
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective separation of amines from continuous processes using automated pH controlled extraction†
Luke A.
Power
 a,
Adam D.
Clayton
a,
William R.
Reynolds
a,
David R. J.
Hose
b,
Caroline
Ainsworth
b,
Thomas W.
Chamberlain
a,
Bao N.
Nguyen
a,
Richard A.
Bourne
a,
Nikil
Kapur
a and
A. John
Blacker
*a
a,
Adam D.
Clayton
a,
William R.
Reynolds
a,
David R. J.
Hose
b,
Caroline
Ainsworth
b,
Thomas W.
Chamberlain
a,
Bao N.
Nguyen
a,
Richard A.
Bourne
a,
Nikil
Kapur
a and
A. John
Blacker
*a
aInstitute of Process Research and Development, School of Chemistry, School of Chemical and Process Engineering, University of Leeds, Leeds, LS2 9JT, UK. E-mail: j.blacker@leeds.ac.uk
bChemical Development, Pharmaceutical Technology and Development, Operations, AstraZeneca, Macclesfield, SK10 2NA, UK
First published on 25th August 2021
Abstract
We present a rapid continuous processing methodology to screen for the optimal, selective, liquid–liquid extraction conditions, from a typical post-reaction mixture of amines, using both inline and online analysis to systematically alter the pH, by controlling the acid addition pump. A mixture of 95% α-methyl-benzylamine, 1, and 5% N-benzyl-α-methyl-benzylamine, 2, simulated a reaction product and impurity, with the former extracted from toluene into water with 92% efficiency and 99% purity. The initial acid concentration and outlet pH (post-extraction), were compared with the amine concentration in each phase. The incorporation of inline, pH and HPLC, monitoring of both the aqueous and organic phases, allowed for detailed analysis of the applied extraction conditions. This produced an autonomous system for exploring the amine extraction conditions: optimal amount of acid and organic-aqueous phase ratio.
Continuous processing within the fine chemicals sector is being widely explored and is showing benefits in reaction selectivity and conversion.1–3 Most papers focus on continuous flow reactions with downstream separation and purification carried out in batch. If the advantages of productivity and consistency, that continuous processing can bring, are to be realised, the removal of impurities and by-products is vital. Adamo et al. have introduced a useful unit for liquid–liquid (L–L) extraction which has been made commercially available. It employs a cast polymeric separation membrane, with diaphragm to internally regulate the pressure and maintain a driving force on the raffinate side.4,5 Several groups have reported additional devices for continuous L–L extraction and used them in the work-up of reaction mixtures.6–8 Acid/base mediated amine extraction has been demonstrated with these units, however, selective extraction, incorporating pH monitoring to control the removal of impurities, has not. Furthermore, there has been little investigation of flow methods to identify optimal extraction conditions, although autonomous optimisation strategies have been employed for batch.9
Dissociation extraction techniques are a subset of reactive extractions, used with, for example, amines or carboxylic acids in pharmaceutical and fine chemical production, fermentation broth extractions and enantioselective extractions.10–15 Although the extractant varies between techniques, from mineral to large aliphatic acids or bases, the function is the same, where ion-pairing alters the organic-aqueous distribution to allow for enhanced extraction. As purification processes account for a significant fraction of process solvent consumption, leading to higher costs and increased environmental impact, a method to more rapidly identify the optimum extraction conditions represents a useful addition to the process chemists repertoire.16–18
The focus of this research is on amine extraction, as 11% of final product pharmaceuticals contain at least one amine in their structure,19 but work on selective extraction of carboxylic acids is on-going and will be reported elsewhere. For the most part, amines have pKaH values that fall within the pH limits of water, which leads to a population of charged and neutral forms, where each species has a significantly different distribution between the aqueous and organic solvent. The selective extraction of one component of a mixture of two similar amines can be achieved if they have sufficiently different pKaH constants (ΔpKaH), e.g. starting material and product or product and impurity.20,21
A modular designed system was assembled, in which an organic and aqueous phase were mixed within a series of fReactor CSTRs and then separated using a Zaiput membrane separator (Fig. 1).4,22 In-line pH and temperature probes were incorporated into the initial acid mixing and final post separation sections by exchanging the lids of two of the CSTRs for faceplates that allow the probes to be mounted. This provided live data monitoring of the initial acid concentration and final post-extraction pH. A series of membranes, volume ratios and total flowrates were screened to determine the optimal system conditions to run the bulk extraction. In an effort to replicate the demands of an industrial process, conditions were selected to maximise the organic![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :aqueous volume ratio which would minimise the water needed. The Zaiput has an optimal volume ratio (organic:aqueous) of 1:1 and this was chosen to minimise the potential for loading (organic phase not passing through the membrane), at a maximum flowrate of 2 mL min−1 and using a PTFE hydrophobic membrane (0.9 μm pore size).
:aqueous volume ratio which would minimise the water needed. The Zaiput has an optimal volume ratio (organic:aqueous) of 1:1 and this was chosen to minimise the potential for loading (organic phase not passing through the membrane), at a maximum flowrate of 2 mL min−1 and using a PTFE hydrophobic membrane (0.9 μm pore size).
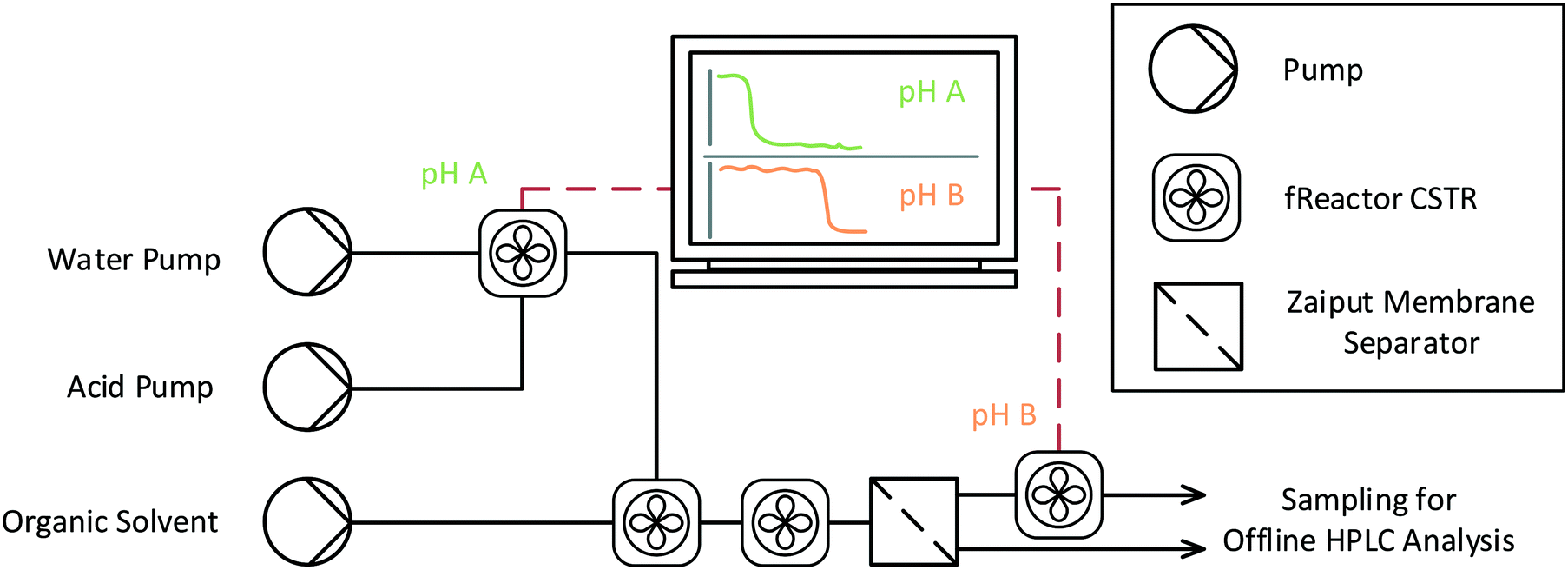 | ||
| Fig. 1 A schematic of the experimental setup for screening extractions in which the water and acid pumps mix a known concentration of acid that is monitored with pH probe A. This is then mixed with the toluene and amines for two CSTR volumes (to reach steady state), before it is separated, and the aqueous pH is further monitored. Samples of both the organic and aqueous streams are taken after that for offline HPLC. | ||
To test the system and mimic process conditions, a combination of a major product (α-methyl-benzylamine 1) and minor impurity (N-benzyl-α-methyl-benzylamine 2) 95:5 mole% at a total concentration of 0.87 M was chosen (Scheme 1). This concentration introduces potential for additional process related deviations, such as phase transfer, that can impact upon the volume and phase ratio, as the species are ∼10% of the liquid volume. The organic solvent was toluene, and the acid concentration was produced by combining the flowrates of a water and dilute hydrochloric acid pump that mix in the initial CSTR.
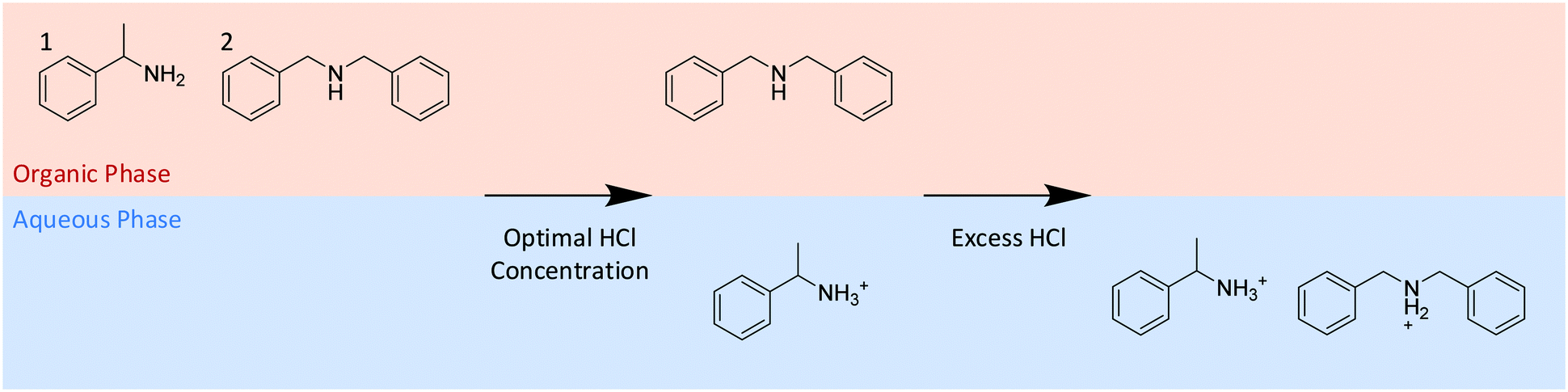 | ||
| Scheme 1 The controlled use of acid to selectively extract α-methyl-benzylamine (1) to the aqueous phase leaving N-benzyl-α-methyl-benzylamine (2) in the toluene phase. | ||
Readings from pH probe A were compared with the calculated acid concentration from the water and acid pumps to act as an initial validation. The correlation, with a linear fitting set to bisect the origin, was R2 = 0.99 and the slope deviated by 2.55% ± 1.4%. This is expected, with minor deviations between pumps and pH probe accuracy decreasing as pH tends towards 0 (1 M acid) due to the exponential increase in proton concentration and further distancing from the calibration points.
The acid concentration, extrapolated from pH probe A, was compared to the aqueous extraction efficiency of the individual amines from the mixture, defined as:20
| Extraction Efficiency = 1/(1 + KD) |
| KD = COrganic/CAqueous |
Fig. 2 illustrates a linear rise in the extraction of 1 into the aqueous phase as the acid concentration is increased until it is completely protonated. The total observed acid concentration required to completely ionise 1 was found to be between 0.78 and 0.84 M, which matches the concentration of 1 in the system (0.82 M). The minor impurity 2 rapidly protonates and transfers across, just as 1 is almost completely transferred. Altogether this forms a linear region where 1 can be isolated in high purity, whilst minimising the presence of 2. Increasing the acid concentration beyond that causes the opposite effect whereby the efficiency of purification rapidly decreases to a minimum.
 | ||
| Fig. 2 The extraction efficiency of each amine compared to the inlet acid concentration found from pH probe A. | ||
This effect can also be explained by the buffer regions that would exist for each of the amines. The titration curve shown in Fig. 3, compares the acid concentration inferred from pH probe A with the final pH probe B for 1 (pKaH = 9.73).23 At low acid concentrations, partition of 1 into the organic phase removes the direct relationship between the pH and pKaH value normally seen by the Henderson–Hasselbach equation.24 However, beyond acid concentrations of 0.7 M, the slope drops rapidly and little buffer region is observed for 2 (pKaH = 7.77), apart from a number of points clustered in a small region after 0.8 M acid, that are due to the lower concentration of the minor component.23 Using this data, the acid concentration for optimal selective extraction is ∼0.79 M, extracting 92% of 1 and minimising the amount of 2 extracted to ∼1%, i.e. 99% pure product in 92% yield.
 | ||
| Fig. 3 Titration curve for the extraction of 1 and 2 comparing acid added and pH after extraction. The buffering effect of 1 is clear, but lower than would be expected due to its partition into the organic phase. The effects of 2 are less apparent due to its lower concentration. | ||
The extraction of the amines into the aqueous phase was related to the pH monitor at position B, Fig. 4. Each amine gives a single stage titration curve as it is protonated and transferred into the aqueous phase (Fig. 4 upper). The difference curve (Fig. 4 lower) was used to highlight the difference in physical-organic behaviours between the two amines, removing the concentration factor present in the extraction efficiency curves. The separation factor is frequently used to compare data of this kind, however, the Δextraction efficiency parameter was chosen in preference with less variance observed in the optimal region (see ESI† Fig. S11 for details of the comparison).25 The pKaH differences that exist between the two species (ΔpKaH between 1 and 2 = 1.96) become apparent, indicating that a region exists where 1 is protonated and extracted, while 2 remains in the organic phase. The Δextraction efficiency curve has fewer datapoints between pH 7 and 4, because, as 2 transfers its lower concentration limits its ability to act as a buffer. The pump used is only able to deliver flow rates 0.01 mL min−1 increments leading to a maximum of 73 experimental points of discrete acid concentration (for the extraction this means there's 69 potential increments for the transfer of 1; and 4 potential increments for the extraction of 2).
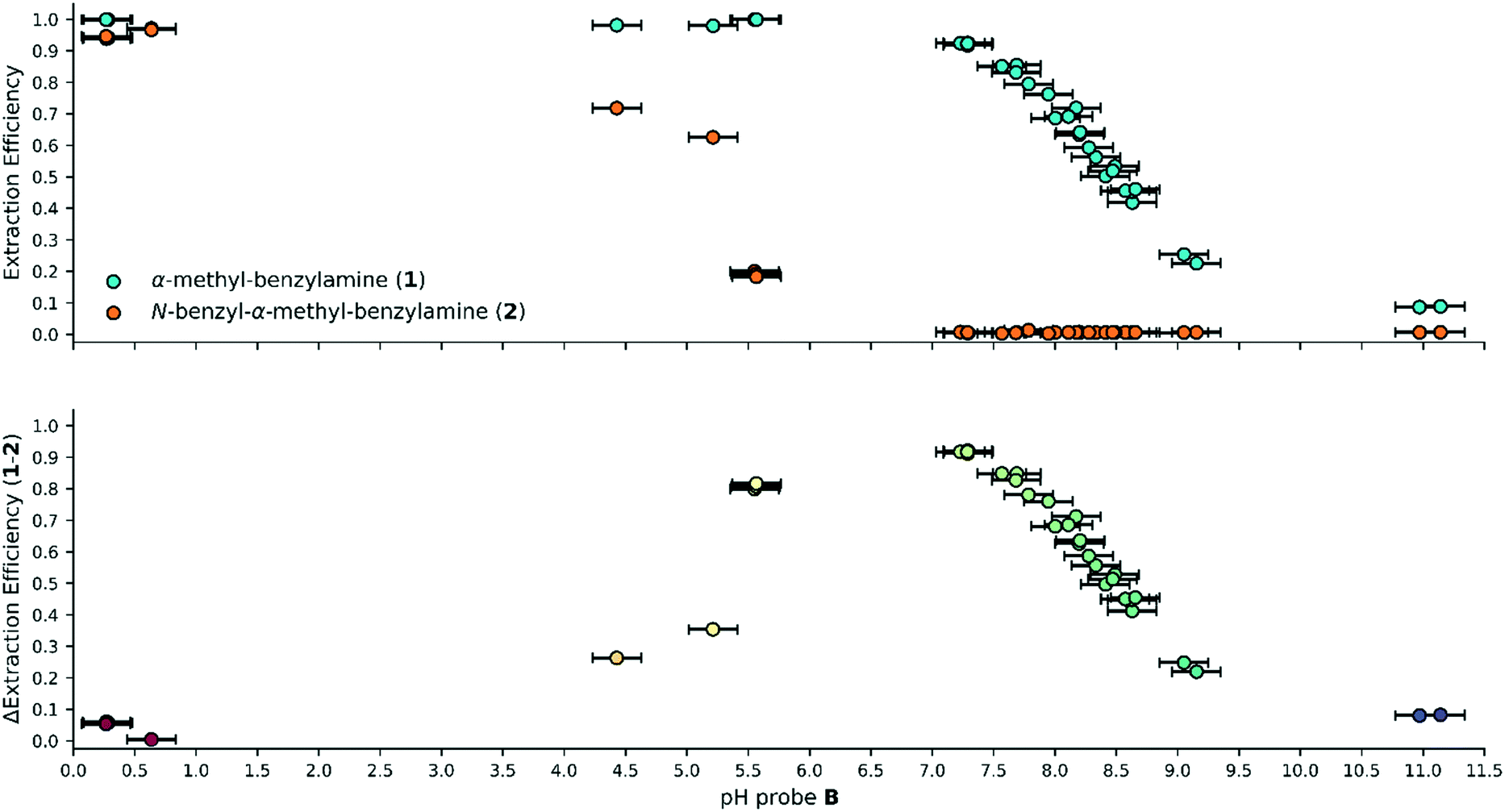 | ||
| Fig. 4 Upper: The extraction efficiency of α-methyl-benzylamine 1 (blue) and N-benzyl-α-methyl-benzylamine 2 (orange) with pH measured from probe B. Lower: The difference in extraction efficiency between 1 and 2, where the colour refers only to the pH, highlighting a normal distribution due to the relationship between the pKaH values. An optimal purification of 1, with 92% selectivity, is seen at pH 7. | ||
With pH screened off-line, an on-line HPLC with computer communicable pumps was incorporated, to enable an autonomous work-up platform. Sample loops were attached to either the aqueous or organic outlets for automated injection into the HPLC. Some deviation in membrane function was observed when the sample loops caused slightly different back pressures to the separator. This overtook the diaphragm's function and led to some instances where loading was observed, but was mitigated by the addition of secondary flowpaths, allowing for a pressure relief.
Given these modifications to the equipment, another extraction variable was included, phase volume ratio. This was varied between 0.9 to 1.1 (volume organic/volume aqueous), wider than this the data reproducibility was found to be poor. A full factorial DOE, with additional points incorporated around the maximum, was designed. These were extrapolated from the optima, determined from the linear experiments. Varying the acid concentration and volume ratio, the effect on the post-extraction pH and extraction efficiency of 1 and 2 are shown in Fig. 5. As the acid concentration is increased, 1 is increasingly transferred to the aqueous phase with a maximum difference between 1 and 2 of 0.85 observed at pH 7.02, similar to the optimum in Fig. 4. The difference in extraction efficiencies rapidly drops after this, as 2 is protonated and also transfers to the aqueous phase. For this system, better extractions are seen with more organic phase. Additional acid is required to titrate a larger mass of amine into the aqueous phase, meaning an overall higher efficiency can be reached when moving to higher volume ratios. The optimal Δextraction efficiency found was 0.85, which compares to 0.92 for the linear screen. The difference is explained by the natural partition of each amine (no pH adjustment).
 | ||
| Fig. 5 A 3D titration graph showing the effect of acid and phase volume ratio on the post extraction pH and the difference in extraction efficiency of amines 1 and 2 is shown by colour. | ||
For fine chemical industries, where most extractive work-ups are carried out in batch, this methodology could turn a 10 stage extraction into a single stage, yielding a high purity, highly extracted product.26 This single stage approach has potential to be expanded to multistage operation, with the same inline and online data acquisition and autonomy, allowing for large data gathering and rapid optimisation.27
Conclusion
A method is reported for screening L–L extraction conditions, for ionisable species, that uses pH monitoring within a continuous flow titration. The study highlights the improvements that can be achieved in extraction by controlling the inlet acid concentration, leading to improvement in extraction efficiency and purification of a product from a minor impurity. The method has removed the link between solute and acid/base concentration so that conditions for selective extraction of mixed species with different pKaH values can be identified. The use of in-line and on-line analytics and computer controlled pumps with a full factorial DOE has been demonstrated to provide similar optimum conditions to more intensive linear methods. Furthermore it has allowed exploration of the phase volume ratio. If an algorithm based approach were also embedded this would further automate the process to provide additional data and evidence for extraction optima.23,28 Further work is looking at separation of mixed carboxylic acids and the effect of additional variables on the extraction efficiency. With a large amount of waste produced through work-up, intensification in this manner can reduce the number of extraction stages or washes required and contribute to improved environmental performance.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the kind support of: University of Leeds, AstraZeneca Ltd and EPSRC for LP (EP/S513829/1).References
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS
.
- M. Baumann, T. S. Moody, M. Smyth and S. Wharry, Org. Process Res. Dev., 2020, 24, 1802–1813 CrossRef CAS
- F. M. Akwi and P. Watts, Chem. Commun., 2018, 54, 13894–13928 RSC
- A. Adamo, P. L. Heider, N. Weeranoppanant and K. F. Jensen, Ind. Eng. Chem. Res., 2013, 52, 10802–10808 CrossRef CAS
-
Zaiput, Zaiput Flow Technologies, https://www.zaiput.com/, (accessed 20 February, 2019) Search PubMed
- A. J. Harvie, J. O. Herrington and J. C. DeMello, React. Chem. Eng., 2019, 4, 1579–1588 RSC
- C. Day, A. Saldarriaga, M. Tilley, H. Hunter, M. G. Organ and D. J. Wilson, Org. Process Res. Dev., 2016, 20, 1738–1743 CrossRef CAS
- M. O'Brien, P. Koos, D. L. Browne and S. V. Ley, Org. Biomol. Chem., 2012, 10, 7031–7036 RSC
- J. A. Selekman, K. Tran, Z. Xu, M. Dummeldinger, S. Kiau, J. Nolfo and J. Janey, Org. Process Res. Dev., 2016, 20, 1728–1737 CrossRef CAS
- G. H. Twigg, Nature, 1949, 163, 1006–1007 CrossRef CAS
- M. M. Anwar, A. S. Arif and D. W. Pritchard, Solvent Extr. Ion Exch., 1995, 13, 127–142 CrossRef CAS
- M. M. Anwar, A. S. Arif and D. W. Pritchard, Solvent Extr. Ion Exch., 1998, 16, 931–950 CrossRef CAS
- K. L. Wasewar, Int. J. Chem. Eng. Appl., 2012, 249–255 CAS
- K. Tang, Y. Wang, P. Zhang, Y. Huang and G. Dai, Sep. Purif. Technol., 2015, 150, 170–178 CrossRef CAS
- B. Schuur, B. J. V. Verkuijl, A. J. Minnaard, J. G. De Vries, H. J. Heeres and B. L. Feringa, Org. Biomol. Chem., 2011, 9, 36–51 RSC
- C. Jiménez-González, P. Poechlauer, Q. B. Broxterman, B.-S. Yang, D. am Ende, J. Baird, C. Bertsch, R. E. Hannah, P. Dell'Orco, H. Noorman, S. Yee, R. Reintjens, A. Wells, V. Massonneau and J. Manley, Org. Process Res. Dev., 2011, 15, 900–911 CrossRef
- A. C. Bédard, A. R. Longstreet, J. Britton, Y. Wang, H. Moriguchi, R. W. Hicklin, W. H. Green and T. F. Jamison, Bioorg. Med. Chem., 2017, 25, 6233–6241 CrossRef
- M. Köckinger, C. A. Hone, B. Gutmann, P. Hanselmann, M. Bersier, A. Torvisco and C. O. Kappe, Org. Process Res. Dev., 2018, 22, 1553–1563 CrossRef
- F. Mao, W. Ni, X. Xu, H. Wang, J. Wang, M. Ji and J. Li, Molecules, 2016, 21, 75 CrossRef PubMed
- I. W. Ashworth and R. E. Meadows, J. Org. Chem., 2018, 83, 4270–4274 CrossRef CAS
- R. G. Robinson and D. Y. Cha, Biotechnol. Prog., 1985, 1, 18–25 CrossRef CAS PubMed
- M. R. Chapman, M. H. T. Kwan, G. King, K. E. Jolley, M. Hussain, S. Hussain, I. E. Salama, C. González Niño, L. A. Thompson, M. E. Bayana, A. D. Clayton, B. N. Nguyen, N. J. Turner, N. Kapur and A. J. Blacker, Org. Process Res. Dev., 2017, 21, 1294–1301 CrossRef CAS
- A. D. Clayton, L. A. Power, W. R. Reynolds, C. Ainsworth, D. R. J. Hose, M. F. Jones, T. W. Chamberlain, A. J. Blacker and R. A. Bourne, J. Flow Chem., 2020, 10, 199–206 CrossRef
- H. N. Po and N. M. Senozan, J. Chem. Educ., 2001, 78, 1499 CrossRef CAS
- V. V. Wadekar and M. M. Sharma, J. Chem. Technol. Biotechnol., 1981, 31, 279–284 CrossRef CAS
- R. Porta, M. Benaglia and A. Puglisi, Org. Process Res. Dev., 2016, 20, 2–25 CrossRef CAS
- R. Lebl, T. Murray, A. Adamo, D. Cantillo and C. O. Kappe, ACS Sustainable Chem. Eng., 2019, 7, 20088–20096 CrossRef CAS
- A. D. Clayton, J. A. Manson, C. J. Taylor, T. W. Chamberlain, B. A. Taylor, G. Clemens and R. A. Bourne, React. Chem. Eng., 2019, 4, 1545–1554 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1re00205h |
| This journal is © The Royal Society of Chemistry 2021 |