
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium catalyzed synthesis of indolizines via the carbonylative coupling of bromopyridines, imines and alkynes†
Sébastien A.
Roy
,
José
Zgheib
 ,
Cuihan
Zhou
and
Bruce A.
Arndtsen
*
,
Cuihan
Zhou
and
Bruce A.
Arndtsen
*
Department of Chemistry, McGill University, 801 Sherbrooke Street W., Montreal, QC H3A 0B8, Canada. E-mail: bruce.arndtsen@mcgill.ca
First published on 22nd December 2020
Abstract
We report herein the development of a palladium-catalyzed, multicomponent synthesis of indolizines. The reaction proceeds via the carbonylative formation of a high energy, mesoionic pyridine-based 1,3-dipole, which can undergo spontaneous cycloaddition with alkynes. Overall, this provides a route to prepare indolizines in a modular fashion from combinations of commercially available or easily generated reagents: 2-bromopyridines, imines and alkynes.
Introduction
Metal catalyzed carbonylations offer an efficient platform to assemble carbonyl-based products from feedstock chemicals.1 In addition to their classical use in carboxylic acid, ester, amide or ketone synthesis, there has been recent research effort directed toward employing carbonylations to generate products that are themselves reactive.2,3 These highlight an additional useful feature of carbon monoxide, its energetics, where its conversion to carboxylic acid derivatives is often exergonic. Carbonylations have been exploited to access various reactive acylating electrophiles and even non-CO containing products.4 Our lab has reported several examples of the latter, wherein the carbonylative formation of 1,3-dipoles (e.g. münchnones) can be coupled with cycloaddition reactions to afford heterocycles (Fig. 1a).4e–g In these, carbon monoxide is initially incorporated into the reactive 1,3-dipole, yet is ultimately converted to CO2 to drive the assembly of heterocycles from combinations of reagents.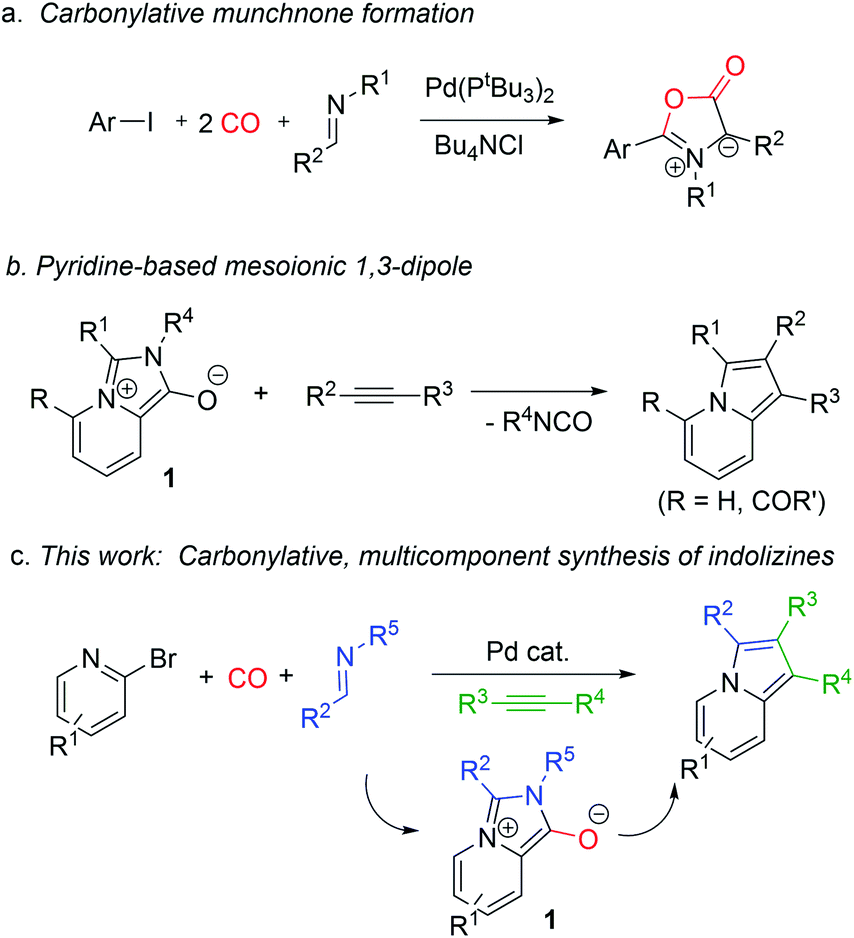 | ||
| Fig. 1 Carbonylative approaches to 1,3-dipoles and their use in multicomponent heterocycle synthesis. | ||
Considering the high value of carbonyl-based building blocks, the use of carbonylation reactions to effectively assemble other classes of reactive substrates could be of synthetic utility. One possibility is the pyridine-based 1,3-dipole 1 (Fig. 1b).51 has been recently described as a reactive version of the mesoionic dye Besthorn's Red,6 and can undergo 1,3-dipolar cycloaddition with alkynes to generate indolizines. These heterocycles, and their reduced derivatives, represent the core of a wide variety of pharmaceutically relevant molecules and natural products,7,8 and their extended conjugation has made them attractive as components in electronic materials.9 Indolizines are classically prepared by cyclizations of substituted pyridines10–12 or pyrroles.13 While some variants of these substrates can be easily generated, they more often require the build-up of the appropriate substituted core for cyclization, which adds synthetic steps, creates waste, and can limit their ease of diversification. Similarly, a limitation of the use of 1 in indolizine synthesis is the initial formation of the 1,3-dipole itself from 2-pyridyl acid chlorides, which must first be synthesized, and, due to their incorporation of both nucleophilic and electrophilic components, have limited scope and stability. Only certain variants of the 1,3-dipole 1 can therefore be accessed.
We hypothesized that carbonylations might provide a solution to these challenges. The mesoionic core of 1 contains a carbonyl-unit, which could in principle be derived by palladium catalyzed carbonylation (Fig. 1c). In addition to representing a new route to exploit carbonylation in synthesis, this would allow the formation of 1,3-dipole 1 from combinations of reagents that are all by themselves stable, functional group compatible, and readily available: halopyridines, imines and carbon monoxide. We describe in this report our development of a palladium catalyzed route to such a synthesis. Coupling the formation of 1 with cycloaddition has opened a new multicomponent synthesis of indolizines, where these heterocycles can now be formed from three simple, easily diversified reagents.
Results and discussion
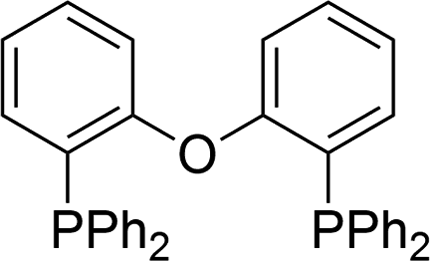
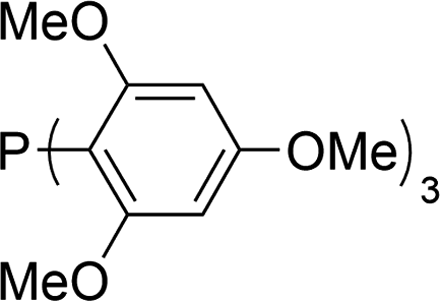
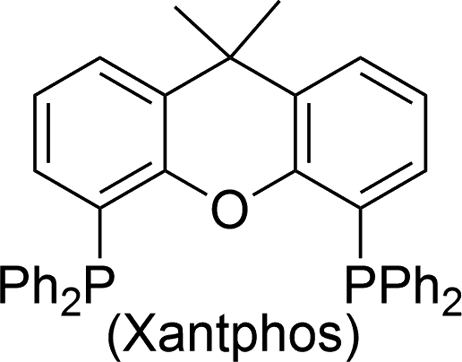
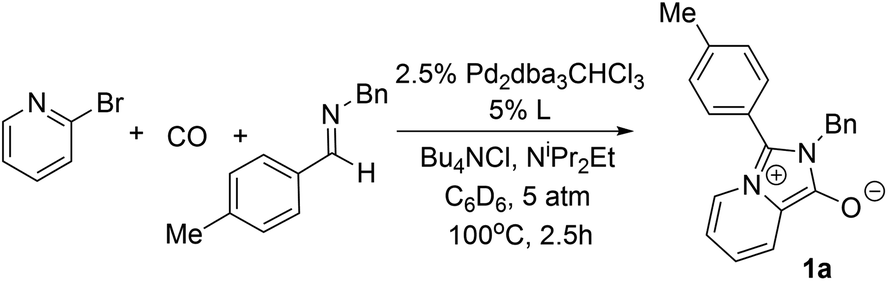
The carbonylative generation of 1,3-dipole 1 presents several design challenges. Imines are rarely employed in carbonylation chemistry due to their weak nucleophilicity and poor reactivity with the palladium–acyl intermediates generated in this chemistry. We envisioned that this might be addressed by instead using carbonylations to build-up in situ acid chloride electrophiles. Recent studies have shown that such a transformation is viable using sterically encumbered phosphines such as PtBu3 on palladium catalysts to favor the challenging reductive elimination of these products.2b However, the carbonylative formation of acid chlorides with coordinating substrates such as 2-bromopyridines has not been previously reported, and even simple aryl bromides require pressing conditions (110°, 20 atm CO) to be converted to acid chloride products.2d The latter could prove problematic for the formation of a reactive 1,3-dipole 1.To probe this potential, we first examined the carbonylative reaction of 2-bromopyridine and the imine p-tolyl(H)C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N(benzyl) in the presence of a chloride source (Bu4NCl, Table 1). Using Pd(PtBu3)2 as catalyst, which was previously noted to allow acid chloride generation,2e does indeed lead to the in situ build-up of dipole 1a in low yield (38%) at 100° (entry 1), but we noted the growth of other decomposition products upon extended reaction. In order to improve the yield of 1a, the influence of ligands on the reaction was examined. The use of Pd2dba3 without added ligand (entry 2) or with various common phosphines (entries 3–6) leads to decreased product yield. Simple bidentate ligands also inhibit catalysis (entries 7 and 8). However, we were pleased to find that large bite angle ligands such as DPE-Phos and xantphos significantly increase catalytic activity, with the latter forming 1a in near quantitative yield (94%, entry 10). Similar yields were noted at 80 °C (entry 11). Xantphos is a rigid, large bite angle bidentate ligand that can create steric strain in Pd(II) and potentially favor reductive elimination (vide infra).14 In addition to the formation of 1,3-dipole 1a, this reaction can be coupled with a cycloaddition. Thus, the palladium catalyzed build-up of 1a, followed by the addition of the electron deficient alkyne dimethylacetylene dicarboxylate (DMAD) leads to the overall one-pot formation of indolizine 2a in 76% yield (Fig. 2). The multicomponent reaction of 2-bromopyridine, imine, carbon monoxide and the less electron deficient alkyne ethyl 3-phenyl-2-propynoate can even be performed in a single operation to access indolizine in good yield (Fig. S1†). While these experiments use imine as the limiting reagent, only slightly diminished yield are observed when a stoichiometric amount of imine is used (entry 12), and 1a can be formed in high yield with 2-bromopyridine as the limiting reagent.
N(benzyl) in the presence of a chloride source (Bu4NCl, Table 1). Using Pd(PtBu3)2 as catalyst, which was previously noted to allow acid chloride generation,2e does indeed lead to the in situ build-up of dipole 1a in low yield (38%) at 100° (entry 1), but we noted the growth of other decomposition products upon extended reaction. In order to improve the yield of 1a, the influence of ligands on the reaction was examined. The use of Pd2dba3 without added ligand (entry 2) or with various common phosphines (entries 3–6) leads to decreased product yield. Simple bidentate ligands also inhibit catalysis (entries 7 and 8). However, we were pleased to find that large bite angle ligands such as DPE-Phos and xantphos significantly increase catalytic activity, with the latter forming 1a in near quantitative yield (94%, entry 10). Similar yields were noted at 80 °C (entry 11). Xantphos is a rigid, large bite angle bidentate ligand that can create steric strain in Pd(II) and potentially favor reductive elimination (vide infra).14 In addition to the formation of 1,3-dipole 1a, this reaction can be coupled with a cycloaddition. Thus, the palladium catalyzed build-up of 1a, followed by the addition of the electron deficient alkyne dimethylacetylene dicarboxylate (DMAD) leads to the overall one-pot formation of indolizine 2a in 76% yield (Fig. 2). The multicomponent reaction of 2-bromopyridine, imine, carbon monoxide and the less electron deficient alkyne ethyl 3-phenyl-2-propynoate can even be performed in a single operation to access indolizine in good yield (Fig. S1†). While these experiments use imine as the limiting reagent, only slightly diminished yield are observed when a stoichiometric amount of imine is used (entry 12), and 1a can be formed in high yield with 2-bromopyridine as the limiting reagent.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | % 1a | Entry | Ligand | % 1a |
| a 2-Bromopyridine (9.5 mg, 0.06 mmol), imine (8.4 mg, 0.04 mmol), NEtiPr2 (6.2 mg, 0.048 mmol), C6D6 (0.75 mL), Bu4NCl (17 mg, 0.06 mmol), Pd2dba3 (1.0 mg, 0.001 mmol), L (0.004 mmol; 0.002 mmol bidentate). b 80 °C. c 0.04 mmol 2-bromopyridine. d 0.04 mmol 2-bromopyridine, 0.06 mmol imine. | |||||
| 1 | PtBu3 | 38 | 7 | dppp | 16 |
| 8 | dppe | 0 | |||
| 2 | — | 15 | 9 |
|
45 |
| 3 | PPh3 | 26 | |||
| 4 | PCy3 | 16 | |||
| 5 |
|
15 | 10 |
|
94 |
| 6 |
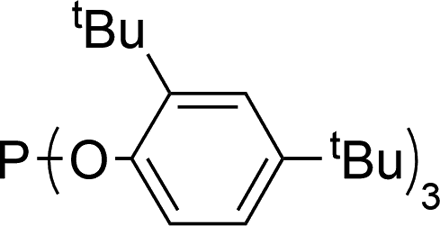
|
15 | 11 | Xantphos | 97b |
| 12 | Xantphos | 89b,c (92)b,d | |||
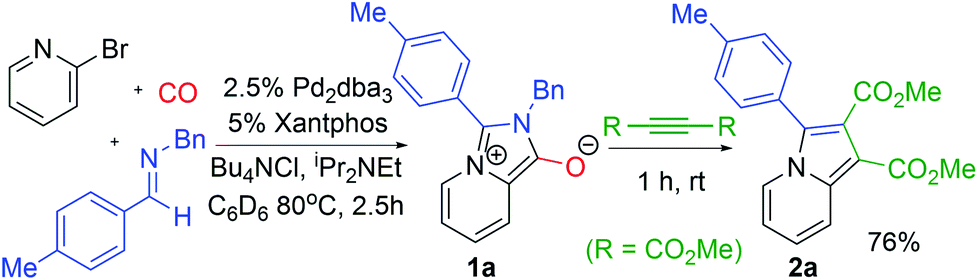 | ||
| Fig. 2 The one-pot, palladium catalyzed synthesis of indolizines. | ||
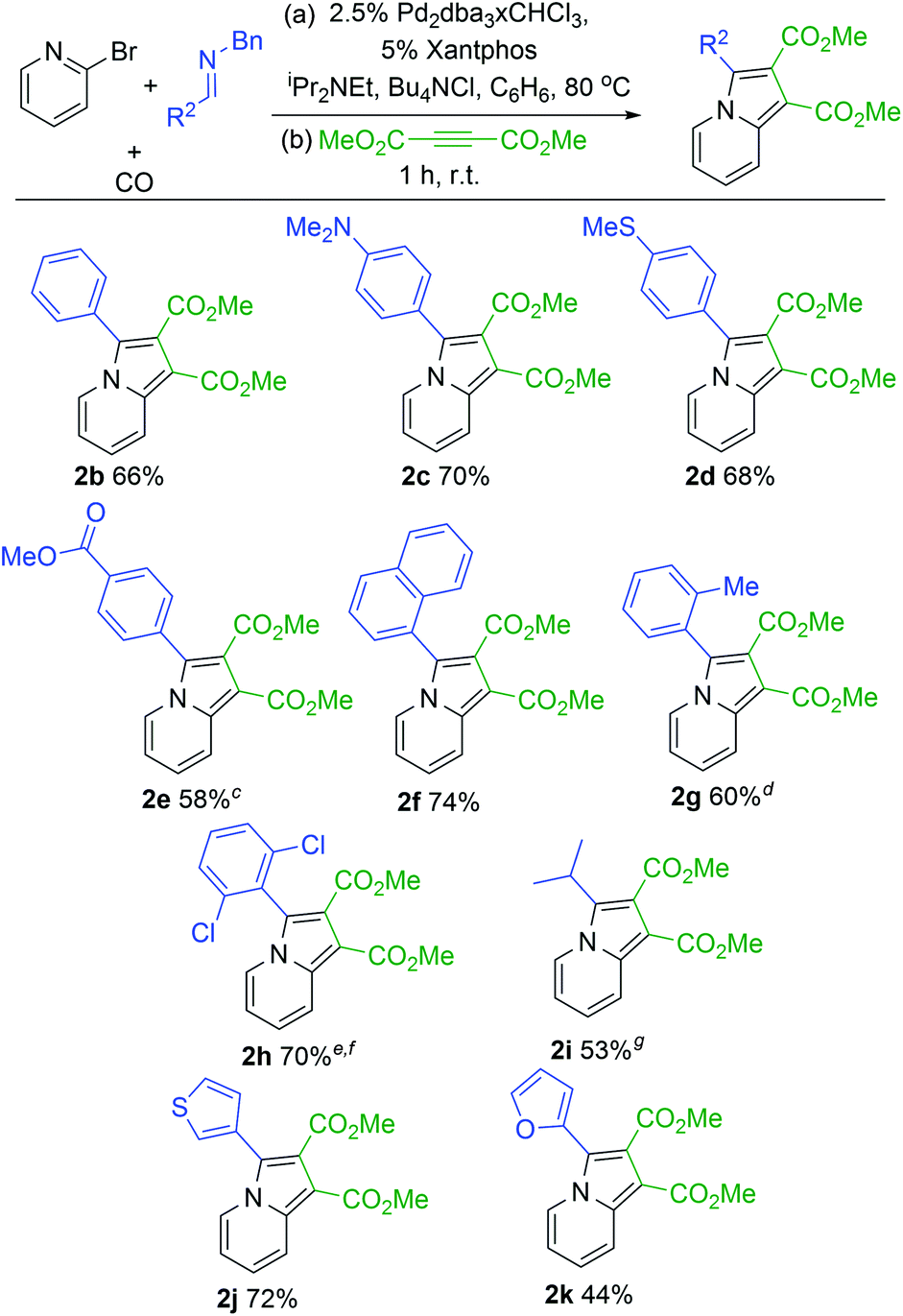
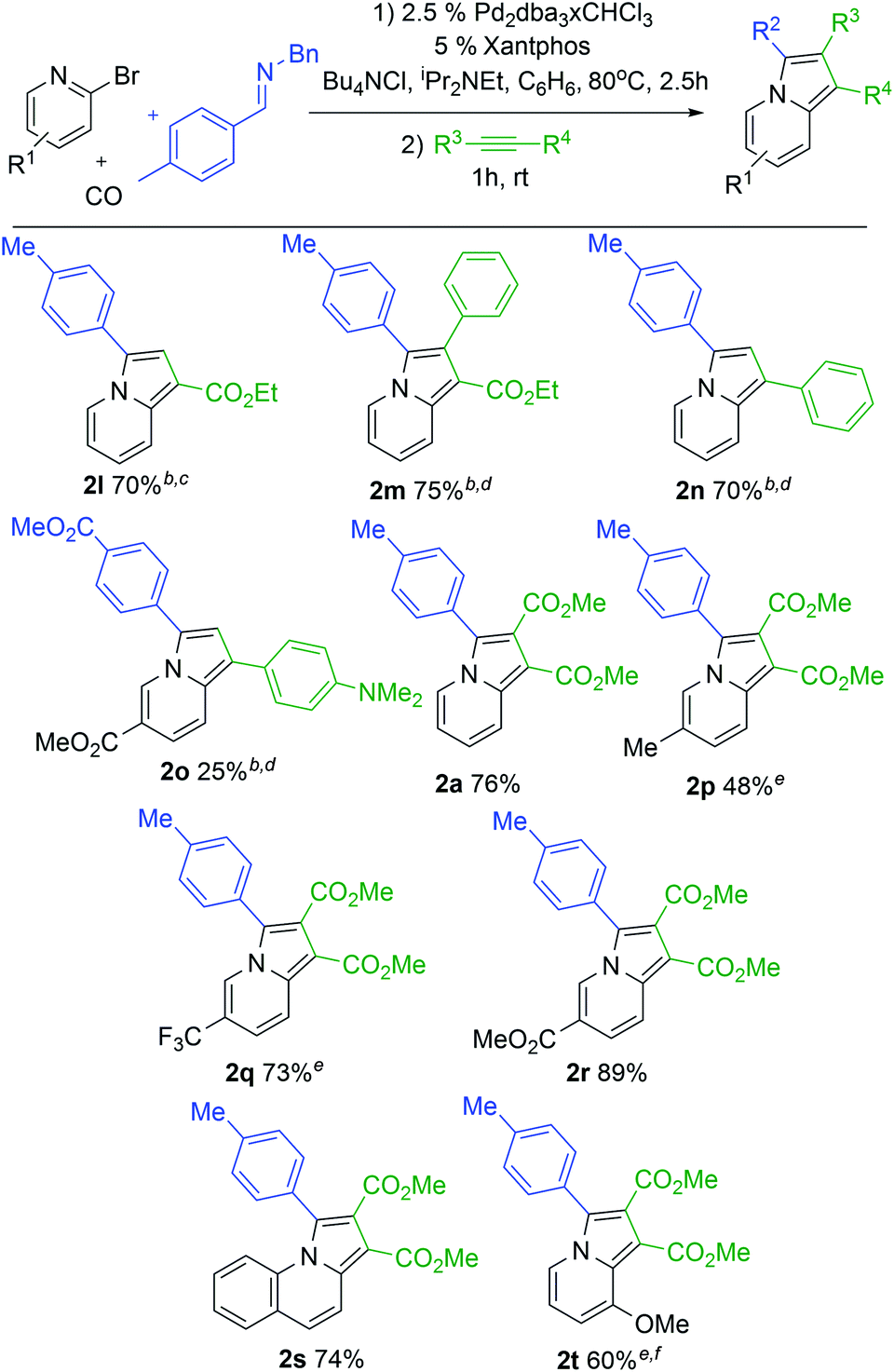
With a modular method to generate indolizines in hand, we next explored if this system could offer access to various indolizine structures. As shown in Table 2, a range of C-aryl substituted imines can be used in this reaction. This includes simple phenyl substituted (2b) and electron-rich (2c,d) imines, which lead to the corresponding indolizine in good yield. Imines with electron withdrawing substituents can also be employed, although these require extended reaction times to build-up the 1,3-dipole (2e). Sterically hindered 2-naphthyl, 2-tolyl and even 2,6-disubstituted imines are similarly viable substrates (2f–h). In the latter two cases, cycloaddition requires elevated temperatures and longer reaction times. Heteroaryl-substituted products are also accessible, such as those with thiophene and furan substituents (2j,k). The reaction can even allow the use of C-alkyl imines, which have proven problematic in related carbonylations due to their ability to readily convert to enamides upon N-acylation.4c However, rapid intramolecular cyclization with the pyridine in the more polar acetonitrile solvent followed by alkyne cycloaddition can afford the isopropyl-substituted indolizine (2i).
| a 2-Bromopyridine (79 mg, 0.50 mmol), imine (0.75 mmol), Pd2dba3·CHCl3 (13 mg, 0.013 mmol); xantphos (14 mg, 0.025 mmol); NiPr2Et (77 mg, 0.6 mmol); Bu4NCl (208 mg, 0.75 mmol); 5 atm CO; 10 mL C6H6. b Dimethylacetylene dicarboxylate (85 mg, 0.6 mmol), 1 h, rt. c 24 h. d Step 2: 12 h. e 100 °C, 3.5 h. f Step 2: 48 h at 80 °C. g 21 h, CH3CN instead of C6H6, 0.25 mmol Bu4NCl. |
|---|
|
In addition to the imine, cycloaddition with variously substituted alkynes can be used to modulate the 1- and 2-indolizine substituents (Table 3). Examples include the terminal alkyne ethyl propiolate (2l) or internal alkynes such as ethyl 3-phenyl-2-propynoate (2m). The more electron rich phenyl acetylene also undergoes cycloaddition with catalytically formed 1 to afford indolizine (2n), as does dimethylamino-substituted phenyl acetylene in lower yield (2o). More pressing conditions are required for the more electron rich alkynes (16 h for 2l, 80° for 2m and 2n) but lead to the formation of the corresponding indolizines in good yields. Notably, only one regioisomeric product is formed with these unsymmetrical alkynes, where the larger substituent is incorporated into the 1-position. This is consistent with steric bias in 1,3-dipole 1 directing the larger alkyne substituent away from R2.5 The 2-bromopyridine structure can also be tuned. Thus, pyridines with donor or electron withdrawing substituents in the 5-position can be incorporated in the reaction (2p–r). The extended conjugation in 2-bromoquinoline is also tolerated, leading to tricyclic product 2s. It is even possible to use a more sterically hindered 3-substituted bromopyridine to generate 8-substituted indolizine 2t. These bromopyridine derivatives are all significantly less expensive and more easily handled than the corresponding acid chlorides or even parent carboxylic acids. Together, this palladium catalyzed carbonylation offers a route to generate indolizines where every substituent can be systematically modulated in a one pot reaction from stable and available reagents.
We have performed several experiments to explore the mechanism of this reaction. Catalysis in the absence of a chloride source significantly diminishes the yield of 1a, and instead leads to the recovery of starting materials (Fig. 3a). Low product yields were also observed upon replacing chloride with other salt additives (e.g. Bu4NOTf: 18%). These observations suggest that chloride is required for an efficient reaction, and are consistent with in situ carbonylative acid chloride formation.15 Competition reaction with two imines varying only in the para-substituent on the C-aromatic ring leads to selective incorporation of the more electron rich imine into the product (Fig. 3b), which supports its role as a nucleophile in the reaction. It is notable, however, that no acid chloride is observed on monitoring the reaction by 1H NMR analysis, nor when performing the reaction in the absence of an imine trap (Fig. S2†). This implies that if acid chloride is generated, it either rapidly adds back to palladium, or, in the presence of an imine trap, is converted to the 1,3-dipole. Carbon monoxide pressure can influence the reaction, where performing the reaction at 1 atm CO leads to lower product yields (Fig. 3c), and consistent with the ability of carbon monoxide ligand to favor reductive elimination and stabilize Pd(0).
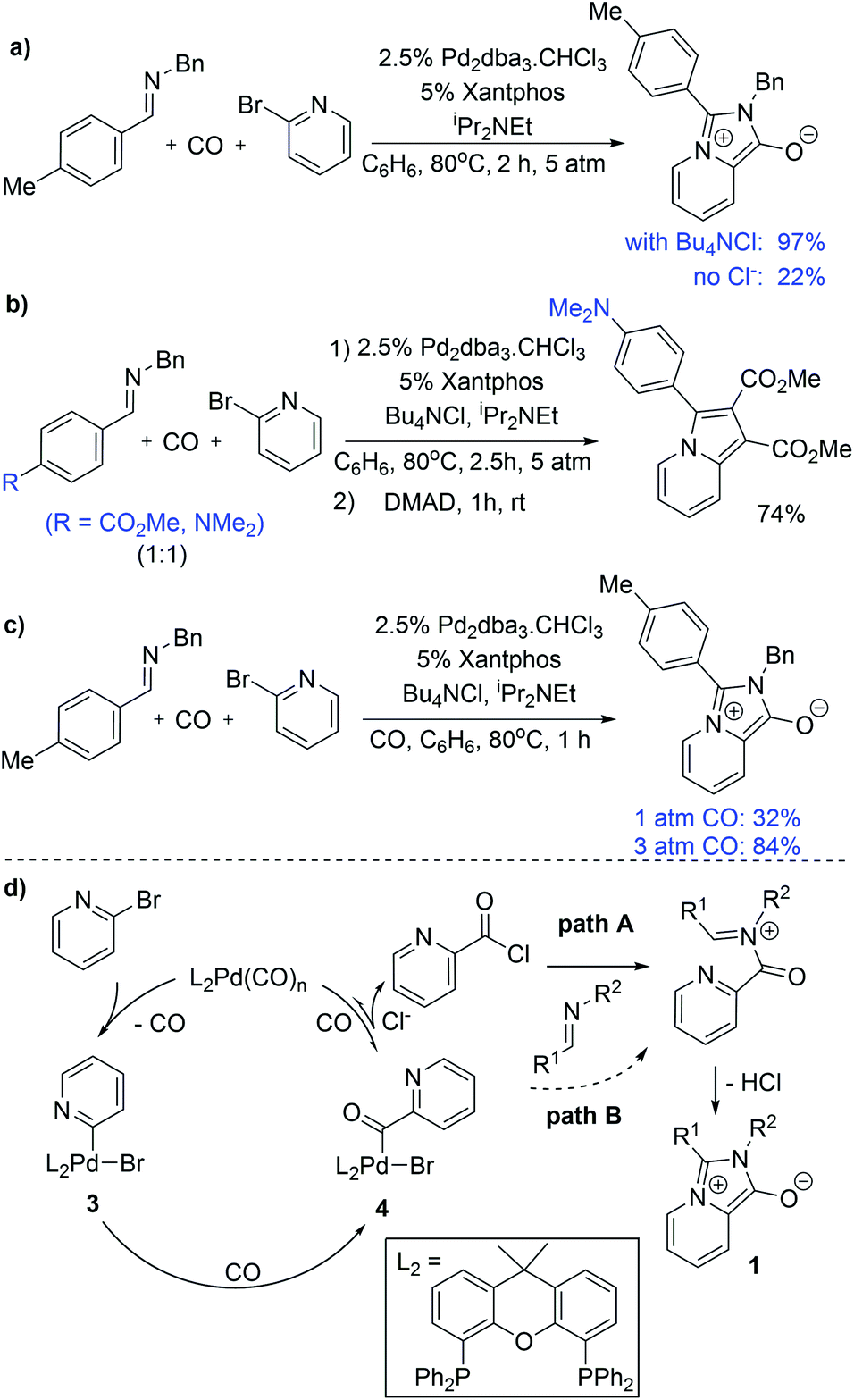 | ||
| Fig. 3 Mechanistic experiments on the palladium catalyzed synthesis of 1,3-dipole 1 and indolizines. (a) Influence of chloride. (b) Competition experiment with varying imines. (c) Influence of CO pressure. (d) Potential reaction mechanism. | ||
On the basis of these experiments, we postulate that the catalytic formation of 1,3-dipole 1 proceeds as shown in Fig. 3d. In this, 2-bromopyridine oxidative addition to Pd(0) followed by CO insertion leads to the formation of the palladium–acyl complex 4. In presence of a chloride source, anion exchange can allow the reversible reductive elimination of acid chloride (path A). The re-addition of acid chloride to Pd(0) is presumably rapid, but can be inhibited by nucleophilic trapping with the imine to generate an N-acyl iminium salt for cyclization to 1,3-dipole 2. The efficiency of the xantphos ligand in catalysis may be tied to its large bite angle (111°),14 which creates significant steric and electronic strain in 4 and can favor reductive elimination of a reactive acid chloride intermediate. Nevertheless, the ability of this system to proceed to product in the absence of chloride implies that the imine can react with other electrophilic intermediates in the reaction, such as the palladium–acyl complex 4 (path B) or potentially an acid bromide, albeit at a slower rate than with acid chloride.
Conclusions
In conclusion, a palladium catalyzed, multicomponent synthesis of indolizines from 2-bromopyridines, CO, imines and alkynes has been developed. In this, carbon monoxide is not incorporated into the final product, but instead serves to first build-up the high energy 1,3-dipole 1, and is then liberated with the nitrogen unit from the imine as an isocyanate. From a synthetic perspective, the reaction has opened a route to prepare indolizines from combinations of stable, tunable reagents, and with the ability to modulate all substituents by variation of the pyridine, imine and alkyne employed. Considering the utility of 1,3-dipoles in synthesis, we anticipate this chemistry could offer a modular route to access a range of fused-ring heterocyclic products.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Natural Sciences and Engineering Research Council of Canada (NSERC), the Canadian Foundation for Innovation (CFI), McGill University (James McGill Research Fund), and the Fonds de recherche du Québec – Nature et Technologies (FQRNT) supported Centre for Green Chemistry and Catalysis for funding this research.Notes and references
- (a) C. F. J. Barnard, Organometallics, 2008, 27, 5402–5422 CrossRef CAS; (b) A. Brennführer, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 4114–4133 CrossRef; (c) M. Beller and X.-F. Wu, Transition Metal Catalyzed Carbonylation Reactions, Springer, 2013 CrossRef; (d) Y. Li, Y. Hu and X.-F. Wu, Chem. Soc. Rev., 2018, 47, 172–194 RSC; (e) Y. Bai, D. C. Davis and M. Dai, J. Org. Chem., 2017, 82, 2319–2328 CrossRef CAS; (f) C. Zhu, J. Liu, M.-B. Li and J.-E. Bäckvall, Chem. Soc. Rev., 2020, 49, 341–353 RSC.
- Recent examples of acyl (pseudo)halides: (a) T. A. Cernak and T. H. Lambert, J. Am. Chem. Soc., 2009, 131, 3124–3125 CrossRef CAS; (b) J. S. Quesnel and B. A. Arndtsen, J. Am. Chem. Soc., 2013, 135, 16841–16844 CrossRef CAS; (c) T. Ueda, H. Konishi and K. Manabe, Org. Lett., 2013, 15, 5370–5373 CrossRef CAS; (d) J. S. Quesnel, L. V. Kayser, A. Fabrikant and B. A. Arndtsen, Chem.–Eur. J., 2015, 21, 9550–9555 CrossRef CAS; (e) J. S. Quesnel, S. Moncho, K. E. O. Ylijoki, G. M. Torres, E. N. Brothers, A. A. Bengali and B. A. Arndtsen, Chem.–Eur. J., 2016, 22, 15107–15118 CrossRef CAS; (f) X. Fang, B. Cacherat and B. Morandi, Nat. Chem., 2017, 9, 1105 CrossRef CAS; (g) Y. H. Lee and B. Morandi, Nat. Chem., 2018, 10, 1016–1022 CrossRef CAS; (h) M. De La Higuera Macias and B. A. Arndtsen, J. Am. Chem. Soc., 2018, 140, 10140–10144 CrossRef CAS; (i) D. R. Gauthier Jr, N. R. Rivera, H. Yang, D. M. Schultz and C. S. Shultz, J. Am. Chem. Soc., 2018, 140, 15596–15600 CrossRef; (j) R. G. Kinney, J. Tjutrins, G. M. Torres, N. J. Liu, O. Kulkarni and B. A. Arndtsen, Nat. Chem., 2018, 10, 193–199 CrossRef; (k) G. M. Torres, Y. Liu and B. A. Arndtsen, Science, 2020, 368, 318–323 CrossRef CAS.
- Other electrophiles: (a) J. R. Martinelli, T. P. Clark, D. A. Watson, R. H. Munday and S. L. Buchwald, Angew. Chem., Int. Ed., 2007, 46, 8460–8463 CrossRef CAS; (b) A. Wiȩckowska, R. Fransson, L. R. Odell and M. Larhed, J. Org. Chem., 2011, 76, 978–981 CrossRef; (c) T. Ueda, H. Konishi and K. Manabe, Org. Lett., 2012, 14, 5370–5373 CrossRef CAS; (d) F. M. Miloserdov and V. V. Grushin, Angew. Chem., Int. Ed., 2012, 51, 3668–3672 CrossRef CAS; (e) M. N. Burhardt, R. H. Taaning and T. Skrydstrup, Org. Lett., 2013, 15, 948–951 CrossRef CAS; (f) F. M. Miloserdov, C. L. McMullin, M. M. N. Belmonte, J. Benet-Buchholz, V. I. Bakhmutov, S. A. Macgregor and V. V. Grushin, Organometallics, 2014, 33, 736–752 CrossRef CAS; (g) J. S. Quesnel, A. Fabrikant and B. A. Arndtsen, Chem. Sci., 2016, 7, 295–300 RSC; (h) Y. Wang and V. Gevorgyan, Angew. Chem., Int. Ed., 2017, 56, 3191–3195 CrossRef CAS; (i) P.-L. Lagueux-Tremblay, A. Fabrikant and B. A. Arndtsen, ACS Catal., 2018, 8, 5350–5354 CrossRef CAS.
- (a) A. M. Schmidt and P. Eilbracht, J. Org. Chem., 2005, 70, 5528 CrossRef CAS; (b) S. T. Staben and N. Blaquiere, Angew. Chem., Int. Ed., 2010, 49, 325 CrossRef CAS; (c) B. A. Arndtsen, Chem.–Eur. J., 2009, 15, 302–313 CrossRef CAS; (d) K. Worrall, B. Xu, S. Bontemps and B. A. Arndtsen, J. Org. Chem., 2011, 76, 170–180 CrossRef CAS; (e) S. Bontemps, J. S. Quesnel, K. Worrall and B. A. Arndtsen, Angew. Chem., Int. Ed., 2011, 50, 8948–8951 CrossRef CAS; (f) G. M. Torres, J. S. Quesnel, D. Bijou and B. A. Arndtsen, J. Am. Chem. Soc., 2016, 138, 7315–7324 CrossRef CAS; (g) J. Tjutrins and B. A. Arndtsen, Chem. Sci., 2017, 8, 1002–1007 RSC; (h) D. C. Leitch, L. V. Kayser, H.-Y. Han, A. R. Siamaki, E. N. Keyzer, A. Gefen and B. A. Arndtsen, Nat. Commun., 2015, 6, 7411 CrossRef CAS.
- H. Erguven, D. C. Leitch, E. N. Keyzer and B. A. Arndtsen, Angew. Chem., Int. Ed., 2017, 129, 6174–6178 CrossRef.
- (a) E. Besthorn and G. Jaeglé, Ber. Dtsch. Chem. Ges., 1894, 27, 907–914 CrossRef; (b) F. Krollpfeiffer and K. Schneider, Justus Liebigs Ann. Chem., 1937, 530, 34–50 CrossRef CAS; (c) B. R. Brown and E. H. Wild, J. Chem. Soc., 1956, 1158–1163 RSC.
- For reviews: (a) T. Uchida and K. Matsumoto, Synthesis, 1976, 209–236 CrossRef CAS; (b) B. Sadowski, J. Klajn and D. T. Gryko, Org. Biomol. Chem., 2016, 14, 7804–7828 RSC.
- (a) G. S. Singh and E. E. Mmatli, Eur. J. Med. Chem., 2011, 46, 5237–5257 CrossRef CAS; (b) V. Sharma and V. Kumar, Med. Chem. Res., 2014, 23, 3593–3606 CrossRef CAS; (c) J. P. Michael, in The Alkaloids: Chemistry and Biology, ed. H.-J. Knölker, Academic Press, 2016, vol. 75, pp. 1–498 Search PubMed; (d) W.-G. Lee, R. Gallardo-Macias, K. M. Frey, K. A. Spasov, M. Bollini, K. S. Anderson and W. L. Jorgensen, J. Am. Chem. Soc., 2013, 135, 16705–16713 CrossRef CAS; (e) W. Huang, T. Zuo, X. Luo, H. Jin, Z. Liu, Z. Yang, X. Yu, L. Zhang and L. Zhang, Chem. Biol. Drug Des., 2013, 81, 730–741 CrossRef CAS.
- (a) E. Kim, Y. Lee, S. Lee and S. B. Park, Acc. Chem. Res., 2015, 48, 538–547 CrossRef CAS; (b) A. J. Huckaba, F. Giordano, L. E. McNamara, K. M. Dreux, N. I. Hammer, G. S. Tschumper, S. M. Zakeeruddin, M. Grätzel, M. K. Nazeeruddin and J. H. Delcamp, Adv. Energy Mater., 2015, 5, 1401629 CrossRef.
- For recent examples: (a) S. Adachi, S. K. Liew, C. F. Lee, A. Lough, Z. He, J. D. S. Denis, G. Poda and A. K. Yudin, Org. Lett., 2015, 17, 5594–5597 CrossRef CAS; (b) S. Tang, K. Liu, Y. Long, X. Gao, M. Gao and A. Lei, Org. Lett., 2015, 17, 2404–2407 CrossRef CAS; (c) X. Wu, P. Zhao, X. Geng, J. Zhang, X. Gong, Y.-D. Wu and A.-X. Wu, Org. Lett., 2017, 19, 3319–3322 CrossRef CAS; (d) H. Li, X. Li, Y. Yu, J. Li, Y. Liu, H. Li and W. Wang, Org. Lett., 2017, 19, 2010–2013 CrossRef CAS; (e) D. Yang, Y. Yu, Y. Wu, H. Feng, X. Li and H. Cao, Org. Lett., 2018, 20, 2477–2480 CrossRef CAS; (f) F. Penteado, C. S. Gomes, G. Perin, C. S. Garcia, C. F. Bortolatto, C. A. Brüning and E. J. Lenardão, J. Org. Chem., 2019, 84, 7189–7198 CrossRef CAS.
- Metal catalyzed examples: (a) V. Mamane, P. Hannen and A. Fürstner, Chem.–Eur. J., 2004, 10, 4556–4575 CrossRef CAS; (b) S. Chuprakov, F. W. Hwang and V. Gevorgyan, Angew. Chem., Int. Ed., 2007, 46, 4757–4759 CrossRef CAS; (c) Y. Liu, Z. Song and B. Yan, Org. Lett., 2007, 9, 409–412 CrossRef CAS; (d) T. Schwier, A. W. Sromek, D. M. L. Yap, D. Chernyak and V. Gevorgyan, J. Am. Chem. Soc., 2007, 129, 9868–9878 CrossRef CAS; (e) B. Yan and Y. Liu, Org. Lett., 2007, 9, 4323–4326 CrossRef CAS; (f) J. Barluenga, G. Lonzi, L. Riesgo, L. A. López and M. Tomás, J. Am. Chem. Soc., 2010, 132, 13200–13202 CrossRef CAS; (g) Y. Yang, C. Xie, Y. Xie and Y. Zhang, Org. Lett., 2012, 14, 957–959 CrossRef CAS; (h) R.-R. Liu, C.-J. Lu, M.-D. Zhang, J.-R. Gao and X.-X. Jia, Chem.–Eur. J., 2015, 21, 7057–7060 CrossRef CAS; (i) L. Zhang, X. Li, Y. Liu and D. Zhang, Chem. Commun., 2015, 51, 6633–6636 RSC; (j) H. Kim, S. Kim, J. Kim, J.-Y. Son, Y. Baek, K. Um and P. H. Lee, Org. Lett., 2017, 19, 5677–5680 CrossRef CAS; (k) M. Meazza, L. A. Leth, J. D. Erickson and K. A. Jørgensen, Chem.–Eur. J., 2017, 23, 7905–7909 CrossRef CAS; (l) T. Jin, Z. Tang, J. Hu, H. Yuan, Y. Chen, C. Li, X. Jia and L. Li, Org. Lett., 2018, 20, 413–416 CrossRef CAS; (m) J. Vaitla, A. Bayer and K. H. Hopmann, Angew. Chem., Int. Ed., 2018, 57, 16180–16184 CrossRef CAS; (n) T. Wu, M. Chen and Y. Yang, J. Org. Chem., 2017, 82, 11304–11309 CrossRef CAS; (o) S. Roy, S. K. Das and B. Chattopadhyay, Angew. Chem., Int. Ed., 2018, 57, 2238–2243 CrossRef CAS; (p) M. D. Rossler, C. T. Hartgerink, E. E. Zerull, B. A. Boss, A. K. Frndak, M. M. Mason, L. A. Nickerson, E. O. Romero, J. E. Van de Burg, R. J. Staples and C. E. Anderson, Org. Lett., 2019, 21, 5591–5595 CrossRef CAS.
- For 1,3-dipolar cycloaddition routes to indolizines: (a) V. Boekelheide and K. Fahrenholtz, J. Am. Chem. Soc., 1961, 83, 458–462 CrossRef CAS; (b) A. V. Gulevskaya and J. I. Nelina-Nemtseva, Chem. Heterocycl. Compd., 2018, 54, 1084–1107 CrossRef CAS.
- For recent examples: (a) D. I. Chai and M. Lautens, J. Org. Chem., 2009, 74, 3054–3061 CrossRef CAS; (b) H. Zhu, J. Stöckigt, Y. Yu and H. Zou, Org. Lett., 2011, 13, 2792–2794 CrossRef CAS; (c) L. H. Phun, J. Aponte-Guzman and S. France, Angew. Chem., Int. Ed., 2012, 51, 3198–3202 CrossRef CAS; (d) J.-R. Huang, Q.-R. Zhang, C.-H. Qu, X.-H. Sun, L. Dong and Y.-C. Chen, Org. Lett., 2013, 15, 1878–1881 CrossRef CAS; (e) M. Kim, Y. Jung and I. Kim, J. Org. Chem., 2013, 78, 10395–10404 CrossRef CAS; (f) M. Kucukdisli and T. Opatz, J. Org. Chem., 2013, 78, 6670–6676 CrossRef CAS; (g) J. H. Lee and I. Kim, J. Org. Chem., 2013, 78, 1283–1288 CrossRef CAS; (h) W. Hao, H. Wang, Q. Ye, W.-X. Zhang and Z. Xi, Org. Lett., 2015, 17, 5674–5677 CrossRef CAS; (i) V. K. Outlaw, F. B. d'Andrea and C. A. Townsend, Org. Lett., 2015, 17, 1822–1825 CrossRef CAS; (j) X. Li, X. Xie and Y. Liu, J. Org. Chem., 2016, 81, 3688–3699 CrossRef CAS; (k) X. Li, J. Zhao, X. Xie and Y. Liu, Org. Biomol. Chem., 2017, 15, 8119–8133 RSC; (l) T. Lepitre, R. Le Biannic, M. Othman, A. M. Lawson and A. Daïch, Org. Lett., 2017, 19, 1978–1981 CrossRef CAS; (m) A. S. Kulandai Raj, K.-C. Tan, L.-Y. Chen, M.-J. Cheng and R.-S. Liu, Chem. Sci., 2019, 10, 6437–6442 RSC.
- P. C. J. Kamer, P. W. N. M. van Leeuwen and J. N. H. Reek, Acc. Chem. Res., 2001, 34, 895–904 CrossRef CAS.
- Attempts at using 2-chloropyridine as replacements for 2-bromopyridine and Bu4NCl were unsuccessful under these catalytic conditions, potentially due to the more challenging oxidative addition of the C–Cl bond.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc03977b |
| This journal is © The Royal Society of Chemistry 2021 |