
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solvent-free manufacture of methacrylate polymers from biomass pyrolysis products†
J.
Ryan
 ,
M. T.
Elsmore
,
E. R.
Binner
,
D. S. A.
De Focatiis
,
D. J.
Irvine
and
J. P.
Robinson
*
,
M. T.
Elsmore
,
E. R.
Binner
,
D. S. A.
De Focatiis
,
D. J.
Irvine
and
J. P.
Robinson
*
Coates Building, Faculty of Engineering, University of Nottingham, NG7 2RD, UK. E-mail: John.robinson@nottingham.ac.uk
First published on 8th December 2020
Abstract
This work demonstrates a novel approach to add value to pyrolysis liquids by exploiting the diverse range of alcohol functional groups present within the mixture to yield a non-energy product, without requiring extensive separation. It is shown that 79.2% of the alcohol functional groups can be converted by esterification and subsequently polymerised (85.7%) to produce a range of polymer products with peak molecular weight (Mp) ranging from 22.9–36.9 kDa. Thermal and rheological properties of the most promising pyrolysis material have been compared with conventional poly(butyl methacrylate) (pBMA) of similar molecular weight, showing viability as a potential replacement owing to similarities in its thermorheological behaviour. A low molecular weight wax component of the novel polymer has been identified as a possible plasticizing agent, causing some decreases in viscosity. Production of the monomer is achieved in one reaction step and without separation or the use of toxic reagents. The overall mass balance and relevance to a biorefinery process is highlighted and strategies to tune the process to vary glass transition temperature (Tg) and Mp are discussed.
Introduction
Worldwide dependence on single-use and commodity plastics results in the production of over 300 million tonnes of polymer materials per year, relying on finite fossil reserves as the primary source of methacrylates.1 With reserves being depleted at a growing rate, there is a drive towards development of alternative feedstocks for polymer production. It has been reported that the EU produces around 26 million tonnes of unrecycled wood waste each year, which could offer a considerable untapped resource for monomer synthesis.2 Pyrolysis techniques can be used to depolymerise the cellulose, hemicellulose and lignin components of woody biomass to produce a liquid mixture containing the condensable decomposition products. This process is employed on an industrial scale, with the Empyro pyrolysis plant in Enschede, Netherlands converting pine into pyrolysis liquid at a throughput of 5 tonnes per hour. This equates to a pyrolysis liquid yield of 65–70 wt% for an annual production of 24 million litres.3,4 Typical pyrolysis liquids have broad composition, low pH and a tendency to undergo crosslinking and/or polymerisation during storage. These characteristics currently make it difficult to employ as a ‘drop in’ replacement feedstock, with extensive separation and/or upgrading processes needed to obtain specific products, as well as the associated challenge of finding markets for the separation and reaction co-products.5–10 An alternative approach is to utilise reactive functional groups within the mixture to yield a spectrum of products, rather than attempt to isolate specific compounds.A broad range of polymeric materials can be obtained from pyrolysis liquid owing to variability in structure of the monomers either found in or derived from the pyrolysis liquid. Works by Qu et al., Mialon et al., Stanzione et al. and Nowakowska et al. have demonstrated the viability of polymer production from tightly-controlled feedstocks such as lignin, vanillin, syringol, guaiacol and eugenol.11–15 It has further been reported that acyl chlorides and anhydrides offer potential for the synthesis of methacrylate monomers from specifically controlled model feedstocks or pyrolysis liquid fractions. These methacrylate polymers show viability in a wide range of applications, from resins and adhesives to bitumen binders.15–21 It has been shown that polymers developed from biomass can be tuned to a wide range of thermoset and thermoplastic applications including bio-based polyurethanes, melt-spinnable fibres and pressure sensitive adhesives.4,13,15,20,22 However, numerous additional steps are required to extract or fractionate useful parts of the pyrolysis oil involving the use of toxic and corrosive solvents and reagents.15,22 Ideally, for its eventual use in any integrated bio-refining process, pyrolysis liquid should be processable to the desired value added product from this impure state.23
Transesterification is an industrially applied technology that can transfer hydroxyls onto to methacrylate monomers.24 Our approach uses a transesterification of an industrially produced pyrolysis liquid to generate monomers. Subsequent free radical polymerisation (FRP) on the functionalised pyrolysis oil should enable phase separation of polymer product and unreacted components of the functionalised pyrolysis liquid, see Fig. 1.
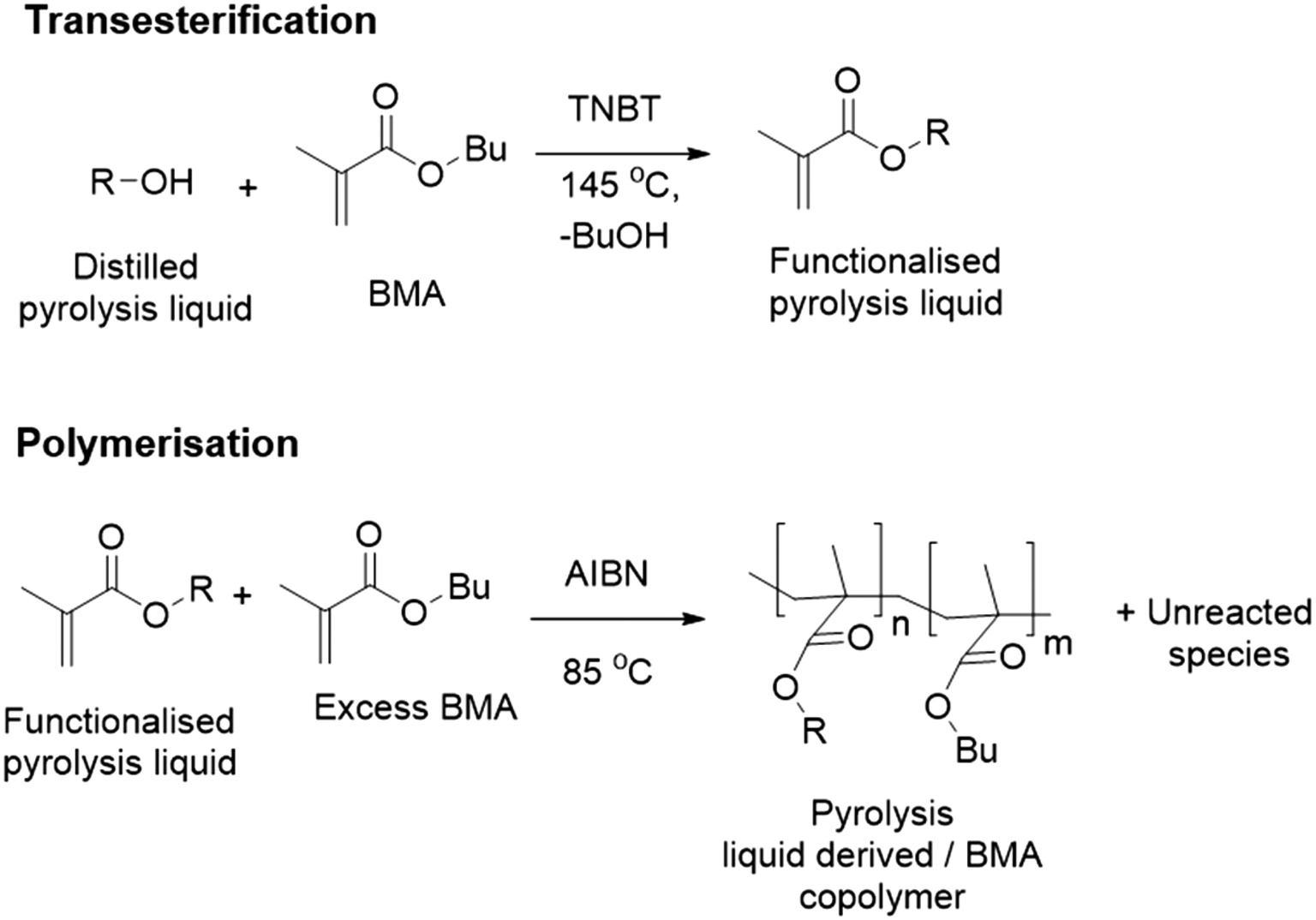 | ||
| Fig. 1 Pyrolysis liquid to methacrylate polymer via transesterification24 and radical polymerisation.25 | ||
Unlike previous studies the entire crude pyrolysis liquid is used as feedstock for synthesis of a sustainably-sourced replacement for methacrylates, without the need for extensive, complex separation stages or significant upgrading. The aim of this work is to investigate the effectiveness of the proposed transesterification–FRP method. The objectives are (1) determine the yield and physical properties of polymer produced from crude pyrolysis oils (2) present an initial mass balance and (3) benchmark the thermal and rheological properties against pBMA.
Experimental
Materials
Azobisisobutyronitrile (AIBN, 97%) was recrystallised from methanol prior to use, titanium butoxide (TNBT, 97%) was used as received. Butyl methacrylate (BMA, 99%) inhibited with 10–55 ppm methyl ether of hydroquinone was filtered through alumina prior to use. Dichloromethane (DCM, HPLC grade) was used as supplied. Tetrahydrofuran (THF, HPLC grade) was used as supplied. Acetone, toluene and petroleum ether, (analytical reagent grade) were used as supplied. Anhydrous pyridine (HPLC grade) was used as supplied, triphenylphosphine oxide (TPPO, 97%) was used as supplied and purity accounted for in calculations, chromium(III) acetylacetonate (Cr(acac)3, 99.9%) was used as supplied, 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (TMDP, 98.2%) was used as supplied. Pyrolysis liquid was obtained commercially from the Empyro plant (Enschede, Netherlands), stored at 3 °C and used as supplied within two months of opening.Characterisation of pyrolysis liquid
The liquid was characterised by gas chromatography mass spectrometry (GCMS), 31P-nuclear magnetic resonance (NMR) and Karl Fischer (KF) prior to transesterification and FRP. The crude polymer yield was established via1H-NMR and, following precipitation, the polymer was characterised by size exclusion chromatography (SEC), thermogravimetric analysis (TGA), differential scanning calorimetry (DSC) and dynamic parallel plate rheometry.GC-MS
The injection port temperature was set at 200 °C and was operated in splitless mode with TG-POLAR capillary column (30 m × 0.25 mm, 0.25 μm stationary phase thickness). Samples were dissolved in DCM (1 mg mL−1) and helium was used as the carrier gas, at a flow rate of 1.5 mL minute−1. The GC oven was heated, after an initial three minute hold, from 40 °C to 260 °C at a rate of 5 °C per minute. The GC interface was held at 240 °C while the MS ion source was heated to 280 °C. Components eluting from the GC were ionized at 70 eV and their mass spectra recorded by the TOF-MS. The area percentage method was used for the quantification of the compounds present in the pyrolysis liquid.5,26 Identification of individual compounds was performed using the NIST library. All peaks below 1% intensity or below 80% match probability were excluded from analysis. Compounds were grouped according to their functionality to produce a composition based on total peak area, it should be noted that absolute values are not presented as this approach is only semi-quantitative.NMR
The 31P NMR data for the pyrolysis liquid and transesterifications were collected following the method and calculations outlined by NREL and prior publication by the authors and outlined in Fig. S1 and Table S1.†27,28 A stock solution was prepared by dissolving 10.90 mg of Cr(acac)3 and 72.28 mg of TPPO, internal standard, in 10 mL of a 1.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 pyridine:CDCl3 solution. This solution was dried using 4 Å molecular sieves. A 20–30 mg sample of the material was dissolved in 0.4 mL stock solution, before adding 50 μL TMDP. The mixture was left to react for 30 minutes at room temperature before filtering through a 25 μm filter into the NMR tube. 31P-NMR data was acquired, using a AV 400 MHz cryoprobe spectrometer. The inverse gated decoupling technique was used with a 10 second delay time and 128 scans. Spectra were referenced in relation to the δTPPO peak at 27.91 ppm, δTMDP = 175.5; δAliphatic OH = 152–145; δAromatic OH = 145–138; δCarboxylic acid OH = 138–134.6; δWater dimer = 133.7–130; δWater = 16.75; and the comparison integral intensities were used for quantification, with reagent purity accounted for. 1H NMR of the polymers generated from the pyrolysis oils was conducted using a 400 MHz cryoprobe in acetone-d6. Polymer conversion calculations are detailed in Fig. S2.† Magnetic resonance data analysis was conducted in Mestrelab Mnova 14.1.1.
:1 pyridine:CDCl3 solution. This solution was dried using 4 Å molecular sieves. A 20–30 mg sample of the material was dissolved in 0.4 mL stock solution, before adding 50 μL TMDP. The mixture was left to react for 30 minutes at room temperature before filtering through a 25 μm filter into the NMR tube. 31P-NMR data was acquired, using a AV 400 MHz cryoprobe spectrometer. The inverse gated decoupling technique was used with a 10 second delay time and 128 scans. Spectra were referenced in relation to the δTPPO peak at 27.91 ppm, δTMDP = 175.5; δAliphatic OH = 152–145; δAromatic OH = 145–138; δCarboxylic acid OH = 138–134.6; δWater dimer = 133.7–130; δWater = 16.75; and the comparison integral intensities were used for quantification, with reagent purity accounted for. 1H NMR of the polymers generated from the pyrolysis oils was conducted using a 400 MHz cryoprobe in acetone-d6. Polymer conversion calculations are detailed in Fig. S2.† Magnetic resonance data analysis was conducted in Mestrelab Mnova 14.1.1.
KF
Moisture content of samples were measured using a Mitsubishi CA-200 coulometric moisture meter. Samples above 0.5 wt% water were diluted with THF, with water content calculated as a weight fraction of the initial sample.Characterisation of pyrolysis liquid polymers
Preparation of rheometry specimens
15 mm diameter and 0.5 mm thick discs were prepared from pure polymer. Ethylene tetrafluoroethylene (ETFE) films were placed between the sample and platen surface during moulding. Specimens were heated to 80 °C and a compressive force of 5 tons applied for 3 minutes, released and re-applied for 3 minutes before cooling to room temperature under compressive pressure.Results and discussion
Pyrolysis liquid transesterification
Temperature, time and BMA concentration, were treated as variables in this study. The general reaction schemes and experimental setups are outlined in Fig. S3–S5.† When the preliminary transesterification conducted with crude pyrolysis liquid at 160 °C, a significant quantity of brown, insoluble solid was produced during the reaction. The low OH content indicated that either the transesterification reaction was successful, or that alcohols were incorporated into the solid. The insolubility of the products suggested that crosslinking; bonds formed between multiple polymer chains, had occurred. Previous work suggested that reducing the degree of esterification could avoid this crosslinking.22 A series of reactions were then conducted to optimise the temperature at which the transesterification was conducted, shown in Table S2,† this table includes quantitative values of alcohol concentration before and after transesterification for a range of reaction temperatures. Prior to transesterification, water and volatile pyrolysis liquid components were removed by vacuum distillation. It was demonstrated that by reducing the temperature, the transesterification yield was reduced. Kinetic experiments, shown in Fig. 2 show that there is no further decrease in OH content after 30 minutes. For this reason, transesterifications using 145 °C and a reaction time of 30 minutes were used in the next part of the study.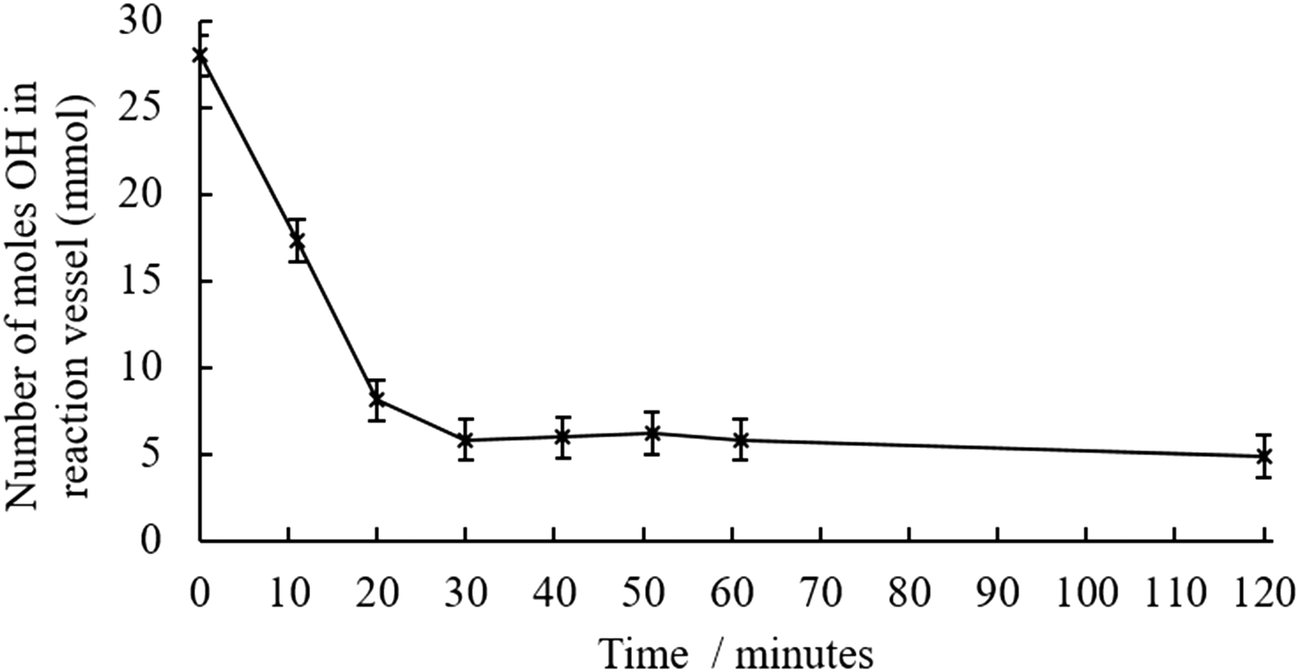 | ||
| Fig. 2 Decrease in hydroxyl content in the distilled pyrolysis liquid during the transesterification reaction at 145 °C along with transesterification reaction shown in the insert. | ||
The concentration of alcohol was measured in the reaction vessel with the butanol (BuOH) by-product being removed by distillation throughout the reaction. Control reactions of distilled pyrolysis liquid and BMA, with TNBT confirm that this decrease in OH is due to transesterification reaction. Consequently, as conversion is less than 100% and an excess of BMA was added there is unreacted BMA blended with functionalised pyrolysis liquid. This means that any polymer produced was likely a copolymer of BMA and pyrolysis liquid methacrylate monomers.
Sustainability and scalability of pyrolysis polymer manufacture via transesterification
A block flow diagram is presented in Fig. 3, with masses and compositions shown in Table 1.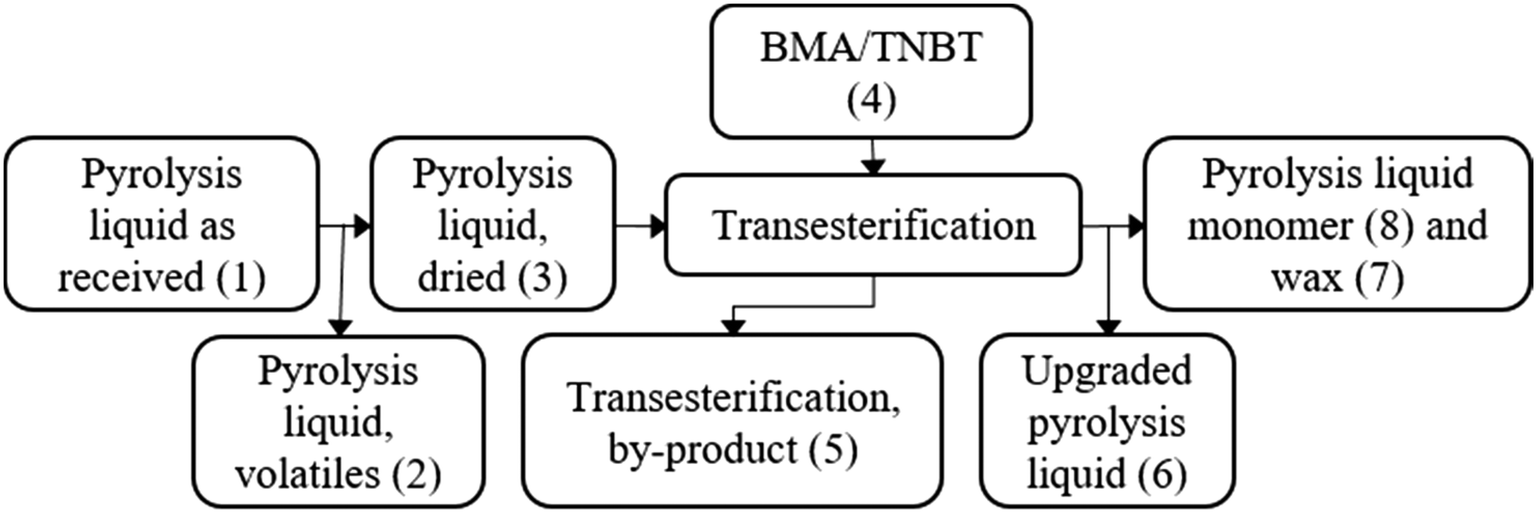 | ||
| Fig. 3 Block flow diagram to outline the streams in the proposed transesterification of pyrolysis liquid. | ||
| Pyrolysis liquid, as received | Pyrolysis liquid, volatiles | Pyrolysis liquid, distilled | BMA & TNBT | Transesterification by-product | Upgraded pyrolysis liquid | Wax | Pyrolysis liquid monomer | ||
|---|---|---|---|---|---|---|---|---|---|
| Stream | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |
| Mass (kg) | 1 | 0.4 | 0.6 | 0.6 | 0.12 | 0.078 | 0.03 | 0.972 | |
| Composition from GC (%) | Acid | 6 | 4 | — | — | 2.3 | 18.8 | 17.1 | |
| Alcohol | 29 | 5 | 63.5 | — | 70.4 | — | 33.4 | ||
| Aldehyde | 4 | 2 | — | — | 4.2 | — | 6.3 | ||
| Aromatic | 3 | 1 | 17.3 | — | 6 | — | 3.2 | ||
| Ester | 1 | 1 | — | — | — | — | 8.1 | ||
| Furans | 26 | 15 | 11.2 | — | 4.8 | — | 3.2 | ||
| Ketone | — | — | — | — | 5.9 | — | 18.1 | ||
| Non identified | 2 | 1 | 8 | — | 6.5 | — | — | ||
| Sugars | — | — | — | — | — | 81.2 | 10.7 | ||
| Excluded | <1 | 1.17 | <1 | — | <1 | <1 | <1 | — | |
| Water content (%) | 29 | 72 | <1 | — | <1 | <1 | <1 | <1 | |
The block flow masses in Table 1 show that the main components of the pyrolysis liquid volatiles, removed in the drying step, are water and furans. Alcohols, aromatics and some non-identified species remain in pyrolysis liquid after distillation. The transesterification by-product is composed of 70% butanol and other species that could be useful in combustion. The upgraded pyrolysis liquid stream, Fig. S6 and S7,† shows an enrichment of acid and sugar derivatives compared to the pyrolysis liquid as supplied, confirmed with analytical standards as acetic acid and levoglucosan. The wax comprises unreacted alcohols, acid, aldehyde, ketone and sugar derivatives miscible with the pyrolysis monomer. Titanium catalyst fate was in fraction 6 confirmed by gravimetric ash measurement of fractions 6 & 8 in 550 °C oven.
Reaction yield is a one dimensional way of measuring the quality of a reaction, there are several inter-dependant steps, so there are mass yields of polymer with respect to the pyrolysis liquid monomer (8), BMA (4) and the pyrolysis liquid, as received (1), these are 85.7%, 138.8% and 83.3% respectively. These mass yields, particularly with respect to BMA, show that pyrolysis liquid is incorporated into the mass of the polymer.
Estimations of how much mass in stream 8 is derived from BMA, itself a derivative of natural gas via methacrylic acid from the ACH process, can also be made from the masses in Table 1. However, bio-based routes to methacrylic acid have now been identified and are in the process of being commercialised, thus sustainability footprint of this process in the near future will exhibit significant improvement.29 With respect to defining an crude estimation of the final copolymer BMA:pyrolysis polymer ratio. The known input of BMA was 0.6 mass equivalents and the quantity of pyrolysis liquid monomer was estimated by taking accounting for the 0.0845 (0.12 × 0.704) equivalents of BuOH removed by distillation. This gave a mass ratio for BMA:pyrolysis liquid monomer of 0.515:0.972 or 53.0 wt%. It should be noted that this value includes both excess BMA and the mass of the methacrylate functional group in the pyrolysis liquid monomer. The addition of a BMA recycling step prior to polymerisation could further reduce the amount of BMA, and hence petrochemical derivatives, in the product. The choice of initiator is interesting grounds for further work, as there is a limited number of thermally triggered radical initiators available to us. Thus, there would be fertile ground in trying to produce more sustainable options. Although such a study would need to reflect on the fact that, as the initiator only accounts for an extremely small percentage of the reaction mass, its influence of the overall sustainability if the process is very limited. Furthermore, as it also determines the reaction parameters that can be used, for instance 4,4-azobis(4-cyanovaleric acid) allows for lower initiation temperatures than AIBN. However, at lower temperatures the viscosity is usually much higher, which may result in the need to use a solvent which would introduce much greater sustainability issues. Fermentation routes to C3–C12 methacrylates have been identified elsewhere that could reduce the mass of natural gas derived methacrylate.30
However, the commercial prospect of pyrolysis polymers produced using this approach will also depend in part on the utilisation of the coproduct streams shown in Table 1. There are many options that can be considered here, e.g. utilisation of the volatiles stream as a source of platform furan chemicals and the upgraded pyrolysis liquid as a fermentation feedstock. Full consideration of co-product utilisation is beyond the scope of this work, however there is an exciting opportunity for future work to use different technologies such as microwave pyrolysis to change the starting chemistry of the pyrolysis liquid5 and subsequently to understand how the pyrolysis polymer properties and the overall mass balance are affected. Future work will also consider catalysts that can tolerate water within the pyrolysis liquids, thereby removing the need to undertake the first distillation step and purification or control agent strategies to increase the molecular weight could improve rheological properties.
Polymerisation of transesterified pyrolysis liquid monomer
The functionalised pyrolysis liquid was used as the starting material for FRP. AIBN was chosen as this is the most widely used thermal Vazo-radical initiator for acrylate monomers. A representative SEC chromatogram for the product is shown in Fig. 4.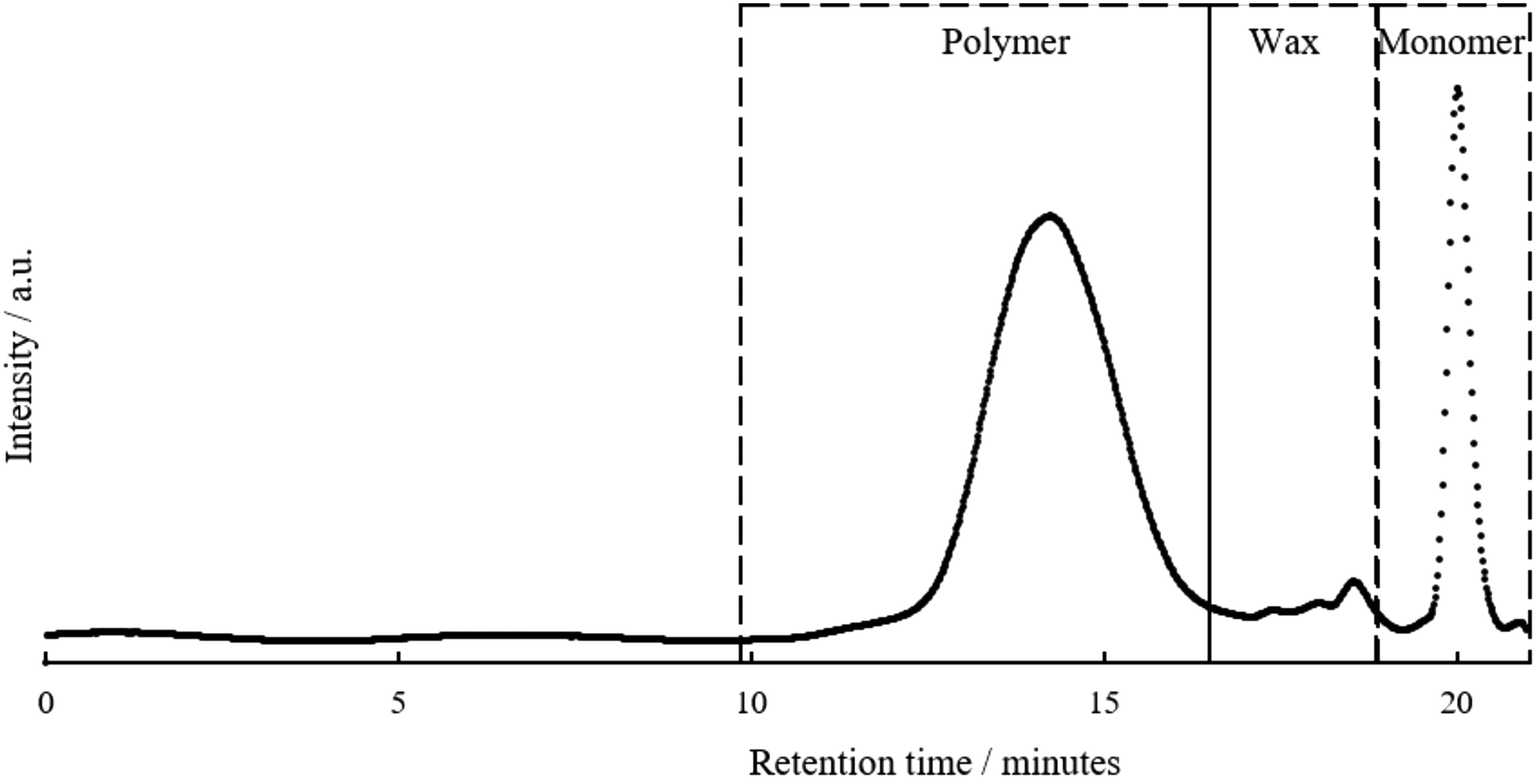 | ||
| Fig. 4 Size exclusion chromatograph of upgraded pyrolysis liquid prior to purification including annotation for pyrolysis polymer, wax and monomer peaks from left to right. | ||
The SEC chromatogram shows that an additional molecular weight species is present in the polymer sample. A kinetic study of the evolution of the polymer peaks given in Fig. S8† demonstrated that this material was present from the outset of the polymerisation and did not increase during reaction. The only peaks noted to change during the reaction are the depletion of the monomer peak and the increase of a monomodal polymer peak. Thus, it is proposed that this additional peak may be an unreactive wax. The molecular weight of this wax, 0.5–2 kDa is similar to that of pyrolytic lignin, the origin of this wax is unclear. SEC analysis of the; (a) pyrolysis liquid as received, (b) pyrolysis liquid after drying, (c) transesterified pyrolysis liquid and (d) transesterification residue are presented in Fig. S9.† The apparent change in the wax concentration/molecular weight after the drying step was attributed to both (a) volatile low molecular weight compounds being removed contributing the wax being a greater percentage of the total mass and (b) a small amount of wax crosslinking as has been reported in the higher temperature experiments within this study. There were instances where there was incomplete baseline separation between polymer and wax peaks, this artificially increased Mn and to a lesser extent Mw values and for this reason Mp was used in this comparison because of its lack of dependence on the distribution of the sample molecular weight.
Additionally, the mono-modal nature of the SEC polymer peak was unexpected. There is such a wide variety of alcohols present in the pyrolysis liquid that it was anticipated that their transesterification would have produced a wide range of different monomers with disparate levels of reactivity. A range of different polymers formed at different rates and resulting in different molecular weight products would give rise to polymers with different hydrodynamic volumes and hence a multi-modal SEC. Analysis of the light scattering derived molecular weight of the pyrolysis polymer is shown in Fig. 5.
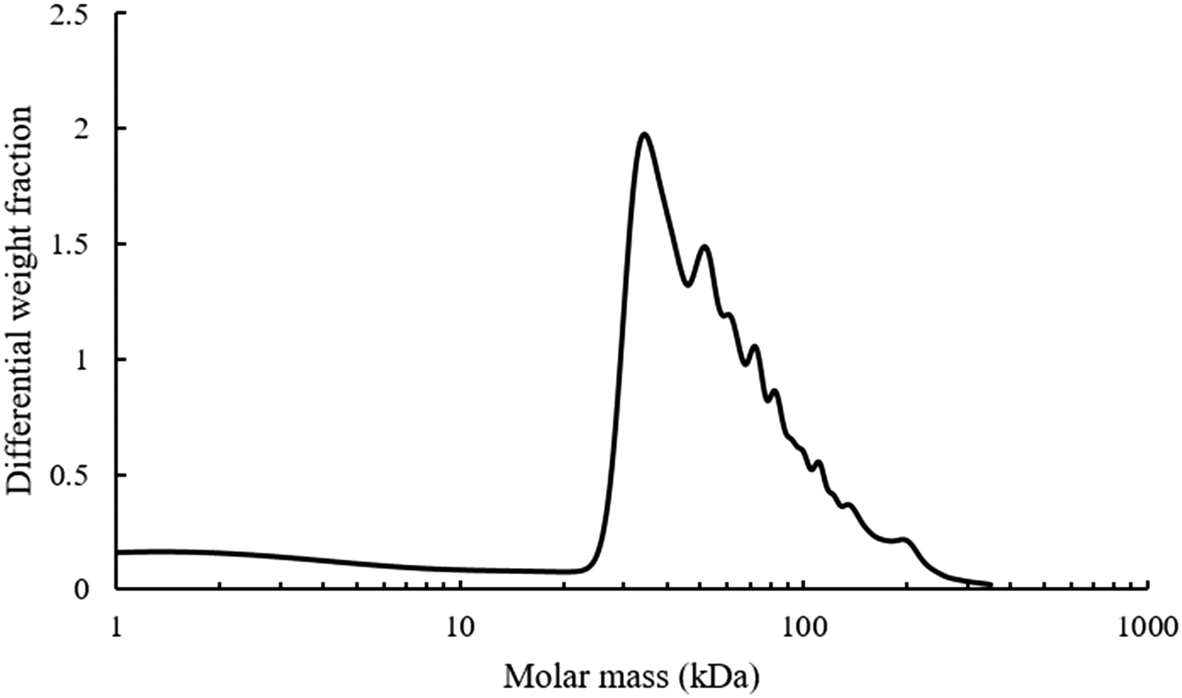 | ||
| Fig. 5 Molecular weight analysis of pyrolysis polymers using light scattering, dn/dc value of 0.087 mL g−1. | ||
The refractive index increment (dn/dc) value of BMA is 0.087 mL g−1 and was used for molecular weight analysis of polymer as BMA is likely to be the most abundant single motif in the system. There are multiple different shoulder peaks apparent in Fig. 5, indicating the presence of other high molecular weight species, distinct from a BMA homopolymer. These components are likely to have a range of different dn/dc values and full characterisation of the components and their respective dn/dc values would be needed to validate these molecular weights. However, the existence and relative position of these peaks is consistent with results elsewhere supporting the idea of multiple copolymers.15
The effect of pyrolysis liquid concentration on the polymerisation was investigated further by varying the molar amount of BMA added to the pyrolysis liquid prior to transesterification. The stoichiometry and nomenclature used is provided in Table S3.† By increasing the amount of residual BMA present in the transesterification step the effect of the pyrolysis liquid monomers on the polymerisation was reduced.
It was not possible to define the exact BMA:pyrolysis liquid monomer ratio in the transesterification product via NMR analysis due to the overlap of coincidental peaks in the spectra. Instead, the mass of butanol condensate distilled out of the reaction vessel in the transesterification step was used as a measure of the amount of pyrolysis monomer formed. This value could then be compared to the amount of BMA in the feed to give an indicative measure of the BMA:pyrolysis liquid monomer ratio. The data used for this calculation is included in Table S3† and show that using the distillate mass to estimate the BMA:pyrolysis liquid monomer ratio overestimates the concentration of BMA in the monomer mixture, this is not the case for 31P-NMR, the difference between the two calculations becomes more pronounced as the amount of distillate increases.
The initiator concentration was varied to investigate the effect of a change in concentration on the polymer peak molecular weight and conversion, Table 2.
| Entry # | Ratio of BMA:pyrolysis liquid |
[AIBN] (wt%) | Conversion (%) | M p (kDa) | M n (kDa) | M w (kDa) | Đ |
|---|---|---|---|---|---|---|---|
| Control | 0.6:1 |
0 | * | * | * | * | * |
| 1 | 0.6:1 |
0.01 | * | * | * | * | * |
| 2 | 0.6:1 |
0.11 | * | * | * | * | * |
| 3 | 0.6:1 |
0.98 | 79.04 | 22.9 | 16.1 | 28.4 | 1.77 |
| Control | 1.2:1 |
0 | * | * | * | * | * |
| 4 | 1.2:1 |
0.02 | 8.26 | 29.8 | 6.99 | 35.7 | 5.11 |
| 5 | 1.2:1 |
0.17 | 10.71 | 27.4 | 9.86 | 33.4 | 3.39 |
| 6 | 1.2:1 |
0.80 | 82.49 | 24.6 | 15.8 | 29.5 | 1.87 |
| 7 | 1.2:1 |
1.00 | 85.12 | 22.9 | 20.9 | 34.3 | 1.64 |
| Control | 2.4:1 |
0 | * | * | * | * | * |
| 8 | 2.4:1 |
0.02 | 7.41 | 36.9 | 19.0 | 40.1 | 2.11 |
| 9 | 2.4:1 |
0.10 | 27.01 | 32.2 | 21.7 | 41.3 | 1.90 |
| 10 | 2.4:1 |
0.62 | 87.03 | 13.4 | 6.57 | 15.5 | 1.65 |
Table 2 shows increasing initiator concentration in the polymerisations resulted in a decrease in Mp, increased conversion and lower Đ. When comparing the initiator concentrations, the effects of different BMA:pyrolysis liquid ratios are most apparent in their effect on Mp. In all cases when initiator concentration was increased the expected decrease in Mp was observed, The differences are more pronounced at higher BMA:pyrolysis liquid ratios. For example, comparing 5 with 7, a 5.9 fold increase in initiator concentration decreased Mp by 17% where in the case of 9 and 10, a 6.2 factor increase in initiator concentration reduced Mp by approximately 40%. For the same examples, the effect on dispersity is even more pronounced, with entries 5 & 7 showing a 52% decrease and 9 & 10 showing only a 13% decrease. Comparing samples with comparable initiator concentrations, entries 4 & 8, similar conversions but entry 8 had higher molecular weight and lower dispersity. This suggests that the wide variety of monomers in the functionalised pyrolysis liquid decrease the conversion and molecular weight and increase polydispersity.
Thermal and rheological properties of pyrolysis polymer and p(BMA)
The elastic storage modulus G′ and viscous loss modulus G′′ responses measured in thermal ramps at constant 1 Hz frequency are shown in Fig. 6 for the pyrolysis polymer and the p(BMA). In order to account for the slightly different Tg, the ordinate axis is offset by Tg. The elastic and viscous parts of the responses of both materials are similar across a broad range of temperature, with only some deviation noted at higher temperature.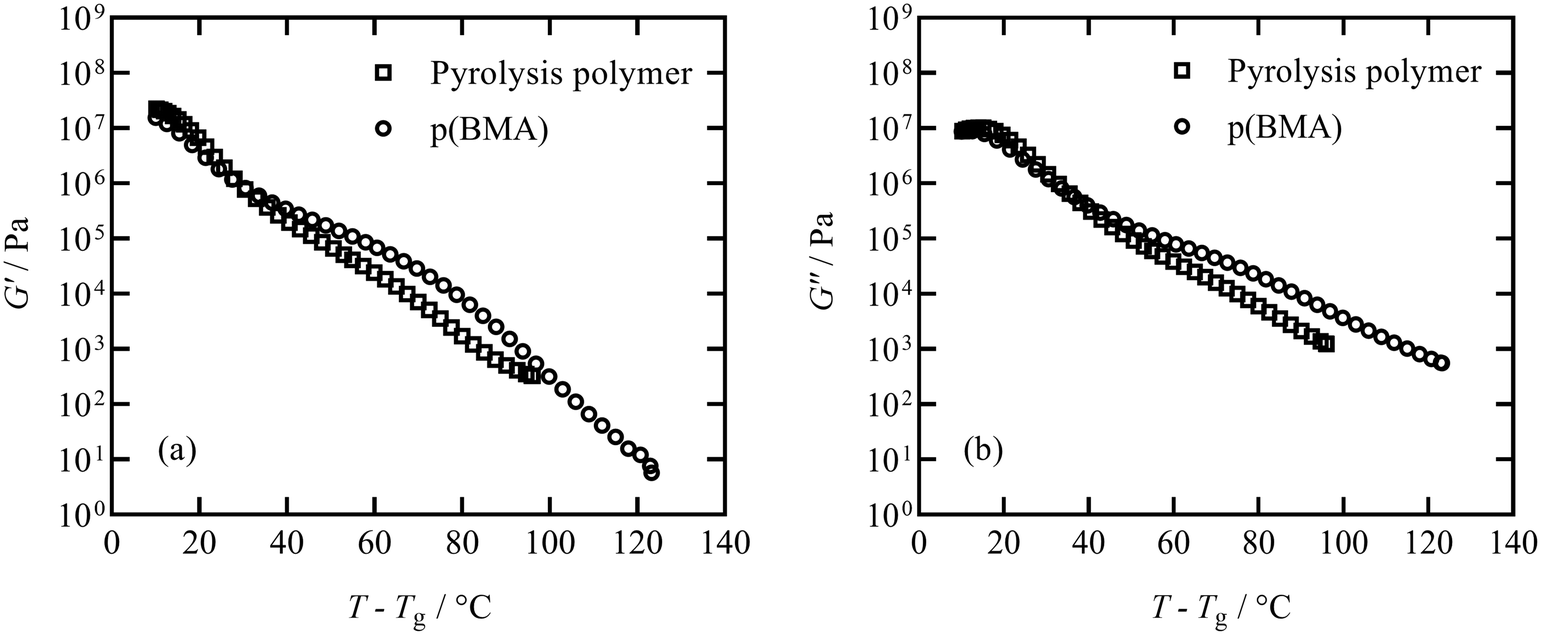 | ||
| Fig. 6 Thermal scan conducted at a cooling rate of 2 °C min−1 shows G′ (a) and G′′ (b) response for the pyrolysis polymer and p(BMA). Close agreement between the two materials is noted for temperatures around Tg, however deviation at higher temperatures is observed. | ||
In order to study this in more detail, isothermal frequency sweeps were carried out at temperature intervals of 30, 35, 40, 45, 55, 65, 80, 100 °C for the pyrolysis polymer and 40, 50, 60, 80, 95, 125 °C for p(BMA), to provide sufficient overlap of individual data curves to form mastercurves. Temperature intervals were predicted using the Williams–Landel–Ferry equation and universal C1 and C2 constants aiming for an overlap of 0.5 decades of frequency.31,32 The individual isothermal curves were manually shifted in frequency to produce the Tg-normalised frequency mastercurves of G′ and G′′ shown in Fig. 7.
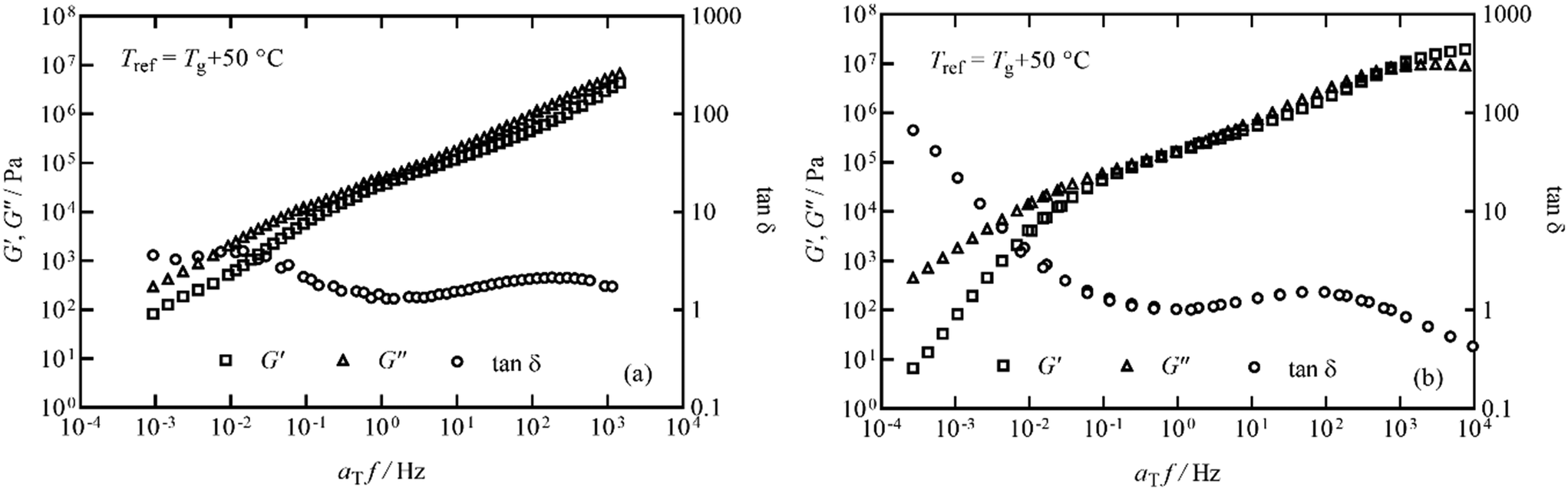 | ||
| Fig. 7 Frequency mastercurves for (a) the pyrolysis polymer and (b) p(BMA) at Tref = Tg + 50 °C. G′, G′′ and tanδ are shown as squares, triangles and circles respectively for both materials. | ||
Although the behaviour is similar, there are two important differences. Firstly, the pyrolysis polymer exhibits lower moduli by approximately an order of magnitude across the temperature range. Secondly, in the low frequency flow region, the p(BMA) exhibits the expected power-law behaviours with gradients very close to 2 and 1 in G′ and G′′ respectively,33 but the pyrolysis polymer does not, particularly in G′. These differences point to the role of the wax component in reducing the elasticity and the viscosity and preventing a clear entanglement plateau. In addition, structural changes are apparent at high temperatures in the pyrolysis polymer in processes related to those that eventually lead to char formation.
TGA data obtained for the pyrolysis polymer and p(BMA) of a similar molecular weight is included in Fig. S10.† A photograph of the pyrolysis polymer is included in Fig. S11.† Temperatures at 1%, 10% and 50% mass loss are reported in Table 3 along with molecular weight information and Tg values established from DSC experiments. Despite the similar molecular weight, the presence of the low molecular weight wax species is the likely cause of the reduction in Tg, the earlier onset of degradation and the residual char in the same manner as found for related bimodal blended systems.14,34
| Property | Pyrolysis polymer | p(BMA) |
|---|---|---|
| M p (kDa) | 35.4 | 37.6 |
| M n (kDa) | 18.6 | 25.1 |
| M w (kDa) | 37.9 | 47 |
| Đ | 2.04 | 1.87 |
| T g (°C) | 13.9 | 26.7 |
| T 1% (°C) | 145.6 | 274.9 |
| T 10% (°C) | 245.8 | 302.2 |
| T 50% (°C) | 363.9 | 329.2 |
| Mass residue, 500 °C (%) | 11.2 | 0 |
| tanδmin |
1.3 | 1.01 |
| f (Hz) | 1.16 | 1.51 |
| τ (s) | 0.87 | 0.66 |
| G′ (kPa) | 39.85 | 217.58 |
| G′′ (kPa) | 51.64 | 220.45 |
| η* (kPa s) | 41.33 | 492.97 |
Table 3 gives a comparison of rheological parameters measured at the frequency where the loss tangent, tanδ, is a minimum for the pyrolysis polymer and p(BMA). Across the relevant processing temperature range, the pyrolysis polymer exhibits moduli that are smaller than those of the p(BMA) by almost an order of magnitude. Similarly, the complex viscosity η* was found to be lower for the pyrolysis polymer by just over an order of magnitude. The pyrolysis polymer's reduced viscosity could be due to the diluting and plasticizing effects of the wax. The frequency at which the loss tangent is closest to unity is comparable for the two polymers, suggesting similar terminal relaxation times, τ. To probe further the rheological behaviour of the pyrolysis polymer at elevated temperature, a 15 mm disc was held at 110 °C and subjected to small angle oscillations of γ = 0.1% and f = 1 Hz for 1 hour. The complex viscosity rose a little, at a constant rate, from 181 Pa s to 193 Pa s, suggesting an overall small increase in molecular weight.35 SEC experiments were carried out before and after the 1 hour test. Although similar, there was an increase in Mp of the waxy peak by a factor of ∼4.7, although remaining oligomeric in nature, and a very small reduction in Mp of the polymer peak by 8.4%. This suggests that there has been some degree of polymerisation of the waxy component, and possibly a small degree of degradation or scission of the polymer.
Based on the results shown, the rheological performance of the pyrolysis polymer is similar to that of the p(BMA) but distinct, likely due to the presence of the oligomeric wax. Further investigation into identifying and controlling the mass fraction and thermal stability of the wax in the pyrolysis polymer may offer a facility for tuning processing performance to desired applications. It has been noted that the rheological performance of this novel material is comparable to that of numerous bitumen binders from crude oil and synthetic sources across similar temperature ranges.36–38 Works published in the road surfacing sector have expressed the need for renewable acrylates and methacrylates for use as synthetic bitumen binders which offers a potential application for this polymer and will be the subject of further work.39,40 As this is a process that stems from the use of naturally occurring reagents, there will likely be variability in the monomer/polymer make up if the source of the bio-reagent is changed. Thus, in this study, pyrolysis liquid from the same batch source was used. Should an industrial outlet be found from a potentially variable feedstock then at that point, a full parametric study would need to be conducted to draw conclusions on the compositions of the products and their effect on the properties with specific relevance to a chosen application.
Conclusions
The process developed here demonstrates a sustainable strategy to produce methacrylate polymers from unrefined pyrolysis liquids that are derived from lignocellulosic industrial scale biomass wastes. A route to polymer directly from pyrolysis liquids has been identified without requiring extensive separation. Production of the monomer is achieved in one step without the use of toxic reagents and 79% of the alcohol functional groups can be converted by esterification with BMA. This process can be integrated into established technologies, involves fewer process steps and avoids toxic thionyl and halogenated wastes generated in other processes.14,15,39 Effect of initiator and BMA on polymerisation was investigated and the most promising route identified and example syntheses are provided. Thermal and rheological properties of this material were found to be similar to those of p(BMA) of comparable molecular weight. Therefore, this material shows potential as a replacement resin, adhesive or binder. The presence of a low molecular weight wax in the pyrolysis polymer leads to a reduced viscosity and glass transition. Through tuning of the pyrolysis liquid production to vary the amount of the wax species present, further opportunities for improvements in molecular weight and control of polymer properties are identified and tuning Tg and viscosity for different application based testing will be the subject of further investigation.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Scholarship funding was awarded to John Ryan from the EPRSC Centre for Doctoral Training in Sustainable Chemistry (University of Nottingham). The authors would also like to thank Peter Licence, Steven Howdle, Ross Denton and James Dowden for the use of laboratory space and William Meredith for assistance with GCMS analysis.References
- PlasticsEurope, Plastics – the Facts, 2018, p. 18, https://www.plasticseurope.org/application/files/6315/4510/9658/Plastics_the_facts_2018_AF_web.pdf, (accessed June 5 2019) Search PubMed.
- M. Borzecka, European Wood Waste Statistics Report for Recipient and Model Regions, (BioREG D1.1), November 2018, pp. 1–48 Search PubMed.
- BTL-BTG, BTG-BTL pyrolysis process, https://www.btg-btl.com/en/technology, (accessed June 5, 2019) Search PubMed.
- BTL-BTG, Mega-order from Finland for Dutch energy technology, https://www.btg-btl.com/en/company/news/news/article?id=134, (accessed Jun 5, 2019) Search PubMed.
- B. J. Shepherd, J. Ryan, M. Adam, D. B. Vallejo, P. Castaño and E. T. Kostas, Microwave Pyrolysis of Biomass within a Liquid Medium, J. Anal. Appl. Pyrolysis, 2018, 134(June), 381–388 CrossRef CAS.
- X. Hu and M. Gholizadeh, Biomass Pyrolysis: A Review of the Process Development and Challenges from Initial Researches up to the Commercialisation Stage, J. Energy Chem., 2019, 39, 109–143 CrossRef.
- A. Dufour, Thermochemical Conversion of Biomass for Energy and Chemicals Production, Wiley-ISTE, 2016, pp. 1–20 Search PubMed.
- J. Robinson, C. Dodds, A. Stavrinides, S. Kingman, J. Katrib, Z. Wu, J. Medrano and R. Overend, Microwave Pyrolysis of Biomass: Control of Process Parameters for High Pyrolysis Oil Yields and Enhanced Oil Quality, Energy Fuels, 2015, 29(3), 1701–1709 CrossRef CAS.
- D. C. Elliott, D. Meier, A. Oasmaa, B. Beld and A. V. Bridgwater, Results of the International Energy Agency Round Robin on Fast Pyrolysis Bio-Oil Production, Energy Fuels, 2017, 31, 5111–5119 CrossRef CAS.
- C. Lindfors, A. Oasmaa, A. Välimäki, T. Ohra-aho, H. Punkkinen, C. Bajamundi and K. Onarheim, Standard Liquid Fuel for Industrial Boilers from Used Wood, Biomass Bioenergy, 2019, 127, 105265 CrossRef CAS.
- L. Mialon, A. G. Pemba and S. A. Miller, Cutting-Edge Research for a Greener Sustainable Future Biorenewable Polyethylene Terephthalate Mimics Derived from Lignin and Acetic Acid †, Green Chem., 2010, 12, 1704–1706 RSC.
- J. F. Stanzione, P. A. Giangiulio, J. M. Sadler, J. J. Scala and R. P. Wool, Lignin-Based Bio-Oil Mimic as Biobased Resin for Composite Applications, ACS Sustainable Chem. Eng., 2013, 1, 419–426 CrossRef CAS.
- M. Nowakowska, O. Herbinet, A. Dufour and P. A. Glaude, Kinetic Study of the Pyrolysis and Oxidation of Guaiacol, J. Phys. Chem. A, 2018, 122, 7894–7909 CrossRef CAS PubMed.
- W. Qu, Y. Xue, Y. Gao, M. Rover and X. Bai, Repolymerization of Pyrolytic Lignin for Producing Carbon Fiber with Improved Properties, Biomass Bioenergy, 2016, 95, 19–26 CrossRef CAS.
- S. Wang, L. Shuai, B. Saha, D. G. Vlachos and T. H. Epps, From Tree to Tape: Direct Synthesis of Pressure Sensitive Adhesives from Depolymerized Raw Lignocellulosic Biomass, ACS Cent. Sci., 2018, 4, 701–708 CrossRef CAS PubMed.
- A. L. Holmberg, J. F. Stanzione, R. P. Wool and T. H. Epps, A Facile Method for Generating Designer Block Copolymers from Functionalized Lignin Model Compounds, ACS Sustainable Chem. Eng., 2014, 2, 569–573 CrossRef CAS.
- A. L. Holmberg, M. G. Karavolias and T. H. Epps, RAFT Polymerization and Associated Reactivity Ratios of Methacrylate-Functionalized Mixed Bio-Oil Constituents†, Polym. Chem., 2015, 6, 5728–5739 RSC.
- A. L. Holmberg, K. H. Reno, N. A. Nguyen, R. P. Wool and T. H. Epps, Syringyl Methacrylate, a Hardwood Lignin-Based Monomer for High-Tg Polymeric Materials, ACS Macro Lett., 2016, 5, 574–578 CrossRef CAS.
- A. L. Holmberg, N. A. Nguyen, M. G. Karavolias, K. H. Reno, R. P. Wool and T. H. Epps, Softwood Lignin-Based Methacrylate Polymers with Tunable Thermal and Viscoelastic Properties, Macromolecules, 2016, 49, 1286–1295 CrossRef CAS.
- A. Llevot, E. Grau, S. Carlotti, S. Grelier and H. Cramail, From Lignin-Derived Aromatic Compounds to Novel Biobased Polymers, Macromol. Rapid Commun., 2016, 37, 9–28 CrossRef CAS PubMed.
- Methacrylates: chemical safety, https://www.chemicalsafetyfacts.org/methacrylates/, (accessed June 16, 2020) Search PubMed.
- W. Qu, Y. Huang, Y. Luo, S. Kalluru, E. Cochran, M. Forrester and X. Bai, Controlled Radical Polymerization of Crude Lignin Bio-Oil Containing Multihydroxyl Molecules for Methacrylate Polymers and the Potential Applications, ACS Sustainable Chem. Eng., 2019, 7(9), 9050–9060 CrossRef CAS.
- J. A. Sullivan and S. Burnham, The Selective Oxidation of Glycerol over Model Au/TiO2 Catalysts - The Influence of Glycerol Purity on Conversion and Product Selectivity, Catal. Commun., 2014, 56, 72–75 CrossRef CAS.
- A. A. Dundas, A. L. Hook, M. R. Alexander, S. W. Kingman and D. J. Irvine, Methodology for the Synthesis of Methacrylate Monomers Using Designed Single Mode Microwave Applicators, React. Chem. Eng., 2019, 4, 1472–1476 RSC.
- C. Barner-Kowollik and S. Perrier, The Future of Reversible Addition Fragmentation Chain Transfer Polymerization, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5715–5723 CrossRef CAS.
- E. T. Kostas, M. Cooper, B. J. Shepherd and J. P. Robinson, Identification of Bio-Oil Compound Utilizing Yeasts Through Phenotypic Microarray Screening, Waste Biomass Valoriz., 2020, 11, 2507–2519 CrossRef CAS.
- M. V. Olarte, S. D. Burton, M. Swita, A. B. Padmaperuma, J. Ferrell and H. Ben, Determination of Hydroxyl Groups in Pyrolysis Bio-Oils Using 31P-NMR, NREL/TP-5100-65887, Golden, CO, USA, 2016 Search PubMed.
- A. R. Goddard, S. Pérez-Nieto, T. M. Passos, B. Quilty, K. Carmichael, D. J. Irvine and S. M. Howdle, Controlled Polymerisation and Purification of Branched Poly(Lactic Acid) Surfactants in Supercritical Carbon Dioxide, Green Chem., 2016, 18, 4772–4786 RSC.
- IUK, Sweet Perpex (103761), https://gtr.ukri.org/projects?ref=103761#/tabOverview, (accessed Aug 27, 2020).
- G. Eastham, Z. Disley, D. Johnson, G. Stephens and M. Waugh, Process for the production of methyl methacrylate, WO2018096326A1, 2019.
- J. Dudowicz, J. F. Douglas and K. F. Freed, The Meaning of the “ Universal ” WLF Parameters of Glass-Forming Polymer Liquids, J. Chem. Phys., 2017, 014905, 1–7 Search PubMed.
- M. L. Williams, R. F. Landel and J. D. Ferry, The Temperature Dependence of Relaxation Mechanisms in Amorphous Polymers and Other Glass-Forming Liquids, J. Am. Chem. Soc., 1955, 77, 3701–3707 CrossRef CAS.
- M. Rubinstein and R. H. Cloby, Polymer Physics, OUP Oxford, 2003, pp. 253–277 Search PubMed.
- W. E. I. Zheng and S. L. Simon, The Glass Transition in Athermal Poly(a-Methyl Styrene)/ Oligomer Blends, J. Polym. Sci., Part B: Polym. Phys., 2008, 46, 418–430 CrossRef CAS.
- S. Porter and F. Johnson, Viscosity of Polyethylenes: Dependence on Molecular Weight and Temperature *, J. Appl. Polym. Sci., 1960, 3(8), 194–199 CrossRef.
- G. D. Airey and M. H. Mohammed, Rheological Properties of Polyacrylates Used as Synthetic Road Binders, Rheol. Acta, 2008, 47, 751–763 CrossRef CAS.
- G. D. Airey, J. R. A. Grenfell, A. Apeagyei, A. Subhy and D. Lo Presti, Time Dependent Viscoelastic Rheological Response of Pure, Mech. Time-Depend. Mater., 2016, 455–480 CrossRef CAS.
- G. D. Airey, J. Wilmot, J. R. A. Grenfell, D. J. Irvine, I. A. Barker and J. El, Rheology of Polyacrylate Binders Produced via Catalytic Chain Transfer Polymerization as an Alternative to Bitumen in Road Pavement Materials, Eur. Polym. J., 2011, 47, 1300–1314 CrossRef CAS.
- G. D. Airey, M. H. Mohammed and C. Fichter, Rheological Characteristics of Synthetic Road Binders, Fuel, 2008, 87, 1763–1775 CrossRef CAS.
- L. P. Ingrassia, X. Lu, G. Ferrotti and F. Canestrari, Renewable Materials in Bituminous Binders and Mixtures: Speculative Pretext or Reliable Opportunity?, Resour., Conserv. Recycl., 2019, 144, 209–222 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0re00419g |
| This journal is © The Royal Society of Chemistry 2021 |