
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Renewable dimethyl carbonate for tertiary amine quaternisation: kinetic measurements and process optimisation
Roel J. T.
Kleijwegt
 a,
Vera C.
Henricks
a,
Wyatt
Winkenwerder
b,
Wim
Baan
c and
John
van der Schaaf
*a
a,
Vera C.
Henricks
a,
Wyatt
Winkenwerder
b,
Wim
Baan
c and
John
van der Schaaf
*a
aLaboratory of Chemical Reactor Engineering, Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, P.O. Box 513, 5600MB, The Netherlands. E-mail: j.vanderschaaf@tue.nl
bNouryon, Brewster, New York 10509, USA
cNouryon, Zutphenseweg 10, 7418 AJ Deventer, The Netherlands
First published on 9th August 2021
Abstract
Quaternary ammonium salts (QAS) are an important part of the increasing surfactant market. Conventional production processes employ toxic alkyl halides in a Menshutkin reaction with a tertiary amine (DMDA). Dimethyl carbonate (DMC) can provide a renewable route, while also leading to more benign, and non-corrosive products. This work aims to use linear ramp-flow in a plug flow reactor (PFR), combined with in-line 1H NMR spectroscopy to determine reaction kinetics. These kinetics will be used to further optimise the production process with a computational model. Solvent effects were first studied in a batch reactor. Methanol (MeOH) was found most suitable as a solvent. Subsequently, the reaction kinetics were measured in a PFR set-up. The used ramp-flow was successfully validated with data from batch and steady-state experiments. Arrhenius parameters were determined with the ramp-flow method, which proved to be an accurate and efficient technique. The kinetic data was implemented in a computational model. After validation of the model with experimental data, it was employed to extrapolate this data and optimise the reaction. The optimum QAS productivity was predicted at 122 kg h−1 L−1, obtained at 270 °C, 0.25 min residence time, and a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5:10 (DMDA:DMC:MeOH). These conditions would provide significant intensification of the QAS production processes.
:2.5:10 (DMDA:DMC:MeOH). These conditions would provide significant intensification of the QAS production processes.
1 Introduction
There is an ever-growing incentive to move towards more sustainable and more efficient processes. Within the chemical industry, process intensification and the use of renewable feedstock can allow for major contributions to this end. The decrease in environmental impact can be further amplified by using less toxic and biodegradable reactants for producing chemicals. Surfactants account for a large share of the chemical industry. Their current total market size is $39.69 billion, which is continuously expanding with approximately 2.5% each year.1Quaternary ammonium salts (QAS) are one of the primary surfactants, with an annual production estimated at millions of tonnes.2,3 These QASs are widely applied, in households as well as in various industries, with applications ranging from detergents3 to phase-transfer catalysts.3,4 Currently, the SARS-CoV-2 pandemic results in an increased demand in QASs, due to their effectiveness as disinfectants.5 Their conventional production is performed according to the Menshutkin reaction, which was first published in 1890.6 Nowadays this reaction is still carried out almost exclusively in large batch reactors. As operation of this type of reactor is generally labour-intensive, costly, and not very scalable, progressing towards a continuous process seems paramount. Other challenges in the production process are introduced by the QAS's high viscosity, and the consecutive thermal degradation reaction. At higher temperatures, QASs are (depending on their configuration and anion) increasingly susceptible for decomposing according to Hofmann elimination.7
In the Menshutkin reaction, a tertiary amine reacts with an alkyl halide (predominately methyl chloride), as shown in Fig. 1.3,8 Under mild temperatures, between 60 and 100 °C, methyl chloride, benzyl chloride, dimethyl sulfate and diethyl sulfate are commonly employed to complete the quaternisation reaction.9,10 The reaction is well-known to occur according to an SN2 mechanism, in which the nature of the leaving group and the nucleophilic strength of the nitrogen base determine the rate of reaction.3 Although these alkylating agents allow for relatively high reaction rates, they are accompanied with high toxicities and environmental impacts.3 Dimethyl sulfate, e.g., has a low LD50 of 133 mg kg−1,11–14 and is also known to be carcinogenic.15 As a result, their allowed concentrations in the final products is constrained to ppm levels to ensure safety, which complicates product purification.16
 | ||
| Fig. 1 The Menshutkin reaction of a tertiary amine with an alkyl halide according to an SN2 mechanism.3,6 | ||
Dimethyl carbonate (DMC) has been posed as a high-potential renewable building block for various chemicals.17,18 It has negligible toxicity, is biodegradable, and can be produced sustainably from methanol (MeOH) and CO or CO2 precursors.13,19,20 For its use as an alkylating agent in QAS production processes, several patents and articles have already been published.21–26 The resulting QAS features no corrosivity, making it eligible for specific demanding and specialty applications. In Fig. 2, an exemplary alkylation reaction with DMC is presented. Besides its high flammability, DMC is not hazardous, as there is no long-term toxicity and it has a high LD50 at 5000 mg kg−1.20 Its high thermal stability up to 390 °C allows for high-temperature operation and reactant recovery.27
 | ||
| Fig. 2 The alkylation of N,N-dimethyldecylamine with DMC according to an SN2 mechanism. | ||
The main disadvantage of DMC is its relatively low reaction rate compared to the aforementioned conventional reagents. For trialkyl amines, for example, the reaction rate constant with DMC at 125 °C (5.5 × 10−6 L mol−1 s−1) is similar to that of methyl chloride at room temperature (6.0 × 10−6 L mol−1 s−1).2 However, as already stated by Friedli16 in 1990, it remains difficult to find literature relating reaction kinetics of particular amines. Often, tertiary amines with equally long side-chains are investigated. Previous studies on DMC alkylation rates in MeOH with various trialkyl amines have shown an activation energy of 79 kJ mol−1 and rate constants from 2.9 × 106 L mol−1 s−1 to 2.1 × 107 L mol−1 s−1 for temperatures of 115 to 155 °C.2 For these trialkyl amines, the activation energy is independent for chain-lengths beyond three carbons.2,16
Alkylation reactions with DMC are also highly susceptible to solvent effects.16,28 Reaction intermediates are known to be best stabilised by polar aprotic solvents, e.g., acetonitrile, acetone, and propylene glycol.29 Nevertheless, polar protic solvents such as MeOH, ethanol, and isopropanol are also typically employed for these types of reactions.2,16,28
Another key advantage of using DMC as an alkylating agent, is the fact that it produces a QAS with an anion that can be readily exchanged according to the scheme in Fig. 3. This reaction results in the formation of carbon dioxide and MeOH which are both highly volatile, favouring the consecutive purification. Any organic or inorganic acid can facilitate this exchange, as long as their pKa value is lower than methyl carbonic acid (pKa 5.612).2 Although ion exchange is also possible after the conventional alkylation process, it is very impractical as it is difficult to remove the resulting salts from the product.30
 | ||
| Fig. 3 The exchange reaction with DMC enables facile substitution of the counterion. | ||
Together with the side-chain length, the anion has a profound influence on the properties of the QAS and thus on its potential applications. Currently, about 500 different QASs are produced and commercially used.31 DMC-based QASs have the potential to be versatile platforms to produce a much wider range of QASs.
This research focuses on optimising the quaternisation of N,N-dimethyldecylamine (DMDA) with DMC (Fig. 2). This tertiary amine's kinetics have not been reported in literature, while it leads to industrially relevant products. First, solvent effects are investigated in a batch reactor, after which the most suitable solvent is used in kinetic experiments in flow. These experiments are carried out using linear ramp-flow under continuous analysis of the reactor outflow with Proton nuclear magnetic resonance (1H NMR) spectroscopy. Similar methods have been applied in other studies.10,32–34 The kinetic parameters and solvent effects will then be used in a computational model of the flow reactor. This model is employed to extrapolate and optimise the operating conditions to maximise the yield and productivity of the reactor.
2 Methods
2.1 Materials
The materials used, their origin, and their purity are listed in Table 1. The DMDA was known to contain traces of other tertiary amines, some secondary amines, and very low amounts of unreacted primary amines. The solvents that are shown were used to determine solvent effects only in batch reactions and their purity was at least 99%. MeOH, however, was also used in the flow experiments. The main source of impurity in the solvents was water.| Chemical | Type | Supplier | Grade |
|---|---|---|---|
| N,N-Dimethyldecylamine | Reactant | TCI | ≥95% purity |
| Dimethyl carbonate | Reactant | Sigma Aldrich | ≥99% purity |
| Methanol | Solvent | VWR Chemicals | Technical grade |
| Ethanol | Solvent | Merck | Absolute grade |
| Isopropanol | Solvent | VWR Chemicals | Technical grade |
| Acetone | Solvent | Merck | Analysis grade |
| Acetonitrile | Solvent | Biosolve | HPLC grade |
| Dimethyl acetamide | Solvent | Alfa Aesar | ≥99% purity |
| Dimethyl sulfoxide | Solvent | Merck | Analysis grade |
| 1,4-Dichlorobenzene (Cl2Bz) | 1H NMR standard | Alfa Aesar | ≥99% purity |
| 1,4-Difluorobenzene (F2Bz) | 1H NMR standard | Sigma Aldrich | ≥99% purity |
| Deuterated chloroform | 1H NMR solvent | VWR Chemicals | ≥99.80% purity |
2.2 Solvent effects in batch
Batch experiments were performed to determine solvent effects on the alkylation rate of DMDA with excess DMC. The batch reactors consisted of a 1.5 mL glass vial, in which the liquid was contained by a membrane crimp cap. These reactors were filled with pre-prepared stock solutions, consisting of the concerning solvent, DMDA, DMC, and 1H NMR standards. The ratio of the amine to DMC was varied between 1:5 and 1:10, while the ratio of the amine to solvent was varied between 1:0 (neat) and 1:15.
The vials were inserted in a heated mantle or an oil bath, where a temperature was set ranging from 80 to 130 °C. For each experiment, a series of six vials were filled with the stock solution. One vial acted as a blank, while the others were inserted into the heating source simultaneously, and removed, one by one, at fixed time intervals. After their removal, the vials were directly subjected to a thermal quench in an ice bath. For the samples that showed segregation, MeOH was added until a single phase was obtained again. This dilution was accounted for by the internal standard present in the mixture.
The resulting time series was used to determine reaction rate constants for the various reaction mixtures and temperatures. These rate constants were determined by computing the conversion of the amine, XA, according to eqn (1), which corresponds with a second-order kinetic equation. Here, the initial concentration of compound i (Ci,0), the reaction rate constant (k), and the residence time (τ) are the required input parameters. The consecutive fitting of the Arrhenius equation allowed for the determination of the desired kinetic parameters.
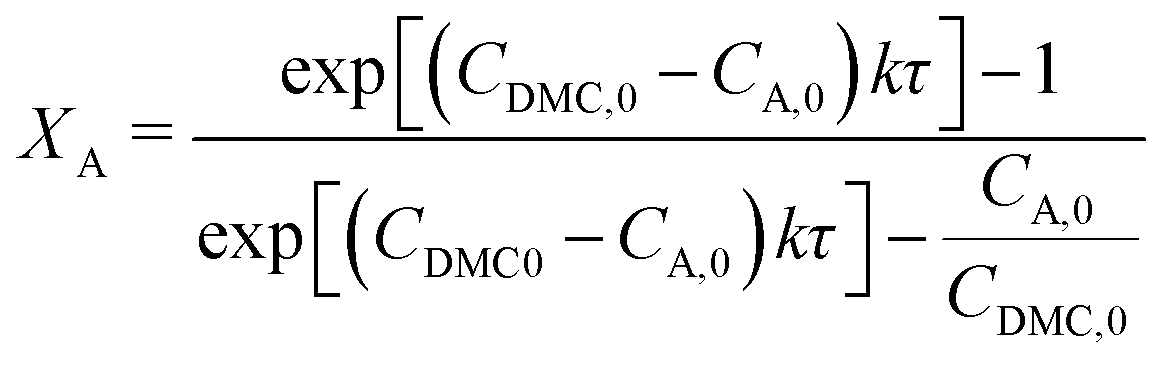 | (1) |
2.3 Reaction kinetics in a plug flow reactor
The kinetic experiments in flow were performed in a PFR set-up, of which a schematic is presented in Fig. 4. For reference, this set-up has also been used and described in previous work.10 Two Shimadzu CL 20 AD HPLC pumps were used to separately feed the two reactant mixtures through a pre-heat loop into the reactor. The pumps had been calibrated for their respective mixture. The first mixture consisted of the solvent, DMDA, and a F2Bz internal standard, while the second mixture contained DMC and a Cl2Bz internal standard. This double internal standard approach allowed for determining individual compound concentration at the back-end of the reactor, and also to verify the flow and mixing ratio of the two inlet streams. Before each experiment, all solutions were stirred, and degassed with a helium purge. | ||
| Fig. 4 Schematics of the continuous flow setup. | ||
The pre-heat loop consisted of two isolated 1/16″ stainless steel tubes (with an inner diameter of 1 mm), enclosed by HM-RD2011SO heat tracing from Mohr & Co, which was regulated with a thermocouple. The reactor was devised from a 1/8″ stainless steel tube (with a wall thickness of 0.711 mm for an inner diameter of 1.75 mm), with a length of 1 m for a total volume of 2.4 mL. Prior to the reactor volume, one of the pre-heat tubes was adapted to the 1/8″, in which the other 1/16″ tube was inserted. The resulting concentric tubes continued for 1 m, after which the inner tube was terminated, the inlet streams mixed, and the reaction volume started. The design allowed for inserting or retracting the inner tube, for a variable effective reactor volume. The entire 2 m 1/8″ tubing was fed through a 1/2″ stainless steel tube, connected to a LAUDA Proline P5 thermostat oil bath, serving as a heat exchanger. This ensured that the reactants were mixed at the desired operating temperature, and that the heat of reaction was removed. Isothermal operation could thus be assumed for the entire reaction volume. As this design made mixing at a precise spatial coordinate possible, the reactor volume can be considered very well-defined. The reactor system of concentric tubes has been schematically depicted in Fig. 5.
 | ||
| Fig. 5 Schematic representation of the reactor system of concentric tubes. The design allowed for a very well-defined and controlled reactor volume. | ||
After the reactor, the tubing was immediately led through an ice bath to cool the reaction mixture to 0 °C. At this temperature, the reaction rate is negligible, and the reaction can thus be considered successfully quenched. Subsequently, the stainless steel tubing was converted to perfluoralkoxy alkane (which does not interfere with the spectrometer), and fed through a Magritek Spinsolve benchtop 43 spectrometer. After the 1H NMR, a back-pressure regulator was installed to have a constant elevated pressure of 30 bar, which ensured a liquid phase for all compounds.
Kinetic experiments were conducted in a range from 130 to 160 °C. After equilibrating and reaching a steady-state in the reactor, a linear decreasing flow ramp was introduced. This resulted in modulation of the residence time. Therefore, a single experiment allowed for measuring reactor outlet concentrations at increasing reaction times. Both HPLC pumps were programmed, and although their ratio remained constant, total flow rates were varied between 0.20 and 1.60 mL min−1. For each temperature, a separate experiment was carried out.
2.4 Ramp-flow and data processing
The ramp-flow method employs a linear decrease in the volumetric flow rate, resulting in an increasing residence time in the reactor, τR, over the duration of the experiment. Similar methods have been described with, e.g. model-based design of experiments, IR analysis, or HPLC analysis.35–38 In this study's method, the experimental time has to be converted into the actual residence time. The required processing has been described in previous work.10 One profound difference in this case, however, is the fact that no reaction occurs in the transport volume (VT). Fig. 6 shows a graphical representation of the set-up, where ti, tf, and tm are the times at which a discrete element flowing through the reactor reaches its entrance, its exit, and the 1H NMR sensor. | ||
| Fig. 6 Graphical representation of the relevant volumes and experimental times in the continuous flow setup. | ||
These times were used to compute the required residence times, according to eqn (2). The different situations in which a discrete element could be travelling through the set-up before, during, and after the flow ramp, are displayed in Fig. 7. The initial volumetric flow rate is represented by F0, while the final flow rate is shown as Fe.
| τR = tf − tiτT = tm − tf | (2) |
 | ||
| Fig. 7 An element travelling through the setup can be in six discrete situations. | ||
Accounting for the ramp of the flow, a, started at t0 and ended at te, leads to the flow rate developing as a function of time as described in eqn (4). Under the assumption of plug-flow, and with the imposed flow ramp, the concerning reaction volumes can be expressed by eqn (3).
 | (3) |
| t < t0 : F(t) = F0t0 ≤ t < te : F(t) = F0 − att > te : F(t) = F0 − ate = Fe | (4) |
Eqn (3) can be integrated and substituted to acquire the desired set of equations for the unknown experimental times, ti and tf. These equations are shown in eqn (5), along with their associated boundary conditions. Subsequent substitution of these equations into eqn (2), results in calculation of the required residence times of the set-up.
 | (5) |
Finally, a representative flow ramp and the resulting residence time profile in the reactor are shown in Fig. 8. Note that this residence time is based on ideal plug-flow behaviour, and is a mere estimation of the actual residence time.
 | ||
| Fig. 8 Example of a flow ramp and the resulting residence time in the reactor for a reactor volume of 2.4 mL, and a transport volume of 15 mL. | ||
2.5 1H NMR spectroscopy
For all kinetic measurements, 1H NMR spectroscopy was used to determine the concentrations of the individual compounds in the reaction mixtures. The batch measurements were carried out offline, in a Bruker-400400 MHz NMR spectrometer. Prior to the analysis, 0.1 mL of sample was diluted with 0.4 mL of deuterated chloroform in a NMR tube, and subjected to 32 scans with a recycle delay of 2 s. Typical spectra before, and after the reaction are shown in Fig. 9c and d, respectively. The assigned protons are elaborated in Fig. 9a. The peak areas in the spectra were determined after an automated phase correction, an automatic third order Bernstein polynomial fit baseline-correction, and one zero-filling step using the forward linear prediction method of Zhu–Bax. An internal standard allowed for the conversion from integrated area to the concentration of the respective species.
 | ||
| Fig. 9 1H NMR spectra of the quaternisation of DMDA with DMC in MeOH. b) shows a spectrum taken during flow reaction at 160 °C with a residence time of 6 minutes, reaching 60% amine conversion. c) and d) show spectra taken before and after batch reaction at 90 °C for 4 hours, also reaching 60% amine conversion. The line thickness for all spectra is the same, but the online spectrum has a lower signal-to-noise ratio. | ||
The analysis for the flow experiments was performed using a Magritek Spinsolve benchtop 43 NMR spectrometer (43 MHz). The 1H NMR was calibrated and relative standard deviations lower than 1.5% were observed. As the reaction mixture was flowed through the NMR, i.e. measured in-line, the data acquisition was automated. At the sensor, every 6.4 s a spectrum was recorded with a 90° pulse angle. A powershim, executed before each experiment with a 0.5 mL min−1 flow rate ensured the spectrometer was locked on the highest intensity peak (3.48 ppm, MeOH). During the experiments, the 1H NMR was re-calibrated at 30 min intervals, to preclude any decay of the signal. The spectrometer could be operated with total flow rates of up to 2.5 mL min−1, approximately. This maximum is dictated by the sensor volume and the acquisition time settings.
2.6 Reactor characterisation
In order to verify if plug-flow can be assumed for the set-up, a reactor characterisation study was conducted. This study included a numerical verification, as well as experimental work. For the numerical characterisation, Reynolds and Péclet dimensionless numbers were used, as shown in eqn (6) and (7), where u and d represent the superficial velocity through, and the inner diameter of the tube, correspondingly. | (6) |
 | (7) |
It is generally accepted that Péclet numbers approaching zero indicate an ideally mixed system, while plug-flow is assumed from Péclet numbers exceeding 100.39,40 The diffusion coefficient, D, is computed according to the Wilke–Chang correlation, shown in eqn (8).41,42 A system of dilute DMC in MeOH is assumed, as its diffusion coefficient in MeOH is expected to be higher than that of the tertiary amine due to its smaller size. Furthermore, the dynamic viscosity, μ, of pure MeOH was calculated according to Xiang et al.,43 and the characteristic length, d, is equal to 1.75 mm (the reactor's inner diameter). Finally, the association factor (x) has been reported to be 1.9.44
 | (8) |
Table 2 shows the resulting dimensionless numbers. For the operated range of temperatures, these have been calculated at the lowest, and highest flow rate. With Reynolds numbers not exceeding 112, all experiments exclusively operated in the laminar regime. Nevertheless, the Péclet numbers clearly demonstrate overall plug-flow behaviour, as only at the lowest temperature and flow, a value lower than 100 is observed.
| T | Low flow | High flow | Low Re | High Re | Low Pe | High Pe |
|---|---|---|---|---|---|---|
| [°C] | [mL min−1] | [mL min−1] | — | — | — | — |
| 130 | 0.05 | 0.20 | 3 | 10 | 59 | 238 |
| 140 | 0.10 | 0.40 | 6 | 23 | 104 | 417 |
| 150 | 0.20 | 0.80 | 12 | 50 | 181 | 723 |
| 160 | 0.40 | 1.60 | 28 | 112 | 309 | 1235 |
The experimental characterisation was performed by means of a residence time distribution (RTD) analysis. The PFR set-up was subjected to a tracer step input, while keeping the total volumetric flow rate constant. Total flow rates between 0.15 and 2.00 mL min−1 were applied with a MeOH solvent and DMDA as the traces. These RTD experiments were thus very representative of the kinetic experiments. The concentration of the tertiary amine was determined as elaborated in the 1H NMR spectroscopy section. The resulting concentration profile was used to generate the corresponding F-curves, and to calculate the corresponding tanks-in-series (TIS), as well as the backmixing time.
3 Modelling approach
3.1 Model structure
A computational model was built in close resemblance to the operated PFR. The model was employed to extrapolate the obtained experimental results into conditions that were infeasible in the PFR. It aims to represent the PFR, and it was validated using the experimental data. Thereafter, optimisation of the system's yield and productivity was carried out.As the reactor characterisation has shown, the reactor operates according to non-ideal plug-flow. In this case, the system can be approached by a dispersion model or a TIS model, which are equably suitable.39 However, the TIS model does not require diffusion rate data, so this model is selected. The model was built in MATLAB, and solved by means of a ODE45 ODE solver. Fig. 10 presents a schematic representation of the model. It consists of n tanks in VR, and m tanks in VT, depending on the operating conditions. Each compound has a mass balance for each cell, which communicates with the adjacent cells. The resulting balance for a compound in tank i is shown in eqn (9). In accordance with the experiments, the reactor was considered to be operated at isothermal conditions.
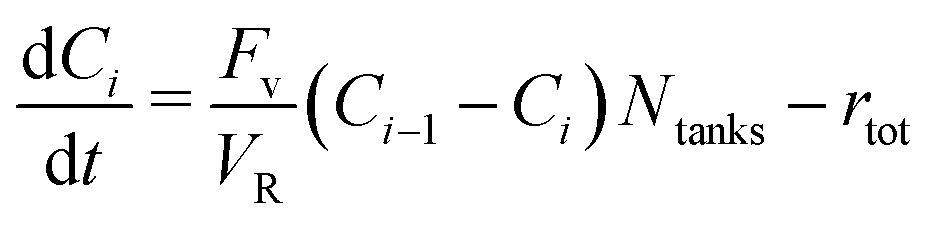 | (9) |
 | ||
| Fig. 10 Schematic representation of the TIS model that was used to simulate the PFR set-up. | ||
The overall reaction term (rtot) consists of the QAS formation reaction, the degradation reaction, or both, depending on the species. All tanks were initialised at the respective initial concentrations, and at t = 0 the flow, heating, and reactions were started. Full convergence is obtained at equilibrium for all concentrations.
3.2 Parameter input
The actual PFR properties were used in the model (VR = 2.4 mL and VT = 15 mL). This ensured that the model could be validated with the experimental data. The reaction rate constants were computed based on the Arrhenius parameters that were determined with the ramp-flow experiments. For the degradation reaction, Arrhenius parameters from previous work were used.7For the validation of the model, the input parameters were matched with the operating conditions. The flow rate was set between 0.05 and 1.60 mL min−1, and the temperature was set between 130 and 160 °C. Resulting concentration and conversion profiles were compared with the respective experimental data.
After the validation was satisfactory, the operating conditions were expanded for the optimisation. Various molar ratios of DMDA:DMC:MeOH were varied, and a temperature of up to 300 °C was investigated. The flow rates were now set between 0.005 and 10 mL min−1, corresponding with residence times of ca. 480 to 0.25 min. The main monitored design specifications of the model were the yield of QAS and the specific productivity of the reactor.
4 Results and discussion
4.1 Solvent effects on quaternisation rate
Reaction kinetics of DMDA with DMC were determined in batch in MeOH, acetonitrile, acetone, and excess DMC. Isopropanol, ethanol, dimethyl acetamide, and dimethyl sulfoxide were also investigated, but their presence caused a side reaction with DMC. These side reactions were confirmed after further testing: DMC was added to all solvents, along with an internal standard and subjected to 130 °C for 28 hours. Side reactions were then indeed only observed in isopropanol, ethanol, dimethyl acetamide, and dimethyl sulfoxide, while the other solvents were confirmed to be stable. In these solvents, reaction rate constants of DMDA quaternisation were successfully determined in different ratios. A subsequent Arrhenius fit was performed and has been displayed in Fig. 11. Variations in concentrations were accounted for according to eqn (1) and this equation was used to fit the k values. As the graphs of different reactant ratios in the same solvent overlap, it appears that the second-order fit assumption is validated.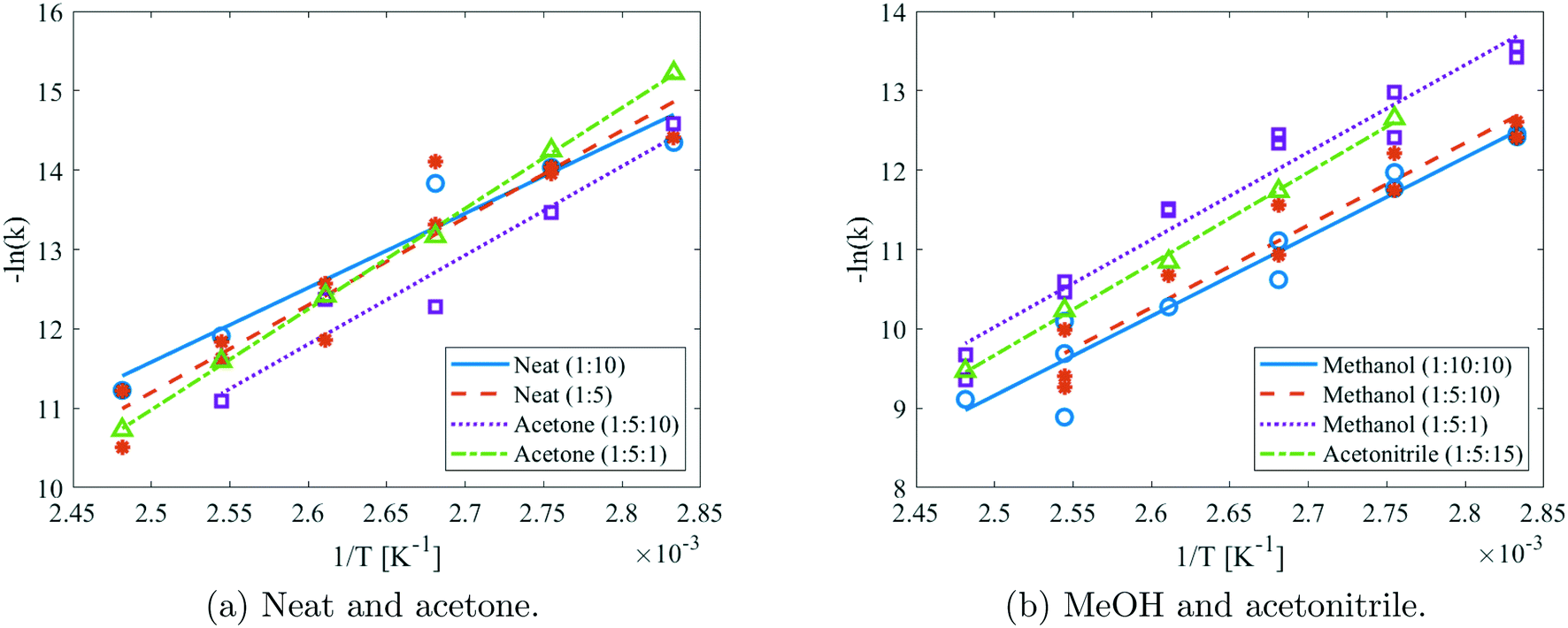 | ||
| Fig. 11 Arrhenius plots of the batch experiments in different solvents and molar ratios. | ||
The Arrhenius parameters that resulted from the fit are presented in Table 3, along with their 95% confidence bounds. For reference purposes, the required reaction time to reach 90% conversion has also been computed. These indicate that MeOH is the preferable solvent, as it features the lowest required reaction times. Therefore, this solvent was selected for further use in the kinetic measurements in flow. Furthermore, at lower temperatures the neat production is outperforming the acetone system, while this is reversed at higher temperatures.
| Reaction mixture DMDA:DMC:solvent |
E a [kJ mol−1] | −lnk0 [—] |
k 0 [107 L mol−1 s−1] | t(X = 90%) at 150 °C [min] |
|---|---|---|---|---|
| MeOH (1:10:10) |
83.2 ± 0.5 | 15.8 ± 0.1 | 0.760 | 16 |
| MeOH (1:5:10) |
86.3 ± 0.5 | 16.7 ± 0.2 | 1.80 | 26 |
| MeOH (1:5:1) |
91.6 ± 0.4 | 17.5 ± 0.1 | 4.06 | 32 |
| Neat (1:10:0) |
77.9 ± 0.8 | 11.8 ± 0.2 | 0.0139 | 133 |
| Neat (1:5:0) |
91.5 ± 0.6 | 16.3 ± 0.2 | 1.22 | 94 |
| Acetone (1:5:10) |
93.3 ± 0.9 | 17.4 ± 0.3 | 3.48 | 116 |
| Acetone (1:5:1) |
105.5 ± 0.1 | 20.7 ± 0.1 | 102 | 67 |
| Acetonitrile (1:5:15) |
95.8 ± 0.2 | 19.1 ± 0.1 | 20.6 | 42 |
4.2 Residence time distribution in the plug flow reactor
The RTD experiments resulted in F-curves for varying operating conditions within the intended range for kinetic experiments. Fig. 12 contains a representative F-curve, acquired at 0.35 mL min−1. All curves showed a sharp breakthrough with a broadening tail, which is indicative of a slight deviation from ideal plug-flow.39 The residence time at the midpoint of the F-curve was used to validate the set-up's total volume at 17.4 mL (of which 2.4 mL for the reactor section). A TIS model was fitted to the F-curves to determine the corresponding number of tanks. All except one of the RTD experiments (at 0.8 mL min−1) resulted in a number of tanks greater than 50, at which plug-flow can be assumed.39,40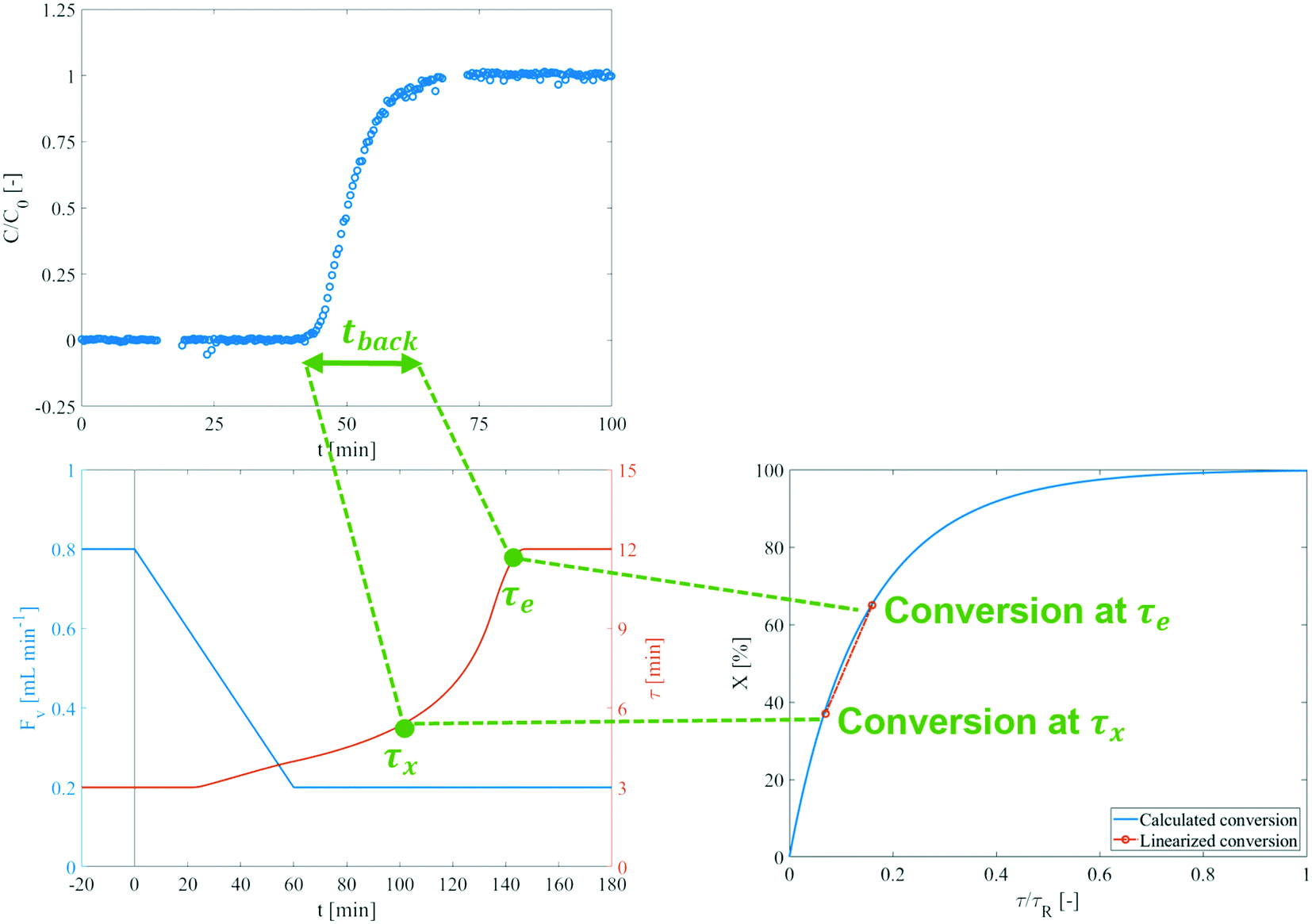 | ||
| Fig. 12 An illustrative explanation of the check on the plug-flow assumption. If the change in conversion over the change in residence time from τx to τe, which correspond to the backmixing time tback from the RTD, can assumed to be linear, the plug-flow assumption can be applied. The used plots are not from experiments at similar conditions, so the time axes do not match. | ||
Experimental validation of the plug-flow assumption was also achieved by estimating the change in conversion over the backmixing time, tback. For an ideal PFR, this value would be 0. However, if the conversion can be approximated to be linear for the concerning range, the distribution of residence times will result in an evenly-averaged instantaneous conversion. The backmixing time as a function of flow was fitted with respect to all RTD experiments, resulting in tback = 9.60/Fv = 9.60·τe/VR (R2 = 0.831). The backmixing will be most severe at lower flow rates, c.q. the highest τ, at the end of the experiment (τe). The residence time of the fluid element that can be backmixed with the flow at τe, τx, is given by eqn (10).
| t(τx) = t(τe) − tback | (10) |
For all flow experiments, the resulting tback, τe, and τx have been compiled in Table 4, along with the respective computed conversions. The change in conversion is the largest for the experiment at 140 °C, with other temperatures showing very similar ranges. Even the largest change in conversion is nearly linear, as shown in Fig. 12. Plug-flow can thus indeed be assumed for all conducted experiments.
| T [°C] | τ e [min] | t back [min] | τ x [min] | X(τe) [%] | X(τx) [%] |
|---|---|---|---|---|---|
| 130 | 48 | 192 | 20.9 | 67 | 40 |
| 140 | 24 | 96 | 10.2 | 65 | 37 |
| 145 | 12 | 48 | 5.2 | 52 | 28 |
| 150 | 12 | 48 | 5.2 | 62 | 36 |
| 155 | 6 | 24 | 2.6 | 49 | 26 |
| 160 | 6 | 24 | 3.4 | 49 | 32 |
4.3 Kinetic parameters in flow
The flow experiments with the linear ramp produced extensive sets of transient data for temperatures ranging from 130 to 160 °C (DMDA:DMC:MeOH = 1:5:10). These conditions and the residence times (Table 5) were selected based on the batch results and with respect to some physical limitations of the set-up. The data was fitted in accordance with eqn (1), resulting in conversion profiles. An example, obtained at 160 °C has been plotted in Fig. 13a. One key advantage of the ramp-flow method is distinctly illustrated here: a large amount of data points is acquired in a single experiment. The development of the experimental conversion clearly follows the trend of the residence time. Furthermore, the fitted conversion, based on the fitted reaction rate constant, is in agreement with the data. In Fig. 13b, the conversion (at 160 °C) has been plotted vs. the investigated residence time.
| T [°C] | τ i [min] | τ e [min] | t ramp [min] | Fitted k [10−4 mole per L s−1] | R adj 2 [—] |
|---|---|---|---|---|---|
| 130 | 12 | 48 | 240 | 1.31 ± 0.021 | 0.8718 |
| 140 | 6 | 24 | 120 | 2.94 ± 0.019 | 0.9780 |
| 145 | 3 | 12 | 60 | 3.73 ± 0.023 | 0.9869 |
| 150 | 3 | 12 | 60 | 4.76 ± 0.031 | 0.9845 |
| 155 | 1.5 | 6 | 30 | 7.21 ± 0.058 | 0.9856 |
| 160 | 1.5 | 6 | 60 | 8.01 ± 0.062 | 0.9946 |
 | ||
| Fig. 13 Example of a flow experiment and its fit. This experiment was run at 160 °C and 1:5:10 ratio DMDA:DMC:MeOH. Gaps in the data are due to shimming. | ||
Subsequently, for each temperature a reaction rate constant was computed. These constants are presented in Table 5, and they are accompanied by the respective operating conditions, confidence intervals, and the Radj2 values. All experiments show high coefficients of determination, except for the k value at 130 °C. Nevertheless, no systemic error was observed in the data at this temperature. Arrhenius fitting then resulted in the determination of the kinetic parameters for the 1:5:10 system. The computed activation energy is 88.5 ± 0.3 kJ mol−1 and the frequency factor is 4.14 ± 0.09 × 107 L mol−1 s−1. These values correspond well with the Arrhenius parameters in batch, presented earlier.
4.4 Method comparison
The linear ramp-flow method certainly has potential as a more efficient technique to acquire accurate kinetic data.10 However, the method is relatively novel and not very established yet. Therefore, validation of the method with respect to conventional methods, e.g. batch and steady-state flow, is paramount. Besides the reported batch and ramp-flow kinetic experiments, steady-state measurements were also conducted. In the Arrhenius plot in Fig. 14, the results acquired from batch, steady-state, and the ramp-flow method can be directly compared. The reaction constants of steady-state and ramp-flow are in very good agreement. And while the batch experiments were conducted at different temperatures, extrapolation of the fitted curve would evidently result in an acceptable fit of the flow data as well. Moreover, the kinetic parameters that were calculated from the steady-state data (Ea = 91.1 ± 0.1 kJ mol−1 and k0 = 8.81 ± 0.11 × 107 L mol−1 s−1), are also in accordance with those from the ramp-flow.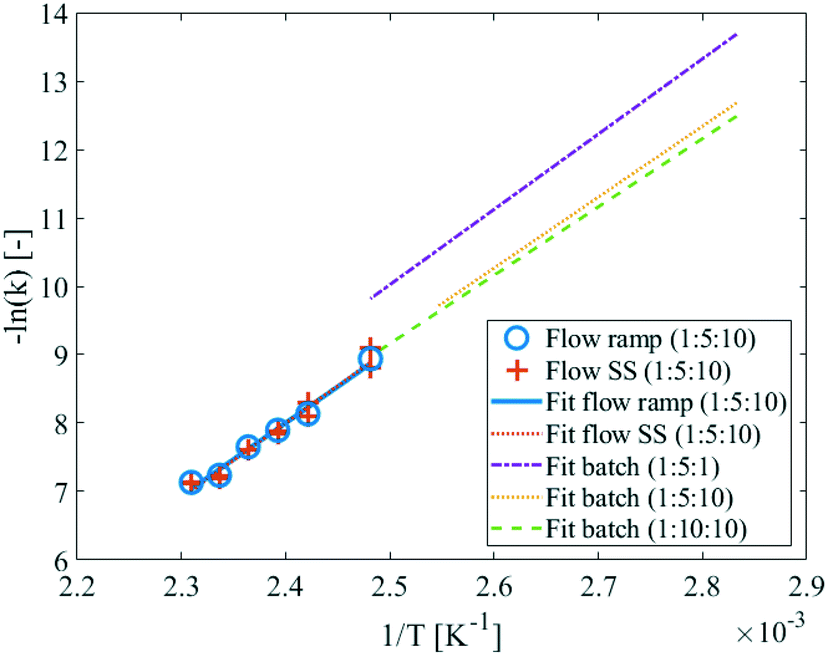 | ||
| Fig. 14 Arrhenius plot for the flow experiments. Arrhenius plots from batch and steady-state flow data show good agreement. The molar ratios (DMDA:DMC:MeOH) provided in the legend. | ||
The comparison between the steady-state and the ramp-flow results has been presented in more detail in Table 6. For all experiments, the reaction rate constants and their relative difference with the corresponding ramp-flow values are shown. Steady-state reaction rate constants were determined at the upper flow rate, as well as at the lower flow rate. Deviations vary from 15.5% to −15.2%, with values as low as −0.7%. In general, the deviations are considered to be acceptable and not systemic. Ultimately, also based on the agreement with the batch results, the ramp-flow method is deemed to be validated.
| Temperature [°C] | Ramp k [10−4 L mol−1 s−1] | Steady-state k | |
|---|---|---|---|
| High flow [10−4 L mol−1 s−1] | Low flow [10−4 L mol−1 s−1] | ||
| 130 | 1.31 ± 0.021 | 1.50 ± 0.020 (15.5%) | 1.11 ± 0.017 (−15.2%) |
| 140 | 2.94 ±0.019 | 3.05 ± 0.018 (3.8%) | 2.50 ± 0.022 (−14.9%) |
| 145 | 3.73 ± 0.023 | 3.80 ± 0.024 (1.8%) | 3.90 ± 0.026 (4.5%) |
| 150 | 4.76 ± 0.031 | 5.00 ± 0.028 (5.0%) | 4.59 ± 0.026 (−3.6%) |
| 155 | 7.21 ± 0.058 | 7.30 ± 0.049 (1.3%) | 7.53 ± 0.052 (4.4%) |
| 160 | 8.01 ± 0.062 | 8.12 ± 0.069 (1.4%) | 7.96 ± 0.066 (−0.7%) |
As mentioned earlier, the ramp-flow method allows for the acquisition of a vast amount of transient data in a singular experiment. With the investigated operating conditions at least 150 data points were generated per run. Similar amounts of data are unfeasible with steady-state methods. Two steady-state equilibrations are required per data set with the ramp-flow method, in contrast to one equilibration for every data point with the steady-state method. So even for a minimum amount of five data points, the time and material savings would be quite significant.
The comparison with batch experiments is less obvious. Approximately 150 mL of chemicals was used per experiment, which amounts to ca. 1 mL per data point. This could be further reduced by decreasing the transport volume to the spectrometer in particular. Nevertheless, this is still achievable in batch. However, the manual labour and time required for preparing and analysing the batch samples is significantly higher. Especially at large-scale experiments that demand vast amounts of data, the ramp-flow method is profoundly superior.
4.5 Model validation
Prior to using the model for optimisation, it was first validated with experimental data. By settings the exact same operating conditions as the ramp-flow experiments, a direct comparison with the model could be made. In total, the results were compared at six different temperatures, in accordance to the ones shown in Tables 5 and 6. Two examples are shown in Fig. 15. A peculiar, yet acceptable deviation is observed at 130 °C, as can be seen in Fig. 15a. At lower times, the model predicts conversions that are a bit too high, though at higher times the model overshoots quite severely. No explanation was found for this remarkable observation which was absent at other temperatures that were used for the validation. In contrast, Fig. 15b, which was recorded at 150 °C, demonstrates excellent agreement. The deviation also appears to be random and not systemic at all. At different temperatures, very reasonable fitting was obtained as well. The model was thus considered validated, and is accurate within a range of approximately 10%. Consequently, it was employed to perform the aforementioned optimisation procedures. | ||
| Fig. 15 Validation of the model by comparison with two experiments at different temperatures. | ||
4.6 Optimisation of a continuous process
The model provided insight into the optimum QAS production conditions, beyond the used PFR's limits. During the optimisation, the residence time was varied from 0.25 to 48 min (without a ramp), and the temperature ranged up to 300 °C. The resulting contour plots for all design specifications are presented in Fig. 16. The ratio between the reactants and MeOH was also varied. Lastly, the performance of a PFR was compared to a continuous stirred tank reactor (CSTR). The results have been summarised in Table 7. | ||
| Fig. 16 Contour plots resulting from an optimisation of the PFR at a molar ratio of 1:2.5:10 (DDMA:DMC:MeOH). | ||
:DMC:MeOH. Optimised variable in bold
| Molar ratio | Reactor | t [min] | T [°C] | X [%] | S [%] | Productivity [g h−1] |
|---|---|---|---|---|---|---|
| 1:2.5:10 |
PFR | 0.25 | 270 | 99.71 | 97.51 | 293 |
| 1:2.5:10 |
PFR | 0.25 | 220 | 64.67 | 99.91 | 194 |
| 1:2.5:10 |
PFR | 48 | 150 | 100.00 | 99.90 | 1.5 |
| 1:5:10 |
PFR | 0.25 | 250 | 99.72 | 99.20 | 251 |
| 1:5:10 |
PFR | 0.25 | 220 | 84.21 | 99.90 | 213 |
| 1:5:10 |
PFR | 48 | 140 | 99.36 | 99.96 | 0.1 |
| 1:10:10 |
PFR | 0.25 | 260 | 99.04 | 98.65 | 132 |
| 1:10:10 |
PFR | 0.25 | 220 | 96.99 | 99.91 | 86 |
| 1:10:10 |
PFR | 48 | 150 | 99.28 | 99.90 | 0.7 |
| 1:2.5:10 |
CSTR | 0.25 | 280 | 89.24 | 95.23 | 256 |
| 1:2.5:10 |
CSTR | 0.25 | 210 | 39.97 | 99.93 | 120 |
| 1:5:10 |
CSTR | 0.25 | 290 | 90.69 | 92.26 | 212 |
| 1:5:10 |
CSTR | 0.25 | 210 | 36.33 | 99.93 | 92 |
The highest reactor productivity of 293 g h−1 was computed at a molar ratio of 1:2.5:10 (DMDA:DMC:MeOH), 270 °C, and with a residence time of 0.25 min. Taking into account the reactor volume (2.4 mL), this translates to a specific productivity of 122 kg h−1 L−1. At these conditions a high conversion of 99.71%, and reasonable selectivity of 97.51% are reached. Conventional QAS production is carried out at approximately 120 °C, so this optimum poses quite a compelling increase in temperature. It demonstrates a clear opportunity for process intensification. It should be noted that the vapour pressure of MeOH is ca. 126 bar at this temperature, so an even higher pressure would be required. This is feasible in a PFR, especially since the high production rate allows for a small reactor volume.
Further temperature increase results in even higher required pressures, and lower selectivities. The reduced residence time this results in seems redundant and will certainly not outweigh the aforementioned drawbacks. For applications where a very high yield (over 99.9%) is desired the temperature has to be lowered to 150 °C. This does increase the reaction time to 48 min, however, and lowers the productivity to 1.5 g h−1. Evidently, there can be an intermediate optimum, depending on the application.
Increasing the relative amount of DMC can also allow for a slight increase in selectivity. However, since this reduces the tertiary amine content of the reaction mixture, this leads to lower QAS production rates. The proposed optimum molar ratios could be further optimised by taking into account the technicals and economics of subsequent separation steps. Finally, Table 7 does allow for the comparison between production in a PFR vs. a CSTR. The results show that the PFR significantly outperforms the CSTR. This was expected, as the consecutive degradation reaction is accelerated here, due to the higher average QAS concentration.
5 Conclusions
Reaction kinetics of DMDA alkylation with DMC were successfully determined in various reactor systems. In batch, solvent effects were elucidated. MeOH was considered to be most eligible for industrial use, compared to acetonitrile, acetone, and solventless production. Isopropanol, ethanol, dimethyl acetamide, and dimethyl sulfoxide were found to be unsuitable because of side reactions.A linear ramp-flow method was employed to measure the reaction kinetics in a PFR with in-line 1H NMR. The Arrhenius parameters were computed (DMDA:DMC:MeOH = 1:5:10) at Ea = 88.5 ± 0.3 kJ mol−1 and k0 = 4.14 ± 0.09 × 107 L mol−1 s−1. The results were compared with the data in batch, and with conventional steady-state experiments. Consistent agreement was ascertained, so the method was considered valid. The ramp-flow method can allow for more efficient kinetic experiments. It saves time, material, and required labour costs compared to both batch, and steady-state techniques.
The PFR was modelled with a TIS model. The kinetic data from the ramp-flow method was used as input, along with degradation kinetic data. Using RTD studies and reactor characterisation, non-ideal plug-flow behavior was confirmed. Thereafter, the model was validated with data obtained in the PFR.
After validation, the model was employed to extrapolate the operating conditions from the experiments. This provided a platform to optimise the QAS production process, and productivity and yield were optimised. A maximum production rate of 122 kg h−1 L−1 was predicted by the model at 270 °C, 0.25 min residence time, and a molar ratio of 1:2.5:10. This shows considerable potential for the intensification of the process.
Conflicts of interest
There are no conflicts of interest to declare.Note added after first publication
This article replaces the version published on 13th August 2021, which contained an error in the temperature in the Abstract.Acknowledgements
This research was carried out within the HighSinc Program – a joint research initiative between Nouryon and the Department of Chemical Engineering and Chemistry at the Eindhoven University of Technology.References
- Acmite Market Intelligence, Market Report: World Surfactant Market, 2017, http://www.acmite.com/market-reports/chemicals/global-surfactant-market.html.
- D. E. Weisshaar, G. W. Earl, E. M. Villa, J. L. Zierke, C. J. Fry, K. Becvar, S. Li and M. C. Schafer, Kinetic study of the reaction of dimethyl carbonate with trialkylamines, Int. J. Chem. Kinet., 2010, 42, 221–225 CrossRef CAS.
- G. W. Earl, D. E. Weisshaar, D. Paulson, M. Hanson, J. Uilk, D. Wineinger and S. Moeckly, Quaternary methyl carbonates: Novel agents for fabric conditioning, J. Surfactants Deterg., 2005, 8, 325–329 CrossRef CAS.
- Z. Zheng, T. Wu and X. Zhou, The synthesis of quaternary ammonium salts from ammonium salts and dialkyl carbonate, Chem. Commun., 2006, 1864–1865 RSC.
- P. I. Hora, S. G. Pati, P. J. McNamara and W. A. Arnold, Increased Use of Quaternary Ammonium Compounds during the SARS-CoV-2 Pandemic and Beyond: Consideration of Environmental Implications, Environ. Sci. Technol. Lett., 2020, 7, 622–631 CrossRef CAS.
- J.-L. M. Abboud, R. Notario, J. Bertrán and M. Solà, Progress in Physical Organic Chemistry, John Wiley & Sons, Ltd, 2007, pp. 1–182 Search PubMed.
- R. Kleijwegt, W. Winkenwerder, W. Baan and J. van der Schaaf, Degradation Kinetics and Solvent Effects of Various Long-Chain Quaternary Ammonium Salts, 2021, DOI:10.26434/chemrxiv.14381540.v1, https://chemrxiv.org/articles/preprint/Degradation_Kinetics_and_Solvent_Effects_of_Various_Long-Chain_Quaternary_Ammonium_Salts/14381540.
- W. H. Saunders, J. Am. Chem. Soc., 2005, 78, 4501–4502 CrossRef.
- C. W. Weston, J. R. Papcun and M. Dery, Kirk-Othmer Encyclopedia of Chemical Technology, American Cancer Society, 2003, ch. 11 Quaternary Ammonium Compounds, vol. 2, pp. 728–753 Search PubMed.
- R. J. Kleijwegt, S. Y. Doruiter, W. Winkenwerder and J. van der Schaaf, Investigating tertiary amine alkylation/benzylation kinetics with ramp-flow in a plug-flow reactor using in-line 1H NMR spectroscopy, Chem. Eng. Res. Des., 2021, 168, 317–326 CrossRef CAS.
- G. R. Hoffmann, Genetic effects of dimethyl sulfate, diethyl sulfate, and related compounds, Mutat. Res., Rev. Genet. Toxicol., 1980, 75, 63–129 CrossRef CAS.
- M. Selva and A. Perosa, Green chemistry metrics: a comparative evaluation of dimethyl carbonate, methyl iodide, dimethyl sulfate and methanol as methylating agents, Green Chem., 2008, 10, 457–464 RSC.
- Y. Ono, Dimethyl carbonate for environmentally benign reactions, Catal. Today, 1997, 35, 15–25 CrossRef CAS , Proceedings of the International Forum on Environmental Catalysis '95 (IFEC '95).
- Agency, E. C., Dimethyl Sulfate, Online Database, 2019, https://echa.europa.eu/nl/brief-profile/-/briefprofile/100.000.963, Accessed on September 8, 2019 Search PubMed.
- A. Vyskocil and C. Viau, Dimethyl sulphate: Review of toxicity, J. Occup. Environ. Med., 1999, 5, 72–82 Search PubMed.
- F. E. Friedli, Rate of Quaternization as a Function of the Alkylating Agent, in Proceedings of World Conference on Oleochemicals, Into the 21st Century, 1990, pp. 296–297 Search PubMed.
- P. Tundo, M. Musolino and F. Aricò, The reactions of dimethyl carbonate and its derivatives, Green Chem., 2018, 20, 28–85 RSC.
- F. Aricò and P. Tundo, Dimethyl carbonate: A modern green reagent and solvent, Russ. Chem. Rev., 2010, 79, 479–489 CrossRef.
- Y. Jiang, T. Geng and Q. Li, Synthesis of Quaternary Ammonium Salts with Novel Counterions, J. Surfactants Deterg., 2012, 15, 67–71 CrossRef CAS.
- E. C. Agency, Dimethyl Carbonate, Online Database, 2019, https://echa.europa.eu/nl/brief-profile/-/briefprofile/100.009.527, Accessed on September 8, 2019 Search PubMed.
- J. Werntz, Monoquaternary Ammonium Carbonates, US Pat., 2635100, 1953 Search PubMed.
- P. Tundo and M. Selva, The Chemistry of Dimethyl Carbonate, Acc. Chem. Res., 2002, 35, 706 CrossRef CAS PubMed.
- S. Mori, I. Ida, M. Ue and U. Makoto, Process for Producing Quaternary Salts, US Pat., 4892944, 1990 Search PubMed.
- F. Rivetti and U. Romano, Alcohol Carbonylation with Palladium(II) Complexes, J. Organomet. Chem., 1979, 174, 221 CrossRef CAS.
- M. Mauri, U. Romano and F. Rivetti, Dimethyl Carbonate: A New Building Block for Organic Chemicals Production, Italian Journal of Chemical Engineering, 1985, 21, 6 CAS.
- U. Romano and P. Zani, Quaternization with Methyl Carbonate, Italian Pat., Appl. 21878A/82, 1982 Search PubMed.
- J. Cross, R. Hunter and V. Stimson, The Thermal Decomposition of Simple Carbonate Esters, Aust. J. Chem., 1976, 29, 1477 CrossRef CAS.
- F. E. Friedli, Effect of chain length on the rate of quaternization of alkyl amines, J. Am. Oil Chem. Soc., 1990, 67, 469–471 CrossRef CAS.
- P. Bruice, Organic Chemistry, Pearson Education, Inc., 7th edn, 2014 Search PubMed.
- Z. Zheng, T. Wu, R. Zheng, Y. Wu and X. Zhou, Study on the synthesis of quaternary ammonium salts using imidazolium ionic liquid as catalyst, Catal. Commun., 2007, 8, 39–42 CrossRef CAS.
- J. Pernak and P. Chwała, Synthesis and anti-microbial activities of choline-like quaternary ammonium chlorides, Eur. J. Med. Chem., 2003, 38, 1035–1042 CrossRef CAS PubMed.
- M. Rubens, J. Van Herck and T. Junkers, Automated Polymer Synthesis Platform for Integrated Conversion Targeting Based on Inline Benchtop NMR, ACS Macro Lett., 2019, 8, 1437–1441 CrossRef CAS.
- P. Sagmeister, J. Poms, J. D. Williams and C. O. Kappe, Multivariate analysis of inline benchtop NMR data enables rapid optimization of a complex nitration in flow, React. Chem. Eng., 2020, 5, 677–684 RSC.
- P. Sagmeister, R. Lebl, I. Castillo, J. Rehrl, J. Kruisz, M. Sipek, M. Horn, S. Sacher, D. Cantillo, J. D. Williams and C. O. Kappe, Advanced Real-Time Process Analytics for Multistep Synthesis in Continuous Flow**, Angew. Chem., Int. Ed., 2021, 60, 8139–8148 CrossRef CAS PubMed.
- C. Waldron, A. Pankajakshan, M. Quaglio, E. Cao, F. Galvanin and A. Gavriilidis, Model-based design of transient flow experiments for the identication of kinetic parameters, React. Chem. Eng., 2020, 5, 112–123 RSC.
- B. M. Wyvratt, J. P. McMullen and S. T. Grosser, Multidimensional dynamic experiments for data-rich process development of reactions in flow, React. Chem. Eng., 2019, 4, 1637–1645 RSC.
- K. C. Aroh and K. F. Jensen, Efficient kinetic experiments in continuous flow microreactors, React. Chem. Eng., 2018, 3, 94–101 RSC.
- C. Waldron, A. Pankajakshan, M. Quaglio, E. Cao, F. Galvanin and A. Gavriilidis, An autonomous microreactor platform for the rapid identification of kinetic models, React. Chem. Eng., 2019, 4, 1623–1636 RSC.
- O. Levenspiel, Chemical Reaction Engineering, John Wiley & Sons, Inc., 3rd edn, 1999 Search PubMed.
- H. S. Fogler, Elements of Chemical Reaction Engineering, Prentice Hall, 5th edn, 2015 Search PubMed.
- C. R. Wilke and P. Chang, Correlation of diffusion coefficients in dilute solutions, AIChE J., 1955, 1, 264–270 CrossRef CAS.
- E. Cussler, Diffusion: Mass Transfer in Fluid Systems, Cambridge University Press, 3rd edn, 2009 Search PubMed.
- H. Xiang, A. Laesecke and M. Huber, A New Reference Correlation for the Viscosity of Methanol, J. Phys. Chem. Ref. Data, 2006, 35, 1597–1620 CrossRef CAS.
- T. M. Aminabhavi and K. Banerjee, Density, Viscosity, Refractive Index, and Speed of Sound in Binary Mixtures of Dimethyl Carbonate with Methanol, Chloroform, Carbon Tetrachloride, Cyclohexane, and Dichloromethane in the Temperature Interval (298.15308.15) K, J. Chem. Eng. Data, 1998, 43, 1096–1101 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |