
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupramolecular adducts between macrocyclic Gd(III) complexes and polyaromatic systems: a route to enhance the relaxivity through the formation of hydrophobic interactions†
Enza
Di Gregorio
 a,
Luciano
Lattuada
b,
Alessandro
Maiocchi
b,
Silvio
Aime
a,
Giuseppe
Ferrauto‡
*a and
Eliana
Gianolio‡
*a
a,
Luciano
Lattuada
b,
Alessandro
Maiocchi
b,
Silvio
Aime
a,
Giuseppe
Ferrauto‡
*a and
Eliana
Gianolio‡
*a
aMolecular Imaging Center, Department of Molecular Biotechnologies and Health Sciences, University of Torino, Via Nizza 52, Torino, 10126, Italy. E-mail: giuseppe.ferrauto@unito.it
bBracco Imaging Spa, Bracco Research Centre, Via Ribes 5, Colleretto Giacosa, TO 10010, Italy
First published on 17th November 2020
Abstract
The set-up of reversible binding interactions between the hydrophobic region of macrocyclic GBCAs (Gadolinium Based Contrast Agents) and SO3−/OH containing pyrene derivatives provides new insights for pursuing relaxivity enhancements of this class of MRI contrast agents. The strong binding affinity allows attaining relaxation enhancements up to 50% at pyrene/GBCA ratios of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. High resolution NMR spectra of the Yb-HPDO3A/pyrene system fully support the formation of a supramolecular adduct based on the set-up of hydrophobic interactions. The relaxation enhancement may be accounted for in terms of the increase of the molecular reorientation time (τR) and the number of second sphere water molecules. This effect is maintained in blood serum and in vivo, as shown by the enhancement of contrast in T1w-MR images obtained by simultaneous injection of GBCA and pyrene derivatives in mice.
:1. High resolution NMR spectra of the Yb-HPDO3A/pyrene system fully support the formation of a supramolecular adduct based on the set-up of hydrophobic interactions. The relaxation enhancement may be accounted for in terms of the increase of the molecular reorientation time (τR) and the number of second sphere water molecules. This effect is maintained in blood serum and in vivo, as shown by the enhancement of contrast in T1w-MR images obtained by simultaneous injection of GBCA and pyrene derivatives in mice.
Introduction
Gd(III) complexes are routinely used in clinical settings as MRI contrast agents (CAs).1–4 Upon i.v. administration, they display vascular and extravascular, and extracellular distribution with a quick renal excretion (>90% after 12 h).5 They are considered among the safest xenobiotics as the number of adverse effects is definitively very low.6 However, in the last few years, there is some growing concern due to observations reporting that tiny amounts of the administered Gd can be retained in the brain and other tissues.5,7–10 Although no clinical evidence could be related to the occurrence of retained Gd, these findings opened a discussion on the potential risks associated with the in vivo release of Gd from the administered CAs. Considerations based on the thermodynamic and kinetic stabilities pointed out that macrocyclic Gd(III) complexes should be preferred to the linear ones in order to minimize the amount of free gadolinium released.11 Besides a good in vivo tolerability, the property of choice for these agents is represented by the relaxivity (r1p), that is, the relaxation enhancement brought to water protons in the presence of 1 mM concentration of the paramagnetic Gd3+ containing chelates.12,13The three commercial macrocyclic Gd(III) contrast agents (Scheme 1) display a relatively similar relaxivity in plasma (r1 = 3.6–5.2 mM−1 s−1 at 37 °C, 1.5 T).14
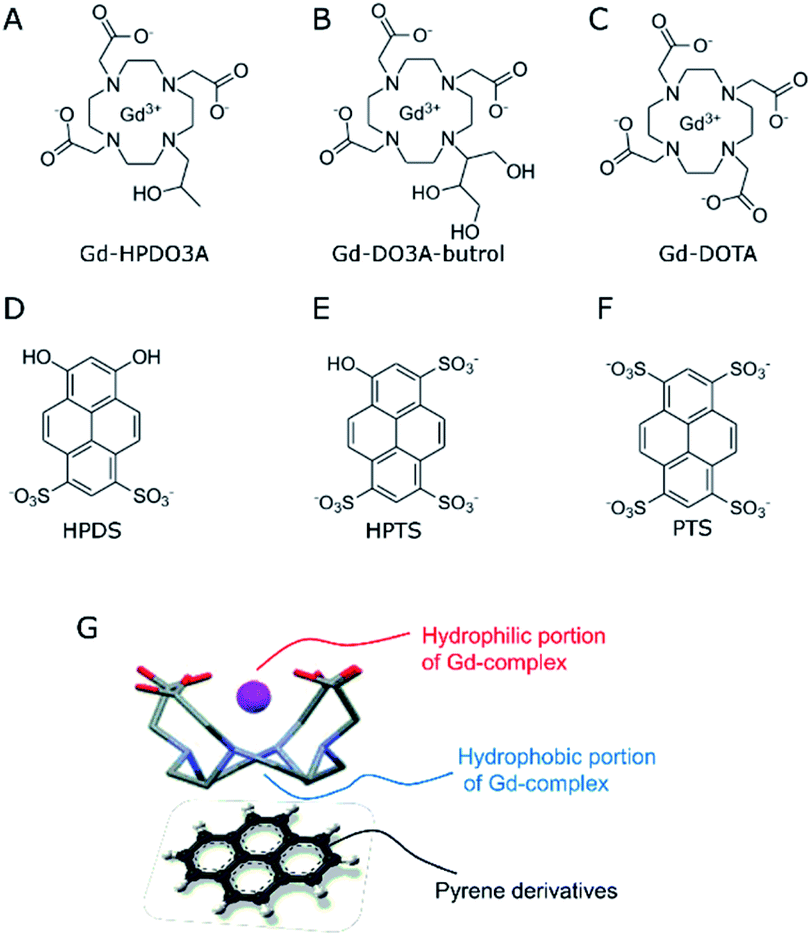 | ||
| Scheme 1 (A–C) Chemical structures of macrocyclic Gd-based contrast agents; (D–F) chemical structures of hydrophilic pyrene derivatives. (G) Sketched structure of the supramolecular adduct between Gd-complexes and pyrene derivatives. | ||
Obviously, the possibility of enhancing the relaxivity would be considered an interesting goal as it would allow the use of lower doses. Much work has been carried out over the years to identify which are the structural and dynamic determinants for improving the relaxivity of Gd-containing agents.15–17 It was shown that, at the magnetic fields of the clinical scanners (i.e. in the 0.5–3 T range), the slowing down of the molecular motion may be a useful way to enhance the relaxivity provided that the exchange rate of the coordinated water molecule is in the suitable range.18 The lengthening of the molecular reorientational time may be pursued through the set-up of reversible binding interactions with suitable interacting species.19–21 The three complexes whose structures are reported in Scheme 1 represent highly hydrophilic systems without any specific ability of binding to endogenous molecules able to affect, in a significant amount, their relaxivities.22 With the aim of exploring possible routes to form supramolecular systems endowed with a lengthening of the reorientational motion with respect to the parent complex, we considered the possibility of exploiting the hydrophobic characteristic of the tetra-aza macrocyclic moiety.
Actually, this region of the macrocyclic Gd(III) complexes is considered not to contribute to the observed relaxivity as its hydrophobicity is expected to push out second sphere water molecules. However, one may envisage its use for designing novel supramolecular architectures. Herein, we report our observations concerning the supramolecular adducts between the three macrocyclic complexes and substituted pyrene derivatives (Scheme 1). The most known to biologists is pyranine (HPTS, 8-hydroxypyrene-1,3,6-trisulfonic acid, Scheme 1E), as it is often used as a pH-sensitive fluorescent dye in cellular studies.23,24
The investigated pyrene derivatives contain on their outer perimeter SO3− and OH moieties that endow them with a good solubility in water. They are sites for gathering second sphere water molecules that may eventually contribute to enhancing the observed relaxivity.
The interaction occurring between macrocyclic lanthanide(III) complexes and pyrene derivatives has been investigated by using both 1H-relaxometry at variable magnetic field and 1H high resolution NMR.
Results and discussion
1H-relaxometry
Fig. 1A reports the increase of relaxivity upon the addition of HPTS to solutions containing six different clinically approved GBCAs: gadoteridol (Gd-HPDO3A), gadobutrol (Gd-DO3A-Butrol), gadoterate (Gd-DOTA), gadopentetate (Gd-DTPA), gadodiamide (Gd-DTPA-BMA) and gadobenate (Gd-BOPTA).The analysis of each titration curve allowed us to extract, on the basis of the assumption of the occurrence of 1:1 adducts, the values for the affinity constant (Ka) and the relaxivity of the adduct (Rb). The binding parameters were calculated by using the well-established Proton Relaxation Enhancement (PRE) technique, i.e. by fitting the observed R1 values vs. substrate concentration binding isotherms.25,26
The obtained results indicated that Gd-DO3A-Butrol showed the highest binding affinity toward HPTS (Ka = 1157 M−1), followed by Gd-HPDO3A (Ka = 628 M−1) and Gd-DOTA (Ka = 174 M−1) (Fig. 1A and Table 1).
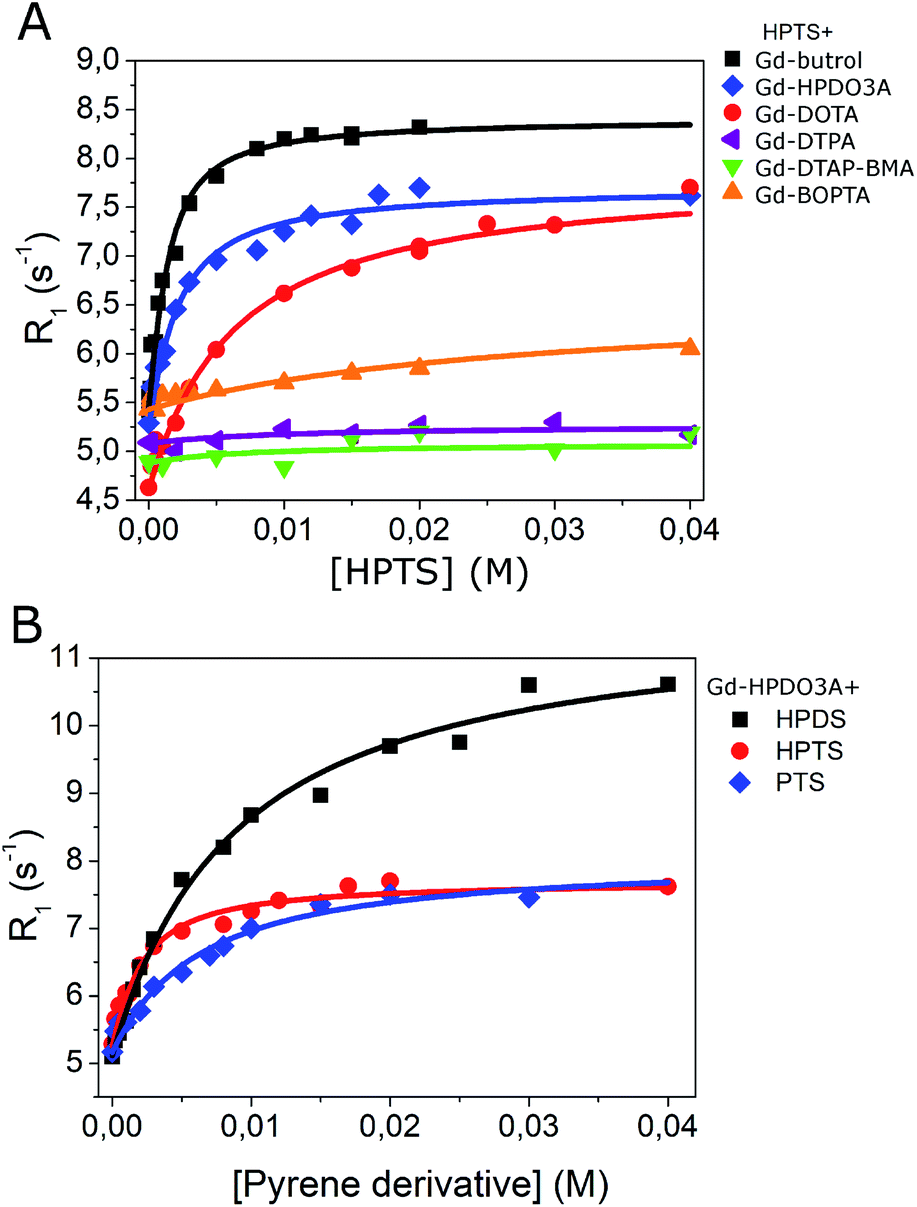 | ||
| Fig. 1 (A) R1 of GBCAs (1 mM) in the presence of HPTS at variable concentration (0–40 mM); (B) R1 of Gd-HPDO3A (1 mM) in the presence of pyrene derivatives at variable concentration (1–40 mM). B0 = 0.5 T, T = 25 °C, pH = 7.0 ± 0.1. | ||
| r 1p (mM−1 s−1) | K a (M−1) | r b (mM−1 s−1) |
R
1 (s−1) GBCA:pyrene (1:3) |
|
|---|---|---|---|---|
| a r 1p = relaxivity for 1 mM Gd-complex in the absence of pyrenes. | ||||
| Gd-HPDO3A/HPTS | 4.6 | 628 | 6.9 | 6.7 |
| Gd-DO3A-Butrol/HPTS | 5.0 | 1157 | 7.9 | 7.6 |
| Gd-DOTA/HPTS | 4.2 | 174 | 7.4 | 5.7 |
| Gd-DTPA/HPTS | No interaction | |||
| Gd-DTPABMA/HPTS | No interaction | |||
| Gd-BOPTA/HPTS | No interaction | |||
| Gd-HPDO3A/HPDS | 4.6 | 124 | 10.9 | 6.9 |
| Gd-HPDO3A/PTS | 4.6 | 177 | 7.3 | 6.1 |
As far as the relaxation enhancement is concerned, besides the Rb values (i.e. the relaxivity values of the supramolecular adducts), also the relaxation enhancements attained when the Gd(III) complex and the pyrene molecule are in the 1:3 molar ratio are reported (Table 1 last column). This ratio was chosen because it seems a good compromise to achieve sufficient formation of the supramolecular adduct without having to increase too much the concentration of HPTS.
Analogous titrations were carried out by using linear GBCAs, i.e. Gd-DTPA, Gd-DTPA-BMA and Gd-BOPTA (chemical structures in Fig. S1†) with HPTS. No increase of the observed relaxation rate was observed, to support the view that the interaction observed in the case of Gd-DO3A-Butrol, Gd-DOTA and Gd-HPDO3A occurs because of the presence of the hydrophobic region in macrocyclic compounds (Fig. 1A and Table 1).
Gd-HPDO3A was also titrated with HPDS (6,8-hydroxypyrene-1,3-disulfonic acid), and PTS (1,3,6,8-pyrenetetrasulfonic acid) (Fig. 1B) obtaining Ka values of 124 M−1 and 177 M−1, respectively (Table 1).
More insights into the understanding of the observed relaxation enhancements were gained by recording 1/T1 NMRD profiles (where 1/T1 = R1). Fig. 2 reports the 1/T1 NMRD profiles acquired over an extended range of magnetic field strengths (from 0.01 to 80 MHz when expressed in terms of proton larmor frequencies) for a solution of Gd-HPDO3A (1 mM) and HPTS (3 mM) compared to that of the parent Gd-HPDO3A alone.
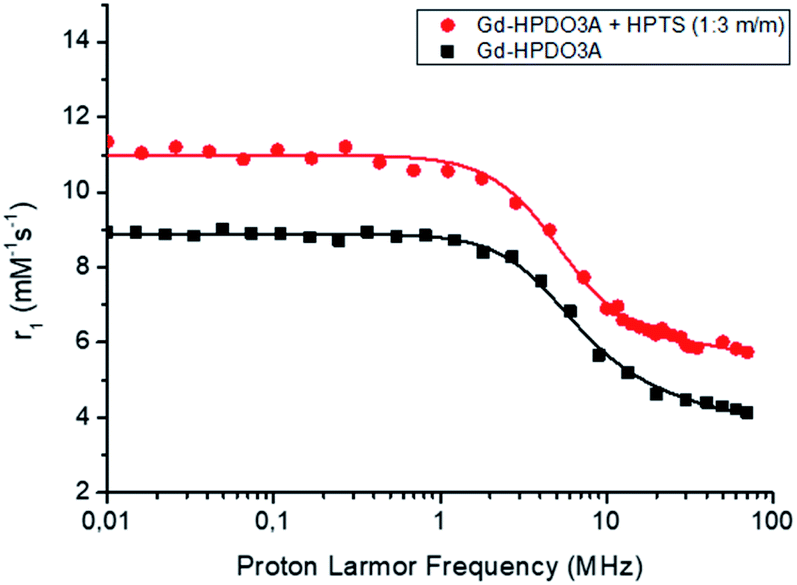 | ||
Fig. 2
1H-NMRD profiles of Gd-HPDO3A alone (■) and in the presence of 3 mM HPTS ( ) ([Gd-complex] = 1 mM, in Hepes/NaCl, pH = 7.0, T = 25 °C). ) ([Gd-complex] = 1 mM, in Hepes/NaCl, pH = 7.0, T = 25 °C). | ||
The obtained profile of the Gd-HPDO3A/HPTS adduct was nicely fitted to the values calculated based on the established Solomon–Bloembergen–Morgan (SBM) theory and is reported in Table 2. When compared with data obtained for the parent Gd-HPDO3A, two parameters appear to be responsible for the relaxation enhancement observed in the presence of HPTS, namely τR that increases from 69 to 94 ps and the number of second sphere water molecules (qss) that moves from 0 to 2.5, respectively. These findings are fully consistent with the reversible formation in solution of a supramolecular adduct between the macrocyclic Gd(III) complex and the substituted pyrene system. The increase of the second sphere term appears reasonable as the charged SO3− moiety and the neutral OH one could act as hydration sites in a region that was not considered to contribute to the observed relaxivity in the parent complex. We surmise that the more cylindrical structure of the adduct may involve changes in the isotropic reorientational motion of Gd-HPDO3A eventually giving more relevance to the motion of the coordinated water molecule along the cylindrical symmetry axis. This property is at the basis of the high relaxivity previously shown by gadolinium complexes of symmetrical tetrasubstituted DOTA derivatives.29–31
| Δ 2 (s−2) (×1019) | τ v (ps) | τ R (ps) | τ M (ns) | q ss | |
|---|---|---|---|---|---|
| a The following parameters were fixed during the fitting procedure: q = 1, rGdH = 3.1 Å, aGdH = 3.8 Å, D = 2.24 × 10−5 cm2 s−1, rGdH (2nd sphere) = 4 Å, τc (2nd sphere) = 60 ps. b τ M was fixed to the value of the derived weighted average of τM for the two SAP/TSAP isomers.27,28 | |||||
| ProHance | 1.31 ± 0.16 | 38 ± 4.1 | 69 ± 1.7 | 450b | 0 |
| ProHance + HPTS | 3.22 ± 0.81 | 22 ± 3.8 | 94 ± 5.5 | 450b | 2.5 |
The supramolecular adduct was also tested for its kinetic stability by measuring the occurrence of transmetallation in the presence of zinc chloride in phosphate buffer according to a previously reported relaxometric procedure.32 To this aim, Gd-HPDO3A 1 mM (in phosphate buffer) was mixed with an excess of ZnCl2 (10 mM). The experiment was carried out for Gd-HPDO3A free or in the presence of HPTS (1:3 Gd-HPDO3A:HPTS). As reported in Fig. S2,† no effect of transmetallation was observed in an experiment carried out at 37 °C over a period of 8 days. Hence, we can assume that no difference in kinetic stability is occurring upon the formation of the supramolecular adduct.
High resolution 1H-NMR spectra
Further support to the proposed supramolecular structure was gained from the high resolution 1H-NMR spectra of Yb-HPDO3A in the presence of HPTS (Fig. 3).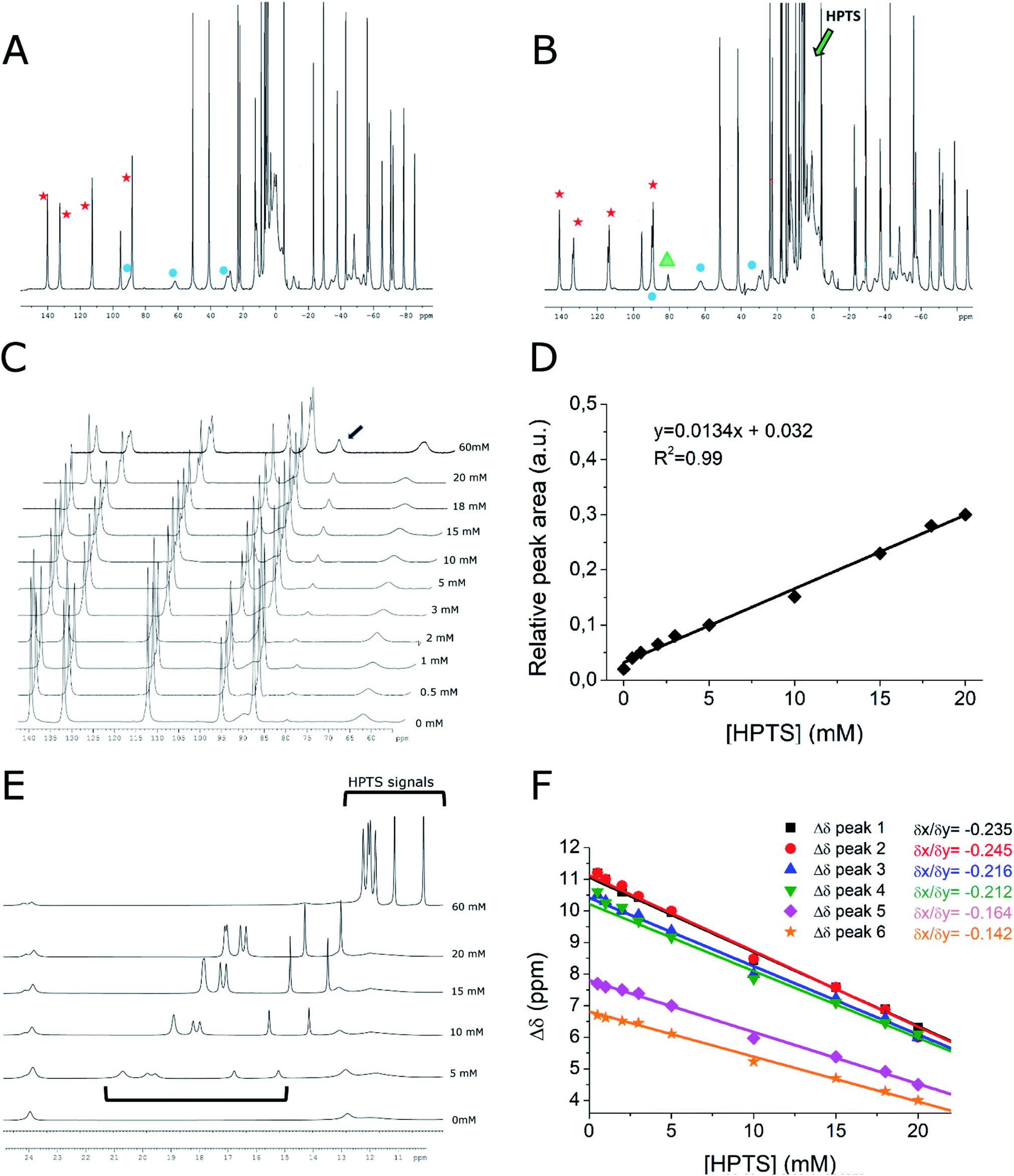 | ||
| Fig. 3 1H-NMR spectra (B0 = 14 T, 15 °C) of Yb-HPDO3A (20 mM, pH = 7) (A) and Yb-HPDO3A (20 mM, pH = 7) + HPTS (20 mM) (B). Red stars indicate axial SAP signals, blue circles indicate axial TSAP signals, green arrow indicates the region of HPTS signals. The green triangle indicates the new occurring signal when Yb-HPDO3A is placed in the presence of HPTS. (C) 1H-NMR spectra of Yb-HPDO3A/HPTS resonances: [Yb-HPDO3A] (20 mM, pH = 7) in the presence of increasing amounts of HPTS (from 0 to 60 mM HPTS). The black arrow indicates the new occurring signal. (D) Relative area of the peak at 81 ppm (the one indicated with an arrow in C) upon increasing the concentrations of HPTS. (E and F) 1H-NMR spectra and shifts of HPTS proton resonances at variable concentrations of HPTS. | ||
In the presence of HPTS, Yb-HPDO3A maintains almost the same proportion of SAP (Square AntiPrism, 70% red stars in Fig. 3) and TSAP (Twisted Square AntiPrism, 30%, blue circles in Fig. 3A and B). Actually, minor shifts are observed and a small splitting of the most shifted (axial) resonances is easily detectable (Fig. 3B and S3†). These effects can be accounted for in terms of the formation of the adduct with HPTS.
In particular, the splitting may be related to the differences that substituted pyrene adopts in its binding mode to the tetra-aza-macrocycle which is asymmetric on its own due to the presence of three acetic and one hydroxypropyl-arms. Interestingly, the titration with HPTS causes the progressive increase of another minor species (Fig. 3C, highlighted in green) which, although previously not noticed, is already present in the spectrum of the parent Yb-HPDO3A and it became more evident in the presence of the binding to HPTS (Fig. 3A and B green triangle).
The increase in the concentration of such species can be easily detected by following the peak at 81 ppm whose relative area increases 15 times by passing from a solution containing only Yb-HPDO3A to the one containing Yb-HPDO3A/HPTS in 1:1 molar ratio (Fig. 3C and D). On the basis of the available evidence, we may surmise that this additional species may represent another isomer (eventually present in a diastereoisomeric pair). At the moment, it does not appear possible to make any suggestion for its structure on the basis of the available data.
Further insights can be extrapolated from the analysis of the line broadening of the shifted 1H-NMR peaks of Yb-HPDO3A upon the formation of the adduct with HPTS.33,34 The increase in the reorientational correlation time (τR) of the supramolecular adduct with respect to the free Yb-complex is, in fact, reflected in the broadening of NMR resonances as a consequence of the decrease of their transversal relaxation time (T2). We measured the increase in signal linewidth (and calculated R2, being equal to πΔν1/2) of selected proton resonances as a function of the addition of increasing amounts of HPTS to Yb-HPDO3A (Fig. S4†). From the analysis of these data, it was possible to estimate the increase in τR obtained upon the formation of the adduct. From this experiment we calculated an increase in R2 of axial SAP protons, and thus in τR, of ca. 40%, which perfectly recalls the increase in τR value obtained from the fitting of NMRD profiles of the adduct Gd-HPDO3A/HPTS (τR = 94 ps) with respect to the free Gd-complex (τR = 69 ps). Very interestingly, the peaks of axial TSAP ring protons are not broadened to the same extent, to suggest a preferential interaction of HPTS toward the SAP structure isomer.
Very instructive was the detection of the six resonances of the HPTS as their chemical shifts move towards the paramagnetic region as the concertation ratio of HPTS/Yb-HPDO3A decreases, as expected in the case of a diamagnetic molecule reversibly interacting with a paramagnetic system (Fig. 3E and F). The observation that all six HPTS resonances behave in an analogous way supports the view that HPTS binding to the tetra-aza-macrocycle occurs at the bottom of the lanthanide(III)-HPDO3A macrocycle through its parallel positioning to the N-4 containing macrocycle (Scheme 1G).
In vivo MRI
As shown above, the good binding affinity between HPTS and Gd-HPDO3A implies that a relatively high percentage (>90%) of the adduct is present already at HPTS/Gd-HPDO3A ratio of 3:1. In serum such a system yielded a 40% relaxation enhancement with respect to the value observed in the absence of HPTS (i.e. r1p of Gd-HPDO3A in serum is 6.5 mM−1 s−1 and r1p of the adduct Gd-HPDO3A/HPTS is 9.2 mM−1 s−1, T = 25 °C, B0 = 0.5 T, Fig. S5†). On this basis it was decided to test the Gd-HPDO3A/HPTS (1:3 mol/mol) adduct on a murine model of subcutaneous cancer.
The model was prepared by inoculating TS/A cells35 under the skin of 10–12 week old female Balb/c mice.36 MRI was carried out 3 weeks after cell injection, when the tumor mass reached a volume of ca. 700 mm3.37
It was found that the signal enhancement in the tumor xenograft, immediately after the i.v. administration of the Gd-HPDO3A/HPTS adduct, was 80% (N = 3). As a comparison, the signal enhancement after the i.v. administration of the same dose of Gd-HPDO3A (w/o HPTS) was 42% (N = 3) (Fig. 4E, S6 and S7†).
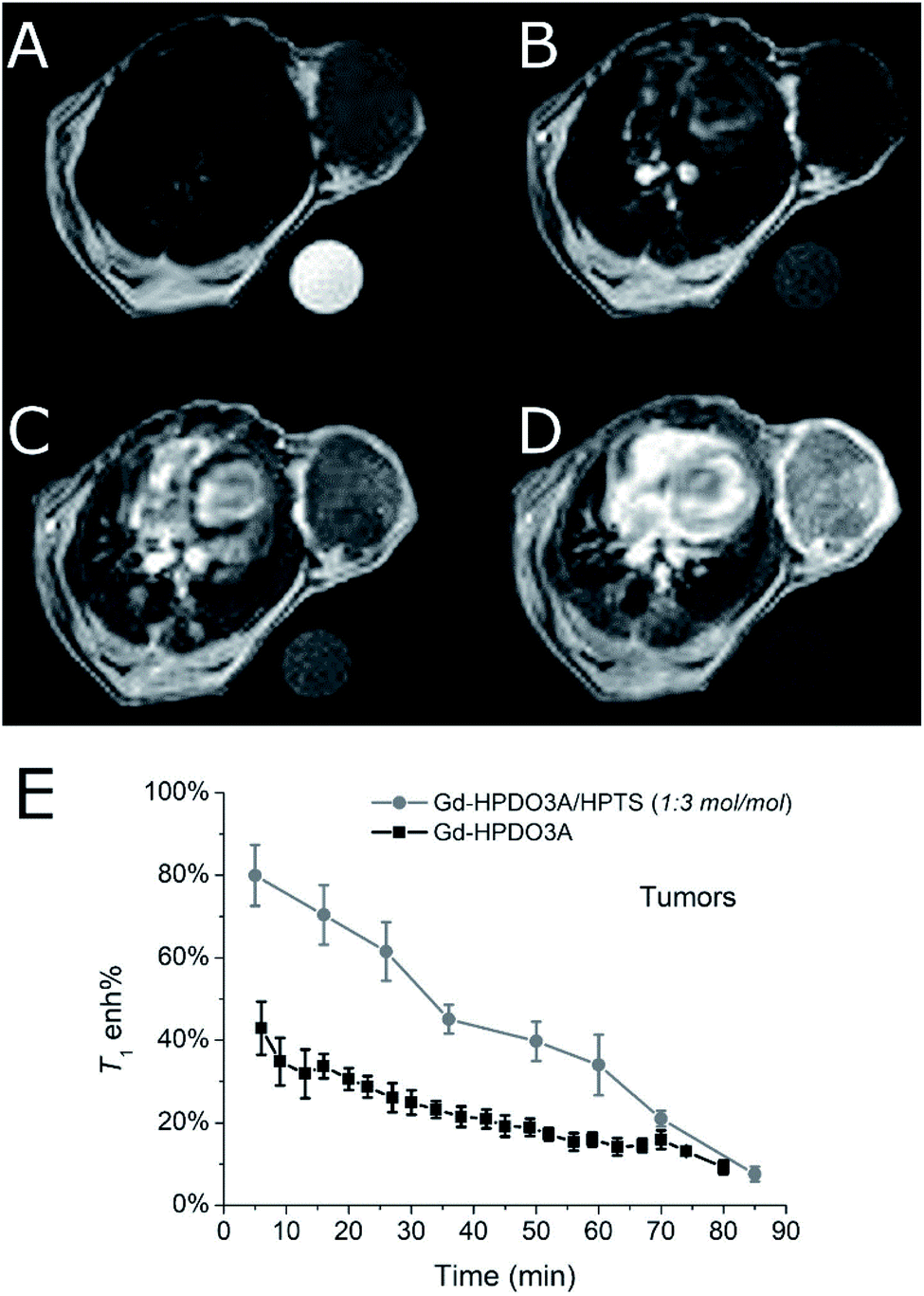 | ||
| Fig. 4 Representative in vivo axial MR images of the tumor region in a Balb/c mouse bearing subcutaneous TS/A tumor. (A) T2w MR image, (B) uncontrasted (pre) T1w MR image, (C) T1w MR image 2 min after the injection of Gd-HPDO3A (0.15 mmol kg−1) and (D) T1w MR image 2 min after the co-injection of Gd-HPDO3A (0.15 mmol kg−1) and HPTS (0.45 mmol kg−1). B0 = 7.1 T, room temperature. (E) Tumor enhancement curves after injection of Gd-HPDO3A (0.15 mmol kg−1) or Gd-HPDO3A/HPTS adduct (0.15 mmol kg−1/0.45 mmol kg−1, 1:3 mol/mol) (mean of 3 animals). | ||
Furthermore, insights into the biodistribution of the adduct have been gained from these preliminary MRI acquisitions. The elimination pathway of the adduct appears to be similar to that of the free Gd-HPDO3A complex, i.e. through kidneys/bladder, with similar kinetics (Fig. S8†).
Cell toxicity
In order to assess the eventual cell toxicity of the supramolecular adduct, MTT assays have been carried out, as previously reported.38TS/A cells were incubated in the presence of (i) Gd-HPDO3A, (ii) Gd-HPDO3A/HPTS, (iii) Gd-HPDO3A/HDPS or (iv) Gd-HPDO3A/PTS in the 1:3 mol/mol stoichiometric ratio, at four different concentrations (Fig. 5). The obtained results show a cell viability higher than 95% for all the tested concentrations and for all the investigated adducts, i.e. well comparable to the one reported upon incubating Gd-HPDO3A alone under the same experimental conditions.
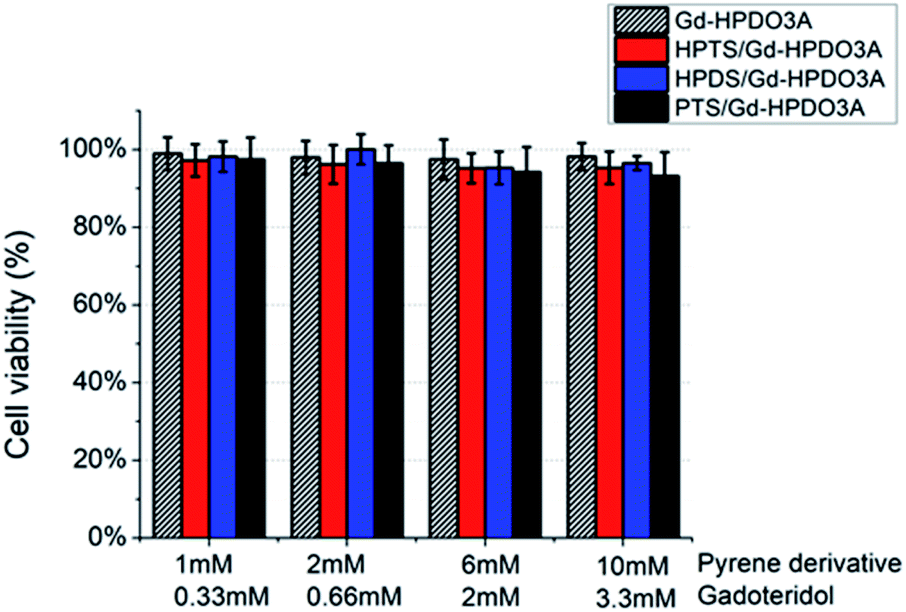 | ||
| Fig. 5 TS/a cell viability (MTT assay) upon incubation with (i) Gd-HPDO3A, (ii) Gd-HPDO3A/HPTS, (iii) Gd-HPDO3A/HDPS or (iv) Gd-HPDO3A/PTS (1:3 mol/mol Gd-HPDO3A/pyrene) derivative at variable concentrations. | ||
Experimental
Chemicals
The following clinical Gd-based contrast agents were employed in this study: (1) gadoteridol (ProHance; Bracco Imaging, Gd-HPDO3A), (2) gadodiamide (Omniscan; GE Healthcare, Gd-DTPA-BMA), (3) gadopentetate dimeglumine (Magnevist; Bayer, Gd-DTPA), (4) gadoterate meglumine (Dotarem; Guerbet, Gd-DOTA), (5) gadobutrol (Gadovist; Bayer, Gd-Butrol) and (6) gadobenate dimeglumine (MultiHance; Bracco Imaging, Gd-BOPTA).Yb-HPDO3A complexes were prepared as previously reported; the metal concentration has been measured by Evan's method,39 the absence of free Yb has been checked by using the xylenol orange method.40
HDPS (6,8-hydroxypyrene-1,3-disulfonic acid), HPTS (8-hydroxypyrene-1,3,6-trisulfonic acid) and PTS (1,3,6,8-pyrenetetrasulfonic acid), sodium chloride, hepes and all other chemicals were purchased from Sigma Aldrich, Co. LLC–Merck KGaA, Darmstadt, Germany and used without further purification. Human serum (Seronorm) was purchased from Sera, Norway. The 1H NMR spectrum of HPTS in D2O (pH 6.0, T = 15 °C) is reported in Fig. S9.†
Cells and animals
Cell media and supplements (RPMI, FBS, glutamine, pen/strep, MycoAlert™ Mycoplasma Detection Kit) were purchased from Lonza Sales AG-EuroClone SpA, Milano (IT).TS/a murine breast cancer cells were derived at the University of Torino from a spontaneous mammary adenocarcinoma which arose in a retired breeder BALB/c female.41
They were grown in RPMI (Roswell Park Memorial Institute) 1064 medium supplemented with 10% heat-inactivated fetal bovine serum (FBS), 2 mM glutamine, 100 U mL−1 penicillin and 100 μg mL−1 streptomycin. Cells were seeded in 75 cm2 flasks at a density of ca. 2 × 104 cells per cm2 in a humidified 5% CO2 incubator at 37 °C. At confluence, they were detached by adding 1 mL of trypsin–EDTA solution (0.25% (w/v) trypsin–0.53 mM EDTA). Cells were negative for mycoplasma as tested by using the MycoAlert™ Mycoplasma Detection Kit. All cell media and supplements were purchased from Lonza Sales AG-EuroClone SpA, Milano (IT).
In vivo experiments were carried out by using 8 week old female Balb/C mice (Charles River Laboratories, Calco, Italy) bred at the Molecular Biotechnology Center of the University of Turin, Italy. Mice were kept in standard housing (12 h light/dark cycle) with rodent chow and water available ad libitum.
Experiments were performed according to the Amsterdam Protocol on Animal Protection and in conformity with institutional guidelines that are in compliance with national laws (D.L.vo 116/92, D.L.vo 26/2014 and following additions) and international laws and policies (2010/63/EU, EEC Council Directive 86/609, OJL 358, Dec 1987, NIH Guide for the Care and Use of Laboratory Animals, U.S. National Research Council, 1996). This study was carried out in the framework of a protocol approved by the Italian Ministry of Health (no. 229/2016-PR).
For the tumor model preparation, ca. 5 × 105 TS/A breast cancer cells were suspended in 0.1 mL of PBS and subcutaneously injected in the flank of 10–12 week old female mice (n = 3, a low number in agreement with 3 Rs principles for the use of animals, by considering that only a proof of concept of the in vivo feasibility of the proposed method).
For the experiments, mice were anesthetized by intramuscular injection of tiletamine/zolazepam (Zoletil 100; Virbac, Milan, Italy) 20 mg kg−1 plus xylazine (Rompun; Bayer, Milan, Italy) 5 mg kg−1 by using a 27-G syringe. A permanent vein access was obtained by inserting a PE10 catheter into the tail vein (27-G needle).
Animals were monitored weekly for changes in tumor size by MRI. PAI was performed 3 weeks after tumor cell implantation when the mean tumour volume was ca. 700 mm3.
Mice were sacrificed after MRI experiments by cervical dislocation in agreement with ethical guidelines.
Relaxometric titration and PRE method
The binding parameters Ka and Rb between the investigated Gd-complexes and the polyarylene additives were calculated by using the well-established Proton Relaxation Enhancement (PRE) technique.42This method consists of measuring the variation in the longitudinal proton relaxation rates (R1 = 1/T1) of the paramagnetic solutions upon increasing the concentration of the interacting substrate. By this method, the binding parameters Ka (the affinity constant) and Rb (the relaxivity of the resulting adducts at saturation) can be determined. To this end, solutions of the Gd-complexes (1 mM) were titrated with the hydrophobic compounds (in the concentration range 1–40 mM, leading to the saturation of the contrast agent), at 21.5 MHz and 298 K, in Hepes/NaCl buffer solution ([hepes] = 3.8 mM, [NaCl] = 150 mM) at pH 7.0–7.2.
Relaxation rates were measured on a Stelar SpinMaster Relaxometer (Stelar, Mede, PV, Italy) operating at 21.5 MHz by means of the inversion recovery method (16 experiments, two scans). The reproducibility of the T1 data was ±0.5%.
The least-squares fit of the R1vs. substrate concentration binding isotherms enable calculation of the affinity constants and the relaxivities of the adducts. Analysis of data was carried out by using Origin 9.0 software.
The effect of Human Serum Albumin (HSA) was investigated by measuring the relaxivity values of Gd-HPDO3A and Gd-HPDO3A/HPTS adduct in serum (Seronorm™ SERO AS, Stasjonsveien 44, 1396 Billingstad, Norway) solution.
NMRD profiles
In order to investigate if the enhancement of the relaxivity is maintained at different magnetic field strengths, the proton 1/T1 NMRD profiles were measured at 298 K on 1 mM GBCA solutions in the absence and in the presence of 40 mM pyrene derivatives. Measurements were carried out on a fast field-cycling Stelar relaxometer over a continuum of magnetic field strengths from 0.00024 to 0.47 T (corresponding to 0.01–20 MHz proton Larmor Frequency). The relaxometer operates under computer control with an absolute uncertainty in 1/T1 of ±0.1%. Additional data points in the range 21.5–70 MHz were obtained on the Stelar SpinMaster relaxometer. The concentration of the solutions used for the relaxometric characterization was determined according to a previously reported relaxometric method.43High resolution 1H-NMR
YbHPDO3A complex was dissolved in D2O (20 mM) in the presence of different amounts of HPTS. The pD was adjusted by addition of DCl or KOD and checked with a glass electrode connected to an AsInstruments pH-meter. The 1H-NMR spectra were recorded at 14.1 T on a Bruker Avance 600 spectrometer. The temperature was controlled with Bruker thermostating units.MRI experiments and data analysis
MR images have been acquired, pre and post injection of GBCA, at 7.1 T by using a Bruker Avance 300 spectrometer equipped with a Micro 2.5 microimaging probe at room temperature (ca. 21 °C). Mice were injected with 0.15 mmol kg−1 of Gd-HPDO3A followed (after 1 h when the wash-out of the GBCA was considered complete) by the injection of a mixture of Gd-HPDO3A and HPTS (0.15 mmol kg−1 Gd-HPDO3A and 0.45 mmol kg−1 HPTS).Tumor volume and morphology were determined by multislice anatomical T2w images. An ROI was manually drawn, and the slice volume was calculated. Mice were used ca. 3 weeks after tumor cell implantation, when the tumor volume was ca. 700 mm3.
T 2w images were acquired by using a standard T2w RARE (Rapid Acquisition with Refocused Echoes) sequence with the following parameters (TR = 5000 ms, TE = 5.5 ms, RARE factor = 32, FOV = 3.5 × 3.5 cm, slice thickness = 1 mm, matrix 128 × 128).
T 1w images were acquired 2 min after the injection of the Gd-HPDO3A or Gd-HPDO3A/HPTS adduct by using a standard T1w-MSME (multislice multiecho) sequence with the following parameters (TR = 200 ms, TE = 3.3 ms, number of averaged image acquisitions = 6, FOV = 3.5 × 3.5 cm, slice thickness = 1 mm, matrix 128 × 128, resolution 0.273 × 0.273 mm per pixel). A glass tube containing a standard solution was used as the internal reference. It was located in the field of view in close proximity to the mouse body.
Cell toxicity
Tests to assess the toxicity of Gd-HPDO3A/pyrene supramolecular adducts were carried out by using TS/a breast cancer cells and applying MTT assay.38 TS/a cells were seeded into 96-well tissue culture plates (104 cells for plate) 24 h before the experiment. Then, they were incubated with fresh complete medium in the presence of (i) Gd-HPDO3A, (ii) Gd-HPDO3A/HPTS, (iii) Gd-HPDO3A/HDPS or (iv) Gd-HPDO3A/PTS. Different concentrations of pyrene derivatives (HPTS, HPDS or PTS) and Gd-HPDO3A were tested, maintaining constant the 1:3 Gd-HPDO3A/pyrene derivative stoichiometric ratio (mol/mol).
After the incubation time (3 h), the medium was removed, cells washed and re-incubated in the presence of fresh medium supplemented with 0.5 mg mL−1 MTT (Thiazolyl Blue Tetrazolium Bromide, Sigma Aldrich) for 4 h in a humidified 5% CO2 incubator at 37 °C. Then, the MTT containing solution was removed and the plates were filled with DMSO (0.1 mL for plate) for 1/2 h at room temperature, under gentle agitation, for allowing solubilization of formazan crystals. The absorbance of the resulting colored solutions was quantified using a 96-multiwell iMark Bio-Rad microplate reader (λ = 570 nm). The percentage of viable cells was calculated on the basis of control blank cells by using the following formula:
| Viable cells, % = (AbsT/Abscnt) × 100 |
Cell experiments were repeated five times and data reported as mean value ± standard deviation. Blank was repeated 10 times.
Conclusions
The herein reported results show that a novel tool to enhance the relaxivity of paramagnetic tetra-aza-dodecane based macrocyclic complexes is available through the formation of supramolecular adducts via the implementation of hydrophobic interactions. This possibility represents an additional route to the several approaches reported in the last three decades mainly based on the set-up of binding interactions between a suitable substituent introduced on the surface of the ligand and an endogenous or exogenous macromolecular substrate. An important caveat from our findings is that the reported relaxation enhancement is attainable for macrocyclic Gd(III)-based CAs already at the clinically approved doses. When the 1:3 stoichiometric ratio between GBCA and pyrene derivatives is used, the i.v. dose of pyrene derivatives is at least one order of magnitude lower than the reported LD50 value.44 This allowed us to work safely with mice and makes us confident that suitable substrates may be found for envisaging possible in vivo translation of the proposed relaxation enhancement procedure.
Finally, the herein reported results can open new routes in the rational design of innovative Gd-based contrast agents whose relaxivity is enhanced in comparison to commercial compounds. For instance, one may propose the synthesis of a new macrocyclic GBCA (e.g. HPDO3A- or DOTA-derivative) bearing on its surface a substituent containing HPTS or related moieties. According to the characteristics of the spacer between the macrocyclic cage and the pyrene moiety one may pursue an intramolecular folding (thus generating relaxivities similar to the ones reported in this work) or an intermolecular interaction that may generate chains with much longer molecular reorientational times and higher relaxivities at the clinical magnetic fields. Of course, suggestions for the replacement of pyrene derivatives with other substrates able to generate analogous interactions represent interesting and desired possibilities have been made.
Identifying possible interactions between GBCAs and endogenous substrates, besides being important for the design of systems endowed with higher relaxivities, is a hot topic also in the context of the issues related to Gd retention. The possibility of showing the occurrence of interactions with macromolecular systems present in plasma or in the extra-cellular/extra-vascular matrix by measuring the relaxation enhancements in the presence of increasing concentrations of a given macromolecular substrate is often used. For instance, Yan Wang et al.45 found that Gd-DTPA and Gd-DTPA-BMA showed relaxation enhancements in the presence of plasma proteins such as albumin and lysozyme. From the acquisition of the NMRD profiles it was shown that the relaxation enhancements are particularly high at the clinical MRI fields and in the presence of protein concentrations much higher than those found in plasma. As far as the highly hydrophilic macrocyclic GBCAs are concerned, there is an interesting observation that Gd-HPDO3A and Gd-DO3A-Butrol display enhancement of relaxivity on going from buffer solutions to plasma, not simply ascribable to the increase in viscosity.46 Basically, the determinants of the binding interactions between GBCAs and proteins may be envisaged in terms of the electrostatic interactions and of H-bonding the GBCAs may form with domains or specific epitopes on the surface of the proteins. The herein reported observations that the hydrophobic region at the tetra-aza-macrocyclic ring of clinically used agents such as Gd-DOTA, Gd-HP-DO3A and Gd-DO3A-Butrol can be the site for binding interactions may add new insights to the search for possible in vivo interactions. Obviously the herein reported pyrene model is far from what we may expect to encounter in biological systems, but it may provide suggestions to envisage the occurrence of binding interactions not previously anticipated.
Conflicts of interest
L. L. and A. M. are Bracco Imaging S.p.A. employees. The Department of molecular biotechnologies and health sciences has received a Research Grant from Bracco Imaging S.p.A. for the topic object of this study.Acknowledgements
The authors acknowledge the Italian Ministry of Research for FOE contribution to the Euro-BioImaging MultiModal Molecular Imaging Italian Node (http://www.mmmi.unito.it). This work has received funding from Regione Piemonte (P.O.R. FESR 2014/2020 Bando IR2 – Industrializzazione dei risultati della ricerca – project “Gadoplus”). Funding was also received from the University of Torino (ex60%, G. F.). This research was performed in the framework of COST Action AC15209 (EURELAX). Bracco Imaging S.p.A. is gratefully acknowledged for providing gadoteridol.Notes and references
- The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging, ed. A. Merbach, L. Helm and É. Tóth, Wiley, New York, 2nd edn, 2013 Search PubMed.
- J. Wahsner, E. M. Gale, A. Rodríguez-Rodríguez and P. Caravan, Chemistry of MRI Contrast Agents: Current Challenges and New Frontiers, Chem. Rev., 2019, 119, 957–1057 CrossRef CAS.
- C. F. G. C. Geraldes and S. Laurent, Classification and basic properties of contrast agents for magnetic resonance imaging, Contrast Media Mol. Imaging, 2009, 4, 1–23 CrossRef CAS.
- V. C. Pierre, M. J. Allen and P. Caravan, Contrast Agents for MRI: 30+ Years and Where Are We Going?, J. Biol. Inorg Chem., 2014, 19, 127–131 CrossRef CAS.
- S. Aime and P. Caravan, Biodistribution of Gadolinium-Based Contrast Agents, Including Gadolinium Deposition, J. Magn. Reson. Imaging, 2009, 30, 1259–1267 CrossRef.
- E. Kanal and M. F. Tweedle, Residual or Retained Gadolinium: Practical Implications for Radiologists and Our Patients, Radiology, 2015, 275, 630–634 CrossRef.
- D. Hao, T. Ai, F. Goerner, X. Hu, V. M. Runge and M. J. Tweedle, MRI contrast agents: basic chemistry and safety, Magn. Reson. Imaging, 2012, 36, 1060–1071 CrossRef.
- E. Di Gregorio, R. Iani, G. Ferrauto, R. Nuzzi, S. Aime and E. Gianolio, Gd accumulation in tusses of healthy mice upon repeated administrations of Gadidiaminde and Gadoteridol, J. Trace Elem. Med. Biol., 2018, 48, 239–245 CrossRef CAS.
- E. Gianolio, E. Di Gregorio and S. Aime, Chemical Insights into the Issues of Gd Retention in the Brain and Other Tissues Upon the Administration of Gd-Containing MRI Contrast Agents, Eur. J. Inorg. Chem., 2019, 2, 137–151 CrossRef.
- M. Le Fur and P. Caravan, The biological fate of gadolinium-based MRI contrast agents: a call to action for bioinorganic chemists, Metallomics, 2019, 11, 240–254 RSC.
- M. Port, J.-M. Idée, C. Medina, C. Robic, M. Sabatou and C. Corot, Efficiency, thermodynamic and kinetic stability of marketed gadolinium chelates and their possible clinical consequences: a critical review, BioMetals, 2008, 21, 469–490 CrossRef CAS.
- L. Helm, J. R. Morrow, C. J. Bond, F. Carniato, M. Botta, M. Braun, Z. Baranyai, R. Pujales-Paradela, M. Regueiro-Figueroa, D. Esteban-Gómez, C. Platas-Iglesias and T. J. Scholl, Gadolinium-based Contrast Agents, in Contrast Agents for MRI: Experimental Methods, ed. V. C. Pierre and M. J. Allen, RSC Editor, ch. 2, 2017 Search PubMed.
- L. M. De León-Rodríguez, A. F. Martins, M. C. Pinho, N. M. Rofsky and A. D. Sherry, Basic MR relaxation mechanisms and contrast agent design, J. Magn. Reson. Imaging, 2015, 42, 545–565 CrossRef.
- M. Rohrer, H. Bauer, J. Mintorovitch, M. Requardt and H.-J. Weinmann, Comparison of Magnetic Properties of MRI Contrast Media Solutions at Different Magnetic Field Strengths, Invest. Radiol., 2005, 40, 715–724 CrossRef.
- P. Hermann, J. Kotek, V. Kubiček and I. Lukeš, Gadolinium(III) complexs as MRI contrast agents: ligand design and properties of the complexes, Dalton Trans., 2008, 3027–3047 RSC.
- P. Caravan, Strategies for increasing the sensitivity of gadolinium based MRI contrast agents, Chem. Soc. Rev., 2006, 35, 512–523 RSC.
- F. Hu, H. M. Joshi, V. P. Dravid and T. J. Meade, High-performance Nanostructured MR Contrast Probes, Nanoscale, 2010, 2, 1884–1891 RSC.
- A. D. Sherry and Y. Wu, The importance of water exchange rates in the design of responsive agents for MRI, Curr. Opin. Chem. Biol., 2013, 17, 167–174 CrossRef CAS.
- S. Laurent, L. Vander Elst, V. Henrotte and R. N. Muller, Noncovalent binding of some new lipophilic gadoliniumDTPA
complexes to human serum albumin. A structure-affinity relationship, Chem. Biodiversity, 2010, 7, 2846–2855 CrossRef CAS.
- V. R. S. Harrison, C. E. Carney, K. W. Macrenaris and T. J. Meade, A Multimeric MR-optical Contrast Agent for Multimodal Imaging, Chem. Commun., 2014, 50, 11469–11471 RSC.
- Y. Song, H. Zong, E. R. Trivedi, B. J. Vesper, E. A. Waters, A. G. M. Barrett, J. A. Radosevich, B. M. Hoffman and T. J. Meade, Synthesis and Characterization of New porphyrazine-Gd(III) Conjugates as Multimodal MR Contrast Agents, Bioconjugate Chem., 2010, 21, 2267–2275 CrossRef CAS.
- G. Ferrauto, E. Di Gregorio, W. Dastru, S. Lanzardo and S. Aime, Gd-loaded-RBCs for the assessment of tumor vascular volume by contrast-enhanced-MRI, Biomaterials, 2015, 58, 82–92 CrossRef CAS.
- S. Chakraborty, S. Nandi, K. Bhattacharyya and S. Mukherjee, Time Evolution of Local pH Around a Photo-Acid in Water and a Polymer Hydrogel: Time Resolved Fluorescence Spectroscopy of Pyranine, ChemPhysChem, 2019, 20, 3221–3227 CrossRef CAS.
- W. Dastrù, V. Menchise, G. Ferrauto, S. Fabretto, C. Carrera, E. Terrreno, S. Aime and D. Delli Castelli, Modulation of the Prototropic Exchange Rate in pH-Responsive Yb-HPDO3A Derivatives as ParaCEST Agents, ChemistrySelect, 2018, 3, 6035–6041 CrossRef.
- G. M. Clore and J. Iwahara, Theory, practice, and applications of paramagnetic relaxation enhancement for the characterization of transient low-population states of biological macromolecules and their complexes, Chem. Rev., 2009, 109, 4108–4139 CrossRef CAS.
- E. Di Gregorio, G. Ferrauto, E. Gianolio, S. Lanzardo, C. Carrera, F. Fedeli and S. Aime, An MRI method to map tumor hypoxia using Red Blood Cells loaded with a pO2-responsive Gd-Agent, ACS Nano, 2015, 9, 8239–8248 CrossRef CAS.
- D. Delli Castelli, M. C. Caligara, M. Botta, E. Terreno and S. Aime, Combined High Resolution NMR and 1H and 17O Relaxometric Study sheds Light on the Solution Structure and Dynamics of the Lanthanide(III) Complexes of HPDO3A, Inorg. Chem., 2013, 52, 7130–7138 CrossRef CAS.
- L. R. Tear, C. Carrera, E. Gianolio and S. Aime, Towards an Improved Design of MRI Contrast Agents: Synthesis and Relaxometric Characterisation of Gd-HPDO3A Analogues, Chem.–Eur. J., 2020, 26, 6056–6063 CrossRef CAS.
- C. Corot, M. Port, I. Raynal, A. Dencausse, M. Schaefer, O. Rousseaux, C. Simonot, L. Devoldere, J. Lin, M. Foulon, P. Bourrinet, B. Bonnemain and D. Meyer, Physical, chemical, and biological evaluations of P760: a new gadolinium complex characterized by a low rate of interstitial diffusion, J. Magn. Reson. Imaging, 2000, 11, 182–191 CrossRef CAS.
- M. Port, C. Corot, O. Rousseaux, I. Raynal, L. Devoldere, J. M. Idee, A. Dencausse, S. Le Greneur, C. Simonot and D. Meyer, P792: a rapid clearance blood pool agent for magnetic resonance imaging: preliminary results, Magma: Magnetic Resonance Materials in Physics, Biology and Medicine, 2001, 12, 121–127 CrossRef CAS.
- D. A. Fulton, M. O'Halloran, D. Parker, K. Senanayake, M. Botta and S. Aime, Efficient relaxivity enhancement in dendritic gadolinium complexes: effective motional coupling in medium molecular weight conjugates, Chem. Commun., 2005, 474–476 RSC.
- S. Laurent, L. Vander Elst, C. Henoumont and R. N. Muller, How to measure the transmetallation of a gadolinium complex, Contrast Media Mol. Imaging, 2010, 5, 305–308 CrossRef CAS.
- S. Aime, L. Barbero, M. Botta and G. Ermondi, Determination of metal-proton distances and electronic relaxation times in lanthanide complexes by nuclear magnetic resonance spectroscopy, J. Chem. Soc., Dalton Trans., 1992, 225–228 RSC.
- S. Aime, G. Ermondi and M. Botta, NMR study of solution structures and dynamics of lanthanide(III) complexes of DOTA, Inorg. Chem., 1992, 31, 4291–4299 CrossRef CAS.
- D. Delli Castelli, G. Ferrauto, E. Di Gregorio, E. Terreno and S. Aime, Sensitive MRI Detection of Internalized T1 Contrast Agents Using Magnetization Transfer Contrast, NMR Biomed., 2015, 28, 1663–1670 CrossRef CAS.
- G. Ferrauto, E. Di Gregorio, D. Delli Castelli and S. Aime, CEST-MRI Studies of cells loaded with lanthanide shift reagents, Magn. Reson. Med., 2018, 80, 1626–1637 CrossRef CAS.
- G. Ferrauto, F. Carniato, E. Di Gregorio, M. Botta and L. Tei, Photoacoustic ratiometric assessment of mitoxantrone release from theranostic ICG-conjugated mesoporous silica nanoparticles, Nanoscale, 2019, 11, 18031–18036 RSC.
- M. Tripepi, G. Ferrauto, P. B. Oronzo, S. Aime and D. Delli Castelli, Multilamellar lipocest agents obtained from osmotic shrinkage of paramagentically loaded giant unilamellar vesicles (GUVs), Angew. Chem., Int. Ed., 2020, 59, 2279–2283 CrossRef CAS.
- D. M. Corsi, C. Platas-Iglesia, H. van Bekkum and J. A. Peters, Detrmination of paramagnetic lanthanide(III) concentrations from bulk magnetic susceptibility shifts in NMR spectra, Magn. Reson. Chem., 2001, 39, 723–726 CrossRef CAS.
- A. Barge, G. Cravotto, E. Gianolio and F. Fedeli, How to determine free Gd and free ligand in solution of Gd chelates. A technical note, Contrast Media Mol. Imaging, 2006, 1, 184–188 CrossRef.
- P. Nanni, C. De Giovanni, P. L. Lollini, G. Nicoletti and G. Prodi, TS/A: a new metastasizing cell line from BALB/c spontaneous mammary adenocarcinoma, Clin. Exp. Metastasis, 1983, 1, 373–380 CrossRef CAS.
- S. Aime, M. Fasano, S. Paoletti, A. Bellelli, M. Coletta and P. Ascenzi, Stabilization of the T-state of ferrous human adult and fetal hemoglobin by Ln(III) complexes: a thermodynamic study, Inorg. Biochem., 1998, 71, 37–43 CrossRef CAS.
- F. Arena, J. Bhagavath, E. Gianolio, R. Stefania and S. Aime, β-Gal Gene Expression MRI Reporter in Melanoma Tumor Cells. Design, Synthesis, and in Vitro and in Vivo Testing of a Gd(III) Containing Probe Forming a High Relaxivity, Melanin-Like Structure upon β-Gal Enzymatic Activation, Bioconjugate Chem., 2011, 22, 2625–2635 CrossRef CAS.
- G. A. Lutty, The acute intravenous toxicity of biological stains, dyes, and other fluorescent substances, Toxicol. Appl. Pharmacol., 1978, 44, 225–229 CrossRef CAS.
- Y. Wang, M. Spiller and P. Caravan, Evidence for weak protein binding of commercial extracellular gadolinium contrast agents, Magn. Reson. Med., 2010, 63, 609–616 CrossRef CAS.
- M. Rohrer, H. Bauer, J. Mintorovitch, M. Requardt and H. J. Weinmann, Comparison of magnetic properties of MRI contrast media solutions at different magnetic field strengths, Invest. Radiol., 2005, 40, 715–724 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary figures. See DOI: 10.1039/d0sc03504a |
| ‡ These authors equally contributed. |
| This journal is © The Royal Society of Chemistry 2021 |