
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceApplication of reactor engineering concepts in continuous flow chemistry: a review
Nicole C.
Neyt
 and
Darren L.
Riley
*
and
Darren L.
Riley
*
Faculty of Natural and Agricultural Sciences, Department of Chemistry, University of Pretoria, 0002, South Africa. E-mail: darren.riley@up.ac.za
First published on 4th May 2021
Abstract
The adoption of flow technology for the manufacture of chemical entities, and in particular pharmaceuticals, has seen rapid growth over the past two decades with the technology now blurring the lines between chemistry and chemical engineering. Current indications point to a future in which flow chemistry and related technologies will be a major player in modern chemical manufacturing and the 4th industrial revolution. In this review we highlight the application of new reactor configurations and designs in the context of either bespoke or commercial flow apparatus specifically related to microwave chemistry, photochemical transformations, electrochemical promoted reactions and multi-phasic reactions. In addition, we look at how 3D printing in reactor design and computer-aided automation is growing within the field and finally describe how innovative solutions are being developed to tackle challenging down-stream processing operations.
Introduction
The past 150 years have been truly phenomenal in the field of synthetic organic chemistry. There has been unprecedented growth, affording researchers with a better understanding of the fundamentals of the discipline, the methodologies employed, and the analytical capabilities required. In addition, chemical manufacturing technologies have also grown substantially but remained largely batch-by-batch focused. These successes have not come without their consequences: environmental pollution and negative socioeconomic impacts, and at this point in time the future still holds enormous challenges for the field in terms of sustainable manufacturing, healthcare provision, lifestyle management and smarter energy solutions.1In the past decade flow chemistry and the application of flow devices has made a fundamental impact on academic research and academic laboratories globally. It is considered a valuable alternative to traditional batch processing primarily because of its ease of use and control. Flow chemistry, microchemistry or continuous flow chemistry is the process of performing chemical reactions in a continual flowing stream.2 Reactive components are pumped together at a mixing junction and flowed through a reactor (temperature controlled) to perform a specific reaction.
The vision of the FDA's pharmaceutical quality initiative for the 21st century is to create a more robust and flexible pharmaceutical sector, and the use of continuous manufacturing is one strategy capable of meeting this vision.3 As such the adoption of the technology is now supported by the Food and Drug Administration and the Green Chemistry Society, and it has been identified as one of the ten most important chemical innovations that will change the world and pave a way for a sustainable future in chemistry.4
The complementary relationship between chemistry and technology development has been highlighted by continuous flow processing over the past few years, with constant advancements in microreactor designs and architecture. This on-going growth from round bottom flasks to flow reactors has led to the integration of new concepts in mixing, dosing, heat transfer and processing in general.5 In many instances, innovative bespoke engineering solutions have been developed in laboratories across the globe greatly expanding the scope of flow chemistry. In this review we have focused on the role of new reactor configurations and designs in the context of either custom-made or commercial apparatus specifically related to microwave chemistry, photochemical transformations, electrochemical promoted reactions, gas–liquid mediations, 3D printing in reactor design, computer-aided automation and down-stream processing operations. These have been the areas with the most advances in reactor designs and equipment, with the primary application range for these technologies being the synthesis of active pharmaceutical ingredients (API's). Several reviews have been written on the various topics discussed in this overview;6–13 as such, we have elected to highlight key applicator concepts with a focus on recent and highly influential articles in the field.
Although continuous-flow chemistry has numerous benefits, it has drawbacks and limitations as well. For example, handling of solids, precipitates and sediments can lead to the blockage of the flow system, however, in many cases these problems are mitigated though the use of solid-supported reagents, appropriate solvent choices and in certain applications through the use of larger channel sizes.
Method development can also be time consuming as a range of different reaction parameters need to be considered such as the temperature, solvent, concentration, pressure and residence time. However, once optimal conditions have been established, they can often be directly applied to similar reactions. Tailing or dispersion of reagents through the flowing solvent stream can also sometimes be a challenge when performing array type chemistry, or multi-step chemistry and finally, the relatively high cost of flow reactor platforms is still considered to be a limiting factor, particularly in academia, though in many instances the use of low-cost bespoke systems is now opening access to the field.
1 Microwave applications under flow conditions
The application of microwave (MW) chemistry in flow was first realized by Strauss14,15 and Chen16 in the early 1990's using a quartz or Teflon based helical tube. The application of MW flow chemistry has since been widely applied to various organic reactions including nucleophilic aromatic substitutions (SNAr), eliminations, decarboxylations, amidations and condensation reactions. Flow technology has offered the unique ability of scaling-up procedures with microwave chemistry which had been lacking under traditional batch conditions. The major drawback associated with microwave chemistry under traditional batch conditions is the depth penetration of microwaves into the absorbing media. This is where flow microwave chemistry offers a unique advantage over batch, only a small amount of material is present at a single point in time with consistent heat transfer into the reaction media and negligible depth of penetration.6 Several types of MW flow reactors are reported in literature: multimode reactors with magnetron, single mode reactors with magnetron and multi-mode reactors with a solid state oscillator. A magnetron is an electron tube designed to amplify the microwaves generated by the external magnetic field.17 Most of the reported MW flow reactors have been constructed by placing a flow path within a commercially available single mode MW reactor. Several different flow paths have been reported and designed by different groups, consisting of either helical, capillary type or straight/packed tube type vessels.Despite early uptake one could argue that MW chemistry in flow is redundant with rapid heating being achievable with suitable reactor modules. Furthermore, no new exciting evolutions have appeared in recent years with regards to the amalgamation of flow chemistry and MW heating.6 That being said, we still feel that it is still pertinent to highlight a few important concepts linked to the historical use of MW's in flow over the past ten years.
 | ||
| Scheme 1 Microwave-assisted continuous-flow organic synthesis (MACOS) utilizing a glass capillary reactor with Pd and/or Ru catalysts. | ||
Similar capillary-based microwave flow reactors have been reported.18–22 A capillary based microreactor was used in the oxidation of benzyl alcohol to benzaldehyde and reported by Varma and co-workers. They made use of a narrow microchannel device consisting of two main parts: an aluminium section and a polytetrafluoroethylene (PTFE) section. The reactor design allowed reactions to be carried out under isothermal conditions while being exposed to MW irradiation. A flow rate of 4 mL min−1 equating to 75 second residence time, produced 96% conversion to benzaldehyde.23
Straight tube inserts. Wood and co-workers developed a simple means of performing microwave assisted continuous flow organic synthesis (MACOS) under flow conditions. They used a Pyrex tube with two drilled frits and a custom metal head fitted with a standard inlet tube and outlet fitting. The tube was filled with sand as a dispersive agent but could also be replaced with a catalyst of choice. With this they investigated the hydrolysis of chloromethylthiazole 12 to give alcohol 13 and the Fischer indole synthesis of 14 from hydrazine 15 and cyclohexanone 16 in glacial acetic acid. Efficient conversions were achieved at 150 °C, processing 1 g in 15–30 min, with products 13 and 14 afforded in 85% and 91% yield respectively (Scheme 2). Finally, they investigated a Bohlmann–Rahtz synthesis of 17 from 18 which was achieved in a 98% yield.24
 | ||
| Scheme 2 Hydrolysis and Fischer indole transformations using microwave assisted continuous flow chemistry. | ||
A silicon carbide (SiC) tubular reactor as well as a borosilicate glass reactor has been developed by WaveCraft AB Ltd. These reactors formed part of their ArrheniusOne system which was used by Larhed and co-workers to synthesize spiro-oxindole dihydroquinazolinones known as insulin regulated aminopeptidase (IRAP) inhibitors.25 The route involved a MW flow synthesis to build a library of spiro-cyclic compounds 19, from a three component reaction involving 20, 21 and 22. These spiro-cycles were synthesized in yields varying from 40–87% yield. This was followed by a Suzuki–Miyaura cross coupling reaction with phenylboronic acid 23 to form the potential IRAP inhibitor 22, the product was formed in 71% yield with Pd-catalyst (Scheme 3). Further publications also demonstrated a range of Suzuki–Miyaura couplings in excellent yields with the same SiC MW flow vessel.26 A similar quartz tubular reactor was designed by Suzuki and co-workers demonstrating rapid heating of solvents at pressures up to 10 MPa. Polar solvents such as water, ethylene glycol and ethanol were heated instantaneously to temperatures above their boiling points by applying pressure. They also demonstrated the rapid synthesis of Cu-nanoparticles by elevating the reaction temperature of ethylene glycol.27
 | ||
| Scheme 3 Microwave heated flow synthesis of spiro-oxindole dihydroquinazolinone based IRAP inhibitors utilizing a borosilicate and SiC MW vessel. | ||
Koloini and co-workers were one of the first groups to perform MW reactions under flow conditions. In 1999 they designed a fixed bed MW reactor packed with a solid supported ion resin to catalyse esterification reactions. They used a conventional microwave unit from Osaka, Japan called the Panasonic NE-1780. The temperature of the outlet was measured with a Ni–Cr thermocouple. The esterification of benzoic acid was performed by passing the reaction mixture through an ion exchange resin in the presence of excess ethanol to form ethyl benzoate. The optimized flow reaction conditions were: 140 °C and 7 atm. Reaction times were significantly decreased, which typically occurred over several days at 80 °C under conventional methods.28 Similarly Sajiki and co-workers demonstrated Mizoroki–Heck reactions with an immobilized Pd-catalyst supported on an anion exchange resin (DIAION WA30). The reaction was conducted in a straight tube reactor, designed by SAIDA FDS Inc. The supported catalyst was housed in this cartridge and exposed to MW irradiation.29 Various iodoarenes were converted to their equivalent cinnamates in excellent yields, while the cartridge could be reused for at least 5 runs without exchange.30
U-tube reactor insert. Tranmer and co-workers developed a method to perform Suzuki–Miyaura couplings under MW irradiation. The method involved the use of a U-tube reactor constructed out of glass and packed with an encapsulated Pd-catalyst (Pd-EnCat). The optimized reaction conditions for the Suzuki–Miyaura couplings were set at: 5 mol% Pd-EnCat, 3 equivalents of potassium carbonate and a solvent mixture of toluene, water and ethanol (4
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1) at elevated temperatures of 120 °C. They studied the simple reaction between phenylboronic acid 24 and bromoanisole 25 and achieved 98% conversion to 26. The reaction time was reduced from 8 h under standard batch conditions to 10 min in flow (Scheme 4).31 A commercial microwave heating system, EMRYS Synthesizer Tm., was used and the U-tube glass applicator was inserted inside the MW cavity. A similar design was shown by Fletcher and co-workers in 2004, with a U-tube glass capillary and a solid supported Pd-catalyst. The catalyst was embedded inside the U-tube glass applicator and lowered inside the MW cavity. Suzuki–Miyaura couplings were performed under several reaction conditions. Optimized conditions were determined as 90 W, 80 °C MW heating, a Pd/Al2O3 catalyst and potassium carbonate as base, with a 99% conversion to the coupled products.32,33
:2:1) at elevated temperatures of 120 °C. They studied the simple reaction between phenylboronic acid 24 and bromoanisole 25 and achieved 98% conversion to 26. The reaction time was reduced from 8 h under standard batch conditions to 10 min in flow (Scheme 4).31 A commercial microwave heating system, EMRYS Synthesizer Tm., was used and the U-tube glass applicator was inserted inside the MW cavity. A similar design was shown by Fletcher and co-workers in 2004, with a U-tube glass capillary and a solid supported Pd-catalyst. The catalyst was embedded inside the U-tube glass applicator and lowered inside the MW cavity. Suzuki–Miyaura couplings were performed under several reaction conditions. Optimized conditions were determined as 90 W, 80 °C MW heating, a Pd/Al2O3 catalyst and potassium carbonate as base, with a 99% conversion to the coupled products.32,33
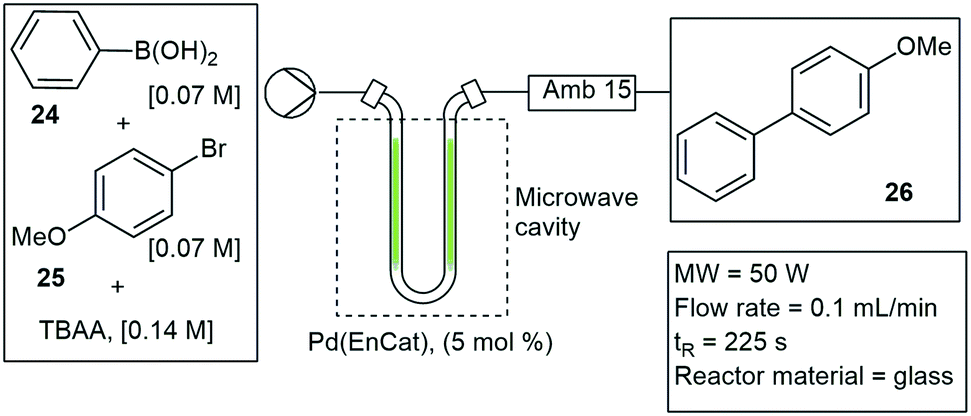 | ||
| Scheme 4 U-tube microwave insert for the application of Suzuki–Miyaura couplings under continuous flow conditions. | ||
Coiled or helical inserts. Coiled or channel-based reactors have also frequently been used. They allow for longer residence times and faster flow rates. Reactor walls are usually coated or simply packed with a catalyst. A microwave assisted Cu-catalyzed Ullmann ether synthesis was developed by Schouten and co-workers in 2012, using a coiled type glass reactor.34 The reactor walls were coated with a Cu/ZnO catalyst or alternatively, packed with a Cu/TiO2 catalyst. The reaction between phenol and 4-chloropyridine was studied. Productivities of the reaction were recorded after 6 h at 28.3 and 55.1 kg h−1 for the Cu/ZnO and Cu/TiO2 respectively. Roth and co-workers followed suit and built a similar coiled glass insert. The reactor consisted of 22, 3 mm internal diameter (ID) borosilicate glass coils encased in a 100 mm × 10 mm protective borosilicate glass sheath, with a total reaction volume of 4 mL. A series of representative reactions were investigated which included a nucleophilic aromatic substitution combining 2-fluoronitrobenzene 27 with 2-phenylethanamine 28 to form 29. An esterification reaction of arylbenzoic acid 30 in methanol (MeOH) to form 31 with a 50% conversion and finally, a Suzuki–Miyaura coupling where 4-bromobenzaldehyde 32 and boric acid 33 were coupled to form 34 with a conversion of 84%. In all cases product yields were greater or equivalent to those produced under conventional heating methods, with the ability to directly scale these reactions (Scheme 5).35
 | ||
| Scheme 5 A practical continuous flow microwave cell for nucleophilic substitutions, esterifications and Suzuki–Miyaura couplings. | ||
In 2015 Akai and co-workers developed a microwave reactor insert that was suitably applied to a wide variety of reactions. This applicator consisted of a wide long helical glass tube with a 3.6 mm ID and an internal volume of 5.5–6 mL. The glass tube was covered with PTFE tubular film for safety and insulation. To validate the function of the MW insert they studied both a Fischer indole reaction and a Diels–Alder reaction. The Fischer indole synthesis involved cyclohexanone 35 and phenylhydrazine 36 treated under MW irradiation to form carbazole 37 on a 100 g scale in 75% yield, with a residence time of 24 s.36
The microwave applicator has since been commercialised by SAIDA FDS Inc., and several reactions including ammonium salt-accelerated hydrazinolysis of unactivated amides,37 Fischer esterification,36 alcohol acetylation reactions,38 Suzuki–Miyaura couplings, dehydrogenative aromatizations,30 Wolff–Staudinger cascade reaction,39 Williamson esterification,40C-alkylations of N-alkylamides41 and a Johnson–Claisen rearrangement42 have all been tested and demonstrated utilizing this insert. One of the more notable reactions performed was the scalable and selective synthesis of C60/indene monoadducts 38 from the C60 fullerene 39 and indene 40.43 The 5.2 mL helical reactor tube was used at a temperature of 180–270 °C, to form the indene C-60 monoadduct and bisadduct 38 in 1.1 g h−1 and 0.5 g h−1 respectively (Scheme 6).
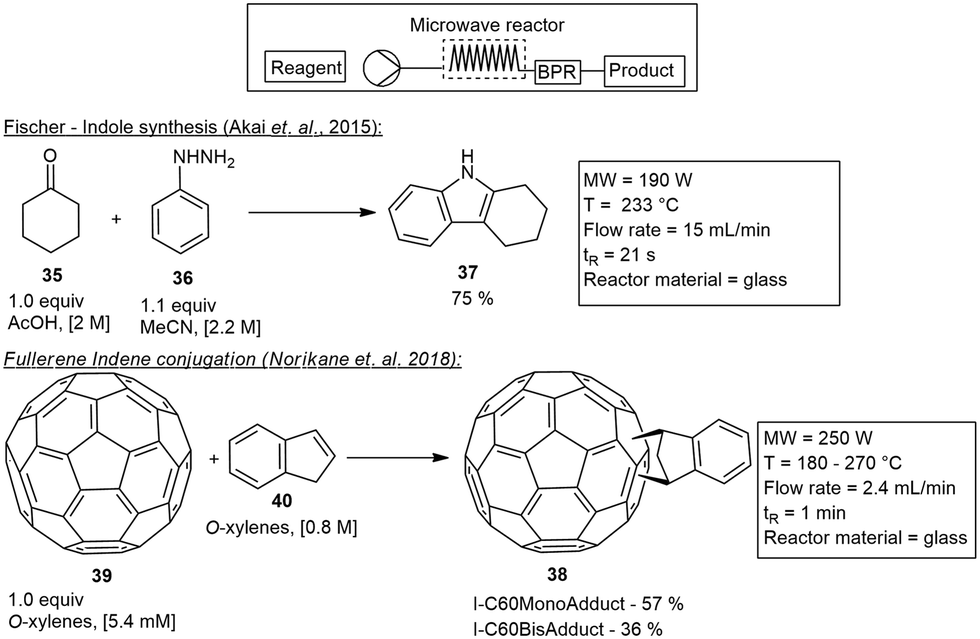 | ||
| Scheme 6 Various reactions demonstrated under continuous microwave conditions utilizing the SAIDA FDS Inc. insert. | ||
Morschhäuser and co-workers reported on another industrial scale MW reactor demonstrating several organic reactions on large scale.47 The industrial reactor was designed to operate at temperature of 310 °C and pressures of 60 bar. It was constructed from a chemically resistant, transparent, and cylindrical material made of γ-Al2O3. The MW reactor operated at 0.6–6 kW and demonstrated a 3.5–6 L h−1 throughput. The first reaction investigated was a simple amide coupling to form 41 from a readily available amine substrate 42 and carboxylic acid derivative 43. The amide 41 was produced in a yield of 93%. Another reaction involved the transformation of carboxylic acid 44 to amide 45 in the presence of dimethylamine. They also demonstrated the formation of a Fischer ester 46 with 2-hydroxypropanoic acid 47 and 2,2-ethyl hexanol 48 in 89% yield. Finally, the formation of imidazole 49 was achieved in 90% yield with aniline 50 and carboxylic acid 51 as substrates (Scheme 7).47
 | ||
| Scheme 7 Microwave assisted continuous flow on industrial scale. | ||
2 Flow photochemistry applications
The use of light in chemical reactions has been dated back to the 18th century, but as with MW reaction, limitations associated with depth of penetration of, in this case photons into reaction media held back the widespread adoption of the technique. Recently applications of light harvesting in continual flowing streams has sparked a renaissance in the photochemistry community, with significant interest being poured into the field with the focus on the design and application of light in flow chemistry. The efforts in this field have further stimulated the development of both novel photocatalysts and photon transport improvements.48 | ||
| Scheme 8 The oxidation and reduction reactions with a milled glass microchip reactor. | ||
Similarly, Reiser and co-workers developed a novel photocatalytic method for the E/Z isomerisation of cinnamyl acetate 57 and translated this to a custom single layer continuous flow setup. The reaction involved the use of an Asia dual pump and custom glass microchip reactor irradiated with blue light emitting diode (LED) light. A novel polyisobutylene tagged iridium catalyst was also synthesized, the catalyst was recycled in a continuous fashion. The photocatalytic isomerisation of cinnamyl acetate 57 to (Z)-3-phenylallyl acetate 58, was investigated, with a Z/E ratio of 82:18 achieved in 4 h (Scheme 9).51
 | ||
| Scheme 9 The isomerisation reaction of cinnamyl acetate under photocatalytic conditions with a glass microchip reactor irradiated with blue LED's. | ||
The multigram production of hypericin was carried out using an inexpensive Pyrex® based microchip photoreactor. The photoreactor unit consisted of four or more modules connected in series. This allowed for the quantitative photocyclization of protohypericin 59 to hypericin 60. The reactor was designed to allow variable light sources to be tested, however they found full conversion to hypericin 60 with red LED's in only 79 min (Scheme 10). Several microchip based photoreactors have been fabricated with similar materials and dimensions.52
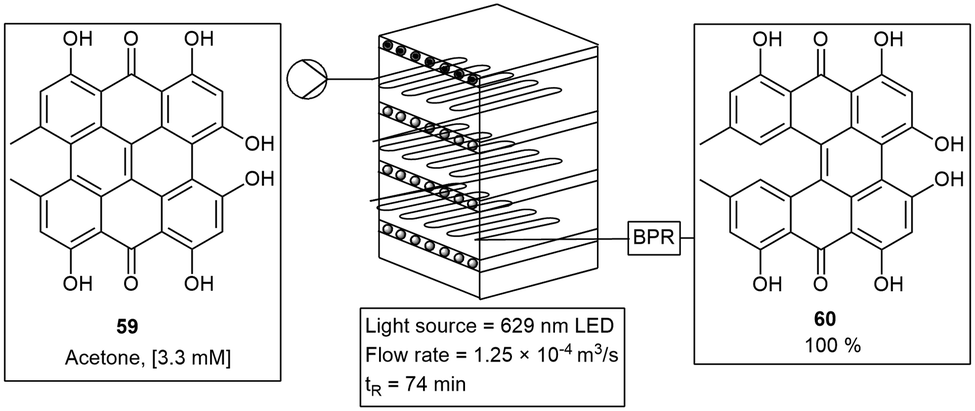 | ||
| Scheme 10 Synthesis of hypericin on multigram scale using a multimode photoreactor. | ||
Coiled channel reactors. Coiled type photoreactors are the most common and accessible photoreactor systems for flow photochemistry applications. Several diverse coiled reactors have been reported in the last decade. Photocatalysis reactions in flow have been gaining interest with most being catalysed by LED's. Seeberger and Bou-Hamdan published a visible light mediated flow paper using a Ru(bpy)32+ complex as a catalyst. The reactions performed with 10 to 15-fold improvement over the conventional batch method. The reactions tested included the reduction of an aromatic azide 61 to the corresponding amine 62, the dehalogenation of α-chlorophenyl acetate 63 to 64, the opening of chalcone-α, β-epoxide 65 to 66 and the bromination of alcohols 67 to the corresponding brominated products 68. All reduction reactions were completed using two 17 W white LED lights, with PTFE tubing wrapped around two vertical metal rods immersed between the two LED lights. Reactions tested were performed in DMF and proceeded with 100% conversions with residence times under ≤30 min (Scheme 11).53
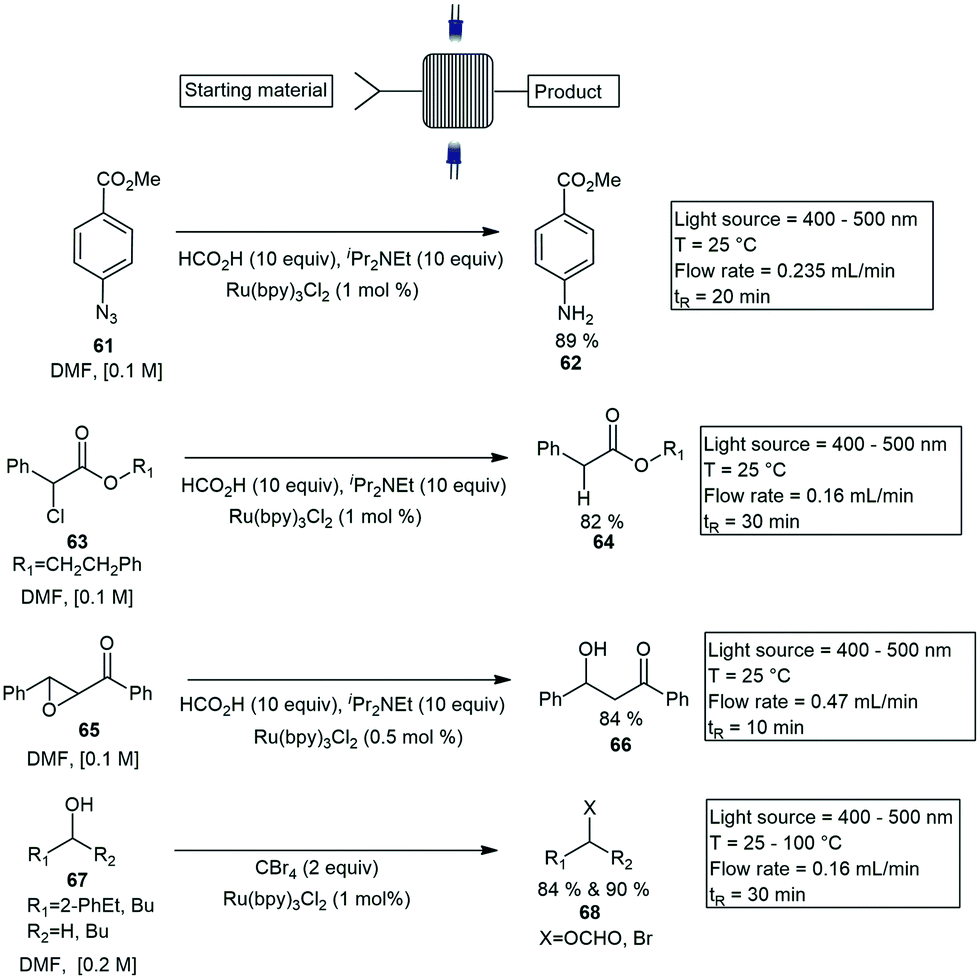 | ||
| Scheme 11 Custom built microchannel reactor for photocatalytic reactions. | ||
Another coil reactor was designed by Telmesani and co-workers in an effort to produce cyclobutanes from the dimerization of various cinnamic acids. The cone shaped photo reactor consisted of fluorinated ethylene propylene (FEP) tubing wrapped around the photoreactor in small grooves that promote heat transfer. The temperature of the reactor was controlled by a circulating chiller. Moderate yields between 19–69% were reported with a residence time of 8 h.54 The oxidative generation of imminium ions 69 from N-tetrahydroisoquinolines 70 using BrCCl3 as terminal oxidant and Ru(bpy)32+ as catalyst has also been reported. The photoreactor design included polyfluoro alkoxy (PFA) tubing wrapped in a figure eight around two glass test tubes with seven blue LED's emitting irradiation on either side. Complete conversion to the desired iminium ion was achieved in less 0.5 min residence time. The iminium ion was trapped and processed with a nucleophile such as nitromethane. Various nucleophiles were used which produced 71–74 in yields ranging between 79–89% (Scheme 12).55 Huvaere and co-workers designed a similar photoreactor, consisting of FEP tubing, of 2.5 mL or 6.0 mL, a 0.8 mm ID, and a 1.6 mm outer diameter (OD) wrapped on a metal frame (8 × 10 cm). A 20 watt LED lamp or a 50 watt LED lamp was positioned at a distance of 0.5 cm from the photoreactor and used for the irradiation of the reaction mixtures.56
 | ||
| Scheme 12 The formation of an imminium ion trapped with various nucleophiles using a custom built photoreactor under flow conditions. Photograph reproduced with permission.55 | ||
Wriedt and co-workers recently published their work on the experimental determination of photo fluxes in a multilayer channel reactor.57 They studied the benefit of adding more layers to the photoreactor using actinometry and the decomposition of [Fe(C2O4)3]3−, carbon dioxide and oxalate. They showed that on average the ratio between the absorbed and available photons in the inner layer was 11%, for the middle layer 3% and 1% for the outer layer. More research has been focused into multilayer capillary photoreactors especially by Elliott and co-workers and Rehm and co-workers, who have already made a significant contribution in the field.58–60
Noel and co-workers constructed a simple photochemical microreactor for gas–liquid photoredox catalysis using visible light. The microreactor was constructed with high-purity PFA capillary tubing having a 0.75 mm ID and 1/16 inch OD and a total volume of 1.1 mL volume, the tubing was coiled around a disposable plastic syringe coated with refractive aluminium tape. An array of LED's was wrapped around the reactor, and the whole assembly was fixed in a larger diameter disposable plastic syringe. They reported the formation of trifluoromethylated heterocycles and thiols.61 Furthermore, trifluoromethylation and hydrotrifluoromethylation protocols were described a year later, for various vinylarene substrates 75. The method involves the use of Fac-Ir(ppy)3, visible light and inexpensive CF3I to produce the trifluoromethylated styrenes 76 (Scheme 13). The use of continuous-flow photochemical reaction conditions allowed for reduced residence times and increased reaction selectivity.62
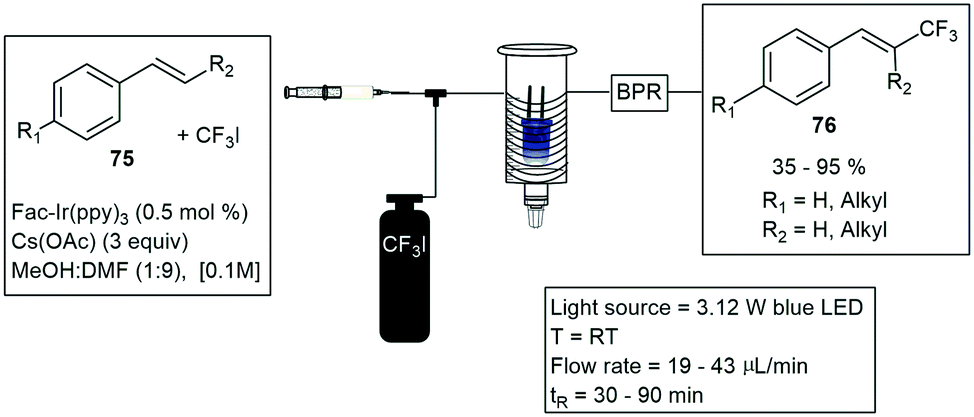 | ||
| Scheme 13 Trifluoromethylations under visible light conditions using a simple syringe to promote a coiled reactor under flow conditions. | ||
A one-pot synthesis of tricyclo-1,4-benzoxazines 78 has been developed via metal free intramolecular cyclization of indole derivatives 77. The reaction utilized visible light photoredox catalysis under flow conditions. Two 20 mL “reaction towers” were connected in series, each contained an LED corn light bulb (34 W) in the centre of the tower, tubing was wrapped around these for optimal light penetration (Scheme 14). The reaction occurred with the use of methylene blue (MB) in 4 mol%, toluene sulfonic acid and oxygen. The reaction proceeded quantitatively in 24 h under batch conditions and 3 h under recycling flow conditions. Various substrates were synthesized and produced in nearly quantitatively yield.63
 | ||
| Scheme 14 The one pot synthesis of tricyclo-1,4-benzoxazines under flow conditions using two photo reaction towers connected in series. | ||
Su and co-workers have conveniently developed a numbering up strategy for the convenient scale up of photochemical reactions. The 8 capillary system was designed to allow gas–liquid photoreactions to occur in parallel. The aerobic oxidation of thiols 79 to disulfides 80, was used as a model reaction. The yield obtained for the numbering up strategy was comparable to the yield obtained in a single device (Scheme 15).64
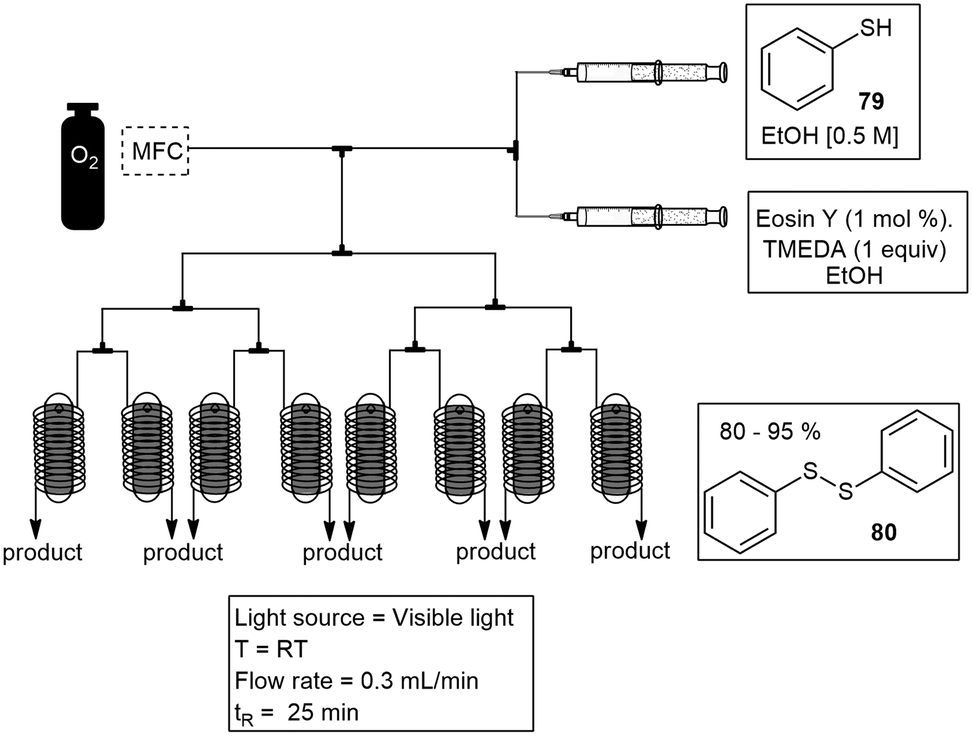 | ||
| Scheme 15 Numbering-up strategy for large scale flow photochemical synthesis. | ||
Recently Smith and co-workers synthesized a library of benzo-[1,3]oxazepines by combining the CH-activation and functionalization with photochemical heterocyclic metamorphosis. The N-oxide substrate was dissolved in toluene and passed through a 10 mL PFA coil, wrapped around a 25 W Exo Terra UVB200 lamp. A 100% conversion to the product was realized in only 6 min, and the product was isolated in near quantitative yield. Ten substrates were investigated with varying aromatic groups and yields between 75 and 99% were obtained within a 10 min residence time.65
Quartz microchannel reactor. Shen and co-workers developed a unique flow photoreactor employing quartz tubing and an aluminium mirror to perform photo-induced electron transfer deoxygenation reactions specifically to produce 2′-deoxy and 2,3′-deoxynucleosides. They showed significant improvement in the efficiency and selectivity of the reaction. Customised quartz coils and a 450 W medium pressure Hg lamp, was used to produce various nucleosides in yields ranging from 62–85%, with residence times that varied from 5–20 min.66
Straight tube reactors. A similar quartz photoreactor was designed by Kim and co-workers. Ten HPFA capillaries were filled with an immobilized eosin catalyst and housed inside the quartz jacket. These capillaries were irradiated by surrounding green LED's wrapped around them. A library of indazole based products were produced by passing a mixture of 2H-indazole and a tetrafluoroborate salt through the capillary photoreactor at a flow rate 2 mL min−1 (0.63 min).67
Harper and co-workers have designed a photochemical reactor with the use of a laser system to promote organic reactions with more selectivity and reactivity. The reactor was fitted with a laser beam focused onto a continuous stirred tank reactor. The reactor operates in a semi-batch fashion, whereby materials are continuously fed into a stirred tank and removed as the reaction progresses. They investigated a C(sp2)–N(sp3) cross coupling reaction between 1-bromo-4-(trifluoromethyl)benzene 81 and pyrrolidine 82 to form 83 (Scheme 16).68
 | ||
| Scheme 16 Laser mediated cross coupling reaction inside a tube reactor. | ||
Elliott and co-workers developed a tubular photo reactor to perform reactions on kilogram scale. They demonstrated several organic reactions. The 120 mL reactor consisted of a succession of quartz tubes connected in series and arranged axially around a variable power mercury lamp. They showed that 3,4,5,6-tetrahydrophthalic anhydride (THPA) 84 and cis-2-butene-1,4-diol can undergo an efficient [2 + 2] cycloaddition–lactonisation sequence to afford tricyclic lactone 85 upon direct UV irradiation with a staggering production of 4 kg in 24 h. They further demonstrated similar a cyclization with productions of kilograms per day (Scheme 17).58
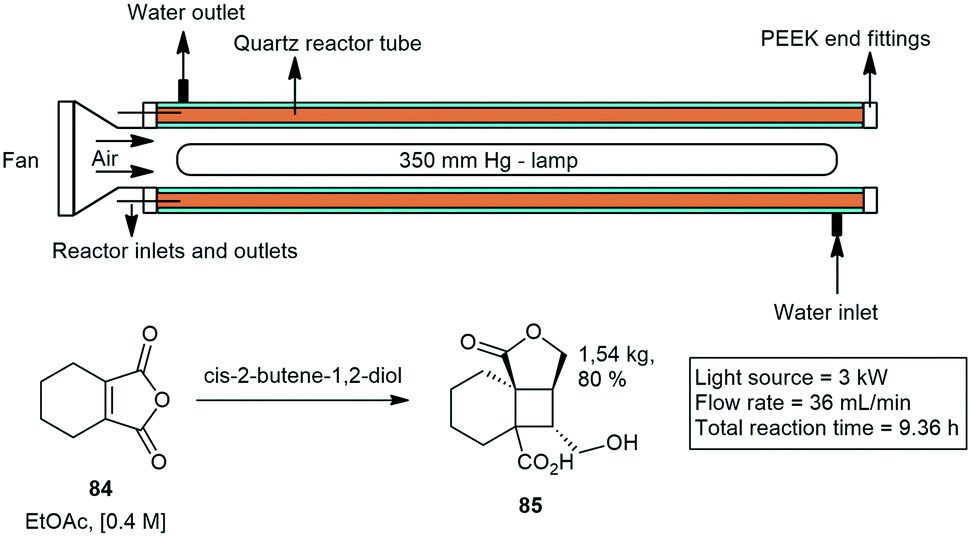 | ||
| Scheme 17 Cyclization reactions demonstrated in a straight tube quartz photo reactor. | ||
 | ||
| Scheme 18 Cyclisation reactions demonstrated in a straight tube quartz photo reactor. | ||
 | ||
| Fig. 1 Luminescent solar concentrator photomicroreactors by Noel and co-workers. Photograph reproduced with permission.72 | ||
Clark and co-workers designed a photoreactor that generates a thin film upon rotation for efficient irradiation. The photoreactor comprises of a rotary evaporator with banks of LED lights mounted above the surface of the rotating evaporation flask. They demonstrated three different photooxidation reactions, and investigated several parameters which included the flask size, volume, rotation speed, and light intensity. The first reaction investigated was a photodesymmetrization of benzene-1,4-diboronic acid 89 to the mono and dihydroxylated products 90 and 91. A 10 min residence time gave nearly quantitative conversion of the starting material 89. The second reaction investigated was the photooxidation of α-terpinene 92 to form ascaridole 93 with the highest yielding and fastest time of 87% yield in 60 seconds. The final reaction investigated was the photooxidation of citronellol 94 to form hydroperoxides which is further reduced to form the corresponding diols followed by the dehydrative cyclisation to yield rose oxide 95. The reaction proceeded to 99% conversion in 420 seconds (Scheme 19).73
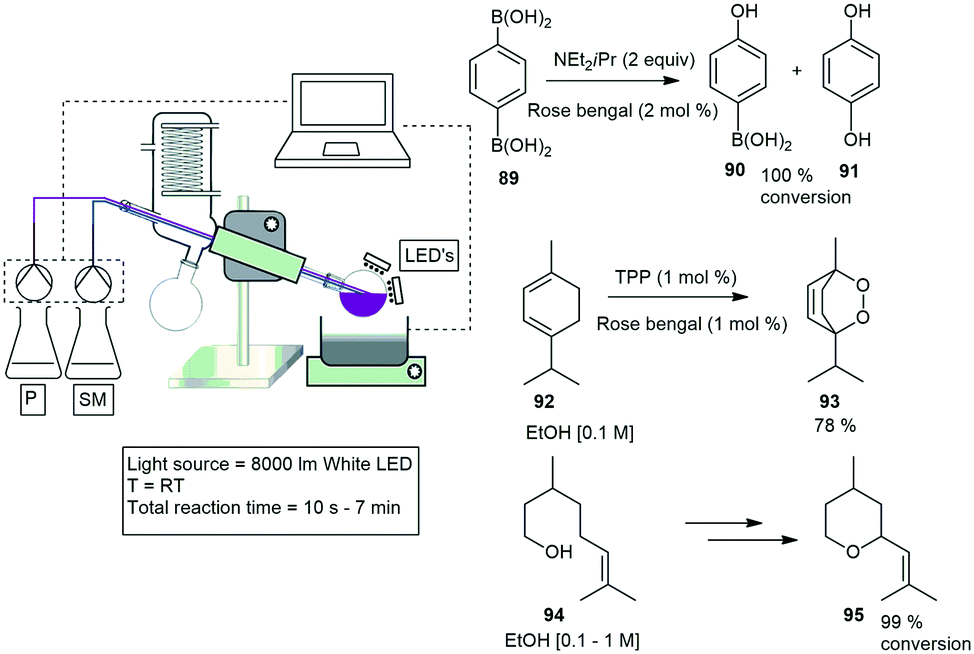 | ||
| Scheme 19 Photo oxidation reactions using a common rotary evaporator. | ||
Ioannou and co-workers made use of a modified continuous flow reactor for biphasic photooxgenation reactions. The reaction solution was nebulized, using either oxygen or air, and the resulting aerosol is irradiated by an LED jacket that surrounds a Pyrex reaction chamber. They showed the formation of complex functionalised γ-lactams in a 4-step synthesis, with the first step involving the photo catalysis reaction, in 49–66% yield from simple furans.
Continuous photo-oxidation reactions have further been reported in vortex reactor systems. The vortex reactor makes use of a rapidly rotating cylinder to generate Taylor vortices for continuous thermal and photochemical reactions. The vortex reactor has been applied to several organic photochemical reactions including the oxidation of α-terpinene 92 to ascaridol 93 in 91%, furfuryl alcohol to furfuraldehyde in 73% as well as the photodeborylisation of phenylboronic acid to phenol in 94% yield. The rotation speed was key to the reaction efficiency, demonstrating the highest efficiency at 4000 rpm, allowing for optimal absorption of air. The reactor was further applied to the oxidation of artemisinin in quantitative conversion and a yield of 50%.74
Another unconventional photoreactor named the photo rotor-stator spinning disk reactor has also been designed by van der Schaaf and co-workers. This conceptually new photochemical reactor was used in the photocatalytic oxidation of α-terpinene 92 to the drug ascaridole 93 with rose bengal as photocatalyst (Scheme 20). Throughputs of over 1 kg per day (270 mmol h−1) were materialised under visible light irradiation. The performance of the reactor directly correlates to various process parameters. Several optimization studies were conducted whereby the authors investigated: rotation speed, liquid flow rate, and catalyst concentration. The conversion and selectivity of the product increased from 37 to 97% and 75 to 90% respectively. This was achieved with an increase of rotation speed from 100 to 2000 RPM. The authors found that the mass transfer was directly related to the rotation speed of the disk and that the reactor operates with negligible pressure drop and allows for facile fine-tuning of the mixing efficiency.75
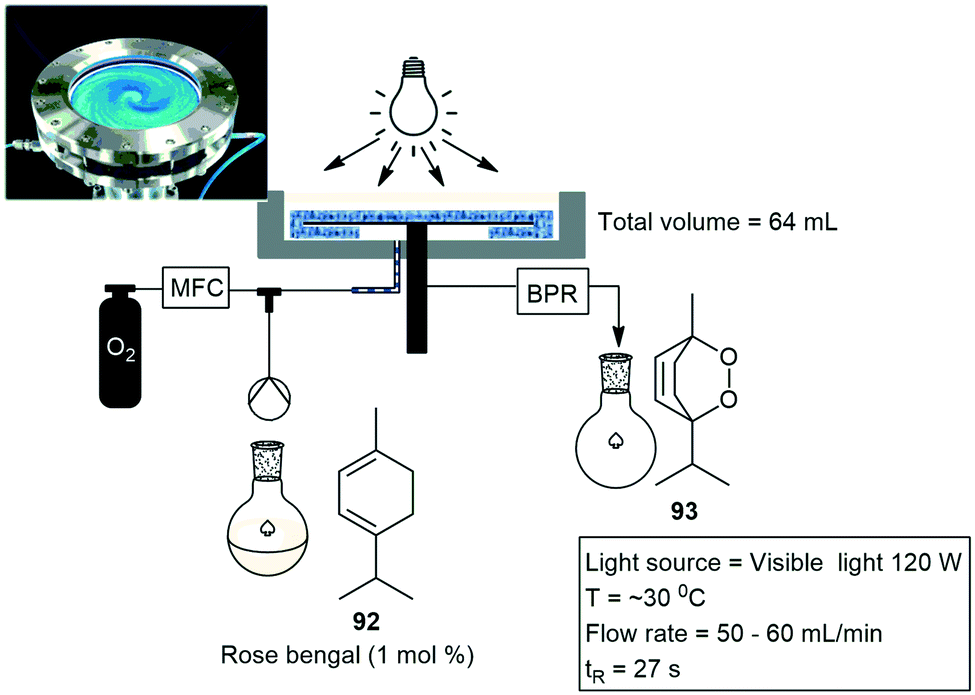 | ||
| Scheme 20 Rotor stator spinning disk reactor. Photograph reproduced with permission.75 | ||
 | ||
| Scheme 21 The synthesis of ibuprofen under photochemical flow chemistry using a Vapourtec UV-150 reactor. | ||
Beatty and co-workers described trifluoromethylation reactions. Trifluoroacetic anhydride 99 and pyridine-N-oxide was used to trifluoromethylate several arene substrates containing both electron-donating and mildly electron-withdrawing groups. The trifluoromethylation of N-Boc pyrrole 100 to the mono- 101 and di-substituted 102 derivatives were achieved in moderate yields (Scheme 22).78 Decarboxylative C(sp3)–C(sp3) cross-coupling reactions utilizing dual photoredox iridium–nickel photocatalysts have been reported by Abdiaj and co-workers, Robinson and co-workers and Lima and co-workers.79–81 In another paper Abdiaj and co-workers developed a method to perform a dual iridium–nickel catalyzed C(sp2)–C(sp3) cross-coupling reaction by circumventing solubility issues associated with potassium trifluoroborate salts. The formation of an intermediate, with a pyridine-derived Lewis base was found to be essential for the photoredox activation of the boronic esters 103. Based on these results they were able to develop a further simplified visible light mediated cross-coupling method using boronic esters 103 and various bromo heteroarenes 104 under flow conditions with a residence time of 50 min, blue LED's and 35 °C, 50–95% yield was reported for the coupled products 105 (Scheme 22).82 The UV-150 photochemical photoreactor has also been employed in the aerobic oxidative coupling of benzylamines 106 to enamine 107, photocatalysed by a novel HOTT–HATN polymer photocatalyst (Scheme 22). In a separate paper the same authors also described arene alkylations in dimethylsulfoxide (DMSO) with a different organic photocatalyst (4-CzIPN) and purple LED's. Reactions proceeded with a residence time of 70 min and yields from 60% (Scheme 23).83,84 Several more organic catalysts have been designed and implemented in the UV-150 photochemical photoreactor.85 Several reactions ranging from photoactivated oxadiazolidones,86 Vilsmeier Haack reactions,87 Negishi cross couplings,88 iron-catalyzed Kumada–Corriu cross couplings89 and several more have been reported with Vapourtec UV-150 photochemical photoreactor. Ley and co-workers published a paper demonstrating both the Vapourtec E-series as well as a prototype UV-reactor from Uniqsis. The reactions that were investigated included the photochemical cross-coupling between boronic esters 108 and carbonyl compounds 109 resulting in the formation of 51 different coupled reactions of which 13 were heterocycles. Reactions were first tested using the Vapourtec UV-150 fitted with a blue LED module 17 W and a 10 mL FEP coil, equating to a residence time of 100 min, a 100% conversion and 70% isolated yield of 110 was obtained. A large scale experiment was carried out using a Uniqsis Photosyn reactor, which operates on a similar principle but is equipped with a more powerful 420 lamp and a bigger 20 mL PFA coil, the residence time was reduced to 60 min and a 65% isolated yield of 110 was produced (Scheme 23).90
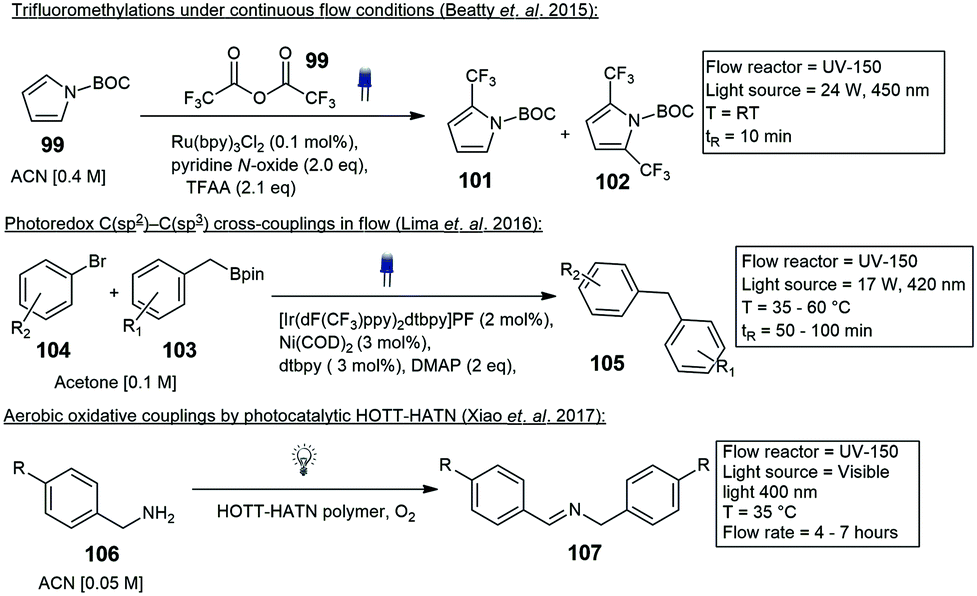 | ||
| Scheme 22 Reactions demonstrated on a commercial Vapourtec E-series photo reactor. | ||
 | ||
| Scheme 23 Commercial application of a photochemical cross-coupling reaction from batch to flow on both a Vapourtec UV-150 reactor and a Uniqsis Photosyn reactor. | ||
3 Flow electrochemistry concepts
Limitations in conventional electrosynthesis can be overcome when using flow electrochemical devices. A literature overview found that under batch conditions, electrolysis is largely based on performing reactions in a glass beaker with counter electrodes and an electrolyte solution. These reactions are largely equated with a poor performance and a very slow rate of conversion. This has changed with continuous flow electrolysis, which is participating in the trend towards innovative reactor designs in flow cells making selective syntheses with high conversions possible in a single pass through the cell. Continuous flow electrochemistry lends itself to improved mixing, the small gap between electrodes is of particular benefit enhancing high mass transfer and the electrode's surface/volume ratio which in-turn reduces energy consumption, fast heat exchange, multistep reaction sequences, improved reaction selectivity, high reproducibility and reliable scale up, with most of these being unattainable under batch conditions. In-addition the electrodes can easily be replaced by another metal electrode.8 | ||
| Scheme 24 Electrochemical synthesis carried out in MeOH in a spiral electrochemical reactor. | ||
 | ||
| Scheme 25 An easy to machine electrochemical microchannel flow cell used to demonstrate the synthesis of various isoindolinones. Photograph reproduced with permission.96 | ||
A similar flow cell was reported by Huang and co-workers demonstrating the synthesis of benzofused S-heterocycles as well as scale-up reactions of these procedures. The custom-built electrochemical flow cell was equipped with a Pt-cathode and an anode made of carbon filled polyvinylidene difluoride (PVDF). The electrochemical cell was manufactured using magnetron sputtering. This method deposits a thin layer of titanium (6 nm) and platinum (200 nm) on a SS plate. The Pt electrode, in turn, was stabilized with titanium, increasing the stability and overall effectiveness of the cell. A range of 1,4-benzoxathins and 1,4-benzothiazines were synthesized in moderate to excellent yields. A flow rate of 0.3 mL min−1 was used which equated to a residence time of 33 minutes, with a current of 35 mA.95
Arai and co-workers have designed an electrochemical flow reactor with platinum foil as both the anode and cathode. These were placed on either side of a FEP channel as shown in Scheme 25. The cell consisted of a single inlet and a single outlet, and with this they demonstrated several di- and trifluoromethylation reactions. The transformation of several acrylates 118 were reported with moderate yields (Scheme 26).96 A year later Arai and co-workers published another electrochemical flow cell with two inlets and a single outlet. The reactor was constructed from glass plates and two platinum Pt electrodes (3 cm width, 3 cm length each). A slit was provided on the cathode plate for introducing the nucleophilic solution into the reactor. They demonstrated an efficient anodic cross-coupling reaction using parallel laminar flow. The model reaction studied proceeded effectively in single flow through operation and the desired product was obtained in much higher yields compared to the performance in a conventional batch cell. Naphthalene 119 was coupled with an alkylbenzyl substrate 120, forming 121 in 85% yield at a rate of 0.47 mL min−1 (Scheme 27).97 This work was similar to that demonstrated by Kuleshova who designed a single channel electrolysis flow cell in 2012 and applied it to the methoxylation of N-formylpyrrolidine in high conversion.98
 | ||
| Scheme 26 Electrolysis of olefins using a single channel electrochemical reactor. | ||
 | ||
| Scheme 27 Anodic C–C cross coupling with naphthalene and a custom built electrochemical flow cell. | ||
Robert Green and co-workers developed a similar flow cell constructed from two rectangular electrode plates (53 mm × 40 mm × 2 mm). The working electrode was constructed with a carbon PVDF blend, and the counter electrode from SS. They described the oxidative conversion of aldehydes 122 to amides 123 mediated through N-heterocyclic carbenes (NHC). This was achieved through the oxidation of Breslow intermediates. After electrochemical oxidation, the reaction intermediate is completed by passage through a heating cell. The flow mixing regimen circumvented the issue of competing imine formation between the aldehyde 122 and amine substrates 124, which otherwise prevented formation of the desired product 123. High yields (71–99%), productivities (up to 2.6 g h−1), and current efficiencies (65–91%) were realized for 19 amides (Scheme 28).99,100 Noël and co-workers have designed a modular and scalable electrochemical flow reactor consisting of an undivided electrochemical cell with a flexible reactor volume (Scheme 29). Their reactor was designed to be used in two different modes: either serial (with volumes ranging from 88 μL to 704 μL), or a parallel mode (numbering up). The electrochemical oxidation of sulfides were studied with the transformation to either sulfoxide or sulfone. Residence time applied voltage and microchannel thickness was studied with an optimal yield of 98% for the desired products. Reaction conditions concluded with a residence time of 10 min and an applied voltage of 2.8 V. Furthermore, the aziridination of alkenes with primary amines using the same electrochemical microreactor with yields ranging from 19–94%, was also studied.101,102
 | ||
| Scheme 28 An electrochemical microreactor designed to produced amides from aldehydes through a Breslow intermediate. | ||
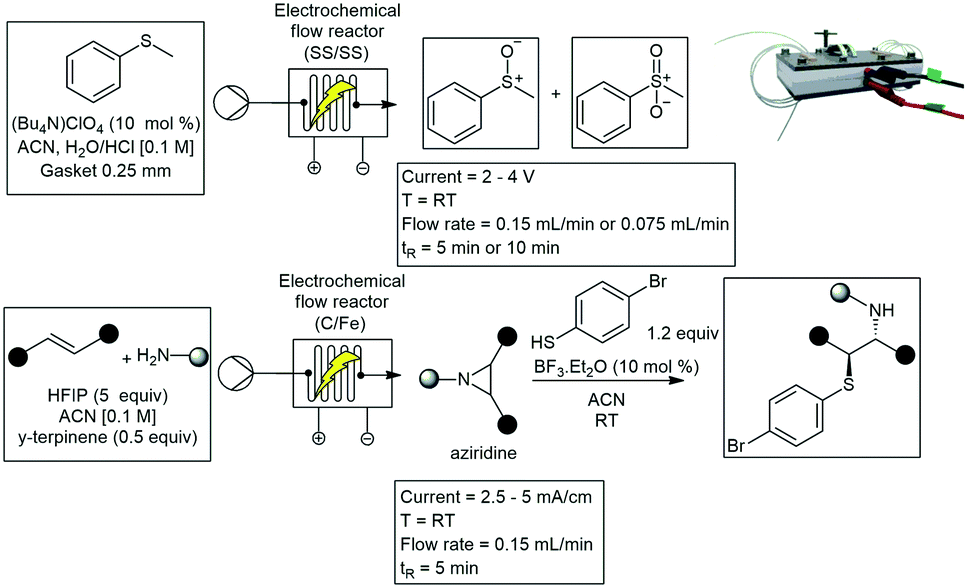 | ||
| Scheme 29 A modular and scalable electrochemical flow reactor consisting of an undivided electrochemical cell with a flexible reactor volume. Photograph reproduced with permission.101 | ||
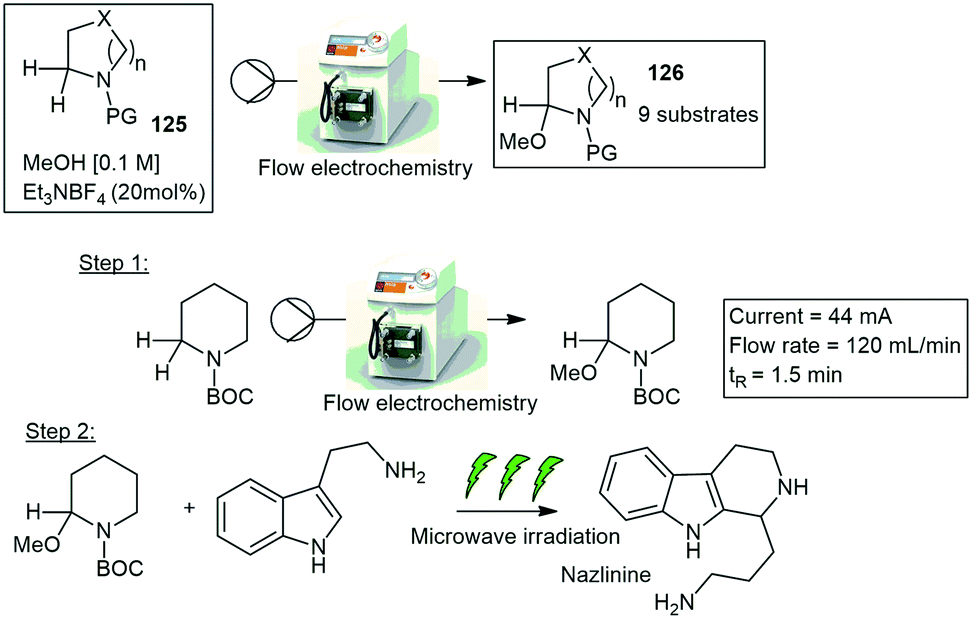 | ||
| Scheme 30 Syrris Asia electrochemical reactor showing the production of nazlinine. | ||
 | ||
| Fig. 2 Commercial electrochemical rectors. | ||
A recent piece of equipment from Vapourtec Ltd., the ion electrochemical flow reactor (Fig. 2), has been used to demonstrate the formation of hypervalent iodine reagents by Elsherbini and co-workers. The reactor (reactor volume = 0.6 mL, spacer 0.5 mm) is fitted with a glassy carbon anode and a platinum cathode. The use of electricity replaces hazardous and costly chemical oxidants generally associated with these types of transformations. Various substrates were oxidised in relatively high yields.104 Furthermore using the same ion electrochemical setup Amri and co-workers demonstrated the efficient mono- and dialkoxylation of pyrrolidine-1-carbaldehyde, with precise control of the reaction conditions either the mono- or the dialkoxylated products were formed.105
Cambridge reactor design (CRD) and Uniqsis supply the Ammonite 8 and 15 electrochemical flow reactors (Fig. 2) which are comprised of a clamshell type channel reactor with a spiral flow path (2 mm × 1.0/0.5 mm) which is designed to maximise the path length/size ratio of the system.104
4 Multiphase chemistry under flow conditions
Several approaches to gas–liquid flow reactions have been studied, most of these involved the use of a biphasic flow regime. In the case of gas–liquid reactions, a high interfacial area is essential for an efficient mass transfer rate. Batch reactions performed in a traditional round bottomed flask have much lower interfacial areas, and this decreases with an increase in the size of the flask. The exact flow regime depends on the gas and liquid flow rates, channel pattern and dimensions, and the physical properties of the fluid and gas composition. The flow regimes observed include segmented flow, bubble flow, stratified flow and annular flow (Fig. 3). Under microfluidic conditions segmented flow, described as bubbles of gas that are separated by slugs of liquid dominates. In contrast annular flow involves a gas and liquid stream concurrently flowing together, arising when a fast flowing high pressured gas stream is introduced into a liquid stream at lower pressures pushing the liquid against the microchannel wall.106 Most gas–liquid reactions in flow chemistry follow a segmented flow regime coupled with a mass flow controller (MFC) to accurately dose the amount of gas into the flow stream, gases utilised include carbon dioxide,107 oxygen,108 nitrogen,109 hydrogen sulphide,110 hydrogen, carbon monoxide111 and ethylene gas.112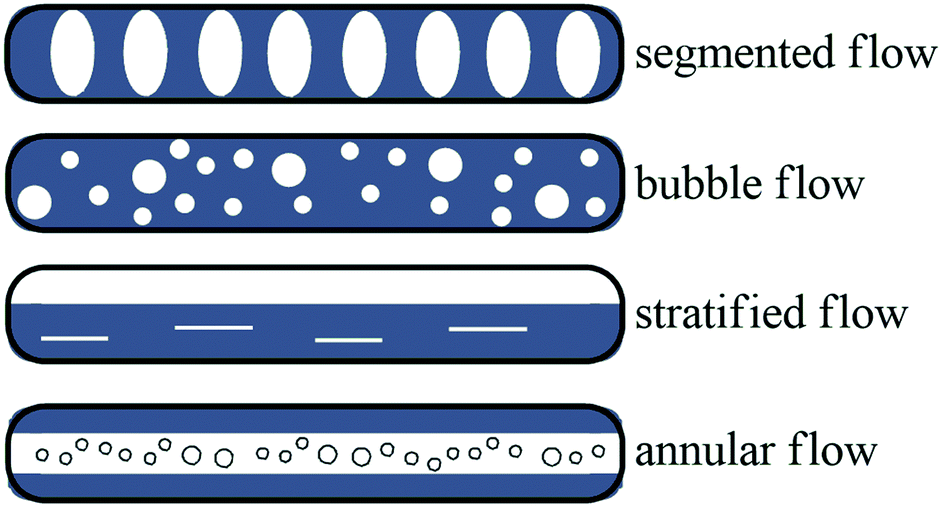 | ||
| Fig. 3 Flow regimes. | ||
 | ||
| Scheme 31 Ozonolysis reactions using a silicon Pyrex microreactor. | ||
Kappe and co-workers printed a SS reactor by selective laser melting. The reactor was designed specifically for difluoromethylation reactions with fluoroform gas and n-BuLi. The reactor was constructed from SS with a 0.8 mm ID and 2.4 m OD. The reaction channel was designed with a slight oval cross section. The specific geometry allowed improved surface quality on the top surface of the reaction channel by reducing the printing area that overhangs into the metal powder. Reactions were performed at −65 °C and 2 min residence time. A 0.5 M solution of diphenylacetonitrile in THF was pumped at a flow rate of 0.8 mL min−1, the n-BuLi was fed into the system at rate of 0.36 mL min−1, a 50 s residence time was obtained in the first zone which was combined with the fluoroform gas at a rate of 26.7 mL min−1. The residence time in the second zone was 1 min, an overall yield of 81% was achieved with a production rate of 79 mg min−1.114
One of the first microchannel reactors plated with an immobilized catalyst was developed by Kobayashi and co-workers in 2003. They conducted several alkene hydrogenations and debenzylations, which proceeded efficiently to afford the desired products 135 in a quantitative yield within 2 minutes. They selected a microchannel reactor having a 200 μm channel width, 100 μm channel depth, and a length of 45 cm. They immobilized a Pd catalyst on the wall of the glass channel with a cross linking polymer. Yields of up to 97% were obtained (Scheme 32).115
 | ||
| Scheme 32 Immobilized palladium catalyst in a microchannel custom built for hydrogenation reactions. | ||
Bendova and co-workers custom built a microfluidic chip for a stereoselective hydrogenation reaction. The microchip consisted of two main inlet ports, a static mixer followed by a residence time loop, a third inlet port for a quencher, a second static mixer, and an outlet port. The total volume was 10 μL/19.5 μL, and provided residence times from 15–120 s for flow rates in the range from 5–20 μL min−1. The reactor was used to transform methylacetoacetate (MAA) to (R)-methylhydroxybutyrate in an ionic liquid [Tf2N]/MeOH/H2O phase, with great success at conversions above 97%. They further used the same microfluidic chip reactor to design the reversible biphasic arrangement for asymmetric hydrogenations of MAA.116
Tsanaktsidis and co-workers custom built a catalytic static mixer (CSM), for the synthesis of a key intermediate of the antimicrobial drug linezolid (Zyvox). The approach combined three-dimensional (3D) printing and additive manufacturing techniques. They obtained a general method for catalytic hydrogenations and the rapid production of 136 from the nitro substrate 137 without the need for catalyst removal or recovery, the reaction productivity proceeded with 317 g L−1 h−1. The CSM's consisted of metal inserts (150 mm × 6 mm), designed for optimal mixing, onto which the metallic catalyst was deposited using an axial flow cell and standard electroplating techniques. The tubular flow reactor itself is approximately the size of a loaf of bread and contains a set of 12 CSM's that were simply and easily inserted into the tubular reactor and operated in series inside the temperature-controlled housing. The reactor volume was approximately 39 mL, and a hydrogen gas line was attached at the front end of the reactor to facilitate gas–liquid mixing with the substrate (Scheme 33).117 They later reported another paper with the same 3D printed CSM, however in this paper the CSM was plated with either palladium or nickel catalyst, and the system was tested on the reductive amination of aldehydes and ketones to amides with moderate to excellent yields.118 Several static mixers have been reported under flow conditions for more details see.118–121
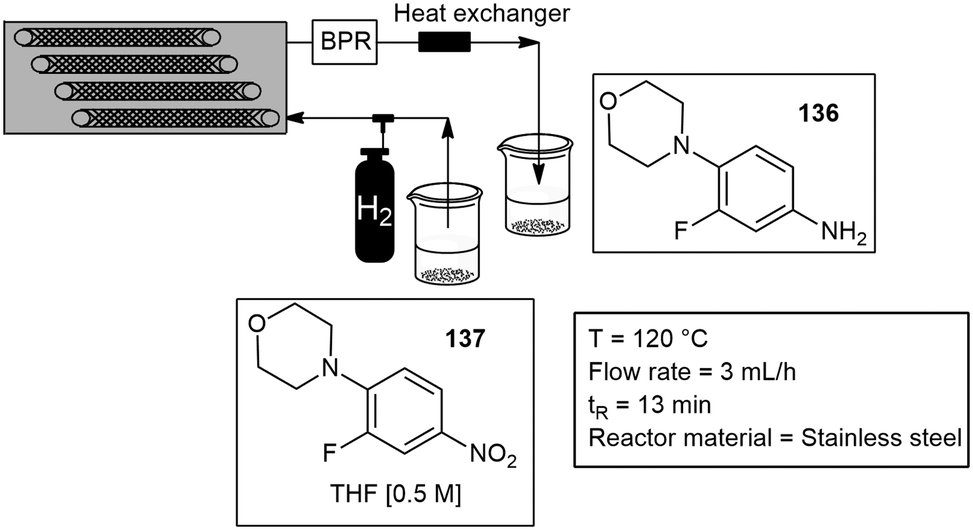 | ||
| Scheme 33 Static mixers for the hydrogenation of a nitro group to an amine. | ||
Microchannel membrane reactors. Kim and co-workers have developed multiple dual and triple channel photoreactors.122,123 The first microreactor consisted of two microfluidic channels separated by a thin PDMS semipermeable membrane, allowing the diffusion of gases from the gas channel into the liquid phase (Scheme 32). They first demonstrated an oxidative Heck reaction making use of arylboronic acids 138 and simple alkenes 139. The products 140 were produced in yields ranging from 75–82%. A year later they published a second reactor model with a triple channel system. The reactor was used for the photosensitized oxygenation of (−)-citronellol 141 to 142 and 143, the microfluidic channels were again separated by the thin PDMS membrane (Scheme 34). Oxygen was introduced in one channel and transported through the membrane towards the liquid channel. The latter channel was irradiated with a white LED light source. Several other membrane reactors with immobilized catalysts have also been developed.124,125
 | ||
| Scheme 34 A PDMS membrane reactor used in a basic Heck reaction as well as the photocatalytic oxygenation of (−)-citronellol. | ||
Tube-in-tube microreactors. The pioneering work by Steven Ley's group demonstrated the first tube-in-tube reactor, designed to deliver gaseous substance into a continuous flowing stream. The first papers were published between 2010 and 2011, demonstrating the tube-in-tube reactor with AF-2400 tubing, a semipermeable tubing allowing the diffusion of gasses into a continual flowing stream. The Teflon AF-2400 tubing was placed within the 1/8′′ OD, PTFE tube. This configuration allowed the flow of a substrate stream within the membrane tubing whilst the gas filled the PTFE outer tubing and diffusion transfers the reactive gas into the substrate stream. The Teflon AF-2400 (1.0 m, 0.28 mL) and the outer PTFE tubing was separated by SS T-pieces (Fig. 4).34,126,127 With this they demonstrated the carboxylation of Grignard reagents under flow conditions. Several more papers have since been published demonstrating this technology.12,126,128–132 Uniqsis Ltd. and Vapourtec Ltd. have also adapted this technology for commercial tube-in-tube reactors.
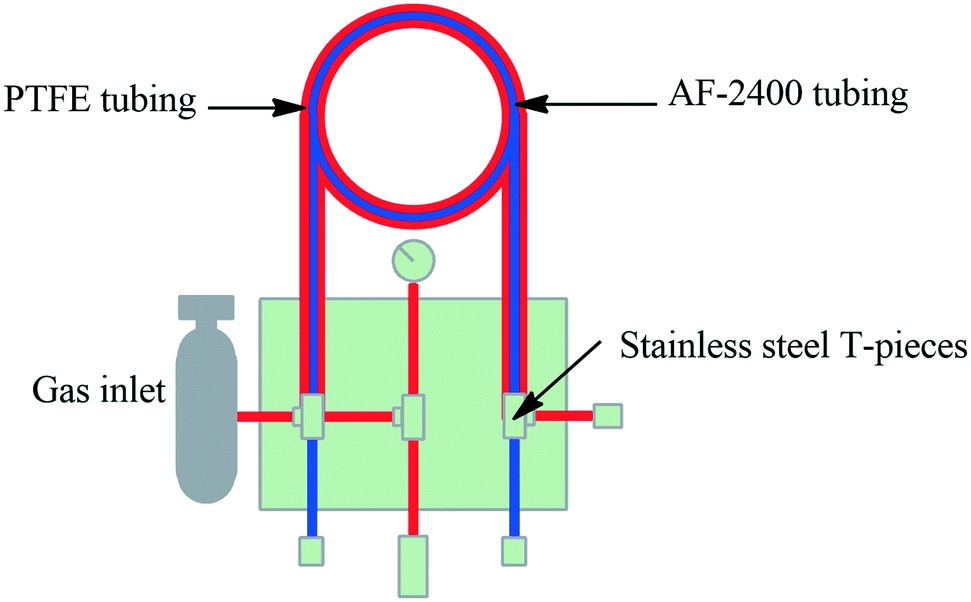 | ||
| Fig. 4 Semipermeable tube-in-tube reactor for gaseous addition to a continuous flowing stream. | ||
Before the innovative design of the tube-in-tube gas module, Steven Ley's group demonstrated the principle with a continuous flow ozonolysis reaction. The semi-permeable Teflon AF-2400 tubing was coiled inside a standard Schott bottle and pressurised with ozone gas. The liquid stream was saturated with ozone gas by means of diffusion. The concept was proven successful by demonstrating the ozonolysis reaction on 11 different alkenes 144 with yields ranging from 57–95% for the equivalent aldehydes and ketones 145 (Scheme 35).130
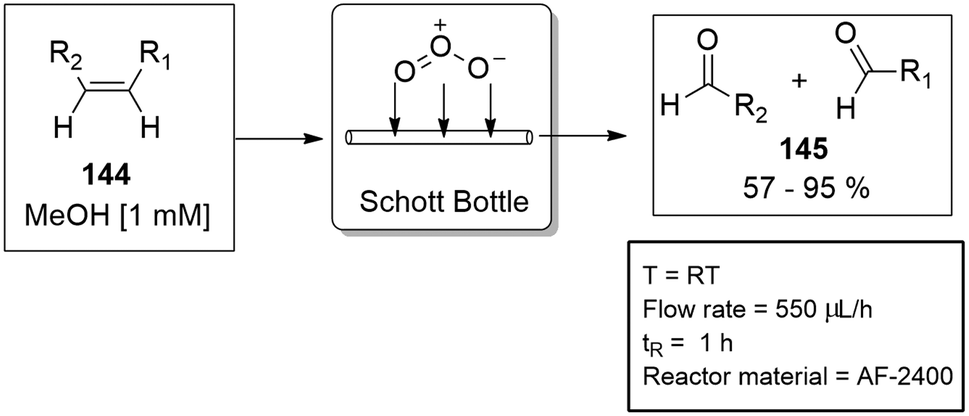 | ||
| Scheme 35 The ozonolysis of various alkenes using AF-2400 semipermeable tubing. | ||
Falling film reactor. The falling film reactor is based on a liquid film forming due to a liquid feed falling under gravity against a solid microstructured surface. The gas phase then flows co-currently or counter-currently in front of the liquid phase. The principle of falling film microreactors have been adapted and utilized by several flow chemistry research groups over the past few years. Recently Chen and co-workers133 designed a novel double-side falling film micro-channel contactor, and investigated the absorption coefficient of carbon dioxide in water. A similar design was shown by Steinfeldt and co-workers134 investigating the cyanide degradation via ozone absorption as well carbon dioxide absorption. The reaction plate was fabricated with 64 microchannels of 100 mm (length), 0.9 mm (width) and 0.5 (depth). The wall separating the channels had a width of 1 mm. Janisch and workers designed a practical falling film reactor, successfully synthesizing the important intermediates of vitamin D analogues 146. The reaction sequence consisted of an ozonolysis step and a subsequent reduction (Scheme 36).
 | ||
| Scheme 36 Vitamin D analogue preparation via ozonolysis in flow. | ||
Recently Rehm and co-workers demonstrated the value of falling film reactors in a photocatalytic application by applying external irradiation to the falling film microreactor. The reactor consisted of a back plate carrying a heat exchanger, as well as an inlet and outlet for the reaction solution which is fed through the falling film by gravity. A front plate with an inspection window constructed out of borosilicate glass and a middle plate (the reaction plate) constructed of microchannels with different geometries. LED's were installed and connected to cover the front inspection window. The microstructured falling film reactor was applied to the dye-sensitized photochemical conversion of 1,5-dihydroxynaphthalene to juglone.135 Several examples of photocatalytic falling film reactors have since been developed and reported on.136–138
In 2018 Krtschil and co-workers developed a falling film microreactor to study heterogeneously catalysed gas–liquid reactions under MW irradiation. They developed a fully automated pilot scale procedure for semi-hydrogenations. The microchannels of the reaction plate were coated with a novel selective catalyst. A special fibre-optical sensor was also developed to control the liquid level in the sump of the microwave reactor. With this they demonstrated the semi-hydrogenation of triple bonds to double bonds, avoiding over-reduction to single bonds (Scheme 37).139
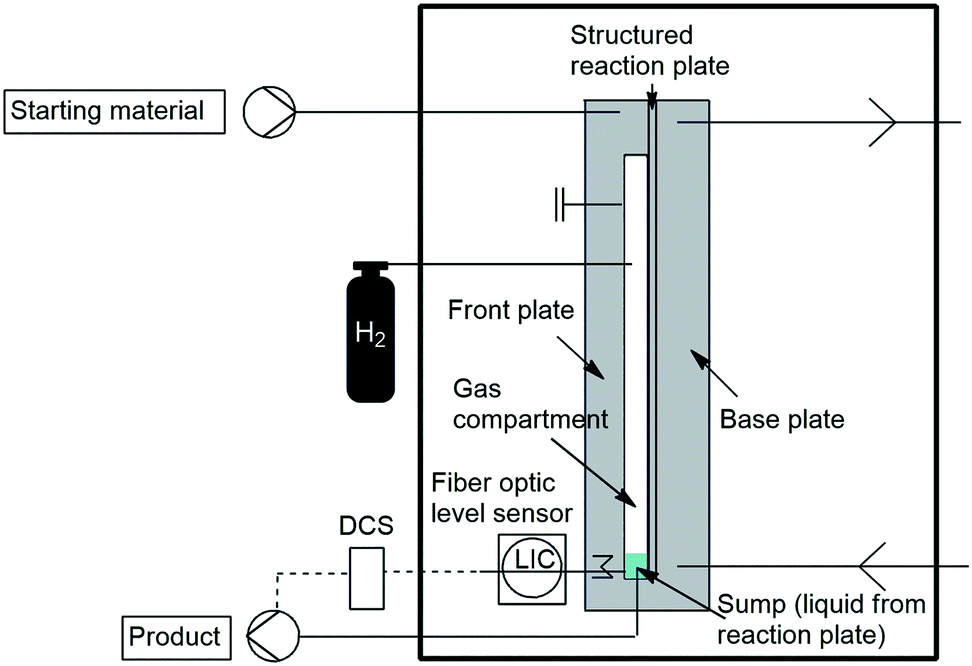 | ||
| Scheme 37 Microwave transparent catalytic falling-film microreactor for automated operation. | ||
 | ||
| Fig. 5 Solid phase quartz reactor design for triphasic reactions. | ||
Wenn and co-workers have used a mesh microreactor design to introduce a gas and liquid flow stream. Reactor enclosures defining chambers on each side of the mesh were formed from milled glass and metal components. They validated their reactor design by performing the hydrolysis of colourless fluorescein diacetate in toluene. Micromeshes with a pore diameter, depth, and spacing each of 5 μm were formed by electrodeposition.141
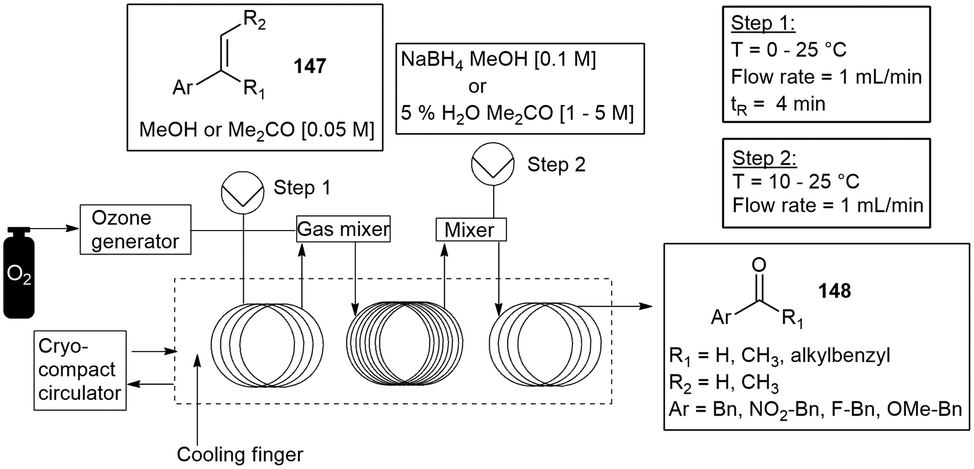 | ||
| Scheme 38 The ozonolysis of various aromatic alkenes with the ThalesNano O-Cube. | ||
The pioneering work of Steven Ley and co-workers set the bar for commercial tube-in-tube reactors which has been quickly adapted by commercial flow companies. Uniqsis Inc. has named their tube-in-tube reactor the gas addition module (GAM), also constructed with AF-2400 tubing, the gas stream and the liquid flow stream flowed in opposite directions, with the gas phase in the inner tube and the liquid in the surrounding tube. Kappe and co-workers identified the GAM as a potential tool to perform safe and scalable gaseous reactions with hazardous diazomethane gas. They performed various reactions which included methylations of various nucleophiles; [2,3]-cycloadditions and cyclopropanation of alkenes. Yields ranging from 59–99% were reported. Brzozowski and co-workers covered an extensive review on various reactions performed under continuous flow conditions with the GAM as well as AF-2400 tube-in-tube reactors.12
Vapourtec Inc. has also launched their version of the tube-in-tube reactor fabricated with AF-2400 tubing. The gas–liquid reactor fits into the standard glass manifold and is seen to be compatible with all existing R-Series systems. The liquid phase is fed through a coil, with a connection for the gas phase pressure from a regulated supply. Huvaere and co-workers developed a method to perform photooxidations and they validated the reactions using a model system, the photooxidation of ethyl 3-(2-furyl)propanoate. The reaction was further optimized for optimal conversion.56
A second gas–liquid reactor was designed by Vapourtec Inc., a mixed flow cooled reactor module, and its efficiency was demonstrated with the ozonolysis of various alkenes. The gas reactor was coupled with a cooled flow cell to perform the ozonolysis of n-decene 149. The reactions were carried at flow rates of 0.25–1 mL min−1, and temperatures of −10 °C. Residence times varied between 1–80 s, with 150 produced in 75–80% yield, and the by-product 151 in 15–20% (Scheme 39).143
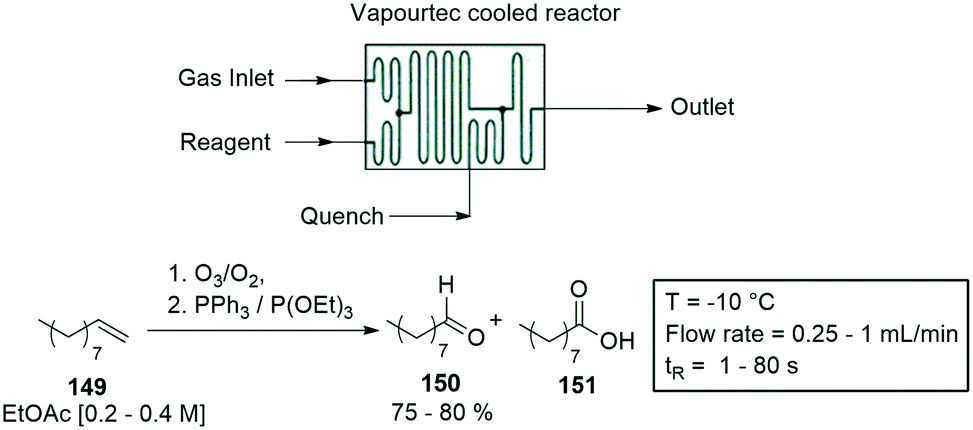 | ||
| Scheme 39 Ozonolysis of alkenes using a commercially available Vapourtec cooled reactor cell. | ||
5 3D printing in flow chemistry
3D printing, also called additive manufacturing (AM) or rapid prototyping (RP), is a layer-by-layer manufacturing method and has been widely used in many areas such as organ printing, aerospace and industrial design. In this section, we focused on the advances of 3D printed microfluidic chips especially modified for use in continuous flow chemistry. Based on a brief overview of different 3D printing methods and materials, we have chosen to highlight a few papers describing some of these method applications in microfluidics here.10,144,145Cronin and co-workers designed two custom made flow systems and utilised two types of organic reactions which included an imine synthesis and an imine reduction. Both reactors consisted of two inlets and a single outlet. The approximate volume of the first reactor was 0.4 mL and was employed in the imine syntheses, while the second reactor had a volume of 0.35 mL and was used in the imine reduction processes. Both reactors were fabricated from polypropylene (PP), a thermopolymer that is inert in a range of organic solvents and organic compounds.144,147
Hilton and co-workers followed with another PP 3D printed continuous flow module. The reactor module was designed as a circular disk reactor CDR to easily fit onto any commercial stirrer plate. The custom-built reactors were 75 mm wide and 7 mm high with an internal 2 mm reactor channel. All of the reactors were printed on an Ultimaker 3, 3D printer using PP filament which resulted in an internal volume of 4.2 mL. The reactor was able to facilitate the formation of a range of ether derivatives 152 from aryl halides 153 and aromatic alcohols 154 with most proceeding with good to excellent yields of the product (Scheme 40). The number of CDR's were increased in series to obtain longer residence times and improved yields, with the flow rate being maintained at 0.33 mL min−1 and the temperature at 65 °C.148
 | ||
| Scheme 40 Circular disk reactor 3D printed using PP. | ||
A photoactive monomer was incorporated into a 3D printing resin, which was used to fabricate a 3D photosensitizing flow reactor, which was incorporated in the singlet oxygen synthesis reaction under visible light irradiation. A solution containing 2-furoic acid was pumped together with oxygen through the photoreactor at a flow rate of 0.1 mL min−1, irradiated at 420 nm and recycled for 5 h.149
Kim and co-workers implemented a numbering-up microreactor system composed of stainless steel and copper was fabricated by CNC machining and 3D printing. The reactor was used in a cost-effective manner in the high throughput synthesis of rufinamide 155, an anticonvulsant medicine. The reactor consisted of 25 coiled copper capillaries configured in parallel to achieve high throughput experimental studies. Total flow rates of 6, 12, and 24 mL min−1 were tested during the optimization of the azide–alkyne cycloaddition reaction. Reactants 2,6-difluorobenzyl azide 156 and propiolamide 157 were passed through the copper capillary reactor along with ascorbic acid 158 to afford rufinamide 155. The numbering-up approach allowed the scale-up of rufinamide 155 achieving a high productivity (31 g h−1) at 12 mL min−1 flow rate and 110 °C, which equated to roughly 25 times more product than that obtained using a single reactor with the same yield. The main body of the reactor was fabricated by CNC machining and the complex baffle disk as shown in Scheme 41 was fabricated by 3D printing.150
 | ||
| Scheme 41 Copper coiled reactor fabricated by using 3D printing and CNC machining. | ||
The use of 3D-printed stainless-steel metal microreactors (3D-PMR's), have also been gaining interest with the same research group Kim and co-workers fabricating a circular metal reactor with different cross-sectional geometries with the aim to control ultrafast intramolecular rearrangement reactions in a comparative manner. The 3D-PMR with circular channel flow paths demonstrates the improved controllability in rapid Fries-type rearrangement reactions. This is mainly due to the superior mixing efficiency to rectangular cross-section microchannels (250 μm × 125 μm) which has been confirmed based on computational flow dynamic simulations. The high mixing efficiency of 159, phenyllithium and the appropriate electrophile created by the 3D- PMR's has resulted in high conversions and yields being achieved for the Fries rearranged products with residence times of 333 μs (Scheme 42).151
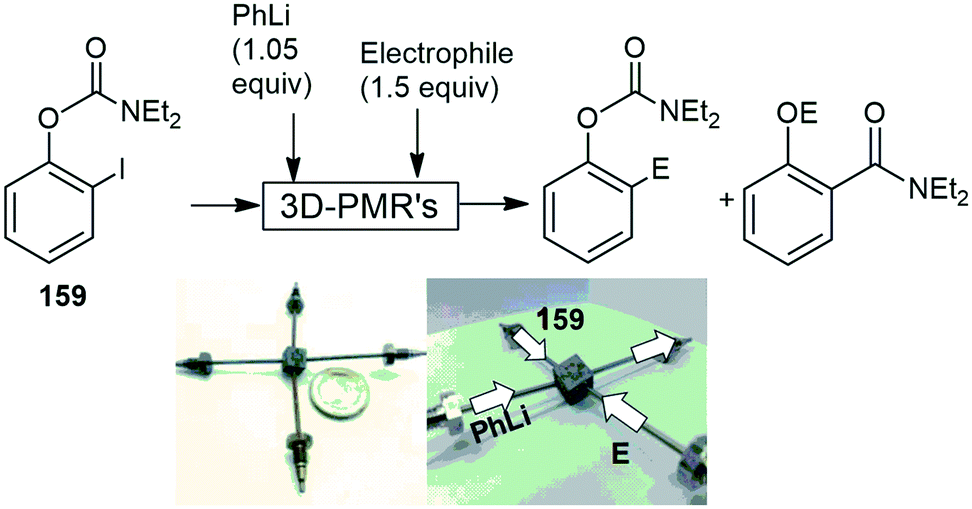 | ||
| Scheme 42 3D-printed stainless steel metal microreactor, utilized in a Fries rearrangement. Photograph reproduced with permission.151 | ||
 | ||
| Scheme 43 Solid phase column reactor printed for the synthesis of heterocycles. | ||
Cronin and co-workers printed a device designed to synthesize supramolecules on microliter scale. Metal-salt coordinating complexes were formed in MeOH. The PP device consisted of 3 inlets and a single outlet which was fed into a T-piece specifically designed to split the stream to collect and separate samples for ESI-MS real time analysis.147 Similarly Gong and co-workers have printed several microreactor components such as valves, pumps and multiplexers from a specially formulated resin consisting of a monomer, photo initiator and absorber, which allows for longer and more durable use.153
6 Advanced computer technology
Advanced computer technologies have led to the integration of software and technologies for performing multi-step synthesis under flow conditions. Notable the Ley group have been key players in this field of research with a particular focus on advocating for machine assisted continuous flow chemistry.154 They have envisioned the many possibilities of this technology and integrated it into synthetic procedures of small organic molecules and API's.To date there are several reports detailing how computer technologies have been integrated in enhancing the processing of small organic molecules and pharmaceutical compounds in end-to-end multistep processes. These include examples of algorithmic chemical search, whereby a range of reaction types could be accessed and, thus, the flexibility of these single, compact flow designs may be revealed. Another example includes automation of flow systems which allow several reactions to take place on the same system, enabling direct comparison of reactions under different conditions. Moreover, the production of new and known target compounds can be made faster and more efficient, the recipes of which can then be stored as digital files. Some of the automating software has employed machine-powered learning to assist the chemist in developing intelligent algorithms and artificial intelligence (AI) driven synthetic route planning. This ultimately produces a continuous flow platform that can design its own viable pathway to synthesize a particular molecule.
Commonly available software platforms used to control reagents, hardware modules and analytics in a user-friendly manner include MATLAB and LabVIEW. These software's allow remote progress monitoring. Which is also compatible with optimisation algorithms, e.g. flexibility and generality.
 | ||
| Scheme 44 Liquid–liquid separations using computer aided technology. | ||
 | ||
| Scheme 45 Automated flow synthesis of piprazine-2-carboxamide. | ||
They further harnessed the power of the cloud and remote servers to demonstrate the synthesis of tramadol, lidocaine and bupropion in a sine automated fashion from across the world.162 This automated control system was shown to be unencumbered by location or time domain. The control system had facilitated the rapid transition from an unexplored route to an optimized and telescoped process within four working days. Bupropion was synthesized at a rate of 2.88 g h−1. O'Brien and co-workers have also successfully demonstrated the use of open-source software technologies in the synthesis of several benzamides, an improved automation system with an autosampling/product-collection system was described. The software was designed to select and load starting materials as well as collect products into separate collection flasks.163
Aka and co-workers have recently described a powerful automated flow-based strategy for identifying and optimizing a cobalt-catalyzed oxidizing system for the bio-inspired dimerization of 2-propenyl phenols. They designed a reconfigurable flow reactor capable of online monitoring and process control. It was implemented as an automated screening platform to evaluate a matrix of 4 catalysts and 5 oxidants (plus the blank) at two different temperatures, resulting in an array of 50 reactions.164
7 In-line purification/downstream processing technologies in flow
Downstream processing and purification under flow conditions is typically characterised by chemical differentiation between reagents, products and by-products, and today the use of scavengers and catch-and-release approaches are commonplace in the field. The translation of more traditional purification techniques such as extraction, recrystallisation, distillation, filtration and chromatography which are largely based on the physical properties of molecules in large continue to prove challenging under flow conditions. The development of engineering solutions to overcome these shortcomings have seen growth in recent years as the drive to develop more integrated and truly continues processes has increased.1 Several pertinent examples of how these processes are being integrated into flow systems are discussed below including, solvent swopping approaches, liquid–liquid extraction devices, in-line crystallizations and triturations, in-line distillations, and in-line filtrations.Ley and co-workers demonstrated an elegant solution through their use of a bespoke meso-fluidic evaporator wherein a reagent stream is aspirated to afford a high surface area spray inside a heated column module which is exposed to a desolvation gas that rapidly removes the solvent (Fig. 6).165 The approach allows for rapid evaporation/concentration of the reagent stream and it can also be utilised to effect solvent swops through the introduction of a secondary solvent prior to entry into the evaporator. In this respect, the selectively removal of the lower vapour pressure solvent can be achieved. The group utilised the approach during the synthesis of the neurotensin receptor-1, Meclinertant (SR48692) in which they were able to successfully swop a toluene for methanol.166
 | ||
| Fig. 6 Ley groups meso-fluidic aspirating evaporator (left) and single stage distillation column (right). Photographs reproduced with permission.160,164 | ||
The Ley group also developed a means of swopping solvents based on their boiling points using a single stage distillation column. In this instance PTFE tubing was wrapped around an Omnifit® column reactor and the end of the tubing was then directed into the top of the column module (Fig. 6).161 The column was mounted into a heated (90 °C) jacket from a VapourTec R4+ unit. A solvent mix, dichloromethane and ethanol in the case reported were pumped into the coiled tubing. The lower boiling point solvent, dichloromethane rapidly boiled in the tubing ejecting the ethanol enriched reaction mixture into the centre of the column. The reaction mixture then exited the column out the bottom end, pumped directly into the next stage using a peristaltic pump.
In a similar vein Gruber-Woelfler and co-workers during the development of an approach for the continuous crystallisation of vitamin D3 developed an evaporative solvent concentrator wherein a reagent stream is allowed to drop into a vertical heated column equipped with an internal thermocouple.167 The column is fitted with a side-arm which is attached to a vacuum controller maintaining an internal pressure of 280 mbar. In this instance introduction of a mixed solvent system allows selective droplet evaporation of the solvent with the lower boiling point.
In the first instance, as described earlier in this review Ley and co-workers have on several occasions demonstrated the use of in-line gravity-based extractors to effect liquid–liquid extractions, the operation of which has been automated using computer vision to track the solvent interface.159 Adamo and co-workers fabricated an automated gravity-based separator that could be used in-line with continuous manufacturing, along with various filtration and crystalizing units. The gravity-based separator consisted of a Dean-Stark tube and a capacitive sensor that monitors the interface between the organic and aqueous streams (Fig. 7). The aqueous stream outlet was controlled to allow for complete separation between the aqueous stream and the organic streams.
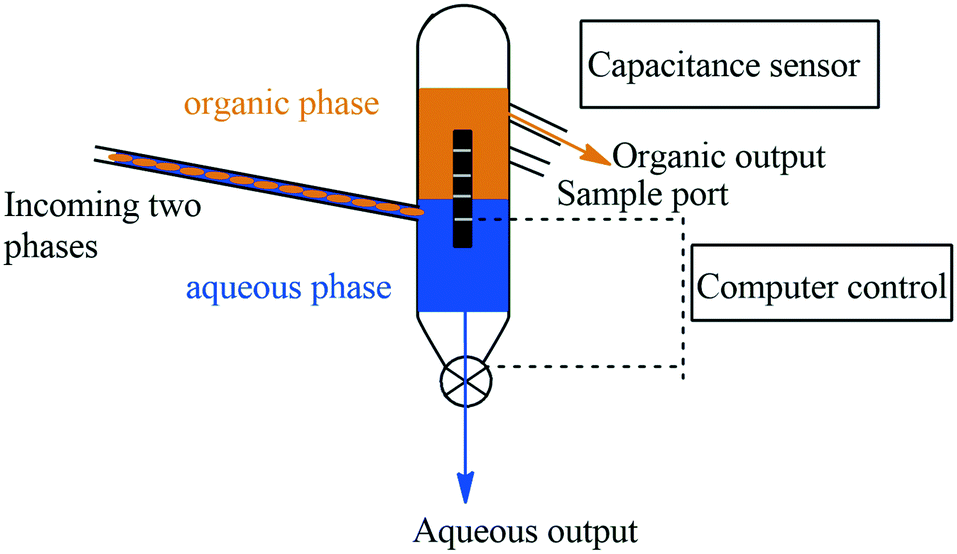 | ||
| Fig. 7 An automated gravity-based separator by Adamo and co-workers. | ||
The group also designed the well-known Zaiput liquid–liquid and gas–liquid in-line membrane-based separator. The separator was originally highlighted during the synthesis of fluoxetine hydrochloride and was designed to be chemically compatible by machining the wetted parts in perfluorinated polymers ethylene tetrafluoroethylene (ETFE) for the internal structure, PTFE for the porous membrane and PFA for the impermeable diaphragm of the pressure controller (Fig. 8).3
 | ||
| Fig. 8 Zaiput membrane separator by Adamo and co-workers. Photograph reproduced with permission.3 | ||
Syrris as part of their ASIA flow reactor series also developed a membrane-based separator called the FLEXX (flow liquid–liquid extraction) in which organic and aqueous phases are mixed and pumped across a membrane allowing diffusion to occur before splitting the stream into their constituent parts.168 The membrane technology employed is notable in that it allows the separation of difficult to separate or miscible solvent systems such as THF and water (Fig. 9).
 | ||
| Fig. 9 Syrris Asia FLEXX. | ||
In most instances the use of miniaturized crystallization tanks allows the convenient crystallisation of material directly out of a flow reactor in a batch or semi-continuous manner.3 Adamo and co-workers developed one such crystallisation device which was implemented in the downstream processing of lidocaine hydrochloride, diazepam, fluoxetine hydrochloride and diphenhydramine hydrochloride. The unit is comprised of a 120 mL jacketed HDPE (high density polyethylene) crystallizer was equipped with a PTFE coated marine impeller. The module is equipped with a level sensor and the temperature is controlled with a thermoelectric liquid cooler (LC-200, TE Technology, Inc.) to provide sufficient cooling capacity ensuring cooling rates below room temperature (Fig. 10).
 | ||
| Fig. 10 In-line crystallization under flow conditions by Adamo and co-workers. Image reproduced with permission.3 | ||
Utilising a similar approach our own research group demonstrated the synthesis of clozapine with in-line purification mediated in a semi-continuous fashion through the use of a trituration/solvent exchanger chamber.171 The process allowed for the trituration of two intermediate products during the four-stage synthesis. The in-line triturator (Fig. 11) was constructed as a double walled glass vessel fitted with a sintered glass frit, the outlet of which was connected to a vacuum pump and the jacketed vessel could be attached to a separate cooling/heating unit. The process was affected through the introduction of the reagent stream to the cooled triturator which was pre-primed with an anti-solvent. Upon completion of the trituration process the filtrate was removed from the vessel under vacuum filtration. The chamber was then re-primed with a solubilising solvent allowing the dissolution of the triturated material prior to it being pumped into the next reaction stage (Scheme 46).
 | ||
| Fig. 11 In-line double walled glass triturator. Photograph and image reproduced with permission.170 | ||
 | ||
| Scheme 46 The synthesis of clozapine with purification/solvent swopping using an in-line triturator. | ||
Scott and co-workers designed and demonstrated a system which allowed inline crystallisation in a truly continuous manner.172 The approach involved the use of a tri-segmented tubular crystallizer (KRAIC) in the form of three coil reactors (1/8′′ ID FEP tubing, 15 m) in which the reaction mixture is segmented with air and an immiscible carrier solvent allowing in-line crystallization. Notably the authors found it necessary to insert a section of jacketed tubing between the segmentor and the KRAIC which was cooled to 10 °C to ensure the rapid formation of large numbers of nuclei. This approach reduced crystallite size and was necessary to prevent blocking. The crystalliser was successfully used to purify pyrazinamide prepared upstream by reaction of pyrazinecarbonitrile with manganese (Scheme 47).
 | ||
| Scheme 47 In-line crystallization of pyrazinamide. | ||
In terms of the application of distillations in flow several purification techniques have been developed to afford solvent swopping under micro-evaporation conditions several of which have been described previously in this article.161,165–167 Hartman and co-workers used a novel approach when they designed a distillation device based upon a silicon microreactor with an integrated PTFE membrane (Fig. 12).173 They demonstrated the distillation device by implementing a Heck transformation with aryl triflates. These transformations are commonly associated with chlorinated solvents. The formation of the triflate was performed in DCM, after which toluene (or DMF) was pumped into the reactor system. The reactor was heated to 70 °C allowing the DCM to vaporize and the aryl triflate along with the toluene to pass through the reactor. Gas–liquid segmented flow was established combining a nitrogen feeding line into the reactor. The device was designed such that the liquid flowed through the integrated PTFE membrane and the vapour did not.
 | ||
| Fig. 12 Distillation device based upon a silicon microreactor with an integrated PTFE membrane. Image and photograph reproduced with permission.172 | ||
Although not an in-line distillation, Baumann and co-workers recently developed a platform that integrates reactive distillation with modern flow chemistry equipment.174 In this instance, a Hickmann distillation apparatus was used prepare pure cyclopentadiene by cracking. The distillate was collected at the rim of the top flask and was subsequently withdrawn via a side port in a continuous fashion. Several tetrahydroquinolines were generated through different aryl imines and cyclopentadiene were synthesized in this manner in yields of 75–90% (Scheme 48).
 | ||
| Scheme 48 The use of an in-line distillate apparatus for the synthesis of quinoline derivatives. | ||
Several reports have been described wherein a flow stream is directed into a series of simulated moving bed chromatography systems,175–178 however, the complexity of such systems and the technical limitations leave one largely restricted with regards to the separation of systems comprised of more than two components. An arguably more attractive approach is the use of centrifugal partition chromatography (CPC), such as was described by Greiner and co-workers wherein they were able to achieve a semi-continuous purification of aniline 169 which was produced in two steps under flow conditions (Scheme 49).179 The final product stream was continuously collected and diluted with appropriate solvents to afford an appropriate biphasic liquid system prior to being pumped into a CPC column in a portion-wise fashion. Fortuitously, aniline 169 could readily be separated from by-products 170 and 171 in both ascending (eluted first) and descending (eluted last) mode. As such the purification protocol was readily automated, an initial sample injection was run in descending mode allowing the by-products to be washed out, thereafter a second sample injection occurred and the process was run in ascending mode allowing the isolation of the product 169 in 99.9% purity. The process was then repeatedly reverse cycled ultimately affording a 91% recovery of 169 after 5 h.
 | ||
| Scheme 49 Inline column chromatography demonstrated with a cross coupling reaction and hydrogenation [S(L) – lower layer, S(U) – upper layer]. | ||
The Ley group in their efforts to purify more complex reaction matrices developed a semi-continuous supercritical fluid chromatography (SFC) system which notably allowed the isolation of material that did not elute either first or last and furthermore the group was able to successfully integrate the purification system between two discrete reactions (Scheme 50).180 The system operated by allowing a crude reaction mixture to be continuously recirculated through the loading loops of four independent Rheodyne injection valves and each valve was in turn connected to a separate column housed in a heating oven. Switching a valve to its injection orientation allowed a SFC system to pump a sample of the crude mixture through the column whereafter a selection valve could be automated to allow the collection of up to five fractions.180 The use of super critical carbon dioxide necessitated the need to separate the reaction mixture or purified fractions from gaseous carbon dioxide at points where system depressurization occurred, as such cyclone separators were inserted within the crude reaction mixture circulating loop directly after the Rheodyne injection valves, and after each of the final selection valves. The system was automated in such a manner as to try stagger the injections between the Rheodyne valves to allow an almost constant output stream of material.
 | ||
| Scheme 50 The use of super critical carbon dioxide for in-flow separations. | ||
8 Specialised flow reactors
Lastly, we report on specialized flow reactors, designed for specific reaction applications or as a general reactor.A few examples of microreactor etching are described below with Newman and Jensen demonstrating Wolff Kishner type reductions using a SiC etched microreactor. The reactor was designed to ensure general applicability to any reaction, which included two inlets, a quench line and an outlet. The cooled inlet and outlet zones were thermally separated from a spiral heated zone by a halo etch. The trapezoid-shaped channels amounted to an approximate 470 μL internal volume. The microreactor demonstrated good corrosion resistance, typically problematic in glass and steel microreactors. Benzophenone 172 was reduced using hydrazine, carbitol and NaOH with a residence time of 5 min obtaining 173 in a 75% yield (Scheme 51).182,183 Several more carbonyl compounds were reduced with yields ranging from 76–96%.184
 | ||
| Scheme 51 Silicon carbide microreactor custom built for general type applications. | ||
Aminolysis of epoxides were performed at high temperatures and pressures with a silicon microreactor fabricated by deep reactive ion etching to form square cross section channels (400 μm on the side), amounting to a reactor volume of 120 μL. The inlet and outlet section of the reactor was compressed in an aluminum fluidic interface. They performed several epoxide aminolysis reactions, starting with the epoxide 174 and aniline 175 to form alcohol 176 (Scheme 52). During this reaction it was found that two other products could also be formed. Reactions were completed in under 5 min with excellent yields and conversions ranging from 66–84%.185,186 The kinetics and scale-up of the epoxide aminolysis was investigated in a separate paper. The kinetic investigations showed that the activation energy for the formation of the desired product is observed to be higher than those for regio isomer formation and for secondary aminolysis, indicating that increasing temperature improves selectivity in addition to accelerating the reaction. A scale-up of this reaction was performed in a 12.5 mL steel tube mesoreactor, and 9 g of the product was produced in 30 min residence time.186
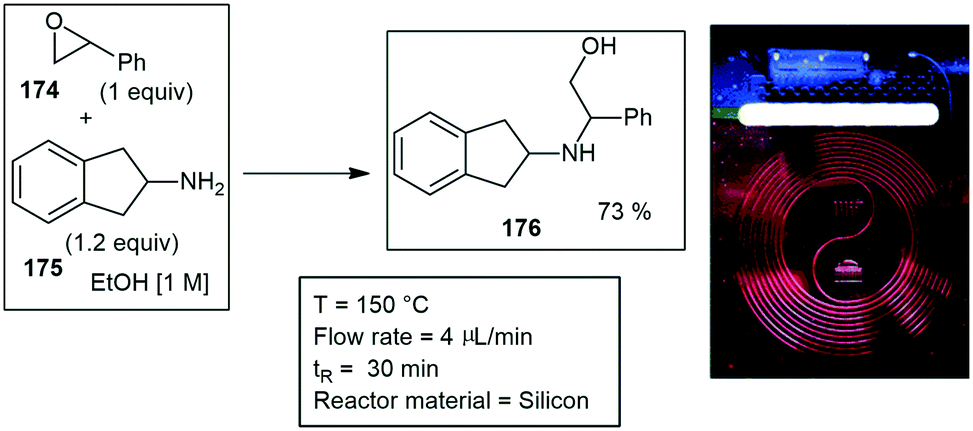 | ||
| Scheme 52 Aminolysis of epoxides and amines. Photograph reproduced with permission.146 | ||
Reports of rapid flow chemistries aimed at performing reactions on millisecond time scales has also lend itself to specialised flow reactors designed to perform these ultra fast reactions. Kim and co-workers have reported a compact reaction module on a pad (CRP) with the following diameters, 170 × 170 × 1.2 mm. The CRP was utilized in the synthesis of α-phosphonyloxy ketone 177, an invaluable drug precursor. The basic architecture of the CRP is a channel for integrated micro-flow circuits between two banks of reactors, each bank containing a number of microreactors. The CRP system was devised by stacking 9 films of the patterned polyimide to integrate micro-flow circuits, combining the functions of the even distribution of feeds, being completely mixed in less than a few milliseconds. The drug precursor was synthesized in a few milli seconds equating to a production rate of 19.2 g h−1 of the drug precursor 177. The following reaction conditions were used: 0.067 M of 178 in toluene with a flow rate of 3 mL min−1 and a residence time of 3.93 s, which resulted in 67% yield of 177 (Scheme 53).187
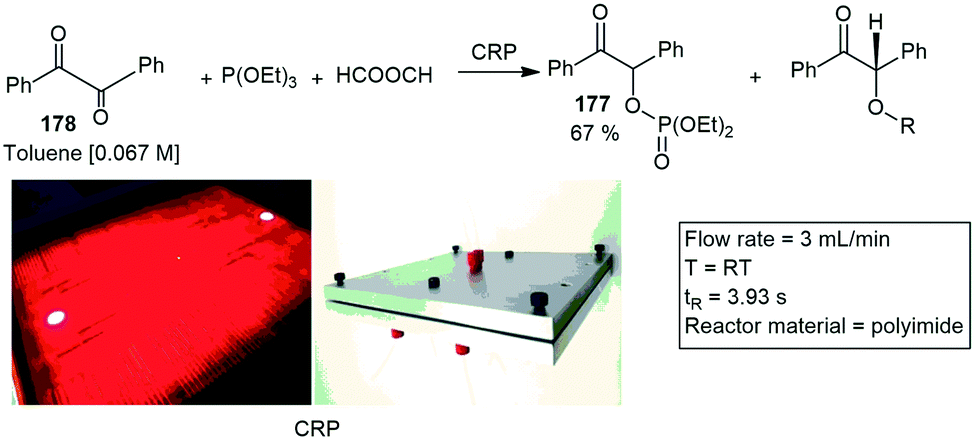 | ||
| Scheme 53 The use of super critical carbon dioxide for in-flow separations. Photograph reproduced with permission.187 | ||
Yoshida and co-workers being most prominent in the field of rapid microflow chemistries has recently synthesized afesal a bioactive molecule utilizing a polymer-based chip microreactor (CMR) with a 3D serpentine microchannel design. The internal volume of the CMR, 25 nl, is provided by the rectangular serpentine channels (width 200 mm, height 125 mm, length 1 mm). The Fries rearrangement was achieved on a sub millisecond time scale, providing a straightforward synthetic route for afesal. The rapid continuous flow nature of the CMR delivered a throughput of the product in 5.3 g h−1. Thus, we can assume by stacking multiple small and portable CMR systems, which individually fit in one hand, would have great potential for scalable industrial chemical synthesis.188
Conclusion
Traditional organic synthesis methods are rapidly changing, and currently environmental concerns and impacts created by chemical manufacturers, and in particular by Big Pharma are being more heavily scrutinized than ever before. In today's world there is a drive for cleaner, safer, economical and conscientious chemical synthesis and manufacturing and this has in part been responsible for the rapid rise of flow chemistry and related technologies.Although flow chemistry, in this the age of the fourth industrial revolution, is often seen as being the next evolutionary step in chemical manufacturing human resources are still just as important as material use, however, new these technologies and machines are transforming the way in which laboratory chemists work. Current evidence suggests a future in which the lines between chemistry and engineering are set to become more and more integrated from early on in process development and where the manufacture of chemical entities under continuous conditions is commonplace. That being said, despite some of the truly outstanding developments in the field much work still needs to take place in order to realise this. In particular, the development of truly continuous multi-step syntheses with integrated downstream processing and automation is today still challenging and by no means straightforward.
We hope that the works reported in this review paint a picture that serves as inspiration to synthetic chemists showing the vast scope of chemistries that now find themselves part of the flow chemistry family and highlight how the adoptation of new technologies can benefit the modern chemist.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of South Africa (grant number UID87893 and UID106959), the University of Pretoria (University, Science Faculty Research Councils and Research and Development Program), South Africa and Pelchem Pty Ltd. Opinions expressed in this publication and the conclusions arrived at, are those of the authors, and are not necessarily attributed to the NRF.References
- S. V. Ley, D. E. Fitzpatrick, R. J. Ingham and R. M. Myers, Organic synthesis: march of the machines, Angew. Chem., Int. Ed., 2015, 54(11), 3449–3464 Search PubMed.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, The hitchhiker's guide to flow chemistry parallel, Chem. Rev., 2017, 117(18), 11796–11893 Search PubMed.
- A. Adamo, R. L. Beingessner, M. Behnam, J. Chen, T. F. Jamison, K. F. Jensen, J. C. Monbaliu, A. S. Myerson, E. M. Revalor, D. R. Snead, T. Stelzer, N. Weeranoppanant, S. Y. Wong and P. Zhang, On-demand continuous-flow production of pharmaceuticals in a compact, reconfigurable system, Science, 2016, 352(6281), 61–67 Search PubMed.
- https://www.worldofchemicals.com/media/iupac-names-top-10-emerging-technologies-in-chemistry/13101.html, (accessed 20-12- 2020).
- A. Macchi, P. Plouffe, G. S. Patience and D. M. Roberge, Experimental methods in chemical engineering: Micro-reactors, Can. J. Chem. Eng., 2019, 97(10), 2578–2587 Search PubMed.
- C. O. Kappe, My twenty years in microwave chemistry: From kitchen ovens to microwaves that aren't microwaves, Chem. Rec., 2019, 19(1), 15–39 Search PubMed.
- K. F. Jensen, Flow chemistry - Microreaction technology comes of age, AIChE J., 2017, 63(3), 858–869 Search PubMed.
- T. Noel, Y. Cao and G. Laudadio, The fundamentals behind the use of flow reactors in electrochemistry, Acc. Chem. Res., 2019, 52(10), 2858–2869 Search PubMed.
- C. Sambiagio and T. Noël, Flow photochemistry: Shine some light on those tubes!, Trends Chem., 2020, 2(2), 92–106 Search PubMed.
- Y. He, Y. Wu, J.-Z. Fu, Q. Gao and J.-J. Qiu, Developments of 3D printing microfluidics and applications in chemistry and biology: A review, Electroanalysis, 2016, 28(8), 1658–1678 Search PubMed.
- C. A. Hone and C. O. Kappe, The use of molecular oxygen for liquid phase aerobic oxidations in continuous flow, Top. Curr. Chem., 2018, 377(1), 2 Search PubMed.
- M. Brzozowski, M. O'Brien, S. V. Ley and A. Polyzos, Flow chemistry: intelligent processing of gas-liquid transformations using a tube-in-tube reactor, Acc. Chem. Res., 2015, 48(2), 349–362 Search PubMed.
- S. V. Ley, Y. Chen, D. E. Fitzpatrick and O. May, A new world for chemical synthesis?, Chimia, 2019, 73(10), 792–802 Search PubMed.
- C. Strauss, A continuous microwave reactor for laboratory-scale synthesis, Chem. Aust., 1990, 186 Search PubMed.
- T. Cablewski, A. F. Faux and C. R. Strauss, Development and application of a continuous microwave reactor for organic synthesis, J. Org. Chem., 1994, 59(12), 3408–3412 Search PubMed.
- S.-T. Chen, S.-H. Chiou and K.-T. Wang, Preparative scale organic synthesis using a kitchen microwave oven, J. Chem. Soc., Chem. Commun., 1990,(11), 807–809 Search PubMed.
- E. Thostenson and T.-W. Chou, Microwave processing: fundamentals and applications, Composites, Part A, 1999, 30(9), 1055–1071 Search PubMed.
- E. Comer and M. G. Organ, A microcapillary system for simultaneous, parallel microwave-assisted synthesis, Chemistry, 2005, 11(24), 7223–7227 Search PubMed.
- E. Comer and M. G. Organ, A microreactor for microwave-assisted capillary (continuous flow) organic synthesis, J. Am. Chem. Soc., 2005, 127(22), 8160–8167 Search PubMed.
- G. Shore, S. Morin and M. G. Organ, Catalysis in capillaries by Pd thin films using microwave-assisted continuous-flow organic synthesis (MACOS), Angew. Chem., Int. Ed., 2006, 45(17), 2761–2766 Search PubMed.
- W. S. Bremner and M. G. Organ, Multicomponent reactions to form heterocycles by microwave-assisted continuous flow organic synthesis, J. Comb. Chem., 2007, 9(1), 14–16 Search PubMed.
- G. Shore, M. Tsimerman and M. G. Organ, Gold film-catalysed benzannulation by microwave-assisted, continuous flow organic synthesis (MACOS), Beilstein J. Org. Chem., 2009, 5, 35 Search PubMed.
- R. J. J. Jachuck, D. K. Selvaraj and R. S. Varma, Process intensification: oxidation of benzyl alcohol using a continuous isothermal reactor under microwave irradiation, Green Chem., 2006, 8(1), 29–33 Search PubMed.
- M. C. Bagley, R. L. Jenkins, M. C. Lubinu, C. Mason and R. Wood, A simple continuous flow microwave reactor, J. Org. Chem., 2005, 70(17), 7003–7006 Search PubMed.
- K. Engen, J. Sävmarker, U. Rosenström, J. Wannberg, T. Lundbäck, A. Jenmalm-Jensen and M. Larhed, Microwave heated flow synthesis of spiro-oxindole dihydroquinazolinone based IRAP inhibitors, Org. Process Res. Dev., 2014, 18(11), 1582–1588 Search PubMed.
- V. Konda, J. Rydfjord, J. Sävmarker and M. Larhed, Safe palladium-catalyzed cross-couplings with microwave heating using continuous-flow silicon carbide reactors, Org. Process Res. Dev., 2014, 18(11), 1413–1418 Search PubMed.
- M. Nishioka, M. Miyakawa, Y. Daino, H. Kataoka, H. Koda, K. Sato and T. M. Suzuki, Single-mode microwave reactor used for continuous flow reactions under elevated pressure, Ind. Eng. Chem. Res., 2013, 52(12), 4683–4687 Search PubMed.
- G. Pipus, I. Plazl and T. Koloini, Esterification of benzoic acid in microwave tubular flow reactor, Chem. Eng. J., 2000, 76(3), 239–245 Search PubMed.
- T. Ichikawa, M. Mizuno, S. Ueda, N. Ohneda, H. Odajima, Y. Sawama, Y. Monguchi and H. Sajiki, A practical method for heterogeneously-catalyzed Mizoroki–Heck reaction: Flow system with adjustment of microwave resonance as an energy source, Tetrahedron, 2018, 74, 1810–1816 Search PubMed.
- T. Ichikawa, T. Matsuo, T. Tachikawa, T. Yamada, T. Yoshimura, M. Yoshimura, Y. Takagi, Y. Sawama, J.-I. Sugiyama, Y. Monguchi and H. Sajiki, Microwave-mediated site-selective heating of spherical-carbon-bead-supported platinum for the continuous, efficient catalytic dehydrogenative aromatization of saturated cyclic hydrocarbons, ACS Sustainable Chem. Eng., 2019, 7(3), 3052–3061 Search PubMed.
- I. R. Baxendale, C. M. Griffiths-Jones, S. V. Ley and G. K. Tranmer, Microwave-assisted Suzuki coupling reactions with an encapsulated palladium catalyst for batch and continuous-flow transformations, Chemistry, 2006, 12(16), 4407–4416 Search PubMed.
- P. He, S. J. Haswell and P. D. Fletcher, Microwave heating of heterogeneously catalysed Suzuki reactions in a micro reactor, Lab Chip, 2004, 4(1), 38–41 Search PubMed.
- P. He, S. J. Haswell and P. D. I. Fletcher, Microwave-assisted Suzuki reactions in a continuous flow capillary reactor, Appl. Catal., A, 2004, 274(1–2), 111–114 Search PubMed.
- F. Benaskar, N. G. Patil, V. Engels, E. V. Rebrov, J. Meuldijk, L. A. Hulshof, V. Hessel, A. E. H. Wheatley and J. C. Schouten, Microwave-assisted Cu-catalyzed Ullmann ether synthesis in a continuous-flow milli-plant, Chem. Eng. J., 2012, 207–208, 426–439 Search PubMed.
- N. S. Wilson, C. R. Sarko and G. P. Roth, Development and applications of a practical continuous flow microwave cell, Org. Process Res. Dev., 2004, 8(3), 535–538 Search PubMed.
- S. Yokozawa, N. Ohneda, K. Muramatsu, T. Okamoto, H. Odajima, T. Ikawa, J.-i. Sugiyama, M. Fujita, T. Sawairi, H. Egami, Y. Hamashima, M. Egi and S. Akai, Development of a highly efficient single-mode microwave applicator with a resonant cavity and its application to continuous flow syntheses, RSC Adv., 2015, 5(14), 10204–10210 Search PubMed.
- M. Noshita, Y. Shimizu, H. Morimoto, S. Akai, Y. Hamashima, N. Ohneda, H. Odajima and T. Ohshima, Ammonium salt-accelerated hydrazinolysis of unactivated amides: Mechanistic investigation and application to a microwave flow process, Org. Process Res. Dev., 2019, 23(4), 588–594 Search PubMed.
- P. Vámosi, K. Matsuo, T. Masuda, K. Sato, T. Narumi, K. Takeda and N. Mase, Rapid optimization of reaction conditions based on comprehensive reaction analysis using a continuous flow microwave reactor, Chem. Rec., 2019, 19(1), 77–84 Search PubMed.
- B. Musio, F. Mariani, E. Śliwiński, M. A. Kabeshov, H. Odajima and S. V. Ley, Combination of enabling technologies to improve and describe the stereoselectivity of Wolff–Staudinger cascade reaction, Synthesis, 2016, 48(20), 3515–3526 Search PubMed.
- H. Egami, T. Sawairi, S. Tamaoki, N. Ohneda, T. Okamoto, H. Odajima and Y. Hamashima, (E)-3-[4-(Pent-4-en-1-yloxy)phenyl]acrylicc Acid, Molbank, 2018, 2018(2), M996 Search PubMed.
- J. P. Barham, S. Tamaoki, H. Egami, N. Ohneda, T. Okamoto, H. Odajima and Y. Hamashima, C-Alkylation of N-alkylamides with styrenes in air and scale-up using a microwave flow reactor, Org. Biomol. Chem., 2018, 16(41), 7568–7573 Search PubMed.
- H. Egami, S. Tamaoki, M. Abe, N. Ohneda, T. Yoshimura, T. Okamoto, H. Odajima, N. Mase, K. Takeda and Y. Hamashima, Scalable microwave-assisted Johnson–Claisen rearrangement with a continuous flow microwave system, Org. Process Res. Dev., 2018, 22(8), 1029–1033 Search PubMed.
- J. P. Barham, S. Tanaka, E. Koyama, N. Ohneda, T. Okamoto, H. Odajima, J.-I. Sugiyama and Y. Norikane, Selective, scalable synthesis of C60-fullerene/indene monoadducts using a microwave flow applicator, J. Org. Chem., 2018, 83(8), 4348–4354 Search PubMed.
- F. Bergamelli, M. Iannelli, J. A. Marafie and J. D. Moseley, A commercial continuous flow microwave reactor evaluated for scale-up, Org. Process Res. Dev., 2010, 14(4), 926–930 Search PubMed.
- J. Moseley and E. Woodman, Scaling-out pharmaceutical reactions in an automated stop-flow microwave reactor, Org. Process Res. Dev., 2008, 12, 967–981 Search PubMed.
- J. Moseley and E. Woodman, Energy efficiency of microwave- and conventionally heated reactors compared at meso scale for organic reactions, Energy Fuels, 2009, 23, 5438–5447 Search PubMed.
- R. Morschhäuser, M. Krull, C. Kayser, C. Boberski, R. Bierbaum, P. A. Püschner, T. N. Glasnov and C. O. Kappe, Microwave-assisted continuous flow synthesis on industrial scale, Green Processes Synth., 2012, 1(3), 281–290 Search PubMed.
- K. Gilmore and P. H. Seeberger, Continuous flow photochemistry, Chem. Rec., 2014, 14(3), 410–418 Search PubMed.
- A. Nakamura, K. Yoshida, S. Kuwahara and K. Katayama, Photocatalytic organic syntheses using a glass-milled microchip, J. Photochem. Photobiol., A, 2016, 322–323, 35–40 Search PubMed.
- Z. Meng, X. Zhang and J. Qin, A high efficiency microfluidic-based photocatalytic microreactor using electrospun nanofibrous TiO2 as a photocatalyst, Nanoscale, 2013, 5(11), 4687–4690 Search PubMed.
- D. Rackl, P. Kreitmeier and O. Reiser, Synthesis of a polyisobutylene-tagged fac-Ir(ppy)3 complex and its application as recyclable visible-light photocatalyst in a continuous flow process, Green Chem., 2016, 18(1), 214–219 Search PubMed.
- R. S. Gonçalves, B. R. Rabello, G. B. César, P. C. S. Pereira, M. A. S. Ribeiro, E. C. Meurer, N. Hioka, C. V. Nakamura, M. L. Bruschi and W. Caetano, An efficient multigram synthesis of hypericin improved by a low power LED based photoreactor, Org. Process Res. Dev., 2017, 21(12), 2025–2031 Search PubMed.
- F. R. Bou-Hamdan and P. H. Seeberger, Visible-light-mediated photochemistry: accelerating Ru(bpy)32+−catalyzed reactions in continuous flow, Chem. Sci., 2012, 3(5), 1612–1616 Search PubMed.
- R. Telmesani, S. H. Park, T. Lynch-Colameta and A. B. Beeler, [2+2] Photocycloaddition of cinnamates in flow and development of a thiourea catalyst, Angew. Chem., Int. Ed., 2015, 54(39), 11521–11525 Search PubMed.
- J. W. Tucker, Y. Zhang, T. F. Jamison and C. R. Stephenson, Visible-light photoredox catalysis in flow, Angew. Chem., Int. Ed., 2012, 51(17), 4144–4147 Search PubMed.
- A. Kouridaki and K. Huvaere, Singlet oxygen oxidations in homogeneous continuous flow using a gas–liquid membrane reactor, React. Chem. Eng., 2017, 2(4), 590–597 Search PubMed.
- B. Wriedt, D. Kowalczyk and D. Ziegenbalg, Experimental determination of photon fluxes in multilayer capillary photoreactors, ChemPhotoChem, 2018, 2(10), 913–921 Search PubMed.
- L. D. Elliott, M. Berry, B. Harji, D. Klauber, J. Leonard and K. I. Booker-Milburn, A small-footprint, high-capacity flow reactor for uv photochemical synthesis on the kilogram scale, Org. Process Res. Dev., 2016, 20(10), 1806–1811 Search PubMed.
- L. D. Elliott, J. P. Knowles, P. J. Koovits, K. G. Maskill, M. J. Ralph, G. Lejeune, L. J. Edwards, R. I. Robinson, I. R. Clemens and B. Cox, Batch versus flow photochemistry: A revealing comparison of yield and productivity, Chem. – Eur. J., 2014, 20(46), 15226–15232 Search PubMed.
- T. H. Rehm, C. Hofmann, D. Reinhard, H.-J. Kost, P. Löb, M. Besold, K. Welzel, J. Barten, A. Didenko and D. V. Sevenard, Continuous-flow synthesis of fluorine-containing fine chemicals with integrated benchtop NMR analysis, React. Chem. Eng., 2017, 2(3), 315–323 Search PubMed.
- N. J. Straathof, Y. Su, V. Hessel and T. Noel, Accelerated gas-liquid visible light photoredox catalysis with continuous-flow photochemical microreactors, Nat. Protoc., 2016, 11(1), 10–21 Search PubMed.
- N. J. Straathof, S. E. Cramer, V. Hessel and T. Noel, Practical photocatalytic trifluoromethylation and hydrotrifluoromethylation of styrenes in batch and flow, Angew. Chem., Int. Ed., 2016, 55(50), 15549–15553 Search PubMed.
- G. Wu, T. Lv, W. Mo, X. Yang, Y. Gao and H. Chen, One-pot synthesis of tricyclo-1,4-benzoxazines via visible-light photoredox catalysis in continuous flow, Tetrahedron Lett., 2017, 58(14), 1395–1398 Search PubMed.
- Y. Su, K. Kuijpers, V. Hessel and T. Noël, A convenient numbering-up strategy for the scale-up of gas–liquid photoredox catalysis in flow, React. Chem. Eng., 2016, 1(1), 73–81 Search PubMed.
- J. S. Babra, A. T. Russell, C. D. Smith and Y. Zhang, Combining C-H functionalisation and flow photochemical heterocyclic metamorphosis (FP-HM) for the synthesis of benzo[1,3]oxazepines, Tetrahedron, 2018, 74(38), 5351–5357 Search PubMed.
- B. Shen, M. W. Bedore, A. Sniady and T. F. Jamison, Continuous flow photocatalysis enhanced using an aluminum mirror: Rapid and selective synthesis of 2′-deoxy and 2′,3′-dideoxynucleosides, Chem. Commun., 2012, 48(60), 7444–7446 Search PubMed.
- S. Jang, S. Vidyacharan, B. T. Ramanjaneyulu, K.-W. Gyak and D.-P. Kim, Photocatalysis in a multi-capillary assembly microreactor: toward up-scaling the synthesis of 2H-indazoles as drug scaffolds, React. Chem. Eng., 2019, 4(8), 1466–1471 Search PubMed.
- K. C. Harper, E. G. Moschetta, S. V. Bordawekar and S. J. Wittenberger, A laser driven flow chemistry platform for scaling photochemical reactions with visible light, ACS Cent. Sci., 2019, 5(1), 109–115 Search PubMed.
- C. J. Kong, D. Fisher, B. K. Desai, Y. Yang, S. Ahmad, K. Belecki and B. F. Gupton, High throughput photo-oxidations in a packed bed reactor system, Bioorg. Med. Chem., 2017, 25(23), 6203–6208 Search PubMed.
- J. M. Tobin, T. J. D. McCabe, A. W. Prentice, S. Holzer, G. O. Lloyd, M. J. Paterson, V. Arrighi, P. A. G. Cormack and F. Vilela, Polymer-supported photosensitizers for oxidative organic transformations in flow and under visible light irradiation, ACS Catal., 2017, 7(7), 4602–4612 Search PubMed.
- D. Cambie, F. Zhao, V. Hessel, M. G. Debije and T. Noel, A leaf-inspired luminescent solar concentrator for energy-efficient continuous-flow photochemistry, Angew. Chem., Int. Ed., 2017, 56(4), 1050–1054 Search PubMed.
- D. Cambié, J. Dobbelaar, P. Riente Paiva, J. Vanderspikken, C. Shen, P. Seeberger, K. Gilmore, M. Debije and T. Noel, Energy-efficient solar photochemistry with luminescent solar concentrator based photomicroreactors, Angew. Chem., Int. Ed., 2019, 58, 14374–14378 Search PubMed.
- C. A. Clark, D. S. Lee, S. J. Pickering, M. Poliakoff and M. W. George, A simple and versatile reactor for photochemistry, Org. Process Res. Dev., 2016, 20(10), 1792–1798 Search PubMed.
- G. I. Ioannou, T. Montagnon, D. Kalaitzakis, S. A. Pergantis and G. Vassilikogiannakis, Synthesis of cyclopent-2-enones from furans using a nebulizer-based continuous flow photoreactor, Org. Biomol. Chem., 2017, 15(48), 10151–10155 Search PubMed.
- A. Chaudhuri, K. P. L. Kuijpers, R. B. J. Hendrix, P. Shivaprasad, J. A. Hacking, E. A. C. Emanuelsson, T. Noël and J. van der Schaaf, Process intensification of a photochemical oxidation reaction using a Rotor-Stator Spinning Disk Reactor: A strategy for scale up, Chem. Eng. J., 2020, 400, 125875 Search PubMed.
- M. Oelgemöller and D. Gurthie, Continuous-flow photochemistry made easy with Vapourtec's photoreactor series, 2020, vol. 97, pp. 38–42 Search PubMed.
- M. Baumann and I. R. Baxendale, Continuous photochemistry: the flow synthesis of ibuprofen via a photo-Favorskii rearrangement, React. Chem. Eng., 2016, 1(2), 147–150 Search PubMed.
- J. W. Beatty, J. J. Douglas, K. P. Cole and C. R. Stephenson, A scalable and operationally simple radical trifluoromethylation, Nat. Commun., 2015, 6, 7919 Search PubMed.
- I. Abdiaj and J. Alcazar, Improving the throughput of batch photochemical reactions using flow: Dual photoredox and nickel catalysis in flow for C(sp(2))C(sp(3)) cross-coupling, Bioorg. Med. Chem., 2017, 25(23), 6190–6196 Search PubMed.
- H.-W. Hsieh, C. W. Coley, L. M. Baumgartner, K. F. Jensen and R. I. Robinson, Photoredox iridium–nickel dual-catalyzed decarboxylative arylation cross-coupling: From batch to continuous flow via self-optimizing segmented flow reactor, Org. Process Res. Dev., 2018, 22(4), 542–550 Search PubMed.
- F. Lima, M. A. Kabeshov, D. N. Tran, C. Battilocchio, J. Sedelmeier, G. Sedelmeier, B. Schenkel and S. V. Ley, Visible light activation of boronic esters enables efficient photoredox C(sp2)–C(sp3) cross-couplings in flow, Am. Ethnol., 2016, 128(45), 14291–14295 Search PubMed.
- I. Abdiaj, A. Fontana, M. V. Gomez, A. de la Hoz and J. Alcazar, Visible-light-induced nickel-catalyzed Negishi cross-couplings by exogenous-photosensitizer-free photocatalysis, Angew. Chem., Int. Ed., 2018, 57(28), 8473–8477 Search PubMed.
- R. Xiao, J. M. Tobin, M. Zha, Y.-L. Hou, J. He, F. Vilela and Z. Xu, A nanoporous graphene analog for superfast heavy metal removal and continuous-flow visible-light photoredox catalysis, J. Mater. Chem. A, 2017, 5(38), 20180–20187 Search PubMed.
- T. C. Sherwood, H. Y. Xiao, R. G. Bhaskar, E. M. Simmons, S. Zaretsky, M. P. Rauch, R. R. Knowles and T. G. M. Dhar, Decarboxylative intramolecular arene alkylation using N-(acyloxy)phthalimides, an organic photocatalyst, and visible light, J. Org. Chem., 2019, 84(13), 8360–8379 Search PubMed.
- C. Bottecchia, R. Martín, I. Abdiaj, E. Crovini, J. Alcazar, J. Orduna, M. J. Blesa, J. R. Carrillo, P. Prieto and T. Noël, De novo design of organic photocatalysts: Bithiophene derivatives for the visible-light induced C−H functionalization of heteroarenes, Adv. Synth. Catal., 2019, 361(5), 945–950 Search PubMed.
- Y. Chen, D. C. Blakemore, P. Pasau and S. V. Ley, Three-component assembly of multiply substituted homoallylic alcohols and amines using a flow chemistry photoreactor, Org. Lett., 2018, 20(20), 6569–6572 Search PubMed.
- Y. Chen, D. Cantillo and C. O. Kappe, Visible light-promoted beckmann rearrangements: Separating sequential photochemical and thermal phenomena in a continuous flow reactor, Eur. J. Org. Chem., 2019, 2019(11), 2163–2171 Search PubMed.
- I. Abdiaj, L. Huck, J. M. Mateo, A. de la Hoz, M. V. Gomez, A. Diaz-Ortiz and J. Alcazar, Photoinduced palladium-catalyzed Negishi cross-couplings enabled by the visible-light absorption of palladium-zinc complexes, Angew. Chem., Int. Ed., 2018, 57(40), 13231–13236 Search PubMed.
- X. J. Wei, I. Abdiaj, C. Sambiagio, C. Li, E. Zysman-Colman, J. Alcazar and T. Noel, Visible-light-promoted iron-catalyzed C(sp2)-C(sp3) Kumada cross-coupling in flow, Angew. Chem., Int. Ed., 2019, 58(37), 13030–13034 Search PubMed.
- Y. Chen, O. May, D. C. Blakemore and S. V. Ley, A photoredox coupling reaction of benzylboronic esters and carbonyl compounds in batch and flow, Org. Lett., 2019, 21(15), 6140–6144 Search PubMed.
- P. Vatankhah and A. Shamloo, Parametric study on mixing process in an in-plane spiral micromixer utilizing chaotic advection, Anal. Chim. Acta, 2018, 1022, 96–105 Search PubMed.
- R. A. Green, R. C. D. Brown, D. Pletcher and B. Harji, A microflow electrolysis cell for laboratory synthesis on the multigram scale, Org. Process Res. Dev., 2015, 19(10), 1424–1427 Search PubMed.
- R. A. Green, K. E. Jolley, A. A. M. Al-Hadedi, D. Pletcher, D. C. Harrowven, O. De Frutos, C. Mateos, D. J. Klauber, J. A. Rincon and R. C. D. Brown, Electrochemical deprotection of para-methoxybenzyl ethers in a flow electrolysis cell, Org. Lett., 2017, 19(8), 2050–2053 Search PubMed.
- A. A. Folgueiras-Amador, K. Philipps, S. Guilbaud, J. Poelakker and T. Wirth, An easy-to-machine electrochemical flow microreactor: Efficient synthesis of isoindolinone and flow functionalization, Angew. Chem., Int. Ed., 2017, 56(48), 15446–15450 Search PubMed.
- C. Huang, X. Y. Qian and H. C. Xu, Continuous-flow electrosynthesis of benzofused S-heterocycles by dehydrogenative C-S cross-coupling, Angew. Chem., Int. Ed., 2019, 58(20), 6650–6653 Search PubMed.
- K. Arai, K. Watts and T. Wirth, Difluoro-and trifluoromethylation of electron-deficient alkenes in an electrochemical microreactor, ChemistryOpen, 2014, 3(1), 23–28 Search PubMed.
- T. Arai, H. Tateno, K. Nakabayashi, T. Kashiwagi and M. Atobe, An anodic aromatic C,C cross-coupling reaction using parallel laminar flow mode in a flow microreactor, Chem. Commun., 2015, 51(23), 4891–4894 Search PubMed.
- J. Kuleshova, J. T. Hill-Cousins, P. R. Birkin, R. C. D. Brown, D. Pletcher and T. J. Underwood, The methoxylation of N-formylpyrrolidine in a microfluidic electrolysis cell for routine synthesis, Electrochim. Acta, 2012, 69, 197–202 Search PubMed.
- R. A. Green, D. Pletcher, S. G. Leach and R. C. D. Brown, N-heterocyclic carbene-mediated oxidative electrosynthesis of esters in a microflow cell, Org. Lett., 2015, 17(13), 3290–3293 Search PubMed.
- R. A. Green, D. Pletcher, S. G. Leach and R. C. D. Brown, N-Heterocyclic carbene-mediated microfluidic oxidative electrosynthesis of amides from aldehydes, Org. Lett., 2016, 18(5), 1198–1201 Search PubMed.
- M. Ošeka, G. Laudadio, N. P. van Leest, M. Dyga, A. D. A. Bartolomeu, L. J. Gooßen, B. de Bruin, K. T. de Oliveira and T. Noël, Electrochemical Aziridination of Internal Alkenes with Primary Amines, Chem, 2021, 7(1), 255–266 Search PubMed.
- J. Yu and J. Wu, Aziridination of Internal Alkenes Using Primary Alkyl Amines in a Microflow Electrocell, Chem, 2021, 7(1), 18–19 Search PubMed.
- M. Kabeshov, B. Musio, P. Murray, D. Browne and S. Ley, Expedient preparation of nazlinine and a small library of indole alkaloids using flow electrochemistry as an enabling technology, Org. Lett., 2014, 16, 4618–4621 Search PubMed.
- M. Elsherbini, B. Winterson, H. Alharbi, A. A. Folgueiras-Amador, C. Genot and T. Wirth, Continuous-flow electrochemical generator of hypervalent iodine reagents: Synthetic applications, Angew. Chem., Int. Ed., 2019, 58(29), 9811–9815 Search PubMed.
- N. Amri, R. A. Skilton, D. Guthrie and T. Wirth, Efficient flow electrochemical alkoxylation of pyrrolidine-1-carbaldehyde, Synlett, 2019, 30(10), 1183–1186 Search PubMed.
- C. T. Crowe, Handbook, Multiphase Flow, Taylor & Francis Group, New-York, 2006 Search PubMed.
- H. Seo, L. V. Nguyen and T. F. Jamison, Using carbon dioxide as a building block in continuous flow synthesis, Adv. Synth. Catal., 2019, 361(2), 247–264 Search PubMed.
- C. A. Hone and C. O. Kappe, Correction to: The use of molecular oxygen for liquid phase aerobic oxidations in continuous flow, Top. Curr. Chem., 2019, 377(2), 7 Search PubMed.
- B. Pieber, M. Shalom, M. Antonietti, P. H. Seeberger and K. Gilmore, Kontinuierliche heterogene photokatalyse in seriellen mikro-batch-reaktoren, Angew. Chem., 2018, 130(31), 10127–10131 Search PubMed.
- D. Cantillo, P. A. Inglesby, A. Boyd and C. O. Kappe, Hydrogen sulfide chemistry in continuous flow: Efficient synthesis of 2-oxopropanethioamide, J. Flow Chem., 2017, 7(2), 29–32 Search PubMed.
- M. Ramezani, M. A. Kashfipour and M. Abolhasani, Minireview: Flow chemistry studies of high-pressure gas-liquid reactions with carbon monoxide and hydrogen, J. Flow Chem., 2020, 10(1), 93–101 Search PubMed.
- C. Battilocchio, G. Iannucci, S. Wang, E. Godineau, A. Kolleth, A. De Mesmaeker and S. V. Ley, Flow synthesis of cyclobutanones via [2 + 2] cycloaddition of keteneiminium salts and ethylene gas, React. Chem. Eng., 2017, 2(3), 295–298 Search PubMed.
- Y. Wada, M. A. Schmidt and K. F. Jensen, Flow distribution and ozonolysis in gas−liquid multichannel microreactors, Ind. Eng. Chem. Res., 2006, 45(24), 8036–8042 Search PubMed.
- B. Gutmann, M. Köckinger, G. Glotz, T. Ciaglia, E. Slama, M. Zadravec, S. Pfanner, M. C. Maier, H. Gruber-Wölfler and C. Oliver Kappe, Design and 3D printing of a stainless steel reactor for continuous difluoromethylations using fluoroform, React. Chem. Eng., 2017, 2(6), 919–927 Search PubMed.
- J. Kobayashi, Y. Mori, K. Okamoto, R. Akiyama, M. Ueno, T. Kitamori and S. Kobayashi, A microfluidic device for conducting gas-liquid-solid hydrogenation reactions, Science, 2004, 304(5675), 1305–1308 Search PubMed.
- P. Kluson, P. Stavarek, V. Penkavova, H. Vychodilova, S. Hejda and M. Bendova, Microfluidic chip reactor and the stereoselective hydrogenation of methylacetoacetate over (R) -Ru-BINAP in the [N 8222 ][Tf 2 N]/methanol/water mixed phase, Chem. Eng. Process.: Process Intesif., 2017, 115, 39–45 Search PubMed.
- J. Gardiner, X. Nguyen, C. Genet, M. D. Horne, C. H. Hornung and J. Tsanaktsidis, Catalytic static mixers for the continuous flow hydrogenation of a key intermediate of linezolid (zyvox), Org. Process Res. Dev., 2018, 22(10), 1448–1452 Search PubMed.
- C. Genet, X. Nguyen, B. Bayatsarmadi, M. D. Horne, J. Gardiner and C. H. Hornung, Reductive aminations using a 3D printed supported metal(0) catalyst system, J. Flow Chem., 2018, 8(2), 81–88 Search PubMed.
- C. H. Hornung, X. Nguyen, A. Carafa, J. Gardiner, A. Urban, D. Fraser, M. D. Horne, D. R. Gunasegaram and J. Tsanaktsidis, Use of catalytic static mixers for continuous flow gas–liquid and transfer hydrogenations in organic synthesis, Org. Process Res. Dev., 2017, 21(9), 1311–1319 Search PubMed.
- A. Avril, C. H. Hornung, A. Urban, D. Fraser, M. Horne, J. P. Veder, J. Tsanaktsidis, T. Rodopoulos, C. Henry and D. R. Gunasegaram, Continuous flow hydrogenations using novel catalytic static mixers inside a tubular reactor, React. Chem. Eng., 2017, 2(2), 180–188 Search PubMed.
- X. Nguyen, A. Carafa and C. H. Hornung, Hydrogenation of vinyl acetate using a continuous flow tubular reactor with catalytic static mixers, Chem. Eng. Process., 2018, 124, 215–221 Search PubMed.
- C. P. Park and D.-P. Kim, Dual-channel microreactor for gas−liquid syntheses, J. Am. Chem. Soc., 2010, 132(29), 10102–10106 Search PubMed.
- R. A. Maurya, C. P. Park and D.-P. Kim, Triple-channel microreactor for biphasic gas–liquid reactions: Photosensitized oxygenations, Beilstein J. Org. Chem., 2011, 7(1), 1158–1163 Search PubMed.
- H. Aran, D. Salamon, T. Rijnaarts, G. Mul, M. Wessling and R. Lammertink, Porous photocatalytic membrane microreactor (P2M2): a new reactor concept for photochemistry, J. Photochem. Photobiol., A, 2011, 225(1), 36–41 Search PubMed.
- H. Aran, H. Klooster, J. Jani, M. Wessling, L. Lefferts and R. G. Lammertink, Influence of geometrical and operational parameters on the performance of porous catalytic membrane reactors, Chem. Eng. J., 2012, 207, 814–821 Search PubMed.
- P. Koos, U. Gross, A. Polyzos, M. O'Brien, I. Baxendale and S. V. Ley, Teflon AF-2400 mediated gas-liquid contact in continuous flow methoxycarbonylations and in-line FTIR measurement of CO concentration, Org. Biomol. Chem., 2011, 9(20), 6903–6908 Search PubMed.
- S. L. Bourne, P. Koos, M. O'Brien, B. Martin, B. Schenkel, I. R. Baxendale and S. V. Ley, The continuous-flow synthesis of styrenes using ethylene in a palladium-catalysed heck cross-coupling reaction, Synlett, 2011, 2011(18), 2643–2647 Search PubMed.
- F. Mastronardi, B. Gutmann and C. O. Kappe, Continuous flow generation and reactions of anhydrous diazomethane using a teflon AF-2400 tube-in-tube reactor, Org. Lett., 2013, 15(21), 5590–5593 Search PubMed.
- M. O'Brien, N. Taylor, A. Polyzos, I. R. Baxendale and S. V. Ley, Hydrogenation in flow: Homogeneous and heterogeneous catalysis using Teflon AF-2400 to effect gas–liquid contact at elevated pressure, Chem. Sci., 2011, 2(7), 1250–1257 Search PubMed.
- M. O'Brien, I. R. Baxendale and S. V. Ley, Flow ozonolysis using a semipermeable teflon AF-2400 membrane to effect gas−liquid contact, Org. Lett., 2010, 12(7), 1596–1598 Search PubMed.
- B. Venezia, L. Panariello, D. Biri, J. Shin, S. Damilos, A. N. Radhakrishnan, C. Blackman and A. Gavriilidis, Catalytic Teflon AF-2400 membrane reactor with adsorbed ex situ synthesized Pd-based nanoparticles for nitrobenzene hydrogenation, Catal. Today, 2020, 104–112 Search PubMed.
- C. Xue, J. Li, J. P. Lee, P. Zhang and J. Wu, Continuous amination of aryl/heteroaryl halides using aqueous ammonia in a Teflon AF-2400 tube-in-tube micro-flow reactor, React. Chem. Eng., 2019, 4(2), 346–350 Search PubMed.
- H. Zhang, J. Yue, G. Chen and Q. Yuan, Flow pattern and break-up of liquid film in single-channel falling film microreactors, Chem. Eng. J., 2010, 163, 126–132 Search PubMed.
- N. Steinfeldt, R. Abdallah, U. Dingerdissen and K. Jähnisch, Ozonolysis of acetic acid 1-vinyl-hexyl ester in a falling film microreactor, Org. Process Res. Dev., 2007, 11, 1025–1031 Search PubMed.
- T. Rehm, S. Gros, P. Lob and A. Renken, Photonic contacting of gas-liquid phases in a falling film microreactor for continuous-flow photochemical catalysis with visible light, React. Chem. Eng., 2016, 1, 636–648 Search PubMed.
- A. Cantu-Perez, D. Ziegenbalg, P. Löb, A. Gavriilidis, V. Hessel and F. Schönfeld, Microstructure-based intensification of a falling film microreactor through optimal film setting with realistic profiles and in-channel induced mixing, Chem. Eng. J., 2012, 179, 318–329 Search PubMed.
- S. Ruwei, Q. Deng, R. Ding and L. Zhang, Generation of ethynyl grignard reagent in a falling film microreactor: An expeditious flow synthesis of propargyl alcohols and analogues, Adv. Synth. Catal., 2014, 356, 2931–2936 Search PubMed.
- D. Lokhat, A. Domah, K. Padayachee, A. Baboolal and D. Ramjugernath, Gas–liquid mass transfer in a falling film microreactor: Effect of reactor orientation on liquid-side mass transfer coefficient, Chem. Eng. Sci., 2016, 155, 38–44 Search PubMed.
- B. K. Vankayala, P. Löb, V. Hessel, G. Menges, C. Hofmann, D. Metzke, U. Krtschil and H.-J. Kost, Scale-up of process intensifying falling film microreactors to pilot production scale, Int. J. Chem. React. Eng., 2007, 5(1), 1–10 Search PubMed.
- M. De Sousa Duarte, M. Rolland, C. Sagnard, D. Suire, F. Flacher, O. Delpoux and C. P. Lienemann, High-pressure high-temperature transparent fixed-bed reactor for operando gas-liquid reaction follow-up, Chem. Eng. Technol., 2019, 42(3), 655–660 Search PubMed.
- D. A. Wenn, J. E. Shaw and B. Mackenzie, A mesh microcontactor for 2-phase reactions, Lab Chip, 2003, 3(3), 180–186 Search PubMed.
- M. Irfan, T. N. Glasnov and C. O. Kappe, Continuous flow ozonolysis in a laboratory scale reactor, Org. Lett., 2011, 13(5), 984–987 Search PubMed.
- M. D. Roydhouse, A. Ghaini, A. Constantinou, A. Cantu-Perez, W. B. Motherwell and A. Gavriilidis, Ozonolysis in flow using capillary reactors, Org. Process Res. Dev., 2011, 15(5), 989–996 Search PubMed.
- V. Dragone, V. Sans, M. H. Rosnes, P. J. Kitson and L. Cronin, 3D-printed devices for continuous-flow organic chemistry, Beilstein J. Org. Chem., 2013, 9, 951–959 Search PubMed.
- C. R. Sagandira, M. Siyawamwaya and P. Watts, 3D printing and continuous flow chemistry technology to advance pharmaceutical manufacturing in developing countries, Arabian J. Chem., 2020, 13(11), 7886–7908 Search PubMed.
- S. Rossi, R. Porta, D. Brenna, A. Puglisi and M. Benaglia, Stereoselective catalytic synthesis of active pharmaceutical ingredients in homemade 3D-printed mesoreactors, Angew. Chem., Int. Ed., 2017, 56, 4290–4294 Search PubMed.
- J. S. Mathieson, M. H. Rosnes, V. Sans, P. J. Kitson and L. Cronin, Continuous parallel ESI-MS analysis of reactions carried out in a bespoke 3D printed device, Beilstein J. Nanotechnol., 2013, 4, 285–291 Search PubMed.
- M. R. Penny, Z. X. Rao, B. F. Peniche and S. T. Hilton, Modular 3D printed compressed air driven continuous-flow systems for chemical synthesis, Eur. J. Org. Chem., 2019, 2019(23), 3783–3787 Search PubMed.
- A. Zhakeyev, J. Tobin, H. Wang, F. Vilela and J. Xuan, Additive manufacturing of photoactive polymers for visible light harvesting, Energy Procedia, 2019, 158, 5608–5614 Search PubMed.
- G.-N. Ahn, T. Yu, H.-J. Lee, K.-W. Gyak, J.-H. Kang, D. You and D.-P. Kim, A numbering-up metal microreactor for the high-throughput production of a commercial drug by copper catalysis, Lab Chip, 2019, 19(20), 3535–3542 Search PubMed.
- H.-J. Lee, R. C. Roberts, D. J. Im, S.-J. Yim, H. Kim, J. T. Kim and D.-P. Kim, Enhanced Controllability of Fries Rearrangements Using High-Resolution 3D-Printed Metal Microreactor with Circular Channel, Small, 2019, 15(50), 1905005 Search PubMed.
- Z. X. Rao, B. Patel, A. Monaco, Z. J. Cao, M. Barniol-Xicota, E. Pichon, M. Ladlow and S. T. Hilton, 3D-printed polypropylene continuous-flow column reactors: exploration of reactor utility in SNAr reactions and the synthesis of bicyclic and tetracyclic heterocycles, Eur. J. Org. Chem., 2017, 2017(44), 6499–6504 Search PubMed.
- H. Gong, B. P. Bickham, A. T. Woolley and G. P. Nordin, Custom 3D printer and resin for 18 mum x 20 mum microfluidic flow channels, Lab Chip, 2017, 17(17), 2899–2909 Search PubMed.
- D. E. Fitzpatrick, M. O'Brien and S. V. Ley, A tutored discourse on microcontrollers, single board computers and their applications to monitor and control chemical reactions, React. Chem. Eng., 2020, 5(2), 201–220 Search PubMed.
- D. L. Browne, B. H. Harji and S. V. Ley, Continuous cold without cryogenic consumables: Development of a convenient laboratory tool for low-temperature flow processes, Chem. Eng. Technol., 2013, 36(6), 959–967 Search PubMed.
- M. O'Brien and D. Cooper, Continuous flow liquid–liquid separation using a computer-vision control system: The bromination of enaminones with N-bromosuccinimide, Synlett, 2016, 27(01), 164–168 Search PubMed.
- S. V. Ley, R. J. Ingham, M. O'Brien and D. L. Browne, Camera-enabled techniques for organic synthesis, Beilstein J. Org. Chem., 2013, 9, 1051–1072 Search PubMed.
- M. O'Brien, D. A. Cooper and J. Dolan, Continuous flow iodination using an automated computer-vision controlled liquid-liquid extraction system, Tetrahedron Lett., 2017, 58(9), 829–834 Search PubMed.
- R. J. Ingham, C. Battilocchio, J. M. Hawkins and S. V. Ley, Integration of enabling methods for the automated flow preparation of piperazine-2-carboxamide, Beilstein J. Org. Chem., 2014, 10, 641–652 Search PubMed.
- D. E. Fitzpatrick, C. Battilocchio and S. V. Ley, A novel internet-based reaction monitoring, control and autonomous self-optimization platform for chemical synthesis, Org. Process Res. Dev., 2015, 20(2), 386–394 Search PubMed.
- D. E. Fitzpatrick and S. V. Ley, Engineering chemistry: integrating batch and flow reactions on a single, automated reactor platform, React. Chem. Eng., 2016, 1(6), 629–635 Search PubMed.
- D. E. Fitzpatrick, T. Maujean, A. C. Evans and S. V. Ley, Across-the-world automated optimization and continuous-flow synthesis of pharmaceutical agents operating through a cloud-based server, Angew. Chem., Int. Ed., 2018, 57(46), 15128–15132 Search PubMed.
- M. O'Brien, A. Hall, J. Schrauwen and J. van der Made, An open-source approach to automation in organic synthesis: The flow chemical formation of benzamides using an inline liquid-liquid extraction system and a homemade 3-axis autosampling/product-collection device, Tetrahedron, 2018, 74(25), 3152–3157 Search PubMed.
- E. C. Aka, E. Wimmer, E. Barre, N. Vasudevan, D. Cortes-Borda, T. Ekou, L. Ekou, M. Rodriguez-Zubiri and F. X. Felpin, Reconfigurable flow platform for automated reagent screening and autonomous optimization for bioinspired lignans synthesis, J. Org. Chem., 2019, 84(21), 14101–14112 Search PubMed.
- B. J. Deadman, C. Battilocchio, E. Sliwinski and S. V. Ley, A prototype device for evaporation in batch and flow chemical processes, Green Chem., 2013, 15(8), 2050–2055 Search PubMed.
- C. Battilocchio, B. J. Deadman, N. Nikbin, M. O. Kitching, I. R. Baxendale and S. V. Ley, A Machine-Assisted Flow Synthesis of SR48692: A Probe for the Investigation of Neurotensin Receptor-1, Chem. – Eur. J., 2013, 19(24), 7917–7930 Search PubMed.
- M. Escriba-Gelonch, V. Hessel, M. C. Maier, T. Noel, M. F. Neira d'Angelo and H. Gruber-Woelfler, Continuous-Flow In-Line Solvent-Swap Crystallization of Vitamin D3, Org. Process Res. Dev., 2018, 22(2), 178–189 Search PubMed.
- 22-12-2020), h. w. s. c. p. a.-f.-c. a.
- B. J. Deadman, D. L. Browne, I. R. Baxendale and S. V. Ley, Back Pressure Regulation of Slurry-Forming Reactions in Continuous Flow, Chem. Eng. Technol., 2015, 38(2), 259–264 Search PubMed.
- D. L. Browne, B. J. Deadman, R. Ashe, I. R. Baxendale and S. V. Ley, Continuous Flow Processing of Slurries: Evaluation of an Agitated Cell Reactor, Org. Process Res. Dev., 2011, 15(3), 693–697 Search PubMed.
- N. Neyt and D. Riley, Batch–flow hybrid synthesis of the antipsychotic clozapine, React. Chem. Eng., 2018, 3(1), 17–24 Search PubMed.
- C. D. Scott, R. Labes, M. Depardieu, C. Battilocchio, M. G. Davidson, S. V. Ley, C. C. Wilson and K. Robertson, Integrated plug flow synthesis and crystallisation of pyrazinamide, React. Chem. Eng., 2018, 3(5), 631–634 Search PubMed.
- R. L. Hartman, J. R. Naber, S. L. Buchwald and K. F. Jensen, Multistep microchemical synthesis enabled by microfluidic distillation, Angew. Chem., Int. Ed., 2010, 49(5), 899–903 Search PubMed.
- M. Baumann, Integrating reactive distillation with continuous flow processing, React. Chem. Eng., 2019, 4(2), 368–371 Search PubMed.
- A. G. O'Brien, Z. Horváth, F. Lévesque, J. W. Lee, A. Seidel-Morgenstern and P. H. Seeberger, Kontinuierliche synthese und aufreinigung durch direkte kopplung eines durchflussreaktors mit “simulated-moving-bed”-chromatographie, Angew. Chem., 2012, 124(28), 7134–7137 Search PubMed.
- J. W. Lee, Z. Horváth, A. G. O'Brien, P. H. Seeberger and A. Seidel-Morgenstern, Design and optimization of coupling a continuously operated reactor with simulated moving bed chromatography, Chem. Eng. J., 2014, 251, 355–370 Search PubMed.
- I. G. T. M. Penders, Z. Amara, R. Horvath, K. Rossen, M. Poliakoff and M. W. George, Photocatalytic hydroxylation of arylboronic acids using continuous flow reactors, RSC Adv., 2015, 5(9), 6501–6504 Search PubMed.
- B. Sreedhar, B. Shen, H. Li, R. Rousseau and Y. Kawajiri, Optimal design of integrated SMB-crystallization hybrid separation process using a binary solvent, Org. Process Res. Dev., 2017, 21(1), 31–43 Search PubMed.
- R. Örkényi, J. Éles, F. Faigl, P. Vincze, A. Prechl, Z. Szakács, J. Kóti and I. Greiner, Continuous synthesis and purification by coupling a multistep flow reaction with centrifugal partition chromatography, Angew. Chem., Int. Ed., 2017, 56(30), 8742–8745 Search PubMed.
- D. E. Fitzpatrick, R. J. Mutton and S. V. Ley, In-line separation of multicomponent reaction mixtures using a new semi-continuous supercritical fluid chromatography system, React. Chem. Eng., 2018, 3(5), 799–806 Search PubMed.
- R. J. Ingham, C. Battilocchio, D. E. Fitzpatrick, E. Sliwinski, J. M. Hawkins and S. V. Ley, A systems approach towards an intelligent and self-controlling platform for integrated continuous reaction sequences, Angew. Chem., 2015, 127(1), 146–150 Search PubMed.
- S. G. Newman, L. Gu, C. Lesniak, G. Victor, F. Meschke, L. Abahmane and K. F. Jensen, Rapid Wolff–Kishner reductions in a silicon carbide microreactor, Green Chem., 2014, 16(1), 176–180 Search PubMed.
- R. L. Hartman, J. R. Naber, N. Zaborenko, S. L. Buchwald and K. F. Jensen, Overcoming the challenges of solid bridging and constriction during Pd-Catalyzed C− N bond formation in microreactors, Org. Process Res. Dev., 2010, 14(6), 1347–1357 Search PubMed.
- D. Znidar, A. O'Kearney-McMullan, R. Munday, C. Wiles, P. Poechlauer, C. Schmoelzer, D. Dallinger and C. O. Kappe, Scalable Wolff–Kishner reductions in extreme process windows using a silicon carbide flow reactor, Org. Process Res. Dev., 2019, 23(11), 2445–2455 Search PubMed.
- M. W. Bedore, N. Zaborenko, K. F. Jensen and T. F. Jamison, Aminolysis of epoxides in a microreactor system: a continuous flow approach to β-amino alcohols, Org. Process Res. Dev., 2010, 14(2), 432–440 Search PubMed.
- N. Zaborenko, M. W. Bedore, T. F. Jamison and K. F. Jensen, Kinetic and scale-up investigations of epoxide aminolysis in microreactors at high temperatures and pressures, Org. Process Res. Dev., 2011, 15(1), 131–139 Search PubMed.
- S. J. Yim, B. T. Ramanjaneyulu, S. Vidyacharan, Y. D. Yang, I. S. Kang and D.-P. Kim, Compact reaction-module on a pad for scalable flow-production of organophosphates as drug scaffolds, Lab Chip, 2020, 20(5), 973–978 Search PubMed.
- H. Kim, K.-I. Min, K. Inoue, D. J. Im, D.-P. Kim and J.-I. Yoshida, Submillisecond organic synthesis: Outpacing Fries rearrangement through microfluidic rapid mixing, Science, 2016, 352(6286), 691 Search PubMed.
| This journal is © The Royal Society of Chemistry 2021 |