quinones are very stable in the solid state and in solution. This fits again well with the already pronounced instability of 4Me4hq and 4nhq which slowly decompose in n-hexane and toluene at ambient temperature and rapidly undergo decomposition in THF. Tracking of the progress of the decomposition by 1H NMR spectroscopy revealed the formation of 1 and other paramagnetic CeIII species which, however, could not be identified. The progressing decomposition can also be seen in the ligand-to-metal charge transfers observed in the UV-Vis spectra (Fig. S68, ESI†). As the spectra of 4hq, 4Cl4hq and 4ddhq show mainly one strong absorption band at around 500 nm (ε > 5060 L mol−1 cm−1), the spectra of 4Me4hq and 4nhq show several absorption bands with significantly lower intensities (ε < 4400 L mol−1 cm−1) indicative of CeIII species and therefore redox decomposition of the compounds.
All attempts to isolate putative 4ahq, derived from the weakest oxidising quinone under study, namely 9,10-anthraquinone (E0 = 89 mV; 2e−/2H+, vs. NHE),5 were unsuccessful with the reaction mixtures showing no colour change immediately after addition of the anthraquinone. However, a colour change from yellow to green occurred after two days and the respective 1H NMR spectrum showed multiple paramagnetic signals.
Quinone oxidation of siloxides [Ce{OSi(OtBu)3}3]2 (2) and [Ce{OSiiPr3}3]2 (3)
Reacting cerous siloxides 2 and 3 with the selected quinones in THF immediately gave a colour change of the reaction mixtures (from colourless to: dark purple (BQ), dark red (Cl4BQ), dark yellow/orange (DDQ), pale purple (tBu2BQ), pale blue (Me4BQ), pale green (NQ)). The ceric compounds [CeL3(thf)]2(μ2-O2C6H4) (5hq, 6hq), [CeL3(thf)]2(μ2-O2C6Cl4) (5Cl4hq, 6Cl4hq), [CeL3(thf)]2(μ2-O2C6Cl2(CN)2) (5ddq, 6ddq), with L = OSi(OtBu)3 or OSiiPr3 derived from quinones with a relatively strong oxidising effect were successfully isolated from these reactions (Scheme 1).However, the weakly oxidizing quinones Me4BQ and NQ did not lead to tetravalent cerium species, as indicated by the detection of only paramagnetic signals in the 1H NMR spectra (for an example of such a 1H NMR spectrum, see Fig. S34 in the ESI;† formation of semiquinolates, vide infra). The accessible complexes 5 and 6 were obtained in moderate to good crystalline yields of 42 to 71% upon recrystallisation from THF or THF/Et2O mixtures. Crystals suitable for XRD analysis were obtained for complexes 5hq, 5Cl4hq, 6Cl4hq, 6ddhq and 6tBu2hq, revealing the same structural motif as complexes 4, that is two CeL3 moieties connected via a hydroquinolato linker (Fig. 2).
Strikingly, the 1H NMR spectrum of 6tBu2hq indicated the existence of an equilibrium similar to that of ceric 4tBu2hq (cf.Scheme 2). However, along with the reactants 3 and tBu2BQ additional signals assignable to distinct dia- and paramagnetic decomposition products were detected. Further, the crystal structures of complexes 5 and 6 show that the cerium atoms are additionally coordinated by THF donor molecules. The Ce1–Osiloxide distances of 2.066(2) to 2.1534(10) (see Table 2 for a complete list of interatomic distances) compare well to other ceric siloxides like Ce{OSi(OtBu)3}4 (2.089(2)–2.157(2) Å26 and 2.084–2.160 Å21d) or Ce{OSiPh3}4(dme) (2.098(1)–2.133(1) Å).29 Also, as seen for the silylamides 4, the Ce1–Ohq distances of 2.1244(10) to 2.2325(16), as well as the C–C and C–O distances underline the formation of an aromatic hq linker.20g,241H NMR spectroscopic measurements also validate the formation of CeIV species, showing a sharp singlet for the tert-butyl groups and a doublet plus a septet for the iso-propyl groups depending on the siloxy co-ligand.
| Complex | 5hq | 5Cl4hq | 5ddq | 6hq | 6Cl4hq | 6ddq |
|---|---|---|---|---|---|---|
| a NMR spectra recorded in THF-d8. b Spectra recorded in toluene. c Spectra recorded in THF. d Determined in THF using c(analyte) = 2 mM and c(electrolyte) = 0.1 M and a scan rate of 50 mV s−1. | ||||||
| Ce1–O1 | 2.1244(10) | 2.184(3) | — | — | 2.207(2) | 2.2325(16) |
| Ce1–O2 | 2.1534(10) | 2.091(3) | — | — | 2.095(2) | 2.0841(16) |
| Ce1–O3 | 2.1334(11) | 2.094(3) | — | — | 2.066(2) | 2.0710(17) |
| Ce1–O4 | 2.1396(11) | 2.104(3) | — | — | 2.080(2) | 2.091(2) |
| C–Carom | 1.390(2)–1.395(2) | 1.373(8)–1.406(5) | — | — | 1.380(4)–1.407(4) | 1.379(5)–1.415(3) |
| C1–O1 | 1.3540(17) | 1.326(4) | — | — | 1.322(3) | 1.316(3) |
| Ce1–O1–C1 | 151.76(10) | 140.0(2) | — | — | 138.54(18) | 144.85(14) |
| 1H NMRa | 1.36 | 1.36 | 1.36 | 1.12/1.05 | 1.12/1.05 | |
| 13C{1H} NMRa | 72.5/32.6 | 72.8/32.4 | 73.0/32.5 | 18.0/14.0 | 19.1/15.1 | |
| 29Si{1H} NMRa | −103.2 | −104.6 | −105.3 | 7.0 | 9.6 | 10.7 |
| μ eff | 0.82 | 0.54 | 0.60 | 0.68 | 0.50 | 0.66 |
| UV-Vis absorption bandb | 369/622 | 493 | 384/450 | 526c | 511 | 381/470 |
| E pc | −1.7855 | −1.414 | −1.353 | −1.116/−1.816 | −1.580 | −1.149/−1.484 |
| E pa | −0.3625 | −0.521 | 0.130 | −1.273/−0.817 | −0.729 | −0.751 |
| ΔE | 1.416 | 0.893 | 1.483 | 0.999 | 0.851 | 0.733 |
Electrochemical investigation of complexes 4xhq, 5xhq and 6xhq
Cyclic voltammetry (CV) measurements of complexes 4xhq, 5xhq and 6xhq have been conducted at ambient temperature in 0.2 mM solutions in THF and 0.1 M [nPr4N][B(C6H3(CF3)2-3,5)4] as a support electrolyte, and referenced vs. Fc/Fc+. Due to the low stability of compounds 4Me4hq, 4tBu2hq and 4nhq in polar solvents CV measurements of these complexes were not feasible. Most of the CV measurements revealed successive quasireversible (4) or irreversible (5/6) CeIV → CeIII reduction steps, but badly resolved (for Epc values see Tables 1 and 2). The detection of two closely adjacent redox events (Epc values) in some cyclic voltammograms may correspond to a successive reduction/oxidation of the cerium centres. Similar features were also described for the hq-bridged Ce(IV)–BINOLate complex II.24b All complexes under study display redox processes with a large separation of Epc and Epa (ΔE ≈ 0.6 V for 4; 1.5 V for 5 and 1.0 V for 6).Representatively, the cyclic voltammograms of the DDQ-functionalized CeIII/CeIV redox couples are depicted in Fig. 3 (top graphic). The silylamide complexes 4 gave reduction potentials similar to those reported for halogenido-functionalised ceric complexes Ce[N(SiMe3)2]3X (E1/2 = −0.56 (X = F), −0.30 (X = Cl), −0.31 (X = Br)) with E1/2 values of −0.46 V for 4Cl4hq and −0.36 V for 4ddhq.30 Only 4hq with E1/2 = −0.76 V gave a significantly higher stabilisation by 0.20 V. The extra-large separation of the reduction/oxidation events observed for the siloxide complexes 5 and 6 had been noticed previously for rare-earth-metal siloxides and was assigned to oxidation-state-dependent ligand reorganisation processes.31
 | ||
| Fig. 3 Top: Stacked cyclic voltammograms of complexes 4ddq (red), 5ddq (blue), 6ddq (black) in THF (v = 50 mV s−1; c(analyte) = 2 mM; c([nPr4N][B(C6H3(CF3)2-3,5)4]) = 0.1 M). Bottom: comparison of the second reduction potentials of complexes 4 (red), 5 (black), and 6 (blue) in THF vs. Fc/Fc+. Squares: complexes with bridging 1,4-hydroquinolates; cycles: complexes with bridging tetrachloro-1,4-hydroquinolates; triangles: complexes with bridging 2,3-dichloro-5,6-dicyano-1,4-hydroquinolates. | ||
Stabilisation of the tetravalent oxidation state of cerium in complexes 4, 5, and 6 increases in the order of N(SiMe3)2 < OSi(OtBu3)3 < OSiiPr3 as co-ligand (Fig. 3/bottom) which is in accordance with previous findings.20f,26,30,31c Surprisingly, the stabilisation of CeIV proceeds in reverse order of the oxidation potential of the 1,4-quinones under study giving the most stable complexes for the hydroquinolato-bridged complexes and the least stable compounds for its 2,3-dichloro-5,6-dicyano-hydroquinolato congeners. A reason for this trend could be the increasingly electron-deficient nature of the aromatic hydroquinolato linkers due to the large −I effect of the substituents. The CeIV oxidation state can be stabilised by increasing donor strength of the ligands.32 Based on this, it seems surprising that isolable complexes 4Me4hq and 4nhq, derived from weakly oxidizing quinones, are not stable in solution at ambient temperature. This might be a result of another reaction pathway preferred after formation of the hydroquinolato-bridged CeIV complexes (like following up redox processes and the formation of CeIII semiquinolates, cf. vide infra).
Reduction of silylamide 4hq with cobaltocene
Having investigated the electrochemical reduction of compounds 4, 5 and 6, the chemical reduction with cobaltocene (CoCp2) (−1.31 V vs. Fc/Fc+ in DME)2a was attempted, as it has already been shown to engage in such reductions.31a,33 Accordingly, treatment of a solution of 4hq in THF with two equivalents of CoCp2 resulted in a colour change from dark brown to pale yellow (Scheme 3). The 1H NMR spectrum of the reaction mixture showed complete consumption of CoCp2 and only broadened signals indicating the formation of a paramagnetic CeIII species. Crystallisation from a concentrated THF-d8 solution at −40 °C gave light brown crystals of the composition [(Ce{N(SiMe3)2}3)2(μ2-O2C6H4)][CoCp2]2 (7) (Fig. 4). | ||
| Scheme 3 Reduction of 4bq with two equivalents of CoCp2. | ||
 | ||
| Fig. 4 Crystal structure of [(Ce{N(SiMe3)2}3)2(μ2-O2C6H4)][CoCp2]2 (7). Ellipsoids are shown at the 50% probability level. Hydrogen atoms, disordering of the cyclopentadienyl ligands and lattice THF are omitted for clarity. Selected interatomic distances [Å]: Ce1–N1 2.420(2), Ce1–N2 2.402(2), Ce1–N3 2.418(2), Ce1–O1 2.202(2), C1–O1 1.344(4), C1–C2 1.391(4), C1–C3 1.392(4), C2–C3′ 1.392(4). | ||
Complex 7 shows the same structural motif as 4hq but is flanked by two cobaltocenium cations. Compared to 4hq, the Ce–N and Ce1–O1 distances are elongated by approximately 0.19 Å as expected for the larger CeIII ion size.34 On the contrary, the bonding parameters within the bridging hydroquinolato linker did not change, further corroborating a cerium-borne redox chemistry. Reacting 4hq with one equivalent of CoCp2 did not lead to a mixed CeIII/IV complex but gave a mixture of 50% of 7 and 50% of unreacted starting material.
The reactions of CoCp2 with other complexes 4 to 6 in THF-d8 showed immediate decolourisation of the solution while the 1H NMR spectra of the reaction mixtures displayed only paramagnetic signals (for an example, see Fig. S33 in the ESI†). However, the isolation of additional reduced species similar to 7 was not successful.
Cerium semiquinolates
A closer look at the reactions of cerous siloxides 2 and 3 with the weakly oxidising quinone Me4BQ (which did not produce any tetravalent cerium species; vide supra) revealed another important detail of the cerium–quinone redox system. Treatment of 2 or 3 with 0.5 equivalents of Me4BQ led to a colour change from colourless to light blue. Upon recrystallisation from THF dark blue crystals suitable for X-ray diffraction could be grown and were identified as cerous semiquinolates [CeL2(thf)2]2(μ2-O2C6Me4)2 (with L = OSi(OtBu)3 (8) or OSiiPr3 (9)) (Scheme 4). | ||
| Scheme 4 Formation of cerous semiquinolates 8 and 9 from the reaction of 2 or 3 with Me4BQ. | ||
Examining the reaction mixtures by 1H NMR spectroscopy in THF-d8 showed, besides paramagnetic signals for 8 and 9, a sharp singlet at 1.39 ppm (for 8) or a doublet plus a septet at 1.13 and 1.04 ppm (for 9), indicating the formation of homoleptic Ce[OSi(OtBu)3]4 or [Ce(OSiiPr3)4], respectively, as a result of the one-electron reduction of Me4bq followed by ligand redistribution. Crucially, such a reaction pathway seems unfeasible for complexes 4, since putative homoleptic “Ce[N(SiMe3)2]4” is unknown.20a Emergent kinetic constraints in the case of ceric complexes 4 were also suggested by the redox behaviour of [Ce{N(SiHMe2)2}3]2 derived from a less bulky silylamido ligand. Accordingly, the cerous bis(dimethylsilyl)amide complex was treated with one equivalent of both BQ and Me4BQ in THF-d8 and C6D6 (see Fig. S38–S41, ESI†). The 1H NMR spectra of these reactions suggest the formation of a tetravalent species of the composition “[Ce{N(SiHMe2)2}3]2(μ2-O2C6R4)”. However, the ceric products appear to be unstable in solution at ambient temperature. In C6D6, the formation of Ce[N(SiHMe2)2]4 was observed in the reaction with BQ as well as other insoluble products. In THF-d8, the resulting product seemed more stable but after 24 h in solution also traces of decomposition products were found. The Me4BQ reaction in C6D6 also indicated successful oxidation, however, after 24 h the 1H NMR spectrum revealed signals for trivalent decomposition products as well as traces of Ce[N(SiHMe2)2]4. In THF-d8, the putatively formed hydroquinolate complex was even less stable, showing signals for trivalent by-products directly after addition of Me4BQ. In addition, the stability of 4bq was investigated in THF-d8 showing small amounts of decomposition products like Ce[N(SiMe3)2]3 after 24 h (Fig. S42, ESI†).
Regrettably, purification of complexes 8 and 9 was impeded by co-crystallisation with the ceric by-products CeL4. The crystal structures of 8 and 9 revealed two six-coordinate cerium atoms surrounded by two siloxy ligands, two THF donor molecules and two bridging tetramethyl semiquinolato moieties (Fig. 5). The Ce1–Osilanolato distances are elongated by about 0.1 Å compared to the respective tetravalent compounds 5 and 6 and in accordance with other CeIII siloxides like 3, Ce{OSi(OtBu)3]3(thf)3 (2.243(2)–2.249(2) Å), [Ce{OSi(OtBu)3}3]2 (Ce–Oterm 2.186(3)–2.202(3) Å),26 Ce{OSiPh3}3(thf)3 (Ce–Oavg 2.222(4) Å)35 and [Ce{OSiPh3}3]2 (Ce–Oterm 2.141(7)–2.184(6) Å).27 As expected for semiquinolato ligands the six-membered rings display two shortened C–C and four elongated C–C bonds. Additionally, the six-membered rings are slightly bent in comparison to the flat aromatic hydroquinolato linkers in complexes 4, 5 and 6 resulting in an angle of 170.34° for 8 and 170.29° for 9, respectively (see Fig. 5, bottom). Notwithstanding, the bridging radicals engage in significant π-stacking as indicated by close semiquinolato–semiquinolato distances of 3.112 Å for 8 and 3.156 Å for 9. Overall, complexes 8 and 9 display the same arrangement of the semiquinolato radical bridges as observed in complexes [LnCl2(THF)3(μ-Me4sq)2]2 (Ln = Y, Gd) (Ct⋯Ct 3.097 Å; Ct = centroid of benzene rings).13
 | ||
| Fig. 5 Top: Crystal structure of [Ce{OSiiPr3}2(thf)2]2(μ2-O2C6Me4)2 (9). Ellipsoids are shown at the 50% probability level. Hydrogen atoms are omitted for clarity. Selected interatomic distances [Å]: Ce1–O1 2.446(7), Ce1–O2 2.422(7), Ce1–O3 2.219(9), Ce1–O4 2.237(6), Ce1–O5 2.648(7), Ce1–O6 2.644 (7), C1–O5 1.306(5), C4–O6 1.307(5), C1–C2 1.458(6), C2–C3 1.401(6), C3–C4 1.449(8), C4–C5 1.463(6), C5–C6 1.388(6), C1–C6 1.463(8). Bottom: side view of [(Ce{OSiiPr3}2(thf)2)2(μ2-O2C6Me4)2] (9). | ||
To investigate the electronic behaviour of the bridging semiquinolates, X-band EPR spectra of compounds 8 and 9 were recorded from a crystal powder sample at 123 K (Fig. 6). For both complexes cw-EPR spectra are composed of two distinct sets of resonances, one that results from the transition within the Kramers doublet corresponding to mj = ±1/2 and one at half-field, H ≈ 160 mT. The transition for the mj = ±1/2 state associates with an axial g tensor with principal components gII = 2.094 and g⊥ = 2.032 for 8 and gII = 2.088 and g⊥ = 2.032 for 9, respectively. The transition locating at half-field gives rise to a very broad dispersion line with gII ≈ 4.359 for 8 and gII ≈ 4.351 for 9, respectively. Notably, an identical line pattern derives from a frozen 2-Me-THF solution at 77 K; cf. Fig. S67, ESI,† for pertinent details. The cw-EPR spectra corroborate the presence of Ce3+, and agree with early work on mononuclear complexes of Ce3+.36 This indicates a radical–radical π-bonding, as it was recently shown for yttrium and gadolinium semiquinolates [LnCl2(thf)2]2(μ2-O2C6Me4)2 (Ln = Y or Gd).13 Additionally and also similar to the yttrium semiquinolate, complex 9 shows a signal with a g value of 1.999, which most likely results from a non-coupled radical impurity.
Note that the reactions of cerium siloxides 2 and 3 with 1,4-naphthoquinone resulted also in the formation of homoleptic ceric siloxides as well as paramagnetic by-products (cf. Fig. S36, ESI†) indicating a similar reactivity as observed for Me4BQ. Unfortunately, any putative semiquinolate complexes could not be isolated.
 | ||
| Fig. 6 X-Band cw-EPR spectra of crystalline [Ce{OSi(OtBu)3}2(thf)2(μ2-O2C6Me4)]2 (8) (black trace) and [Ce(OSiiPr3)2(thf)2)(μ2-O2C6Me4)]2 (9) (red trace). | ||
Conclusions
Cerium(III) silylamides and siloxides are suitable reagents for assessing the oxidising power/reducibility of differently substituted 1,4-quinones in non-aqueous solutions. The cerium–quinone redox matching is revealed by the ease of formation of CeIV hydroquinolates [CeL3]2(μ2-O2C6R4), in the case of the parent 1,4-benzoquinone (BQ) or when R represents electron-withdrawing groups (Cl, CN). Depending on their reduction potential, alkyl-substituted BQs engage in redox equilibria, with the CeIV hydroquinolate species being preferentially stable in the solid state, but also afford semiquinolates via redox ligand redistribution. The structurally characterized siloxide semiquinolate complexes [(CeL2(thf)2)(μ2-O2C6Me4)]2 (L = OSi(OtBu)3, OSiiPr3) exhibit a molecular arrangement, recently detected for [LnCl2(THF)3(μ-Me4sq)2]2 (Ln = Y, Gd).13 The stabilisation of the tetravalent oxidation state in hydroquinolato-bridged complexes [CeIVL3]2(μ2-O2C6R4) was examined by electrochemical measurements, as well as NMR and UV/Vis spectroscopies. In accordance with previous findings,16,18–20 the stability of the ceric complexes increases in the order of N(SiMe3)2 < OSi(OtBu3)3 < OSiiPr3 as supporting ligand, but surprisingly drops in reverse order of the oxidation potential of the 1,4-quinones, being the least stable for the 2,3-dichloro-5,6-dicyano-hydroquinolato congener. The preferred formation of hydroquinolato-bridged silylamides [Ce{N(SiMe3)2}3]2(μ2-O2C6R4) seems kinetically favoured. Finally, the electrochemical reduction of the hydroquinolato-bridged ceric complexes [CeIVL3]2(μ2-O2C6R4) can be mimicked by chemical reduction with cobaltocene, as shown for the isolation and structural characterisation of cerous [(Ce{N(SiMe3)2}3)2(μ2-O2C6H4)][CoCp2]2. The cerium–quinone redox matching and tuning might be used as a role-model in tetravalent praseodymium and terbium chemistry.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Institute of Organic Chemistry at EKUT for EPR measurements.Notes and references
- N. El-Najjar, H. Gali-Muhtasib, R. A. Ketola, P. Vuorela, A. Urtti and H. Vuorela, Phytochem. Rev., 2011, 10, 353–370 CrossRef CAS.
- (a) N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS; (b) D. H. Evans, Chem. Rev., 2008, 108, 2113–2144 CrossRef CAS.
- (a) A. Osyczka, C. C. Moser, F. Daldal and P. L. Dutton, Nature, 2004, 427, 607–612 CrossRef CAS; (b) J. F. Allen and W. Martin, Nature, 2007, 445, 610–612 CrossRef CAS; (c) J. L. Nevarez, A. Turmo, J. Hu and R. P. Hausinger, ChemCatChem, 2020, 12, 4242–4254 CrossRef CAS.
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. G. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS.
- M. T. Huynh, C. W. Anson, A. C. Cavell, S. S. Stahl and S. Hammes-Schiffer, J. Am. Chem. Soc., 2016, 138, 15903–15910 CrossRef CAS.
- C. Frontana, Á. Vázquez-Mayagoitia, J. Garza, R. Vargas and I. González, J. Phys. Chem. A, 2006, 110, 9411–9419 CrossRef CAS.
- (a) D. Walker and J. D. Hiebert, Chem. Rev., 1967, 67, 153–195 CrossRef CAS; (b) S. B. Bharate, Synlett, 2006, 3, 496–497 CrossRef.
- (a) K. Ohkubo, A. Fujimoto and S. Fukuzumi, J. Am. Chem. Soc., 2013, 135, 5368–5371 CrossRef CAS; (b) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS.
- (a) M. Oh, G. B. Carpenter and D. A. Sweigart, Acc. Chem. Res., 2004, 37, 1–11 CrossRef CAS; (b) S. U. Son, S. B. Kim, J. A. Reingold, G. B. Carpenter and D. A. Sweigart, J. Am. Chem. Soc., 2005, 127, 12238–12239 CrossRef CAS.
- (a) S. Kitagawa, Coord. Chem. Rev., 2002, 224, 11–34 CrossRef CAS; (b) S. Masaoka, G. Akiyama, S. Horike, S. Kitagawa, T. Ida and K. Endo, J. Am. Chem. Soc., 2003, 125, 1152–1153 CrossRef CAS.
- (a) B. F. Abrahams, J. Coleiro, B. F. Hoskins and R. Robson, Chem. Commun., 1996, 603–604 RSC; (b) S. Halis, A. K. Inge, N. Dehning, T. Weyrich, H. Reinsch and N. Stock, Inorg. Chem., 2016, 55, 7425–7431 CrossRef CAS.
- J. O. Moilanen, A. Mansikkamäki, M. Lahtinen, F.-S. Guo, E. Kalenius, R. A. Layfield and L. F. Chibotaru, Dalton Trans., 2017, 46, 13582–13589 RSC.
- T. Han, J. B. Petersen, Z.-H. Li, Y.-Q. Zhai, A. Kostopoulos, F. Ortu, E. J. L. McInnes, R. E. P. Winpenny and Y.-Z. Zheng, Inorg. Chem., 2020, 59, 7371–7375 CrossRef CAS.
- (a) J. Yuasa, T. Suenobu and S. Fukuzumi, J. Am. Chem. Soc., 2003, 125, 12090–12091 CrossRef CAS; (b) J. Yuasa and S. Fukuzumi, J. Am. Chem. Soc., 2007, 129, 12912–12913 CrossRef CAS.
- For examples of other structurally characterised semiquinone complexes, see: (a) J.-M. Lü, S. V. Rosokha, I. S. Neretin and J. K. Kochi, J. Am. Chem. Soc., 2006, 128, 16708–16719 CrossRef; (b) H. Nakamori, T. Matsumoto, T. Yatabe, K.-S. Yoon, H. Nakai and S. Ogo, Chem. Commun., 2014, 50, 13059–13061 RSC; (c) I.-R. Jeon, B. Negru, R. P. Van Duyne and T. D. Harris, J. Am. Chem. Soc., 2015, 137, 15699–15702 CrossRef CAS.
- Â. Domingos, I. Lopes, J. C. Waerenborgh, N. Marques, G. Y. Lin, X. W. Zhang, J. Takats, R. McDonald, A. C. Hillier, A. Sella, M. R. J. Elsegood and V. W. Day, Inorg. Chem., 2007, 46, 9415–9424 CrossRef.
- (a) F. Sroor and F. Edelmann, in Cerium: Molecular Structure, Technological Applications and Health Effects, Nova Science Publishers, Hauppage, N.Y., 2012, pp. 73–106 Search PubMed; (b) N. A. Piro, J. R. Robinson, P. J. Walsh and E. J. Schelter, Coord. Chem. Rev., 2014, 260, 21–36 CrossRef CAS; (c) Y.-M. So and W.-H. Leung, Coord. Chem. Rev., 2017, 340, 172–197 CrossRef CAS; (d) R. Anwander, M. Dolg and F. T. Edelmann, Chem. Soc. Rev., 2017, 46, 6697–6709 RSC.
- (a) V. Nair, J. Mathew and J. Prabhakaran, Chem. Soc. Rev., 1997, 26, 127–132 RSC; (b) V. Nair and A. Deepthi, Tetrahedron, 2009, 65, 10745–10755 CrossRef CAS.
- Y. Qiao and E. J. Schelter, Acc. Chem. Res., 2018, 51, 2926–2936 CrossRef CAS.
- (a) O. Eisenstein, P. B. Hitchcock, A. G. Hulkes, M. F. Lappert and L. Maron, Chem. Commun., 2001, 1560–1561 RSC; (b) M. D. Walter, R. Fandos and R. A. Andersen, New J. Chem., 2006, 30, 1065 RSC; (c) P. Dröse, A. R. Crozier, S. Lashkari, J. Gottfriedsen, S. Blaurock, C. G. Hrib, C. Maichle-Mössmer, C. Schädle, R. Anwander and F. T. Edelmann, J. Am. Chem. Soc., 2010, 132, 14046–14047 CrossRef; (d) A. R. Crozier, A. M. Bienfait, C. Maichle-Mössmer, K. W. Törnroos and R. Anwander, Chem. Commun., 2013, 49, 87–89 RSC; (e) D. Schneider, T. Spallek, C. Maichle-Mössmer, K. W. Törnroos and R. Anwander, Chem. Commun., 2014, 50, 14763–14766 RSC; (f) U. J. Williams, J. R. Robinson, A. J. Lewis, P. J. Carroll, P. J. Walsh and E. J. Schelter, Inorg. Chem., 2014, 53, 27–29 CrossRef CAS; (g) D. Schneider, N. Harmgarth, F. T. Edelmann and R. Anwander, Chem.– Eur. J., 2017, 23, 12243–12252 CrossRef CAS; (h) L. A. Solola, P. J. Carroll and E. J. Schelter, J. Organomet. Chem., 2018, 857, 5–9 CrossRef CAS; (i) N. T. Rice, J. Su, T. P. Gompa, D. R. Russo, J. Telser, L. Palatinus, J. Bacsa, P. Yang, E. R. Batista and H. S. La Pierre, Inorg. Chem., 2019, 58, 5289–5304 CrossRef CAS.
- (a) M. Gregson, E. Lu, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2013, 52, 13016–13019 CrossRef CAS; (b) V. Lorenz, B. M. Schmiege, C. G. Hrib, J. W. Ziller, A. Edelmann, S. Blaurock, W. J. Evans and F. T. Edelmann, J. Am. Chem. Soc., 2011, 133, 1257–1259 CrossRef CAS; (c) J. A. Bogart, C. A. Lippincott, P. J. Carroll, C. H. Booth and E. J. Schelter, Chem.– Eur. J., 2015, 21, 17850–17859 CrossRef CAS; (d) R. P. Kelly, L. Maron, R. Scopelliti and M. Mazzanti, Angew. Chem., Int. Ed., 2017, 56, 15663–15666 CrossRef CAS.
- (a) P. B. Hitchcock, M. F. Lappert and A. V. Protchenko, Chem. Commun., 2006, 3546–3548 RSC; (b) M. P. Coles, P. B. Hitchcock, A. V. Khvostov, M. F. Lappert, Z. Li and A. V. Protchenko, Dalton Trans., 2010, 39, 6780–6788 RSC.
- (a) I. J. Casely, S. T. Liddle, A. J. Blake, C. Wilson and P. L. Arnold, Chem. Commun., 2007, 5037–5039 RSC; (b) P. L. Arnold, I. J. Casely, S. Zlatogorsky and C. Wilson, Helv. Chim. Acta, 2009, 92, 2291–2303 CrossRef CAS; (c) D. Werner, G. B. Deacon, P. C. Junk and R. Anwander, Dalton Trans., 2017, 46, 6265–6277 RSC; (d) U. Bayer, L. Bock, C. Maichle-Mössmer and R. Anwander, Eur. J. Inorg. Chem., 2020, 101–106 CrossRef CAS.
- (a) A. Sen, H. A. Stecher and A. L. Rheingold, Inorg. Chem., 1992, 31, 473–479 CrossRef CAS; (b) J. R. Robinson, C. H. Booth, P. J. Carroll, P. J. Walsh and E. J. Schelter, Chem.– Eur. J., 2013, 19, 5996–6004 CrossRef CAS; (c) D. Werner, G. B. Deacon, P. C. Junk and R. Anwander, Chem.– Eur. J., 2014, 20, 4426–4438 CrossRef CAS.
- The reactivity of a series of methyl-substituted 1,4-benzoquinones toward π-allylnickel bromide was probed (yielding ring-allylation in 2- or 6-position), revealing enhanced reactivity of the more easily reduced BQs: L. S. Hegedus and E. L. Waterman, J. Am. Chem. Soc., 1974, 96, 6789–6791 CrossRef CAS.
- J. Friedrich, C. Maichle-Mössmer and R. Anwander, Chem. Commun., 2017, 53, 12044–12047 RSC.
- W. J. Evans, R. E. Golden and J. W. Ziller, Inorg. Chem., 1991, 30, 4963–4968 CrossRef CAS.
- H. A. Stecher, A. Sen and A. L. Rheingold, Inorg. Chem., 1989, 28, 3280–3282 CrossRef CAS.
- P. S. Gradeff, K. Yunlu, A. Gleizes and J. Galy, Polyhedron, 1989, 8, 1001–1005 CrossRef CAS.
- U. J. Williams, P. J. Carroll and E. J. Schelter, Inorg. Chem., 2014, 53, 6338–6345 CrossRef CAS.
- (a) J. Friedrich, Y. Qiao, C. Maichle-Mössmer, E. J. Schelter and R. Anwander, Dalton Trans., 2018, 47, 10113–10123 RSC; (b) C. T. Palumbo, I. Zivkovic, R. Scopelliti and M. Mazzanti, J. Am. Chem. Soc., 2019, 141, 9827–9831 CrossRef CAS; (c) A. R. Willauer, C. T. Palumbo, R. Scopelliti, I. Zivkovic, I. Douair, L. Maron and M. Mazzanti, Angew. Chem., Int. Ed., 2020, 59, 3549–3553 CrossRef CAS.
- P. G. Eller and R. A. Penneman, J. Less-Common Met., 1987, 127, 19–33 CrossRef CAS.
- (a) U. Kilimann, R. Herbst-Irmer, D. Stalke and F. T. Edelmann, Angew. Chem., Int. Ed. Engl., 1994, 33, 1618–1621 CrossRef; (b) H. Fang, B. E. Cole, Y. Qiao, J. A. Bogart, T. Cheisson, B. C. Manor, P. J. Carroll and E. J. Schelter, Angew. Chem., Int. Ed., 2017, 56, 13450–13454 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., 1976, 32, 751–767 CrossRef.
- P. S. Gradeff, K. Yunlu, T. J. Deming, J. M. Olofson, R. J. Doedens and W. J. Evans, Inorg. Chem., 1990, 29, 420–424 CrossRef CAS.
- K. D. Karlin and S. J. Lippard, Progress in Inorganic Chemistry, John Wiley and Sons, New York, 1959 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2022461, 2022462, 2022463, 2022464, 2022465, 2022466, 2022467, 2022468, 2022469, 2022470, 2022471, 2022472, 2022473, 2022474, 2022475 and 2022476. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc04489j |
| This journal is © The Royal Society of Chemistry 2021 |