
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of ethane and ethylene on catalytic non oxidative coupling of methane†
Rolf S.
Postma
 and
Leon
Lefferts
*
and
Leon
Lefferts
*
Catalytic Processes and Materials Group, Faculty of Science and Technology, MESA+ Institute for Nanotechnology, University of Twente, PO Box 217, Enschede, 7500 AE, Netherlands. E-mail: l.lefferts@utwente.nl
First published on 30th September 2021
Abstract
The effect of addition of ethane and ethylene (C2) on methane coupling at 1000 °C was investigated. A Fe/SiO2 catalyst was used to determine the contributions of catalytic as well as C2 initiated methane activation. The catalyst load as well as the residence times at 1000 °C downstream of the catalyst bed were varied. C2 addition significantly increases methane conversion rates, similarly for both ethane and ethylene, although ethylene is more effective when operating with long residence times in the post-catalytic volume. Methane activation via C2 addition proceeds dominantly in the gas-phase whereas catalytic C2 activation is negligible. The catalyst has no effect on methane conversion when the feed contains more than 2 vol% C2. Product selectivity distribution as well as total hydrocarbon yield at 10% conversion is not influenced by C2 addition, but is influenced by the amount of catalyst as well as residence time in the post-catalytic volume at high temperature. It is proposed that C2 impurities in natural gas change from a nuisance to an advantage by enhancing methane conversion and simplifying purification of the natural gas feed. A process is proposed in which ethylene is recycled back into the reactor to initiate methane coupling, leading to a process converting methane to aromatics.
Introduction
Natural gas, consisting typically of 90 vol% CH4 and 5 vol% C2 hydrocarbons,1 is considered as a highly interesting resource for the production of olefins and aromatics.2–4 Traditionally, these products are by-products of crude oil refining.5,6 It is, however, predicted that raw oil extraction will reach peak production in the coming decades,7 and hence the increase of global olefins and aromatics demand needs to be met via another process. Natural gas is already used on large industrial scale for the synthesis of base chemicals and fuels.8–10 These processes convert natural gas to specific hydrocarbons with high carbon efficiencies to olefins and aromatics over crude oil refining.11,12Current industrial methods for converting natural gas into base chemicals are multistep processes starting from methane steam-reforming to obtain syngas.13 The syngas can be converted into paraffins in the Fischer–Tropsch (FT) process8 or used for methanol synthesis, followed by methanol to gasoline (MTG)14 or methanol to olefins (MTO).12 The large number of process steps at different temperatures and pressures makes the processes energy intensive and only viable at large installed capacities.15
Direct conversion of methane to higher hydrocarbons is hence receiving a growing interest over the last decades, as a more efficient alternative to the indirect routes mentioned before. Three main research directions can be distinguished, i.e. methane dehydro-aromatization (MDA),16 oxidative coupling of methane (OCM)17 and non-oxidative coupling of methane (NOCM) at high temperature.18 Both MDA as well as OCM suffer from low single pass conversion as well as low product yields.16,17 Guo et al.19 reported in 2014 that a Fe/SiO2 catalyst is able to couple methane non-oxidatively to olefins and aromatics at high conversion levels and without coke formation. Measurements were carried out at temperatures in excess of 950 °C. Follow-up research showed that methane conversion can be increased by in situ hydrogen removal,20 by using a catalytic wall reactor21 and by increasing the residence time in the reactor at high temperature downstream the catalyst bed (post-catalytic volume).22,23 However, all these studies reported coke formation and lower catalytic activity19–25 compared to the original work of Guo et al.19 It is generally accepted that the catalyst initiates methane conversion, through formation of methyl radical, followed by free radical chain reactions and coupling reactions in gas phase, determining the product distribution.19,22,25 The publications concerning the Fe/SiO2 catalyst have focused on a pure methane feed, even though natural gas contains a significant fraction of C2 hydrocarbons.1
Ethane and ethylene readily form radicals that participate in the autocatalytic cycle of methane pyrolysis at reaction temperature, i.e. above 950 °C.26–29 Addition of ethane can significantly reduce the induction period during non-catalytic methane pyrolysis as reported in early work by Germain et al.30 Methane conversion rates can thus be significantly increased by introduction of small amounts of C2 hydrocarbons, up to 3%, into the reactant mixture as reported in early work by Schneider29 and Rokstad et al.31 Ogihara et al.32 reported recently that methane is activated by ethane addition even at relatively low temperatures 700–800 °C in absence of any catalyst. Guo et al.19 reported a significant increase in methane conversion upon addition of 1–5% C2H6 at 900 °C using the Fe/SiO2 catalyst, although ethane addition caused coke formation, which crucially was not observed in their experiments using pure methane. SABIC23 patented a concept where post-catalytic ethane injection would quench the free radical coupling reaction, stabilizing the formed olefin products. The effect of addition of C2 hydrocarbons in presence or absence of the Fe/SiO2 catalyst on methane conversion and especially product distribution has not been reported, to the best of our knowledge.
This study reports for the first time on the interaction between catalytic methane activation and activation via the addition of free-radical initiators, i.e. ethane and methane conversion, product selectivity distribution and deposit formation.
Experimental
Catalyst synthesis
The Fe/SiO2 catalyst is synthesized according to the method described in ref. 19 and details concerning the catalyst synthesis as well as characterization can be found in ref. 22. In short, inhouse synthesized Fe2SiO4 is mixed with quartz and ball-milled in N2 atmosphere overnight using a zirconia milling jar. The resulting powder is fused for 6 h at 1700 °C in air. The resulting slab is crushed and sieved, the fraction between 250–500 μm is used in the experiments. The sieved catalyst particles are leached for 2 h in 0.5 M HNO3, rinsed and dried to obtain the final catalyst.Reactor setup
A modular three-zone oven is used for catalytic testing. Each zone is thermally insulated from the others and from the environment, allowing for steep temperature gradients, as presented in Fig. 1. The pre-heater is always operated at 400 °C and the reactor-zone, containing the catalyst, at 1000 °C. The post-heater is either operated at 400 °C, cooling down the gas-stream directly after the reactor-zone, or at 1000 °C to create an extended residence time at higher temperature, increasing the conversion as reported in our previous work.22 Both temperature profiles, for operating the poster-heater at 400 °C or 1000 °C have been included in Fig. 1. Gas flowrates are controlled using digital mass flow controllers. Product gasses are analyzed using a three-channel Varian CP-3800 in-line gas chromatograph, the tubing between the reactor and the GC is heated to 200 °C to minimize hydrocarbon condensation. Further details concerning the reactor setup are presented in ref. 22. | ||
Fig. 1 Temperature profile inside the reactor measured with an empty reactor tube b) zoom in on Fig. 1a; gas-flow rates of 10 ml min−1 N2; vertical bars represent the insulating layers between the 3 different zones.  Pre- and post-heater at 400 °C; reactor-zone at 1000 °C; Pre- and post-heater at 400 °C; reactor-zone at 1000 °C;  post-heater at 1000 °C.22 post-heater at 1000 °C.22 | ||
Experimental procedure
The experimental procedure is adapted from ref. 22. The catalyst is placed at the desired position in the quartz reactor according to Fig. 2, held in position by a small quartz wool plug. The reactor is flushed for 10 minutes with 10 ml min−1 N2. The catalyst is heated with 20 °C min−1 to 900 °C under 10 ml min−1 N2. The catalyst is then activated in 90 vol% CH4 in N2 at 1000 ml gcat−1 h−1 during two hours, following the procedure of Bao c.s.33 After activation, the reactor is flushed for 10 minutes at 900 °C with N2 10 ml min−1. The temperature is increased to the desired reaction temperature, with a heating rate of 20 °C min−1 and the experiment is started by feeding 90 vol% CH4 balanced with N2. Ethane or ethylene are added while reducing the amount of nitrogen in the feed, keeping the methane concentration as well as the total flowrate constant. Catalyst,19 gas-phase27 and reactor-wall34 all contribute to methane conversion so that space velocity is not useful as a descriptor; therefore the total gas flowrate was kept constant at 16.6 ml min−1 to allow for a fair comparison. The product gas is analyzed using an online three channel gas chromatograph (Varian CP-3800), measuring CH4 and N2 on the first channel, H2 on the second channel and all hydrocarbon products on a third FID channel. At the end of an experiment, the reactor is flushed for 10 minutes with N2 at 10 ml min−1 after which the reactor ovens are turned off and left to cool, maintaining the N2 flow while cooling. The catalyst is removed from the reactor and analyzed for coke deposition using TGA (Mettler Toledo TGA/DSC 3+ Star System). Details on the temperature-programmed-oxidation measurement in the TGA can be found in ref. 22. The online GC samples every 27 minutes and all data points presented are based on at least 3 measurements in steady state conditions. | ||
| Fig. 2 3 ways to position the catalyst inside the 6 cm zone of oven segment 2. Situations 1 (S0) contains no catalyst, 2 (S1) contains a 1 cm high bed (160 mg), situation 3 (S3) contains a catalyst bed 3 cm height (420 mg), situation 4 (S6) a catalyst bed height of 6 cm (820 mg), leaving no free volume at reaction temperature. | ||
Methane conversion is calculated according:
 | (1) |
P X: partial pressure of compound X (bar).
Methane conversion is corrected for any change in the molar flow rate based on change in the nitrogen tracer concentration according eqn (1).19,35 In the same way, the conversion of the C2 reactant is calculated, according eqn (2). Note that C2 conversion as calculated is determined by both C2 fed to the reactor and formation of the same C2 species in the reactor, therefore this value can also become negative when formation dominates over conversion.
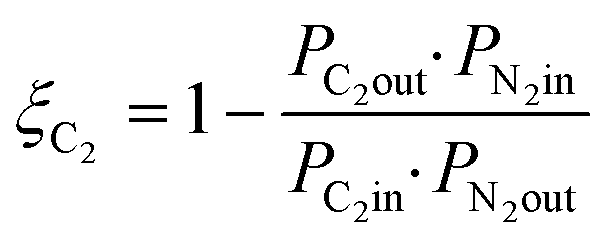 | (2) |
Product selectivity is calculated on carbon base, corrected for any change in the molar flow rates based on the concentration of the N2 tracer, eqn (3).19,35 The selectivity calculation takes into account conversion of both methane and C2, adjusting for stoichiometry.
 | (3) |
The measured hydrogen signal was used to validate the calculated conversion and selectivity distribution and closes to within 5%.
Experiments were performed over a maximum period of 8 h, a stability test over 16 h showed a deactivation of 10% methane conversion in a test using pure methane at 1.8% conversion.
Results
Effects on methane and C2 conversion
Fig. 3 shows the effect of adding small amounts of ethane and ethylene to the reactant mixture, varying the amount of catalyst and post-catalytic volume. Addition of ethane and ethylene significantly increases CH4 conversion. Interestingly, there is no discernible difference between the methane conversion when adding ethane (Fig. 3a and b) or ethylene (Fig. 3c). The effect of C2 addition on methane conversion is decreases with increasing catalyst amount, to the extent that catalyst has no effect when at least 2% ethane is added. (Fig. 3b) Fig. 3d shows a linear correlation, including the origin, between the free-volume in the reactor-zone and the linear acceleration in methane conversion rate by addition of C2 molecules, as calculated based on Fig. 3a and c. | ||
| Fig. 3 Effect of C2 addition on methane conversion, varying the amount of catalyst inside the reactor-zone: (a) ethane addition at lower concentration and b) higher ethane concentration; (c) ethylene addition; (d) the effect of free volume inside the reactor on the slope of increase in methane conversion when increasing C2 addition, as shown figures (a) and (c) (free volume is estimated based on the free volume surrounding the catalyst particles as well as the free volume downstream of the catalyst bed at packing density of 0.45); conditions: 90% CH4 N2 balance; 16.6 ml min−1 total flowrate. Reactor-zone at 1000 °C, pre-heater and post-heater at 400 °C. | ||
Fig. 4 shows the overall ethane and ethylene conversion in all experiments. Ethane is close to completely converted in all cases although conversion is somewhat lower when employing a large catalyst bed. Ethylene conversion on the other hand is always lower than 50%. Interestingly, ethylene production dominates over conversion when using the post-heater and when using a large catalyst bed (S6), i.e. conditions resulting in high methane conversion. Note that the C2 conversions presented in Fig. 4 are the result of C2 conversion and C2 production in the reactor.
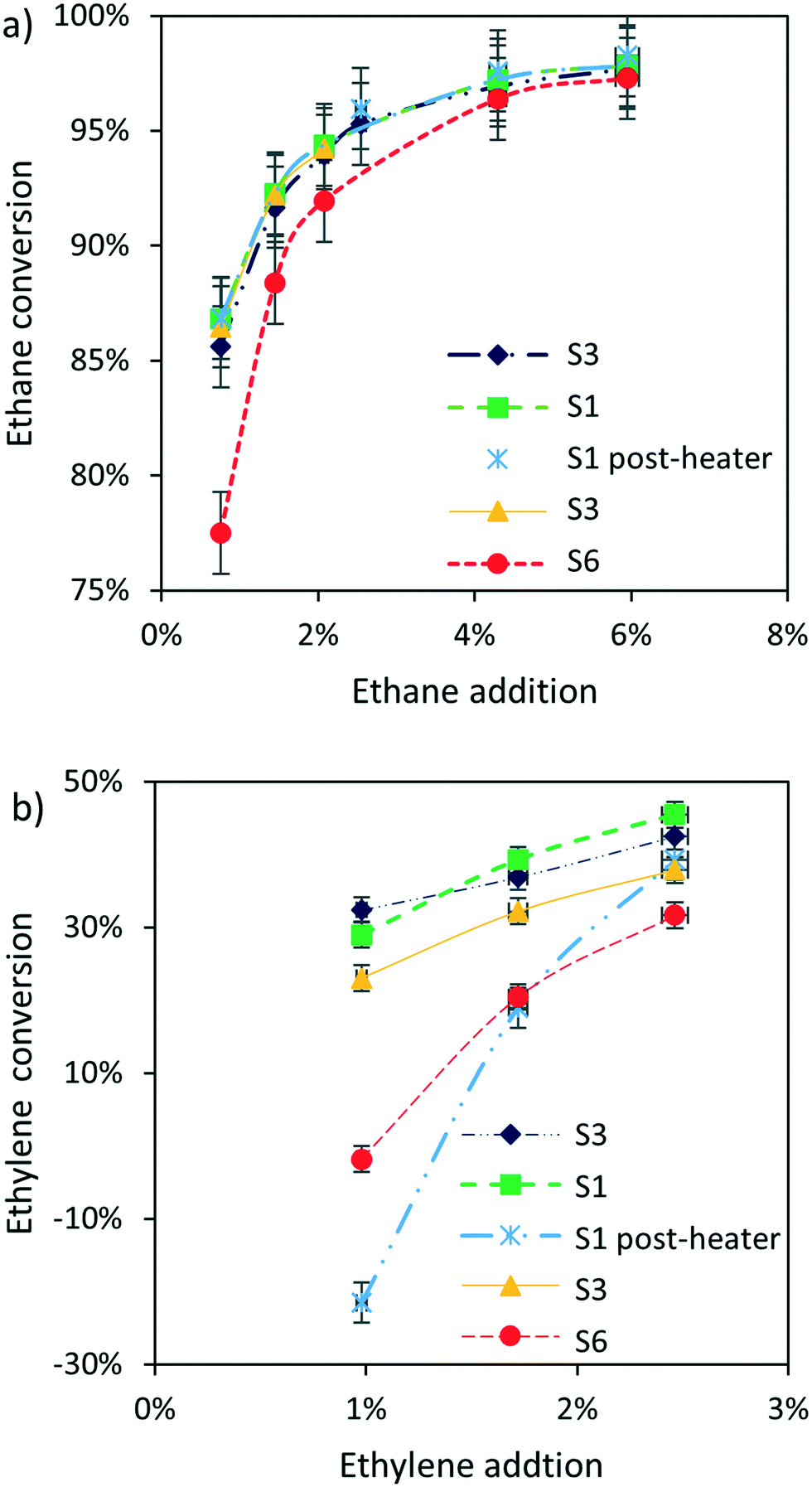 | ||
| Fig. 4 C2 conversion as function of C2 addition, (a) ethane, (b) ethylene; 90% CH4 N2 balance; 16.6 ml min−1 total flowrate. Reactor-zone at 1000 °C, pre-heater and post-heater at 400 °C, except for S1 post-heater having Tpost-heater at 1000 °C. | ||
The post catalytic residence time at high temperature can also be increased by using the post-heater as shown in Fig. 1. Fig. 5 shows a significant increase in methane conversion, keeping the level of ethane addition constant, when operating the post-heater at the same temperature as the reactor, i.e. 1000 °C. Fig. 5 also shows that ethylene addition results in the highest methane conversion when compared with ethane addition at the same concentration. Note that the difference in post-catalytic free volume between the S1 case with or without post-heater is a factor of 3.7, significantly larger than the increase in conversion observed in Fig. 5, showing a diminishing return of C2 addition at longer residence time.
 | ||
| Fig. 5 Effect of the post-heater on methane conversion with ethane or ethylene added, compared the effect of ethane addition without the post-heater (identical to S1 case Fig. 3b). 90% CH4, N2 balance; 16.6 ml min−1 total flowrate. Reactor-zone and post-heater at 1000 °C (except for S1 ethane case), pre-heater at 400 °C. | ||
Fig. 6 shows the selectivity towards the 3 major product groups: C2 hydrocarbons, C3–5 hydrocarbons and aromatics, keeping the conversion constant at 10%. This conversion level was achieved by either addition of ethane, addition of ethylene, or by changing the flow rate and consequently space-velocity without any addition of C2. The selectivity data at 10% conversion of hydrocarbon in the feed stream, assuming full conversion of the added C2 species, are obtained by interpolation as presented in the ESI,† Fig. S1 and S2. It is assumed that the added C2 compounds are completely converted whereas any ethane and ethylene detected in the product is assigned to formation in the reactor. In Fig. 6, data are interpolated to 10% conversion of hydrocarbons in the feed stream including full conversion of the added C2 compounds. Fig. S5† presents the same type of analysis at 10% methane conversion, disregarding the conversion of C2 compounds, resulting in very similar trends. Fig. 6 shows that, within the error margin, the selectivity to the various product groups is constant, independent of the method to enhance CH4 conversion to 10%, i.e. C2 addition or decreasing space velocity. The formation of aromatic products is somewhat suppressed when ethylene is added, as well as when a full-catalyst bed is used (S6). The product distribution of all hydrocarbon products is given in ESI† Fig. S4.
 | ||
| Fig. 6 Product selectivity distribution for different methods of increasing methane conversion, by ethane addition or ethylene addition to the reactant mixture or by decreasing space velocity. The results have been linearly interpolated at 10% total hydrocarbon conversion as explained in the experimental section. Similarly, selectivity is also calculated based on total hydrocarbon conversion. Reactor-zone at 1000 °C; pre-heater and post-heater are both at 400 °C, except for the S1-post-heater in that case Tpost-heater = 1000 °C. The total flowrate for the cases with ethane or ethylene addition is 16.6 ml min−1 90% CH4, N2 as balance. The graphs used for the interpolation can be found in ESI† Fig. S1 and S2. | ||
Fig. 7 shows the product yield to the three main product groups as function of conversion obtained with the small catalysts bed (S1) with and without the post-heater at high temperature, as a result of adding ethane (Fig. 7a) and ethylene (Fig. 7b). C2 yield and C3–5 yield are independent of the use of the post-heater, in contrast the aromatics yield which increased when using the post-heater.
 | ||
| Fig. 7 Effect on product yield when using the post-heater in the S1 case, (a) ethane, (b) ethylene; 90% CH4 N2 balance; 16.6 ml min−1 total flowrate. Reactor-zone at 1000 °C, pre-heater and post-heater at 400 °C, except for S1 post-heater having Tpost-heater at 1000 °C. | ||
Discussion
Addition of ethane
Methane conversion is significantly increased on ethane addition, apparent from Fig. 3a and b. The explanation for this significant enhancement in methane conversion is found in the studies of Roscoe and Thompson27 as well as Dean28 and schematically presented in Fig. 8. Formation of higher hydrocarbons from ethane, via ethyl radicals, releases hydrogen radicals which react with methane according to CH4 + H → CH3 + H2, which is the dominant reaction for activation of methane. The methyl radicals react to ethane, which maintains the cycle in Fig. 8, known as methane auto-catalysis.22,27,28 In the presence of catalyst, methyl radicals form on the Fe active sites while the hydrogen atoms stay on the active site, to combine to form di-hydrogen, according to DFT calculations.19,25 | ||
| Fig. 8 Proposed reaction paths for methane activation and product formation, based on ref. 26 and 27. | ||
A similar enhancement in methane conversion is also observed in non-catalytic operation in literature.29–31 Germain et al.30 and Rokstad et al.31 observed decreasing effectiveness of ethane addition on methane conversion when the ethane concentration is above 2% in absence of catalyst, very similar to the observation with catalyst in Fig. 3b, which Germain attributed to scavenging of free radical by formed aromatic species.
The catalyst has no effect on methane conversion at ethane concentrations above 2%. This shows that ethyl radicals and consequently H radicals mainly form in gas phase whereas catalytic ethane activation is negligible. This is further supported by the linear correlation between the enhancement of methane conversion due to ethane addition and the free volume available in the reactor, as shown in Fig. 3d, as well as by the observation that a large amount of catalyst reduces the ethane conversion, as shown in Fig. 4a. Methane conversion is not influenced by the presence of catalyst when at least 2% ethane is added, as shown in Fig. 3b. Apparently, enhancement of methane conversion on the catalyst is compensated by a decrease is methane conversion by decreasing the free volume when catalyst is introduced.
Fig. 5 shows that the increase in methane conversion due to an extended post-catalytic residence time22 is also observed when adding ethane, although the effect is relatively mild when compared with the 5-fold enhancement in absence of ethane. The activity for methane conversion without C2 addition in the post-catalytic free-volume22 is attributed largely to C2 hydrocarbon formation via catalytic methane activation.
The selectivity distribution over the main product groups at 10% conversion of hydrocarbons in the feed in Fig. 6 is obtained by interpolation, based on the data in Fig. S1 and S2 in the ESI.† The selectivity to ethane is included by assuming that the ethane in the feed is completely converted, as observed in Fig. 4a especially when the concentration of ethane added is high. The relatively small amount of ethane in the product stream is therefore counted as a product. Fig. S4† shows the selectivity for all hydrocarbon compounds detected. Product selectivity at 10% conversion level is independent from the method applied to achieve 10% conversion, i.e. catalytic or by ethane addition (Fig. 6). Fig. 6 shows that the total hydrocarbon selectivity decreases with increasing the amount of catalyst (S3, S6) in presence of ethane, probably because of the increased formation of coke-on-catalyst with increasing catalyst amount as observed in Fig. S7.† The observed influence of the amount of catalyst on coke formation on the catalyst is well in line with our previous work.22 The highest total hydrocarbon selectivity is achieved using the post-heater, in which the 10% conversion level is achieved via a significantly increased residence time at 1000 °C, rather than via addition of C2.
The invariance of the product selectivity of C2 addition is also reported by Ogihara et al.32 for non-catalytic pyrolysis of mixtures of ethane–methane between 700 and 800 °C. The results in the supporting information of Guo et al.19 confirm that product selectivity is unaffected by C2H6 addition, in good agreement with our results. Aromatic products dominate when the conversion is high by adding more C2 as shown in Fig. 7, which also shows that product yield is mainly determined by the conversion level. The main C2 specie measured at high conversion levels is ethylene whereas benzene and naphthalene are the dominant aromatic species (Fig. 3), in agreement with the observations by Guo et al.19 at high conversion.
Addition of ethylene
Ethylene has a very similar effect on methane conversion as ethane, shown in Fig. 3c and even better exemplified in Fig. 5 as the effects of ethane and ethylene are the same within experimental error. This is in agreement with Roscoe and Thompson27 as well as Dean,28 reporting that ethylene participates in methane activation in the same way as ethane. Ethylene causes a slightly higher methane conversion compared to ethane when operating with a long post-catalytic residence time as presented in Fig. 5. This is speculatively attributed to the higher thermal stability of ethylene compared to ethane,36 releasing radicals in a more dosed manner, continuing in the post-heater at relatively long residence times. In contrast, ethane is likely to decompose more readily, forming radicals only in the first part of the reactor and increasing the probability of unproductive termination by coupling of two hydrogen radicals to form H2. It must also be noted that likely a significant fraction of ethane dehydrogenates to ethylene, giving an alternative explanation for the similarity in effectiveness between ethylene and ethane addition.C2 compounds can consecutively dehydrogenate, from ethane via ethylene to acetylene to finally coke.22,37,38 C2H5 radical formed from ethane can release a hydrogen radical to form ethylene, in addition to the pathways presented in Fig. 8. Furthermore, ethylene can decompose to C2H3˙ via hydrogen abstraction by a methyl radical, followed by a similar cycle as described for ethane in Fig. 8. These consecutive dehydrogenation reactions consume one methyl radical and form one hydrogen radical and thus have no net impact on the free radical propagation.27,28
Note that ethane and ethylene need to react to form higher hydrocarbons in order to propagate the cycle in Fig. 8, limiting the maximum C2 yield when CH4 activation via gas phase autocatalysis is dominant. The auto-catalytic activation of methane will be discussed in more detail based on ongoing work.
Based on the similarity of the effect of ethane and ethylene on the conversion of methane and the fact that ethane converts almost completely, we assume that also all added ethylene is converted. In other words, the limited ethylene conversion in Fig. 4b is attributed to production of ethylene as a result of methane conversion and the product distribution presented in Fig. 6 is calculated based on the same assumption. Fig. 6 as well as Fig. S4† show that ethylene addition has no significant effect on the product distribution at 10% conversion, similar to ethane addition. Increasing the methane conversion by ethylene addition results in slightly lower selectivity to aromatics (Fig. 6), which is attributed to enhanced consecutive formation of deposits-downstream.
Industrial relevance
Addition of C2 significantly increases the methane conversion rate in the reactor (Fig. 3) while maintaining product selectivity (Fig. 6), thus increasing productivity per unit reactor volume. C2+ hydrocarbons are often present in natural gas with typical concentrations similar to the concentrations used in this study, for example Gulf-coast natural gas contains up to 4.9 vol% C2 hydrocarbons1 (details in Table S1†). Our study implies that removal of ethane from natural gas is not required, simplifying the purification of natural gas. The catalyst becomes obsolete at C2 concentrations above 2 vol% as shown in Fig. 3. Thus, the reaction can be operated at high productivity without requiring a catalyst, simplifying the reactor design and operation. Deposit build-up on the catalyst can be prevented and catalyst purchasing, handling, disposal and plant-stops for catalyst replacement are all not required. Rapid control of reactor capacity would be possible by tuning the concentration of the C2 initiator in the reactor feed.Overall ethylene conversion is typically low or even negative, meaning net-production at high methane conversion levels as shown in Fig. 4b. Ethylene can be recycled back into the reactor, to generate a process with a net-zero consumption of ethylene, as presented in the schematic process diagram in Fig. 9. In this scheme, the aromatic products are separated from the olefins, which are recycled back to the reactor, after separation of hydrogen. Fig. S3† shows that the yields of ethane as well as C3–5 hydrocarbons becomes negligible at high methane conversion levels. The activity increase in the reactor can be achieved both by recycled ethylene as well as ethane in the feed. Fig. 4 shows that overall ethylene conversion is positive at higher added ethylene concentrations and negative, i.e. net-production, at lower ethylene concentrations, demonstrating an internal feedback-loop that will prevent both ethylene accumulation as well as depletion in the process. This results in a process converting methane to aromatics, also alleviating the energy intensive cryogenic recovery of ethylene. The highest single pass methane conversion achieved in this paper, 25%, with 16% aromatics yield is already higher than benchmark performance of MDA, around 15% conversion and 11% aromatics yield.16 On the other hand, it must be noted that the main product is naphthalene compared to more valuable benzene produced in the MDA reaction and additional conversion might be required. Also, this scheme is obviously not producing any ethylene as originally targeted with methane pyrolysis.
 | ||
| Fig. 9 Basic schematic concept process for natural gas to aromatics based on NOCM including C2 recycle. | ||
Conclusion
The effect of C2 addition on non-oxidative methane coupling at high temperature was investigated. Addition of ethane and ethylene (C2) up to 6 vol% in methane significantly increases methane conversion during both catalytic as well as non-catalytic NOCM. The activation of methane through C2 addition is a dominated by homogenous gas-phase reaction and catalytic ethane or ethylene conversion is negligible. Product selectivity at constant conversion is unaffected by C2 addition and is influenced by catalyst amount and residence time in the hot zone of the reactor, both within and downstream of the catalyst bed, in line with our previous work. The catalyst has no effect on methane conversion at C2 concentrations above 2%, showing that an optimal reaction design involves either no catalyst at all or a minimal amount of catalyst. The catalyst is needed to initiate methane coupling via radical chain reactions only when less then 2 vol% of ethane or ethylene are present in the feed. C2 addition during NOCM significantly increases productivity and allows simplification of purification of the reactor-feed. A process design is envisioned in which the produced ethylene is recycled back into the reactor to initiate the methane conversion, resulting in a process converting methane to aromatics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support from the Dutch National Science Foundation (NWO) and the industrial partners: SABIC, Sasol and BASF. We thank ing. B. Geerdink, K. J. Altena, ing. T.L.M. Velthuizen and ir. T. Lubbers for their experimental and analytical support.References
- Uniongas, Chemical Composition of Natural Gas, https://www.uniongas.com/about-us/about-natural-gas/chemical-composition-of-natural-gas Search PubMed.
- C. Mesters, Annu. Rev. Chem. Biomol. Eng., 2016, 7, 223–238 CrossRef PubMed.
- C. Karakaya and R. J. Kee, Prog. Energy Combust. Sci., 2016, 55, 60–97 CrossRef.
- W. Taifan and J. Baltrusaitis, Appl. Catal., B, 2016, 198, 525–547 CrossRef CAS.
- H. Zimmermann and R. Walzl, Ethylene, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2009, DOI:10.1002/14356007.a10_045.pub3.
- H. O. Folkins, Benzene, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2000, DOI:10.1002/14356007.a03_475.
- Z. Caineng, Z. Qun, Z. Guosheng and X. Bo, Nat. Gas Ind., 2016, 3, 1–11 Search PubMed.
- A. D. Klerk, Journal, 2013, 1–20, DOI:10.1002/0471238961.fiscdekl.a01.
- H. A. Wittcoff, B. G. Reuben and J. S. Plotkin, Chemicals from Methane, Wiley & Sons, 2012, ch. 12, DOI:10.1002/9781118229996.ch12, ISBN: 9780470537435.
- M. Appl, Ammonia, in Ullmann's Encyclopedia of Industrial Chemistry, 2011, DOI:10.1002/14356007.a02_143.pub3.
- J. H. Lunsford, Catal. Today, 2000, 63, 165–174 CrossRef CAS.
- P. Tian, Y. Wei, M. Ye and Z. Liu, ACS Catal., 2015, 5, 1922–1938 CrossRef CAS.
- A. Abdulrasheed, A. A. Jalil, Y. Gambo, M. Ibrahim, H. U. Hambali and M. Y. S. Hamid, Renewable Sustainable Energy Rev., 2019, 108, 175–193 CrossRef CAS.
- E. Kianfar, S. Hajimirzaee, S. Mousavian and A. S. Mehr, Microchem. J., 2020, 156, 104822 CrossRef CAS.
- S. Mohajerani, A. Kumar and A. O. Oni, Energy, 2018, 150, 681–693 CrossRef.
- A. Galadima and O. Muraza, Catal. Surv. Asia, 2019, 23, 149–170 CrossRef CAS.
- I. Amghizar, L. A. Vandewalle, K. M. V. Geem and G. B. Marin, Engineering, 2017, 3, 171–178 CrossRef CAS.
- P. Schwach, X. Pan and X. Bao, Chem. Rev., 2017, 117, 8497–8520 CrossRef CAS PubMed.
- X. Guo, G. Fang, G. Li, H. Ma, H. Fan, L. Yu, C. Ma, X. Wu, D. Deng, M. Wei, D. Tan, R. Si, S. Zhang, J. Li, L. Sun, Z. Tang, X. Pan and X. Bao, Science, 2014, 344, 616–619 CrossRef CAS PubMed.
- M. Sakbodin, Y. Wu, S. C. Oh, E. D. Wachsman and D. Liu, Am. Ethnol., 2016, 55, 16149–16152 CAS.
- S. C. Oh, E. Schulman, J. Zhang, J. Fan, Y. Pan, J. Meng and D. Liu, Am. Ethnol., 2019, 58, 7083–7086 CAS.
- R. S. Postma and L. Lefferts, ChemCatChem, 2021, 13(4), 1157–1160 CrossRef CAS.
- D. A. Nagaki, Z. Zhao, M. N. Zin Myint, I. Lengyel, A. Mamedov, C. W. Gundlach, K. Sankaranarayanan and D. Falcone, US Pat., US9902665B2, SABIC Global Technologies, B.V., 2018 Search PubMed.
- S. J. Han, S. W. Lee, H. W. Kim, S. K. Kim and Y. T. Kim, ACS Catal., 2019, 9, 7984–7997 CrossRef CAS.
- S. K. Kim, H. W. Kim, S. J. Han, S. W. Lee, J. Shin and Y. T. Kim, Commun. Chem., 2020, 3, 58 CrossRef CAS.
- C. J. Chen and M. H. Back, Can. J. Chem., 1976, 54, 3175–3184 CrossRef CAS.
- J. M. Roscoe and M. J. Thompson, Int. J. Chem. Kinet., 1985, 17, 967–990 CrossRef CAS.
- A. M. Dean, J. Phys. Chem., 1990, 94, 1432–1439 CrossRef CAS.
- I. A. Schneider, Z. Phys. Chem., 1963, 223, 234–248 CrossRef CAS.
- J. Germain and C. Vaniscotte, Bull. Soc. Chim. Fr., 1958, 964–967 CAS.
- O. A. Rokstad, O. Olsvik and A. Holmen, Natural Gas Conversion, 1991, pp. 533–539 Search PubMed.
- H. Ogihara, H. Tajima and H. Kurokawa, React. Chem. Eng., 2020, 5, 145–153 RSC.
- X. Bao, Personal Communication, 2016 Search PubMed.
- W. E. Slater, J. Chem. Soc., Trans., 1916, 109, 160–164 RSC.
- S. Liu, L. Wang, R. Ohnishi and M. Ichikawa, J. Catal., 1999, 181, 175–188 CrossRef CAS.
- C. Guéret, M. Daroux and F. Billaud, Chem. Eng. Sci., 1997, 52, 815–827 CrossRef.
- M. S. Khan and B. L. Crynes, Ind. Eng. Chem., 1970, 62, 54–59 CrossRef CAS.
- A. Holmen, O. A. Rokstad and A. Solbakken, Ind. Eng. Chem. Process Des. Dev., 1975, 15, 439–444 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1re00261a |
| This journal is © The Royal Society of Chemistry 2021 |