
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Surface site density and utilization of platinum group metal (PGM)-free Fe–NC and FeNi–NC electrocatalysts for the oxygen reduction reaction†
Fang
Luo
 a,
Stephan
Wagner
b,
Ichiro
Onishi
c,
Sören
Selve
d,
Shuang
Li
e,
Wen
Ju
a,
Huan
Wang
a,
Julian
Steinberg
a,
Arne
Thomas
e,
Ulrike I.
Kramm
*b and
Peter
Strasser
*a
a,
Stephan
Wagner
b,
Ichiro
Onishi
c,
Sören
Selve
d,
Shuang
Li
e,
Wen
Ju
a,
Huan
Wang
a,
Julian
Steinberg
a,
Arne
Thomas
e,
Ulrike I.
Kramm
*b and
Peter
Strasser
*a
aThe Electrochemical Catalysis, Energy and Materials Science Laboratory, Department of Chemistry, Technische Universität Berlin, Straße des 17. 10623, Berlin, Germany. E-mail: pstrasser@tu-berlin.de
bDepartment of Chemistry and Department of Materials and Earth Sciences, Catalysts and Electrocatalysts Group, Technical University of Darmstadt, Otto-Berndt-Str. 3, 64287 Darmstadt, Germany. E-mail: kramm@ese.tu-darmstadt.de
cJEOL Ltd, 3-1-2 Musashino, Akishima, Tokyo, Japan
dTechnische Universität Berlin, Center for Electron Microscopy (ZELMI), Straße des 17. Juni 135, 10623 Berlin, Germany
eFunctional Materials, Department of Chemistry, Technical Universität Berlin, Hardenbergstr. 40, Berlin, 10623, Germany
First published on 13th October 2020
Abstract
Pyrolyzed iron-based platinum group metal (PGM)-free nitrogen-doped single site carbon catalysts (Fe–NC) are possible alternatives to platinum-based carbon catalysts for the oxygen reduction reaction (ORR). Bimetallic PGM-free M1M2–NC catalysts and their active sites, however, have been poorly studied to date. The present study explores the active accessible sites of mono- and bimetallic Fe–NC and FeNi–NC catalysts. Combining CO cryo chemisorption, X-ray absorption and 57Fe Mössbauer spectroscopy, we evaluate the number and chemical state of metal sites at the surface of the catalysts along with an estimate of their dispersion and utilization. Fe L3,2-edge X-ray adsorption spectra, Mössbauer spectra and CO desorption all suggested an essentially identical nature of Fe sites in both monometallic Fe–NC and bimetallic FeNi–NC; however, Ni blocks the formation of active sites during the pyrolysis and thus causes a sharp reduction in the accessible metal site density, while with only a minor direct participation as a catalytic site in the final catalyst. We also use the site density utilization factor, ϕSDsurface/bulk, as a measure of the metal site dispersion in PGM-free ORR catalysts. ϕSDsurface/bulk enables a quantitative evaluation and comparison of distinct catalyst synthesis routes in terms of their ratio of accessible metal sites. It gives guidance for further optimization of the accessible site density of M–NC catalysts.
1. Introduction
The growing concerns about our current unsustainable energy infrastructure and its environmental effects have fueled research and development into alternative power supply technologies such as hydrogen fuel cells. Platinum-based catalysts have been identified as the most efficient catalysts for fuel cell cathodes and anodes. However, the high catalyst cost and their durability have remained hurdles for a wider commercialization of fuel cell technology. Carbon materials doped with non-noble transition metals and nitrogen, that is, PGM-free materials, are promising alternative catalysts for the electrochemical conversion of molecular oxygen; they already show promising performance in fuel cells, metal air batteries, and the chlor-alkali industry.1–6 The most important family of PGM-free ORR catalysts is the class of carbon-embedded, nitrogen-coordinated single metal site catalysts, typically referred to as metal–nitrogen–carbon (MNC) catalysts.7,8 Their state-of-the-art catalytic reactivity rivals that of state-of-the-art Pt nanoparticle catalysts.1,9 Particular attention was paid to a specific family of monometallic MNC catalysts derived from Co, Fe, and Mn in the active M–N4 moiety. Many studies confirmed their favorable catalytic activity for the ORR, as well as bimetallic M1M2–NC derived from them.1,9–18 Other PGM-free metals, such as Ni, and its bimetallic M1M2–NC ORR catalysts, however, have remained largely unexplored.9,12,13,19–22 Based on the design one tandem catalyst where combination a stronger O* binding sites on Fe and a weaker *OOH/*OH binding site on Ni,23–28 the co-existence of the atomic Fe and Ni single atom sites may results in an average chemisorption energy of ORR intermediates, as well as promising results of iron and nickel containing catalysts for the oxygen evolution reaction (OER), that it could be an interesting combination with potential promise for metal air battery applications and bifunctional ORR/OER capabilities.Based on this, in this contribution, we analyze and contrast the fundamental reactivity of monometallic and bimetallic MNC ORR catalysts in previously unavailable ways. We address four distinct ORR catalysts: two mono-metallic Fe–NC and Ni–NC catalysts, the corresponding bimetallic FeNi–NC catalyst, and, as a reference, a “metal-free” (that is, metal content below the detection limit) NC catalyst. To study the effects of the combined presence of both metals on the physico–chemical and catalytic properties of these catalysts, we correlate their electrochemical ORR performance and X-ray absorption spectra (XAS) with their CO chemisorption and desorption behavior and their Mössbauer spectra. CO pulse chemisorption is a quite recent means to probe the total number of available metal active sites and thus yields conservative estimates of previously unavailable, intrinsic catalytic ORR turnover frequencies in alkaline and acidic electrolytes. Mössbauer analysis yielded the total site density in the bulk, from which a site utilization factor ϕSDsurface/bulk was derived and discussed.
2. Results and discussion
2.1. Synthesis, XPS, XAS and STEM analysis
Three MNC catalysts, a Fe–NC, a Ni–NC, and the corresponding bimetallic FeNi–NC ORR catalysts were synthesized using polyaniline (PANI) as a nitrogen precursor and metal salts as metal precursors. A metal-free N–C catalyst was prepared and served as a reference material. The catalyst precursors were subjected to a multi-step preparation involving pyrolysis steps (at 900 °C) and acid leaching steps.To characterize the chemical state of the metal atoms and nitrogen atoms of the prepared catalysts, high-resolution X-ray photoelectron spectroscopy (XPS) and X-ray absorption spectroscopy (XAS) were used to investigate the N 1s region and the Fe and Ni L3,2 edge region (Fig. 1 and S1 and Table S1 and S2†), respectively. Sorption techniques were used to learn about the porosity characteristics of the catalysts (Table S3†). The N1s XPS spectrum was fitted using eight individual spectral components that cover typical binding energy ranges of N species or motifs typically present in NC and MNC materials. The individual eight spectral component peaks were clustered into 4 BE-ranges. BE-range 1 includes Peak 1 (398.1 eV) and Peak 2 (398.7 eV) covering the N atom bonded to two sp2-hybridized carbons and –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–C double bond motifs (e.g. imine N, pyridinic N or triazinic N). BE-range 2 includes Peak 3 (399.3 eV) and Peak 4 (399.8 eV) covering the sp2 hybridized N atom in metal–N coordination, OC–NH–C partial double bonds, or multiple graphitic N motifs in a single aromatic ring (e.g. M–Nx, amides). BE-range 3 includes Peak 5 (400.7 eV), Peak 6 (401.8 eV), and Peak 7 (403 eV) covering N atoms bonded with 1 H (in-plane hydrogenated N), isolated graphitic N, out-of-plane hydrogenated/protonated N and hydrogenated graphitic N (e.g. pyrrolic or protonated pyridinic and protonated graphitic N). BE-range 4 includes Peak 8 (405 eV) covering oxidized N (e.g. CN–O or other oxidized N).10,11,29–33 The positions of the binding energies were chosen from DFT and experimental-DFT combinatorial studies on MNC and NC materials performed by Artyushkova et al.9,28–30,33–37 We note that this N-species assignment is unambiguous, yet a more accurate alternative evaluation of the relative contribution of N species is currently elusive,33,38 but requires probably an in-depth theoretical study that also considers the typical Fe/C ratios found in MNC catalysts. Our analysis in Fig. 1 and S1c and Table S2† suggested quite distinct relative concentrations of BE-range 2 in the PGM-free ORR catalysts: while the peak area is the lowest for FeNi–NC. Comparing the Fe and Ni L3,2-edge spectra of the Fe–NC and FeNi–NC catalysts (Fig. 1c and d) revealed further details about the chemical states of Fe and Ni.39Fig. 1c evidences Fe2+ and Fe3+ to be present in both Fe–NC and FeNi–NC, while the nitrogen-coordinated motif appeared to give rise to a characteristic “double resonance” feature in the 708 to 710 eV range. The presence of this feature indicated a very similar chemical state of the Fe–Nx sites in both catalysts. In FeNi–NC, there is no evidence of zero-valent Ni. Instead, the main absorption peak is shifted to higher photon energies and is assigned to a Ni3+ state with a minor Ni2+ state contribution (Fig. 1d).39 This was further supported by XPS (Fig. S1d and e†). The narrow-scan Ni 2p spectrum of FeNi–NC featured a broad peak in the 2p3/2 region at ∼854.2 eV, and a relatively weak satellite located at ∼861 eV indicating the Ni2+ oxidation state (Fig. S1d†). Difference spectra between the experimental FeNi–NC and expected envelopes of the Ni2+ compound based on the satellite feature suggested the presence of Ni3+, possibly in the carbon-embedded N-coordinated Ni–Nx format with axial oxygen ligands (Fig. S1e†).28,40–46
N–C double bond motifs (e.g. imine N, pyridinic N or triazinic N). BE-range 2 includes Peak 3 (399.3 eV) and Peak 4 (399.8 eV) covering the sp2 hybridized N atom in metal–N coordination, OC–NH–C partial double bonds, or multiple graphitic N motifs in a single aromatic ring (e.g. M–Nx, amides). BE-range 3 includes Peak 5 (400.7 eV), Peak 6 (401.8 eV), and Peak 7 (403 eV) covering N atoms bonded with 1 H (in-plane hydrogenated N), isolated graphitic N, out-of-plane hydrogenated/protonated N and hydrogenated graphitic N (e.g. pyrrolic or protonated pyridinic and protonated graphitic N). BE-range 4 includes Peak 8 (405 eV) covering oxidized N (e.g. CN–O or other oxidized N).10,11,29–33 The positions of the binding energies were chosen from DFT and experimental-DFT combinatorial studies on MNC and NC materials performed by Artyushkova et al.9,28–30,33–37 We note that this N-species assignment is unambiguous, yet a more accurate alternative evaluation of the relative contribution of N species is currently elusive,33,38 but requires probably an in-depth theoretical study that also considers the typical Fe/C ratios found in MNC catalysts. Our analysis in Fig. 1 and S1c and Table S2† suggested quite distinct relative concentrations of BE-range 2 in the PGM-free ORR catalysts: while the peak area is the lowest for FeNi–NC. Comparing the Fe and Ni L3,2-edge spectra of the Fe–NC and FeNi–NC catalysts (Fig. 1c and d) revealed further details about the chemical states of Fe and Ni.39Fig. 1c evidences Fe2+ and Fe3+ to be present in both Fe–NC and FeNi–NC, while the nitrogen-coordinated motif appeared to give rise to a characteristic “double resonance” feature in the 708 to 710 eV range. The presence of this feature indicated a very similar chemical state of the Fe–Nx sites in both catalysts. In FeNi–NC, there is no evidence of zero-valent Ni. Instead, the main absorption peak is shifted to higher photon energies and is assigned to a Ni3+ state with a minor Ni2+ state contribution (Fig. 1d).39 This was further supported by XPS (Fig. S1d and e†). The narrow-scan Ni 2p spectrum of FeNi–NC featured a broad peak in the 2p3/2 region at ∼854.2 eV, and a relatively weak satellite located at ∼861 eV indicating the Ni2+ oxidation state (Fig. S1d†). Difference spectra between the experimental FeNi–NC and expected envelopes of the Ni2+ compound based on the satellite feature suggested the presence of Ni3+, possibly in the carbon-embedded N-coordinated Ni–Nx format with axial oxygen ligands (Fig. S1e†).28,40–46
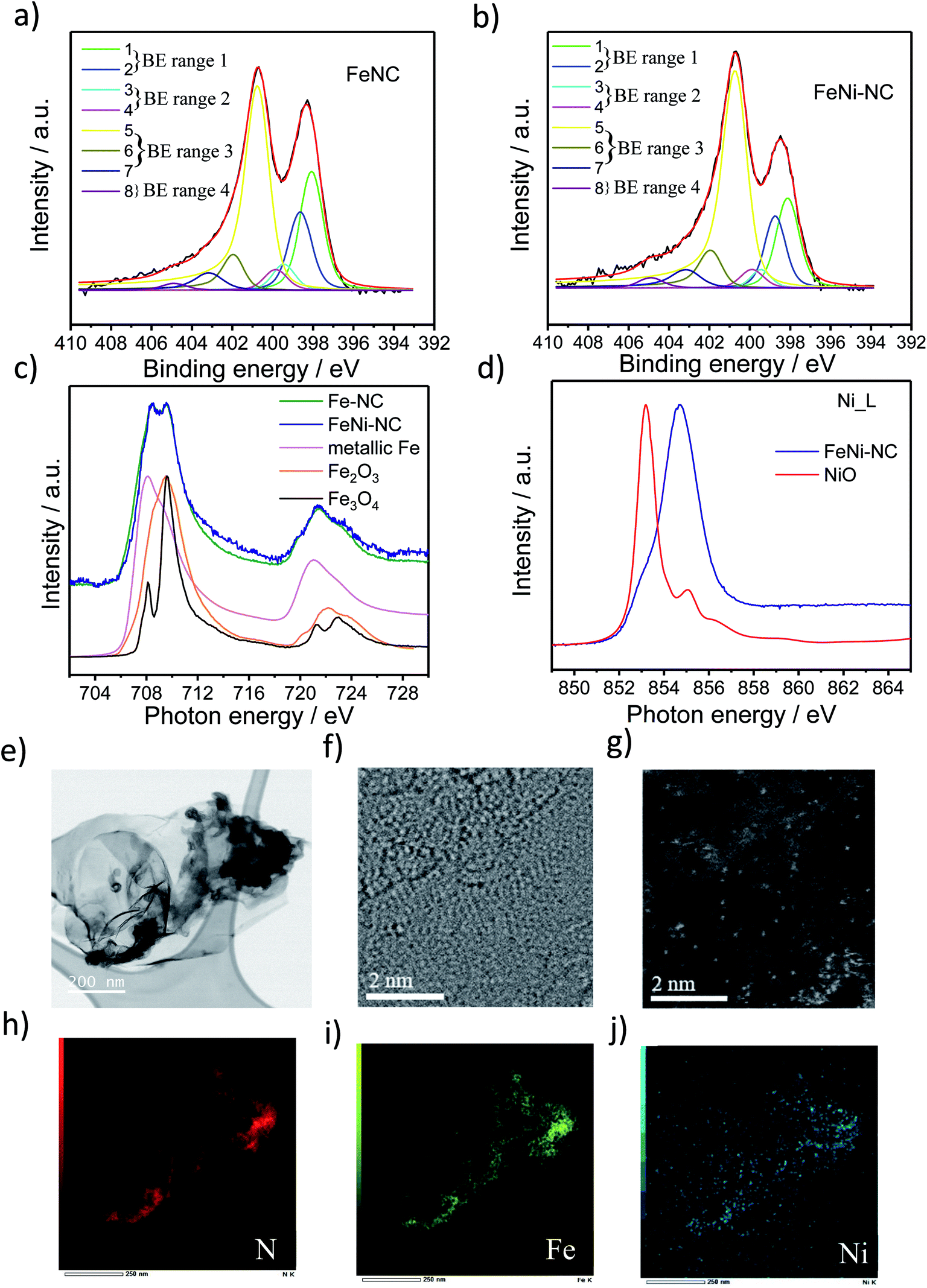 | ||
| Fig. 1 Characterization of the chemical states of N, Fe and Ni speciation using X-ray spectroscopy, low voltage probe-corrected scanning transmission electron microscopy (Cs-corr. STEM) and electron energy-loss spectroscopy (EELS) elemental maps. High-resolution N 1s XPS spectra of (a) Fe–NC and (b) FeNi–NC. The N1s XPS spectrum was fitted using eight individual spectral components that cover typical binding energy ranges of N species or motifs typically present in NC and MNC materials. The individual eight spectral component peaks were clustered into 4 BE-ranges. (c) Fe L3,2-edge spectra of catalysts and reference compounds. (d) Ni L-edge spectra of FeNi–NC and reference compounds. (e and f) Bright-field (BF) and high-resolution (HR)-STEM image of FeNi–NC catalysts. (g) High-angle annular dark-field (HAADF)-STEM image showing bright Fe and Ni atoms atomically dispersed in or on graphene layers. Element mapping image of the (h) N and (i) Fe and (j) Ni in FeNi–NC catalyst extracted from energy dispersive X-ray (EDX) spectroscopy, the mapping are related to (e). | ||
Carbon-hosted FeN4 single metal sites were recently directly imaged for the very first time using AC-STEM.47 The AC-STEM technique was used here to reveal the atomic-level structure and chemistry of a bimetallic FeNi–NC catalyst (Fig. 1e–j). The catalysts consisted primarily of fibrous carbon, graphene and amorphous carbon sheets (Fig. 1e, f and S2†). The dense fibrous carbon particles were composed of randomly oriented, intertwined, turbostratic graphitic domains on the order of a few nanometers in size (Fig. 1f, g and S2†). This indicated a very small graphitic domain size coupled with turbostratic graphene basal planes. Single metal atoms were dispersed homogeneously and randomly across the carbon sheets (Fig. 1g). The elemental mapping images by using EDX mapping with respect to N (red in Fig. 1), Fe (green), and Ni (blue) suggested that the Fe and Ni single metal sites were uniformly and homogeneously distributed over the entire carbon basal planes (Fig. 1h–j). The local co-existence of N and both Fe and Ni metal atoms provided evidence for the existence of coordinatively stabilized M–Nx motifs. The elemental mapping images thus confirmed our conclusions from XAS and XPS that single Fe and Ni metal atoms were successfully coordinated by N across the carbon domains. This is the direct observation of single atom catalysts with M–Nx type sites in bimetallic M1M2–NC catalysts, the N/M ratio could however not be quantified by STEM/EDX, as this resulted in the displacement of single heavy metal atom under an electronic beam, while the atomic ratio of Fe and Ni to N in FeNi–NC was, as expected, about the factor of 10, respectively (0.4 and 0.4 at% to 4.3 at% in Table S1,† and 1.74 wt%/56 and 2.44 wt%/59 to 4.3 wt%/14 in Table S3,† respectively).
To summarize this section, our XAS and XPS results revealed that the nitrogen-coordinated Fe-centers in Fe–NC and FeNi–NC appear chemically identical. However, the site density of BE-range 2 moieties in the bimetallic FeNi–NC catalyst appeared to have decreased sharply upon introducing Ni as a second metal (Fig. S1c†). As we will show below, this has consequences on the electrocatalytic reactivity.
2.2. Electrocatalytic oxygen reduction activity
We used the rotating-ring disk electrode (RRDE) technique to obtain the catalytic oxygen reduction reactivity in alkaline and acidic electrolytes. Fig. 2a and b display the measured linear sweep voltammograms during the ORR, characterized by their kinetic, mixed, and diffusion-limited regions (<∼0.4 VRHE). The number of transferred electrons (Fig. S3†) is a measure of the ratio of the direct 4e− reduction and the 2e− peroxide pathway48 for each catalyst. The highest ORR activity was observed for Fe–NC in either pH range. Fe–NC showed around 50 mV and 90 mV lower over-potentials (at 0.5 mA cm−2) than FeNi–NC in KOH and HClO4, respectively. FeNi–NC and the N–C reference showed comparable activities in KOH, while, in the acid electrolyte, the N–C reference catalyst showed a sharply lower ORR activity (increased overpotential, see Fig. 2b). Fe–NC showed favorable, small H2O2 yields of 4% in alkaline and 8% in acid electrolytes (at 0.1 VRHE). FeNi–NC showed a clearly higher H2O2 yield, followed by the N–C catalyst. Despite a comparable total (Fe + Ni) metal content of about 4 wt% (Table S3†) for both the Fe–NC and the FeNi–NC catalysts, the bi-metallic FeNi–NC catalyst showed a significantly lower catalyst mass-based ORR activity (Table S4†). We attribute this to the sharply reduced Fe wt% in the bimetallic catalyst. Considering the identical synthesis procedure and conditions, we are inclined to conclude that the competition between Ni and Fe metal sites during the bimetallic FeNi–NC catalyst formation process resulted in fewer Fe–Nx sites and a lower overall Fe concentration in the surface and bulk (Tables S1–S3†). Indeed, the calculated energy of formation of Ni–N4 sites is lower and thus more favorable in comparison to that of Fe–Nx.49 In addition, we hypothesize that the generated Ni–Nx moieties exhibit a lower intrinsic ORR reactivity than Fe–Nx sites. This hypothesis is indeed supported by studies of MNC ORR catalysts.17,50,51 In that study, Zagal reported that the Fe(III)/Fe(II) redox process provides the driving force for the catalytic ORR thanks to the nearly half-filled d-energy levels of Fe; in contrast, such d characteristics are lost in metals such as Cu and Ni.46 Also, Fig. 2 directly confirms our hypothesis that Ni–NC sites display a very low catalytic ORR activity; under alkaline conditions, even worse than the N–C reference catalyst. Among the MNC materials, the Ni–NC catalyst showed the lowest ORR activity in both acid and alkaline electrolytes. Its catalyst mass activity, jm, dropped by a factor of ∼85× in the acid compared to that of Fe–NC, while its half wave potential was up to 340 mV higher than that of Fe–NC (Tables S4 and S5†).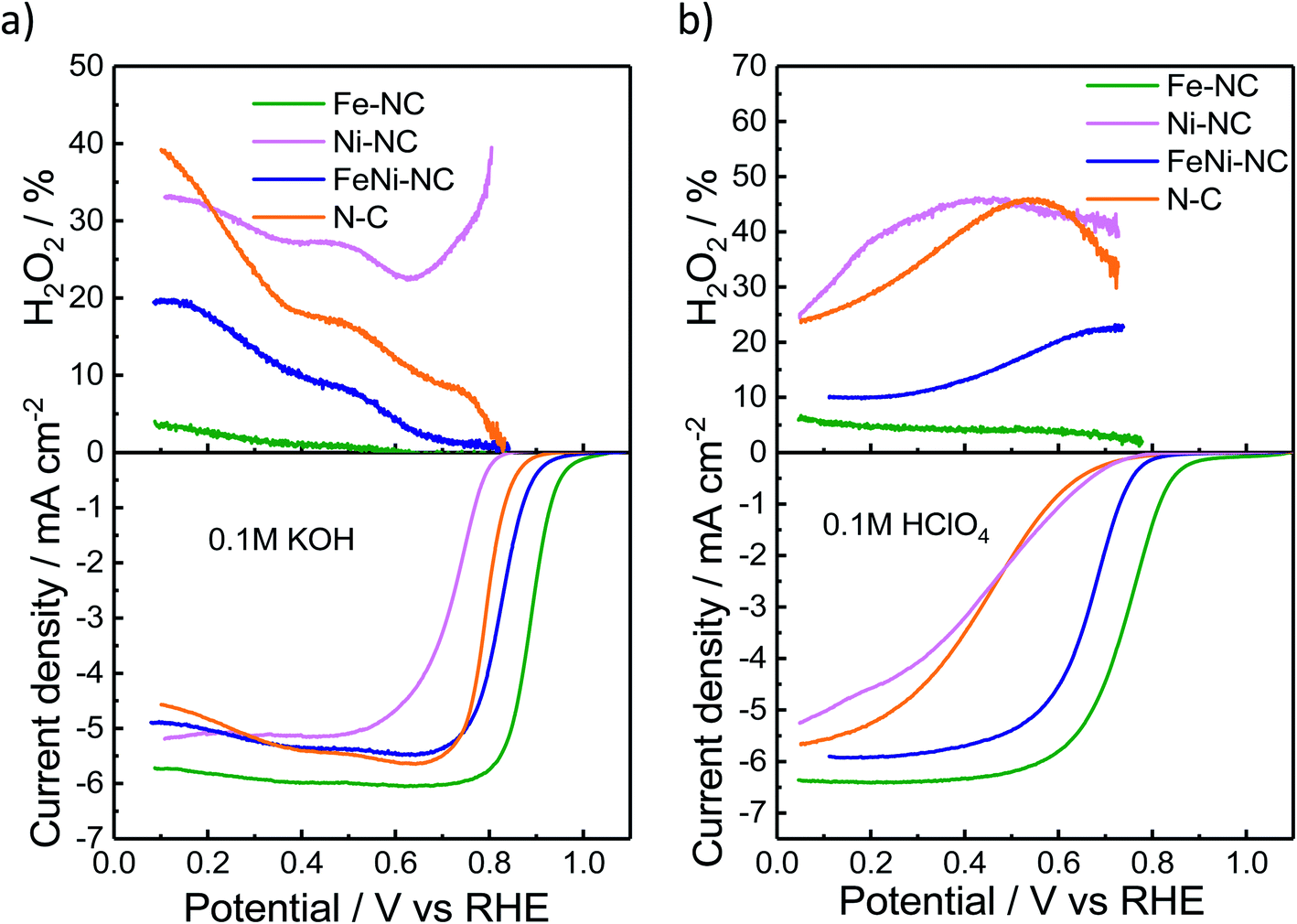 | ||
| Fig. 2 Electrochemical activity evaluation. H2O2 percentage and disk current density from rotating-ring disk electrode (RRDE) experiments of MNC catalysts at 1600 rpm in oxygen-saturated (a) alkaline and (b) acidic electrolytes. | ||
The electrochemical Tafel curves in Fig. S3c and d† illustrate the effect of the applied electrode potential on the observed catalytic ORR current density. In an ideal one-electron transfer approximation, the Tafel slope is viewed as a measure of the kinetic exponent of the electrons in the kinetic rate law derived from the rate-determining elementary step. However, in more realistic multi-step reaction mechanisms that include bond making and bond breaking between adsorbed intermediates, such as in the ORR, the Tafel slope becomes a function of many different factors, such as the potential-dependent coverage of oxygenated intermediates. The potential dependence of the surface intermediate coverage may follow a simple Langmuir, a Temkin, or, in more complex cases, even a Frumkin isotherm.52 As such, the Tafel slope may vary dramatically with applied potential, even though the rate-determining step remains the same. This is why an interpretation of the Tafel slopes in terms of a simple sequence of chemical or electrochemical elementary steps is mostly difficult, and this is why we refrain from an oversimplified interpretation of the Tafel slope values.53 That said, Fig. S3c and d† reveal low Tafel slopes of 57 and 85 mV dec−1 for Fe–NC in KOH and HClO4, respectively. The FeNi–NC catalyst showed rather similar Tafel slopes of 68 and 87 mV dec−1 in KOH and HClO4, respectively. Here, it may be safe to assume that the ORR rate-determining step remained unchanged under either pH conditions. A remarkable difference in the Tafel slope was observed for the N–C reference catalysts in the acid compared to alkaline media (ca. 160 mV dec−1vs. ca. 61 mV dec−1). This is in line with earlier reports on the distinctly different purely chemical reaction step between the first and second electron transfer step.53 Fe–NC displayed an electron transfer number of close to 4, while N–C showed the lowest electron transfer number coupled with the highest H2O2 yield during the ORR (Fig. S3†). According to a recent model of the ORR pathway (Fig. 3 and S4 in ref. 53†), together with the results regarding the selectivity, electron transfer number and Tafel slopes, the 4e− ORR pathway is most favorable on the Fe–NC catalyst, while the FeNi–NC catalyst appears to support a mixture of 2e− and 4e− pathways, in accordance with its mixed Fe–Nx and Ni–Nx sites, while the N–C and Ni–NC catalysts are prone to an almost pure 2e− oxygen reduction pathway.
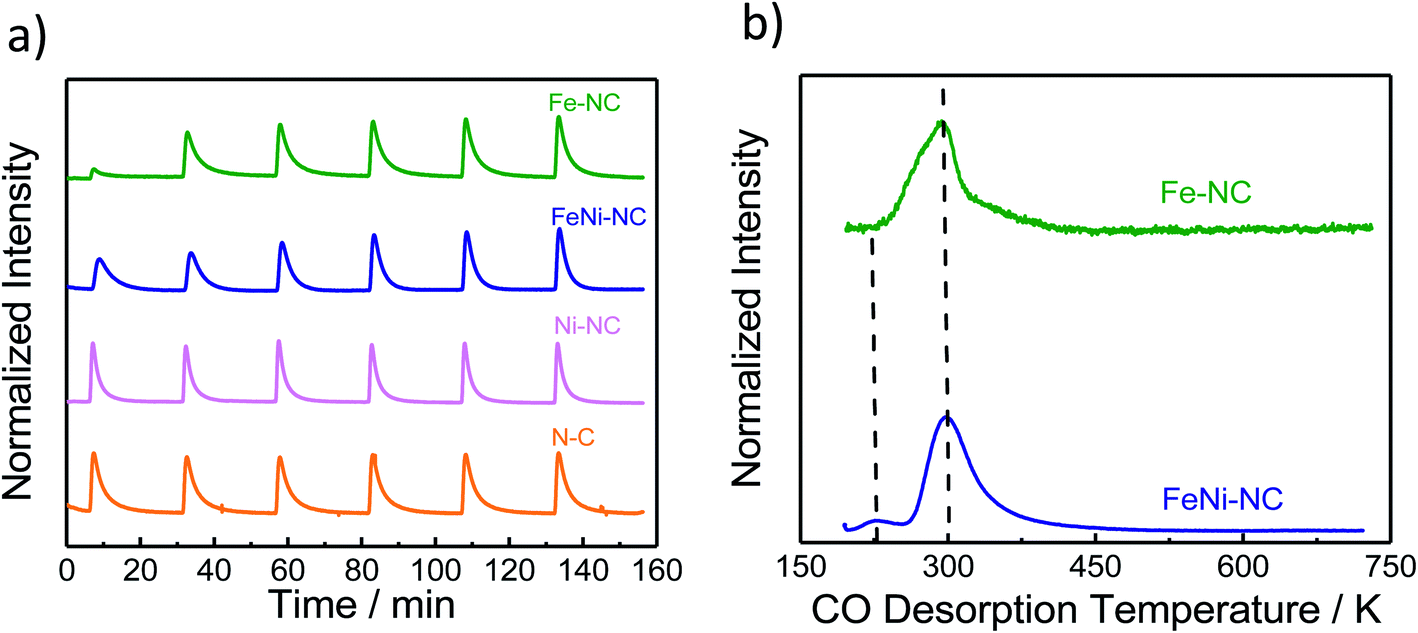 | ||
| Fig. 3 The molar CO uptake and TPD results. (a) CO pulse chemisorption and (b) normalized temperature-programmed desorption (TPD) profiles of MNC catalysts. | ||
2.3. CO cryo-adsorption and temperature-programmed desorption (TPD)
Carbon monoxide (CO) has long been proposed as a blocking probe molecule of the catalytically active sites of FeNC ORR electrocatalysts, because CO can form strong metal–ligand coordination bonds.54,55 As such, it would inhibit adsorption and reaction on accessible metal sites that catalyze the oxygen reduction reaction.55–57 However, CO adsorption and stripping under electrochemical conditions have remained elusive. Recently, however, it was reported that CO cryo adsorption on accessible FeNx sites of pyrolyzed Fe–N–C ORR catalysts at temperatures at around −80 °C following careful thermal annealing protocols does, in fact, allow an accurate quantification of the gas-accessible Fe–Nx sites on the surface and hence an evaluation of the accessible metal site density.9,58,59Fig. 3a shows the adsorption trajectory of six consecutive CO adsorption pulses for each MNC catalyst. The gradual increase in the apparent CO pulse area indicated CO adsorption by the accessible metal sites of the catalyst until full saturation levels were reached in the 4th or 5th pulse. While Fe–NC showed the largest CO uptake, FeNi–NC showed clearly reduced CO uptake (reduced by a factor around three). In contrast, no measureable adsorption of CO was detected on the N–C reference or the Ni–NC ORR catalyst, despite the highest abundance of Ni–Nx moieties. Clearly, the nature and the abundance of the metal centers play a key role in the CO pulse chemisorption process for MNC ORR catalysts.40,41 Ni–Nx sites are unable to bind CO strongly enough, even at low temperatures.The molar CO uptake of the Fe–NC and the FeNi–NC catalysts was converted into a catalyst mass-based accessible site density on the surface, SDsurface, with the unit site g−1 (Table S6†). Fig. 4 (solid bars) compares the resulting SDsurface values of the monometallic Fe–NC and the FeNi–NC catalysts.
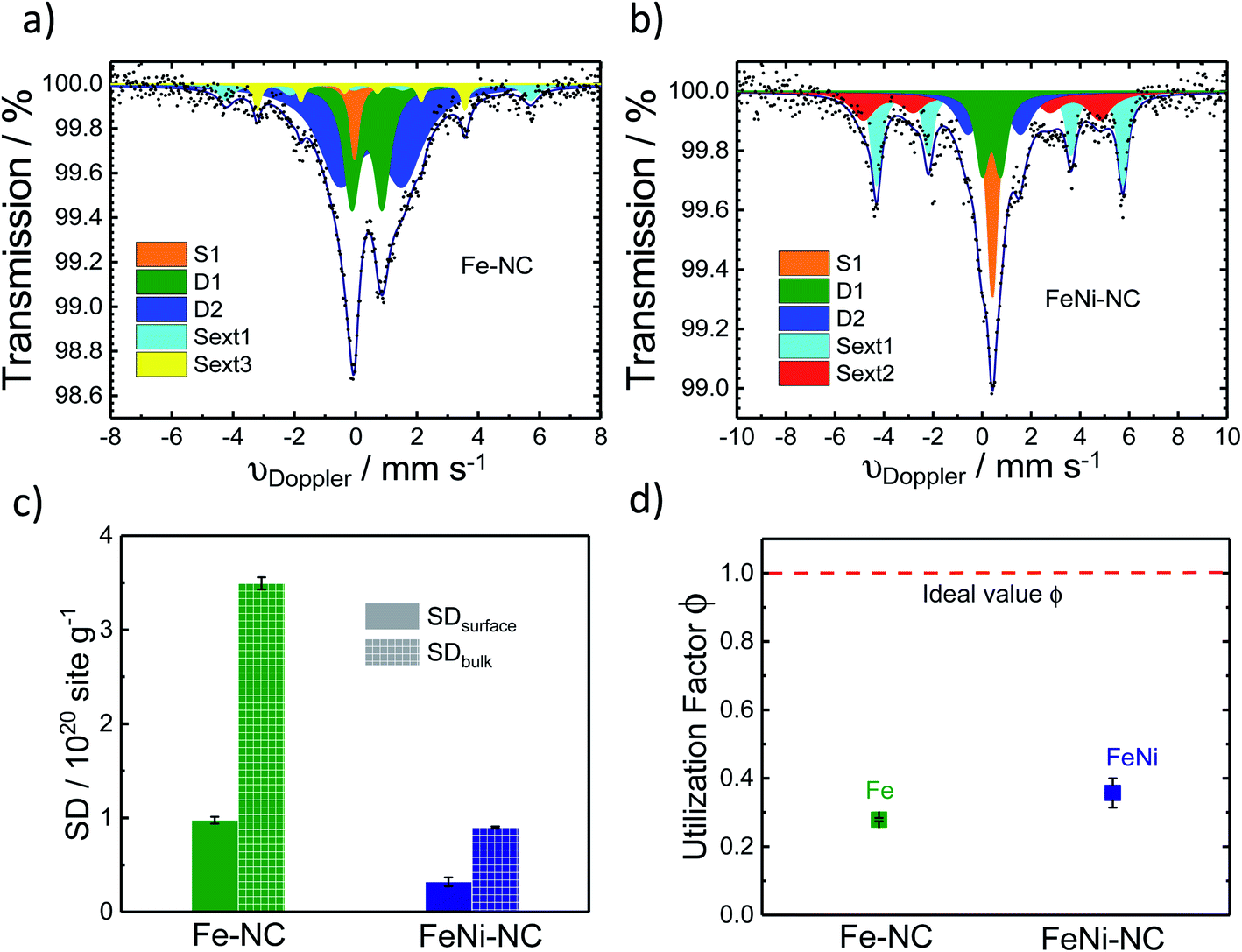 | ||
| Fig. 4 57Fe Mössbauer spectra of (a) Fe–NC and (b) FeNi–NC catalysts. (c) Comparison of SDsurfacce and SDbulk derived from CO cryo chemisorption and Mössbauer spectroscopy experiments (all FeN4 species, D1 + D2), respectively. (d) The active-site utilization factor ϕSDsurface/bulk as defined in eqn (3) is plotted for the Fe–NC and FeNi–NC, and the red dotted line means ideal utilizationϕSDsurface/bulk = 1. | ||
The sharply lower CO uptake for the Ni-containing bi-metallic sample is consistent with the lower electrocatalytic ORR reactivity of the FeNi–NC catalysts and is in excellent agreement with our physico-chemical analysis, which suggested a reduced Fe content in the bimetallic catalyst. The presence of Ni precursors, combined with the rapid formation of Ni–Nx sites at the surface limits or partially suppresses the generation of Fe–Nx active sites in FeNi–NC, resulting in lower CO chemisorption. This is the first time of an experimental confirmation of the prediction already made by theory (compare Fig. S7 in ref. 49†). Moreover, the Ni–NC catalyst showed the lowest ORR activity, which, judging from its CO uptake, may be due to the insufficiently strong bond between the Ni central atom and reactive oxygenated intermediates. This is why Ni–Nx is unable to split the O–O bond and favors the 2e− oxygen reduction pathway to H2O2.
As shown in Fig. 3b, following the CO cryo uptake, both the Fe–NC and the FeNi–NC catalysts showed a major CO desorption peak around 300 K. However, the shape of Fe–NC also indicates an additional contribution probably from a second-type of Fe–Nx site. This shoulder is not as intensive for FeNi–NC, while the main peak associated with most of the FeNx sites is identical. This may suggest similar CO bond strengths and desorption rates from the CO-adsorbing Fe–Nx sites, and ultimately a similar nature of the adsorbing Fe–Nx sites of these two catalysts. This finding is in line with our results from XAS (Fig. 1c) where the Fe L3,2-edge spectra suggested a close chemical similarity between both catalysts. However, the number of active Fe–Nx sites, that is, the active site density, was quite different for the two catalysts, as unambiguously revealed by the CO chemisorption experiments. Considering the identical initial molar amounts of Fe precursors at the beginning of the synthesis and the identical synthesis procedures, we are inclined to conclude that the bimetallic FeNi–NC catalysts displayed fewer active Fe–Nx surface sites due to the presence of Ni as a second metal precursor. We speculate that the Ni precursor affects the formation rate of active Fe–Nx during the pyrolysis process, or else affects the dispersion of the Fe–Nx sites of the resulting catalyst material.60
In summary, the data above suggest that the CO adsorption and CO desorption are strongly governed by the chemical nature and accessible density of the N/C-coordinated central metal ion site. Fe-sites show strong uptake, while Ni-sites show weak to none. The FeNi–NC catalyst displayed a sharply reduced CO uptake which is in agreement with the significantly smaller content of nitrogen associated with Fe–Nx sites. The presence of Ni during the synthesis and Ni–Nx centers implanted to this catalyst appears to negatively impact the formation of catalytic active Fe–Nx sites on the surface, which accounts for the reduced CO uptake.
2.4. The active site density in bulk and the utilization factor
57Fe Mössbauer spectroscopy was used to estimate the number of Fe–Nx sites of the catalyst (Fig. 4a and b). It is a powerful fingerprinting technique to determine different electronic and chemical iron states.61In the Fe–NC sample five different iron species can be identified (Fig. 4a). Beside a superparamagnetic iron phase (S1, δiso = −0.04 mm s−1)62 two doublets and two singlets were assigned. The doublet D1 (δiso = 0.37 mm s−1, ΔEQ = 0.99 mm s−1) is found for all Fe–NC catalysts investigated by Mössbauer spectroscopy so far62–69 and represents a ferrous FeN4 site in the low-spin state. As discussed by Kramm et al., its overall coordination environment is most likely six-fold with a nitrogen ligand and the oxygen molecule in axial directions.60,61,70,71 This site was previously assigned as the ORR active site by us and others;63,68,72–74 however it should be noted that small iron oxide nanoparticles can also lead to a Mössbauer doublet with similar parameters.71,75 The doublet D2 (δiso = 0.50 mm s−1, ΔEQ = 2.04 mm s−1) shows an isomer shift with a ferrous intermediate spin site. However, the quadrupole splitting is almost in-between the values found for iron porphyrins and iron phthalocyanine (FePc).61 The large quadrupole splitting for FePc in relation to iron porphyrins is caused by a pseudo-six fold coordination of the FeN4 sites with the above and underlining aza-bridges of the stacked FePcs.76 Based on this, the intermediate quadrupole splitting observed for the D2 doublet in this work might still be caused by an interaction in the axial direction, which is possibly not as much pronounced in relation to that of the phthalocyanine. The catalyst further showed two inorganic iron components assigned as iron sulphide (sext1, δiso = 0.74 mm s−1)77 and iron carbide (sext3, δiso = 0.18 mm s−1).78 Both phases are formed frequently during the heat treatment of iron based catalysts.77 The relative absorption areas are given in Table 1. Following previous studies, we assume the D1 site to be the primary catalytically active site of our catalysts.68,73,79 The overall amount of inorganic iron species amounts to less than 20%, whereas more than 80% of the Fe atoms are in fourfold coordination with nitrogen atoms.
| δ/mm s−1 | ΔEQ/mm s−1 | H 0/T | fwhm/mm s−1 | Rel. area/% | |
|---|---|---|---|---|---|
| Fe–NC | |||||
| D1 | 0.37 | 0.99 | 0.61 | 27.4 | |
| D2 | 0.50 | 2.04 | 1.59 | 55.5 | |
| Singlet1 | −0.04 | 0.34 | 4.9 | ||
| Sext1 | 0.74 | 30.86 | 0.58 | 7.0 | |
| Sext3 | 0.18 | 21.01 | 0.27 | 5.2 | |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||
| FeNi–NC | |||||
| D1 | 0.39 | 0.75 | 0.61 | 15.8 | |
| D2 | 0.50 | 2.15 | 1.1 | 18.5 | |
| Singlet1 | 0.42 | 0.52 | 15.1 | ||
| Sext1 | 0.72 | 31.22 | 0.47 | 30.2 | |
| Sext2 | −0.02 | 29.98 | 1.09 | 20.4 | |
For the nickel-containing FeNi–NC catalyst sample (Fig. 4b) a different behavior was observed. The Mössbauer spectral fit identified a singlet with an isomer shift (δiso = 0.42 mm s−1) that is unusual for paramagnetic iron phases. We are not able to assign this singlet now to a known species only based on Mössbauer parameters. Low-temperature Mössbauer spectroscopy might be used in the future to give further insights. As for the “sext2” species a mere Mössbauer parameter-based assignment to both alpha-Fe and a FeNi alloy with 30% Ni is possible.80–83 Beside these inorganic phases, the sample FeNi–NC contains the mentioned D1 and D2 sites with absorption areas of 15.8% and 18.5%, respectively. The experimentally determined trend in the abundance of the D1 sites (Table 1) agrees well with that in the electrocatalytic activity of Fe–NC and FeNi–NC (note: beside a larger relative fraction in terms of absorption the iron content in FeNi–NC is also lower). The higher abundance of the catalytically active D1 sites in the Fe–NC catalyst (Table 1) is responsible for the higher electrochemical activity of the Fe–NC catalyst and is consistent with the trends in the Fe–Nx site bulk density (SDbulk) values discussed below. Hence, while FeNi–NC features active Fe–Nx sites at the surface with a nearly identical chemical nature, its Fe–Nx surface site density is lower than that of the Fe–NC catalyst and it contains more inorganic Fe-based phases. We consider the larger formation of inorganic Fe-based phases in the FeNi–NC catalyst compared to Fe–NC to be the consequence of the presence and competing reactivity of the Ni precursor during the synthesis.
As the TPD data indicate that not only one single type of FeNx site adsorbs the CO, for the bulk-related site density we considered the overall iron assigned to the doublets D1 and D2. However, as recently shown, other ORR inactive iron speciation might overlay in the central part of the Mössbauer spectrum, so D1 and D2 might be overestimated.71 This must be considered and taken into account. Thus, the value determined from Mössbauer spectroscopy needs to be considered as the upper limit of possibly present FeNx sites.
Active site utilization factors provide useful guidelines for a directed synthesis of catalyst materials with optimized active site dispersion on the surface. Surface site utilization factors are measures for the ratio of the total number of Fe–Nx sites that are located on the surface of the catalyst. Efficient catalysts are characterized by large site utilization factors. For example, in our previous work, the utilization factor was strongly dependent on the number of heat-treatment and preparation steps.9 For the purpose of evaluating surface site utilization factors, we point out the fundamental difference between the CO chemisorption experiments and the Mössbauer spectroscopy. CO chemisorption is a method to quantify the accessible Fe–Nx metal sites on the surface. In contrast, Mössbauer spectroscopy is a bulk method probing the maximum density of the active Fe–Nx sites. For a catalyst where all active sites are surface accessible the number of sites identified by CO sorption should be identical to the number from Mössbauer spectroscopy.
From the Mössbauer parameters the catalyst mass-based Fe–Nx site density in the catalyst bulk, SDbulk, was derived and is plotted in Fig. 4c (hatched bars). For the calculation of SDbulk both doublets D1 and D2 were considered, thus reflecting the maximum possible number of FeNx sites9,58,81,84,85 and it was evaluated using the following relation:
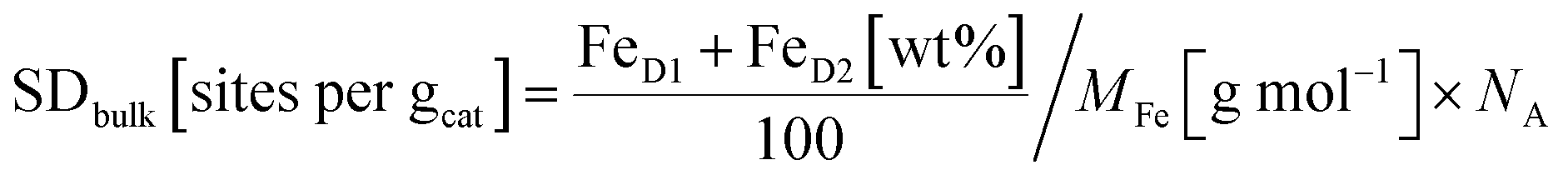 | (1) |
The CO-adsorption data were used to quantify the catalytically active adsorption sites on the catalyst surface. SDsurface, on the other hand, was estimated according to:
| SDsurface [sites per gcat] = nCO [mol g−1] × NA | (2) |
Fig. 4c shows that both SDsurface and SDbulk of the Fe–NC catalyst are significantly larger than those of FeNi–NC (Table S7†), which is in accordance with the observed trends in electrocatalytic ORR reactivity and our conclusions above.
Combining the SDsurface and SDbulk values, a site utilization factor can be derived according to:9
| ϕSDsurface/bulk = SDsurface/SDbulk | (3) |
The general importance of the site utilization factor ϕSDsurface/bulk lies in its guiding nature for the synthetic optimization of the surface site density (SDsurface). An optimized synthesis generates MNC catalysts exhibiting ϕSDsurface/bulk = 1, that is, all Fe–Nx sites are located at the surface and can potentially contribute to the surface catalytic reaction process. Comparison of ϕSDsurface/bulk values obtained from varying synthesis pathways allows the selection and prioritization of synthesis pathways and identification of synthetic variables that favor dispersion of sites on the surface. Once ϕSDsurface/bulk = 1, the synthesis pathway is optimal, and further efforts to improve the catalytic reactivity can focus on improving the intrinsic turnover frequency.
2.5. Catalytic turnover frequency (TOF)
The TOF is an intrinsic reactivity descriptor of a catalyst. It describes the number of catalytic turnover events per catalytically active site and second. In electrochemistry, TOF values are often normalized to the number of electrons transferred per active site and second.To evaluate electrocatalytic TOF values, RRDE-derived, mass-transport corrected, kinetic current densities are normalized by the number of catalytic surface sites. Here, we estimate mean TOF values from the molar CO uptake, nCO, and the kinetic catalyst mass-based current density, jm, evaluated at 0.85 V vs. RHE in alkaline and 0.8 V vs. RHE in acidic media.
The CO-derived TOF values, TOF, were calculated using the relation:
| TOF [e− s−1 site−1] = jm [e− s−1 g−1]/nCO [site g−1] | (4) |
Fig. 5 displays the relevant nCOvs. Jm relation, the slopes of which are the respective TOF values. The catalytic TOF values of the Fe–NC catalyst were ∼3× larger than those of the bimetallic FeNi–NC catalyst for both acid and alkaline electrolytes (Table S9†). This is mainly because the kinetic catalyst mass-based ORR activity, jm (at 0.85 VRHE in KOH and 0.8 VRHE in HClO4, respectively) of FeNi–NC was markedly smaller, in particular by a factor of around 8×. This sharp drop in kinetic activity was not offset by the roughly 3× smaller SDsurface value of the FeNi–NC catalyst, which is why bimetallic FeNi–NC appeared as an intrinsically less active ORR catalyst compared to the Fe–NC catalyst (Fig. 5c). This seems at odds with the discussion above: based on our analysis of the chemical state of the Fe–Nx sites in Fe–NC and FeNi–NC, we concluded that both Fe–Nx sites display very similar physico-chemical characteristics; hence, we would expect similar intrinsic catalytic TOF values, regardless of the varying absolute number of active sites at the surface (SDsurface). There are different reasons that could contribute to the observed discrepancy between the TOF values of Fe–NC and FeNi–NC. Firstly, we point to the larger ratio of micropores in the bimetallic FeNi–NC catalyst (Table S3†). While active Fe–Nx sites on the inner walls of micropores are probed by the CO cryo chemisorption technique, electrolyte flooding combined with diffusional oxygen transport limitations and ionomer poisoning could lower the apparent catalyst mass-based ORR activity of the FeNi–NC catalyst (see Fig. 5a and b). Secondly, the ORR mechanism and rate-limiting step could be different during the ORR for FeNi–NC compared to Fe–NC (Fig. 2 and S3†). The higher H2O2 yields of the FeNi–NC catalyst appear to support the co-existence of a 2e− and 4e− pathway, where the enhanced exposure of FeNi–NC to H2O2 may affect the reactivity of the catalytic sites.53 Thirdly, it was shown before that additional nitrogen atoms in the carbon frame can enhance the TOF value.69 As indicated in Tables S1 and S3† the nitrogen content of Fe–NC is much higher compared to that of FeNi–NC. Thus, a larger contribution of nitrogen functional groups and consequently TOF-increasing effects is likely for Fe–NC. While we cannot discriminate the individual contributions of these possibilities, as a result, the slopes of the FeNi–NC catalyst and the TOF values are smaller than those of Fe–NC (Fig. 5c).
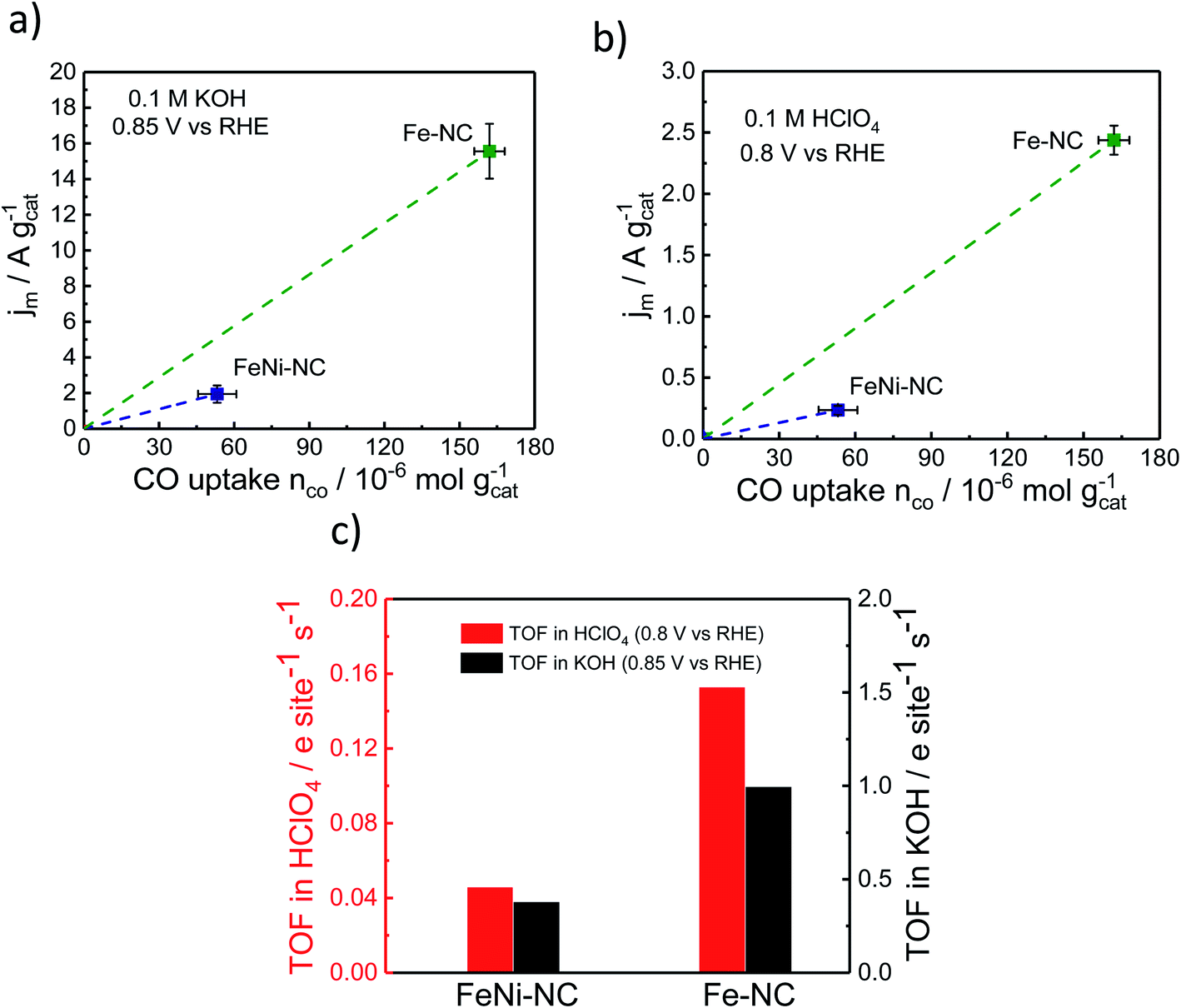 | ||
| Fig. 5 ORR catalyst kinetic mass activity jm at (a) 0.85 V vs. RHE in alkaline and (b) 0.8 V vs. RHE in acidic media, respectively as a function of CO uptake. (c) Comparison of the turn-over frequency values as derived from CO chemisorption and RRDE experiments in acid and alkaline electrolytes. | ||
3. Conclusions
The objective of the present study was to achieve a deeper understanding of the chemical nature, surface site density, and intrinsic reactivity (turnover numbers) of the atomically dispersed, catalytically active Fe–Nx sites located at the surface of a bimetallic PGM-free FeNi/N-doped carbon (“FeNi–NC”) ORR catalyst and of the monometallic Fe–NC and Ni–NC analogues. We provided evidence that Fe–Nx sites and not Ni–Nx sites are responsible for the kinetic ORR reactivity. XAS and Mössbauer spectroscopy as well as CO desorption data suggested that the chemical nature and bonding characteristics of the Fe–Nx sites are very similar for the Fe–NC and FeNi–NC catalysts. CO cryo chemisorption experiments and Mössbauer spectroscopy, on the other hand, yielded an estimate of the metal Fe–Nx site densities in the surface (SDsurface) and in the bulk (SDbulk) of Fe–NC and FeNi–NC. Contrasting these site density values for each catalyst and between the two catalysts, we concluded that the co-generation and/or presence of Ni–Nx sites in the bimetallic catalyst partially suppressed the generation of catalytically active Fe–Nx sites, which, in turn, reduced the resulting SD values of FeNi–NC by a factor of 3. The reduction in the corresponding kinetic ORR reactivity of the bimetallic FeNi–NC catalyst, however, sharply exceeded this factor (8×) and might have different origins as changes in the contribution of pore regimes and the higher exposure of active sites to H2O2 could affect their reactivity or higher intrinsic TOF of Fe–NC by nitrogen doping of the graphene planes. As a result, the intrinsic TOF values of FeNi–NC fell short of those of Fe–NC, despite chemical identity.The site density values further allowed the estimation of a site utilization factor ϕSDsurface/bulk. ϕSDsurface/bulk offers rational guidelines for the design and synthesis of PGM-free M–NC catalysts toward optimal metal site dispersion on the surface. By means of the ϕSDsurface/bulk, catalyst development can address the increase in the dispersion of catalytic surface sites and the increase in the catalytic TOF values independently.
4. Experimental methods
4.1. Catalyst synthesis
Aniline (Sigma-Aldrich) was first dissolved in 0.5 M HCl solution. Thereafter, metal precursors (6.05 g FeCl3·6H2O for Fe–NC, 6.05 g FeCl3·6H2O and 1.26 g NiCl2·6H2O for FeNi–NC, and 2.60 g NiCl2·6H2O for Ni–NC) were added into the solution and kept stirring for 15 min to be fully mixed with aniline. After the complete dissolution/mixing of the precursor with aniline, ammonium peroxydisulfate (APS, (NH4)2S2O8, Sigma-Aldrich) was dissolved in the solution as an oxidant to form poly-aniline, followed by adding activated carbon which was pretreated with 0.5 M HCl and 67% HNO3. The polymer suspension was kept at room temperature as long as no color change was observed. Carbon supported metal/poly-aniline was refluxed for 5 hours at 80–90 °C and then dried at 80 °C overnight by using an oil bath. The subsequently dried powder was ball milled and pyrolyzed at 900 °C for 1 hour in N2 (blowing N2 through the quartz tube at room temperature for 2 hours, then 5 °C min−1 temperature ramp for heating up to 900 °C, holding 1 hour under N2 and subsequent cooling overnight under N2). After the first pyrolysis step, one typical acid leaching step was used to remove all unreacted residues: the well-ground powder was dispersed in a diluted acidic solution and refluxed under a N2 atmosphere at 90 °C for 8 hours. After acid leaching, the samples were washed to neutralize them and then dried. The acid leached powders went through a second heat-treatment at 900 °C for 1 hour (2nd pyrolysis) followed by a second acid leaching/washing/drying step. Finally, the third pyrolysis (900 °C/1 h/N2) step was performed to achieve the final catalyst.4.2. Physical characterization
Elemental analysis for determining the catalysts' composition was performed by using a Thermo Electron, Flash EA 1112 analyzer. The N2-physisorption measurements were performed on an Autosorb-1-C from the Quantachrome Company. The samples were first degassed by heating for 24 hours at 60 °C in a vacuum. Before the Brunauer–Emmett–Teller (BET) measurements, the samples were cooled using a flow of liquid nitrogen and continuously loaded with nitrogen during the measurements. After saturation of N2 on the catalyst surface, the gas pressure was reduced which results in the desorption of N2 molecules from the surface. The BET-specific surface area was calculated using Quantachrome Software and a DFT model was used to determine the pore size distribution of the carbon material. ICP-OES was carried out on a Varian 715-ES. Therefore, a certain amount of the sample was digested in a mixture of 2 ml sulfuric (98 wt%), 2 ml nitric (69 wt%), and 6 ml chloric (37 wt%) acids in a microwave (CEM SP-D Discover) until it became transparent. XPS was carried out on a K-Alpha™ + X-ray Photoelectron Spectrometer (XPS) System (Thermo Scientific), with a Hemispheric 180° dual-focus analyzer with a 128-channel detector and the X-ray monochromator was micro focused Al-Kα radiation. N1s spectra were fitted with eight Gauss–Lorenz (FWHM: 1.6 ± 0.05 eV, GL ratio: 0.65) and Gauss convoluted Doniach–Sunjic (FWHM: 1.45 ± 0.05 eV) functions after testing various combinations of fit parameters (Peaks 1–8). The eight peak functions were then clustered into 4 binding energy (BE) ranges and denoted as BE-range 1–4. BE-range 1 includes Peak 1 (398.1 eV) and Peak 2 (398.7 eV) covering N bonded with 2 sp2 carbon atoms and –CN–C double bond motifs (e.g. imine N, pyridinic N, or triazinic N). BE-range 2 includes Peak 3 (399.3 eV) and Peak 4 (399.8 eV) covering sp2 hybridized N in metal–N coordination, OC–NH–C partial double bonds, or multiple graphitic N motifs in a single aromatic ring (e.g. M–Nx, amides). BE-range 3 includes Peak 5 (400.7 eV), Peak 6 (401.8 eV), and Peak 7 (403 eV) covering hybridized N bonded with 1 H (in-place hydrogenated N), isolated graphitic N, out-of-plane hydrogenated/protonated N, and hydrogenated graphitic N (e.g. pyrrolic or protonated pyridinic and protonated graphitic N). BE-range 4 includes Peak 8 (405 eV) covering oxidized N (e.g. CN–O or other oxidized N). The Gauss convoluted Doniach–Sunjic function was employed for Peak 5 (400.7 eV), Peak 6 (∼401.8 eV), and Peak 7 (∼403.0 eV) to account for the asymmetry on the high binding energy side of the major spectral peak. Fe, Ni L-edge XAS data were collected at the Innovative Station for In Situ Spectroscopy (ISISS) beamline of BESSY II of the Helmholtz–Zentrum Berlin at RT under ultra-high vacuum conditions.46 The Fe, Ni L-edge spectra were recorded in the total electron yield mode detected with a Faraday cup. STEM images and energy-dispersive X-ray (EDX) spectroscopy elemental maps were acquired with a probe-corrected (Cs-corrected for the illumination system) JEM-ARM200F NeoARM scanning transmission electron microscope (JEOL Ltd.), operated at 80 kV using a cold-FEG, equipped with a Dual-EDX-System with 2 × 100 mm2 detectors. Specimens were prepared by drop-casting 3 μl of the sonicated dispersion onto a lacey carbon TEM grid and dried in air. 57Fe Mössbauer spectroscopy was performed in transmission mode at room temperature. A CMCA-550 (WissEI) equipped with a constant electronic drive system and triangular reference wave form (Halder Electronics) combined with a 57Co/Rh source was used. For calibrating the velocity range the isomer shift δiso of the sextet lines of α-Fe foil (25 μm thick, 99.99% purity) within a velocity range of ± 8 mm s−1 and ± 10 mm s−1 was used for Fe–NC and FeNi–NC, respectively. The Mössbauer spectra were fitted with MossWinn 4.0i.
4.3. Electrochemical measurements of ORR activity and selectivity
Rotating-ring disk electrode (RRDE) measurements were performed to determine the ORR activity of PANI-derived non-noble metal catalysts. Therefore, 15.7 mg catalyst powder, 750 μl de-ionized water, 190 μl ethanol and 60 μl Nafion were used to prepare an ink and sonicated for 15 min. The required aliquot of the ink was dropped on a glassy carbon disk (0.2475 cm2) to achieve a loading of 800 μg cm−2. A carbon rod and a reversible hydrogen electrode (RHE) were used as the counter electrode and reference electrode, respectively. As electrolytes O2-saturated aqueous solutions of 0.1 M KOH and 0.1 M HClO4 were used. The potential range of the disk was set from 0.05 to 1.1 V vs. RHE in an alkaline environment, and 0 to 1.1 V vs. RHE for the acidic electrolyte by choosing 5 mV s−1 scan rate at 800, 900, 1200, 1600 and 2000 rpm. The potential for the ring was kept at 1.2 V vs. RHE. The collection efficiency of the RRDE electrode used was 0.37 ± 0.01.The kinetic current density (jk), kinetic mass activity (jm), Tafel slope (mV per decade), fractional hydrogen peroxide (H2O2) yield (%) and electron transfer number are based on the following equations:
Kinetic current density (jk): combining the diffusion and kinetic controlled region, the total current can be determined using the Koutecky–Levich equation, which is expressed as follows:
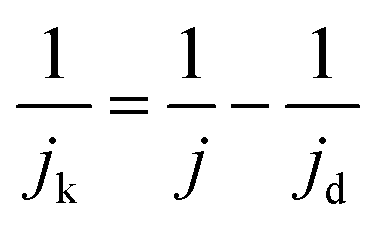 | (5) |
The kinetic mass activity (jm) for the MNC catalysts in this work is defined as:
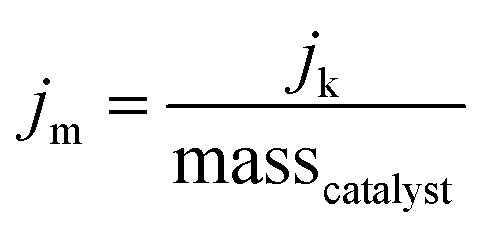 | (6) |
Hydrogen peroxide yield (%): H2O2 (%) was calculated based on the following equation:
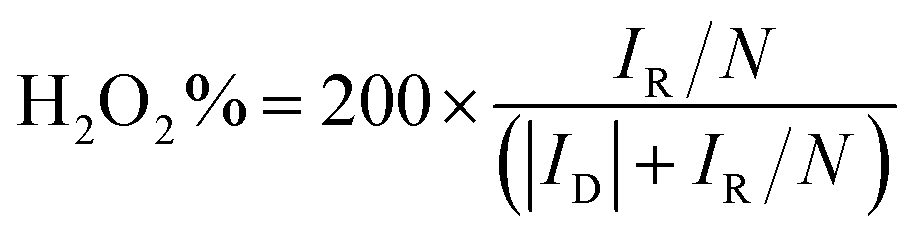 | (7) |
The electron transfer number was calculated based on the following equation:
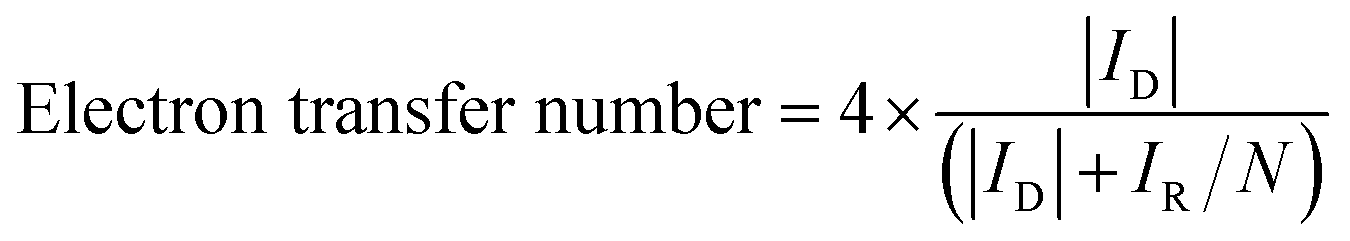 | (8) |
4.4. CO cryo chemisorption and temperature programmed desorption (TPD)
CO cryo chemisorption experiments and TPD were performed using a Thermo Scientific TPD/R/O 1110 instrument under a 20 ml min−1 flow of helium as the carrier gas. 100–150 mg of catalysts was added into a U-shaped quartz reactor and underwent the three-step analysis. (i) Thermal cleaning pretreatment involved heating the sample from room temperature to 600 °C with a ramp rate of 10 °C min−1 under a continuous 20 sccm 5 N helium flow, followed by a hold of 15 min at 600 °C, and then followed by subsequent convective cooling back to room temperature. (ii) CO pulse chemisorption involved subsequent cooling of the pretreated catalysts to −80 °C, using a mixture of dry ice and acetone, followed by passing six consecutive 0.338 ml CO pulses, dosed at 25 min intervals, through the catalyst samples, and the amount of CO retained in the catalysts was monitored and quantified using a TCD detector. (iii) A temperature-programmed CO desorption (TPD) was performed by ramping the temperature from −80 °C up to 600 °C with a ramp rate of 10 °C min−1 under a continuous 20 sccm 5 N helium flow, and the CO desorption temperature peak profiles were monitored for each catalyst.Author contributions
F. L., P. S. and U. I. K. conceived, designed and coordinated the study. F. L. carried out the materials synthesis, characterization and electrochemical evaluations. S. W. carried out the Mössbauer spectroscopy, I. O. and S. S. carried out the STEM., S. L. and A. T. carried out the XPS, and H. W. carried out the BET measurements. J. S. and J. W. participated in the discussion of the electrochemical results section. All the authors discussed the results and commented on the manuscript. F. L. wrote the manuscript with the contribution of all the co-authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge Dr Detre Teschner and Dr Tobias Reier for assistance with XPS and CO pulse chemisorption experiments, Dr.-Ing. Ralph Krähnert, and Dr Denis Bernsmeier for their help with the nitrogen physisorption experiments, and Dr Ichiro Onishi and the JEOL Team for their support with microsopy. U. I. Kramm and S. Wagner would like to acknowledge financial support from the BMBF via contract 05K16RD1 and by the Graduate School of Excellence Energy Science and Engineering (GRC1070). The authors would also like to acknowledge financial support from BMBF “KorrZellKat” (Grant number: 03XP0251) project. The research leading to the present results has received funding from the Fuel Cells and Hydrogen 2 Joint Undertaking under grant agreement no.779366. This Joint Undertaking receives support from the European Union's Horizon 2020 research and innovation program, Hydrogen Europe and Hydrogen Europe research. Fang Luo also thanks Berlin International Graduate School for Nature Sciences and Engineering, China Scholarship Council for the financial support.References
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443–447 CrossRef CAS.
- M. Lefevre, E. Proietti, F. Jaouen and J. P. Dodelet, Science, 2009, 324, 71–74 CrossRef CAS.
- E. Proietti, F. Jaouen, M. Lefevre, N. Larouche, J. Tian, J. Herranz and J. P. Dodelet, Nat. Commun., 2011, 2, 416 CrossRef.
- D. H. Zhongwei Chen, A. Yu, L. Zhang and J. Zhang, Energy Environ. Sci., 2011, 4, 3167–3192 RSC.
- Z. L. Wang, D. Xu, J. J. Xu and X. B. Zhang, Chem. Soc. Rev., 2014, 43, 7746–7786 RSC.
- O. Spalek, J. Appl. Electrochem., 1994, 24, 751–757 CrossRef CAS.
- M. Lefèvre, E. Proietti, F. Jaouen and J.-P. Dodelet, Science, 2009, 324, 71–74 CrossRef.
- F. Jaouen, E. Proietti, M. Lefèvre, R. Chenitz, J.-P. Dodelet, G. Wu, H. T. Chung, C. M. Johnston and P. Zelenay, Energy Environ. Sci., 2011, 4, 114–130 RSC.
- N. R. Sahraie, U. I. Kramm, J. Steinberg, Y. Zhang, A. Thomas, T. Reier, J. P. Paraknowitsch and P. Strasser, Nat. Commun., 2015, 6, 8618 CrossRef CAS.
- S. Kattel, P. Atanassov and B. Kiefer, Phys. Chem. Chem. Phys., 2013, 15, 148–153 RSC.
- S. Kattel, P. Atanassov and B. Kiefer, J. Phys. Chem. C, 2012, 116, 17378–17383 CrossRef CAS.
- J. Wang, Z. Huang, W. Liu, C. Chang, H. Tang, Z. Li, W. Chen, C. Jia, T. Yao, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 17281–17284 CrossRef CAS.
- X. Fu, Y. Liu, X. Cao, J. Jin, Q. Liu and J. Zhang, Appl. Catal. B Environ., 2013, 130–131, 143–151 CrossRef CAS.
- S. H. Noh, M. H. Seo, J. Kang, T. Okajima, B. Han and T. Ohsaka, NPG Asia Mater., 2016, 8, e312 CrossRef CAS.
- C. Li, M. Wu and R. Liu, Appl. Catal. B Environ., 2019, 244, 150–158 CrossRef CAS.
- N. A. Galiote, F. E. R. Oliveira and F. H. B. Lima, Appl. Catal. B Environ., 2019, 253, 300–308 CrossRef CAS.
- I. Martinaiou, A. Shahraei, F. Grimm, H. Zhang, C. Wittich, S. Klemenz, S. J. Dolique, H.-J. Kleebe, R. W. Stark and U. I. Kramm, Electrochim. Acta, 2017, 243, 183–196 CrossRef CAS.
- H. Tributsch, U. I. Koslowski and I. Dorbandt, Electrochim. Acta, 2008, 53, 2198–2209 CrossRef CAS.
- S. Li, L. Zhang, J. Kim, M. Pan, Z. Shi and J. Zhang, Electrochim. Acta, 2010, 55, 7346–7353 CrossRef CAS.
- M. Kodali, C. Santoro, S. Herrera, A. Serov and P. Atanassov, J. Power Sources, 2017, 366, 18–26 CrossRef CAS.
- L. Yang, X. Zeng, D. Wang and D. Cao, Energy Storage Materials, 2018, 12, 277–283 CrossRef.
- M. Kuang, Q. Wang, P. Han and G. Zheng, Adv. Energy Mater., 2017, 7, 1700193 CrossRef.
- F. Luo, A. Roy, L. Silvioli, D. A. Cullen, A. Zitolo, M. T. Sougrati, I. C. Oguz, T. Mineva, D. Teschner, S. Wagner, J. Wen, F. Dionigi, U. I. Kramm, J. Rossmeisl, F. Jaouen and P. Strasser, Nat. Mater., 2020 DOI:10.1038/s41563-020-0717-5.
- Y. Sun, L. Silvioli, N. R. Sahraie, W. Ju, J. Li, A. Zitolo, S. Li, A. Bagger, L. Arnarson, X. Wang, T. Moeller, D. Bernsmeier, J. Rossmeisl, F. Jaouen and P. Strasser, J. Am. Chem. Soc., 2019, 141, 12372–12381 CrossRef CAS.
- J. R. J. K. Nørskov, A. Logadottir and L. Lindqvist, J. Phys. Chem., 2004, 108 Search PubMed.
- S. Yang, A. Verdaguer-Casadevall, L. Arnarson, L. Silvioli, V. Čolić, R. Frydendal, J. Rossmeisl, I. Chorkendorff and I. E. L. Stephens, ACS Catal., 2018, 8, 4064–4081 CrossRef CAS.
- F. Calle-Vallejo, J. I. Martínez, J. M. García-Lastra, E. Abad and M. T. M. Koper, Surf. Sci., 2013, 607, 47–53 CrossRef CAS.
- F. Calle-Vallejo, J. I. Martinez and J. Rossmeisl, Phys. Chem. Chem. Phys., 2011, 13, 15639–15643 RSC.
- K. Artyushkova, B. Kiefer, B. Halevi, A. Knop-Gericke, R. Schlogl and P. Atanassov, Chem. Commun., 2013, 49, 2539–2541 RSC.
- K. Artyushkova, A. Serov, S. Rojas-Carbonell and P. Atanassov, J. Phys. Chem. C, 2015, 119, 25917–25928 CrossRef CAS.
- S. Kattel, P. Atanassov and B. Kiefer, Phys. Chem. Chem. Phys., 2014, 16, 13800–13806 RSC.
- Y. Qian, P. Du, P. Wu, C. Cai and D. F. Gervasio, J. Phys. Chem. C, 2016, 120, 9884–9896 CrossRef CAS.
- K. Artyushkova, J. Vac. Sci. Technol., A, 2020, 38, 031002 CrossRef CAS.
- D. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Science, 2016, 351, 361–365 CrossRef CAS.
- J. Herranz, F. Jaouen, M. Lefèvre, U. I. Kramm, E. Proietti, J.-P. Dodelet, P. Bogdanoff, S. Fiechter, I. Abs-Wurmbach, P. Bertrand, T. M. Arruda and S. Mukerjee, J. Phys. Chem. C, 2011, 115, 16087–16097 CrossRef CAS.
- E. Vayner and A. B. Anderson, J. Phys. Chem. C, 2007, 111, 9330–9336 CrossRef CAS.
- J. Masa, W. Xia, M. Muhler and W. Schuhmann, Angew. Chem., 2015, 54, 10102–10120 CrossRef CAS.
- M.-R. Travis, L. Nicole J., W. Alexandra T., A. Kevin, R. Nathan, D. V. V. Troy, M. Jeffrey T. and S. Yogesh, A Pyridinic Fe-N4 Macrocycle Effectively Models the Active Sites in Fe/N-Doped Carbon Electrocatalysts, ChemRxiv, 2020 Search PubMed.
- M. Al Samarai, A. W. Hahn, A. Beheshti Askari, Y. T. Cui, K. Yamazoe, J. Miyawaki, Y. Harada, O. Rudiger and S. DeBeer, ACS Appl. Mater. Interfaces, 2019, 11, 38595–38605 CrossRef CAS.
- W. Ju, A. Bagger, G. P. Hao, A. S. Varela, I. Sinev, V. Bon, B. Roldan Cuenya, S. Kaskel, J. Rossmeisl and P. Strasser, Nat. Commun., 2017, 8, 944 CrossRef.
- T. Möller, W. Ju, A. Bagger, X. Wang, F. Luo, T. Ngo Thanh, A. S. Varela, J. Rossmeisl and P. Strasser, Energy Environ. Sci., 2019, 12, 640–647 RSC.
- M. Chhetri, S. Sultan and C. N. R. Rao, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 8986–8990 CrossRef CAS.
- X. Li, M. Xin, S. Guo, T. Cai, D. Du, W. Xing, L. Zhao, W. Guo, Q. Xue and Z. Yan, Electrochim. Acta, 2017, 253, 302–310 CrossRef CAS.
- B. P. Payne, M. C. Biesinger and N. S. McIntyre, J. Electron Spectrosc. Relat. Phenom., 2009, 175, 55–65 CrossRef CAS.
- W. M. R. C. D. Wanger, L. E. Davis, J. F. Moulder and G. E. Muilenberg Perkin-Elmer Corp, Surf. Interface Anal., 1981, 3, 0142–2421 CrossRef.
- A. Knop-Gericke, E. Kleimenov, M. Hävecker, R. Blume, D. Teschner, S. Zafeiratos, R. Schlögl, V. I. Bukhtiyarov, V. V. Kaichev, I. P. Prosvirin, A. I. Nizovskii, H. Bluhm, A. Barinov, P. Dudin and M. Kiskinova, in Advances in Catalysis, Academic Press, 2009, vol. 52, pp. 213–272 Search PubMed.
- H. T. Chung, D. A. Cullen, D. Higgins, B. T. Sneed, E. F. Holby, K. L. More and P. Zelenay, Science, 2017, 357, 479–484 CrossRef CAS.
- F. d. r. Jaouen and J.-P. Dodelet, J. Phys. Chem. C, 2009, 113, 15422–15432 CrossRef CAS.
- A. Shahraei, A. Moradabadi, I. Martinaiou, S. Lauterbach, S. Klemenz, S. Dolique, H.-J. Kleebe, P. Kaghazchi and U. I. Kramm, ACS Appl. Mater. Interfaces, 2017, 9, 25184–25193 CrossRef CAS.
- N. Ramaswamy, U. Tylus, Q. Jia and S. Mukerjee, J. Am. Chem. Soc., 2013, 135, 15443–15449 CrossRef CAS.
- J. Zagal, Coord. Chem. Rev., 1991, 119(1992), 89–136 Search PubMed.
- N. Eliaz and E. Gileadi, Physical Electrochemistry: Fundamentals, Techniques and Applications, Wiley-VCH, Weinheim, 2018 Search PubMed.
- C. H. Choi, H.-K. Lim, M. W. Chung, G. Chon, N. Ranjbar Sahraie, A. Altin, M.-T. Sougrati, L. Stievano, H. S. Oh, E. S. Park, F. Luo, P. Strasser, G. Dražić, K. J. J. Mayrhofer, H. Kim and F. Jaouen, Energy Environ. Sci., 2018, 11, 3176–3182 RSC.
- A. J. Moody, R. Mitchell, A. E. Jeal and P. R. Rich, Biochem. J., 1997, 324, 743–752 CrossRef CAS.
- L. Birry, J. H. Zagal and J.-P. Dodelet, Electrochem. Commun., 2010, 12, 628–631 CrossRef CAS.
- N. Guo, B. R. Fingland, W. D. Williams, V. F. Kispersky, J. Jelic, W. N. Delgass, F. H. Ribeiro, R. J. Meyer and J. T. Miller, Phys. Chem. Chem. Phys., 2010, 12, 5678–5693 RSC.
- L. Liu, F. Zhou, L. Wang, X. Qi, F. Shi and Y. Deng, J. Catal., 2010, 274, 1–10 CrossRef CAS.
- N. D. Leonard, S. Wagner, F. Luo, J. Steinberg, W. Ju, N. Weidler, H. Wang, U. I. Kramm and P. Strasser, ACS Catal., 2018, 8, 1640–1647 CrossRef CAS.
- F. Luo, C. H. Choi, M. J. M. Primbs, W. Ju, S. Li, N. D. Leonard, A. Thomas, F. Jaouen and P. Strasser, ACS Catal., 2019, 9, 4841–4852 CrossRef CAS.
- J. Li, L. Jiao, E. Wegener, L. L. Richard, E. Liu, A. Zitolo, M. T. Sougrati, S. Mukerjee, Z. Zhao, Y. Huang, F. Yang, S. Zhong, H. Xu, A. J. Kropf, F. Jaouen, D. J. Myers and Q. Jia, J. Am. Chem. Soc., 2020, 142, 1417–1423 CrossRef CAS.
- U. I. Kramm, L. Ni and S. Wagner, Adv. Mater., 2019, 31, e1805623 CrossRef.
- H. Schulenburg, S. Stankov, V. Schünemann, J. Radnik, I. Dorbandt, S. Fiechter, P. Bogdanoff and H. Tributsch, J. Phys. Chem. B, 2003, 107, 9034–9041 CrossRef CAS.
- M. T. Sougrati, V. Goellner, A. K. Schuppert, L. Stievano and F. Jaouen, Catal. Today, 2016, 262, 110–120 CrossRef CAS.
- J. L. Kneebone, S. L. Daifuku, J. A. Kehl, G. Wu, H. T. Chung, M. Y. Hu, E. E. Alp, K. L. More, P. Zelenay, E. F. Holby and M. L. Neidig, J. Phys. Chem. C, 2017, 121, 16283–16290 CrossRef CAS.
- J. Maruyama and I. Abe, J. Electrochem. Soc., 2007, 154, B297 CrossRef CAS.
- J. A. R. v. Veen, J. F. v. Baar and K. J. Kroes, J. Chem. Soc. Faraday Trans. I, 1981, 77, 2827–2843 RSC.
- A. L. Bouwkamp-Wijnoltz, W. Visscher, J. A. R. v. Veen, E. Boellaard, A. M. v. d. Kraan and S. C. Tang, J. Phys. Chem. B, 2002, 106, 12993–13001 CrossRef CAS.
- U. I. Koslowski, I. Abs-Wurmbach, S. Fiechter and P. Bogdanoff, J. Phys. Chem. C, 2008, 112, 15356–15366 CrossRef CAS.
- U. I. Kramm, I. Abs-Wurmbach, I. Herrmann-Geppert, J. Radnik, S. Fiechter and P. Bogdanoff, J. Electrochem. Soc., 2011, 158, B69–B78 CrossRef CAS.
- A. Zitolo, V. Goellner, V. Armel, M.-T. Sougrati, T. Mineva, L. Stievano, E. Fonda and F. Jaouen, Nat. Mater., 2015, 14, 937–942 CrossRef CAS.
- S. Wagner, H. Auerbach, C. E. Tait, I. Martinaiou, S. C. N. Kumar, C. Kubel, I. Sergeev, H. C. Wille, J. Behrends, J. A. Wolny, V. Schunemann and U. I. Kramm, Angew. Chem., 2019, 58, 10486–10492 CrossRef CAS.
- U. I. Kramm, M. Lefèvre, P. Bogdanoff, D. Schmeißer and J.-P. Dodelet, J. Phys. Chem. Lett., 2014, 5, 3750–3756 CrossRef CAS.
- A. Zitolo, V. Goellner, V. Armel, M. T. Sougrati, T. Mineva, L. Stievano, E. Fonda and F. Jaouen, Nat. Mater., 2015, 14, 937–942 CrossRef CAS.
- I. Herrmann, U. I. Kramm, J. Radnik, S. Fiechter and P. Bogdanoff, J. Electrochem. Soc., 2009, 156, B1283 CrossRef CAS.
- E. König, S. Hüfner and E. Steichele, Z. Naturforsch., 1967, 22, 1543–1550 Search PubMed.
- E. Kuzmann, A. Nath, V. Chersky, S. Li, Y. Wei, X. Chen, J. Li, Z. Homonnay, M. Gál, V. K. Garg, Z. Klencsár and A. Vertes, Hyperfine Interact., 2002, 139/140, 631–639 CrossRef CAS.
- M. Ferrandon, A. J. Kropf, D. J. Myers, K. Artyushkova, U. Kramm, P. Bogdanoff, G. Wu, C. M. Johnston and P. Zelenay, J. Phys. Chem. C, 2012, 116, 16001–16013 CrossRef CAS.
- U. I. Kramm, I. Herrmann-Geppert, S. Fiechter, G. Zehl, I. Zizak, I. Dorbandt, D. Schmeisser and P. Bogdanoff, J. Mater. Chem. A, 2014, 2, 2663–2670 RSC.
- J. Li, S. Ghoshal, W. Liang, M.-T. Sougrati, F. Jaouen, B. Halevi, S. McKinney, G. McCool, C. Ma, X. Yuan, Z.-F. Ma, S. Mukerjee and Q. Jia, Energy Environ. Sci., 2016, 9, 2418–2432 RSC.
- H. N. Ok and M. S. Han, J. Appl. Phys., 1973, 44, 1932–1933 CrossRef CAS.
- U. I. Kramm, M. Lefèvre, P. Bogdanoff, D. Schmeißer and J.-P. Dodelet, J. Phys. Chem. Lett., 2014, 5, 3750–3756 CrossRef CAS.
- C. E. Johnson, M. S. Ridout and T. E. Cranshaw, Proc. Phys. Soc., 1963, 81, 1079–1090 CrossRef CAS.
- J. F. Petersen, M. Aydin and J. M. Knudsen, Phys. Lett. A, 1977, 62, 192–194 CrossRef.
- I. A.-W. U. I. Koslowski, S. Fiechter and P. Bogdanoff, J. Phys. Chem. C, 2008, 112, 15356–15366 CrossRef.
- U. I. Kramm, J. Herranz, N. Larouche, T. M. Arruda, M. Lefevre, F. Jaouen, P. Bogdanoff, S. Fiechter, I. Abs-Wurmbach, S. Mukerjee and J. P. Dodelet, Phys. Chem. Chem. Phys., 2012, 14, 11673–11688 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc03280h |
| This journal is © The Royal Society of Chemistry 2021 |