
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnterobactin- and salmochelin-β-lactam conjugates induce cell morphologies consistent with inhibition of penicillin-binding proteins in uropathogenic Escherichia coli CFT073†
Artur
Sargun
 a,
Timothy C.
Johnstone
a,
Hui
Zhi
b,
Manuela
Raffatellu
bcd and
Elizabeth M.
Nolan
*a
a,
Timothy C.
Johnstone
a,
Hui
Zhi
b,
Manuela
Raffatellu
bcd and
Elizabeth M.
Nolan
*a
aDepartment of Chemistry, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. E-mail: lnolan@mit.edu; Tel: +1-617-452-2495
bDivision of Host-Microbe Systems and Therapeutics, Department of Pediatrics, University of California San Diego, La Jolla, CA 92093, USA
cCenter for Microbiome Innovation, University of California San Diego, La Jolla, CA 92093, USA
dChiba University-UC San Diego Center for Mucosal Immunology, Allergy, and Vaccines, La Jolla, CA 92093, USA
First published on 13th January 2021
Abstract
The design and synthesis of narrow-spectrum antibiotics that target a specific bacterial strain, species, or group of species is a promising strategy for treating bacterial infections when the causative agent is known. In this work, we report the synthesis and evaluation of four new siderophore-β-lactam conjugates where the broad-spectrum β-lactam antibiotics cephalexin (Lex) and meropenem (Mem) are covalently attached to either enterobactin (Ent) or diglucosylated Ent (DGE) via a stable polyethylene glycol (PEG3) linker. These siderophore-β-lactam conjugates showed enhanced minimum inhibitory concentrations against Escherichia coli compared to the parent antibiotics. Uptake studies with uropathogenic E. coli CFT073 demonstrated that the DGE-β-lactams target the pathogen-associated catecholate siderophore receptor IroN. A comparative analysis of siderophore-β-lactams harboring ampicillin (Amp), Lex and Mem indicated that the DGE-Mem conjugate is advantageous because it targets IroN and exhibits low minimum inhibitory concentrations, fast time-kill kinetics, and enhanced stability to serine β-lactamases. Phase-contrast and fluorescence imaging of E. coli treated with the siderophore-β-lactam conjugates revealed cellular morphologies consistent with the inhibition of penicillin-binding proteins PBP3 (Ent/DGE-Amp/Lex) and PBP2 (Ent/DGE-Mem). Overall, this work illuminates the uptake and cell-killing activity of Ent- and DGE-β-lactam conjugates against E. coli and supports that native siderophore scaffolds provide the opportunity for narrowing the activity spectrum of antibiotics in clinical use and targeting pathogenicity.
Introduction
Infections caused by Gram-negative bacteria can be difficult to treat due to the semipermeable outer-membrane (OM) that serves as an efficient barrier against most antibiotics.1,2 The overuse of broad-spectrum antibiotics in the treatment of such bacterial infections facilitates the selection of resistant strains, which causes antibiotics to lose effectiveness over time.3,4 Moreover, broad-spectrum antibiotics can damage the commensal microbiota and trigger life-threatening secondary infections such as those caused by Clostridioides difficile.5,6 Coupled with the scarcity of new antibiotics in the drug pipeline, society is faced with the reality that bacterial infections that were once of minor concern can become lethal.7 To address this emerging public health crisis, novel therapies that exhibit narrow-spectrum activity must be developed and implemented in combination with rapid diagnostics.8–11 Such pathogen-selective antibiotics are predicted to reduce the occurrence of antibiotic resistance in microbial populations and have smaller impact on the host microbiome.To date, various strategies of narrowing the activity spectrum of antibiotics in clinical use have been explored.9,12–14 In this regard, essential nutrient transporters located in the OM of Gram-negative bacteria provide opportunities for selective recognition and intracellular delivery of antibacterial molecules.15,16 Iron is an essential nutrient for the vast majority of bacterial species; thus, acquiring adequate levels of iron is important for survival and host colonization.17–19 To scavenge iron from the host, many bacteria biosynthesize small-molecule Fe3+ chelators called siderophores. These secondary metabolites are biosynthesized in the cytoplasm, exported to the extracellular space to coordinate Fe3+ and then returned to the bacterial cell via specialized OM receptors.20–23 Several decades of work showed that the siderophores and siderophore uptake machinery of Gram-negative bacteria can be leveraged to deliver toxic cargos into the bacterial periplasm and cytoplasm.15,24–33 Recent examples demonstrated that the activity of broad-spectrum antibiotics (e.g. β-lactams and fluoroquinolones) can be narrowed by targeting select species or a group of strains within a species through covalent attachment to a siderophore.34–36
Enterobactin (Ent) is a tris-catecholate siderophore produced by Gram-negative species including Escherichia coli, Salmonella enterica and Klebsiella pneumoniae.37,38 The high Fe3+-binding affinity of Ent (Ka ∼ 1049 M−1)39,40 allows bacteria to scavenge Fe3+ from the host environment in the form of [Fe(Ent)]3−.41 In E. coli, [Fe(Ent)]3− is recognized by the OM receptors FepA and IroN, and is transported into the periplasm utilizing the energy provided by the TonB–ExbB–ExbD complex (Fig. S1†). In addition to producing Ent, a number of Gram-negative pathogens including uropathogenic E. coli and Salmonella enterica also biosynthesize salmochelins, C-glucosylated analogs of Ent. For instance, salmochelin S4, hereafter named diglucosylated Ent (DGE), is a C5,C5′-diglucosylated analogue of Ent.42 DGE is deployed by pathogenic bacteria to acquire iron and evade the Ent-scavenging host-defense protein lipocalin-2.43–46 [Fe(DGE)]3− is transported into the periplasm by IroN.43,47 Whereas all E. coli produce Ent and express FepA, the production of salmochelins and expression of IroN are mostly pathogen-associated and require the iroA gene cluster.42,47
In prior work, we attached the aminopenicillins ampicillin (Amp) and amoxicillin (Amx) to the Ent scaffold via a polyethylene glycol (PEG3) linker installed at the C5 position of one catechol ring.34 These Ent-β-lactam conjugates afforded 100–1000-fold increases in antimicrobial activity against E. coli expressing FepA compared to the unmodified β-lactams. We then demonstrated that C-glucosylation of the two remaining catechol rings, yielding DGE-β-lactam conjugates, afforded selective killing of E. coli strains that harbor the iroA gene cluster.35 The antibacterial activity of the Ent/DGE-β-lactam conjugates was attributed to the β-lactam cargos because β-lactam hydrolysis of the Ent congeners abrogated this activity.34
Although it is accepted that the β-lactam cargo of siderophore-β-lactam conjugates is responsible for bacterial cell death, studies addressing the mechanism of antimicrobial activity of siderophore-β-lactam conjugates are limited.48–50 Generally, β-lactam antibiotics enter Gram-negative bacteria via OM porins, bind to penicillin-binding proteins (PBPs) in the periplasm and thereby inhibit cell wall (CW) biosynthesis.51,52 In bacteria, there are numerous PBP enzymes, which are classified according to their enzymatic function. In E. coli, class A PBPs such as PBP1a and PBP1b catalyze both transglucosylation (polymerization) of the peptidoglycan and transpeptidation (cross-linking) of the glycan strands. Class B enzymes such as PBP2 and PBP3 are monofunctional transpeptidases with roles in elongation and cell division, respectively. Class C enzymes, which include PBP4, PBP4b, PBP5, PBP6, PBP6b, and AmpH, carry out various peptidase reactions that facilitate maturation, remodeling, and metabolism of PG.53,54 Consequently, inhibition of essential PBPs by β-lactams induces a diverse array of cellular morphologies, reflecting the roles that the enzymes play in coordinating cell division and elongation. Thus, studying the cellular morphologies that occur as a result of exposing E. coli to siderophore-β-lactam conjugates is likely to provide insight into the observed antibacterial activity and cellular target(s).
In this work, we report the synthesis of four new siderophore-β-lactam conjugates harboring the antibiotics cephalexin (Lex) and meropenem (Mem) (Fig. 1). These conjugates are based on our original Ent/DGE-β-lactam design (e.g. Ent/DGE-Amp) and extend the scope of antibiotic cargos that can be effectively attached to Ent/DGE. These molecules show siderophore receptor-dependent antibacterial activity against E. coli. Importantly, Ent-Mem also retains antibacterial activity against a serine β-lactamase-producing strain. For the first time, we obtain microscopic evidence that Ent/DGE-β-lactam conjugates exert antibacterial activity by inhibiting essential PBP enzymes. This study further demonstrates that the Ent and DGE scaffolds can render broad-spectrum antibiotics selective towards pathogenic bacteria on the basis of siderophore-uptake machineries.
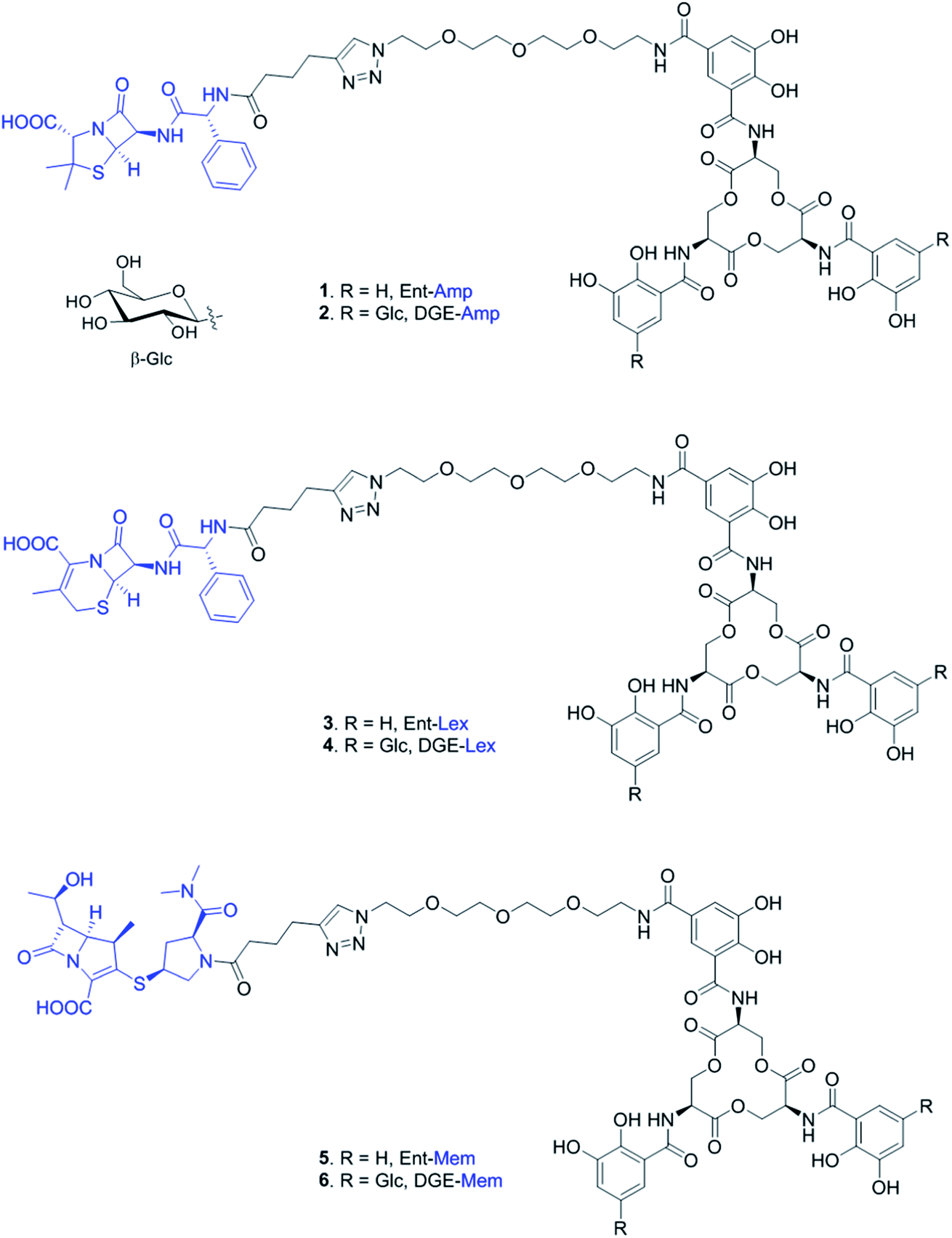 | ||
| Fig. 1 Chemical structures of the Ent- and DGE-β-lactam conjugates with Amp (1, 2), Lex (3, 4), and Mem (5, 6). Glc, glucose. The curved line on the β-glycosidic bond of glucose (β-Glc) represents the point of attachment to the catechol. | ||
Results and discussion
Design and synthesis of Ent/DGE-β-lactam conjugates harboring Lex and Mem
We prepared and evaluated the Ent/DGE conjugates 3–6 carrying two additional broad-spectrum β-lactam antibiotics, Lex and Mem (Fig. 2A). Lex is a first-generation cephalosporin analog of Amp, whereas meropenem is a carbapenem with enhanced resistance to extended-spectrum β-lactamases and cephalosporinases.55–60 From a structural standpoint, Amp, Lex and Mem differ by the size and nature of the ring fused with the β-lactam core: thiazolidine (Amp), dihydrothiazine (Lex), and thiazoline (Mem) (Fig. 1). Compared to Amp, the 4,6-fused ring system of Lex provides enhanced stability to acid hydrolysis,55,61 whereas the 4,5-ring system of Mem, in which the endocyclic S atom of the 5-membered ring is replaced by C, confers superior resistance to serine β-lactamases.58–60 Additionally, these structural differences (i) determine the rate with which Amp, Lex, and Mem diffuse through bacterial porins62 and (ii) induce variations in β-lactam reactivity.61The syntheses of conjugates 3–6 were carried out in one step from either Ent-PEG3-N310 or DGE-PEG3-N311 and one of the acylated β-lactams 7–9, using the methods developed for the assembly of Ent/DGE-Amp/Amx (Schemes 1 and S1†).34,35 Each β-lactam was acylated with hex-5-ynoyl chloride to provide the alkyne functionality for the coupling reactions with azides 10 and 11 (Scheme S2†). The preparation of Lex-alkyne 8 was similar to that developed for Amp-alkyne 7 due to the comparable hydrophilicities of Amp and Lex (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) PAmp = 1.35, logPLex = 0.65),63–65 which allows for an efficient extraction of 7 and 8 from acidified solution. Because Mem is more hydrophilic and difficult to extract from an aqueous phase (logP = −0.6),66 the acylation reaction of Mem was optimized to avoid acid-catalyzed hydrolysis of Mem-alkyne 9 during extraction from an acidic solution. Thus, the water–acetone system employed for preparation of Amp/Lex-alkyne 7, 8 was replaced by MeOH. Removal of MeOH under reduced pressure eliminated the need for extraction from acidified solvent, which avoided hydrolysis of Mem-alkyne 9 during workup.
PAmp = 1.35, logPLex = 0.65),63–65 which allows for an efficient extraction of 7 and 8 from acidified solution. Because Mem is more hydrophilic and difficult to extract from an aqueous phase (logP = −0.6),66 the acylation reaction of Mem was optimized to avoid acid-catalyzed hydrolysis of Mem-alkyne 9 during extraction from an acidic solution. Thus, the water–acetone system employed for preparation of Amp/Lex-alkyne 7, 8 was replaced by MeOH. Removal of MeOH under reduced pressure eliminated the need for extraction from acidified solvent, which avoided hydrolysis of Mem-alkyne 9 during workup.
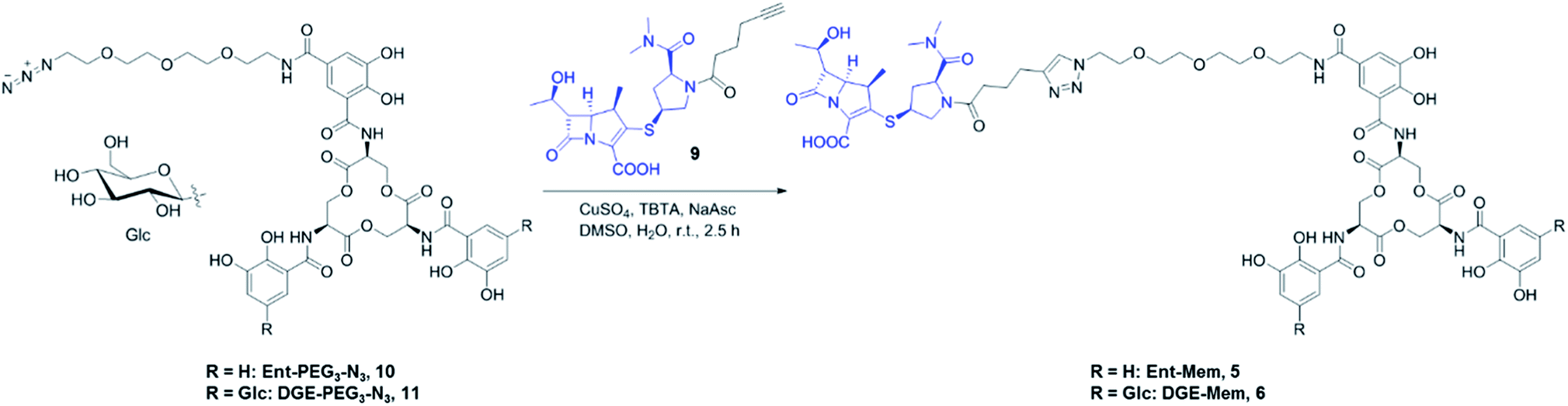 | ||
| Scheme 1 Synthesis of Ent/DGE-Mem 5, 6. The curved line on the β-glycosidic bond of glucose (β-Glc) represents the point of attachment to the catechol. Syntheses for the Amp and Lex conjugates are provided in Scheme S1.† | ||
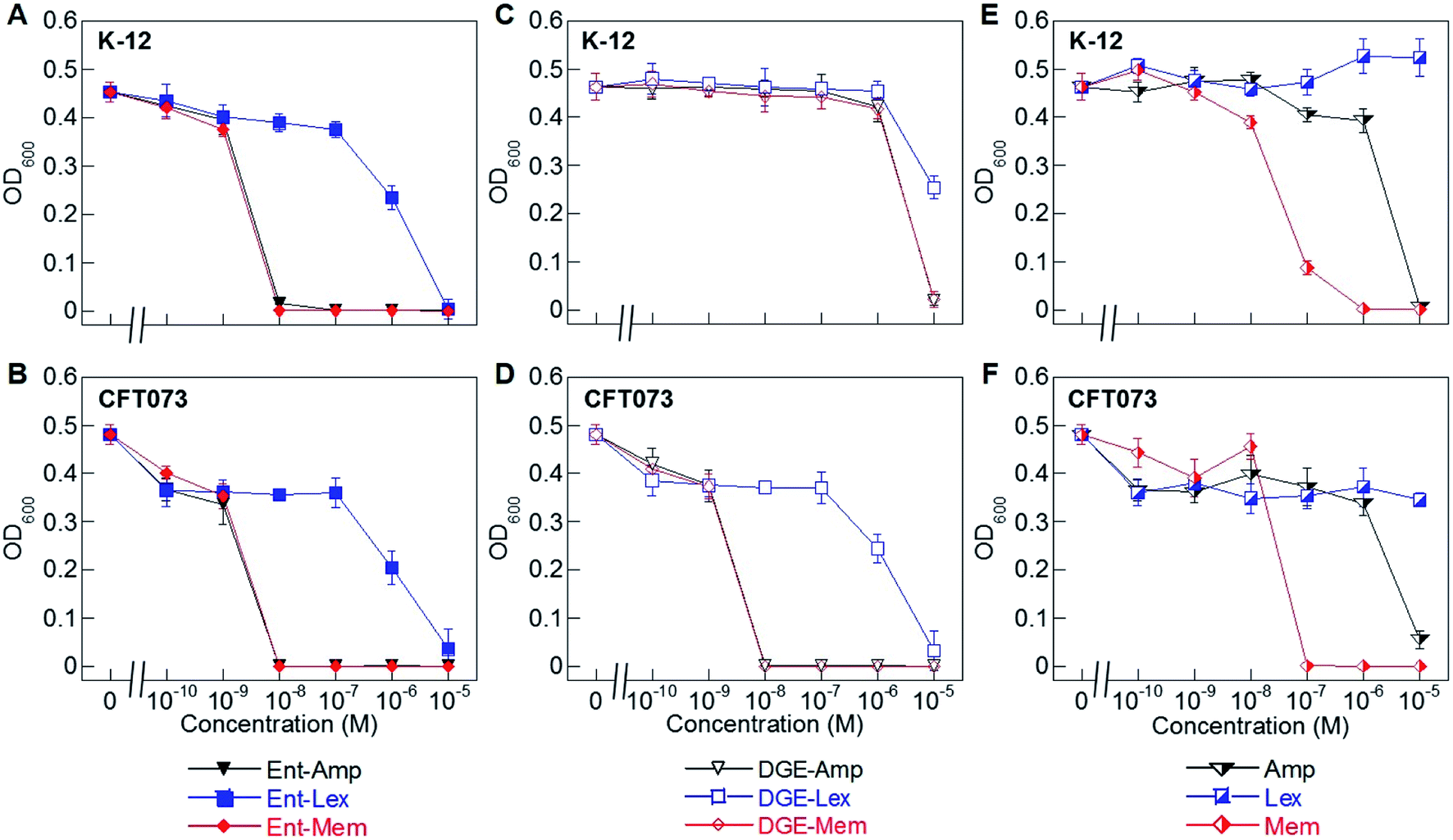 | ||
| Fig. 2 Antibacterial activity of Ent/DGE-β-lactam conjugates 1–6 against E. coli K-12 and CFT073. (A) and (B) Activity of Ent-Amp/Lex/Mem 1, 3, and 5; (C) and (D) activity of DGE-Amp/Lex/Mem 2, 4, and 6; (E) and (F) activity of the parent antibiotics Amp, Lex and Mem. All assays were performed in modified M9 medium (20 h, 30 °C; mean ± standard deviation, n = 3). Data for E. coli UTI89 are presented in Fig. S2.† A summary of all MIC values for conjugates 1–6 is provided in Table S4.† | ||
The resulting β-lactam alkynes 7–9 were coupled to Ent- or DGE-PEG3-N310 and 11via copper-catalyzed azide–alkyne cycloaddition (Schemes 1 and S1†). TBTA (tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine) was used in all cycloaddition reactions to chelate copper and thereby prevent copper-mediated decomposition of the β-lactam ring.34,35 The azide–alkyne cycloaddition reactions yielded 40–70% Ent/DGE-β-lactam conjugates on a milligram scale.
Conjugation to Ent/DGE enhances the antimicrobial activity of parent β-lactams and affords selectivity towards pathogenic E. coli independent of β-lactam structure
To evaluate the antimicrobial activity (AMA) of the Ent/DGE-β-lactam conjugates against E. coli, we employed three E. coli strains: K-12, CFT073, and UTI89. E. coli K-12 (ref. 67) is a non-pathogenic laboratory strain that lacks the iroA gene cluster and relies on FepA for Ent-mediated iron uptake. Uropathogenic E. coli CFT073 (ref. 68–70) and UTI89 (ref. 71) harbor the iroA gene cluster and thus biosynthesize DGE and express IroN.72–75 CFT073 also harbors the iha gene, which encodes an outer membrane receptor, Iha, that has been shown to transport [Fe(Ent)]3−.76,77We performed AMA assays using a 10-fold dilution series to compare the AMA of Ent/DGE-Amp/Lex/Mem 1–6 and the parent antibiotics Amp/Lex/Mem (Fig. 2A–F and S2†). In these assays, the bacteria were treated with the apo siderophore-β-lactam conjugates, which can scavenge iron from the culture medium. These assays were conducted in modified M9 medium that provides iron-limiting conditions (∼0.3 μM Fe, Table S2†), causing E. coli to express the Ent/DGE uptake machineries.
Under iron limitation, the three E. coli strains were more susceptible to Ent-Amp/Lex/Mem (Fig. 2A, B and S2†) than to the corresponding parent antibiotics (Fig. 2E and F). Ent-Amp 1 showed minimum inhibitory concentration (MIC) values that were lower than Amp (10−5 M) by 1000-fold for both K-12 and CFT073, and by 100-fold for UTI89 (Fig. S2†). These findings are in overall agreement with our prior results obtained in MHB medium supplemented with the iron chelator 2,2′-bipyridine (Bpy).34 Ent-Mem 5 afforded an MIC value of 10−8 M in K-12, CFT073 and UTI89 (Fig. 2A, B and S2†), which represents a 10-fold reduction when compared to Mem (10−7 M) (Fig. 2E and F). In contrast to the Ent-Amp and -Mem conjugates, Ent-Lex 3 exhibited a relatively modest enhancement in the AMA compared to parent Lex (Fig. 2A, B and S2†). This fact may be attributed to the lower potency of Lex against the strains employed in this study.78 It is also possible that the cell filamentation readily induced by Lex (vide infra) may impact the relationship between OD600 and cell viability.
The AMA of the DGE-β-lactams 2, 4 and 6 paralleled that of the corresponding Ent-β-lactams against E. coli CFT073 (Fig. 2D) and UTI89 (Fig. S2†). For instance, in CFT073 and UTI89, DGE-Mem 6 afforded an MIC value of 10−8 M, and the MIC values of DGE-Amp 2 were 10−8 M in CFT073 and 10−7 M in UTI89, whereas DGE-Lex 4 showed a modest MIC value (10−5 M) consistent with that of Ent-Lex 3 (Fig. 2B, D and S2†). In contrast, the DGE-β-lactam conjugates, especially DGE-Amp 2 and DGE-Mem 6, exhibited attenuated AMA against K-12 (Fig. 2C). This result agrees with our prior observations for DGE-Amp/Amx35 and, taken with the results for CFT073, indicates that expression of IroN is required for DGE-β-lactam conjugates to exert AMA. We attribute the growth inhibition observed at 10−5 M DGE-Amp/Lex/Mem to iron limitation that results from DGE-β-lactams sequestering Fe3+ in the growth medium.
Ent-β-lactam conjugates retain β-lactamase stability of parent β-lactams
Hydrolysis of β-lactams by β-lactamases is a major mechanism of resistance to these antibiotics.51,79–81 Of the three β-lactams considered in this work, Mem is the most resistant to β-lactamases as it is only a substrate for carbapenemases (metallo-β-lactamases).59,60,82–84 In contrast, Amp and Lex are readily hydrolyzed by serine β-lactamases.85,86 To determine whether conjugation to the Ent scaffold affects the susceptibility of the β-lactam cargos to hydrolysis by a serine β-lactamase, we evaluated the AMA of Ent-Amp/Lex/Mem 1, 3, 5 against E. coli ATCC 35218, a strain that expresses a class A serine β-lactamase (Fig. 3A and B). As expected, neither Amp nor Lex displayed AMA against E. coli ATCC 35218, whereas Mem retained its activity (MIC = 10−7 M) (Fig. 3B). In agreement with prior work,34 Ent-Amp 1 exhibited negligible AMA against ATCC 35218 (Fig. 3A). Similarly to Ent-Amp 1, Ent-Lex 3 showed no AMA against E. coli ATCC 35218 (MIC > 10−5 M) (Fig. 3A and B). In contrast, Ent-Mem 5 retained potent AMA against E. coli ATCC 35218, showing a 10-fold enhancement of the MIC value relative to Mem (10−8 M and 10−7 M, respectively) (Fig. 3A and B). Thus, conjugation of Mem to Ent has negligible effect on the stability of the warhead to a class A serine β-lactamase. A genome search indicated that E. coli ATCC 35218 does not harbor iroN. In agreement with this analysis, none of the DGE-β-lactams inhibited the growth of this strain over the concentration range tested, presumably due to lack of uptake (Table S4†). Taken together with the potent AMA of the Ent/DGE-Mem conjugates 5, 6, we conclude that Mem would be the most promising broad-spectrum β-lactam cargo for further in vitro and in vivo studies.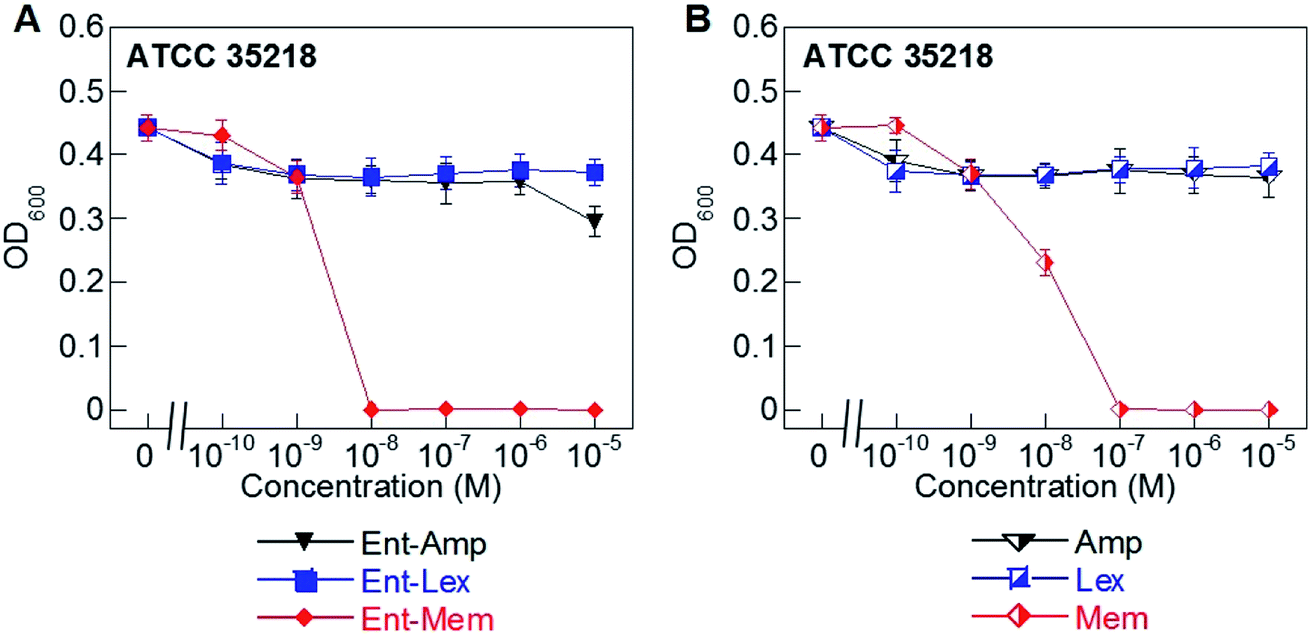 | ||
| Fig. 3 Antibacterial activity of (A) Ent-Amp 1, -Lex 3, and -Mem 5 and (B) Amp, Lex, and Mem against the class A serine β-lactamase producer E. coli ATCC 35218. Assays were performed in modified M9 medium (20 h, 30 °C; mean ± standard deviation, n = 3). A summary of all MIC values, including those for the DGE-β-lactam conjugates, are provided in Table S4.† | ||
Ent/DGE-β-lactam conjugates are transported across the OM by Ent and salmochelin receptors
To ascertain the contributions of the Ent and salmochelin OM transporters for the uptake of the Ent/DGE-β-lactam conjugates, we examined the AMA of Ent/DGE-β-lactam conjugates 1–6 against E. coli CFT073. We selected E. coli CFT073 as a case study because this strain exhibits high susceptibility to all six conjugates and expresses FepA, IroN, and Iha.76,77 We took a genetic approach using six OM and IM receptor mutants. Namely, mutants in fepA, iroN, fepA iroN, and fepA iroN iha were employed to probe conjugate recognition and transport across the OM (Fig. 4 and S3†). A fepC mutant (ATPase) and a fepDG double mutant (IM translocase) were used to investigate whether transport across the IM affects the AMA of the Ent/DGE-β-lactams (Fig. S4†).AMA assays under iron limitation revealed that single deletion of either fepA or iroN has negligible effect on the AMA of Ent-Mem 5 (Fig. 4A). This result is in agreement with our expectations and is explained by uptake of the Ent-based conjugate through both FepA and IroN.87 In the case of DGE-Mem 6, deletion of fepA had no effect on the MIC value, whereas deletion of iroN attenuated the AMA by 1000-fold (Fig. 4B). This result indicates that IroN is essential for transport of the DGE-β-lactam conjugates. Overall, these results are in agreement with prior siderophore competition studies that indicated that both FepA and IroN provide transport of Ent-Amp/Amx into the periplasm, whereas DGE-Amp/Amx can only be transported through IroN.35
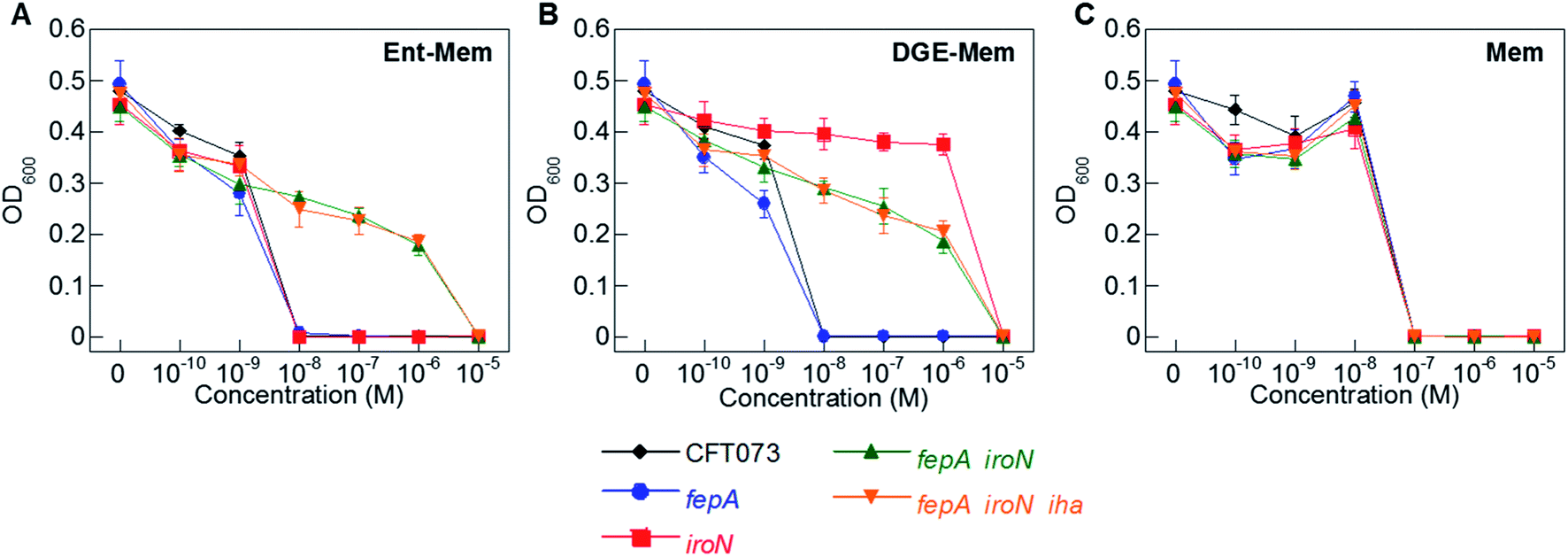 | ||
| Fig. 4 Antibacterial activity of (A) Ent-Mem 5, (B) DGE-Mem 6 and (C) unmodified Mem against uropathogenic E. coli CFT073 wild-type and its isogenic OM receptor mutants in fepA, iroN, fepA iroN, and fepA iroN iha. All assays were performed in modified M9 medium (20 h, 30 °C; mean ± standard deviation, n = 3). The AMA plots for the OM receptor mutants treated with Ent/DGE-Amp/Lex 1–4 are shown in Fig. S3,† whereas the IM receptor mutants fepC and fepDG are shown in Fig. S4.† | ||
The AMA of both Ent- and DGE-Mem conjugates 5 and 6 (Fig. 4A and B) was attenuated against a fepA iroN double mutant as well as a fepA iroN iha triple mutant. Moreover, the similar susceptibility of both mutants to the conjugates suggests that Iha is not involved in conjugate transport. The origin of the residual AMA for Ent/DGE-Mem is unclear. One possible explanation is that these mutant strains are more susceptible to conjugate-mediated Fe3+ sequestration from the growth medium compared to the parent strain because they lack multiple siderophore receptors. Alternatively, E. coli CFT073 might express an additional OM receptor that can transport the Ent/DGE conjugates across the membrane (e.g. IreA),88 albeit less efficiently than FepA and IroN. Lastly, it is also possible that CFT073 secretes hydrolases that linearize Ent/DGE and subsequently imports the resulting iron-bound hydrolyzed conjugates into the periplasm via Cir or Fiu. These possibilities warrant further investigation, which could uncover yet-unknown pathways of Ent/DGE-β-lactam uptake that explain the increased sensitivity of CFT073.
As expected, unmodified Mem exhibited potent AMA (MIC of 10−7 M) against the six CFT073 OM mutants (Fig. 4C). This activity is consistent with porin-mediated uptake of unmodified β-lactams, which is unaffected by siderophore receptor expression.
Next, we investigated whether conjugate transport into the cytoplasm affects the antibacterial activity of Ent/DGE-Mem 5 and 6. Both the carrier and the cargo moieties of the Ent- and DGE-β-lactam conjugates have binding partners in the periplasm. The β-lactam warheads target PBPs and form covalent acyl-enzyme species with an active site serine residue, whereas the siderophore moiety can be captured by FepB for delivery to the IM transporter FepCDG. To determine whether the presence of FepCDG influences the antibacterial activity of the conjugates, we examined the susceptibility of a fepC mutant and a fepDG mutant. Neither deletion of fepC nor fepDG affected the antibacterial activity of the Ent- and DGE-Mem 5, 6 conjugates (Fig. S4†). These results indicate that, if any transport of the conjugates through FepCDG occurs, this process has negligible impact on AMA against E. coli CFT073. Therefore, we presume that the majority of the molecules are being trapped in the periplasm following covalent attachment to the PBPs. This conclusion agrees with our prior analysis of the E. coli K-12 fepC mutant, which showed comparable susceptibility to Ent-Amp/Amx as the parent strain.34
Siderophore conjugation accelerates the killing of E. coli to a greater extent for Amp than for Mem
In previous studies, we found that conjugation of Amp/Amx to either Ent or DGE accelerated the rate of killing of E. coli CFT073 and UTI89 relative to the unmodified β-lactams.34,35 We therefore sought to determine whether siderophore modification also enhances the time-kill kinetics of other β-lactams, and compared the time-kill kinetics of the conjugates with the lowest MIC values, Ent/DGE-Amp 1, 2 and Ent/DGE-Mem 5, 6, against E. coli CFT073.Of the two β-lactams, Mem is more hydrophilic than Amp (vide supra for logP values) and is transported by OM porins more readily.89–92 Indeed, the time-kill curves of Amp (Fig. 5A) and Mem (Fig. 5B) show that Mem reduces OD600 of E. coli CFT073 almost to baseline within 1 h, whereas Amp requires 5 h to reach a similar OD600 value. Conjugation to the Ent/DGE scaffolds changes the uptake pathway of the β-lactam warhead from passive diffusion through porins to TonB-powered active transport, which results in acceleration of killing for Ent/DGE-Amp 1, 2. Consequently, Ent/DGE-Amp/Mem 1, 2, 5, and 6 reduce the OD600 of CFT073 to almost baseline within 1–2 h (Fig. 5A and B). Hence, these results suggest that the rate of bacterial cell killing will be accelerated to a greater extent for more lipophilic β-lactams such as Amp.
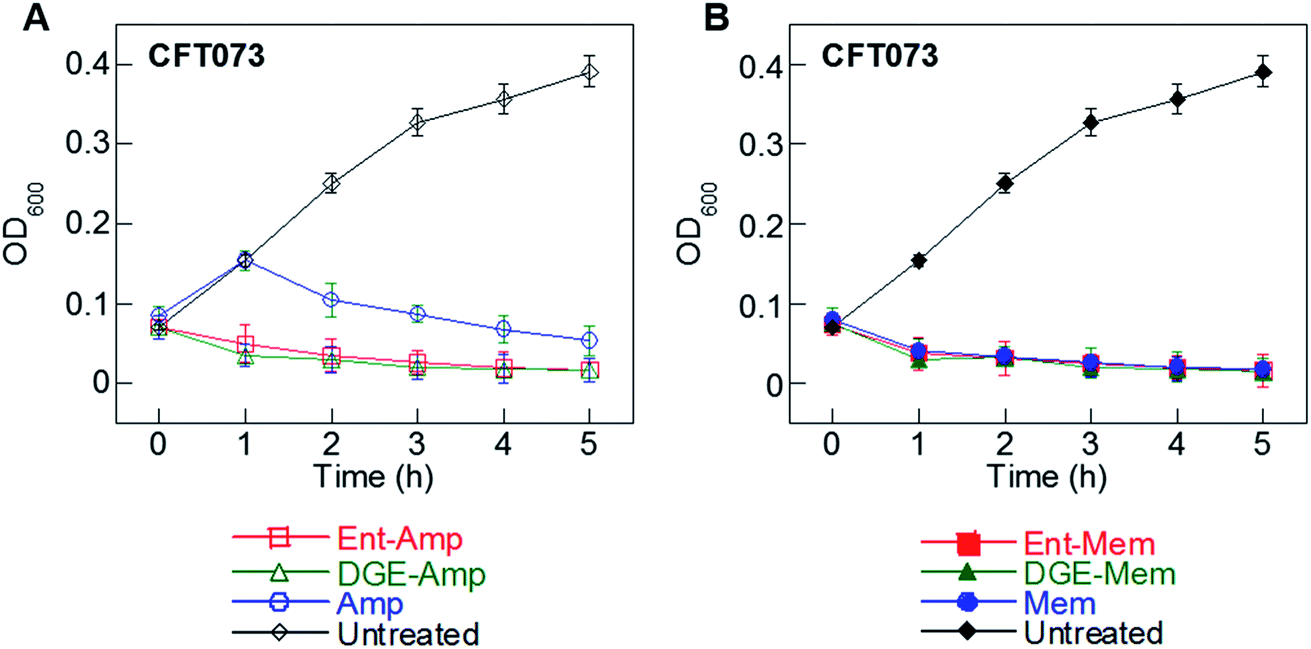 | ||
| Fig. 5 Time-kill kinetics of (A) Ent/DGE-Amp 1, 2 and (B) Ent/DGE-Mem 5, 6 against E. coli CFT073 (∼108 CFU mL−1) treated with 50 μM Ent/DGE-β-lactams and 100 μM unmodified Amp/Mem. All assays were performed in modified M9 medium (37 °C; mean ± standard deviation, n = 3). | ||
Ent/DGE-β-lactam conjugates selectively kill E. coli in co-culture with S. aureus
To investigate species selectivity of the new Ent-β-lactams, we performed mixed-species assays to ascertain if the conjugates selectively kill E. coli CFT073 in co-culture with another bacterial species. We employed phase-contrast microscopy and selected to co-culture E. coli CFT073 with the Gram-positive pathogen Staphylococcus aureus ATCC 25923. The cell shape of E. coli (rod) is distinct from the cell shape of S. aureus (coccus), which facilitates microscopic differentiation of these species. Since S. aureus did not grow in the modified M9 medium, we performed these assays in 50% MHB containing 100 μM Bpy, which provides iron limitation and promotes expression of the siderophore transport machinery. The co-cultures were treated with MIC doses of the Ent/DGE-β-lactams or parent antibiotics adjusted with respect to the cell density used in each imaging experiment. In the co-cultures treated with the unmodified antibiotics (Fig. S6†), both E. coli and S. aureus were killed. In contrast, the Ent/DGE-β-lactam conjugates selectively killed E. coli as evidenced by the presence of cocci and absence of rod-shaped bacteria remaining in cultures following treatment (Fig. 6 and S7†). Together with LIVE/DEAD viability assays (vide infra, Fig. S8†), these results indicate that Ent/DGE conjugation narrows the AMA spectrum of Amp, Lex and Mem and attenuates the AMA of these β-lactams against S. aureus. The latter outcome is in agreement with our previous studies on HardyCHROM™ UTI plates that showed both E. coli selective killing and attenuated AMA against S. aureus ATCC 25923 for Ent-Amp/Amx.34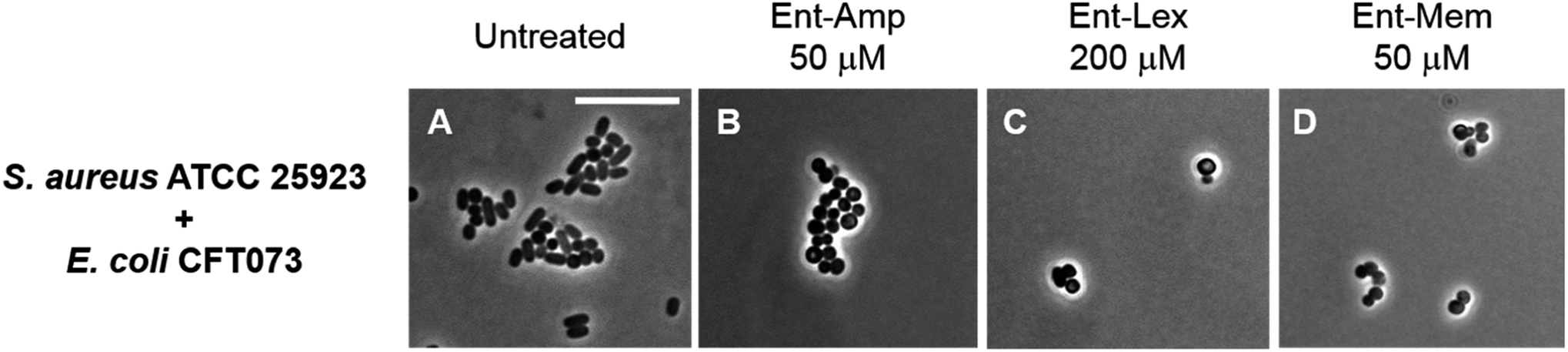 | ||
| Fig. 6 Phase-contrast micrographs of mixed cultures of E. coli CFT073 (rods) with S. aureus ATCC 25923 (cocci) treated with MIC doses of Ent-β-lactam conjugates. All cell cultures were incubated with the conjugates in 50% MHB in the presence of 100 μM Bpy for 20 h at 30 °C prior to the microscopy imaging. Scale bar: 10 μm. The micrographs of the mixed cultures treated with DGE-β-lactam conjugates are provided in Fig. S7.† | ||
Ent/DGE-Amp/Lex induce filamentous growth, whereas Ent/DGE-Mem cause formation of spheroplasts, in E. coli
The three β-lactam antibiotics used as cargos in this study target different class B high-molecular-weight PBPs when administered at sub-MIC doses, inducing different bacterial cell shapes as a result of inhibition of the corresponding PBPs.53,54 Amp and Lex primarily bind to PBP3 that participates in cell division,93–96 whereas Mem targets PBP2 that participates in cell elongation.93,97,98 Consequently, at sub-MIC quantities, Amp and Lex induce bacterial cell filamentation, whereas Mem induces the formation of spheroplasts, which are CW-deficient cells. At quantities equal to or greater than the MIC, all three β-lactams induce cell lysis due to concomitant inhibition of multiple PBPs.54,99,100 To further examine the consequences of Ent- and DGE-β-lactam treatment on E. coli and thereby inform mechanism, we employed phase-contrast microscopy to visualize the cellular morphologies caused by these conjugates (Fig. 7).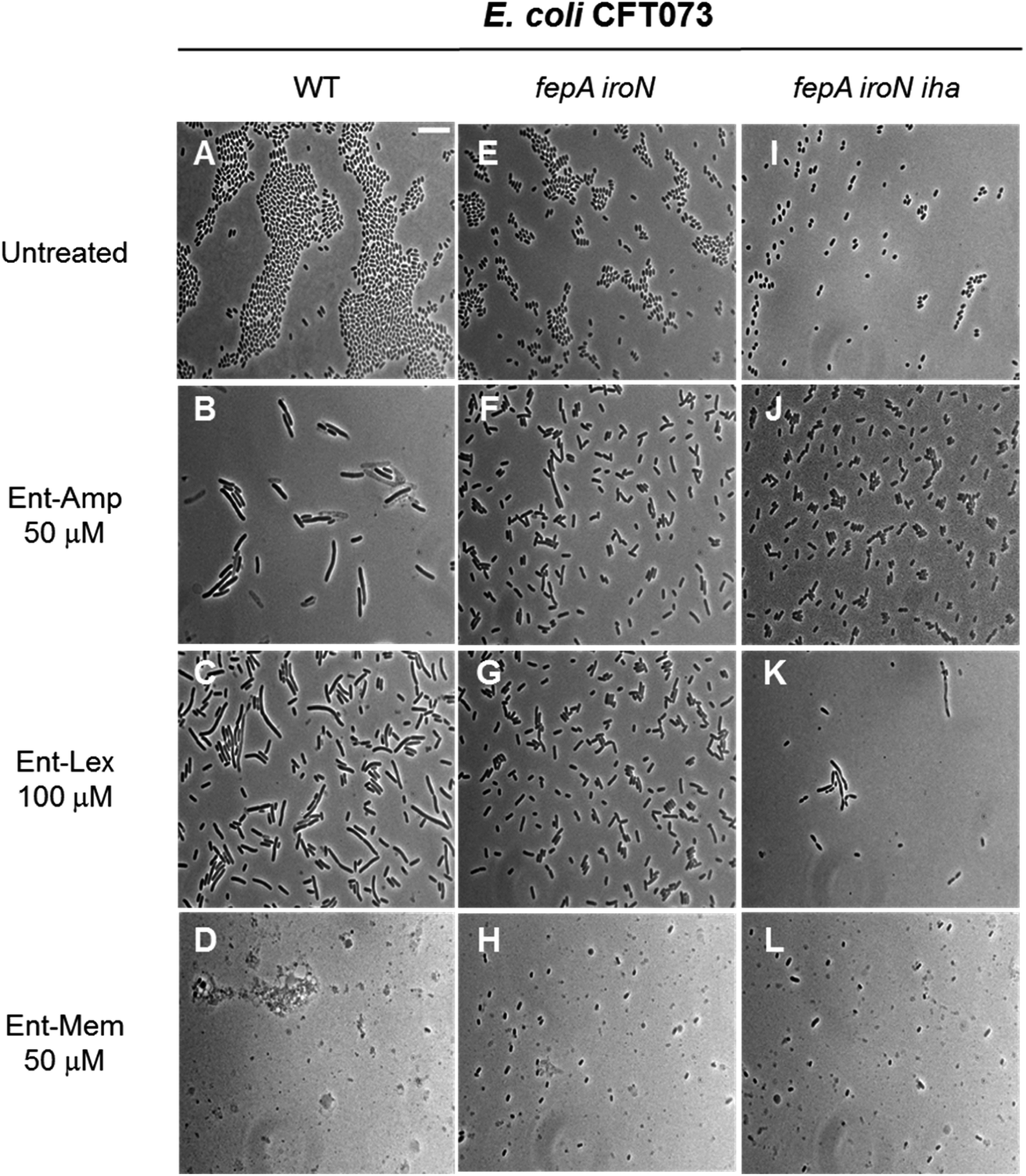 | ||
| Fig. 7 Full-field view micrographs of E. coli CFT073 WT (A–D) and the indicated outer-membrane receptor mutants (E–L) treated with sub-MIC amounts of Ent-β-lactams in modified M9 medium for 20 h at 30 °C. Scale bar: 10 μm. The micrographs of E. coli and outer-membrane receptor mutants treated with sub-MIC amounts of DGE-β-lactams are shown in Fig. S9.† | ||
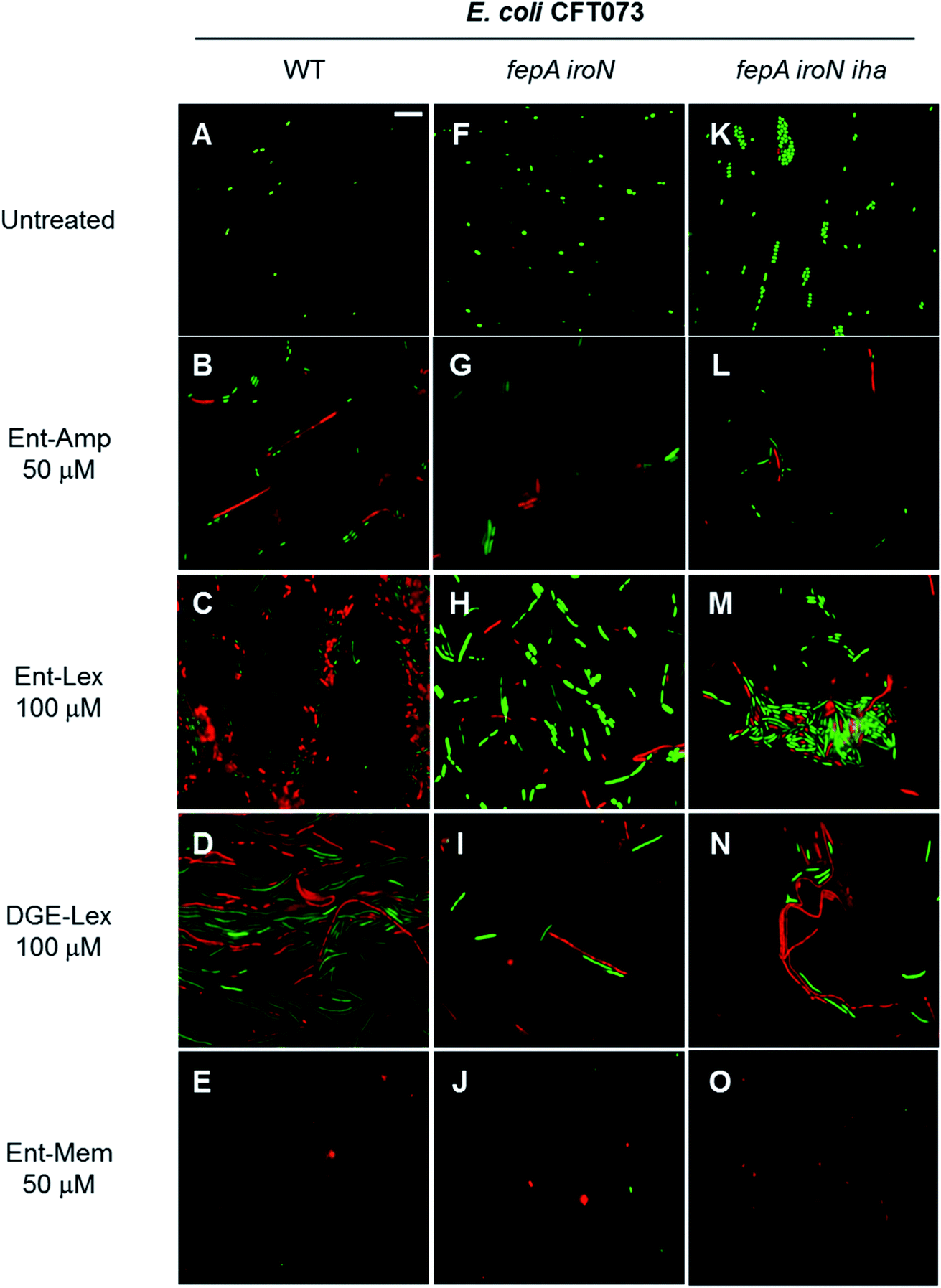
E. coli CFT073 and its fepA iroN and fepA iroN iha mutants were incubated with the Ent-Amp/Lex/Mem conjugates 1, 3, 5 at a high cell density (∼108 CFU mL−1) to facilitate microscopic visualization of the bacterial cells. To probe the effect of the conjugates at sub-MIC quantities, the bacteria were treated with 50 μM of Ent-Amp/Mem 1 and 5 and 100 μM of Ent-Lex 3. Significant cell elongation was observed for E. coli CFT073 treated with Ent-Amp 1 and Ent-Lex 3 (Fig. 7B and C) compared to the untreated control (Fig. 7A). In contrast, E. coli CFT073 cells treated with 50 μM Ent-Mem 5 transformed into round or ovoid-shaped spheroplasts (Fig. 7D). These observations are consistent with the inhibition of the primary PBP targets of the three β-lactams. Treatment of the double and triple mutants fepA iroN and fepA iroN iha with sub-MIC concentrations of the Ent-β-lactam conjugates showed that some of the cells exhibit slight elongation following exposure to Ent-Amp/Lex 1, 3 (Fig. 7F, G, J and K) and that some cells form spheroplasts following exposure to Ent-Mem 5 (Fig. 7H and L). The observation of these morphologies is consistent with the weak AMA observed for these mutants in the AMA assays (Fig. 4), which might suggest the existence of alternative pathways of Ent-β-lactam uptake in E. coli CFT073.
When the DGE-β-lactam conjugates 2, 4, and 6 were added to bacteria at sub-MIC doses, similar morphologies were observed as for the cells treated with Ent-β-lactams 1, 3, and 5 (Fig. S9†). Thus, the β-lactam warhead induces specific morphological changes to the bacterial cell shape independent of the carrier to which it is attached. Curiously, the triple-mutant fepA iroN iha appears to be more sensitive to the action of DGE-β-lactams (Fig. S9J–L†) than to the corresponding Ent-β-lactams (Fig. 7J–L). Further investigation into this finding may uncover additional mechanisms of Ent/DGE uptake and better explain the enhanced sensitivity of E. coli CFT073 to the Ent/DGE conjugates.
Next, we treated E. coli CFT073 with an estimated MIC dose of Ent-Amp or DGE-Amp (100 μM) determined by extrapolating to ∼108 CFU mL−1 of the MIC values obtained in the AMA assays. This concentration of the conjugates resulted in cell lysis, indicated by the cell debris observed in the micrographs (Fig. 8B and C). Moreover, the presence of enlarged cells in the bacterial cultures treated with Ent/DGE-Amp 1, 2 (Fig. 8B and C) is consistent with the concomitant inhibition of multiple essential PBPs by Amp at doses that are equal or exceeding the MIC.54 Similar cell shapes and debris were observed at MIC doses of Ent/DGE-Lex 3, 4 and Ent/DGE-Mem 5, 6 (Fig. S10†).
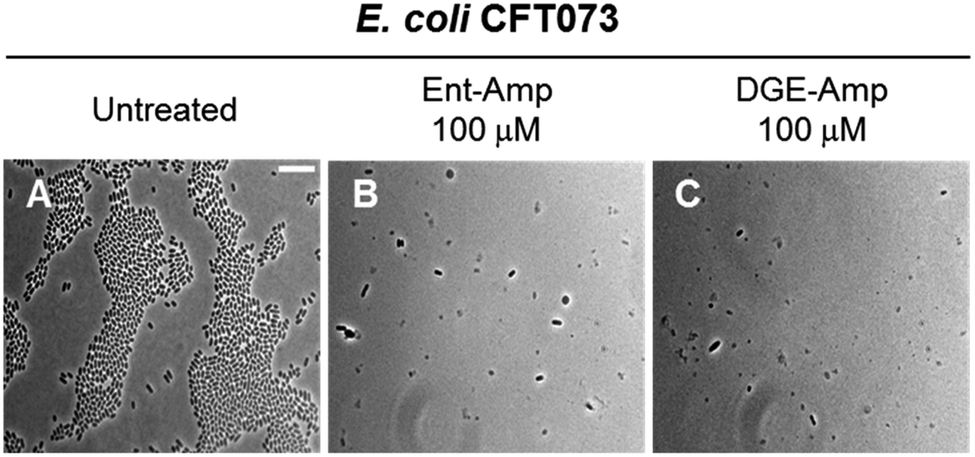 | ||
| Fig. 8 Full-field view micrographs of E. coli CFT073 WT (A–C) treated with MIC amounts of Ent/DGE-Amp 1, 2 in modified M9 medium for 20 h at 30 °C. Scale bar: 10 μm. The micrographs of E. coli CFT073 treated with MIC amounts of Ent/DGE-Lex/Mem 3–6 are shown in Fig. S10.† | ||
Overall, phase-contrast microscopy imaging corroborated the AMA assays and illuminated the differences in cell morphologies due to variable affinities of the β-lactam warheads towards PBP2 and PBP3. It should be noted, however, that sub-MIC doses of β-lactam antibiotics can also inhibit nonessential PBPs (e.g. PBP4)101 in Gram-positive and Gram-negative bacterial strains.54,101–105 Inhibition of nonessential PBPs leads to more subtle morphological effects than inhibition of PBP1, 2 and 3;106–109 thus, further studies are needed to elucidate whether the Ent/DGE-β-lactam conjugates also inhibit nonessential PBPs similar to the parent β-lactams.
Inhibition of PBP2 and PBP3 by the Ent/DGE-β-lactam conjugates concludes with cell lysis
Next, we employed the LIVE/DEAD viability assay to examine the fate of the bacterial cell filaments and spheroplasts formed as result of Ent/DGE-β-lactam treatment. This assay utilizes fluorescent dyes to distinguish between cells with intact OM (stain green with SYTO 9) and cells with compromised OM (stain red with propidium iodide, PI).110As expected, elongation/filamentation was detected in all strains treated with sub-MIC quantities of the PBP3 inhibitors Ent-Amp/Lex 1, 3 and DGE-Amp/Lex 2, 4 (Fig. 9B–D and S11B†), whereas the PBP2 inhibitors Ent/DGE-Mem 5, 6 led to spheroplast formation (Fig. 9E and S11C†). Similarly to the phase-contrast microscopy and the AMA assays, filamentation (Fig. 9G–I, L–N and S11E, H†) and spheroplasts formation (Fig. 9J, O and S11F, I†) were also observed, to different degrees, in the fepA iroN and fepA iroN iha mutants.
 | ||
| Fig. 9 Fluorescence micrographs acquired after incubating E. coli CFT073 and the fepA iroN and fepA iroN iha mutants with the LIVE/DEAD dyes at 30 °C for 15 min. The cell cultures were incubated with the Ent/DGE conjugates in modified M9 for 20 h at 30 °C prior to staining. Scale bar: 10 μm. The micrographs acquired after incubation of DGE-Amp/Mem are shown in Fig. S11.† | ||
All untreated cells stained green with SYTO 9, indicating viability (Fig. 9A, F and K). In contrast, addition of sub-MIC quantities of the conjugates lead to a mixture of green and red cells (Fig. 9B–E, G–J, L–O and S11†). The latter indicated that cell lysis occured for a subset of the cells under these conditions. Thus, after undergoing morphological change as determined by inhibition of either PBP3 (Ent-Amp/Lex 1, 3 and Ent/DGE-Lex 2, 4) (Fig. 9B–D, G–I, L–N and S11B†) or PBP2 (Ent/DGE-Mem 5, 6) (Fig. 9E, J, O and S11C†), the bacterial cells lose OM integrity and can be considered dead based on the PI staining. Lastly, the partial red staining of the fepA iroN (Fig. 9G–J) and fepA iroN iha (Fig. 9L–O) cells is another indication that the conjugates exert some AMA against these cells and are likely transported into the periplasm.
Overall, the LIVE/DEAD assay showed that the treatment of E. coli CFT073 and its mutants with sub-MIC quantities of conjugates leads to the death of both filamentous and CW-deficient cells as result of OM rupture. Therefore, the β-lactam warhead is responsible for the bactericidal effect of the Ent/DGE-β-lactam conjugates 1–6 as it inhibits the target PBPs causing characteristic phenotypes and compromises OM integrity conducive to cell lysis.
Ent-Mem induces explosive cell lysis in E. coli
To obtain a visual depiction of the course of cell killing by the Ent/DGE conjugates, we added MIC amounts of Ent/DGE-Amp 1, 2 and Ent-Mem 5 to E. coli CFT073 and acquired time-lapse images of the process at 37 °C. The Ent/DGE-Lex 3, 4 conjugates were not studied due to their relatively high MIC values. The images of CFT073 cells treated with 100 μM Ent/DGE-Amp 1, 2 show that the cells ceased to grow and divide. Moreover, these cells did not elongate or undergo explosive lysis over the course of 4 h. These observations indicate that non-explosive rupture of the cell membranes occurs and results in cell lysis without bursting (Fig. 10) similar to the red-stained cells in the LIVE/DEAD assay (Fig. 9).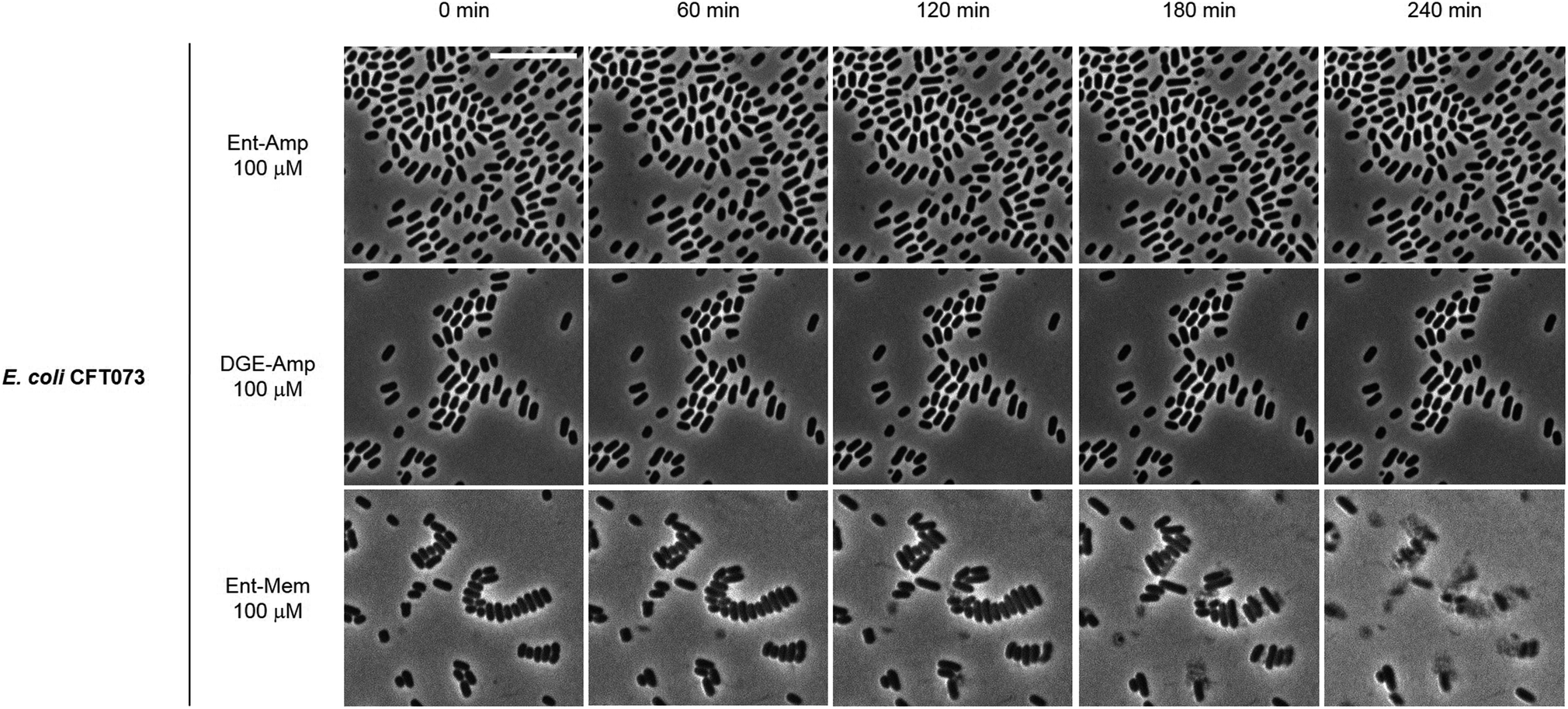 | ||
| Fig. 10 Time-lapse phase-contrast micrographs of E. coli CFT073 incubated with 100 μM Ent/DGE-Amp 1, 2 and Ent-Mem 5 in modified M9 medium (37 °C). Scale bar: 10 μm. | ||
In contrast to Ent/DGE-Amp 1, 2, the treatment of E. coli CFT073 with 100 μM Ent-Mem 3 (Fig. 10) is accompanied by explosive lysis that starts within the first 60 min and is virtually complete by 4 h. Thus, targeting of different primary PBPs by the Ent/DGE-β-lactam conjugates determines not only the morphologies of the cells treated with sub-MIC doses, but also the character of lysis when MIC amounts are reached.
Perspectives on siderophore-β-lactam conjugates
The growing problem of antibiotic resistance and the insufficient amount of new antibiotics in the drug pipeline pose a serious threat to public health. Development of narrow-spectrum antibiotics that kill pathogenic species and spare commensal microbes is considered a promising solution in the face of infections caused by multidrug-resistant Gram-negative bacteria. Leveraging the essential nutrient uptake machineries of such bacteria for a targeted delivery of antibiotics has been reported for a number of different compounds.111–113 Recently, FDA approved the first antibiotic that is reported to hijack Fe3+ uptake machinery to deliver a cephalosporin cargo to the periplasm of Gram-negative bacteria in the treatment of complicated urinary tract infections.114–116Our approach consists of using the native siderophore scaffolds Ent and DGE as antibiotic carriers.34–36 The Ent/DGE-Lex/Mem conjugates synthesized and studied in this work expand the arsenal of clinically-approved broad-spectrum antibiotics that can be endowed with selectivity towards pathogenic strains that express IroN. This selectivity is achieved by targeting the enterobactin/salmochelin uptake machinery of E. coli and is not affected by the β-lactam structure. Moreover, conjugation to Ent/DGE further enhances the MIC of the parent β-lactams by 10–1000-fold. The time-kill kinetics of the conjugated β-lactams is enhanced to a greater extent for the more lipophilic antibiotics such as Amp due to the change of the uptake route from porins (Amp) to TonB-powered FepA or IroN (Ent-Amp and DGE-Amp). These results indicate that the Ent/DGE scaffold could serve as a versatile tool for pharmaceutical development of antibacterial therapeutics that target IroN-expressing strains (e.g. DGE conjugates), enhancing the MIC values and decreasing the time needed to exert its bactericidal effect.
The three β-lactams, Amp, Lex, and Mem, conjugated to the Ent/DGE scaffolds in this work, exert AMA by inhibiting class B high-molecular-weight PBP2 and PBP3 as indicated by the phase-contrast microscopy and LIVE/DEAD staining assays. These results along with the AMA assays of the OM receptor mutants demonstrated that the β-lactam cargo is indeed responsible for the killing of bacteria while the uptake of the Ent/DGE conjugates with different β-lactam cargos takes place via FepA and IroN. In prior work, we hypothesized that Iha may contribute to the sensitivity of E. coli CFT073 to the Ent/DGE-β-lactams. However, the AMA assays and microscopy study of the fepA iroN and fepA iroN iha mutants indicated that Iha does not play an important role in the transport of either Ent- or DGE-β-lactams. Nevertheless, we have observed weak AMA and signs of uptake of the conjugates by the fepA iroN and the fepA iroN iha mutants, which suggests that CFT073 possesses other yet-unknown routes of Ent/DGE uptake.
Although uptake through the OM siderophore receptors allows the β-lactam cargos of the Ent/DGE conjugates to exert antibacterial activity, there may be additional mechanisms in place that explain the enhancement of the MIC values of conjugates relative to parent antibiotics. For instance, it has been shown that accumulation of apo Ent in the periplasm of E. coli leads to deleterious effects for the bacterial cells.117 However, under the assay conditions employed in this work, we did not observe any of the abnormal morphologies that were attributed to the accumulation of Ent in the bacterial periplasm.117
Of the four β-lactam cargos we have thus far conjugated to Ent/DGE – Amp, Amx,34,35 Lex, and Mem – Mem seems to be the most promising for further evaluation. The exceptional stability of the carbapenem ring to the action of extended spectrum β-lactamases, its enhanced MIC upon conjugation to Ent/DGE, and induction of explosive cell lysis at nanomolar concentrations make Mem an outstanding warhead for siderophore conjugation. However, use of Mem as warhead could be limited by the spread of carbapenemase-producing strains, which render Mem inactive.80,82 Likewise, the selectivity afforded by conjugation of antibiotics to Ent/DGE could be compromised by the existence of commensals that can cross-feed using Ent/DGE as xenosiderophores.118 Therefore, further studies are needed to better understand the host microbiome and evaluate the potential of Ent/DGE antibiotic conjugates as pathogen-selective therapeutics.
Conclusion and outlook
In closing, this work further demonstrates that conjugation of β-lactam antibiotics to the Ent/DGE scaffolds is an effective strategy to convert broad-spectrum antibiotics into compounds that selectively target bacteria expressing the catecholate siderophore receptors FepA and IroN. Furthermore, the siderophore-β-lactam conjugates presented in this work efficiently kill pathogenic E. coli regardless of the structure and polarity of the β-lactam cargo. Targeting pathogenesis with such conjugates could reduce the onset of secondary infections and slow the evolution of antibiotic resistance. In future work, studies using mouse models will aid the understanding of the stability and efficacy of such siderophore-β-lactam conjugates in vivo. Lastly, it will be important to determine whether this strategy is applicable to other bacterial pathogens such as Salmonella enterica that cause human disease and utilize both Ent and DGE for Fe3+ acquisition.Experimental section
Synthetic reagents
Anhydrous N,N-dimethylformamide (DMF) and dichloromethane (DCM) were purchased from VWR. Anhydrous dimethyl sulfoxide (DMSO) was purchased from Millipore Sigma and used as received. All other chemicals were purchased from Millipore Sigma or VWR and used as received. The syntheses of Ent-PEG3-N310,34,35,119 DGE-PEG3-N311,35 Amp-alkyne 7,34,35 Ent-Amp 1,34,35 and DGE-Amp 2 (ref. 35) are reported elsewhere.General methods and instrumentation
EMD TLC silica gel 60 F254 plates were used for analytical thin layer chromatography. EMD PLC silica gel 60 F254 plates of 2 mm thickness were used for preparative TLC. Sigma-Aldrich silica gel (70–230 mesh, 60 Å) was used for flash column chromatography.1H NMR spectra were collected on a Bruker Avance III DPX 400 MHz spectrometer operated at ambient probe temperature (293 K). Q-LC/MS and Q-ToF MS analyses were carried out on an Agilent 6125B mass spectrometer attached to an Agilent 1260 Infinity LC and a high-resolution Agilent 6545 mass spectrometer, respectively. For all LC/MS analyses, solvent A was 0.1% formic acid/H2O and solvent B was 0.1% formic acid/MeCN (LC/MS grade, Millipore Sigma). The samples were analyzed using a solvent gradient of 5–95% B over 6 min with a flow rate of 0.4 mL min−1.
Analytical and semi-preparative HPLC were performed using an Agilent 1200 series HPLC system outfitted with a Clipeus reversed-phase C18 column (5 μm pore size, 4.6 × 250 mm, Higgins Analytical) at a flow rate of 1 mL min−1 and an Agilent Zorbax reversed-phase C18 column (5 μm pore size, 9.4 × 250 mm) at a flow rate of 4 mL min−1. The multi-wavelength detector was set to detect absorbance at 220, 280, and 316 nm. HPLC-grade acetonitrile (MeCN) and trifluoroacetic acid (TFA) were purchased from Millipore Sigma. For HPLC analysis, solvent A was 0.1% v/v TFA/H2O and solvent B was 0.1% v/v TFA/MeCN. The percentage of TFA in the HPLC solvents used for antibiotic conjugate purification was 0.005% to avoid β-lactam ring hydrolysis. The HPLC solvents were prepared with HPLC grade MeCN and TFA, with Milli-Q water (18.2 MΩ cm), and filtered through a 0.2 μm filter before use. To evaluate conjugate purity by analytical HPLC, the entire portion of each HPLC-purified compound was dissolved in a 1:1 mixture of MeCN/H2O and an aliquot was taken for HPLC analysis. The remaining solution was subsequently lyophilized.
Optical absorption spectra were recorded on a Beckman Coultier DU800 spectrophotometer (1 cm quartz cuvettes, Starna). A BioTek Synergy HT plate reader was used to record absorbance at 600 nm (OD600) for antimicrobial activity assays and time-kill kinetics.
Synthesis of Lex-alkyne, 8
Hex-5-ynoyl chloride (0.352 g, 2.70 mmol) was added dropwise to a 0 °C solution of Lex sodium salt (0.500 g, 1.35 mmol) and NaHCO3 (0.567 g, 6.75 mmol) in 6.75 mL of water under vigorous stirring. The reaction mixture was warmed to room temperature and left to react for 1 h under stirring. Then, 5 mL of water was added to the solution and the resulting mixture was washed with EtOAc (2 × 10 mL). The aqueous layer was acidified to pH 2 using 1 M HCl and the product extracted with EtOAc (3 × 10 mL). The organic layer was dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure to yield a yellow solid. The crude product was triturated with hexane, then purified by semi-preparative HPLC (0–100% B over 30 min, 4 mL min−1; 0.005% TFA). Lyophilization of the HPLC fractions afforded 0.390 g (65%) of Lex-alkyne, 8, as an off-white powder. Analytical HPLC Rt = 17.5 min (0–100% B over 30 min, 1 mL min−1; 0.005% TFA).1H NMR (DMSO-d6, 400 MHz): δ, ppm 13.20 (br, 1H), 9.25 (d, J = 8.5 Hz, 1H), 8.56 (d, J = 8.1 Hz, 1H), 7.44 (d, J = 7.6 Hz, 2H), 7.25–7.36 (m, 3H), 5.68 (d, J = 8.2 Hz, 1H), 5.62 (dd, J = 4.7, 8.2 Hz, 1H), 4.96 (d, J = 4.7 Hz, 1H), 3.47 (d, J = 18.9 Hz, 1H), 3.28 (d, J = 18.2 Hz, 1H), 2.78 (s, 1H), 2.31 (t, J = 7.3 Hz, 2H), 2.13–2.22 (m, 2H), 1.99 (s, 3H), 1.67 (m, 2H). QToF-MS (m/z): mass calcd for (C22H23N3O5S + Na+) = 464.1256; found 464.1253.Synthesis of Mem-alkyne, 9
Hex-5-ynoyl chloride (0.101 g, 0.78 mmol) was added dropwise to a 0 °C solution of Mem (0.150 g, 0.39 mmol) and Et3N (1.522 mL, 1.105 g, 10.92 mmol) in 4 mL of MeOH under vigorous stirring. The reaction mixture was warmed to room temperature and left to react for 1 h under stirring. Then, the solvent was removed under reduced pressure and the crude was purified by semi-preparative HPLC (0–100% B over 30 min, 4 mL min−1; 0.005% TFA). Lyophilization of the HPLC fractions containing product afforded 0.093 g (50%) of Mem-alkyne, 9, as a yellow powder. Analytical HPLC Rt = 14.0 min (0–100% B over 30 min, 1 mL min−1; 0.005% TFA). The 1H NMR (CD3OD, 400 MHz): δ, ppm 4.24 (t, J = 9.1 Hz, 1H), 4.07–4.17 (m, 1H), 3.82–3.92 (m, 1H), 3.53–3.64 (m, 1H), 3.48 (t, J = 10.2 Hz, 1H), 3.32 (dd, J = 2.7, 7.1 Hz, 2H), 3.19 (s, 3H), 3.05 (s, 1H), 2.98 (s, 3H), 2.73–2.84 (m, 1H), 2.33–2.60 (m, 2H), 2.24–2.31 (m, 4H), 1.77–1.87 (m, 2H), 1.31 (d, J = 6.7 Hz, 3H), 1.28 (d, J = 7.2 Hz, 3H). QToF-MS (m/z): mass calcd for (C23H31N3O6S + H+) = 478.2012; found 478.2010.General synthesis of Ent/DGE-Amp, Ent/DGE-Lex, Ent/DGE-Mem (1–6)
β-lactam alkyne 7–9 (50 μL of a 50 mM solution in DMSO, 2.5 μmol) and Ent/DGE-PEG3-N310, 11 (73 μL of an 11.3 mM solution in DMSO, 0.825 μmol) were combined and 100 μL of DMSO was added. An aliquot of aqueous CuSO4 (50 μL of a 90 mM solution, 4.5 μmol) and TBTA (100 μL of a 50 mM solution in DMSO, 5 μmol) were combined to give a blue solution, to which sodium ascorbate (NaAsc, 100 μL of a 180 mM solution in water, 18.0 μmol) was added. The color of the solution immediately changed from blue to pale yellow (reduction of Cu2+ to Cu+) and the mixture was added to the alkyne/azide solution. The reaction was gently mixed on a bench-top rotator for 2.5 h at 25 °C (150 rpm). Then, the reaction mixture was diluted with 1:1 MeCN/water (3× the volume of solution) and purified by semi-preparative HPLC (0–100% B over 30 min, 4 mL min−1; 0.005% TFA). Lyophilization of the HPLC fractions afforded the corresponding conjugates (1–6) as white or off-white powders. Analytical data and the yields of the Ent/DGE conjugates are provided in Table S3.†
Storage and handling of siderophores and siderophore-β-lactam antibiotic conjugates
All precursors and Ent/DGE conjugates were stored as either powders or DMSO stock solutions at −20 °C. The stock solution concentrations for the Ent/DGE conjugates ranged between 1–2 mM. These values were determined by diluting the DMSO stocks in MeOH and using the reported extinction coefficient for enterobactin in MeOH (ε316 = 9500 M−1 cm−1).39 To minimize multiple freeze–thaw cycles, the resulting solutions were divided into 50 μL aliquots and stored at −20 °C. The β-lactam antibiotic cargos as well as Ent/DGE are prone to hydrolysis, and aliquots were routinely analyzed by HPLC to confirm the integrity of the samples.General microbiology materials and methods
Information pertaining to all bacterial strains used in this study is listed in Table S1.† Freezer stocks of all E. coli strains were prepared from single colonies in 25% glycerol/lysogeny broth (LB) medium. Lysogeny broth (tryptone 10 g L−1, yeast extract 5 g L−1, NaCl 10 g L−1), modified M9 (Na2HPO4 6.8 g L−1, KH2PO4 3 g L−1, NaCl 0.5 g L−1, NH4Cl 1 g L−1, 0.4% glucose, 0.2% casein amino acids, 2 mM MgSO4, 0.1 mM CaCl2, 16.5 μg mL−1 thiamine hydrochloride)36 and agar were obtained from Becton Dickinson (BD). Mueller–Hinton broth (MHB; beef extract powder 2 g L−1, casein hydrolysate 17.5 g L−1, and soluble starch 1.5 g L−1) was purchased from Sigma-Aldrich. The iron chelator 2,2′-bipyridine (Bpy) was purchased from Sigma-Aldrich. All growth media and Milli-Q water used for bacterial cultures or for preparing stock solutions were sterilized in an autoclave. The modified M9 medium was filter-sterilized through a 0.22 μm filter. A 200 mM Bpy stock solution was prepared in anhydrous DMSO and utilized in bacterial growth inhibition assays. Working solutions of Ent and Ent/DGE-antibiotic conjugates were prepared by 10-fold serial dilutions in 10% DMSO/H2O. For all microbiology assays, the final cultures contained 1% v/v DMSO. Sterile polypropylene culture tubes and sterile polystyrene 96-well plates used for culturing were purchased from VWR and Corning Inc., respectively. The optical density at 600 nm (OD600) was recorded on a Beckmann Coulter DU800 spectrophotometer or by using a BioTek Synergy HT plate reader.General procedure for antimicrobial activity (AMA) assays
Overnight bacterial cultures were prepared in 15 mL polypropylene tubes by inoculating 5 mL of growth medium (modified M9) with the appropriate freezer stock. The overnight cultures were then incubated at 37 °C for 16–18 h on a rotating wheel set at 150 rpm. Each overnight culture was diluted in a v/v ratio of 1:100 into 5 mL of fresh modified M9 medium and incubated at 37 °C and 150 rpm until OD600 reached ∼0.6 (mid-log phase). Each culture was subsequently diluted into fresh modified M9 to achieve a final OD600 of 0.001. A 90 μL aliquot of the diluted culture was combined with a 10 μL aliquot of a 10× solution of the antibiotic or antibiotic conjugate in a 96-well plate. The covered plate was wrapped in a wet paper towel and Saran wrap, then incubated at 30 °C with shaking at 150 rpm for 20 h in a tabletop incubator. Bacterial growth was determined by measuring OD600 (end point analysis) on a BioTek Synergy HT plate reader. Each well condition was prepared in duplicate (technical replicate) and at least three independent replicates (biological replicate) utilizing at least two synthetic batches of each conjugate were conducted on different days. The resulting mean OD600 values are reported, and the error bars are the standard deviation of the mean (SDM) obtained from the biological replicates.
Time-kill kinetics
Overnight cultures were prepared by inoculating 5 mL of modified M9 medium with bacterial freezer stocks. The overnight cultures were diluted 1:100 into 5 mL of fresh modified M9 medium and incubated at 37 °C with shaking at 150 rpm until OD600 reached ∼0.3. The culture was centrifuged (3000 rpm × 10 min) and the resulting pellet was re-suspended in modified M9 and centrifuged (3000 rpm × 10 min). The resulting pellet was re-suspended again in fresh modified M9 and the OD600 adjusted to 0.3. A 90 μL aliquot of the resulting culture was combined with a 10 μL aliquot of a 10× solution of parent antibiotic or Ent/DGE conjugate in a 96 well plate, which was wrapped in Saran film and incubated at 37 °C with shaking at 150 rpm. The OD600 values were recorded at 1 h intervals. Each well condition was prepared in duplicate (technical replicate) and at least three independent replicates (biological replicate) utilizing at least two synthetic batches of each conjugate were conducted on different days. The resulting mean OD600 values are reported, and the error bars are the standard deviation of the mean (SDM) obtained from the biological replicates.
Calculation of Ent/DGE-β-lactam MIC for microscopy assays
The MIC values (Table S4†) of the Ent/DGE-Amp/Lex/Mem conjugates 1–6 were determined in modified M9 medium for mid-log phase (OD600 ∼ 0.6) cell cultures diluted to OD600 = 0.001 (vide supra). Plating of the diluted cultures onto LB agar plates gave an average count of 5 × 104 CFU mL−1 (n > 3). Thus, at 5 × 104 CFU mL−1, the MIC of Ent/DGE-Amp/Mem 1, 2, 5, and 6 for E. coli CFT073 is equal to 1 × 10−8 M (Table S4†). For microscopy experiments, 5 × 108 CFU mL−1 of mid-log phase CFT073 cultures were incubated with the Ent/DGE-β-lactams 1, 2, 5, and 6. Hence, the MIC of Ent/DGE-Amp/Mem 1, 2, 5, and 6 per 5 × 108 CFU mL−1 was estimated to be 100 μM. No cells were detected after incubating 5 × 108 CFU mL−1 of E. coli CFT073 with >100 μM of the Ent/DGE-Amp/Mem 1, 2, 5, and 6. For Ent/DGE-Lex 3 and 4, MIC = 1 × 10−5 M per 5 × 104 CFU mL−1. To observe filamentous growth, 100 μM of Ent/DGE-Lex 3, 4 was incubated per 5 × 106 CFU mL−1 (sub-MIC).Phase-contrast microscopy
Phase-contrast microscopy imaging was carried out at the W. M. Keck Biological Imaging Facility of the Whitehead Institute (Cambridge, Massachusetts). The incubation of cell cultures with the Ent/DGE-β-lactams was performed on 96-well plates (100 μL sample per well) using 5 × 108 CFU mL−1 and 50 or 100 μM of Ent/DGE conjugates. For the Ent/DGE-Lex conjugates, the initial cell density was 5 × 106 CFU mL−1 to account for the higher MIC values of the Lex conjugates and use less compound. Following a 20 h incubation (30 °C, 150 rpm), a 5 μL sample from each culture was pipetted on an agarose (Bio-Rad, PCR grade) pad (1% w/w agarose/Milli-Q water) positioned on a microscope slide. The sample was then covered with a coverslip and imaged using a Zeiss Axioplan2 upright microscope equipped with a 100× oil-immersion objective lens. For each type of microscopy experiment, each condition was repeated in at least three independent replicates on different days. Representative micrographs for each condition are shown in the figures.Mixed-species microscopy assays
All bacterial strains were cultured in 50% MHB in the presence of 100 μM of 2,2′-bipyridine to an OD600 ∼ 0.6 (mid-log phase) (S. aureus ATCC 25923 does not grow in the modified M9 medium). Bacterial suspensions were then adjusted to 1 × 107 CFU mL−1, mixed in a 1:1 ratio (107:107 CFU mL−1) and incubated with 50 μM Ent/DGE-Amp/Mem per 100 μL sample in 96-well plates (20 h, 30 °C, 150 rpm). For Ent/DGE-Lex, bacterial suspensions were adjusted to 1 × 106 CFU mL−1 initial cell density and incubated with 200 μM Ent/DGE-Lex. A 5 μL sample from each culture was pipetted on an agarose pad (1% w/w agarose/Milli-Q water) positioned on a microscope slide. The specimen was then covered with a coverslip and imaged using a Zeiss Axioplan2 upright microscope equipped with a 100× oil-immersion objective lens.
Time-lapse microscopy
The time-lapse studies were carried out using poly-D-lysine coated MatTek glass-bottom Petri dishes of 35 mm, with a 14 mm microwell and a No. 1.5 cover glass. The untreated bacteria (5 × 108 CFU mL−1) were mixed on 96-well plates (100 μL sample per well) to give a final concentration of 100 μM Ent/DGE conjugate. Once the conjugate was added to the culture, a 5 μL sample from each suspension was pipetted out onto the 14 mm microwell and covered with an agarose pad (1% w/w agarose/Milli-Q water). The Petri dish with the sample was then immediately covered with the No. 1.5 coverslip and placed on the microscope stage for imaging at 37 °C. Image acquisition was carried out at 5 min intervals over 4.5–5 h. The time-lapse images were collected on a Nikon-TE 2000 U wide-field inverted microscope equipped with a 100× oil-immersion objective lens.LIVE/DEAD assays
All bacterial strains were cultured in the modified M9 growth medium up to an OD600 ∼ 0.6 (mid-log phase). Bacterial suspensions were then adjusted to 5 × 108 CFU mL−1 and incubated with 50 or 100 μM Ent/DGE conjugates for 20 h (30 °C, 150 rpm). Subsequently, the cells were pelleted and re-suspended in 0.85% NaCl (centrifugation at 10000g for 10 min). These NaCl-suspended cells were further incubated in 96-well plates wrapped in aluminum foil with the SYTO 9-propidium iodide (PI) dye mixture (1:5, 12 μM SYTO 9:60 μM PI) for 15 min (30 °C, 150 rpm). Following incubation, 5 μL of cell cultures were pipetted on agarose pads (1% w/w agarose/Milli-Q water) placed on microslides and covered with glass coverslip. Fluorescence microscopy images were recorded on the Zeiss Axioplan2 microscope. The Texas Red (λEx = 532–587 nm; λEm = 608–683 nm) and GFP (λEx = 457–487 nm; λEm = 502–538 nm) channels were utilized to obtain the images of the DEAD and LIVE cells, respectively.
Image analysis
The microscopy images were processed using the FIJI software. For fluorescence images, fluorescence background subtraction was performed using a rolling ball method with a radius of 150 pixels. 8-bit image types were analyzed and the brightness intensity was set between 0 and 350.Conflicts of interest
E. M. N. and M. R. hold patents related to this work.Acknowledgements
This work was supported by the NIH (R01 5R01AI114625 and R21 AI126465 to E. M. N and M. R.). NMR and MS instrumentation is housed by the MIT Department of Chemistry Instrumentation Facility. The ICP-MS instrument at MIT is maintained by the MIT Center for Environmental Health Sciences (NIH P30-ES002109). We thank Dr Wendy Salmon for assistance with microscopy experiments. Work in M. R. lab is also supported by Public Health Service Grants AI126277, AI114625, AI145325, by the Chiba University-UCSD Center for Mucosal Immunology, Allergy, and Vaccines, and by the UCSD Department of Pediatrics. M. R. also holds an Investigator in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund.References
- T. Silhavy, D. Kahne and S. Walker, Cold Spring Harbor Perspect. Biol., 2010, 2, 1–16 CrossRef.
- H. I. Zgurskaya, C. A. López and S. Gnanakaran, ACS Infect. Dis., 2016, 1, 512–522 CrossRef.
- M. Blaser, Nature, 2011, 476, 393–394 CrossRef CAS.
- I. Cho and M. J. Blaser, Nat. Rev. Genet., 2012, 13, 260–270 CrossRef CAS.
- A. E. Clatworthy, E. Pierson and D. T. Hung, Nat. Chem. Biol., 2007, 3, 541–548 CrossRef CAS.
- A. K. Barczak and D. T. Hung, Curr. Opin. Microbiol., 2009, 12, 490–496 CrossRef CAS.
- Wolrd Health Organization, Antimicrobial resistance. Global report on surveillance, 2014 Search PubMed.
- PCAST Antibiotic Resistance Working Group, Report to the President on combating antibiotic resistance, 2014 Search PubMed.
- K. Lewis, Nat. Rev. Drug Discovery, 2013, 12, 371–387 CrossRef CAS.
- E. L. Tsalik, E. Petzold, B. N. Kreiswirth, R. A. Bonomo, R. Banerjee, E. Lautenbach, S. R. Evans, K. E. Hanson, J. D. Klausner, R. Patel, A. Caliendo, C. Chiu, R. Humphries, M. Miller and C. Woods, Clin. Infect. Dis., 2017, 64, S41–S47 CrossRef CAS.
- E. L. Tsalik, R. A. Bonomo and V. G. Fowler, Annu. Rev. Med., 2018, 69, 379–394 CrossRef CAS.
- V. Van Giau, S. S. A. An and J. Hulme, Drug Des., Dev. Ther., 2019, 13, 327–343 CrossRef.
- W. Gao, Y. Chen, Y. Zhang, Q. Zhang and L. Zhang, Adv. Drug Delivery Rev., 2018, 127, 46–57 CrossRef CAS.
- A. E. Paharik, H. L. Schreiber, C. N. Spaulding, K. W. Dodson and S. J. Hultgren, Genome Med., 2017, 9, 110 CrossRef.
- T. A. Wencewicz and M. J. Miller, Top. Med. Chem., 2018, 26, 151–183 Search PubMed.
- K. Schauer, D. A. Rodionov and H. de Reuse, Trends Biochem. Sci., 2008, 33, 330–338 CrossRef CAS.
- J. E. Cassat and E. P. Skaar, Cell Host Microbe, 2013, 13, 509–519 CrossRef CAS.
- N. C. Andrews and P. J. Schmidt, Annu. Rev. Physiol., 2007, 69, 69–85 CrossRef CAS.
- V. Braun and K. Hantke, Curr. Opin. Chem. Biol., 2011, 15, 328–334 CrossRef CAS.
- L. D. Palmer and E. P. Skaar, Annu. Rev. Genet., 2016, 50, 67–91 CrossRef CAS.
- M. Miethke and M. A. Marahiel, Microbiol. Mol. Biol. Rev., 2007, 71, 413–451 CrossRef CAS.
- R. C. Hider and X. Kong, Nat. Prod. Rep., 2010, 27, 637–657 RSC.
- B. C. Chu, A. Garcia-Herrero, T. H. Johanson, K. D. Krewulak, C. K. Lau, R. S. Peacock, Z. Slavinskaya and H. J. Vogel, BioMetals, 2010, 23, 601–611 CrossRef CAS.
- C. Ji, R. E. Juárez-Hernández and M. J. Miller, Future Med. Chem., 2012, 4, 297–313 CrossRef CAS.
- M. Ghosh, P. A. Miller and M. J. Miller, J. Antibiot., 2020, 73, 152–157 CrossRef CAS.
- M. J. Miller, A. J. Walz, H. Zhu, C. Wu, G. Moraski, U. Möllmann, E. M. Tristani, A. L. Crumbliss, M. T. Ferdig, L. Checkley, R. L. Edwards and H. I. Boshoff, J. Am. Chem. Soc., 2011, 133, 2076–2079 CrossRef CAS.
- T. A. Wencewicz, T. E. Long, U. Möllmann and M. J. Miller, Bioconjugate Chem., 2013, 24, 473–486 CrossRef CAS.
- T. A. Wencewicz, U. Möllmann, T. E. Long and M. J. Miller, BioMetals, 2009, 22, 633–648 CrossRef CAS.
- C. Ji and M. J. Miller, BioMetals, 2015, 28, 541–551 CrossRef CAS.
- J. M. Rosenberg II, Y.-M. Lin, Y. Lu and M. J. Miller, Curr. Med. Chem., 2000, 7, 159–197 CrossRef.
- Y. M. Lin, M. Ghosh, P. A. Miller, U. Möllmann and M. J. Miller, BioMetals, 2019, 32, 425–451 CrossRef CAS.
- M. Ghosh, Y. M. Lin, P. A. Miller, U. Möllmann, W. C. Boggess and M. J. Miller, ACS Infect. Dis., 2018, 4, 1529–1535 CrossRef CAS.
- R. Liu, P. A. Miller, S. B. Vakulenko, N. K. Stewart, W. C. Boggess and M. J. Miller, J. Med. Chem., 2018, 61, 3845–3854 CrossRef CAS.
- T. Zheng and E. M. Nolan, J. Am. Chem. Soc., 2014, 136, 9677–9691 CrossRef CAS.
- P. Chairatana, T. Zheng and E. M. Nolan, Chem. Sci., 2015, 6, 4458–4471 RSC.
- W. Neumann, M. Sassone-Corsi, M. Raffatellu and E. M. Nolan, J. Am. Chem. Soc., 2018, 140, 5193–5201 CrossRef CAS.
- J. H. Crosa and C. T. Walsh, Microbiol. Mol. Biol. Rev., 2002, 66, 223–249 CrossRef CAS.
- M. A. Fischbach, H. Lin, D. R. Liu and C. T. Walsh, Nat. Chem. Biol., 2006, 2, 132–138 CrossRef CAS.
- L. D. Loomis and K. N. Raymond, Inorg. Chem., 1991, 30, 906–911 CrossRef CAS.
- K. N. Raymond, E. A. Dertz and S. S. Kim, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3584–3588 CrossRef CAS.
- W. Neumann, A. Gulati and E. M. Nolan, Curr. Opin. Chem. Biol., 2017, 37, 10–18 CrossRef CAS.
- M. A. Fischbach, H. Lin, L. Zhou, Y. Yu, R. J. Abergel, D. R. Liu, K. N. Raymond, B. L. Wanner, R. K. Strong, C. T. Walsh, A. Aderem and K. D. Smith, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16502–16507 CrossRef CAS.
- S. I. Müller, M. Valdebenito and K. Hantke, BioMetals, 2009, 22, 691–695 CrossRef.
- D. H. Goetz, M. A. Holmes, N. Borregaard, M. E. Bluhm, K. N. Raymond and R. K. Strong, Mol. Cell, 2002, 10, 1033–1043 CrossRef CAS.
- M. Raffatellu, M. D. George, Y. Akiyama, M. J. Hornsby, S. P. Nuccio, T. A. Paixao, B. P. Butler, H. Chu, R. L. Santos, T. Berger, T. W. Mak, R. M. Tsolis, C. L. Bevins, J. V. Solnick, S. Dandekar and A. J. Bäumler, Cell Host Microbe, 2009, 5, 476–486 CrossRef CAS.
- Y. R. Chan, J. S. Liu, D. A. Pociask, M. Zheng, T. A. Mietzner, T. Berger, T. W. Mak, M. C. Clifton, R. K. Strong, P. Ray and J. K. Kolls, J. Immunol., 2009, 182, 4947–4956 CrossRef CAS.
- A. J. Bäumler, T. L. Norris, T. Lasco, W. Voigt, R. Reissbrodt, W. Rabsch and F. Heffron, J. Bacteriol., 1998, 180, 1446–1453 CrossRef.
- A. Brochu, N. Brochu, T. I. Nicas, T. R. Parr, A. A. Minnick, E. K. Dolence, J. A. McKee, M. J. Miller, M. C. Lavoie and F. Malouin, Antimicrob. Agents Chemother., 1992, 36, 2166–2175 CrossRef CAS.
- S. S. Jean, S. C. Hsueh, W. Sen Lee and P. R. Hsueh, Expert Rev. Anti-Infect. Ther., 2019, 17, 307–309 CrossRef CAS.
- R. K. Shields, Antimicrob. Agents Chemother., 2020, 64, e00059-20 CrossRef.
- K. F. Kong, L. Schneper and K. Mathee, APMIS, 2010, 118, 1–36 CrossRef CAS.
- H. Zhao, V. Patel, J. D. Helmann and T. Dörr, Mol. Microbiol., 2017, 106, 847–860 CrossRef CAS.
- E. Sauvage, F. Kerff, M. Terrak, J. A. Ayala and P. Charlier, FEMS Microbiol. Rev., 2008, 32, 234–258 CrossRef CAS.
- W. J. Godinez, H. Chan, I. Hossain, C. Li, S. Ranjitkar, D. Rasper, R. L. Simmons, X. Zhang and B. Y. Feng, ACS Chem. Biol., 2019, 14, 1217–1226 CrossRef CAS.
- C. C. Sanders, Antimicrob. Agents Chemother., 1989, 33, 1313–1317 CrossRef CAS.
- B. Vilanova, J. Frau, J. Donoso, F. Muñoz and F. García Blanco, J. Chem. Soc., Perkin Trans. 2, 1997, 2439–2444 RSC.
- C. K. Das and N. N. Nair, Phys. Chem. Chem. Phys., 2017, 19, 13111–13121 RSC.
- N. Masuda and S. Ohya, Antimicrob. Agents Chemother., 1992, 36, 1847–1851 CrossRef CAS.
- Y. Yang, N. Bhachech and K. Bush, J. Antimicrob. Chemother., 1995, 35, 75–84 CrossRef CAS.
- F. Fonseca, E. I. Chudyk, M. W. Van Der Kamp, A. Correia, A. J. Mulholland and J. Spencer, J. Am. Chem. Soc., 2012, 134, 18275–18285 CrossRef CAS.
- P. Proctor, N. P. Gensmantel and M. I. Page, J. Chem. Soc., Perkin Trans. 2, 1982, 1185–1192 RSC.
- R. O'Shea and H. E. Moser, J. Med. Chem., 2008, 51, 2871–2878 CrossRef.
- J. Sangster, J. Phys. Chem. Ref. Data, 1989, 18, 1111–1229 CrossRef CAS.
- Ampicillin – DrugBank, https://www.drugbank.ca/drugs/DB00415, accessed 30 June 2020.
- Cephalexin – DrugBank, https://www.drugbank.ca/drugs/DB00567, accessed 30 June 2020.
- Meropenem – DrugBank, https://www.drugbank.ca/drugs/DB00760, accessed 30 June 2020.
- T. Baba, T. Ara, M. Hasegawa, Y. Takai, Y. Okumura, M. Baba, K. A. Datsenko, M. Tomita, B. L. Wanner and H. Mori, Mol. Syst. Biol., 2006, 2, 2006.0008 CrossRef.
- H. L. Mobley, D. M. Green, A. L. Trifillis, D. E. Johnson, G. R. Chippendale, C. V. Lockatell, B. D. Jones and J. W. Warren, Infect. Immun., 1990, 58, 1281–1289 CrossRef CAS.
- A. Majumdar, V. Trinh, K. J. Moore, C. R. Smallwood, A. Kumar, T. Yang, D. C. Scott, N. J. Long, S. M. Newton and P. E. Klebba, J. Biol. Chem., 2020, 295, 4974–4984 CrossRef.
- S. K. Buchanan, B. S. Smith, L. Venkatramani, D. Xia, L. Esser, M. Palnitkar, R. Chakraborty, D. Van Der Helm and J. Deisenhofer, Nat. Struct. Biol., 1999, 6, 56–63 CrossRef CAS.
- M. A. Mulvey, J. D. Schilling and S. J. Hultgren, Infect. Immun., 2001, 69, 4572–4579 CrossRef CAS.
- J. B. Kaper, J. P. Nataro and H. L. T. Mobley, Nat. Rev. Microbiol., 2004, 2, 123–140 CrossRef CAS.
- E. C. Hagan and H. L. T. Mobley, Infect. Immun., 2007, 75, 3941–3949 CrossRef CAS.
- E. C. Garcia, A. R. Brumbaugh and H. L. T. Mobley, Infect. Immun., 2011, 79, 1225–1235 CrossRef CAS.
- V. S. Forsyth, S. D. Himpsl, S. N. Smith, C. A. Sarkissian, L. A. Mike, J. A. Stocki, A. Sintsova, C. J. Alteri and H. L. T. Mobley, mBio, 2020, 11, e00555-20 CrossRef.
- E. C. Hagan and H. L. T. Mobley, Infect. Immun., 2007, 75, 3941–3949 CrossRef CAS.
- S. Léveillé, M. Caza, J. R. Johnson, C. Clabots, M. Sabri and C. M. Dozois, Infect. Immun., 2006, 74, 3427–3436 CrossRef.
- A. I. Hartstein, K. E. Patrick, S. R. Jones, M. J. Miller and R. E. Bryant, Antimicrob. Agents Chemother., 1977, 12, 93–97 CrossRef CAS.
- D. T. King, S. Sobhanifar and N. C. J. Strynadka, Protein Sci., 2016, 25, 787–803 CrossRef CAS.
- J. Rodriguez-Baño, B. Gutiérrez-Gutiérrez, I. Machuca and A. Pascual, Clin. Microbiol. Rev., 2018, 31, 1–42 CrossRef.
- R. Huwaitat, A. P. McCloskey, B. F. Gilmore and G. Laverty, Future Microbiol., 2016, 11, 955–972 CrossRef CAS.
- C.-C. Sheu, C.-C. Sheu, Y.-T. Chang, S.-Y. Lin, Y.-H. Chen, Y.-T. Chang, S.-Y. Lin, Y.-H. Chen, Y.-H. Chen, P.-R. Hsueh and P.-R. Hsueh, Front. Microbiol., 2019, 10, 80 CrossRef.
- D. M. Livermore and N. Woodford, Curr. Opin. Microbiol., 2000, 3, 489–495 CrossRef CAS.
- A. M. Queenan, W. Shang, R. Flamm and K. Bush, Antimicrob. Agents Chemother., 2010, 54, 565–569 CrossRef CAS.
- A. L. Koch, Crit. Rev. Microbiol., 2000, 26, 205–220 CrossRef CAS.
- K. Poole, Cell. Mol. Life Sci., 2004, 61, 2200–2223 CrossRef CAS.
- H. Celia, N. Noinaj and S. K. Buchanan, Int. J. Mol. Sci., 2020, 21, 375 CrossRef CAS.
- T. A. Russo, U. B. Carlino and J. R. Johnson, Infect. Immun., 2001, 69, 6209–6216 CrossRef CAS.
- A. H. Delcour, Biochim. Biophys. Acta, Proteins Proteomics, 2009, 1794, 808–816 CrossRef CAS.
- E. M. Nestorovich, C. Danelon, M. Winterhalter and S. M. Bezrukov, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9789–9794 CrossRef CAS.
- H. Nikaido and E. Y. Rosenberg, J. Bacteriol., 1983, 153, 241–252 CrossRef CAS.
- F. Yoshimura and H. Nikaido, Antimicrob. Agents Chemother., 1985, 27, 84–92 CrossRef CAS.
- O. Kocaoglu and E. E. Carlson, Antimicrob. Agents Chemother., 2015, 59, 2785–2790 CrossRef CAS.
- B. G. Spratt, J. Bacteriol., 1977, 131, 293–305 CrossRef CAS.
- D. Bramhill, Annu. Rev. Cell Dev. Biol., 1997, 13, 395–424 CrossRef CAS.
- J. Pogliano, K. Pogliano, D. S. Weiss, R. Losick and J. Beckwith, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 559–564 CrossRef CAS.
- E. Gordon, N. Mouz, E. Duée and O. Dideberg, J. Mol. Biol., 2000, 299, 477–485 CrossRef CAS.
- G. Zhao, T. I. Meier, S. D. Kahl, K. R. Gee and L. C. Blaszczak, Antimicrob. Agents Chemother., 1999, 43, 1124–1128 CrossRef CAS.
- H. Cho, T. Uehara and T. G. Bernhardt, Cell, 2014, 159, 1310–1311 Search PubMed.
- J. Errington, R. A. Daniel and D.-J. Scheffers, Microbiol. Mol. Biol. Rev., 2003, 67, 52–65 CrossRef CAS.
- O. Kocaoglu, H. C. T. Tsui, M. E. Winkler and E. E. Carlson, Antimicrob. Agents Chemother., 2015, 59, 3548–3555 CrossRef CAS.
- O. Kocaoglu and E. E. Carlson, Antimicrob. Agents Chemother., 2015, 59, 2785–2790 CrossRef CAS.
- M. Park, J. B. Sutherland and F. Rafii, Anaerobe, 2020, 62, 102179–102186 CrossRef CAS.
- O. Kocaoglu, R. A. Calvo, L. T. Sham, L. M. Cozy, B. R. Lanning, S. Francis, M. E. Winkler, D. B. Kearns and E. E. Carlson, ACS Chem. Biol., 2012, 7, 1746–1753 CrossRef CAS.
- G. Zhao, T. I. Meier, S. D. Kahl, K. R. Gee and L. C. Blaszczak, Antimicrob. Agents Chemother., 1999, 43, 1124–1128 CrossRef CAS.
- J. M. Santos, M. Lobo, A. P. A. Matos, M. A. De Pedro and C. M. Arraiano, Mol. Microbiol., 2002, 45, 1729–1740 CrossRef CAS.
- A. S. Ghosh, C. Chowdhury and D. E. Nelson, Trends Microbiol., 2008, 16, 309–317 CrossRef CAS.
- D. E. Nelson and K. D. Young, J. Bacteriol., 2001, 183, 3055–3064 CrossRef CAS.
- B. M. Meberg, A. L. Paulson, R. Priyadarshini and K. D. Young, J. Bacteriol., 2004, 186, 8326–8336 CrossRef CAS.
- Invitrogen Molecular Probes, LIVE/DEAD BacLight Bacterial Viability Kits, http://probes.invitrogen.com/media/pis/mp07007.pdf, https://www.thermofisher.com/document-connect/document-connect.html?url=https%3A%2F%2Fassets.thermofisher.com%2FTFS-Assets%2FLSG%2Fmanuals%2Fmp07007.pdf%26title=TElWRSYjNDc7REVBRCAmbHQ7aSZndDtCYWMmbHQ7L2kmZ3Q7TGlnaHQgQmFjdGVyaWFsIFZpYWJpbGl0eSBLaXRz, accessed 19 June 2020.
- U. Möllmann, L. Heinisch, A. Bauernfeind, T. Köhler and D. Ankel-Fuchs, BioMetals, 2009, 22, 615–624 CrossRef.
- B. R. Wilson, A. R. Bogdan, M. Miyazawa, K. Hashimoto and Y. Tsuji, Trends Mol. Med., 2016, 22, 1077–1090 CrossRef CAS.
- M. G. P. Page, Ann. N. Y. Acad. Sci., 2013, 1277, 115–126 CrossRef CAS.
- K. H. Negash, J. K. S. Norris and J. T. Hodgkinson, Molecules, 2019, 24, 3314 CrossRef CAS.
- T. Katsube, T. Wajima and R. Echols, Clin. Infect. Dis., 2019, 69, S552–S558 CrossRef CAS.
- Shionogi Inc., Cefiderocol Briefing Document NDA # 209445, 2019 Search PubMed.
- D. E. Vega and K. D. Young, Mol. Microbiol., 2014, 91, 508–521 CrossRef CAS.
- W. Zhu, M. G. Winter, L. Spiga, E. R. Hughes, R. Chanin, A. Mulgaonkar, J. Pennington, M. Maas, C. L. Behrendt, J. Kim, X. Sun, D. P. Beiting, L. V. Hooper and S. E. Winter, Cell Host Microbe, 2020, 27, 376–388.e8 CrossRef CAS.
- T. Zheng, J. L. Bullock and E. M. Nolan, J. Am. Chem. Soc., 2012, 134, 18388–18400 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary experimental, Tables S1–S4, Schemes S1 and S2, Fig. S1–S13 and supplementary references. See DOI: 10.1039/d0sc04337k |
| This journal is © The Royal Society of Chemistry 2021 |