
Electrochemical reduction of CO2 towards multi-carbon products via a two-step process
Xianbiao
Fu
,
Jiahao
Zhang
and
Yijin
Kang
 *
*
Institute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, Chengdu, 610054, China. E-mail: kangyijin@uestc.edu.cn
First published on 5th February 2021
Abstract
The electrochemical conversion of carbon dioxide (CO2) towards clean fuels and chemicals powered by renewable energy is a promising strategy to realize the closing of the loop of carbon footprint. However, the direct reduction of CO2 to multi-carbon (C2+) products suffers from low activity in non-alkaline electrolyte or electrolyte degradation problem caused by carbonate formation in alkaline electrolyte. The two-step process for CO2 electrocution can circumvent such problems by converting CO2 to CO (the first step) in the non-alkaline electrolyte and promote the rate of carbon–carbon coupling for CO-to-C2+ conversion (the second step) in alkaline electrolytes. We summarize the recent progress of CO-selective catalysts, C2+-selective catalysts, tandem catalysts, and tandem reaction systems, which aim to achieve the efficient production of C2+ products with high selectivity. The two-step route of CO2 reduction pushes the chemical production from environmentally abundant molecules closer to the practical application, offering a promising replacement in the petrochemical industry for chemical production under hydrogen economy in the future.
 Xianbiao Fu | Xianbiao Fu is a Ph.D. candidate at the University of Electronic Science and Technology of China, under the supervision of Prof. Yijin Kang. He was a visiting graduate student at Northwestern University for two years and Johns Hopkins University for one year. He obtained his B.S. degree from Central South University in 2016. His research focuses on the electroreduction of CO2/CO into fuels/chemicals, controllable synthesis of model nanocatalysts, and flow-cell/MEA reactor design for electrochemical systems. |
 Jiahao Zhang | Jiahao Zhang received his B.S. degree from Southwest Petroleum University in 2019. He is currently working on his M.S. degree under the supervision of Prof. Yijin Kang at the Institute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China. His research interests include carbon dioxide reduction reaction (CO2RR), oxygen evolution reaction (OER), and polymer electrolyte membrane (PEM) electrolyzers. |
 Yijin Kang | Yijin Kang is currently a Professor at the Institute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China. He was also a Visiting Professor at the Institute for Sustainability and Energy, Northwestern University and an Adjunct Professor at the Department of Chemical Engineering, University of Illinois at Chicago, United States. He received his B.S. (2005) at Fudan University, Ph.D. (2012) from the University of Pennsylvania, and did postdoctoral research at Argonne National Laboratory. His research focuses on energy conversion and storage, including fuel cell technology, battery technology, and chemical upgrade using catalytic processes such as hydrogenation, selective oxidation, CO2/CO reduction, and ammonia synthesis. |
1. Introduction
The electrochemical carbon dioxide reduction reaction (CO2RR) is an alternative route for valuable fuels and chemical production with the plummeting cost of renewable electricity.1–5 In general, CO2 molecules can be converted into carbon monoxide (CO), methane (CH4), methanol (CH3OH), formic acid (HCOOH), ethylene (C2H4), ethanol (CH3CH2OH), acetate acid (CH3COOH), propanol (n-CH3CH2CH2OH), and chemicals with even longer carbon chains. Among all CO2RR electrocatalysts, only copper-based catalysts have been shown to have the unique capability to convert CO2/CO into multicarbon (C2+) products with acceptable selectivities.6–9 Many efforts have been devoted towards regulating the selectivity of C2+ products, such as optimizing crystal facets,6,10 modifying oxidation state,11,12 introducing defects,13 alloying,14,15 functionalizing the catalyst surface,16–18 and engineering the electrolyzer.19,20 However, further improving the selectivity of C2+ products suffers from fundamental problems and industrially relevant challenges. Recently, some progress has been made to improve the selectivity and formation rates of C2+ products via the use of alkaline electrolytes; however, the CO2 electrolyzer is an electrolyte-consuming device due to the formation of carbonate.21,22 In addition, the energy required to regenerate CO2 from aqueous carbonate is much larger than |ΔG°|, making CO2RR in alkaline electrolytes an energy-expensive device. A more detailed discussion of the problem of carbonate formation in alkaline CO2 electrolyzer can be seen in the comments from Matthew W. Kanan.22 Therefore, a two-step route for CO2 electrochemical reduction to C2+ products was proposed to circumvent the carbonate formation in an alkaline CO2 electrolyzer.23 As shown in Fig. 1, CO2 was first reduced to CO in a non-alkaline electrolyte and the produced CO was subsequently converted into C2+ products as the second step in an alkaline electrolyte with significantly improved C2+ selectivity and activity. There is no problem of carbonate formation between CO and hydroxide, and the use of alkaline electrolytes can indeed improve the reactivity of CO in electrolysis. The two-step electroreduction of CO2 to C2+ products with high selectivity and production rates of industrial relevance provides an alternative route for converting CO2 to fuels and chemicals.24,25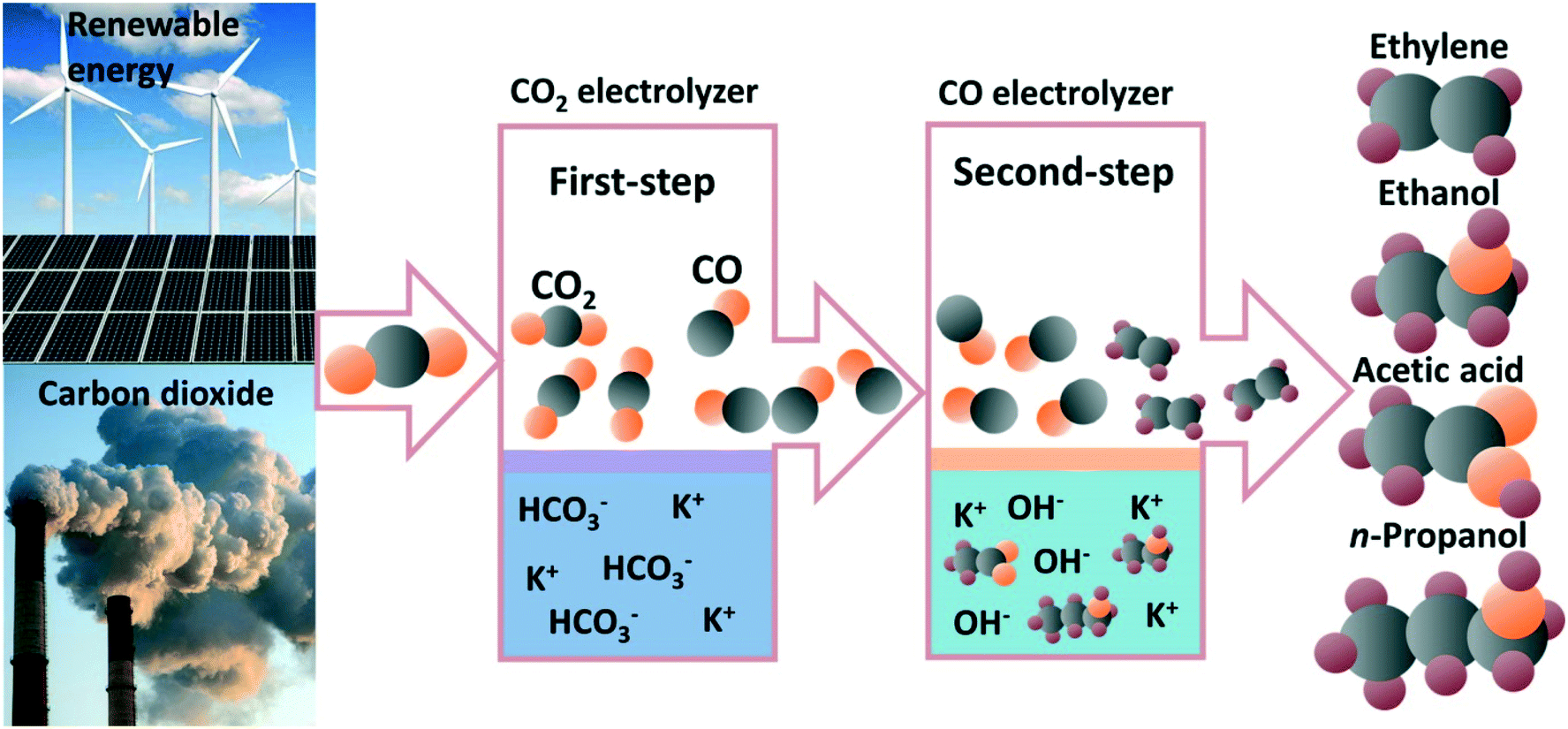 | ||
| Fig. 1 Schematic illustration of the two-step CO2 electrochemical reduction to multicarbon products. | ||
The electrochemical CO reduction reaction (CORR) to C2+ products is the key to the two-step conversion process, which enhances the reaction rate and selectivity. In addition to the fact that the CORR process can be carried out in alkaline electrolytes to reduce the overpotential, CORR holds several other advantages over direct CO2RR.25 It is known that CO is the key intermediate to C2+ products in CO2 electrolysis. The direct use of CO as the reactant can increase C–C coupling rates to improve the formation rates of C2+ products.6,23 Moreover, the inherent reactivity of CO is higher than that of CO2. It is proved that for C2+ production from CO, the reactant higher current densities can be achieved at lower overpotential compared to from CO2 as the reactant.24 The adsorption of CO on the catalyst surface is stronger than CO2, which helps to suppress the competitive hydrogen evolution reaction (HER), which in turn is beneficial for vastly improving the current efficiency for C2+ product formation.26 The electrochemical reduction of CO2 to CO has been achieved with high faradaic efficiencies (FE) at high current densities, which provides sustainable feedstock (CO2-derived CO) in CO electrolysis.
A two-step route for CO2 electroreduction can be achieved via the tandem electrode structure, bimetallic catalysts, and tandem reaction systems, as shown in Fig. 2. Tandem catalysis strategy was obtained from thermal heterogeneous catalysis to break the linear scaling relations of the binding strength for intermediates in CO2RR.27,28 The tandem electrode structure is achieved by two different catalysts for CO2-to-CO and CO-to-C2+ production, respectively. Moreover, the development of the catalyst with two sites for the production of CO and C2+, respectively, has also attracted considerable attention. For example, bifunctional catalysts enable in situ CO production and consumption, making the high-cost gas separation step unnecessary. As shown in Fig. 2b, tandem reaction systems are also a promising alternative to be explored. Many research efforts have been focused on the second step with pure CO as the feedstock but the interdependence of both steps is rarely considered. From the angle of either catalysis science or electrolyzer engineering, there is plenty of room for tandem catalysis to improve the selectivity and production rate of C2+ products. This review offers a perspective regarding the two-step route for the electrochemical reduction of CO2 to C2+ products.
 | ||
| Fig. 2 The two-step CO2 electroreduction strategies. (a) Tandem catalysts reduce CO2 to multicarbons. (b) Tandem reaction systems for converting CO2 to multicarbons. | ||
2. The first step: reduce CO2 to CO
The electrochemical reduction of CO2 to CO with high selectivity and current density is a prerequisite for the two-step route of CO2 electroreduction. In general, Au, Ag, and their alloys are commonly used electrocatalysts for the specific reduction of CO2 to CO at low temperature.29–31 These catalysts with medium hydrogen overpotential and weak CO adsorption can break the carbon–oxygen bond in CO2 and allow the produced CO to readily desorb from the surface of the catalysts. Although many strategies have been developed for improving the properties of noble metal catalysts, such as nanostructuring,32,33 alloying,34,35 phase engineering,30 grain boundary engineering,29 and crystal facet engineering,36,37 the practical applications of noble metal catalysts are still hindered by the high cost and scarcity. However, developing the electrocatalysts based on earth-abundant elements for the electroreduction of CO2 to CO remains challenging.Atomically dispersed earth-abundant metals anchored in nitrogen-doped carbon matrices (M–N–C) are promising candidates for CO2-to-CO conversion, with projected superior activity and selectivity.38–40 Wang and co-works reported a facile ion adsorption process to synthesize Ni single-atom catalysts on commercial carbon black (Ni-NCB) (Fig. 3a).41 The CO2RR tested in an anion membrane electrode assembly (MEA) (Fig. 3b) demonstrated that the Ni-NCB catalyst exhibited a current density up to 130 mA cm−2 (Fig. 3c) and nearly 100% CO faradaic efficiency (FE(CO)) (Fig. 3d). As shown in Fig. 3e, a maximum CO/H2 ratio of 113.8 was achieved at a current density of 74 mA cm−2. The results reveal that earth-abundant catalysts with remarkable CO production performance are promising to replace the noble metal catalysts (e.g., Au and Ag). Liu and co-workers prepared a single Ni-atom catalyst by pyrolyzing a mixture of amino acid, melamine, and nickel acetate in argon.42 Aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) was used to confirm the dispersion of Ni atoms on the graphene (Fig. 3f). In a traditional H-type electrolyzer, the A-Ni-NSG (the Ni atoms atomically dispersed on N-doped graphene prepared by the addition of L-cysteine) catalyst achieved a maximum FE(CO) of about 97% at about −0.5 V (vs. RHE) (Fig. 3g). Noteworthily, the A-Ni-NSG catalyst exhibited a high intrinsic CO2 reduction activity. The turnover frequency (TOF) of A-Ni-NSG reached 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 h−1 at an overpotential of 0.61 V (Fig. 3h), which is much larger than that of some noble metal catalysts and molecular catalysts. Operando X-ray absorption and photoelectron spectroscopy reveal that the monovalent Ni(I) atomic center as the active site can reduce the energy barrier for CO2RR via spontaneous charge transfer, which is responsible for the high intrinsic CO2 reduction activity.
800 h−1 at an overpotential of 0.61 V (Fig. 3h), which is much larger than that of some noble metal catalysts and molecular catalysts. Operando X-ray absorption and photoelectron spectroscopy reveal that the monovalent Ni(I) atomic center as the active site can reduce the energy barrier for CO2RR via spontaneous charge transfer, which is responsible for the high intrinsic CO2 reduction activity.
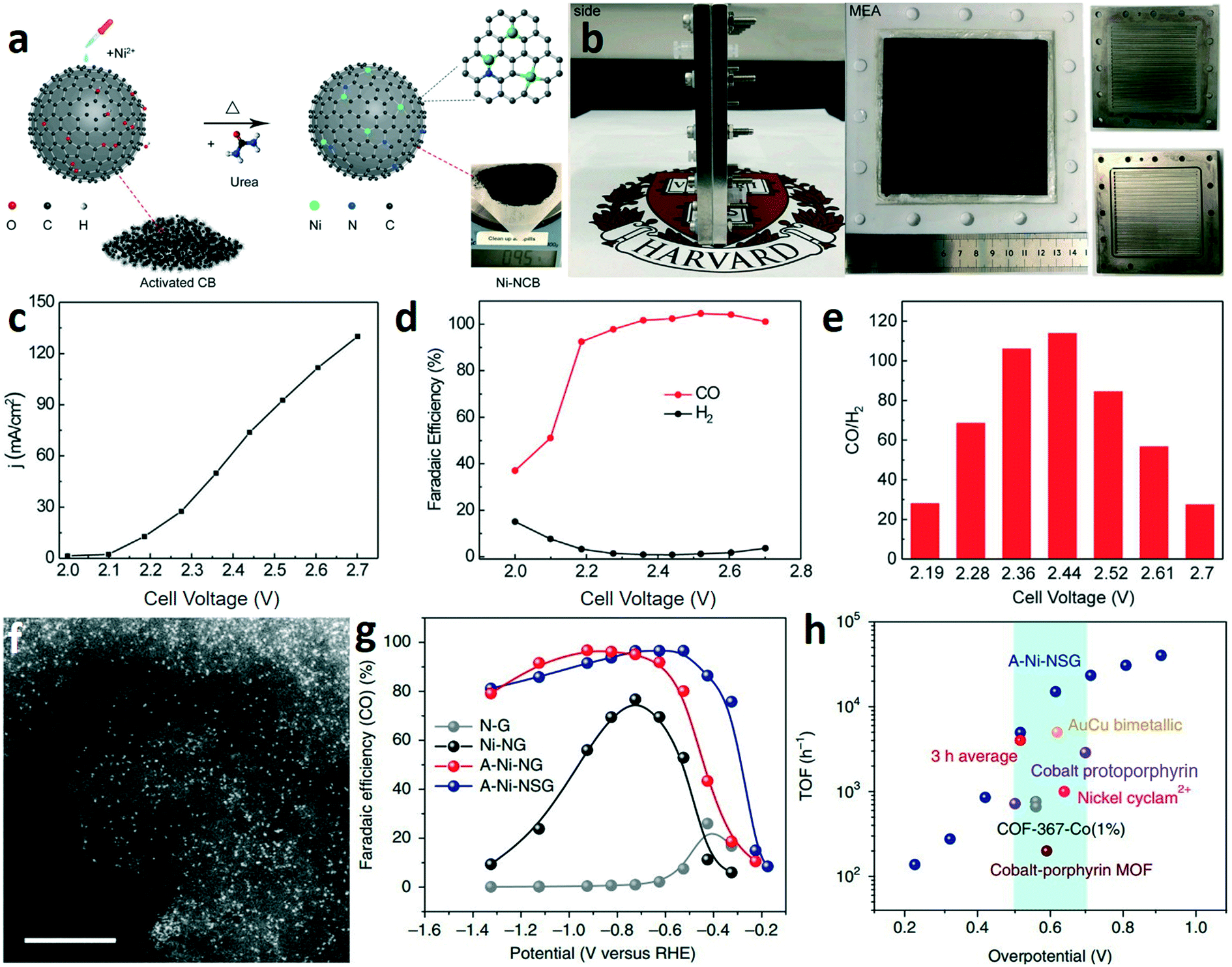 | ||
| Fig. 3 (a) Schematic illustration for the formation process of Ni-NCB. (b) Photographs of the membrane electrode assembly and individual cell components. (c–e) The current densities (c), the corresponding CO faradaic efficiency (d), and the CO/H2 ratio (e) of Ni-NCB. Reproduced with permission.41 Copyright 2019, Elsevier. (f) HAADF-STEM image of A-Ni-NG, scale bar: 5 nm. (g) CO faradaic efficiency at various applied potentials. (h) TOF of A-Ni-NSG compared with those of other state-of-the-art CO2-to-CO catalysts. Reproduced with permission.42 Copyright 2018, Nature Publishing Group. | ||
Molecular catalysts, such as metal phthalocyanine (MPc) and metal tetraphenylporphyrin (MTPP), have attracted considerable interests as electrocatalysts for CO2-to-CO conversion due to their uniform and tunable active sites, which are beneficial for revealing the structure–activity relationships and in turn to guide the rational design of the catalyst.43–45 Due to their poor conductivity, the molecular catalysts are usually supported on conductive materials to afford higher current densities.46 The selectivity and activity of the molecular catalyst can be regulated via the metal center, ligands, and ligand substituents. As shown in Fig. 4a, Liangand co-workers designed a series of nickel phthalocyanine molecules anchored on the side-walls of carbon nanotubes to control the performance of CO2 reduction by pendant group functionalization.47 Among the nickel phthalocyanine (NiPc) molecularly dispersed electrocatalysts (MDEs), NiPc-OMe achieved a CO selectivity of >99% in a wide current density range (i.e., −10 to −400 mA cm−2), which outperforms that of the pyrolyzed Ni–N–C catalyst and noble metal catalysts (Fig. 4b). Based on in situ X-ray absorption spectroscopy (XAS) and density functional theory (DFT) calculations, the electron-donating groups (–OMe) can increase the electron density on the Ni atoms and hence enhance the Ni–N bond strength to improve their electrochemical stability in CO2 electrolysis, which is the reverse for the electron-withdrawing group (–CN). Moreover, NiPc-OMe can stabilize the *COOH intermediate to improve the selectivity towards CO2-to-CO conversion. Berlinguette and co-workers reported that commercially available CoPc (Fig. 4c) achieved a current density of 150 mA cm−2 with CO selectivity >95% in a flow cell (Fig. 4d) and a CO partial current density of 175 mA cm−2 in a two-electrode cell.45 Using phenol as an additive to the CoPc catalysts can maintain the CO selectivity at a high current density due to the function of phenol as a local pH buffer (i.e., providing protons at a high current density) that reduces the overpotential of CO2RR and slows the formation of inactive bicarbonate at the electrode interface (Fig. 4d). This report shows that the molecular catalyst could be a competitive candidate for the electrocatalytic reduction of CO2 to CO with high selectivity at the practical current densities. The performance of the electrocatalysts based on earth-abundant elements for electrochemical CO2-to-CO conversion in the non-alkaline electrolyte is summarized in Table 1.
 | ||
| Fig. 4 (a) Schematic illustration for NiPc molecules anchored on the CNTs. (b) FE(CO) of NiPc MDEs at different applied potentials. Reproduced with permission.47 Copyright 2020, Nature Publishing Group. (c) Chemical structure of CoPc. (d) Diagram of the membrane electrode assembly and the cell components. (e) FE(CO) and Ecell as a function of current density, with and without phenol additive. Reproduced with permission.45 Copyright 2019, American Association for the Advancement of Science (AAAS). | ||
| Electrocatalysts | Electrolyte | Reactor | FE (%) | J (CO) (mA cm−2) | Ref. |
|---|---|---|---|---|---|
| A-Ni-NSG | 1.0 M KHCO3 | H-Cell | 97 | 22 | 42 |
| Cu-APC | 0.2 M NaHCO3 | H-Cell | 92 | 8.6 | 48 |
| NiPc-OMe | 1.0 M KHCO3 | Flow-cell | 99.5 | 300 | 47 |
| Co-TTCOFs | 0.5 M KHCO3 | H-Cell | 91.3 | 1.84 | 49 |
| Fe3+–N–C | 0.5 M KHCO3 | Flow-cell | 90 | 94 | 50 |
| C–Cu/SnO2-0.8 | 0.5 M KHCO3 | H-Cell | 93 | 4.6 | 51 |
| Ni–N4–C | 0.5 M KHCO3 | H-Cell | 99 | 24.8 | 52 |
| Ni-CNT-CC | 0.5 M KHCO3 | H-Cell | 99 | 32.3 | 53 |
| C–Zi1Ni4-ZIF-8 | 1.0 M KHCO3 | H-Cell | 98 | 71.5 | 54 |
| Co–N2 | 0.5 M KHCO3 | H-Cell | 94 | 18.1 | 55 |
| Ni/Fe–N–C | 0.5 M KHCO3 | H-Cell | 98 | 9.5 | 56 |
| Cu/Ni(OH)2 | 0.5 M KHCO3 | H-Cell | 92 | 4.3 | 57 |
| ZnNx/C | 0.5 M KHCO3 | H-Cell | 95 | 13.62 | 58 |
| (Cl, N)–Mn/G | 0.5 M KHCO3 | H-Cell | 97 | 10 | 59 |
| CoPc | Anion exchange membrane | MEA | 95 | 150 | 45 |
| Ni-NCB | Anion-exchange membrane | MEA | 99 | 130 | 41 |
3. The second step: convert CO to C2+
The direct electroreduction of CO2 into C2+ products suffers from low activity in non-alkaline electrolyte and poor selectivity.60 As summarized in section 2, in the first step, CO2-derived CO provides sustainable feedstock in CO electrolysis to enhance the performance for producing C2+ products and to circumvent the reaction between the alkaline electrolyte and CO2. The Cu-based electrocatalyst is the only known material with the ability to reduce CO to C2+ products with appreciable performance. Many strategies have been developed towards engineering Cu-based catalysts, such as grain boundary engineering,61,62 phase engineering,63 crystal facet engineering,6,64 and nanostructuring,65,66 to improve the product selectivity and catalytic activity.Among the mentioned strategies, much attention has been paid to grain boundary engineering for developing oxide-derived Cu catalysts (OD-Cu) due to their ability of improving the selectivity of C2+ products in CO electrolysis.61,62 Kanan and co-workers reported that OD-Cu catalysts can improve the intrinsic activity towards C2+ production in CO electrolysis.62 OD-Cu electrocatalysts were prepared by annealing polycrystalline Cu foil in air at 500 °C to form a Cu2O layer on the Cu foil surface and subsequently reducing this oxide layer to metallic Cu. OD-Cu 1 was prepared by the electroreduction of Cu2O in an aqueous solution at ambient temperature; OD-Cu 2 was obtained via the reduction of Cu2O in H2 atmosphere at 130 °C. As shown in Fig. 5a, the high-resolution TEM image of the OD Cu 1 catalyst showed distinct grain boundaries. Benefiting from the highly active sites at the grain boundary, OD-Cu achieved multi-carbon oxygenates (ethanol, acetate, and n-propanol) with a faradaic efficiency of 57% in CO-saturated alkaline electrolyte (Fig. 5b). Therefore, engineering the grain boundary is a promising strategy to change the intrinsic catalytic property of nanocrystalline materials toward C2+ production.
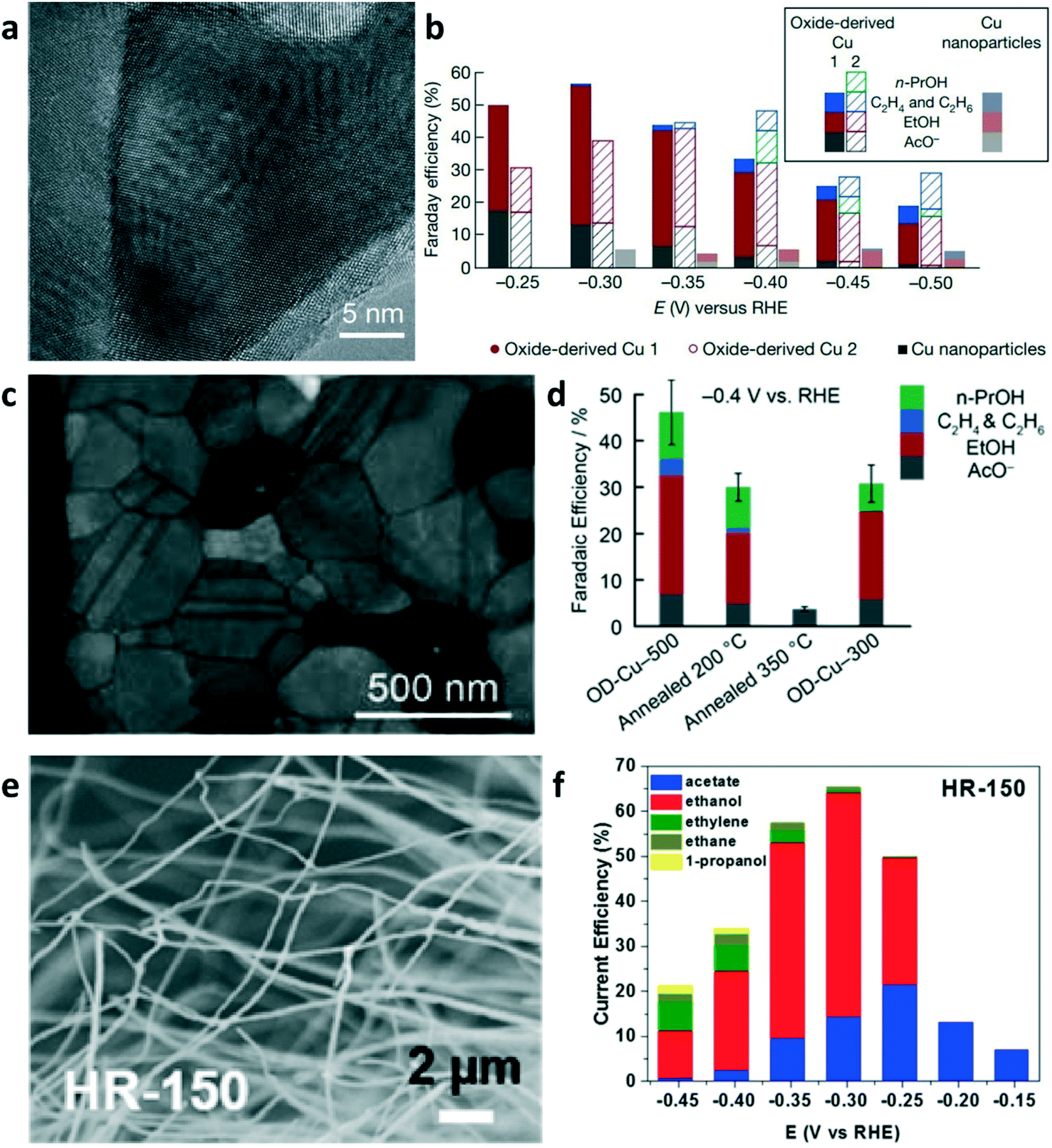 | ||
| Fig. 5 (a) High-resolution TEM image of the oxide-derived (OD) Cu 1 electrode. (b) FEs of C2+ products at various applied potentials. Reproduced with permission.62 Copyright 2014, Nature Publishing Group. (c) TEM image of OD-Cu-500. (d) FEs of C2+ products at −0.4 V vs. RHE in CO-saturated 0.1 M KOH. Reproduced with permission.61 Copyright 2015, American Chemical Society. (e) SEM image of Cu nanowires produced by hydrogen reduction at 150 °C (HR-150). (f) Product distribution of Cu nanowires (HR-150) depending on the electrode potential in CO electrolysis. Reproduced with permission.68 Copyright 2017, American Chemical Society. | ||
To reveal the effect of the annealing temperature of OD-Cu electrodes, scientists annealed the OD-Cu-500 catalyst (an OD-Cu catalyst obtained by 350 °C air oxidation) under N2 at 200 and 350 °C in order to promote grain coalescence at a different degree.61 The TEM image of OD-Cu-500 shows the distribution of grain boundaries in a dense polycrystalline network (Fig. 5c). Upon annealing, the intrinsic activity of OD-Cu for CO reduction was significantly reduced (Fig. 5d). The electrode annealed at 350 °C showed less than 5% faradaic efficiency for CO reduction. The temperature-programmed desorption of CO on OD-Cu revealed the presence of strong CO binding sites on the OD-Cu surface. Strong CO binding may increase the CO coverage on the catalyst surface to enhance the rate of CO reduction. The thermal annealing of OD-Cu electrodes can reduce the proportion of strong-binding sites on the surface, which is responsible for low activity in CO reduction. Xu et al. employed operando attenuated total reflection surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS) to identify the CO adsorption band on OD-Cu at 2058 cm−1, which is different from those on polycrystalline copper but similar to the CO adsorption band on Cu(100).67 OD-Cu may have more Cu(100) facets, which are regarded as sites for C–C coupling in CO2/CO reduction. Unlike the use of Cu2O as the precursor, Wang and co-worker prepared Cu nanowires by the thermal reduction of CuO nanowires in H2 at 150–300 °C.68Fig. 5e shows the SEM image of Cu nanowires produced by hydrogen reduction at 150 °C (HR-150). The HR-150 Cu nanowires achieved an ethanol FE of 50% and a total FE of 65% for CO reduction at −0.3 V vs. RHE (Fig. 5f). The electrosorption of hydroxide (OHa), temperature-programmed CO desorption, and DFT calculations suggest that the high ethanol selectivity is attributed to the coordinately unsaturated (110) surface sites on the Cu nanowires. Although OD-Cu catalysts can enhance the activity and selectivity of the C2+ products, a full picture of the structure–activity relationship that can guide the rational design of catalysts is still missing.
Most studies on CO reduction have been performed in the H-cell with electrocatalysts immersed in CO-saturated aqueous electrolyte, which suffer from mass transport limitations due to the extremely low solubility of CO in aqueous solution (1 mM in 1 bar saturated H2O).69 The flow-cell reactor with gas diffusion electrodes (GDE) is designed to circumvent mass transport limitations by directly feeding gaseous reactants to the electrode–electrolyte interface. As shown in Fig. 6a, the triple-phase boundary was formed at the catalyst particles, liquid electrolyte, and gaseous reactants, which can achieve high rates of CO reduction. Jiao et al. first used a flow system to achieve high rates for CO reduction using commercial copper particles and OD-Cu particles as the electrocatalysts.23 The morphology and particle size of commercial micrometer copper and OD-Cu particles are shown in the SEM images (Fig. 6b and c). The CO reduction performances are summarized in Fig. 6d and e. OD-Cu catalysts achieved a C2+ selectivity of 91% and a partial current density over 635 mA cm−2 in electrochemical CO reduction. This work also compared the performance of CO2RR and CORR to illustrate the advantages of CO electrolysis for C2+ production. Kanan and co-workers employed a membrane electrode assembly cell with GDE and achieved a current density over 100 mA cm−2 for the formation of C2+ as well as produced 1.1 M acetate at a cell potential of 2.4 V after 24 h electrolysis.69 To further illustrate the advantages of CO over CO2 as the reactant for C2+ production at high current densities, O. Hinrichsen and co-workers reported the performances of CO2RR and CORR on gas diffusion electrodes using commercial Cu-powders as the catalyst layer in a flow cell.25 The results showed that higher current densities for C2+ products can be achieved at lower working electrode potentials in CORR compared to CO2RR. Cu nanoparticles (NPs, 40–60 nm) achieved a C2+ FE of 89% at the current density of 300 mA cm−2 in galvanostatic CO electrolysis, while 34% FE (300 mA cm−2) was obtained in direct CO2 electroreduction. Fig. 6f and g show the FE of ethylene on Cu NPs of different sizes at various current densities for CO2RR and CORR. In CORR, the maximum ethylene FE of 54% was achieved using Cu NPs (40–60 nm) but in CO2RR, a maximum FE of ethylene of only 32% on Cu NPs (60–80 nm) was achieved. These results demonstrate that the two-step conversion of CO2 to C2+ is definitely deserves to be further explored for any potential application in the industry.
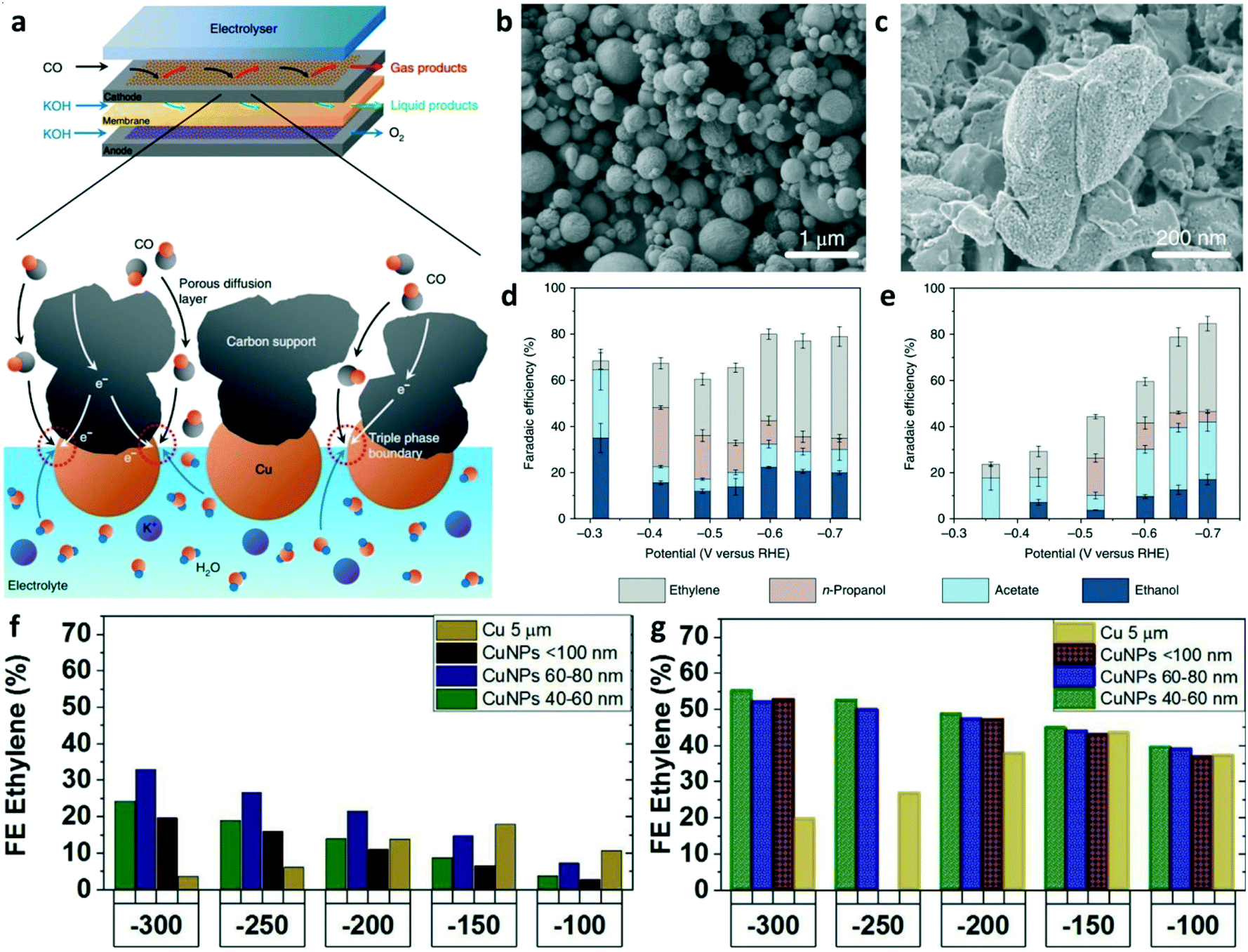 | ||
| Fig. 6 (a) Schematic illustration of the flow-cell electrolyzer and the triple-phase boundary in the flow-cell reactor. (b) and (c) SEM images of commercial micrometer copper and OD-Cu particles, respectively. (d) and (e) FEs of C2+ for OD-Cu and micrometer copper, respectively. Reproduced with permission.23 Copyright 2018, Nature Publishing Group. (f) FE of ethylene on different sizes of Cu NPs at various current densities for CO2RR and CORR (g). Reproduced with permission.25 Copyright 2019, Elsevier. | ||
To date, although high FEs of C2+ with high current density have been achieved in CO electrolysis, the high selectivity for single multicarbon oxygenate production is still an open challenge. Our group reported freestanding high-quality Cu nanosheets synthesized through solution-phase synthesis.6 As shown in Fig. 7a, the Cu nanosheets present a two-dimensional (2D) triangular shape morphology with an average edge length of 1.7 ± 0.5 μm. The HRTEM image in Fig. 7b shows that Cu nanosheets selectively expose the Cu(111) surface. As a model catalyst for the electroreduction of CO, highly selective CO-to-acetate conversion was achieved on the Cu nanosheets, with the FE of acetate as high as 48% at an acetate partial current density up to 131 mA cm−2 in CO electrolysis (Fig. 7c). Further experimental analysis and computational studies suggested that the high acetate selectivity is due to the suppression of ethylene and ethanol formation, and the acetate formation goes through a ketene intermediate. Jaramillo and co-worker proposed that increasing the roughness factor (RF) of Cu catalysts can be used to increase the rates of C2+ production.70 CuO nanosheets were prepared by oxidizing the surface of a polished Cu foil. The as-synthesized CuO nanosheets were reduced to metallic Cu nanosheets under electrochemical conditions. The TEM images of CuO and Cu nanosheets are shown in Fig. 7d and e, respectively. As shown in Fig. 7f, a maximum FE of 60% was achieved for ethanol formation at −0.33 V vs. RHE, which was believed to be benefited by increasing the RF of the Cu catalyst to suppress hydrocarbon and hydrogen production.
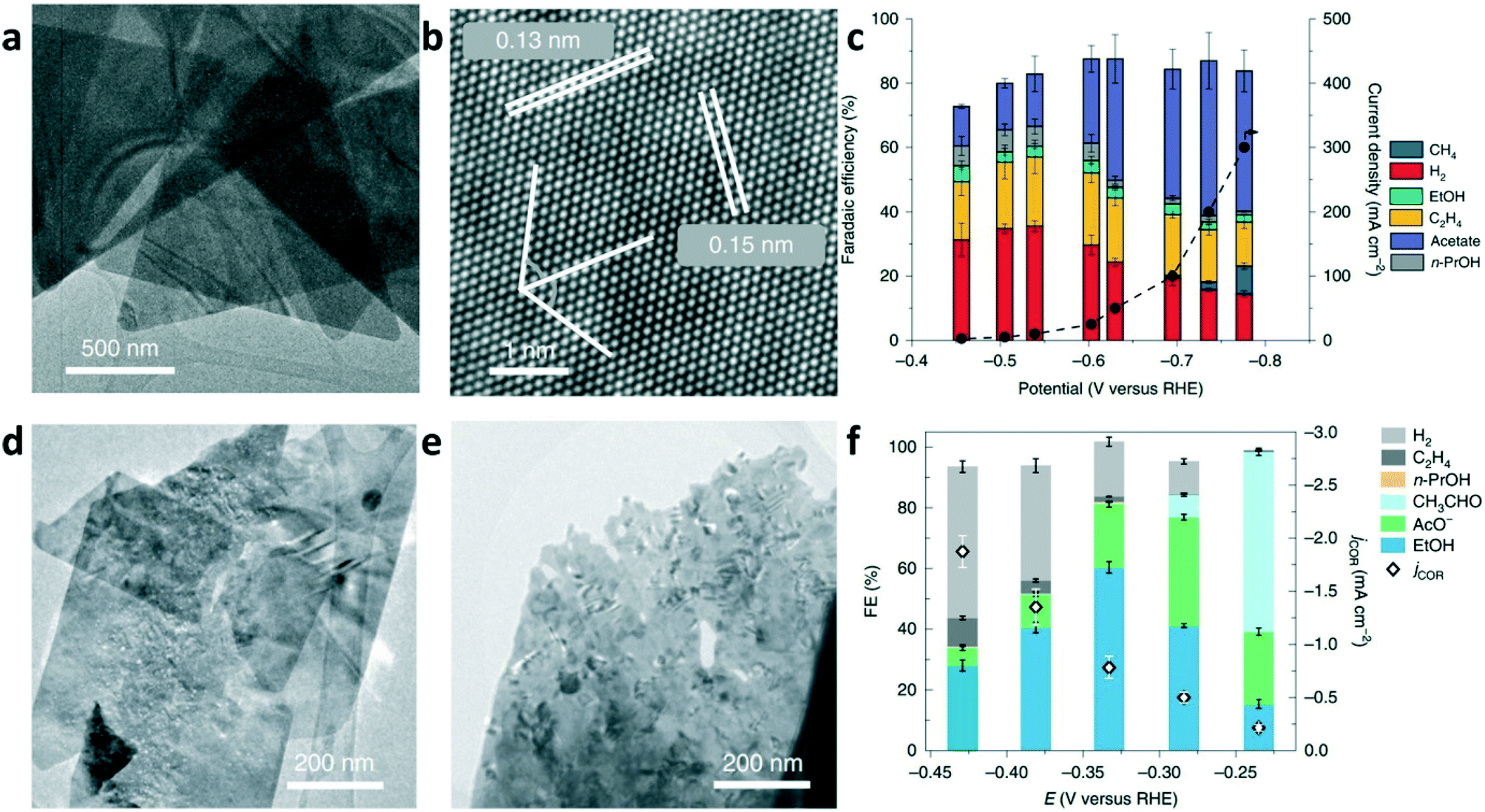 | ||
| Fig. 7 (a) TEM image and (b) HRTEM of the Cu nanosheets selectively exposing the {111} facets. (c) Total current density and FEs versus various applied potentials for CO electrolysis on Cu nanosheets in 2 M KOH. Reproduced with permission.6 Copyright 2019, Nature Publishing Group. (d) TEM image of representative CuO nanosheets. (e) TEM image of Cu nanosheet after the reduction of CuO nanosheets. (f) FE of products and the geometric current densities for CORR as functions of the applied potential. Reproduced with permission.70 Copyright 2019, Nature Publishing Group. | ||
The development of catalysts that can convert CO to C2+ products with high selectivity at commercially relevant current densities are highly desired, offering more choices for tandem catalyst design in the two-step route of CO2 to C2+ reduction. Undoubtedly, more mechanistic investigations are necessary for the rational design of the catalysts available. The optimization of reactor configurations and increasing the intrinsic activity and selectivity of the electrocatalysts are effective routes for improving the overall performance of CO2 to C2+ conversion.
4. Tandem catalysts
In recent years, tandem catalysis has been extensively explored in thermal heterogeneous catalysis. Tandem catalysis can be achieved on dual catalyst systems or multifunctional catalysts that have more than one type of active site.27 It is widely accepted that CO is the key intermediate to C2+ products in CO2 electroreduction. Therefore, tandem catalysis can be used in CO2 electrolysis where CO2 was converted to CO on the first catalyst or the active site and subsequently reduced to C2+ products by the second catalyst or the active site.24 Cascade (domino) catalysis avoids product separation and stop-and-go operation. Tandem catalysts for CO2 electrolysis can have a vertically aligned layer-by-layer structure with two catalysts or a single catalyst with more than one type of active site. As discussed in the previous two section, the consideration for CO production and for C–C coupling are necessary to design tandem catalysts.To rationally control the catalytic selectivity in CO2RR, Cu–Ag composites with two types of active sites were employed for shifting the selectivity toward the C2+ products.71,72Operando Raman spectroscopy confirmed CO formation on the Ag sites and subsequent CO spillover on the Cu surface for hydrogenation.71 The Cu–Ag tandem catalysts were prepared by oxide-derived Cu nanowires mixed with Ag nanoparticles. As shown in Fig. 8a, CO was produced on Ag nanoparticles and then spilled to Cu–Ag boundaries for C–C coupling to selectively generate ethanol.72 Electrolysis was performed in 0.1 M KHCO3. Compared with the FEs of CO2RR products on OD-Cu NW (Fig. 8b) and Ag-20 (meaning, Ag NPs with diameter of 20 nm, Fig. 8c), Cu(Ag-20)20 (meaning, Cu nanowire/Ag NPs composite at the Ag/Cu nominal molar ratio of 20, Fig. 8d) showed a 5-fold improvement in the ethanol current density. The computational study showed the selective generation of ethanol via Langmuir–Hinshelwood *CO + *CHx (x = 1, 2) coupling at the Cu–Ag boundaries. Sun and co-worker prepared Au–bipy–Cu tandem catalysts by assembled Au NPs on Cu nanowires using 4,4′-bipyridine (bipy) as the linker (Fig. 8e).73 The TEM image shows that the Au NPs were uniformly dispersed on Cu nanowires (Fig. 8f). CO2-Saturated aqueous 0.1 M KHCO3 was used as the electrolyte. In addition to the tandem catalytic effect to improve the C2+ formation, bipy also promotes the catalytic performance in CO2 electroreduction. Au–bipy–Cu-1/2 (Au/Cu = 1/2) achieved a CH3CHO FE of 25%, which is the highest aldehyde selectivity ever reported for CORR and CO2RR (Fig. 8g). The assembly strategy offers plenty of room for designing tandem catalysts with tunable catalytic selectivity in CO2RR. Tandem catalysis can also be achieved on multicomponent catalysts with more than one type of active site. Ag1–Cu1.1 (meaning that the mass ratio of Cu to Ag is 1.1) nanodimers (NDs) were prepared by a seeded-growth approach using Ag NPs as the nucleation seeds.74 The HAADF-STEM image (Fig. 8h) reveals the nanoparticles containing two domains, Ag and Cu. The EDS mapping confirmed a segregated distribution of Ag and Cu in the same nanoparticle (Fig. 8i). The CO2RR was performed in CO2-saturated 0.1 M KHCO3 solution. The C2H4 FE of ∼40% (Fig. 8j) was obtained on the Ag1–Cu1.1 NDs because of tandem catalysis and electronic effects. This work demonstrated that tandem catalysis can be achieved by designing multicomponent catalysts.
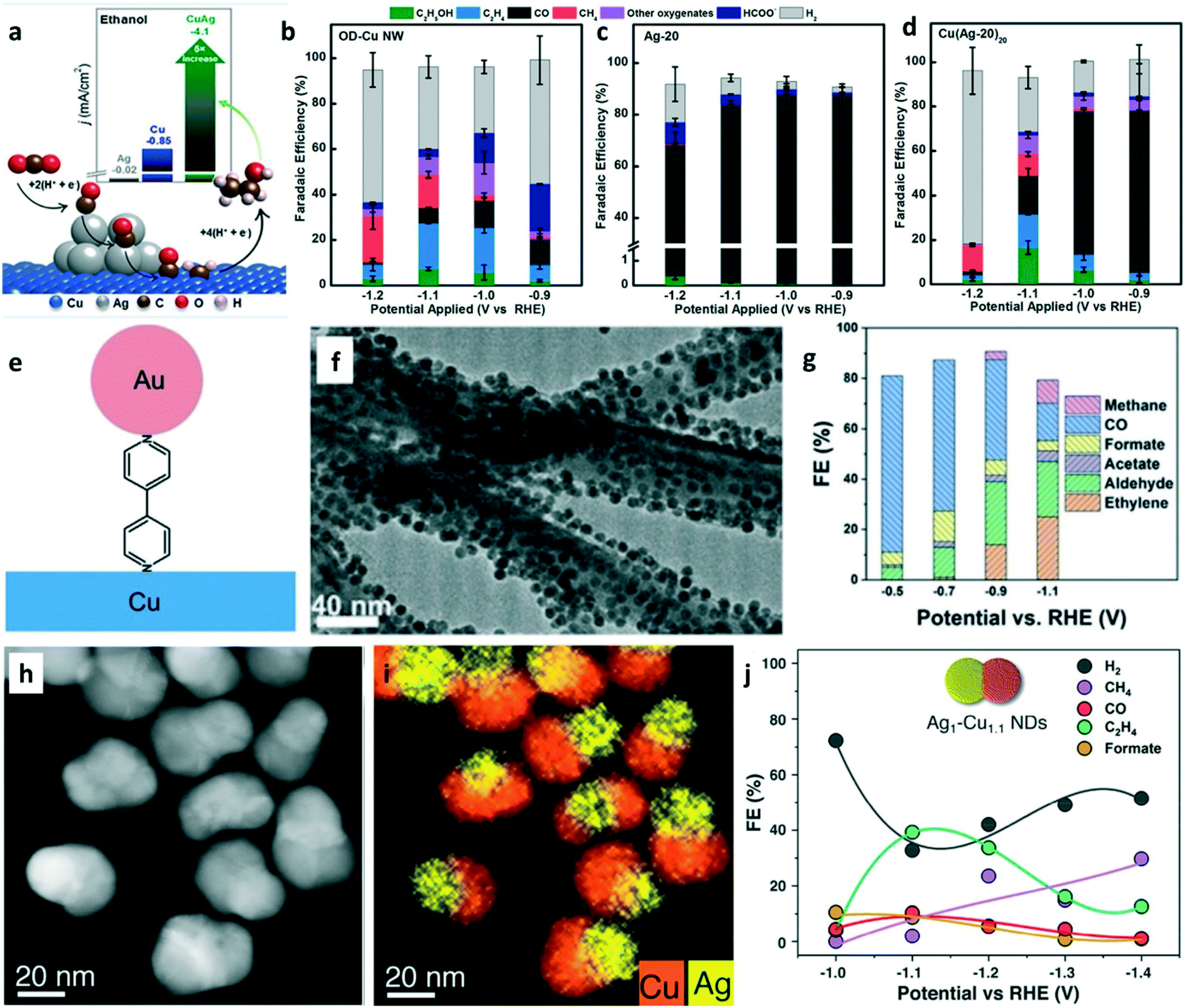 | ||
| Fig. 8 (a) Schematic illustration of the copper–silver composite for enhancing the selectivity of ethanol. FEs of CO2RR products on (b) OD-Cu NW, (c) Ag-20, and (d) Cu(Ag-20)20. Reproduced with permission.72 Copyright 2020, American Chemical Society. (e) Schematic of the Au–bipy–Cu composite catalyst. (f) TEM image of Au–bipy–Cu-1/1. (g) FEs of the products of Au–Cu–bipy catalysts for CO2RR at various potentials. Reproduced with permission.73 Copyright 2019, John Wiley and Sons. (h) HAADF-STEM image of Ag1–Cu1.1 nanodimers (NDs). (i) EDX elemental map of Ag1–Cu1.1 NDs. (j) FEs of the CO2RR products on Ag1–Cu1.1 NDs. Reproduced with permission.74 Copyright 2019, American Chemical Society. | ||
Due to its relatively low cost (vs. precious metals), commercial ZnO has been explored as a CO-selective catalyst. Inspired by the higher efficiency of the plug flow reactor, Wu and co-workers designed layer-by-layer tandem catalysts with the ZnO catalyst layered on top of the Cu catalyst layer, as shown in Fig. 9a.75 The Cu–ZnO tandem electrodes were used in a flow-cell with 1 M KOH electrolyte; the CO2-to-CO conversion occurred on the ZnO catalyst layer and resulted in CO being subsequently reduced to C2+ products in the Cu catalyst layer. The rates of C2+ products on Cu–ZnO tandem catalysts were superior to those on bare Cu electrode and other Cu- and Zn-based bimetallic catalysts (Fig. 9b). The optimal Cu1.0/ZnO0.20 (meaning, the loading of 1.0 mg cm−2 Cu and 0.20 mg cm−2 ZnO) tandem catalysts exhibited a maximum FE of 78% for C2+ products with C2+ partial current density up to 466 mA cm−2. The enhanced CO2-to-C2+ yield was attributed to the breaking of the linear scaling relations because of the segregated active sites. This work highlighted the importance of spatial management in tandem catalyst design. Tandem gold on copper electrocatalyst (Fig. 9c) was explored to improve the activity and selectivity towards alcohols in CO2RR.76 Electrochemical experiments were performed in 0.1 M KHCO3. As shown in Fig. 9d, the Au/Cu tandem catalyst favored the production of alcohols over hydrocarbons. The rate of CO2RR to >2e− products (i.e., excluding HCOOH, CO, and H2) on the Au/Cu tandem catalyst was over 100 times higher than that on copper at low overpotentials, which may be due to the high local concentration of CO (derived from CO2–CO conversion on neighboring Au) on the copper surface to accelerate the rate of C–C coupling. Remarkably, the local concentration of CO near the tandem catalyst surface reached its saturation limit, which is difficult to achieve in CO reduction.
 | ||
| Fig. 9 (a) Schematic illustration of the layered structure of the tandem catalysts in the two-step conversion of CO2 to C2+. (b) FEs and current densities of C2+ on Cu/ZnO tandem catalyst and Cu and ZnO combination catalysts. Reproduced with permission.75 Copyright 2020, Elsevier. (c) SEM image of the gold nanoparticles on a polycrystalline copper foil (Au/Cu) catalyst, scale bars: 100 nm, insets: 20 nm. (d) Rate of CO2 reduction to >2e− products of Au/Cu, copper, and gold catalysts. Reproduced with permission.76 Copyright 2018, Nature Publishing Group. | ||
As mentioned in section 2, molecular electrocatalysts (e.g., metal phthalocyanine and metal tetraphenylporphyrin) have attracted great attention for CO2-to-CO conversion. The catalytic performance can be tuned to the electronic structure of the active sites by the ligands and the central metal atoms. As shown in Fig. 10a, the molecule–metal composite was designed by anchoring the molecular complex on the Cu surface.77 The porphyrin-based metallic complex can efficiently convert CO2 to CO, which creates the CO-rich local environment for facilitating C–C coupling and even optimizes the reaction pathway for ethanol. Therefore, ethanol FE of 41% (Fig. 10b) and partial current density of 124 mA cm−2 (Fig. 10c) were achieved on the FeTPP[Cl]/Cu (meaning, 5,10,15,20-tetraphenyl-21H,23H-porphine iron(III) chloride was immobilized on the Cu electrode) tandem catalysts at −0.82 V (vs. RHE) using 1 M KHCO3 as the electrolyte. This work demonstrated the potential of molecular-complex/copper tandem catalysts to selectively produce C2+ products via enriched intermediates at the interfaces. Unlike the composites tandem catalysts with multi-sites at the nanoscale mentioned earlier, the layer-by-layer and physical mixture tandem catalysts were also extensively explored in CO2 electrolysis. The Cu500Ag1000 (meaning, the Cu–Ag tandem catalyst with 500 μg cm−2 Cu and 1000 μg cm−2 Ag) catalyst was prepared by a physical mixture of Cu and Ag nanopowders.78 The cross-section SEM image of Cu500Ag1000 (Fig. 10d) shows that the thickness of the catalyst layer on carbon paper was several microns. The partial current densities of C2+ products on the Cu500Ag1000 tandem catalysts is 160 mA cm−2 at −0.7 V using 1.0 M KOH as the electrolyte, while the partial current densities of C2+ products on the Cu500 catalysts are only about 37 mA cm−2 (Fig. 10e). Interestingly, the partial current densities of C2+ products on Cu500Ag1000 were much higher than that of Cu500 and Ag1000. To analyze the distribution of the C2+ product (Fig. 10f), the enhancement factor (EF) was defined by the partial current on tandem Cu500Ag1000 divided by that on the Cu500 catalyst. Oxygenate products (ethanol and acetate) were favored over ethylene on the Cu500Ag1000 tandem catalyst, which was consistent with the previous reports.31,51 Wu and co-worker designed layer-by-layer Cu–Ag tandem electrodes for efficiently converting CO2 to C2+ products.79 The structure of the Cu/Ag tandem electrodes is shown in Fig. 10g; the top Ag layer and bottom Cu layer are presented in the EDS elemental maps. The cross-section SEM image shows that the thickness of the catalyst layer is about 2 μm. The performance of CO2RR was evaluated in 1.0 M KOH. The Cu/Ag (0.8 mg cm−2/0.2 mg cm−2) tandem catalysts achieved the highest C2+ FE of 87% at −0.78 V among all the Cu/Ag tandem electrodes and pure Cu electrode (Fig. 10h). It has been proved that the Ni–N–C catalysts are highly active for the production of CO from CO2 reduction. Therefore, Ni–N–C was used as CO-selective catalyst layers in the tandem electrodes. Fig. 10i shows the cross-sectional SEM image and the EDS mapping images for Cu/Ni–N–C tandem catalysts. The maximum C2+ selectivity of Cu/Ni–N–C tandem catalysts is lower than that of Cu/Ag (Fig. 10j). But the optimized Cu/Ni–N–C tandem catalysts achieved an FE of 62% for C2H4 with a partial current density of 415 mA cm−2. The Cu-based/molecular-complex and Cu/M–N–C tandem catalysts have plenty of room to explore for improving the selectivity and activity of C2+ products in CO2 electroreduction.
 | ||
| Fig. 10 (a) Schematic illustration of Cu-molecular complexes tandem catalysts for ethanol production. (b) FEs of ethanol on FeTPP[Cl]/Cu and Cu catalysts at various applied potentials. (c) Partial current densities of ethanol on FeTPP[Cl]/Cu and the Cu catalysts. Reproduced with permission.77 Copyright 2019, Nature Publishing Group. (d) Cross-sectional SEM image of the Cu500Ag1000 catalyst supported on carbon paper. (e) The partial current density of C2+ products versus various applied potentials on Cu500Ag1000, Cu500, and Ag1000 catalysts. (f) An enhancement factor of total C2+ and individual products at various applied potentials on the tandem Cu500Ag1000. Reproduced with permission.78 Copyright 2020, Elsevier. (g) Cross-sectional SEM image (scale bars: 4 μm) and EDS mapping (scale bars: 5 μm) of the Cu/Ag tandem catalyst. (h) FEs of C2+ products on various Cu/Ag tandem catalysts. (i) Cross-sectional SEM image and EDS mapping of the Cu/Ni–N–C tandem catalyst. (j) FEs of C2+ products on various Cu/Ni–N–C tandem catalysts. Reproduced with permission.79 Copyright 2020, Elsevier. | ||
Besides CO2-to-C2+ conversion, CO2-to-methane (CH4) production can also be achieved on tandem catalysts. The electrochemical reduction of CO2 to CH4 is another promising route for carbon utilization. Recently, Wang and co-workers developed a CoPc/Zn–N–C tandem catalyst for CH4 production.80 The HAADF-STEM image (Fig. 11a) of Zn–N–C shows that the single Zn atoms were distributed in the N–C framework. For the CoPc/Zn–N–C tandem catalyst toward CO2RR, the CO2 is first reduced into CO on CoPc and then CO spills over onto Zn–N–C for further reduction to CH4. The CH4 FE of 18.3% (Fig. 11b) and maximum CH4 current density of 44.3 mA cm−2 (Fig. 11c) were achieved on the tandem catalysts at −1.24 V vs. RHE using 1 M KOH as the electrolyte. The high CH4 production rate was attributed to the CoPc-enhanced availability of *H over adjacent N sites in Zn–N4.
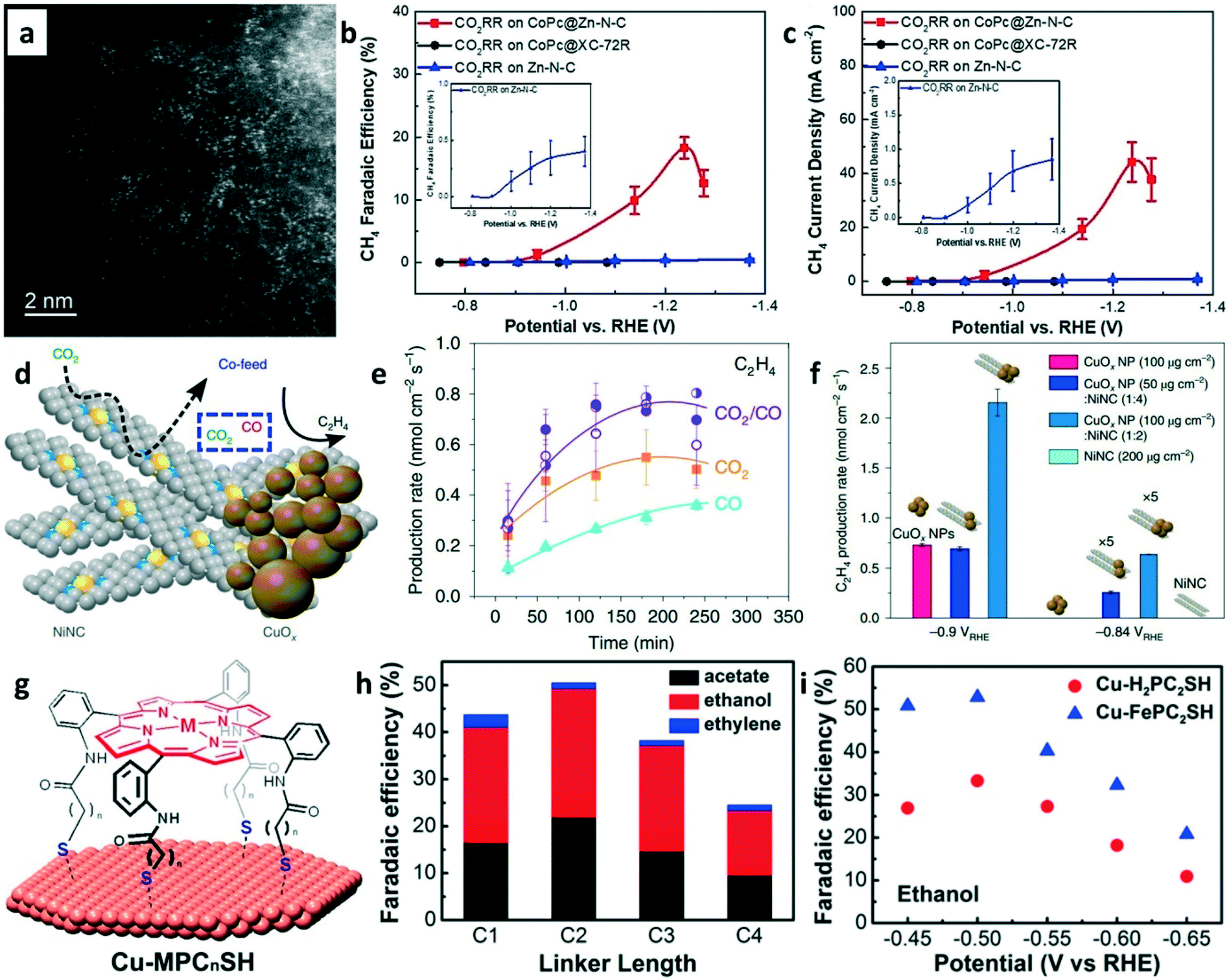 | ||
| Fig. 11 (a) HAADF-STEM image of Zn–N–C. (b) FEs of CH4 on different catalysts at various applied potentials. (c) The partial current density of CH4 on different catalysts at various applied potentials. Reproduced with permission.80 Copyright 2019, John Wiley and Sons. (d) Schematic of CuOx–Ni–N–C tandem catalysts for ethylene production. (e) Production rates of ethylene with a different feed ratio of CO2/CO. (f) Ethylene production rate on different catalysts at various applied potentials. Reproduced with permission.81 Copyright 2019, Nature Publishing Group. (g) Schematic of the functionalization of Cu surfaces with porphyrin cages. (h) FEs of C2+ products on Cu-porphyrin with different linker lengths in CO electrolysis. (i) FEs of ethanol on Cu–H2PC2SH and Cu–FePC2SH in CO-saturated 0.1 M KOH (aq). Reproduced with permission.82 Copyright 2017, American Chemical Society. | ||
CO2 and CO co-exist at the C–C coupling site of the tandem catalyst but whether the presence of CO promotes the coupling rate of CO2 is unknown. CuOx/Ni–N–C tandem catalysts (Fig. 11d) were explored for ethylene production in 0.1 M KHCO3 electrolyte.81 As shown in Fig. 11e, the C2H4 production rates were significantly enhanced when the CO2/CO mixture was used as the feedstock. The CuOx/Ni–N–C tandem catalysts were used for CO2RR by mimicking the CO2/CO mixture feeding. Interestingly, enhanced C2H4 production rates (Fig. 11f) were achieved on CuOx/Ni–N–C tandem catalysts compared to pure CuOx NPs. The reason is well-known, i.e., high local CO concentration increases the *CO coverages on the catalysts surface for enhancing the dimerization rates. A modular synthetic approach was proposed to design the tandem catalysts for CO reduction in 0.1 M KOH electrolyte. Thiol-terminated metalloporphyrins were self-assembled on copper electrodes to construct the molecule–metal interfaces (Fig. 11g).82 The FE of ethanol and acetate in CO reduction can be regulated via the linker lengths of porphyrin (Fig. 11h). The highest ethanol FE of 52% was obtained on the Cu/FePC2SH catalyst at −0.5 V vs. RHE (Fig. 11i). Although this tandem catalyst was used in CORR, it could be an important model for designing tandem catalysts towards CO2RR.
The tandem catalysis for CO2-to-C2+ conversion is a powerful approach to close the anthropogenic carbon cycle and the production of chemicals. The tandem catalysis can be achieved on one single particle with more than one type of active site (e.g., Cu–Ag nanodimers), layer-by-layer tandem electrode, physical mixing of different catalysts, and assembly composites (e.g., Au–bipy–Cu). Developing tandem catalysts to improve the formation rates of overall C2+ products or a specific C2+ product at commercially relevant current densities with high FE are desired for the production of chemicals and storage of renewable electricity. To date, most tandem catalysts have been employed in H-cell and flow-cell for CO2RR. The MEA-based electrochemical cell uses a solid electrolyte instead of a liquid electrolyte, thus avoiding the further separation of products from the liquid electrolyte. Therefore, the tandem catalysis strategy applied in the MEA electrolyzer may enable CO2RR implementation in industries.
5. Tandem reaction systems
Unlike the tandem catalysts where the two-step reactions occur in one electrochemical cell, tandem reaction systems were designed to facilitate two-step CO2 electrochemical reductions in two separate electrochemical cells. As shown in Fig. 12a, CO2-to-CO conversion occurred in the first flow cell with the Ag catalyst layer, and then the output stream of CO, H2, and CO2 (unreacted in the previous step) was separated through the absorption column.24 The gaseous product output (CO and H2) from the absorption column was fed into the second electrolyzer using Cu as the catalyst for the production of C2+. The two-step reactions were performed in two separated electrolyzers using different electrolytes, the first one (CO2-to-CO) with a non-alkaline electrolyte and the second one (CO-to-C2+) with an alkaline electrolyte, thus circumventing the problem of consuming hydroxide to form carbonate by CO2 when an alkaline electrolyte is used for CO2RR. Whether or not an absorption column is used between two electrolyzers can change the overall performance of CO2RR. With a CO2 absorption column, the FE of the C2+ product was increased twice compared to that without the absorption step (Fig. 12b). After two hours of electrolysis, the FE of C2H4 was slightly decreased while the FEs of ethanol and n-propanol remained stable compared with the one hour electrolysis (Fig. 12c). Moreover, CO2-to-CO conversion at the first step required a higher working electrode potential than that required by CO-to-C2+ conversion at the second step. The overall FE of the C2+ products obtained from the two-step electrolysis in the tandem reactors was nearly two times higher than that obtained from the single-step electrochemical reduction of pure CO2 (Fig. 12d).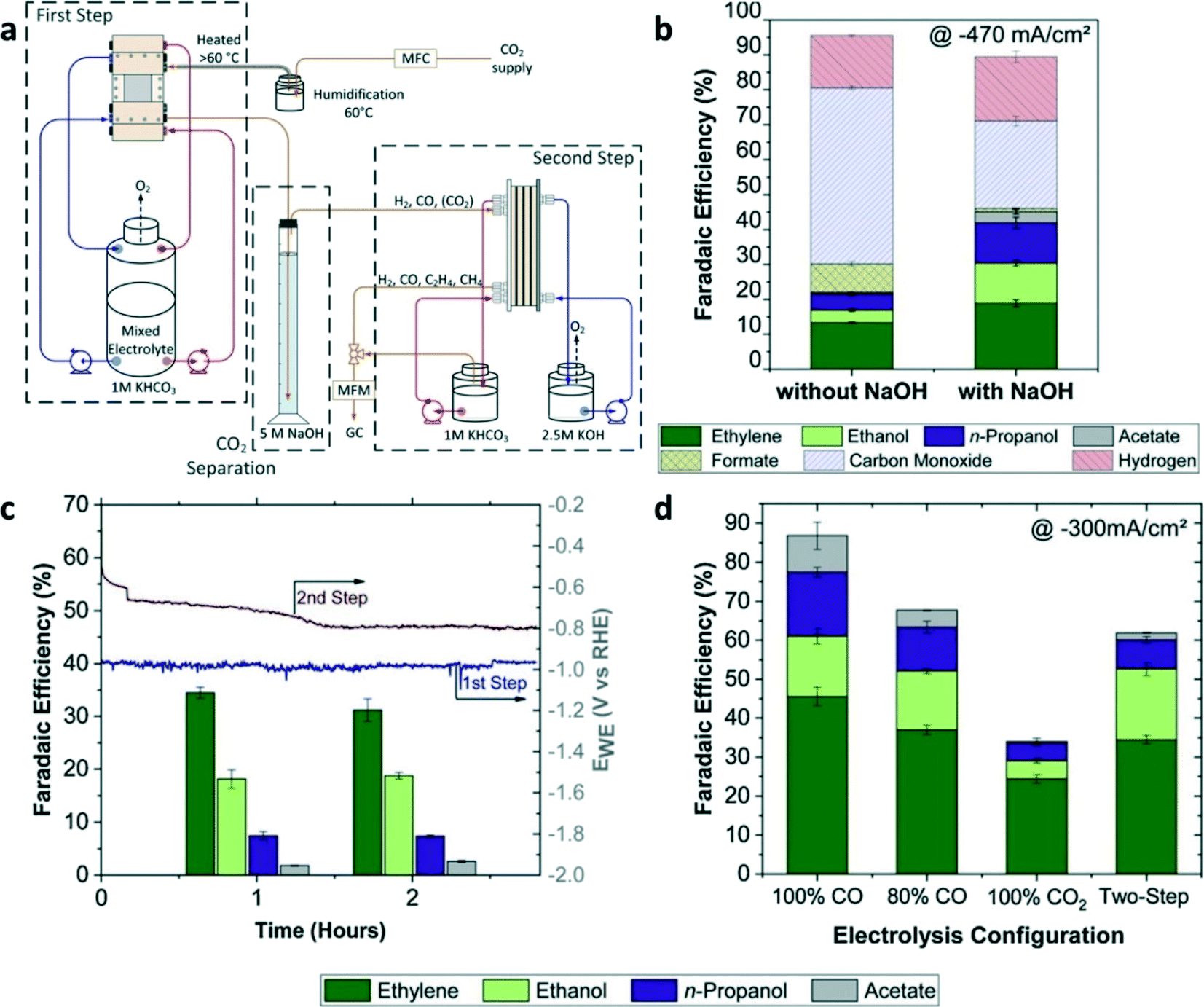 | ||
| Fig. 12 (a) Schematic illustration of the tandem reaction systems for CO2RR. (b) FE of the products with or without CO2 absorption using NaOH as the absorber at a total current density of −470 mA cm−2. (c) FEs of C2+ products at the first and second step after 1 and 2 h of CO2 electrolysis with a total current density of −300 mA cm−2. (d) FEs of C2+ products from single-step electrolysis with a different feed ratio of CO2/CO at a total current density of −300 mA cm−2 compared to tandem reaction systems. Reproduced with permission.24 Copyright 2020, Elsevier. | ||
It was proved that the alkaline electrolyte can improve the selectivity and formation rates of C2+ products in CO2RR and CORR. Therefore, the second step for the reduction of CO to C2+ can be performed in alkaline electrolytes to improve the overall performance of CO2RR in tandem reaction systems. To increase the concentration of CO in the output of the first step, CO-selective catalysts with high single-pass conversion are desired, which can reduce the separation cost and improve the overall performance. Jiao and co-workers optimized the test conditions and achieved maximum single-pass CO2 conversion. The maximum amount of CO2 being reduced to CO was limited to 43% in the flow cell.83
6. Conclusion and outlook
Herein, we discussed the advantages of the two-step approach for CO2RR as well as many applicable strategies (e.g., nanostructuring, alloying, phase engineering, grain boundary engineering, and crystal facet engineering) to improve the intrinsic activity of the catalysts for CO2RR/CORR.21 To enable the two-step CO2RR, both tandem catalysis in a single reactor and tandem reactors have been explored. The most important thing is that the two-step approach can avoid the problem of carbonate formation in the alkaline electrolyte, which is a major hurdle in the direct CO2RR to C2+, while taking the advantage of alkaline electrolytes for promoting the rate of C–C coupling in CO-to-C2+ conversion. To push CO2RR closer to commercialization, we would like to emphasize the following directions: (1) development of stable copper-based catalysts that can work during long-term operation, (2) optimizing the microstructure of the tandem catalyst and the structure of the tandem electrode for the effective production of C2+, (3) increasing the single-pass CO2-to-CO conversion, (4) incorporation of a tandem catalysis strategy into MEA-based electrochemical cells to avoid product separation and concentration processes from liquid electrolytes, (5) reactor engineering, and (6) possible coupling of other useful reactions with CO2RR to produce chemicals.Chemical production through CO2RR is not yet competitive to be employed in the petrochemical industry. Due to the fact that fossil fuels on Earth are depleting, the most promising alternative energy system, hydrogen economy, however, does not have the capability to directly provide the necessary daily chemicals that the petrochemical industry is providing. In that case, CO2RR may be our only option for the sustainable production of necessary chemicals.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the support from the National Natural Science Foundation of China, under Award 21972016 and 21773023.References
- D. F. Gao, R. M. Aran-Ais, H. S. Jeon and B. Roldan Cuenya, Nat. Catal., 2019, 2, 198–210 CrossRef CAS.
- Y. Y. Birdja, E. Perez-Gallent, M. C. Figueiredo, A. J. Gottle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS.
- L. M. Wang, W. L. Chen, D. D. Zhang, Y. P. Du, R. Amal, S. Z. Qiao, J. W. Bf and Z. Y. Yin, Chem. Soc. Rev., 2019, 48, 5310–5349 RSC.
- Y. J. Sa, C. W. Lee, S. Y. Lee, J. Na, U. Lee and Y. J. Hwang, Chem. Soc. Rev., 2020, 49, 6632–6665 RSC.
- W. Luc, X. B. Fu, J. J. Shi, J. J. Lv, M. Jouny, B. H. Ko, Y. B. Xu, Q. Tu, X. B. Hu, J. S. Wu, Q. Yue, Y. Y. Liu, F. Jiao and Y. J. Kang, Nat. Catal., 2019, 2, 423–430 CrossRef CAS.
- Y. Hori, R. Takahashi, Y. Yoshinami and A. Murata, J. Phys. Chem. B, 1997, 101, 7075–7081 CrossRef CAS.
- Y. Hori, A. Murata and Y. Yoshinami, J. Chem. Soc., Faraday Trans., 1991, 87, 125–128 RSC.
- C. T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. G. de Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Q. Zou, R. Quintero-Bermudez, Y. J. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS.
- A. Loiudice, P. Lobaccaro, E. A. Kamali, T. Thao, B. H. Huang, J. W. Ager and R. Buonsanti, Angew. Chem., Int. Ed., 2016, 55, 5789–5792 CrossRef CAS.
- S. L. Chu, X. P. Yan, C. Choi, S. Hong, A. W. Robertson, J. Masa, B. X. Han, Y. S. Jung and Z. Y. Sun, Green Chem., 2020, 22, 6540–6546 RSC.
- W. Zhang, C. Q. Huang, Q. Xiao, L. Yu, L. Shuai, P. F. An, J. Zhang, M. Qiu, Z. F. Ren and Y. Yu, J. Am. Chem. Soc., 2020, 142, 11417–11427 CrossRef CAS.
- B. X. Zhang, J. L. Zhang, M. L. Hua, Q. Wan, Z. Z. Su, X. N. Tan, L. F. Liu, F. Y. Zhang, G. Chen, D. X. Tan, X. Y. Cheng, B. X. Han, L. R. Zheng and G. Mo, J. Am. Chem. Soc., 2020, 142, 13606–13613 CrossRef CAS.
- M. Zhong, K. Tran, Y. M. Min, C. H. Wang, Z. Y. Wang, C. T. Dinh, P. De Luna, Z. Q. Yu, A. S. Rasouli, P. Brodersen, S. Sun, O. Voznyy, C. S. Tan, M. Askerka, F. L. Che, M. Liu, A. Seifitokaldani, Y. J. Pang, S. C. Lo, A. Ip, Z. Ulissi and E. H. Sargent, Nature, 2020, 581, 178–184 CrossRef CAS.
- S. Ma, M. Sadakiyo, M. Heima, R. Luo, R. T. Haasch, J. I. Gold, M. Yamauchi and P. J. A. Kenis, J. Am. Chem. Soc., 2017, 139, 47–50 CrossRef CAS.
- S. H. Zhong, X. L. Yang, Z. Cao, X. L. Dong, S. M. Kozlov, L. Falivene, J. K. Huang, X. F. Zhou, M. N. Hedhili, Z. P. Lai, K. W. Huang, Y. Han, L. Cavallo and L. J. Li, Chem. Commun., 2018, 54, 11324–11327 RSC.
- X. Wei, Z. L. Yin, K. J. Lyu, Z. Li, J. Gong, G. W. Wang, L. Xiao, J. T. Lu and L. Zhuang, ACS Catal., 2020, 10, 4103–4111 CrossRef CAS.
- D. Wakerley, S. Lamaison, F. Ozanam, N. Menguy, D. Mercier, P. Marcus, M. Fontecave and V. Mougel, Nat. Mater., 2019, 18, 1222–1228 CrossRef CAS.
- D. M. Weekes, D. A. Salvatore, A. Reyes, A. X. Huang and C. P. Berlinguette, Acc. Chem. Res., 2018, 51, 910–918 CrossRef CAS.
- C. M. Gabardo, C. P. O'Brien, J. P. Edwards, C. McCallum, Y. Xu, C. T. Dinh, J. Li, E. H. Sargent and D. Sinton, Joule, 2019, 3, 2777–2791 CrossRef CAS.
- M. Jouny, G. S. Hutchings and F. Jiao, Nat. Catal., 2019, 2, 1062–1070 CrossRef CAS.
- J. A. Rabinowitz and M. W. Kanan, Nat. Commun., 2020, 11, 5231 CrossRef CAS.
- M. Jouny, W. Luc and F. Jiao, Nat. Catal., 2018, 1, 748–755 CrossRef CAS.
- N. S. R. Cuellar, C. Scherer, B. Kackar, W. Eisenreich, C. Huber, K. Wiesner-Fleischer, M. Fleischer and O. Hinrichsen, J. CO2 Util., 2020, 36, 263–275 CrossRef.
- N. S. R. Cuellar, K. Wiesner-Fleischer, M. Fleischer, A. Rucki and O. Hinrichsen, Electrochim. Acta, 2019, 307, 164–175 CrossRef.
- H. Xiao, T. Cheng, W. A. Goddard and R. Sundararaman, J. Am. Chem. Soc., 2016, 138, 483–486 CrossRef CAS.
- T. L. Lohr and T. J. Marks, Nat. Chem., 2015, 7, 477–482 CrossRef CAS.
- M. J. Climent, A. Corma, S. Iborra and M. J. Sabater, ACS Catal., 2014, 4, 870–891 CrossRef CAS.
- R. G. Mariano, K. McKelvey, H. S. White and M. W. Kanan, Science, 2017, 358, 1187–1191 CrossRef CAS.
- Y. X. Wang, C. Y. Li, Z. X. Fan, Y. Chen, X. Li, L. Cao, C. H. Wang, L. Wang, D. Su, H. Zhang, T. Mueller and C. Wang, Nano Lett., 2020, 20, 8074–8080 CrossRef CAS.
- S. Zhao, R. X. Jin and R. C. Jin, ACS Energy Lett., 2018, 3, 452–462 CrossRef CAS.
- J. A. Trindell, J. Clausmeyer and R. M. Crooks, J. Am. Chem. Soc., 2017, 139, 16161–16167 CrossRef CAS.
- C. Rogers, W. S. Perkins, G. Veber, T. E. Williams, R. R. Cloke and F. R. Fischer, J. Am. Chem. Soc., 2017, 139, 4052–4061 CrossRef CAS.
- K. Sun, T. Cheng, L. N. Wu, Y. F. Hu, J. G. Zhou, A. Maclennan, Z. H. Jiang, Y. Z. Gao, W. A. Goddard and Z. J. Wang, J. Am. Chem. Soc., 2017, 139, 15608–15611 CrossRef CAS.
- S. Lamaison, D. Wakerley, J. Blanchard, D. Montero, G. Rousse, D. Mercier, P. Marcus, D. Taverna, D. Giaume, V. Mougel and M. Fontecave, Joule, 2020, 4, 395–406 CrossRef CAS.
- S. B. Liu, H. B. Tao, L. Zeng, Q. Liu, Z. G. Xu, Q. X. Liu and J. L. Luo, J. Am. Chem. Soc., 2017, 139, 2160–2163 CrossRef CAS.
- S. Mezzavilla, S. Horch, I. E. L. Stephens, B. Seger and I. Chorkendorff, Angew. Chem., Int. Ed., 2019, 58, 3774–3778 CrossRef CAS.
- M. H. Li, H. F. Wang, W. Luo, P. C. Sherrell, J. Chen and J. P. Yang, Adv. Mater., 2020, 32, 2001848 CrossRef CAS.
- Y. Cheng, S. Z. Yang, S. P. Jiang and S. Y. Wang, Small Methods, 2019, 3, 1800440 CrossRef.
- H. L. Liu, Y. T. Zhu, J. M. Ma, Z. C. Zhang and W. P. Hu, Adv. Funct. Mater., 2020, 30, 1910534 CrossRef CAS.
- T. T. Zheng, K. Jiang, N. Ta, Y. F. Hu, J. Zeng, J. Y. Liu and H. T. Wang, Joule, 2019, 3, 265–278 CrossRef CAS.
- H. B. Yang, S. F. Hung, S. Liu, K. D. Yuan, S. Miao, L. P. Zhang, X. Huang, H. Y. Wang, W. Z. Cai, R. Chen, J. J. Gao, X. F. Yang, W. Chen, Y. Q. Huang, H. M. Chen, C. M. Li, T. Zhang and B. Liu, Nat. Energy, 2018, 3, 140–147 CrossRef CAS.
- N. Corbin, J. Zeng, K. Williams and K. Manthiram, Nano Res., 2019, 12, 2093–2125 CrossRef CAS.
- L. Sun, V. Reddu, A. C. Fisher and X. Wang, Energy Environ. Sci., 2020, 13, 374–403 RSC.
- S. X. Ren, D. Joulie, D. Salvatore, K. Torbensen, M. Wang, M. Robert and C. P. Berlinguette, Science, 2019, 365, 367–369 CrossRef CAS.
- Y. S. Wu, Z. Jiang, X. Lu, Y. Y. Liang and H. L. Wang, Nature, 2019, 575, 639–643 CrossRef CAS.
- X. Zhang, Y. Wang, M. Gu, M. Y. Wang, Z. S. Zhang, W. Y. Pan, Z. Jiang, H. Z. Zheng, M. Lucero, H. L. Wang, G. E. Sterbinsky, Q. Ma, Y. G. Wang, Z. X. Feng, J. Li, H. J. Dai and Y. Y. Liang, Nat. Energy, 2020, 5, 684–692 CrossRef CAS.
- J. Q. Jiao, R. Lin, S. J. Liu, W. C. Cheong, C. Zhang, Z. Chen, Y. Pan, J. G. Tang, K. L. Wu, S. F. Hung, H. M. Chen, L. R. Zheng, Q. Lu, X. Yang, B. J. Xu, H. Xiao, J. Li, D. S. Wang, Q. Peng, C. Chen and Y. D. Li, Nat. Chem., 2019, 11, 222–228 CrossRef CAS.
- H. J. Zhu, M. Lu, Y. R. Wang, S. J. Yao, M. Zhang, Y. H. Kan, J. Liu, Y. Chen, S. L. Li and Y. Q. Lan, Nat. Commun., 2020, 11, 497 CrossRef CAS.
- J. Gu, C. S. Hsu, L. C. Bai, H. M. Chen and X. L. Hu, Science, 2019, 364, 1091–1094 CrossRef CAS.
- Q. Li, J. J. Fu, W. L. Zhu, Z. Z. Chen, B. Shen, L. H. Wu, Z. Xi, T. Y. Wang, G. Lu, J. J. Zhu and S. H. Sun, J. Am. Chem. Soc., 2017, 139, 4290–4293 CrossRef CAS.
- X. G. Li, W. T. Bi, M. L. Chen, Y. X. Sun, H. X. Ju, W. S. Yan, J. F. Zhu, X. J. Wu, W. S. Chu, C. Z. Wu and Y. Xie, J. Am. Chem. Soc., 2017, 139, 14889–14892 CrossRef CAS.
- S. Liu, H. B. Yang, S. F. Hung, J. Ding, W. Z. Cai, L. H. Liu, J. J. Gao, X. N. Li, X. Y. Ren, Z. C. Kuang, Y. Q. Huang, T. Zhang and B. Liu, Angew. Chem., Int. Ed., 2020, 59, 798–803 CrossRef CAS.
- C. C. Yan, H. B. Li, Y. F. Ye, H. H. Wu, F. Cai, R. Si, J. P. Xiao, S. Miao, S. H. Xie, F. Yang, Y. S. Li, G. X. Wang and X. H. Bao, Energy Environ. Sci., 2018, 11, 1204–1210 RSC.
- X. Q. Wang, Z. Chen, X. Y. Zhao, T. Yao, W. X. Chen, R. You, C. M. Zhao, G. Wu, J. Wang, W. X. Huang, J. L. Yang, X. Hong, S. Q. Wei, Y. Wu and Y. D. Li, Angew. Chem., Int. Ed., 2018, 57, 1944–1948 CrossRef CAS.
- W. H. Ren, X. Tan, W. F. Yang, C. Jia, S. M. Xu, K. X. Wang, S. C. Smith and C. Zhao, Angew. Chem., Int. Ed., 2019, 58, 6972–6976 CrossRef CAS.
- L. Dai, Q. Qin, P. Wang, X. J. Zhao, C. Y. Hu, P. X. Liu, R. X. Qin, M. Chen, D. H. Ou, C. F. Xu, S. G. Mo, B. H. Wu, G. Fu, P. Zhang and N. F. Zheng, Sci. Adv., 2017, 3, e1701069 CrossRef.
- F. Yang, P. Song, X. Z. Liu, B. B. Mei, W. Xing, Z. Jiang, L. Gu and W. L. Xu, Angew. Chem., Int. Ed., 2018, 57, 12303–12307 CrossRef CAS.
- B. X. Zhang, J. L. Zhang, J. B. Shi, D. X. Tan, L. F. Liu, F. Y. Zhang, C. Lu, Z. Z. Su, X. N. Tan, X. Y. Cheng, B. X. Han, L. R. Zheng and J. Zhang, Nat. Commun., 2019, 10, 2980 CrossRef.
- X. Y. Liu, P. Schlexer, J. P. Xiao, Y. F. Ji, L. Wang, R. B. Sandberg, M. Tang, K. S. Brown, H. J. Peng, S. Ringe, C. Hahn, T. F. Jaramillo, J. K. Norskov and K. R. Chan, Nat. Commun., 2019, 10, 32 CrossRef CAS.
- A. Verdaguer-Casadevall, C. W. Li, T. P. Johansson, S. B. Scott, J. T. McKeown, M. Kumar, I. E. L. Stephens, M. W. Kanan and I. Chorkendorff, J. Am. Chem. Soc., 2015, 137, 9808–9811 CrossRef CAS.
- C. W. Li, J. Ciston and M. W. Kanan, Nature, 2014, 508, 504–508 CrossRef CAS.
- Y. Chen, Z. X. Fan, J. Wang, C. Y. Ling, W. X. Niu, Z. Q. Huang, G. G. Liu, B. Chen, Z. C. Lai, X. Z. Liu, B. Li, Y. Zong, L. Gu, J. L. Wang, X. Wang and H. Zhang, J. Am. Chem. Soc., 2020, 142, 12760–12766 CrossRef CAS.
- K. J. P. Schouten, E. P. Gallent and M. T. M. Koper, ACS Catal., 2013, 3, 1292–1295 CrossRef CAS.
- Y. X. Wang, D. Raciti and C. Wang, ACS Catal., 2018, 8, 5657–5663 CrossRef CAS.
- E. Bertheussen, T. V. Hogg, Y. Abghoui, A. K. Engstfeld, I. Chorkendorff and I. E. L. Stephens, ACS Energy Lett., 2018, 3, 634–640 CrossRef CAS.
- A. S. Malkani, M. Dunwell and B. J. Xu, ACS Catal., 2019, 9, 474–478 CrossRef CAS.
- D. Raciti, L. Cao, K. J. T. Liv, P. F. Rottmann, X. Tang, C. Y. Li, Z. Hicks, K. H. Bowen, K. J. Hemker, T. Mueller and C. Wang, ACS Catal., 2017, 7, 4467–4472 CrossRef CAS.
- D. S. Ripatti, T. R. Veltman and M. W. Kanan, Joule, 2019, 3, 240–256 CrossRef CAS.
- L. Wang, S. Nitopi, A. B. Wong, J. L. Snider, A. C. Nielander, C. G. Morales-Guio, M. Orazov, D. C. Higgins, C. Hahn and T. F. Jaramillo, Nat. Catal., 2019, 2, 702–708 CrossRef CAS.
- J. Gao, H. Zhang, X. Y. Guo, J. S. Luo, S. M. Zakeeruddin, D. Ren and M. Gratzel, J. Am. Chem. Soc., 2019, 141, 18704–18714 CrossRef CAS.
- L. R. L. Ting, O. Pique, S. Y. Lim, M. Tanhaei, F. Calle-Vallejo and B. S. Yeo, ACS Catal., 2020, 10, 4059–4069 CrossRef CAS.
- J. J. Fu, W. L. Zhu, Y. Chen, Z. Y. Yin, Y. Y. Li, J. Liu, H. Y. Zhang, J. J. Zhu and S. H. Sun, Angew. Chem., Int. Ed., 2019, 58, 14100–14103 CrossRef CAS.
- J. F. Huang, M. Mensi, E. Oveisi, V. Mantella and R. Buonsanti, J. Am. Chem. Soc., 2019, 141, 2490–2499 CrossRef CAS.
- T. Y. Zhang, Z. Y. Li, J. F. Zhang and J. J. Wu, J. Catal., 2020, 387, 163–169 CrossRef CAS.
- C. G. Morales-Guio, E. R. Cave, S. A. Nitopi, J. T. Feaster, L. Wang, K. P. Kuhl, A. Jackson, N. C. Johnson, D. N. Abram, T. Hatsukade, C. Hahn and T. F. Jaramillo, Nat. Catal., 2018, 1, 764–771 CrossRef CAS.
- F. W. Li, Y. G. C. Li, Z. Y. Wang, J. Li, D. H. Nam, Y. Lum, M. C. Luo, X. Wang, A. Ozden, S. F. Hung, B. Chen, Y. H. Wang, J. Wicks, Y. Xu, Y. L. Li, C. M. Gabardo, C. T. Dinh, Y. Wang, T. T. Zhuang, D. Sinton and E. H. Sargent, Nat. Catal., 2020, 3, 75–82 CrossRef CAS.
- C. B. Chen, Y. F. Li, S. Yu, S. Louisia, J. B. Jin, M. F. Li, M. B. Ross and P. D. Yang, Joule, 2020, 4, 1688–1699 CrossRef CAS.
- X. She, T. Zhang, Z. Li, H. Li, H. Xu and J. Wu, Cell Rep. Phys. Sci., 2020, 1, 100051 CrossRef.
- L. Lin, T. Liu, J. Xiao, H. Li, P. Wei, D. Gao, B. Nan, R. Si, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2020, 59, 22408–22413 CrossRef CAS.
- X. L. Wang, J. F. de Araujo, W. Ju, A. Bagger, H. Schmies, S. Kuhl, J. Rossmeisl and P. Strasser, Nat. Nanotechnol., 2019, 14, 1063–1070 CrossRef CAS.
- M. Gong, Z. Cao, W. Liu, E. M. Nichols, P. T. Smith, J. S. Derrick, Y. S. Liu, J. J. Liu, X. D. Wen and C. J. Chang, ACS Cent. Sci., 2017, 3, 1032–1040 CrossRef CAS.
- E. Jeng and F. Jiao, React. Chem. Eng., 2020, 5, 1768–1775 RSC.
| This journal is © The Royal Society of Chemistry 2021 |