
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConformational editing of intrinsically disordered protein by α-methylation†
Valentin
Bauer‡
 a,
Boris
Schmidtgall‡
a,
Gergő
Gógl
bc,
Jozica
Dolenc
d,
Judit
Osz
b,
Yves
Nominé
bc,
Camille
Kostmann
bc,
Alexandra
Cousido-Siah
bc,
André
Mitschler
bc,
Natacha
Rochel
b,
Gilles
Travé
bc,
Bruno
Kieffer
b and
Vladimir
Torbeev
*a
a,
Boris
Schmidtgall‡
a,
Gergő
Gógl
bc,
Jozica
Dolenc
d,
Judit
Osz
b,
Yves
Nominé
bc,
Camille
Kostmann
bc,
Alexandra
Cousido-Siah
bc,
André
Mitschler
bc,
Natacha
Rochel
b,
Gilles
Travé
bc,
Bruno
Kieffer
b and
Vladimir
Torbeev
*a
aInstitut de Science et d'Ingénierie Supramoléculaires (ISIS), International Center for Frontier Research in Chemistry (icFRC), University of Strasbourg, CNRS, UMR 7006, Strasbourg, France. E-mail: torbeev@unistra.fr
bDepartment of Integrated Structural Biology, Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC), INSERM (U1258), University of Strasbourg, CNRS, UMR 7104, Illkirch, France
cÉquipe Labellisée Ligue contre le cancer, France
dChemistry|Biology|Pharmacy Information Center, ETH Zurich, Zurich, Switzerland
First published on 4th November 2020
Abstract
Intrinsically disordered proteins (IDPs) constitute a large portion of “Dark Proteome” – difficult to characterize or yet to be discovered protein structures. Here we used conformationally constrained α-methylated amino acids to bias the conformational ensemble in the free unstructured activation domain of transcriptional coactivator ACTR. Different sites and patterns of substitutions were enabled by chemical protein synthesis and led to distinct populations of α-helices. A specific substitution pattern resulted in a substantially higher binding affinity to nuclear coactivator binding domain (NCBD) of CREB-binding protein, a natural binding partner of ACTR. The first X-ray structure of the modified ACTR domain - NCBD complex visualized a unique conformation of ACTR and confirmed that the key α-methylated amino acids are localized within α-helices in the bound state. This study demonstrates a strategy for characterization of individual conformational states of IDPs.
Introduction
Intrinsically disordered proteins (IDPs) and protein domains complement the functions of well-folded proteins.1–3 They are particularly abundant in eukaryotes, where they contribute to the higher complexity of biological organization and regulatory processes.4 Intrinsic disorder confers distinctive molecular recognition and structural properties compared to well-folded proteins.5 These properties include high affinity even if the complexes remain unstructured,6 multivalent interactions that may lead to phase separation,7 and promiscuity in forming complexes with distinct structurally-unrelated targets.8The gene transcription machinery of eukaryotes is particularly rich in IDPs9 where intrinsic disorder plays a key role, providing high structural and compositional diversity in protein complexes that regulate gene expression.10–12 More than half of eukaryotic transcription factors are predicted to contain highly disordered regions, especially within their activation domains.13 Thus, being able to target IDPs could represent an attractive strategy to systematically interfere with biological processes at the level of gene transcription.14
However, a lack of well-structured hydrophobic binding pockets makes it challenging to identify molecules, either by design or high-throughput screening, that bind IDPs specifically and with high affinity.15 In general, folding of IDPs upon complex formation leads to a large loss of conformational entropy.16 Introduction into the IDP of conformational constraints that minimize such losses is one approach to enhance overall affinity. For example, side-chains in peptide fragments of the IDP that correspond to the most important site at the protein–protein interaction interface can be covalently tethered. The successful development of stapled peptides as inhibitors of protein–protein interactions is illustrative of this strategy.17 However, such methods are best suited to protein complexes where binding energy is localized over a few proximal residues and not dispersed over several distant regions of the protein surface.
As an alternative, more general approach, we are exploring backbone conformational constraints as a means to modulate population of conformers in the conformational ensemble of IDPs and consequently modify their binding affinities in the corresponding protein–protein interactions (Fig. 1). Conformational ensembles contain multiple distinct conformational states,18 however, only a subset of conformations in the ensemble may play essential functional roles (e.g., binding-competent conformation to a particular cellular target). Conformational editing of IDPs through targeted site-specific modifications of the corresponding protein sequences can help populating such functionally relevant conformations. This can be achieved with the help of natural amino acids with different propensities for particular elements of secondary structure (i.e., helices, sheets, turns)19 or using a large variety of non-canonical residues that can mimic elements of secondary or tertiary structure.20 The latter modifications can be advantageous due to stronger localized effects on protein conformation resulting in enhanced control of the induced changes in the conformational ensemble.
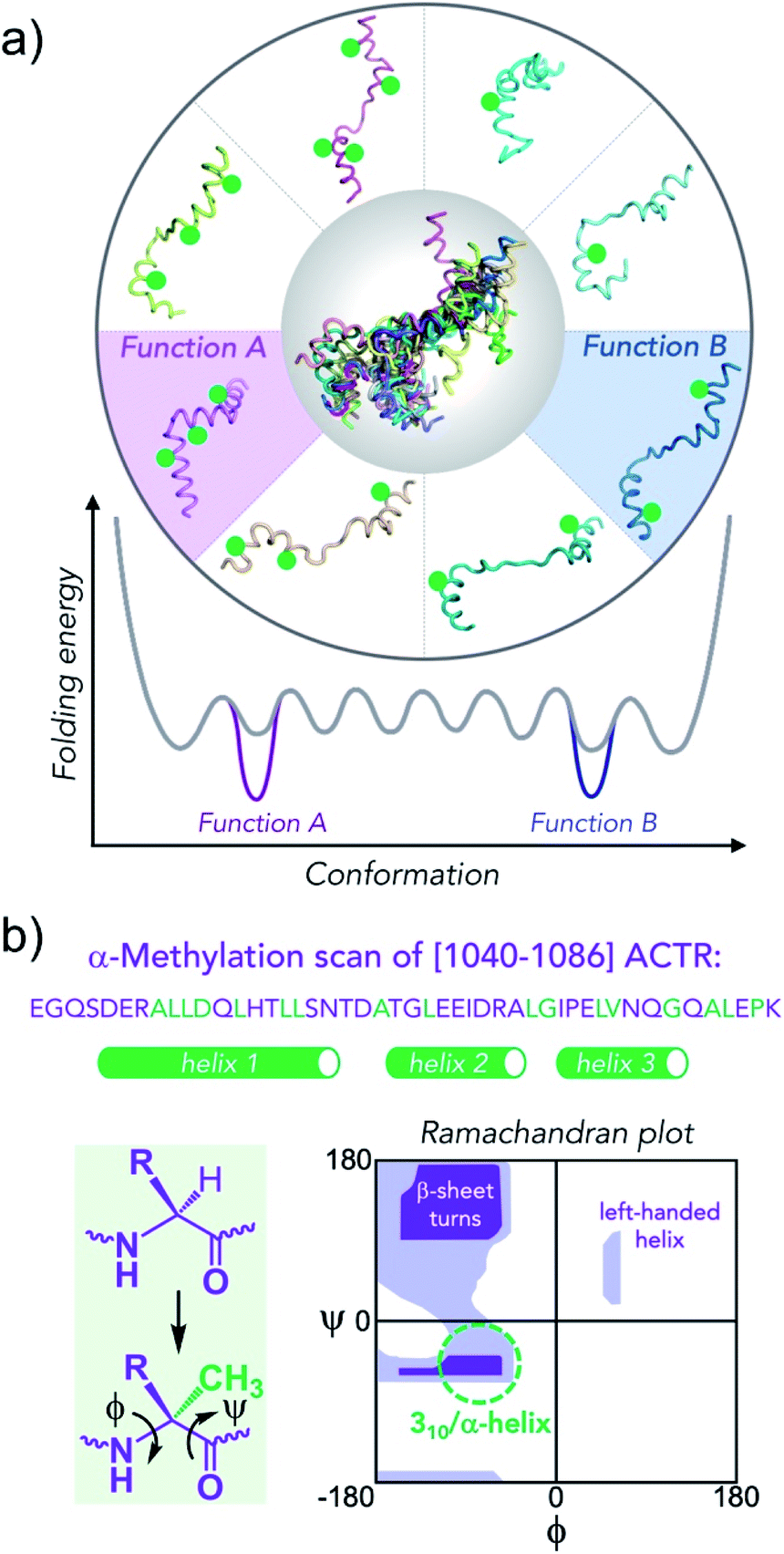 | ||
| Fig. 1 Conformational editing of an intrinsically disordered protein conformational ensemble by site-specific incorporation of residues that modify the population of secondary structure elements. (a) Green circles stabilize helical structures, altering the folding free energy landscape. Knowing which conformation is enhanced in the ensemble together with functional characterization enables the unambiguous assignment of conformation to function. (b) Sequence of [1040–1086]-fragment of ACTR that binds to NCBD of CBP/p300 with residues that were replaced by α-methylated amino acids in green (the helical segments in 1KBH NMR structure are shown as green tubes). Extra methyl group attached at α-carbon constrains backbone (ϕ/ψ)-dihedral angles to values that correspond to helical secondary structures (green dashed circle) in lower, left quadrangle of the Ramachandran plot. | ||
To validate the potential utility of this strategy, we chose a complex formed by domains from two transcriptional coactivators, CBP (CREB-binding protein)21,22 and ACTR (activator for thyroid hormone and retinoid receptors; also called SRC3 – steroid receptor coactivator 3 from p160 family).23 Both CBP (and its paralog p300) and ACTR function as cellular integrators of various signalling pathways that transduce these signals into fine-tuned transcriptional outputs.21–23 In its isolated state, the nuclear coactivator binding domain (NCBD) of CBP has the properties of a molten globule with residual helical structure,24 whereas the activation domain 1 (AD1) of ACTR is highly disordered.25 Together, they undergo coupled binding and folding to form a complex that is predominantly α-helical.26
One efficient way to stabilize helical conformations involves introduction of α-methylated amino acids as it was demonstrated for many short peptides.27,28 The additional α-methyl group increases steric bulk in proximity to polypeptide backbone and leads to conformational constraints reflected in a narrow distribution of Ramachandran (ϕ/ψ)-dihedral angles (Fig. 1b). Importantly, the native side-chains that are essential for protein–protein interactions are left unmodified. In our previous study we demonstrated chemical synthesis of 47-residue isoforms of AD1-ACTR and NCBD, the feasibility of incorporation of α-methylated amino acids into the AD1-ACTR sequence and provided support for induced fit in the complex formation mechanism.29 Here, we present the results for a combinatorial library of nearly 50 different α-methylated AD1-ACTR variants including the detailed biophysical studies to characterize their binding to NCBD. This effort led to the discovery of a set of variants with enhanced affinity for the isolated NCBD as well as for the full length, 265 kDa CBP.
Results and discussion
Combinatorial library of α-methylated variants
Previously, the structure of an AD1-ACTR/NCBD complex in solution had been determined by NMR and showed that the two proteins each contribute three α-helices to a tight hydrophobic interface.26 The seventeen sites in the [1040–1086] fragment of AD1-ACTR that we chose for α-methylation are depicted in Fig. 1b, and the corresponding sequences are listed in Fig. 2 and ESI Table S1†. | ||
| Fig. 2 Chemical synthesis of 50 variants of (1040–1086)-ACTR that were studied in this work by combinatorial ligation of 27 shorter peptide segments. NCL = native chemical ligation. Desulfurization was used to perform Cys-to-Ala conversion.29 Sequences are aligned and shown from N- to C-termini. Sites of modifications are depicted in red. For more details about modifications and numbering of sequences see ESI Table S1†. | ||
The conformational constraints of peptides that enhance helical structures not always lead to the strengthening of binding to their targets.30 Higher stability of the unbound state due to pre-folding or unfavourable packing interactions at the binding interface are among the factors resulting in the reduction of binding affinity. Achiral α-methyl-alanine that has no preference to be in either right- or left-handed helical conformation can lead to alternative folding.31 Furthermore, for α-methylated amino acids with the bulky side chains unfavourable interactions of side chain and backbone atoms can occur.28 As these deleterious effects should depend on sequence context and are hard to predict, we performed exploratory sequence scan to identify the positions where the unique conformational properties of α-methylated amino acids lead to enhancement of binding affinities. The substituted residues were chosen to be located in α-helical segments or in loop regions, at the buried AD1-ACTR/NCBD protein interface, and on the solvent-accessible surface of the protein complex.
Single and multiple substitutions were made, with up to six α-methylated amino acids introduced simultaneously. In total, we prepared 17 single, 22 double, 2 triple, 4 tetra, 1 penta and 1 hexa α-methylated analogues.§ In addition, we have synthesized two variants containing canonical amino acids with different helical propensities [S1043M; D1050E; T1054Q] and [A1047G] previously reported to have distinct binding properties to NCBD.32 To assemble the library of variants of AD1-ACTR (Fig. 2, ESI Tables S1 and S2†), we used combinatorial native chemical ligation of N-terminal and C-terminal peptide precursor segments following procedures described in our previous report.29 In a few cases, the entire polypeptide chain was assembled by microwave-assisted solid-phase peptide synthesis (ESI Materials and methods†).
α-Methylation modifies binding affinities
Isothermal titration calorimetry was used to characterize the formation of complexes between α-methylated variants of AD1-ACTR and NCBD (the [2066–2112]-fragment of CBP) to obtain Gibbs free energy (ΔG) as well as the enthalpy (ΔH) and entropy (−TΔS) changes of binding. The results of measurements at T = 304 K are summarized in Fig. 3 and ESI Table S3† (binding diagrams are depicted in ESI Fig. S1†). In most cases α-methylation caused a decrease in complex stability despite the helix-inducing tendencies of α-methylated amino acids. The greatest binding destabilization (ΔΔG ∼ 1.5–2.0 kcal mol−1) occurred for variants in which Leu1071 was replaced by its α-methylated analogue. Other substitutions that resulted in significant destabilization were 1056meLeu, 1064meLeu, 1049meLeu and 1050meAsp. | ||
| Fig. 3 α-Methylation modulates the thermodynamics of AD1-ACTR binding to NCBD. (a) AD1-ACTR variants are arranged along x-axis according to their binding affinities (from low to high) to NCBD obtained by ITC measurements. In y-axis Gibbs free energy of binding for wild type is subtracted from the corresponding values for each variant (ΔΔG). The corresponding sites of α-methylation and numbering of AD1-ACTR variants are listed below (**variants 1, 4, 7, 11, 13, 15, 19, 20, 21, 23, 25, 33, 36 and 38 were first reported in our previous communication).29 All studied complexes were additionally characterized by CD spectroscopy (ESI Table S6, Fig. S3 and S4b†). The stabilities of the selected complexes determined by thermal denaturation (ESI Table S4 and Fig. S5†) are in agreement with the ITC data. (b) Different magnitudes of enthalpy–entropy compensation reflect the modifications to folding pathways upon α-methylation. | ||
Nevertheless, we identified several sites where α-methylation enhanced binding. Complexes with the highest stability were obtained when 1055meLeu was introduced (ΔΔG = −0.61 kcal mol−1), whereas the 1047meAla-containing variant exhibited slightly increased binding affinity for NCBD (ΔΔG = −0.21 kcal mol−1). Given its exceptionally favourable effect on overall affinity (ΔG) of complexation to NCBD, residue Leu1055 can be regarded as a ‘hot-spot’ revealed by the α-methylation scan. An AD1-ACTR variant with two α-methylations ([1055meLeu; 1076meLeu]) was found to be the best NCBD binder in the entire library (variant 34 in Fig. 3 and ESI Table S1†). According to ITC, it exhibits approximately 5-fold stronger affinity (KD = 0.042 μM) at 304 K than WT (KD = 0.206 μM). The 1076meLeu substitution alone had nearly no effect on the binding of AD1-ACTR to NCBD but contributed to enhanced affinity when combined with 1055meLeu.
Shorter peptide fragments corresponding to the largest structured N-terminal portion of AD1-ACTR,32 including a peptide containing meLeu at the 1055 ‘hot spot’, did not show appreciable binding to NCBD (ESI Materials and methods†). For efficient binding, the full-length AD1-ACTR sequence is apparently essential.
Enthalpy–entropy compensation
The binding of wild-type AD1-ACTR to NCBD is enthalpy-driven at T = 304 K.33 Formation of favourable inter-residue contacts in the resulting complex presumably compensates for the loss of entropy due to coupled folding. Incorporation of α-methylated amino acids resulted in considerable changes in the enthalpic (ΔH) and entropic (−TΔS) contributions to binding depending on the position and number of substituted residues (Fig. 3b). Thus, the AD1-ACTR analogue with ‘hot spot’ 1055meLeu in the N-terminal helix exhibits an anomalously favourable ΔH, partially compensated by a larger unfavourable −TΔS compared to the wild type protein domain. Similarly, higher enthalpy–entropy compensation was observed for variants with substitutions in the C-terminal helix (e.g., 1076meLeu, 1077meVal, 1080meAla, 1083meAla). In contrast, compensation with the opposite sign (enthalpy became less favourable while entropy more favourable) was observed for the highly destabilized complexes containing 1071meLeu or 1056meLeu.Increasing the number of α-methylated residues reinforced the observed enthalpy–entropy compensation. The elevated ΔH and −TΔS contributions are consistent with the formation of more stable electrostatic or hydrogen bonding interactions, leading to gains in ΔH that are counter-balanced by losses in conformational degrees of freedom (−TΔS) resulting from reduced accessible values of the Ramachandran (ϕ/ψ)-dihedral angles. The most elevated values of −TΔΔS (7.3–8.6 kcal mol−1) relative to wild type are seen for the tetra-, penta- and hexa-substituted variants, which would correspond to folding of approximately 5 to 6 additional residues assuming the theoretically estimated value for entropy changes upon folding of 1.4 kcal mol−1 per residue.34
The large set of variants in the library containing two α-methylations enabled assessment of coupling energies (cross-talk) between the modified sites in the sequence. Standard “double-mutant cycle” analyses were performed according to the relationship ΔΔΔG = ΔΔGvariant-X + ΔΔGvariant-Y − ΔΔGdouble-XY, where ΔΔGvariant is the variation in Gibbs free energy for a singly modified ACTR variant and ΔΔGdouble-XY is the variation in Gibbs free energy for the doubly α-methylated protein, compared to the wild-type.35 The Gibbs free energy for most of the analogues (ca. 70%) containing two modifications displays positive cooperativity (ΔΔΔG > 0 in the 0.12–1.23 kcal mol−1 range); for several variants it is nearly 0; and only one, [1047meAla; 1072meAla] shows significant negative cooperativity (ΔΔΔG = −1.04 kcal mol−1) (ESI Fig. S2†). The positive cooperativity observed in several doubly substituted variants despite the unfavourable contribution of individual modifications for the complex formation suggests the existence of long-range structure-inducing effects for folding of the individual AD1-ACTR chain, however, such induced conformations may not be always compatible with the tight interface needed to form stable complexes with NCBD.
α-Methylation induces helical structure in the free ACTR activation domain
Circular dichroism measurements indicated an increase of helical content of free AD1-ACTR upon α-methylation for nearly all members of the library judging from the ratio of ellipticities at 222 and 199 nm (θ222/θ199), characterizing the respective contributions of α-helix and random coil (ESI Table S5, Fig. S3 and S4a†). Moreover, α-methylation of the ‘hot spot’ residue Leu1055 had a greater effect on α-helical content than modification of any other residue. The θ222/θ199 value for [1055meLeu]AD1-ACTR is significantly higher than for wild type (0.44 versus 0.23) and progressively increases when other α-methylated residues are added. For comparison, a previously reported triple mutant [S1043M; D1050E; T1054Q] that was engineered to have enhanced α-helicity by incorporating residues with high helical propensities32 has a lower α-helical content (θ222/θ199 = 0.33) than [1055meLeu]AD1-ACTR, showing that α-methylated amino acids are superior to canonical amino acids at inducing helical structure. This also correlates with the triple mutant's lower affinity for NCBD (KD for [S1043M; D1050E; T1054Q] variant is 0.188 μM versus 0.204 μM for WT and 0.075 μM for [1055meLeu]AD1-ACTR as measured by ITC at 304 K).NMR analysis provided residue-specific insights into the structural changes induced by α-methylation of residues Leu 1055 and 1076. Assignment of Hα, Cα and Cβ resonances was achieved for the wild type domain and two variants, [1055meLeu]AD1-ACTR and [1055meLeu;1076meLeu]AD1-ACTR using a combination of 1H-TOCSY, 1H-NOESY, 1H–13C HSQC, 1H–13C HSQC-TOCSY and HMBC experiments (ESI, Tables S7–S9, Fig. S6–S8†). The backbone chemical shifts in the two variants were compared with random coil values.36 In both cases, positive deviations of 13Cα chemical shifts from random coil reference values indicated the presence of helical conformation in the vicinity of 1055meLeu and 1076meLeu (Fig. 4a). Moreover, local enhancement of helicity at position 1055 propagates towards the N-terminus, resulting in stabilization of the helical structure via long-range effects.
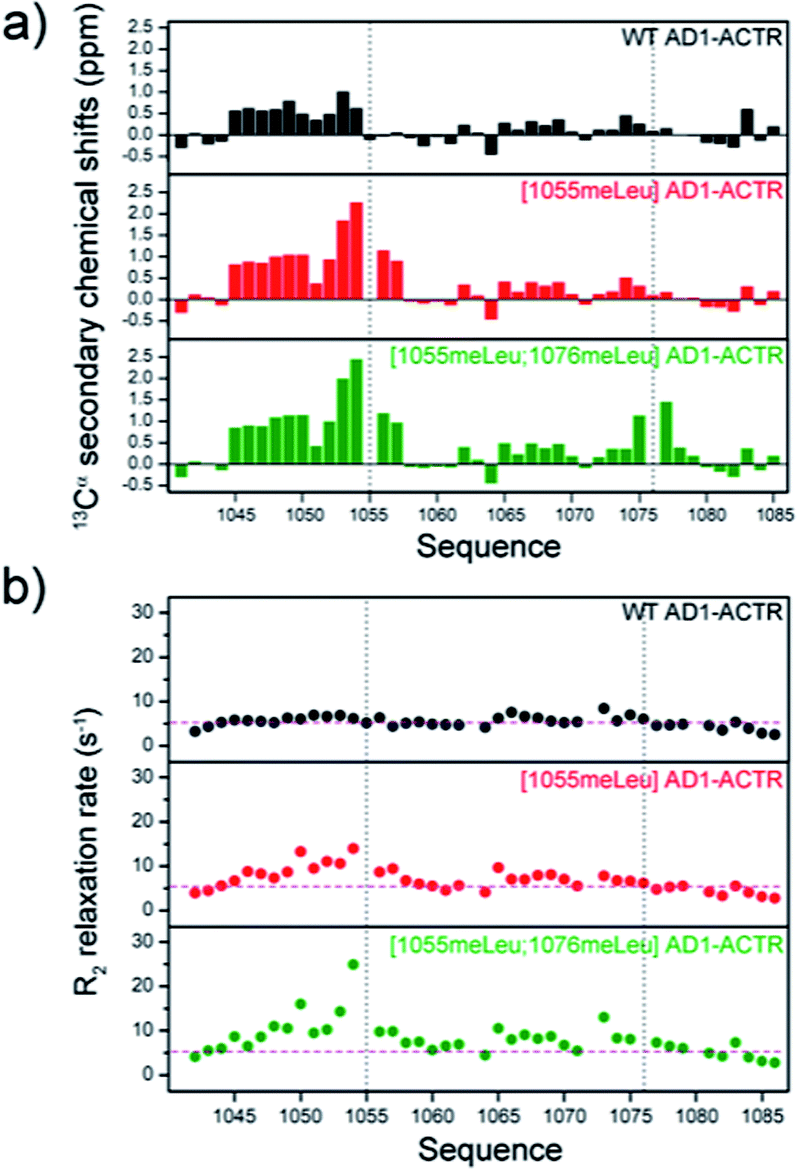 | ||
| Fig. 4 α-Methylation enhances helical structures in free ACTR activation domain. (a) 13Cα secondary chemical shift analysis indicates higher helical content in the proximity to the sites of incorporated α-methylated amino acids 1055 and 1076 (indicated by black dashed line). Furthermore, substitution at 1055 position results in higher population of helical structure at N-terminus (segment 1045–1054). (b) Cα transverse relaxation rates (R2) display increased values upon α-methylation, especially in double methylated [1055meLeu;1076meLeu]AD1-ACTR variant, suggesting the presence of conformational exchange between disordered and structured states. The R2 measurement uncertainties correspond to ∼10% of the R2 values (within the dimension of the circles in the plot). The average R2 value calculated for all residues in WT AD1-ACTR is included as pink dashed line in all three panels to highlight the overall R2 increase observed upon α-methylation. | ||
Although longitudinal relaxation rates (R1) were not affected by α-methylation (ESI Table S10 and Fig. S9†), elevated values for transverse relaxation rates (R2) were observed, particularly for residues near 1055meLeu (Fig. 4b and ESI Table S11†). These results likely reflect the contribution of conformational exchange (Rex) between locally folded and unfolded states, usually a very fast dynamic process that is presumably slowed down to the chemical shift time scale (μs to ms) by α-methylation.
X-ray structure of the [1055meLeu;1076meLeu]AD1-ACTR complex with NCBD
In an attempt to obtain high-resolution structural information, we tethered NCBD to a crystallization-prone mutant of the maltose binding protein (MBP).37 While the complex of wild-type AD1-ACTR with MBP-fused NCBD did not yield crystals, we succeeded in obtaining crystals and solving an X-ray structure of the analogous complex with the best binder, [1055meLeu;1076meLeu]AD1-ACTR (Fig. 5a, ESI Table S12 and Fig. S11–S15†). The X-ray diffraction data were collected at 2.28 Å resolution and the structure was solved by molecular replacement using a high resolution MBP crystal structure and the previously reported NMR structure of AD1-ACTR.26 In [1055meLeu;1076meLeu]AD1-ACTR, both meLeu residues adopt canonical α-helical backbone conformations with (ϕ/ψ)-angles of −58.2°, −65.3° for meLeu1055 and −39.9°, −57.0° for meLeu1076, respectively (Fig. 5a). This result further supports our hypothesis that α-methylation can be used to stabilize α-helical structures in IDPs.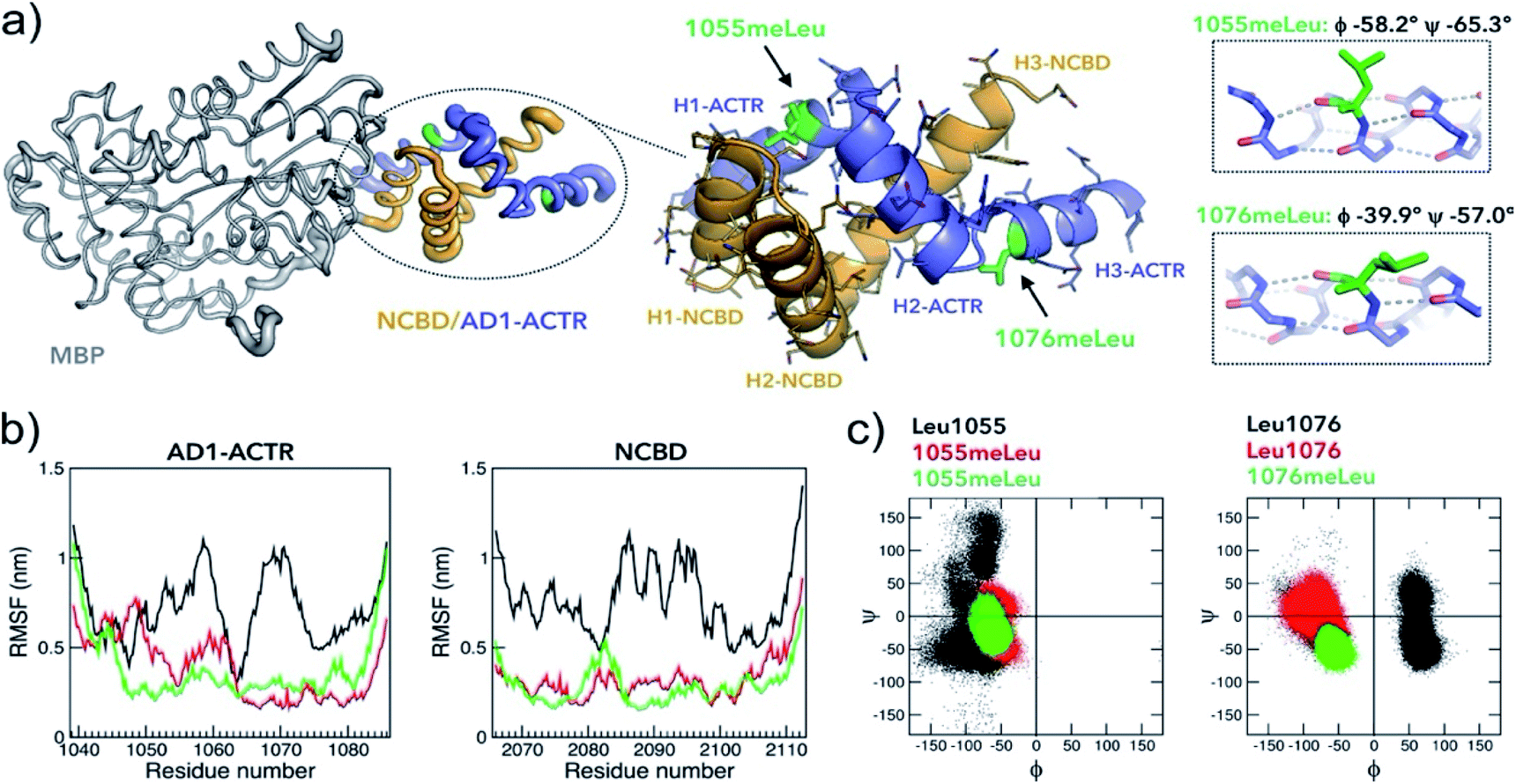 | ||
| Fig. 5 Properties of the complex with the highest binding affinity. (a) An X-ray structure of the α-methylated [1055meLeu;1076meLeu]AD1-ACTR variant bound to NCBD (PDB ID: 6SQC). To facilitate the crystallization, NCBD was fused to maltose-binding protein (MBP, depicted in grey). The [1055meLeu;1076meLeu]AD1-ACTR (in purple-blue) and NCBD (in orange) have elevated thermal B-factors (thicker tube) in comparison to MBP reflecting higher amplitudes of atomic displacements suggesting enhanced conformational dynamics. Enlarged structure of the complex illustrates the arrangement of six α-helices (H1–H3 for ACTR and NCBD) and the positions of α-methylated Leu residues. Both 1055meLeu and 1076meLeu adopt α-helical conformations (corresponding helices are on the right). The side chain of 1055meLeu adopts [trans (t, χ1 = −167.4°), trans (t, χ2 = 153°)] rotamer conformation, while 1076meLeu has its side chain in [gauche- (g-, χ1 = −59.3°), trans (t, χ2 = 137°)] conformation. Stereochemical Newman projections (ESI Fig. S13†) illustrate how α-methyl groups avoid steric clashes with Leu isobutyl side chains. (b) Root-mean square fluctuations (RMSF) in MD simulations (GROMOS, 200 ns) of three complexes (WT in black, [1055meLeu]AD1-ACTR/NCBD in red and [1055meLeu;1076meLeu]AD1-ACTR/NCBD in green) indicating that α-methylation rigidifies motions in both AD1-ACTR and NCBD (additional analysis is in ESI Fig. S16 and S17†). (c) The corresponding Ramachandran plots depict well-defined helical distributions of (ϕ/ψ)-dihedral angles for α-methylated residues 1055meLeu and 1076meLeu in contrast to less defined conformations for non-methylated Leu residues. | ||
A comparison of the crystallized complex with the two NMR structures of activation domain of ACTR and NCBD, as well as other structures of NCBD, is provided in ESI Fig. S12†. The overall arrangement of the three AD1-ACTR helices in the crystal structure is, in most respects, similar to the NMR structure with PDB ID: 1KBH.26 Although, globally the two structures are similar, significant local structural differences are observed in the loops connecting helices, the conformations of side-chains and the hydrogen-bonding network. Moreover, the interaction interface with NCBD in the crystal is less tight than in the NMR structure, with an overall buried surface area of 1066 Å as compared to 1655 Å observed for the NMR structure (1KBH) (ESI Fig. S14†). The most prominent difference is seen in the C-terminal helix, which no longer forms a tight interface with NCBD in the X-ray structure. Analysis of crystal packing did not reveal any interactions with symmetry related molecules (ESI Fig. S15†), suggesting that this is not a crystal packing artefact. Pairwise analysis of helix–helix interactions using generalized Crick parameters and knob-into-hole packing revealed that the helical interfaces are not optimal when compared to ideal geometries (ESI Discussion, Table S15 and Fig. S18†).
A comparison with another available NMR structure of the complex (PDB ID: 6ES7),38 shows much larger deviations including a completely different orientation of the AD1-ACTR C-terminal helix. The same is true for the NMR structure of the complex of NCBD with a homologous activation domain from SRC1 isotype of p160 (PDB ID: 2C52)39 in which only the NCBD chain and N-terminal helix of SRC1 can be reasonably superimposed, whereas the remaining part of SRC1 adopts a completely different conformation (ESI Fig. S12†). Evidently, α-methylation at sites 1055 and 1076 resulting in most potent binding affinity originates from stabilization of a unique conformer of AD1-ACTR trapped in the crystal and this result corroborates our initial idea that site-specific α-methylation can modify conformational ensemble of IDPs.
Attenuated dynamics in high affinity complexes
To better understand the stabilizing effects of 1055meLeu and 1076meLeu substitutions, we performed explicit-solvent all-atom molecular dynamics (MD) simulations for 200 ns for the wild-type AD1-ACTR/NCBD complex as well as for two complexes with the α-methylated analogues [1055meLeu]AD1-ACTR and [1055meLeu;1076meLeu]AD1-ACTR. As can be seen from the reduced RMSF (root-mean-square fluctuations) of backbone atom positions in the complexes with the variants compared to wild type (Fig. 5b), α-methylation at positions Leu1055 and Leu1076 causes a substantial decrease in the simulated amplitudes of motion. Moreover, the overall shapes of the α-methylated complexes were more compact as judged by their radii of gyration (ESI Fig. S16b†). In addition, the (ϕ/ψ)-backbone conformations were clearly constrained by α-methylation for the α-methylated amino acids as well as for nearby residues (Fig. 5c and ESI Fig. S16d†).The higher stability of the α-methylated complexes can be inferred from the overall higher occurrence of predicted hydrogen bonds in the corresponding complexes than in the wild type complex (ESI Table S13†). A salt bridge analysis showed different patterns of populated salt bridges (ESI Table S14†). The structural differences between the average structure of wild type, [1055meLeu] and [1055meLeu;1076meLeu]AD1-ACTR variants complexed with NCBD were further confirmed by observing different chemical shifts in the 1H–13C HSQC spectra of the corresponding complexes, especially for the methyl resonances of aliphatic side chains indicating non identical hydrophobic protein–protein interfaces (ESI Fig. S10†).
Insights into stabilization versus destabilization due to α-methylation
The X-ray structure allows to explain the observed effects of α-methylation at other positions on the stability of AD1-ACTR/NCBD complex. Our data suggest that the stabilization or destabilization does not simply correlate with amino acid secondary structure propensities, since replacing a helix-favouring Leu amino acid with its α-methylated counterpart at different sites in the sequence (i.e., 1049, 1052, 1056 versus 1055 located in H1-ACTR helix) gave opposing effects. Furthermore, there is no obvious correlation with the position of the modification in the helical segment or in the loop, solvent exposed or in the hydrophobic core. Instead, the resulting stabilizing or destabilizing effects are due to alternative side-chain rotamers, compatibility of the substituted and nearby residues with conformational constraints imposed by α-methylation and long-range interactions. Presumably, for the destabilizing variants, where α-methylation is introduced in the hydrophobic core, it leads to alternative conformations of side-chains which may result in steric clashes. This explanation particularly applies for residues that participate in the well-packed complementary knob-into-hole interactions (e.g., Leu residues 1048, 1049, 1052 between helices H1-ACTR and H1-NCBD, see ESI Fig. S18†) or Leu1064 in H2-ACTR helix positioned next to highly important Asp1068⋯Arg2105 salt bridge.33 Indeed, the side-chain rotamers in 1055meLeu and 1076meLeu are distinct from the rotamers observed for other non-methylated Leu residues.Leu1071 as well as Leu1056, where α-methylation leads to the most pronounced destabilization, share a property that they are located near the C-termini of the respective H2-ACTR and H1-ACTR helices. It is known that C-termini of α-helices require unique conformational properties. For instance, the C-capping residue (first non-helical at C-terminus) is often Gly commonly adopting D-amino acid conformation.40,41
Leu1071 (ϕ = −84.5°, ψ = −2.7°) is next to Gly1072 in D-amino acid conformation (ϕ = +71.3°, ψ = +21.0°). Our MD simulations showed that residues flanking α-methylated amino acids also get constrained (ESI Fig. S16†). Thus, constraining Leu1071 via α-methylation may lead to D-to-L conformational switch of Gly1072 resulting in a complete remodelling of the corresponding loop. The lesser degree of folding of all complexes containing 1071meLeu is reflected in less favourable changes of enthalpy (ΔΔH) and more favourable entropy (−TΔΔS) than for wild type complex (Fig. 3b). The same effect observed for Leu1056 can be due to the modifications of loop structure between H1-ACTR and H2-ACTR helices. Interestingly, the substitution of Gly1072 by α-methyl-alanine (meAla) results in a moderate stabilization of the complex. Residue meAla is achiral as Gly and, although it is conformationally constrained, it can adopt dihedral angles corresponding to the positive quadrangle on the Ramachandran map.
Among singly methylated AD1-ACTR variants, substitution of Leu with meLeu at position 1055 in AD1-ACTR provided with the largest increase in binding affinity for NCBD. In the crystal structure, the residue 1055meLeu is partially buried and partially solvent exposed. It serves as a side residue of the hole in one of the knob-into-hole interactions between H1-ACTR and H1-NCBD helices (ESI Fig. S18†). The fact that it is not entirely buried at the helix interface like other leucines in the H1-ACTR helix can explain why the modified side-chain rotamer upon α-methylation can be accommodated at this site and the overall result is the stabilization of the complex.
In this case, α-methylation results in a “gain-of-function”. This contrasts with a previous study where Leu1055 was replaced by alanine leading to a weaker binding, constituting a “loss-of-function” due to substitution of a bulky isobutyl moiety with a smaller methyl group.42 Notably, this was the only site in the protein that adopts a near native conformation in the folding transition state.42 Consistent with this interpretation, NMR and CD measurements showed that α-methylation of Leu1055 leads to partial folding of the N-terminal helix in free AD1-ACTR protein (Fig. 4 and ESI Fig. S4a†), highlighting the role of binding partner preorganization in the complex formation of AD1-ACTR/NCBD.
Targeting NCBD domain in full length 265 kDa CBP
CBP (like its paralog p300) is a large multi-domain protein containing folded and unstructured domains, including NCBD.21 To test molecular recognition of methylated AD1-ACTR variants by CBP in comparison to isolated NCBD and whether these modified sequences bind CBP more potently than wild-type protein, we performed surface plasmon resonance (SPR) experiments (Fig. 6). The C-termini of wild-type AD1-ACTR and [1055meLeu;1076meLeu]AD1-ACTR were conjugated to biotin via a polyethyleneglycol spacer, and the proteins were immobilized on the chip via biotin–streptavidin approach. Singly α-methylated variants [1055meLeu]AD1-ACTR, [1076meLeu]AD1-ACTR and a weak binder [1064meLeu;1071meLeu]AD1-ACTR were prepared in the same way as additional experimental points and a negative control, respectively. The binding to NCBD were measured by SPR at five different temperatures (from 20 to 31 °C). The [1055meLeu;1076meLeu]AD1-ACTR analogue proved to be the best binder at all temperatures, with a binding affinity (KD) strengthened by 11–14 fold in comparison to wild-type (the tightest binding at ∼15 nM was observed at 20 °C, ESI Table S17†). Remarkably, the higher binding affinity of [1055meLeu;1076meLeu]AD1-ACTR was also confirmed for full-length CBP (apparent KD improved from ∼1.2 μM to ∼60 nM at 10 °C) (Fig. 6), suggesting that conformational editing of an intrinsically disordered domain by α-methylation can be used to derive a selective and high affinity binder with the ability to target a multi-domain binding partner.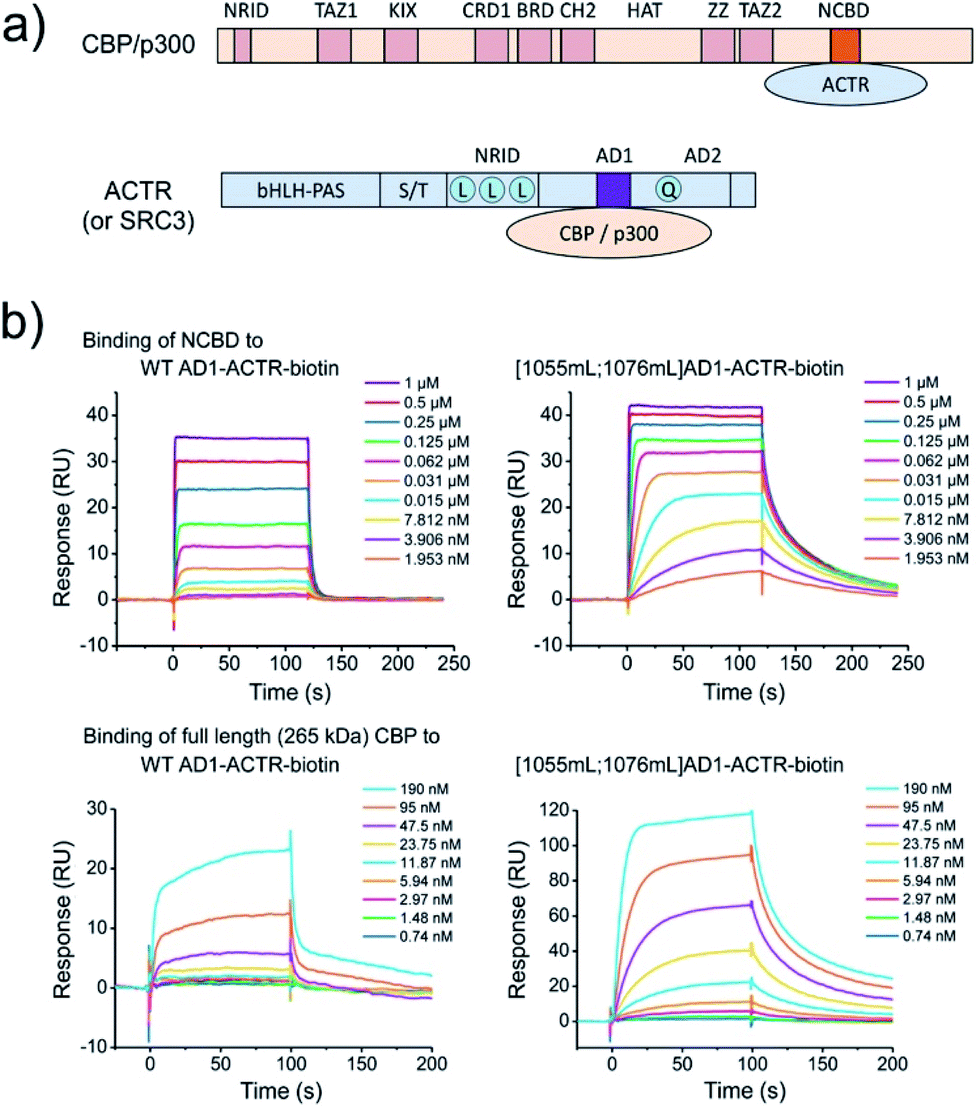 | ||
| Fig. 6 α-Methylation enhances binding to full length (265 kDa) CBP. (a) Domain organization of CBP/p300 and ACTR (or SRC3), which interact with each other via NCBD (in orange) and AD1 (in violet) domains. For clarification of domain abbreviations, see ref. 22 and 23. (b) Surface plasmon resonance (SPR) binding curves of NCBD to wild type AD1-ACTR and [1055meLeu;1076meLeu]AD1-ACTR variant at 20 °C. Steady state analysis reveals ∼10-fold enhanced binding constant for dimethylated variant (data at other temperatures and the estimated KD values are in ESI Tables S16, S17 and Fig. S19, S20†). The same effect is observed for full length 265 kDa CBP at 10 °C with apparent KD values obtained via steady-state analysis (ESI Fig. S21†) indicating an order of magnitude improvement for the [1055mL;1076mL]AD1-ACTR-variant: ∼60 nM versus 1.2 μM for wild-type (complementary fluorescence polarization measurements are consistent with SPR data, shown in ESI Fig. S22;† negative control with weak-binding [1064mL;1071mL] variant is shown in ESI Fig. S23†). | ||
Both CBP and ACTR have been implicated in various diseases (neurological, metabolic disorders and cancers), and regulating their function has been proposed as a possible means of therapeutic intervention.43,44 Indeed, potent small molecule inhibitors have been developed for the well-structured histone acetyltransferase (HAT) domain in CBP/p300.45 However, intrinsically disordered domains in large, complex proteins such as CBP and ACTR are often considered to be ‘undruggable’ by small molecules.15 Our results establish the feasibility of an alternative approach that is based on conformational editing of the IDP domain itself, utilizing backbone conformational constraints to enhance the native structural features and facilitate binding to the protein target.
In order to validate the interaction between the α-methylated AD1-ACTR variant and the CBP protein in a cellular context, we performed pull-down experiment using MCF7 cell line and the biotin conjugated [1055meLeu;1076meLeu]AD1-ACTR variant as a bait. The result of the pull-down experiment analyzed by SDS-PAGE, Western-blot and mass-spectrometry confirmed the specific interaction (ESI Fig. S24†).
Conclusions
In the work reported here, we showed that the free energy landscape of coupled binding and folding of the intrinsically disordered transcription coactivator ACTR activation domain can be efficiently modulated by α-methylation. The combinatorial library approach was essential for exploring position- and context-dependence as well as long-range interactions between distant residues. Overall, approximately 30% of the synthesized variants were found to bind the NCBD domain of CBP more tightly than wild-type AD1-ACTR, whereas the remaining 70% bound more weakly.For the best binding variant in the library, [1055meLeu;1076meLeu]AD1-ACTR, we obtained the first X-ray structure of this intrinsically disordered transcriptional activator in complex with NCBD. Remarkably, the conformation of the [1055meLeu;1076meLeu]AD1-ACTR variant in the complex is distinct from previously reported NMR structures and confirms that α-methylation can be used to isolate a distinct functional conformation of an IDP. As our failure to crystallize the more dynamic wild-type complex attests, crystallizing complexes resulting from the mutual interactions of IDPs can be difficult, so targeted α-methylation could conceivably become a valuable tool for structurally characterizing the complexes formed by many other IDPs. So-called “fuzzy” complexes that exhibit substantial binding affinities and specificities despite enhanced conformational dynamics would be particularly interesting in this regard.46
α-Methylation is bioorthogonal and represents an irreversible modification that leads to enhancement of polypeptide stability against degradation by proteases.47 Previously, the feasibility of biosynthetic incorporation of α-methylated amino acids into proteins via stop codon suppression using aminoacylated tRNA was demonstrated,48 therefore, the conformational editing of IDPs can in principle be performed via an expanded genetic code.49 Thus, we anticipate that modified α-methylated interacting domains of IDPs will be useful for the development of molecular probes to dissect the diverse roles of CBP/p300 and ACTR (SRC3) as well as other disordered proteins in signalling and regulation pathways.
Experimental
Protein production
Samples of wild type and α-methylated variants of ACTR activation domain, biotinylated and fluorescently-labelled constructs as well as NCBD were prepared by chemical protein synthesis (procedures are described in ESI Materials and methods; analytical data are summarized in ESI Table S2 and Fig. S25†). The construct of maltose-binding protein (MBP) tethered to NCBD was recombinantly expressed in BL21(DE3) E. coli cells, whereas CBP was expressed in Sf21 insect cells (detailed procedures are provided in ESI Materials and methods†).Biophysical characterization
CD measurements were performed in 185–280 nm wavelength range for protein samples at 25 mM concentration (ESI Materials and methods, Tables S5, S6 and Fig. S3, S4†). Thermal denaturation of protein complexes was monitored by CD using ellipticity at 222 nm as a function of temperature from 20 to 90 °C (ESI Materials and methods, Table S4 and Fig. S5†). NMR measurements were performed on a 700 MHz Bruker spectrometer equipped with a cryo-probe for 1.4 mM protein samples at 31 °C. Resonance assignment for wild type and two α-methylated variants of ACTR domain were carried out based on 1H–13C HSQC, 1H–1H TOCSY, 1H–1H NOESY, 1H–13C HSQC-TOCSY and 1H–13C HMBC experiments. R1, R2 relaxation rates were measured using standard pulse sequences. Experimental details and data analysis are provided in ESI Materials and methods, Tables S7–S11 and Fig. S6–S10.† All-atom MD simulations were carried out using GROMOS software (http://www.gromos.net) for 200 ns using force–field parameter set 54A7 (ESI Materials and methods, Tables S13, S14 and Fig. S16†).X-ray crystallography
A crystal of MBP-NCBD construct complexed with the best binder [1055meLeu;1076meLeu]AD1-ACTR was obtained by the sitting-drop vapor-diffusion method after 3 weeks of incubation and was flash-frozen in liquid nitrogen using 30% ethylene glycol solution as a cryoprotectant. Data collection was performed at the Swiss Light Source synchrotron. The crystal structure was solved by molecular replacement and refined to an R-factor of 22.6% at 2.28 Å resolution (experimental details and complete refinement statistics are provided in ESI Materials and methods and Table S12†). Coordinates of X-ray structure and associated structure factors have been deposited with the PDB under accession code 6SQC.Protein binding studies
Experimental settings and data analysis for ITC, SPR and fluorescence polarization measurements characterizing binding of wild type and α-methylated ACTR domain variants to NCBD or full length CBP are detailed in ESI Materials and methods, Tables S3, S16, S17 and Fig. S1, S2, S19, S20, S21, S22 and S23.† Pull down experiments were carried out with MCF7 cell extract using Strep-Tactin resin and the biotin conjugated best binder [1055meLeu;1076meLeu]AD1-ACTR variant as a bait. The SDS-PAGE, Western-blot and label-free quantitative mass-spectrometry were used to identify CBP (ESI Materials and methods, Fig. S24†).Conflicts of interest
There are no conflicts to declare.Acknowledgements
X-ray data collection was performed on the PXIII beamline at the Swiss Light Source synchrotron, P. Scherrer Institute, Villigen, Switzerland. We thank V. Olieric and C.-Y. Huang for their help on the beamline. We also thank IGBMC staff members P. Poussin-Courmontagne, A. McEwen and C. Birck from structural and biophysics platforms, B. Morlet from proteomics platform and Prof. S. Riniker from the ETH Zurich for providing CPU time. This work has been funded by the European Research Council (ERC-2016-StG, grant number 715062-HiChemSynPro), the LabEx CSC (ANR-10-LABX-0026_CSC), the ATIP-Avenir 2015 grant (to V. T.), the CNRS, the INSERM, the University of Strasbourg (IDEX program R701/W15RPE19), the Région Alsace and the Hôpital Civil de Strasbourg. The work was also supported by the Ligue Contre le Cancer (Équipe Labellisée 2015 to G. T.). G. G. was supported by the Post-doctorants en France program of the Fondation ARC. The authors acknowledge the support and the use of resources of the French Infrastructure for Integrated Structural Biology FRISBI ANR-10-INBS-05 and of Instruct-ERIC.Notes and references
- R. van der Lee, et al. , Chem. Rev., 2014, 114, 6589 CrossRef CAS.
- C. J. Oldfield and A. K. Dunker, Annu. Rev. Biochem., 2014, 83, 553 CrossRef CAS.
- P. E. Wright and H. J. Dyson, Nat. Rev. Mol. Cell Biol., 2015, 16, 18 CrossRef CAS.
- R. Pancsa and P. Tompa, PLoS One, 2012, 7, e34687 CrossRef CAS.
- V. N. Uversky, Biochim. Biophys. Acta, 2013, 1834, 932 CrossRef CAS.
- A. Borgia, M. B. Borgia, K. Bugge, V. M. Kissling, P. O. Heidarsson, C. B. Fernandes, A. Sottini, A. Soranno, K. J. Buholzer, D. Nettels, B. B. Kragelund, R. B. Best and B. Schuler, Nature, 2018, 555, 61 CrossRef CAS.
- S. F. Banani, H. O. Lee, A. A. Hyman and M. K. Rosen, Nat. Rev. Mol. Cell Biol., 2017, 18, 285 CrossRef CAS.
- A. K. Dunker, M. S. Cortese, P. Romero, L. M. Iakoucheva and V. N. Uversky, FEBS J., 2005, 272, 5129 CrossRef CAS.
- M. Fuxreiter, P. Tompa, I. Simon, V. N. Uversky, J. C. Hansen and F. J. Asturias, Nat. Chem. Biol., 2008, 4, 728 CrossRef CAS.
- E. Martinez, Plant Mol. Biol., 2002, 50, 925 CrossRef CAS.
- T. Nakagawa, M. Yoneda, M. Higashi, Y. Ohkuma and T. Ito, Genes Cells, 2018, 23, 808 CrossRef CAS.
- V. Perissi and M. G. Rosenfeld, Nat. Rev. Mol. Cell Biol., 2005, 6, 542 CrossRef CAS.
- J. Liu, N. B. Perumal, C. J. Oldfield, E. W. Su, V. N. Uversky and A. K. Dunker, Biochemistry, 2006, 45, 6873 CrossRef CAS.
- A. K. Dunker and V. N. Uversky, Curr. Opin. Pharmacol., 2010, 10, 782 CrossRef CAS.
- C. V. Dang, E. P. Reddy, K. M. Shokat and L. Soucek, Nat. Rev. Cancer, 2017, 17, 502 CrossRef CAS.
- T. Flock, R. J. Weatheritt, N. S. Latysheva and M. M. Babu, Curr. Opin. Struct. Biol., 2014, 26, 62 CrossRef CAS.
- L. D. Walensky and G. H. Bird, J. Med. Chem., 2014, 57, 6275 CrossRef CAS.
- A. Bhowmick, D. H. Brookes, S. R. Yost, H. J. Dyson, J. D. Forman-Kay, D. Gunter, M. Head-Gordon, G. L. Hura, V. S. Pande, D. E. Wemmer, P. E. Wright and T. Head-Gordon, J. Am. Chem. Soc., 2016, 138, 9730 CrossRef CAS.
- V. Muñoz and L. Serrano, Proteins, 1994, 20, 301 CrossRef.
- W. S. Horne and T. N. Grossmann, Nat. Chem., 2020, 12, 331 CrossRef CAS.
- N. Vo and R. H. Goodman, J. Biol. Chem., 2001, 276, 13505 CrossRef CAS.
- H. J. Dyson and P. E. Wright, J. Biol. Chem., 2016, 291, 6714 CrossRef CAS.
- J. Xu, R.-C. Wu and B. W. O'Malley, Nat. Rev. Cancer, 2009, 9, 615 CrossRef CAS.
- M. Kjaergaard, K. Teilum and F. M. Poulsen, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 12535 CrossRef CAS.
- M.-O. Ebert, S.-H. Bae, H. J. Dyson and P. E. Wright, Biochemistry, 2008, 47, 1299 CrossRef CAS.
- S. J. Demarest, M. Martinez-Yamout, J. Chung, H. Chen, W. Xu, H. J. Dyson, R. M. Evans and P. E. Wright, Nature, 2002, 415, 549 CrossRef CAS.
- R. Kaul and P. Balaram, Bioorg. Med. Chem., 1999, 7, 105 CrossRef CAS.
- M. Crisma and C. Toniolo, Biopolymers, 2014, 104, 46 CrossRef.
- B. Schmidtgall, O. Chaloin, V. Bauer, M. Sumyk, C. Birck and V. Torbeev, Chem. Commun., 2017, 53, 7369 RSC.
- J. A. Miles, D. J. Yeo, P. Rowell, S. Rodriguez-Marin, C. M. Pask, S. L. Warriner, T. A. Edwards and A. J. Wilson, Chem. Sci., 2016, 7, 3694 RSC.
- K. T. O'Neil and W. F. DeGrado, Science, 1990, 250, 646 CrossRef.
- V. Iesmantavicius, J. Dogan, P. Jemth, K. Teilum and M. Kjaergaard, Angew. Chem., Int. Ed., 2014, 53, 1548 CrossRef CAS.
- S. J. Demarest, S. Deechongkit, H. J. Dyson, R. M. Evans and P. E. Wright, Protein Sci., 2004, 13, 203 CrossRef CAS.
- M. C. Baxa, E. J. Haddadian, J. M. Jumper, K. F. Freed and T. R. Sosnick, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 15396 CrossRef CAS.
- A. Horovitz and A. R. Fersht, J. Mol. Biol., 1992, 224, 733 CrossRef CAS.
- K. Tamiola, B. Acar and F. A. A. Mulder, J. Am. Chem. Soc., 2010, 132, 18000 CrossRef CAS.
- K. Zanier, S. Charbonnier, A. O. Sidi, A. G. McEwen, M. G. Ferrario, P. Poussin-Courmontagne, V. Cura, N. Brimer, K. O. Babah, T. Ansari, I. Muller, R. H. Stote, J. Cavarelli, S. Vande Pol and G. Travé, Science, 2013, 339, 694 CrossRef CAS.
- P. Jemth, E. Karlsson, B. Vögeli, B. Guzovsky, E. Andersson, G. Hultqvist, J. Dogan, P. Güntert, R. Riek and C. N. Chi, Sci. Adv., 2018, 4, eaau4130 CrossRef CAS.
- L. Waters, B. Yue, V. Veverka, P. Renshaw, J. Bramham, S. Matsuda, T. Frenkiel, G. Kelly, F. Muskett, M. Carr and D. M. Heery, J. Biol. Chem., 2006, 281, 14787 CrossRef CAS.
- J. Zou, B. Song, C. Simmerling and D. Raleigh, J. Am. Chem. Soc., 2016, 138, 15682 CrossRef CAS.
- D. Bang, A. V. Gribenko, V. Tereshko, A. A. Kossiakoff, S. B. Kent and G. I. Makhatadze, Nat. Chem. Biol., 2006, 2, 139 CrossRef CAS.
- J. Dogan, X. Mu, Å. Engström and P. Jemth, Sci. Rep., 2013, 3, 2076 CrossRef.
- D. M. Lonard and B. W. O'Malley, Clin. Cancer Res., 2016, 22, 5403 CrossRef CAS.
- M. E. Breen and A. K. Mapp, Curr. Opin. Chem. Biol., 2018, 45, 195 CrossRef CAS.
- L. M. Lasko, et al. , Nature, 2017, 550, 128 CrossRef CAS.
- M. Fuxreiter and P. Tompa, Adv. Exp. Med. Biol., 2012, 725, 1 CrossRef CAS.
- H. M. Werner, C. C. Cabalteja and W. S. Horne, ChemBioChem, 2016, 17, 712 CrossRef CAS.
- D. Mendel, J. Ellman and P. G. Schultz, J. Am. Chem. Soc., 1993, 115, 4359 CrossRef CAS.
- J. W. Chin, Nature, 2017, 550, 53 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc04482b |
| ‡ These authors equally contributed to this work. |
| § Throughout manuscript α-methylated amino acids are abbreviated as mX or meXaa, where X and Xaa correspond to one-letter or three-letter abbreviations of the corresponding standard amino acids. |
| This journal is © The Royal Society of Chemistry 2021 |