
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A high-temperature continuous stirred-tank reactor cascade for the multistep synthesis of InP/ZnS quantum dots†
Ioannis
Lignos
 a,
Yiming
Mo
a,
Loukas
Carayannopoulos
a,
Matthias
Ginterseder
b,
Moungi G.
Bawendi
b and
Klavs F.
Jensen
*a
a,
Yiming
Mo
a,
Loukas
Carayannopoulos
a,
Matthias
Ginterseder
b,
Moungi G.
Bawendi
b and
Klavs F.
Jensen
*a
aDepartment of Chemical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, U.S.A. E-mail: kfjensen@mit.edu
bDepartment of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, U.S.A
First published on 3rd February 2021
Abstract
The multistep and continuous production of core–shell III–V semiconductor nanocrystals remains a technological challenge. We present a newly designed high-temperature and miniature continuous stirred-tank reactor cascade, for the continuous and scalable synthesis of InP/ZnS core–shell quantum dots with a safer aminophosphine precursor comparing to standard protocols involving (TMS)3P. The resulting InP/ZnS QDs exhibit emissions between 520 and 610 nm, narrow emission linewidths in the range of 46–64 nm and photoluminescence quantum yields up to 42%.
Colloidal quantum dots (QDs) have received huge attention as luminescent materials for a wide range of applications including, optoelectronic devices,1,2 biological imaging,3,4 photocatalysis,5,6 and photovoltaics,7 driven by their unique and synthetically tunable optical and electronic properties.8,9 In the past twenty years, many of the reaction pathways added to the synthetic toolbox for nanocrystals have involved toxic heavy metals, e.g., Cd, Pb and Hg.10,11 Typical examples are II–VI and IV–VI QDs including CdX- and PbX-based materials (where, X = S, Se, Te).10 The three main reasons for the development of Cd- and Pb-based QDs are: a) the moderate reaction conditions for their synthesis with relatively short reaction times, b) the well-studied reaction mechanisms and c) the variety of available sophisticated chemical strategies.12
The main less toxic alternatives include the III–V semiconductor QDs, and in particular indium phosphide (InP) QDs.12 To enhance the photoluminescence (PL) properties of III–V semiconductor QDs, it is common to design core/shell heterostructures such as InP/ZnE (E = S, Se),13–16 or double shell heterostructures, such as InP/ZnSe/ZnS.17–20 Such nano-heterostructures exhibit PL quantum yields (QYs) in the range of 20–95% and emission linewidths in between 35–80 nm.12 Although there are noteworthy advances in their synthesis, their adoption in various applications lags behind II–VI and IV–VI QDs. One of the main challenges in the synthesis of III–V compounds is obtaining a wider size range of particles exhibiting quantum confinement effects and high QYs, while keeping the size distribution low. This difficulty is in part resulting from their strongly covalent nature, requiring highly reactive precursors and high temperatures.21
To date, the majority of studies for the synthesis of InP QDs have been focused on the use of a highly reactive phosphorus precursor, tris(trimethylsilyl)phosphine ((TMS)3P).12,21 However, (TMS)3P is a highly pyrophoric and expensive compound. Moreover, a highly toxic gas, phosphine, is released upon contact with moist air.20,22 Consequently, recent studies have explored replacing the (TMS)3P precursor with other economic and “greener” precursors, including P4,23,24 PCl3 (ref. 25) and tris(dialkylamino)phosphines.22,26–28 In particular, aminophosphines have emerged as safer and more economical precursors to produce InP-based QDs with similar PL properties as those prepared by standard protocols involving (TMS)3P. The combination of tris(dimethylamino)phosphine [(DMA)3P] with InX3 (Cl, Br, I) in oleylamine (coordinating solvent) and the subsequent surface passivation with wider bandgap materials, such as ZnS, ZnSe or ZnSeS/ZnS, leads to the formation of core–shell structures with narrow emission linewidths (37–63) and relatively high PL QYs (20–70% for core–shell QDs,22,26,28 and 87% for green double-shell InP/ZnSeS/ZnS QDs29). Although such processes provide “greener” routes to the generation of nanomaterials, their potential scale-up from mostly laboratory batch procedures remains a challenge.30 To fully exploit synthetic methodologies, which are sustainable and at the same time employing “green” chemistry considerations, it will be necessary to design alternative continuous processes.
Flow chemistry has attracted significant interest in the area of chemical31 and material synthesis.30 Various flow chemistry systems have been proposed for the synthesis of a wide range of molecules and materials, primarily based on continuous or droplet-based formats.30,32 Compared to flasks, flow reactors allow for rapid heat and mass transfer, reduced sample/reagent consumption, operational safety and product reproducibility due to the inherent control of reaction parameters.30–32 In particular, modular flow chemistry platforms allow for the rapid screening of reaction parameters, reaction optimization, handling of hazardous reactions and scalable production of chemicals and various types of nanomaterials.33 In this direction, different reactor configurations have been constructed based on commercial tubes and fluidic connectors or microfluidic chips for the production of numerous types of semiconductor nanocrystals.33 However, most flow systems only consider single step reactions and are limited by issues related to pressure drop (low flow rates of precursors, viscous solutions) and occasionally to the formation of gaseous side products (inconsistent flow distribution).32
There are a few reports proposing flow chemistry platforms for QD synthesis with multistep capabilities, but they are mostly limited to chip-based or tube-based flow reactors.34–39 A recent example is the use of a series of microfluidic devices with complex architectures to prevent secondary nucleation of particles, while minimizing pressure drop.40 The latter strategy was recently proposed for the synthesis of III–V nano-heterostructures, using (TMS)3P as the phosphorous precursor. A more recent example for the safer and economic synthesis of InP QDs with aminophosphines was demonstrated by Akdas et al., where the authors used a continuous flow reactor.41 The synthesis of InP QDs was possible, however issues related to the formation of gas by-products and the need of longer reaction times limited the synthetic potential of the microfluidic platform.
Miniature continuous stirred tank reactor (CSTR) cascades have been proposed as an additional tool in the library of flow configurations for chemical transformations.42–44 Such flow formats have advantages comparing to tube- or chip-based reactors, including solid handling, active mixing, facile multi-dosing of reagents, applicability to reactions requiring long residence times, versatility in multiphasic reactions and flexibility for reaction scale-up.45 Recently, we demonstrated the efficiency of 3D printed miniature CSTR cascades in synthesizing gluconolactone-capped polyethylenimine-modified silica NPs in a multistage and fully continuous manner.46 At the same time, it was possible to handle highly viscous solutions, decrease processing time down to 10 min (compared to several hours per days) and reproducibly control surface properties.
Herein, we introduce a series of micro-sized CSTRs for the multistep and high-temperature colloidal synthesis of InP/ZnS QDs using the safer and cost-efficient aminophosphine-derived QD synthesis. Importantly, the novel flow chemistry platform allows for an independent control of temperature and stirring rate of precursors in each CSTR unit. This is essential for designing processes with various heating and mixing needs. The synthesized QDs exhibit PL QYs reaching 42% and emission linewidths are in the range of 54–62 nm, which are in agreement with well-established flask-based methodologies. In addition, their emission spectra can be tuned between 520 nm to 610 nm, by a systematic variation of halide content. We demonstrate the efficiency of the platform in monitoring the effect of reaction time, temperature, molar ratios of precursors and shell thickness. With this approach, we aim to deliver a safer, cost-efficient, and scalable continuous process for the formation of indium-based QDs for their subsequent use in optoelectronic applications.
The CSTR flow platform is illustrated in Fig. 1 (see Fig. S1† for an image of the experimental setup and additional details for the experimental procedure). As shown in Fig. 1b, in the first CSTR unit, there were two feeds. Prior to the first feed, there were two T-junctions (P-727, IDEX Health & Science LLC, USA) and two shut-off valves (ethylene tetrafluoroethylene, P-733, IDEX Health & Science LLC, USA), to combine and select the input streams. The input streams consist of (a) a liquid stream (precursor solutions – indium and zinc sources) for conducting reactions, together with a pure solvent – octadecene for cleaning the CSTR cascade after reaction completion and (b) a gas stream (argon) for ensuring an inert atmosphere inside the CSTR units. On and off position of the valves defined the stream injected into the CSTR cascade. For the second feed (Fig. 1b), a liquid stream of tris(diethylamino)phosphine was injected into the CSTR unit for initializing the reaction. After purging the platform with argon for 1–2 minutes, octadecene was used for filling it up prior to the synthetic procedure. At that point, the platform was rotated vertically to eliminate the potential accumulation of argon bubbles in the reaction units. The solutions of InCl2/ZnCl2, P and ZnI2 were then injected into the first CSTR. The mixing performance of the CSTR cascade was assessed by comparing the outlet concentration profile to the residence time distribution (RTD) of the ideal CSTRs in a series model.45 Fig. S2† illustrates the experimental system for the RTD measurements. The ideal and measured RTD profiles show high consistency for both horizontal and vertical orientation of the setup and at various flow rates, similar to the those used for the experiments (Fig. S3†).
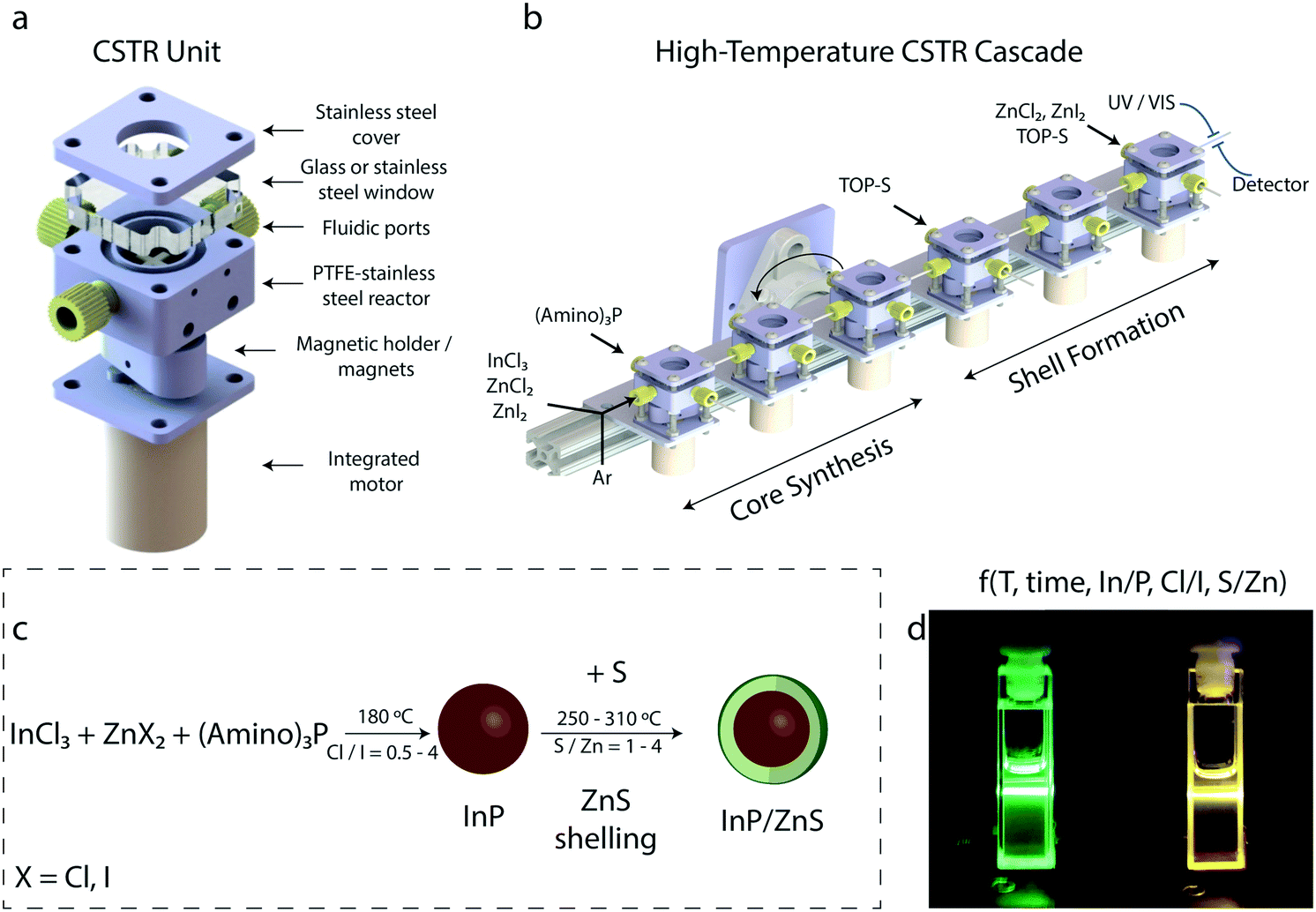 | ||
| Fig. 1 (a) Exploded view CAD drawing of a CSTR unit. From top to bottom, it consists of a stainless-steel cover, a borosilicate glass window or another stainless-steel cover, fluidic ports, a polytetrafluoroethylene (PTFE) stirring bar, a PTFE coated-stainless steel chamber, a magnetic holder and two magnets, and an integrated motor. (b) Sketch of the miniature CSTR cascade design, with multiple CSTRs in series attached on an aluminum block. The aluminum block can be rotated for altering platform's orientation (vertical and horizontal). Various precursor solutions, solvents and argon stream can be injected at different stages of the process. A UV-vis detector is attached at the reactor outlet for reaction monitoring. (c) The synthetic protocol describing the synthesis of the InP cores and the subsequent surface passivation with ZnS. (d) Image of InP/ZnS QD solutions, showing bright emission under UV excitation (405 nm). | ||
For the synthesis of InP cores, the number of the CSTRs was altered between three and five depending on reaction time and synthetic process (core-only synthesis or core/shell synthesis). The synthesis of InP cores was conducted through a variation of reaction times (5–60 min) and operating temperatures (160–200 °C). Again, depending on the synthetic procedure, a solution of sulfur was injected between the 3rd and 6th CSTR for the formation of InP/ZnS QDs. The total number of CSTRs used for the synthesis of InP/ZnS QDs were between six and eight. For the synthesis of InP/ZnS nanocrystals, reaction time was varied between 10–60 min and temperature between 240–320 °C. The synthetic procedure and some indicating reaction parameters are demonstrated in Fig. 1c. Solutions of InP/ZnS emitting in the green and yellow region of the visible spectrum are depicted in Fig. 1d.
InP QDs were synthesized based on a modified synthetic protocol.22,47 The size of the synthesized QDs is strongly dependent on the halide source. For our purpose, InCl3 was used as the indium precursor. ZnCl2 was also used to facilitate InP growth and at the same time to further tune the size of the synthesized InP/ZnS QDs (by introducing ZnI2 as the additional Zn source for the overcoating and for size tuning). Initially, the potential of running reactions at lower precursor concentrations than those reported in the literature,22,26 would be useful for reaction screening and minimizing material consumption. However, it is known that decreasing the precursor concentrations leads to broader particle size distributions.22 To explore this issue, different batch experiments were conducted at 180 °C for 30 min, where precursor concentrations were decreased by 2–4 times and aminophosphine is diluted by 3–5 times using octadecene. By comparing the absorbance spectra of the produced InP QDs (Fig. S4†) with those obtained from standard synthetic protocols,22 there is no observable effect on the excitonic peak. For this reason, we decided to conduct all our experiments with a 2-times decrease of precursor concentrations and 5-times dilution of the aminophosphine precursor.
With the intent to monitor reaction parameters, which could be applicable for the flow synthesis of InP QDs, we used the high-temperature CSTR cascade with 3 or 6 CSTR units. The number of CSTRs was relevant to the targeted reaction times. In-to-P molar ratio, reaction time and temperature were initial parameters to monitor. The CSTR cascade was rotated vertically for all the experiments during InP QD synthesis because of the formation of gaseous side-products from the redox reactions occurring during the formation of InP cores.22,28 Rotating the platform vertically prevented any accumulation of gas side-products inside the CSTR units, which could otherwise reduce residence time and adversely affect product quality. QDs grown in the horizontal position showed wider emission linewidths that those synthesized in the vertical arrangement (see Fig. S5†). By adjusting temperature between 160 and 220 °C, reaction time in the range of 5–30 min and In-to-P molar ratio between 2.1 and 4.9 (Fig. 2 and S6†), it was possible to successfully synthesize InP QDs exhibiting absorption spectra between 550–590 nm. The optimal parameters for generating InP QDs with satisfactory size distributions (qualitative assessment based on the absorption exciton peaks, Fig. 2a) were In-to-P molar ratio of 3.6 and a temperature of 180 °C (see Fig. S7† for absorbance spectra of InP QDs with the extracted optimal parameters and a transmission electron microscopy (TEM) image of the synthesized particles). Reaction time could be adjusted for size tuning of the InP cores (Fig. 2b). With an indium concentration of 0.05 M and a typical reaction time of 25–30 min, the throughput of InP QDs is 1.2 g h−1.
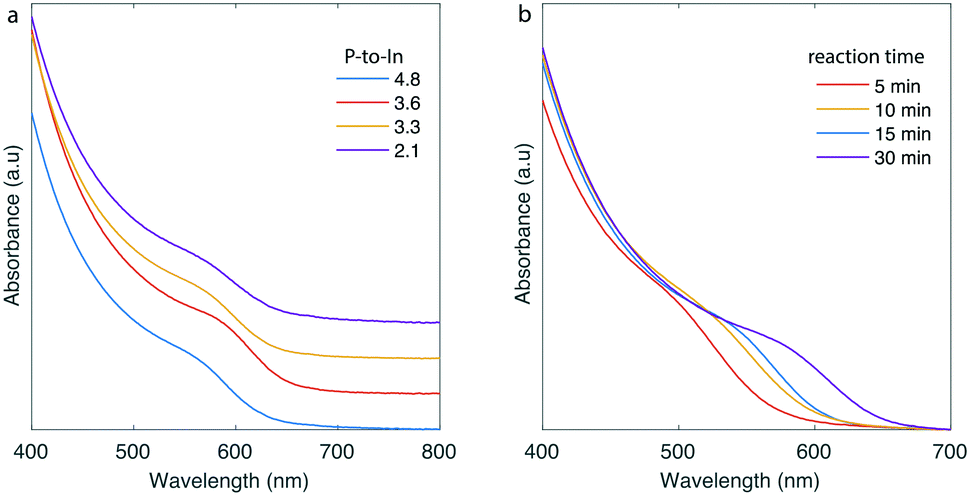 | ||
| Fig. 2 (a) Absorption spectra of InP QDs at different P-to-In molar ratios. Other parameters were fixed: T = 180 °C, reaction time = 30 min. (b) Absorption spectra of InP QDs at different reaction times. Other parameters were fixed: T = 180 °C, P-to-In = 3.6. | ||
To enhance the PL properties of the synthesized InP QDs, we further used the CSTR cascades for the formation of InP/ZnS core/shell morphologies. InP cores with various controlled diameters, either by reaction time adjustment or by halide content, were continuously flowed into the shell formation CSTR units without incorporating any additional purification process. For these experiments, it was necessary to use 3–4 CSTR units for the core formation and then between 3 and 5 CSTR units for the shell formation. To allow the overcoating of ZnS shell, ZnI2 was injected together with ZnCl2 as the Zn source. The Cl-to-I ratio was used as a tuning parameter for particle size. In general, increasing Cl content leads to the red-shift of absorption and PL spectra.22,47 In addition, sulfur was further injected after core formation for initiating the ZnS shell formation. The number of injections and position could be altered based on the experimental needs (time for shell formation, sulfur content and temperature of injection).
Using the high-temperature multistage CSTR cascade, we successfully synthesized InP/ZnS core/shell nanostructures with emission colors from blue to red. The flow reactor allows for continuous generation of core/shell morphologies with different sizes and PL properties avoiding issues related to undesired back-flow. In a typical synthesis, the temperature of the shell-formation CSTR units was fixed between 250–320 °C, and the residence times were altered between 15 and 30 minutes depending on the flowrate of the solution containing the InP cores (the reaction time of the core synthesis was altered between 10–30 min). In addition, the effect of Cl-to-I and S-to-Zn molar ratios on PL and absorption spectra was thoroughly investigated. The product output was monitored by a customized optical device (see ESI† for additional details) to characterize the absorption and emission of the InP/ZnS QDs in-line. In addition, the formation of InP/ZnS QDs was confirmed by TEM characterization (see Fig. S8 and S9†), indicating the high-crystallinity of the synthesized QDs with lattice fringes of 0.32 nm. Fig. 3a shows the emission spectra of three InP/ZnS core/shell QDs. Another critical parameter during InP/ZnS synthesis is the S-to-Zn molar ratio, which was adjusted between 1 and 4 (see ESI† for additional figures, Fig. S10 and S11†) and its effect was examined on the synthesized particles. After synthesizing InP/ZnS QDs with different sulfur content, we concluded that an S-to-Zn molar ratio equal to 2 is necessary for achieving optimum optical spectra. By varying the Cl-to-I content, it was possible to tune the PL peaks of those samples at different positions, i.e., at 526 nm, 552 nm and 578 nm. By introducing higher contents of iodide in the reaction system, the PL peaks were blueshifted (see ESI,† Fig. S12 and S13 for additional PL spectra). The full-width-at-half-maximum (fwhm) of those peaks were 46, 56 and 58 nm, respectively. The narrow emission linewidths reported are comparable to the state-of-the-art emission linewidths using batch methodologies and indicate the advantages of the CSTR cascade on handling long residence times and reactions generating gaseous-side products.
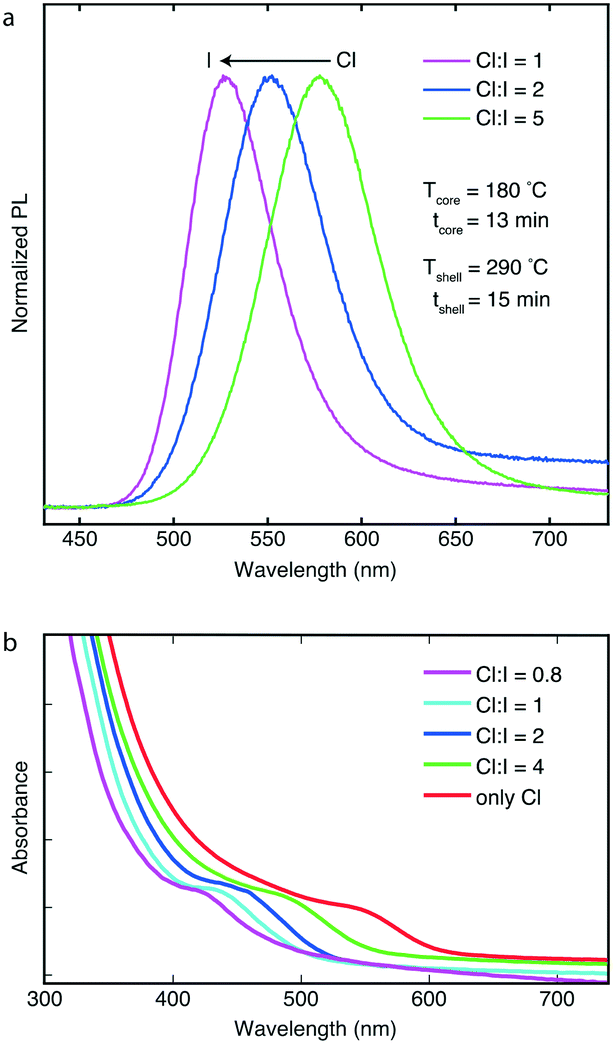 | ||
| Fig. 3 (a) PL spectra of InP/ZnS QDs while varying the Cl-to-I molar ratio. The PL peaks were positioned at 526 nm, 552 nm and 578 nm. (b) Absorption spectra of InP/ZnS QDs by changing iodide and chloride content. | ||
Fig. 3b also indicates the blueshift of absorption excitonic peaks of four other core/shell samples with higher iodide contents (molar ratios in the range of 0.8 to no iodide), while keeping temperatures (Tcore = 180 °C and Tshell = 290 °C) together with reaction time (tcore = 10 min and tshell = 11 min) fixed. The absorption peaks were positioned at 428, 439, 458, 495 and 548 nm.
To gain further insights of the reaction process, we evaluated the effect of shell formation temperature on product characteristics (PL and QYs) and particularly on shell thickness. By precisely maintaining the shell formation temperature between 270 °C and 310 °C and by keeping all other reaction parameters constant (Tcore = 180 °C, tcore = 13 min, tshell = 15 min, S-to-Zn = 2 and Cl-to-I = 6), we successfully obtained InP/ZnS QDs with different shell thicknesses using the same flowing InP cores (Fig. 4). At the same time, we investigated the effect of temperature on PL peak position, emission linewidths and QY. As shown in Fig. 4a, when the temperature is increased, the PL peak position slightly blueshifts from 563 nm to 558 nm (Fig. S14† demonstrates the PL peak position change by increasing shell formation temperature) providing evidence of ZnS monolayer addition. During ZnS deposition on InP core, the addition of sulfur (TOP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :S) at elevated temperatures did not alter significantly PL peak position, meaning that surface etching (PL peak blue shift) was not significant. This is in line with previous studies reporting the synthesis of InP/ZnS with TOP:S.48 In addition, due to relatively low reactivity of zinc precursors, InP/ZnS QDs synthesized at 270 °C and 280 °C exhibit low energy tails, which indicate trap emission.48 Such low energy tails suggest incomplete ZnS shell overcoating and it is in direct correlation with the corresponding emission linewidths and QYs (below 25%) of the samples (Fig. 4b and c).
:S) at elevated temperatures did not alter significantly PL peak position, meaning that surface etching (PL peak blue shift) was not significant. This is in line with previous studies reporting the synthesis of InP/ZnS with TOP:S.48 In addition, due to relatively low reactivity of zinc precursors, InP/ZnS QDs synthesized at 270 °C and 280 °C exhibit low energy tails, which indicate trap emission.48 Such low energy tails suggest incomplete ZnS shell overcoating and it is in direct correlation with the corresponding emission linewidths and QYs (below 25%) of the samples (Fig. 4b and c).
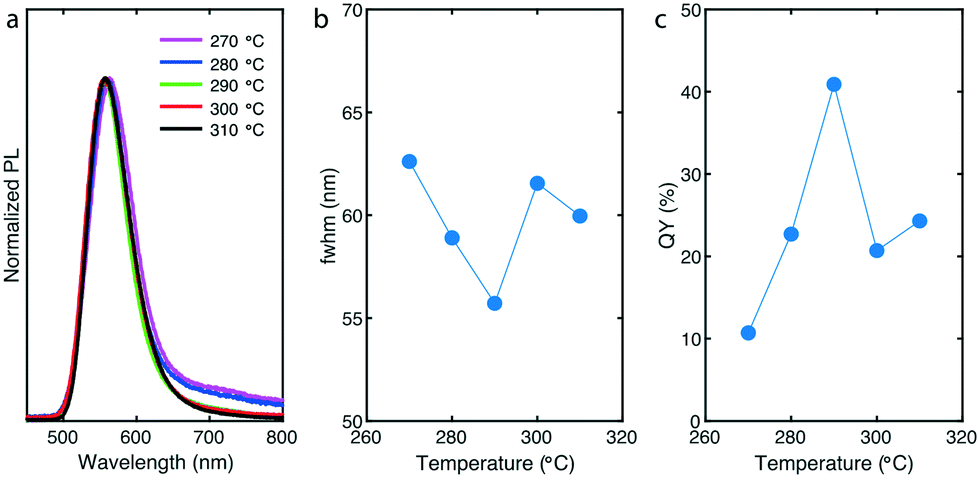 | ||
| Fig. 4 (a) Normalized PL spectra, (b) emission linewidths, and (c) QY, of InP/ZnS QDs at different shell formation temperatures between 270–310 °C. Other reaction parameters we remained constant (Tcore = 180 °C, tcore = 13 min, tshell = 15 min, S-to-Zn = 2 and Cl-to-I = 6). | ||
When a thicker shell was formed on the InP cores (290 °C), the tail almost disappeared, fwhm reached its minimum at 56 nm and QY of the synthesized samples reached its maximum at 42% (Fig. 4b and c). This behavior was also reflected by higher PL intensities. However, when the temperature was increased up to 310 °C (thicker shells), the emission linewidths and QYs were decreased and remained relatively stable at ∼60 nm and ∼22%, respectively. Similar to a previous study,48 this effect is supposedly due to the strain effects (higher lattice mismatch of InP/ZnS – 7.8% comparing to InP/ZnSe – 4.9%) and surface defects after a specific number of monolayers deposited on the surface of InP cores. A more effective strategy for enhanced passivation of InP cores would require a double-shell strategy, first by depositing a ZnSe or ZnSeS shell and then by a subsequent deposition of a thin ZnS shell.
Conclusions
In summary, we presented a high-temperature CSTR cascade able to perform multistep reactions to synthesize InP/ZnS QDs, using a “greener route” and in a fully continuous manner. Importantly, the proposed methodology allows for the controlled injection of multiple precursors, handling of long reaction times (in the range of several minutes to hours) and at the same time it overcomes limitations associated with the generation of gaseous side products (they can be deleterious for the product quality). In addition, due to the enhanced mixing and heating efficiencies in each CSTR unit, it is possible to intensify the synthetic process shortening the total processing time from several hours to approximately 30 min. This newly-designed platform allowed for the continuous synthesis of InP/ZnS QDs exhibiting emissions between 520–610 nm and featuring emission linewidths with a fwhm in the range of 46–64 nm. Such optical properties are comparable to those obtained from state-of-the-art batch synthetic protocols using (TMS)3P or aminophosphine precursors. Finally, by precisely controlling the reaction parameters and subsequently the deposition of ZnS on InP cores, we managed to form core/shell nanostructures exhibiting QYs up to 42%. Parallelizing this continuous process will further increase production throughput to be commensurate with that of state-of-the art flask reactor methodologies. To further improve the PL QY of III–V core/shell structures in a continuous and “greener” route, further studies would include the incorporation of an inner shell such as ZnSeS, which can effectively address issues related to the large lattice mismatch between InP and ZnS.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Science Foundation (EECS-1449291). I. L. thanks the Swiss National Foundation Grant P2EZP2_172127, Y. M. is grateful for the Chyn Duog Shiah Memorial Fellowship from MIT. M. G. thanks the National Science Foundation under grants no. DMR-1905164. The authors declare no competing financial interest. The authors would like to acknowledge Travis Hart for assistance on electronics development.References
- V. L. Colvin, M. C. Schlamp and A. P. Alivisatos, Nature, 1994, 370, 354–357 CrossRef CAS.
- Y. Jiang, S.-Y. Cho and M. Shim, J. Mater. Chem. C, 2018, 6, 2618–2634 RSC.
- S. M. Ng, M. Koneswaran and R. Narayanaswamy, RSC Adv., 2016, 6, 21624–21661 RSC.
- P. D. Howes, R. Chandrawati and M. M. Stevens, Science, 2014, 346, 1247390 CrossRef.
- G. Gao, Q. Xi, H. Zhou, Y. Zhao, C. Wu, L. Wang, P. Guo and J. Xu, Nanoscale, 2017, 9, 12032–12038 RSC.
- C. Huang, X.-B. Li, C.-H. Tung and L.-Z. Wu, Chem. – Eur. J., 2018, 24, 11530–11534 CrossRef CAS.
- K. R. T. Kumar, M. Ramakrishna and G. D. Sukumar, Int. J. Energy Res., 2018, 42, 2305–2319 CrossRef.
- A. P. Alivisatos, Science, 1996, 271, 933–937 CrossRef CAS.
- H. Weller, Angew. Chem., Int. Ed. Engl., 1993, 32, 41–53 CrossRef.
- M. V. Kovalenko, L. Manna, A. Cabot, Z. Hens, D. V. Talapin, C. R. Kagan, V. I. Klimov, A. L. Rogach, P. Reiss, D. J. Milliron, P. Guyot-Sionnnest, G. Konstantatos, W. J. Parak, T. Hyeon, B. A. Korgel, C. B. Murray and W. Heiss, ACS Nano, 2015, 9, 1012–1057 CrossRef CAS.
- D. V. Talapin, J.-S. Lee, M. V. Kovalenko and E. V. Shevchenko, Chem. Rev., 2010, 110, 389–458 CrossRef CAS.
- P. Reiss, M. Carrière, C. Lincheneau, L. Vaure and S. Tamang, Chem. Rev., 2016, 116, 10731–10819 CrossRef CAS.
- L. Li and P. Reiss, J. Am. Chem. Soc., 2008, 130, 11588–11589 CrossRef CAS.
- E. Ryu, S. Kim, E. Jang, S. Jun, H. Jang, B. Kim and S.-W. Kim, Chem. Mater., 2009, 21, 573–575 CrossRef CAS.
- X. Yang, D. Zhao, K. S. Leck, S. T. Tan, Y. X. Tang, J. Zhao, H. V. Demir and X. W. Sun, Adv. Mater., 2012, 24, 4180–4185 CrossRef CAS.
- V. Chandrasekaran, M. D. Tessier, D. Dupont, P. Geiregat, Z. Hens and E. Brainis, Nano Lett., 2017, 17, 6104–6109 CrossRef CAS.
- K. Kim, H. Lee, J. Ahn and S. Jeong, Appl. Phys. Lett., 2012, 101, 073107 CrossRef.
- Y.-H. Won, O. Cho, T. Kim, D.-Y. Chung, T. Kim, H. Chung, H. Jang, J. Lee, D. Kim and E. Jang, Nature, 2019, 575, 634–638 CrossRef CAS.
- Y. Li, X. Hou, X. Dai, Z. Yao, L. Lv, Y. Jin and X. Peng, J. Am. Chem. Soc., 2019, 141, 6448–6452 CrossRef CAS.
- P. Ramasamy, N. Kim, Y.-S. Kang, O. Ramirez and J.-S. Lee, Chem. Mater., 2017, 29, 6893–6899 CrossRef CAS.
- S. Tamang, C. Lincheneau, Y. Hermans, S. Jeong and P. Reiss, Chem. Mater., 2016, 28, 2491–2506 CrossRef CAS.
- M. D. Tessier, D. Dupont, K. De Nolf, J. De Roo and Z. Hens, Chem. Mater., 2015, 27, 4893–4898 CrossRef CAS.
- S. Carenco, M. Demange, J. Shi, C. Boissière, C. Sanchez, P. L. Floch and N. Mézailles, Chem. Commun., 2010, 46, 5578–5580 RSC.
- E. Bang, Y. Choi, J. Cho, Y.-H. Suh, H. W. Ban, J. S. Son and J. Park, Chem. Mater., 2017, 29, 4236–4243 CrossRef CAS.
- Z. Liu, A. Kumbhar, D. Xu, J. Zhang, Z. Sun and J. Fang, Angew. Chem., Int. Ed., 2008, 47, 3540–3542 CrossRef CAS.
- W.-S. Song, H.-S. Lee, J. C. Lee, D. S. Jang, Y. Choi, M. Choi and H. Yang, J. Nanopart. Res., 2013, 15, 1750 CrossRef.
- M. D. Tessier, K. De Nolf, D. Dupont, D. Sinnaeve, J. De Roo and Z. Hens, J. Am. Chem. Soc., 2016, 138, 5923–5929 CrossRef CAS.
- A. Buffard, S. Dreyfuss, B. Nadal, H. Heuclin, X. Xu, G. Patriarche, N. Mézailles and B. Dubertret, Chem. Mater., 2016, 28, 5925–5934 CrossRef CAS.
- J.-H. Jo, D.-Y. Jo, S.-H. Lee, S.-Y. Yoon, H.-B. Lim, B.-J. Lee, Y. R. Do and H. Yang, ACS Appl. Nano Mater., 2020, 3, 1972–1980 CrossRef CAS.
- K. Abdel-Latif, F. Bateni, S. Crouse and M. Abolhasani, Matter, 2020, 3, 1053–1086 CrossRef.
- R. Porta, M. Benaglia and A. Puglisi, Org. Process Res. Dev., 2016, 20, 2–25 CrossRef CAS.
- I. Lignos, R. Maceiczyk and A. J. deMello, Acc. Chem. Res., 2017, 50, 1248–1257 CrossRef CAS.
- Y. Pu, F. Cai, D. Wang, J.-X. Wang and J.-F. Chen, Ind. Eng. Chem. Res., 2018, 57, 1790–1802 CrossRef CAS.
- J. Baek, P. M. Allen, M. G. Bawendi and K. F. Jensen, Angew. Chem., Int. Ed., 2011, 50, 627–630 CrossRef CAS.
- M. S. Naughton, V. Kumar, Y. Bonita, K. Deshpande and P. J. A. Kenis, Nanoscale, 2015, 7, 15895–15903 RSC.
- A. Yashina, I. Lignos, S. Stavrakis, J. Choo and A. J. deMello, J. Mater. Chem. C, 2016, 4, 6401–6408 RSC.
- A. Vikram, V. Kumar, U. Ramesh, K. Balakrishnan, N. Oh, K. Deshpande, T. Ewers, P. Trefonas, M. Shim and P. J. A. Kenis, ChemNanoMat, 2018, 4, 943–953 CrossRef CAS.
- L. Xie, D. K. Harris, M. G. Bawendi and K. F. Jensen, Chem. Mater., 2015, 27, 5058–5063 CrossRef CAS.
- A. M. Nightingale, T. W. Phillips, J. H. Bannock and J. C. de Mello, Nat. Commun., 2014, 5, 3777 CrossRef CAS.
- J. Baek, Y. Shen, I. Lignos, M. G. Bawendi and K. F. Jensen, Angew. Chem., Int. Ed., 2018, 57, 10915–10918 CrossRef CAS.
- T. Akdas and P. Reiss, J. Phys.: Conf. Ser., 2019, 1323, 012007 CrossRef CAS.
- Y. Mo and K. F. Jensen, React. Chem. Eng., 2016, 1, 501–507 RSC.
- M. R. Chapman, M. H. T. Kwan, G. King, K. E. Jolley, M. Hussain, S. Hussain, I. E. Salama, C. González Niño, L. A. Thompson, M. E. Bayana, A. D. Clayton, B. N. Nguyen, N. J. Turner, N. Kapur and A. J. Blacker, Org. Process Res. Dev., 2017, 21, 1294–1301 CrossRef CAS.
- Y. Mo, H. Lin and K. F. Jensen, Chem. Eng. J., 2018, 335, 936–944 CrossRef CAS.
- O. Levenspiel, Ind. Eng. Chem. Res., 1999, 38, 4140–4143 CrossRef CAS.
- I. Lignos, H. Ow, J. P. Lopez, D. McCollum, H. Zhang, J. Imbrogno, Y. Shen, S. Chang, W. Wang and K. F. Jensen, ACS Appl. Mater. Interfaces, 2020, 12, 6699–6706 CrossRef CAS.
- S. Yu, X.-B. Fan, X. Wang, J. Li, Q. Zhang, A. Xia, S. Wei, L.-Z. Wu, Y. Zhou and G. R. Patzke, Nat. Commun., 2018, 9, 4009 CrossRef.
- R. Toufanian, A. Piryatinski, A. H. Mahler, R. Iyer, J. A. Hollingsworth and A. M. Dennis, Front. Chem., 2018, 6, 567 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0re00454e |
| This journal is © The Royal Society of Chemistry 2021 |