
Computational modelling of adsorption and diffusion properties of CO2 and CH4 in ZIF-8 for gas separation applications: a density functional theory approach†
Hari P.
Paudel
*,
Wei
Shi
,
David
Hopkinson
,
Janice A.
Steckel
and
Yuhua
Duan
 *
*
National Energy Technology Laboratory, United States Department of Energy, Pittsburgh, Pennsylvania 15236, USA. E-mail: Hari.Paudel@netl.doe.gov; yuhua.duan@netl.doe.gov
First published on 13th January 2021
Abstract
Understanding of zeolitic imidazolate framework-8 (ZIF-8) interaction with different gas molecules is crucial when ZIF-8 is used in gas separation. Computational studies based on density functional theory (DFT) can be used to investigate gas interactions and diffusion mechanisms that can be directly correlated with experimental observations. Here we present our studies based on DFT calculations on CO2 and CH4 gas adsorption and diffusion in the bulk of ZIF-8. We evaluate the structural and electronic properties of bulk ZIF-8, and determine the most stable adsorption sites and the corresponding diffusion barriers for CO2 and CH4 molecules. Our calculations incorporate long-range dispersion interactions to describe the weak interactions between adsorbate molecules and the framework. We analyze the adsorption and diffusion properties in relation with the material's volume expansion. We find that the CO2 and CH4 adsorption energies at the most stable adsorption sites are 5.01 and 4.47 kcal mol−1, respectively. The diffusivity of CO2 is found to be about two times that of CH4. Our calculated diffusion coefficients were found to have the same order of magnitude with the experimental results. Furthermore, our calculations indicate that CO2 and CH4 diffusivities in fixed ZIF-8 (all ZIF-8 framework atoms are fixed) are 5–9 times lower than the corresponding diffusion values in flexible ZIF-8 (all the framework atoms are allowed to move).
1. Introduction
Polymeric membranes are commercially used in gas separation applications such as to remove carbon dioxide (CO2) from natural gas, and hydrogen (H2) from mixtures of nitrogen (N2) or hydrocarbons in petrochemical processing applications.1–5 However, these materials possess a fundamental problem that arises due to a tradeoff between the gas separation factor and its permeability for the more permeable component in the gas mixture.6–8 Practically, a high separation factor with high permeability that yields an upper bound in the logarithmic relationship of these trade-off parameters (separation factor and permeability) is desirable for gas separation.9 In order to achieve an optimal material performance with high upper bound and overcome the problem of low selectivity and permeability with polymeric compounds, membranes made of zeolites or combinations of these with polymeric systems have been proposed to be used to separate the mixture of gases such as CO2 and CH4.10–13There have been significant efforts devoted to characterizing zeolite imidazolate frameworks (ZIFs) in order to efficiently and economically use them in membrane technology to separate gas components from mixtures. ZIFs have selective adsorption properties that enable them to discriminate on a particular type of gas molecule to be adsorbed more than others.13–16 When compared to traditional chemical sorbent materials (mostly amines), ZIFs have (a) high CO2 loading capacity, (b) lower energy consumption, (c) high thermal and chemical stability, and (d) lower corrosion rates.17–20 Due to these properties and the presence of permanent porosity, ZIFs outperform typical metal organic frameworks (MOFs) for gas separation. One of the most studied systems within the family of zeolitic imidazolium frameworks is ZIF-8. This system has a sodalite topology and a Zn(mim)2 stoichiometric representation, where mim represents 2-methylimidazolate.21 Within the same topology, ZIFs can be synthesized with different metal centers such as Zn, Co, Cu and Fe that are tetrahedrally coordinated and linked by imidazolate ligands.22,23 Importantly, the strength of the bonding between the metal ions and the ligands is thought to provide a high chemical stability.21,24 The structure of ZIF-8 consists of a central nanopore with a 11.6 Å diameter which is surrounded by six and four window pores of 3.4 and 0.8 Å diameters, respectively.25,26 The size of 3.4 Å is comparable to the kinetic diameters of CO2 (3.3 Å) and CH4 (3.8 Å) molecules, making ZIF-8 an ideal material for gas mixture separations. A total of 17 polyforms of Zn(mim)2 have been synthesized to date but many of them do not meet the optimal requirements needed for gas separation. In particular, they either lack the optimal pore size, collapse as they interact with guest molecules or are mechanically weak.27
In addition to gas separation applications, ZIF-8 has a number of other applications such as removal of oil from a water surface,28 on-demand drug delivery,29 heterogenous catalysts,30 chemical vapors and gas sensing and detection.31 ZIF-8 can be integrated with sensitive transducers to achieve high sensitivity in the microwave wavelength regime for CO2 and CH4 detection.32 A review on ZIF-8 with areas of applications is given in ref. 33.
By using high resolution transmission electron microscopy (HRTEM), Zhu et al. revealed experimentally the surface and interfacial structures of ZIF-8.14 In their experiment, an armchair type surface with a (110) index was found to be stable with Zn2+ ions capped by 2-mim as viewed along the [110] direction. This result was shown to be supported by theoretical calculations obtained by using a first principles density functional theory-tight binding (DFT-TB) approach.14 While Zhu et al. work provides an important step towards modelling the surface and interface of ZIF-8, an understanding of the adsorption and diffusion properties at the atomistic scale within the bulk, from the bulk to the surface, and MOF/polymer interfaces is still lacking. Semino et al. work on ZIF-8 interfaced with polymer of intrinsic microporosity-1 (PIM-1) provides a comprehensive theoretical model for the surface and interface while simulating a composite structure.34 By integrating experiment and theory, Hobday et al. revealed the most important adsorption sites of CH4, O2, N2 and Ar gas molecules within the framework of ZIF-8.20 While the later work provides gas adsorption properties at different pressures, the mechanism of diffusion and its changes upon loading of different gas molecules are still unexplained. Molecular properties can be altered greatly by applying an external electric field. Knebel et al. measured the gas permeances for H2, CO2 and CH4 under an applied electric field with a strength of 500 V mm−1.35 The authors performed a comparative study of single-gas permeances with and without the application of an in situ-applied field perpendicular to the MOF layer. Their results indicate that ZIF-8 undergoes a crystallographic structural transformation into polyforms under the applied field, altering the value of permeances. There are several experimental and theoretical studies performed to understand the relaxation in size of pores called “gate opening” to allow gas molecules with size larger than the equilibrium pore diameter of the framework to permeate smoothly through the openings. On the theoretical side, most of the quantitative reports on gate opening and diffusions of gas molecules rely upon molecular dynamics and Monte-Carlo simulations,16,36–40 and some upon DFT and DFTB especially for adsorption energy calculations.20,41,42 Garberoglio et al. have used DFTB to calculate diffusion of H2, CO2 and CH4 molecules in MOF materials with several hundred atoms in a unit cell.43 Recently, Verploegh et al. have introduced an accurate force field which is based on DFT parameterization for ZIFs.44 This work inherently considers the framework flexibility unlike in the cases where generic force fields are empirically tuned and fitted to sets of structural data obtained from the experiment.45–47 Fischer and Bell used DFT with periodic boundary conditions including Grimme's D2 dispersion to identify H2 and CO2 adsorption sites in ZIF-8 and other isostructural systems.48,49 The average interaction energies between H2 and the framework, and between CO2 and the framework were found to be −10.4 and −24.6 kJ mol−1. By combining ab initio MD with classical MD calculations, improved results could be achieved for framework flexibility while CO2 and CH4 diffuse in the pore.50 There are other similar studies on ZIF structures by using different flavors of DFT exchange–correlation functionals.51–53
While there are studies on experiments and theory-simulations that unravel adsorption and diffusion kinetics for gas molecules such as CO2 and CH4 in ZIF-8 materials at the atomistic-level, a deeper understanding of the diffusion of these gas molecules in relation to adsorption and framework expansion using fundamental concepts could help in improving the membrane design and optimal performance. Calculations of surface and interface properties of ZIF-8 relevant to gas mixture separation using theoretical modelling based entirely on quantum mechanics can be challenging due to the size of the framework unit cell. Nevertheless, it is possible to obtain a deeper insight into adsorption and diffusion kinetics such as diffusion pathways, energy profiles and energy hierarchy of adsorption sites for different gas molecules in the bulk structure from quantum mechanical calculations. Diffusion barrier profiles with and without atomic position relaxation allow us to quantitatively explain the orientation dependent interaction of the guest molecule with the framework and its structural changes at pore sites.
There are force field parameters available for ZIF-8 in the open literature and one can naturally think of using the MD/MC method over DFT for such a system.54 In ref. 55, DFT based force field parameters were developed for MOF materials including ZIF-8. Therefore, using the DFT method it is desirable to understand the fundamentals of adsorption and diffusion properties of ZIF-8. This opens a possibility of extending the DFT method to many other systems for which the development of a classical force field is typically a lengthy and daunting work. Therefore, in systems for which force field parameters generally lack, DFT will be an alternative method with relatively low computational cost because developing reasonable classical force field parameters is a time-consuming task. In this work, we show that the DFT method can predict the gas diffusivity and gas saturated loading in ZIF-8.
In this study, we perform first principles DFT calculation with Grimme D3 dispersion corrections to study the adsorption and diffusion properties of CO2 and CH4 in ZIF-8. Using the nudged elastic band (NEB) approach we calculate the energy profile for gas molecules while they diffuse in the bulk structure.56–58 We identify the hierarchy of adsorption sites for gas molecules interacting with the bulk framework atoms. The bond angle of CO2 remains unaltered in ZIF-8, which confirms that the interaction between the framework and guest molecule is mainly of the van der Waals type. The calculated maximum loading amount identified in this work can be compared with the experimental value42 obtained at the saturation limit at high pressure limit. The diffusion energy profiles we presented in this work are for single molecules of CO2 or CH4 under dynamic equilibrium in the DFT framework. CO2 and CH4 are linearly and spherically symmetrical molecules, whose center of mass coincide with the geometrical center of the molecule. There are several possible diffusion pathways for the guest molecule within the framework. We provided only the pathways with the minimum diffusion barriers. The response of the framework to the guest molecule is constrained as well as relaxed to see the impact of the change in framework–guest molecule interaction on the diffusion barriers. We observe that there is about 0.88 kcal mol−1 change in the barrier energy for CO2 and 1.25 kcal mol−1 for CH4 while relaxing both the framework and molecule and the molecule only. This is evidence of framework flexibility in porous materials like ZIF-8, where there are different geometrical pore sizes and not all of them allow for gas diffusion. The size description of pores is used in this work to identify the migration pathways while mapping out the images along a particular path. In fact, the probability of diffusing the molecule through a geometrical pore size of 0.8 Å is close to zero. We calculate the diffusivities corresponding to the obtained energy barriers. The order of magnitude of our calculated diffusion coefficients agrees with the experimental results measured at 298 K.42 We find that membranes made from ZIF-8 have good adsorption and diffusion selective properties for CO2 and CH4 molecules, making these membranes useful in gas separation processes.
2. Computational methodology
Calculations performed in this work are based on a first principles density functional theory (DFT) approach. The orbitals, density and potential are expanded on a plane-wave basis set and the electron–ion interactions are described using the projector-augmented-wave (PAW) method. Periodic boundary conditions are imposed in three dimensions. The Vienna ab initio simulation package (VASP)59,60 was employed to calculate the bulk properties, adsorption energies and diffusion pathways. All calculations were performed using the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional.61,62 Plane wave basis sets were used with a cutoff energy of 520 eV. With this value of cutoff energy, the atomic Hellman–Feynman force was minimized to a value less than 0.01 eV Å−1. Due to the large size of the unit cell, reciprocal space integration was employed with a Monkhorst–Pack grid of 1 × 1 × 1 (k-point sampling in the Brillouin zone with a reciprocal grid length of 0.06 Å−1). The self-consistency loop for the electronic relaxation was considered converged when the energy change was less than 10−6 eV per cell. To account for van der Waals interaction, we used Grimme's zero damping D3 dispersion correction.49,63 In this approach, the total energy of the system is obtained as,| EDFT–D = EKS–DFT + EDisp | (1) |
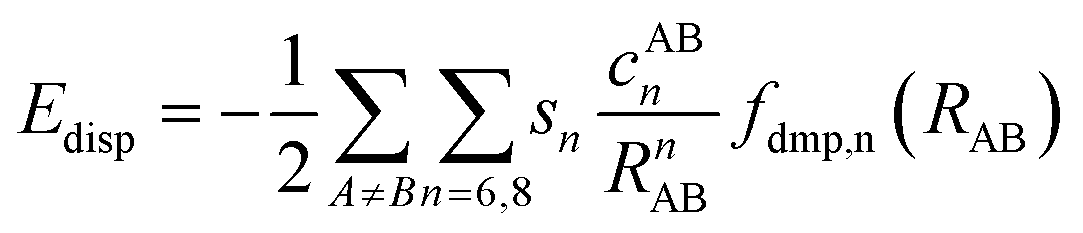 | (2) |
The ZIF-8 structure obtained from the experiment was cleaned to remove disorder and relaxed without any symmetry constraints to allow full relaxation of both the lattice parameters and atomic positions. In order to calculate the diffusion energy profile, the nudged elastic band (NEB) approach was used to map the migration pathways of CO2 and CH4 molecules between two local minima.58 The NEB approach has been shown to predict energy barriers with high accuracy in many solid systems.64,65 The initial and final states were confirmed to be minima based on the lack of imaginary frequency modes.
3. Results and discussion
3.1. Structural and electronic properties
The material geometry and electronic structure calculations are provided in the ESI.† The lattice parameters and bond lengths of bulk ZIF-8 show excellent agreement with the available experimental results as shown in Table S1 and demonstrated in Fig. S1.† In addition, the calculated density of states including van der Waal interaction show that the ZIF-8 has an electronic band gap of 4.38 eV, which is 85% of the corresponding experimental value (Fig. S2†). It is to be noted that the generalized gradient approximation (GGA) level of DFT calculations considered in this study typically underestimates the band gap due to its inherent limitation of properly accounting the exchange correlation effect. We use the above optimized geometry for the adsorption and diffusion property calculations. In this section, we explore the equilibrium adsorption sites and adsorption energies of CO2 and CH4 gas molecules and compare the obtained properties to experimental values where available. The sampling of molecular adsorbing sites was done by relaxing the molecule and framework in different locations in several steps closer to the possible adsorption sites such as the imidazolium ring and pores. The obtained results were compared with the available results from experiments and MD simulation to make sure that we obtained one of the highest energy adsorption sites. More details on the adsorption energies and sites are provided in the ESI.†3.2. CO2 adsorption
In Fig. 1, we show the optimized locations of low-energy adsorption sites of one CO2 molecule in ZIF-8 calculated by fixing the volume of the unit cell while relaxing the positions of all atoms. When more than one CO2 molecule is present, the lowest-energy adsorption sites within the framework are first occupied. The remaining adsorption sites are occupied according to a hierarchy of binding strength of CO2 with the framework. The adsorption energy per CO2 molecule in ZIF-8 can be calculated as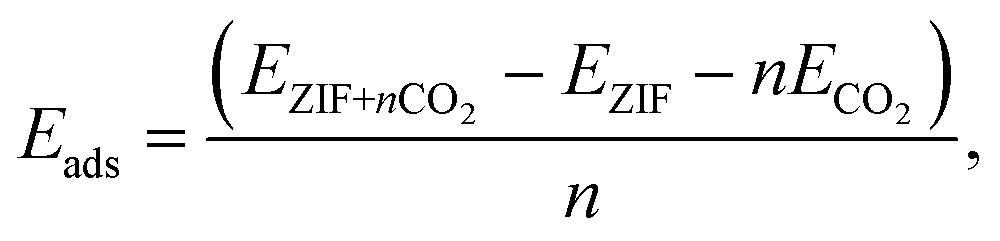 | (3) |
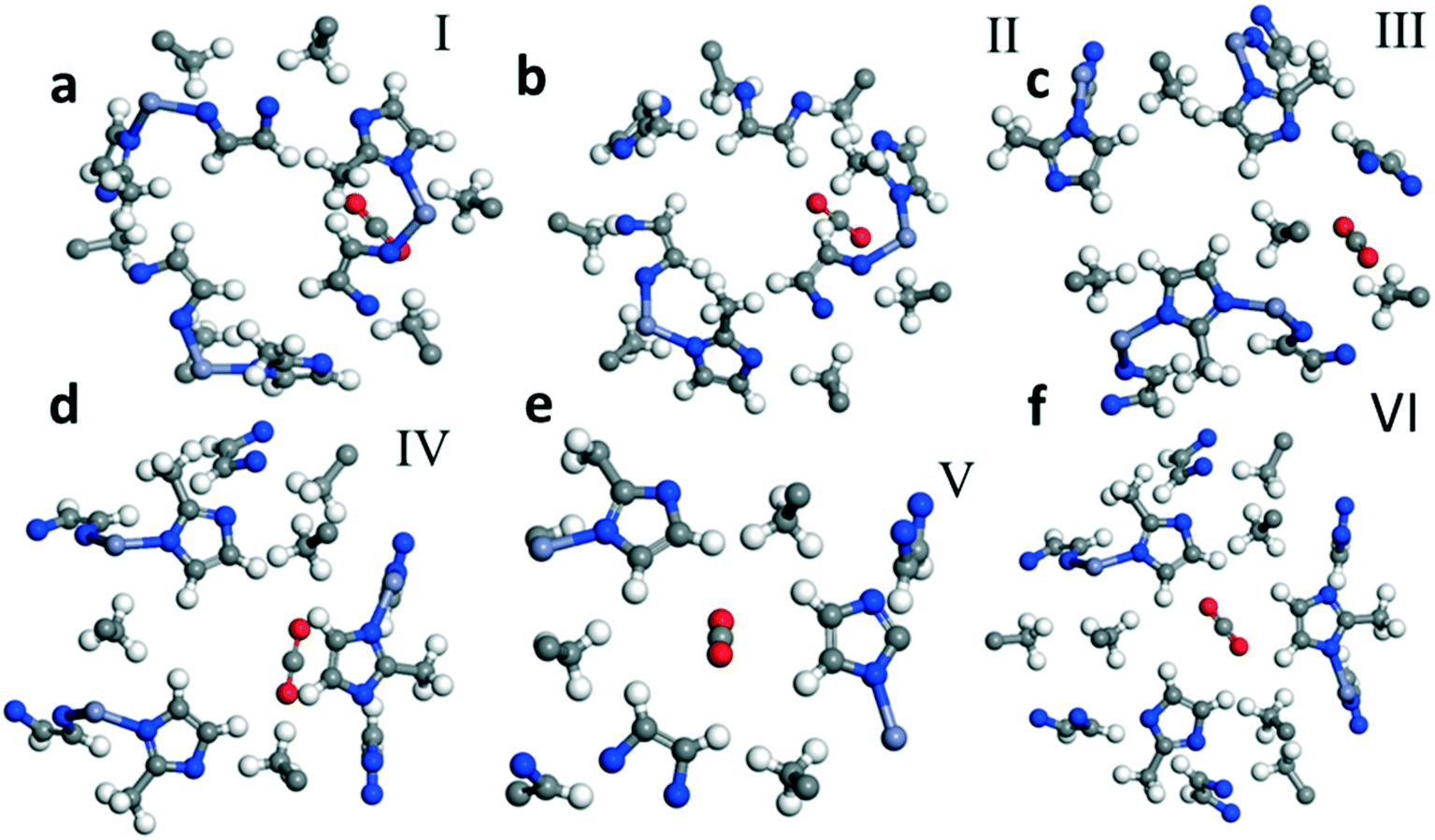 | ||
| Fig. 1 Optimized geometry of CO2 adsorption sites in ZIF 8. Fragments a–f which are labeled I, II, III, IV, V and VI, respectively, show six different representative sites for CO2 adsorption. | ||
We calculate the most favorable binding site of the CO2 molecule inside the framework unit cell (see the ESI†). The calculated value of −5.01 kcal mol−1 for the adsorption energy is comparable with the experimentally reported value for the isosteric heat of adsorption for the CO2 molecule at low coverage limit. In Fig. 1, we also show six different binding sites for CO2. For example, site I lies close to the metal center whereas site III is near the pore of size 0.8 Å. In site I, due to a strong repulsion from the framework's linker atoms we observe that the CO2 molecule is bent by an angle of 3.5° from its original linear structure, indicating that a slight charge transfer occurs between the framework atoms and the CO2 molecule. Detailed information on the adsorption of CO2 is provided in the ESI.†
To understand the change in adsorption energy because of the increase in coverage of the framework–molecule volume, we calculate the adsorption energy per CO2 for an increasing number of adsorbed CO2 molecules. The adsorption energy is calculated both by allowing a change in the volume of the unit cell and by holding the volume fixed. As the loading of molecules increases, the adsorption energy per CO2 molecule fluctuates slightly until 23 molecules are loaded in the pore. After reaching this loading, the adsorption energy per molecule rapidly decreases (in absolute value) as shown in Fig. 2a. Perez-Pellitero et al. reported an experimental loading saturation for CO2 of 8.5 mmol g−1 (∼23 CO2 per unit cell) at 303 K at a pressure of around 30 bar.66 The peak loading amount before the decrease in the DFT-calculated adsorption energy per molecule in Fig. 2a can be considered as an upper limit of the loading amount seen experimentally. After the dotted line in Fig. 2a, the adsorption energy falls in average with a scale of 0.38 kcal per mole per molecule added. Overall, the volume of the unit cell is found to increase noticeably only after the loading of 16 molecules as shown in Fig. 2b where we show the percentage volume change and adsorption energy as a function of the number of CO2 molecules in the cage. It is interesting to note from Fig. 2b that there is a significant change in the volume of the unit cell before the start of the rapid fall in adsorption energy. Our calculations show that the expansion in lattice parameters is uniform in all directions and the change in unit cell angles is negligibly small. In addition, we find that the change of volume by about more than 2% effects the loading amount noticeably as the adsorption energy per molecule rapidly falls after that.
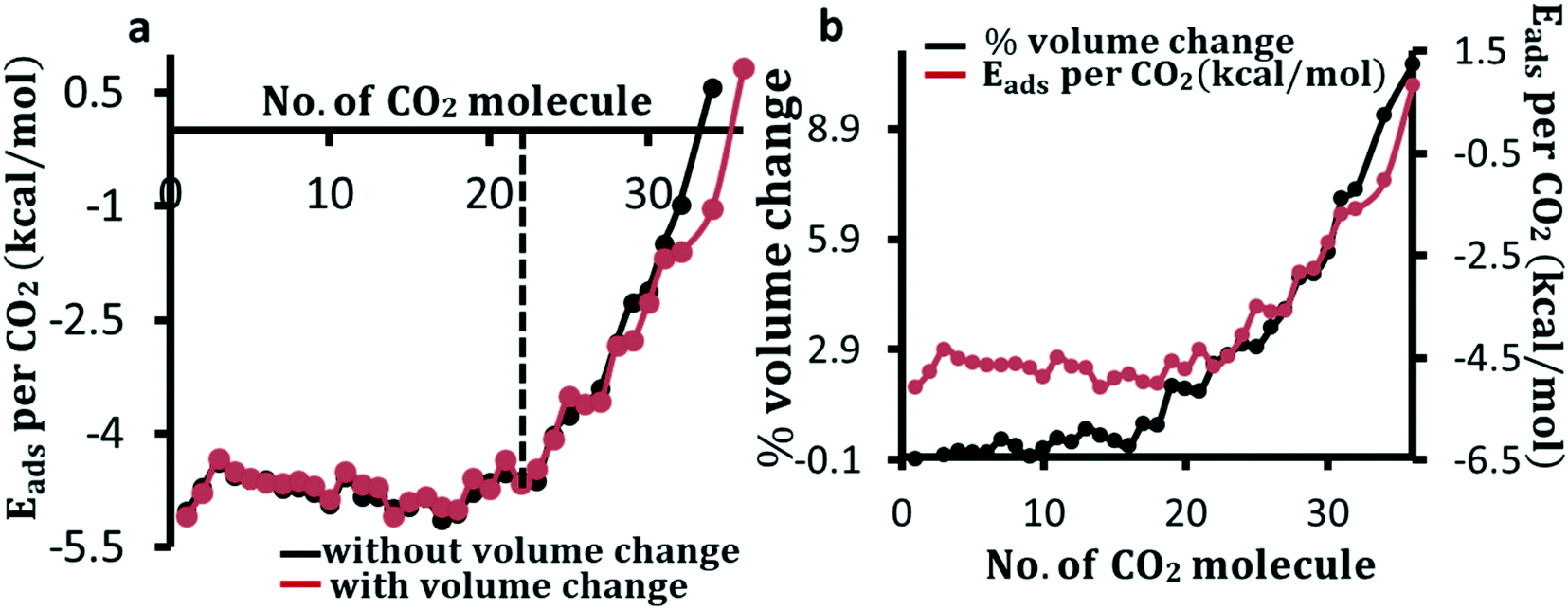 | ||
| Fig. 2 CO2 loading limit in ZIF-8. Adsorption energy, Eads, per CO2 molecule calculated with (red curve) and without (black curve) the change in unit cell volume of the composite system as a function of the number of molecules (a). As the loading amount increases, the framework volume changes negligibly until about 16 molecules are entrapped in the cage. As the number of molecules increases from 16 to 23, the framework volume change rises steeply (b). There is a rapid decrease in adsorption energy per CO2 loading after about 25 molecules. The dotted line in a shows a loading limit after which the adsorption energy decreases rapidly. | ||
3.3. CH4 adsorption
To understand the preferential molecular adsorption sites in ZIF-8, as in the case of CO2, we optimize the composite structure of CH4 and ZIF-8 by adsorbing CH4 in several different locations within the framework. We show the most stable adsorption sites of the CH4 molecule in Fig. S4 in the ESI.† The calculated value of CH4 adsorption energy of −4.50 kcal mol−1 is comparable with the experimentally observed value for the isosteric heat of adsorption. In Fig. 3, we show four representative adsorption sites for CH4 adsorption in the framework, which provide insight into the interaction strength at different sites near the imidazolium ring. The calculated adsorption energy is given in Fig. S4b in the ESI.†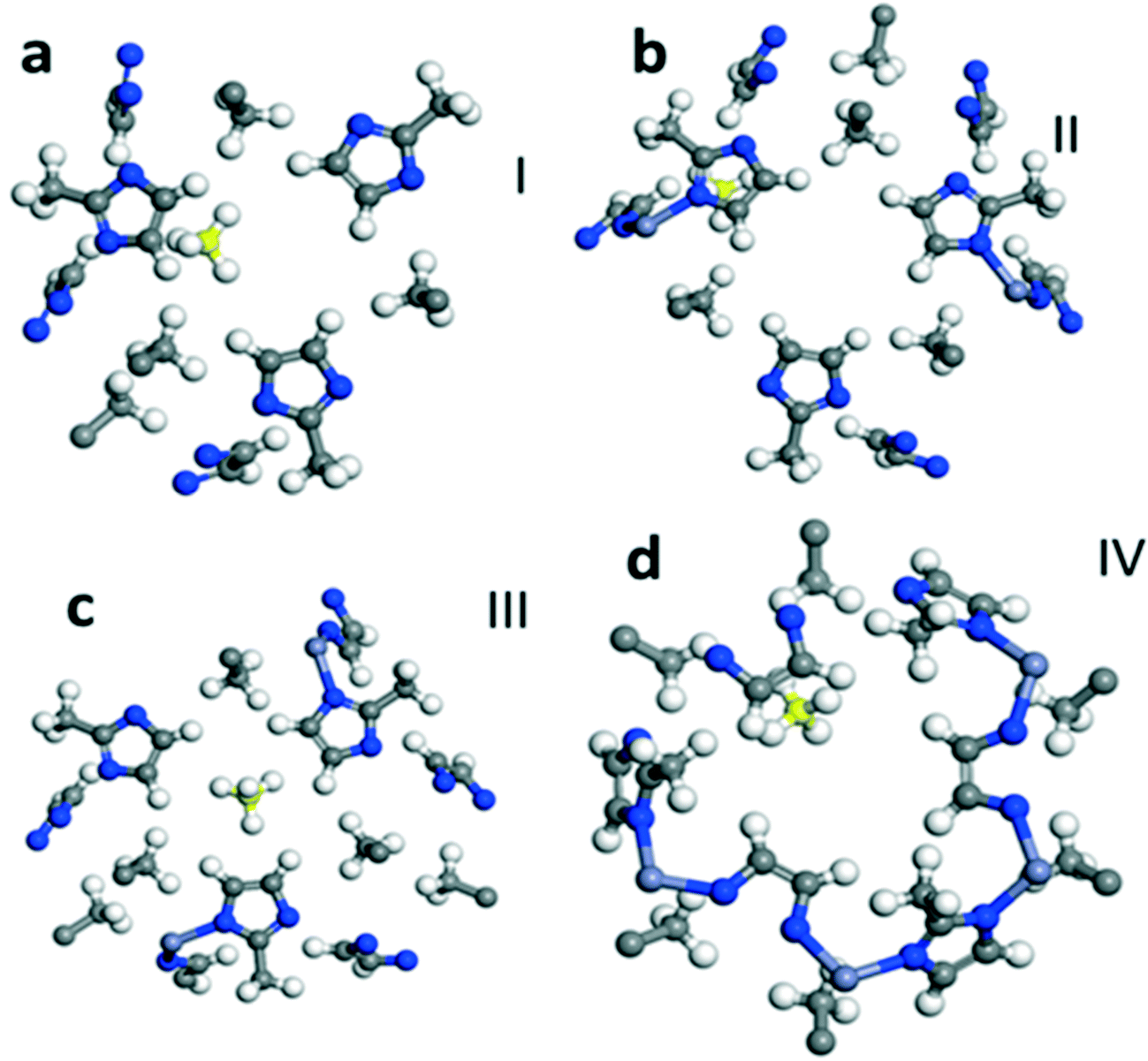 | ||
| Fig. 3 CH4 adsorption sites in ZIF-8. Fragments a to d show the different sites where the CH4 binding energies are calculated. The ‘center’ represents the binding of CH4 near the center of the cage. The C atom of CH4 is marked yellow (online). | ||
As the loading of CH4 molecules increases in ZIF-8, the change in adsorption energy per molecule is found to be within in 0.1 kcal mol−1 until about 17 molecules (∼6.2 mmol g−1) are adsorbed by the framework for both cases of fixed and relaxed volume as shown in Fig. 4a. After this loading, the adsorption energy weakens with an average of 0.18 kcal mol−1 for each molecule added. This attenuation is more than two times steeper than the one observed in the case of CO2. Perez-Pellitero et al. measured the adsorption of CH4 at 303 K, and observed about 4 mmol g−1, but saturation was not achieved in their measurements.66 The application of sufficiently high pressure in certain directions in the experiment results in an anisotropic volume expansion, dilation along the applied force and contraction perpendicular to it. This results in a higher number of gas molecules coming closer to the framework's wall in the perpendicular direction. This results in a different loading amount from the situation of uniform expansion in all directions. Knebel et al.35 have demonstrated changes in lattice parameters upon electric field poling with a strength of 0.02 eV Å−1 followed by a change in phase transformation of ZIF-8 into different polyforms which subsequently reduced the separation selectivity for molecules like H2, CO2 and CH4. There is a difference of less than 0.1 kcal per mole in the adsorption energy at low loading when the volume of the unit cell is relaxed compared to when the unit cell volume is fixed. As we allow changes in the atomic positions and lattice parameters, for the adsorption of the first 5 CH4 molecules, the volume is found to decrease by 0.15% as shown in Fig. 4b. After the adsorption of 17 molecules, the average rate of increase of volume expansion is 13.6 Å3 per molecule for CH4 whereas for CO2 it was 15.5 Å3 per molecule. Therefore, at higher loading, the change in volume and adsorption energy as a function of the number of molecules is found to be larger in the case of CO2 as compared to the case of CH4.
 | ||
| Fig. 4 CH4 loading limit in ZIF-8. Adsorption energy, Eads, per CH4 molecule calculated with (red curve) and without (black curve) the change in unit cell volume of the composite system as a function of the number of molecules (a). As the loading amount increases, the framework volume changes negligibly until about 17 molecules in the cage (b). There is a rapid decrease in adsorption energy per CH4 loading after adsorbing 17 molecules. The dotted line in a shows a loading limit after which the adsorption energy decreases rapidly. | ||
As the number of molecules inside the cage increases, the interactions among the gas molecules and between the gas molecules and the framework increase significantly, and so the expansion in volume occurs.67 The orientation of CO2 molecules becomes important at higher loading. The adsorption energy changes faster with the number of molecules in the framework for CH4 than for CO2. This sets up the lower loading limit for CH4 than for CO2. As the loading increases, the available space within the cage is smaller for CH4 due to its larger size than for CO2 for an equal amount of loading. In such a case, there is a repulsion between two molecules, which is stronger for CH4 that effectively reduces the loading limit for CH4 than for CO2.
3.4. Diffusion properties
In previous sections, we described the adsorption properties and their dependence on the change in volume of the unit cell. Next, we study the diffusion properties of CO2 and CH4 molecules in ZIF-8. Unlike a molecular dynamics (MD) approach,16,41,47,68 here we study these properties from the quantum mechanical perspective that captures the details of the atomic interactions. We use the NEB approach to map the minimum energy pathways along which CO2 and CH4 migrate in ZIF-8. First, we identify the most stable initial and final states of the molecule and map the path between them to find their lowest possible energy barriers.Under the NEB approach, the migration path of the molecule is relaxed in every step and is mapped out vectorially along the direction of the final image. The different vibrational modes of the framework and molecule may give inconsistent results due to the quasi-static responses of their motion under the NEB assumption. To avoid that while calculating the barrier energy, in addition to the entire material relaxation, we fix the MOF and relax the molecule only and vice versa. Fixing the framework and relaxing the molecule results in overall a rather coherent vibrational mode than the ones resulting from the quasi-static responses.
In Fig. 5 we show the initial, transition and final states of diffusing molecules of CO2 (Fig. 5a–c) and CH4 (Fig. 5d–f). For distinct visualization of diffusing molecules, the position of each image within the unit cell is identified with reference to the (111) and (110) Miller planes. The initial image of CO2 lies in the (110) Miller plane near the 6-MR window. The (110) plane bisects the 6-MR ring along the [100] direction as shown in Fig. 5a. The minimum energy diffusion pathway for CO2 lies along the [110] direction. Similarly, the initial state is located slightly off the [110] plane near the 6-MR window as shown in Fig. 5d. The minimum energy diffusion pathway for CH4 is shown to intersect the (110) and (111) planes, which can be seen from Fig. 5d and e. From the initial state, in which the CO2 (CH4) molecule is located within the large pore, the minimum energy path takes the molecule through the 6-MR window and into the next large pore, wherein lies the final state. Unlike the CH4 case, the energetic pathway for CO2 is sensitive to the orientation of the molecule. The barrier value is minimized when the CO2 axis is perpendicular to the plane of the 6-MR window.
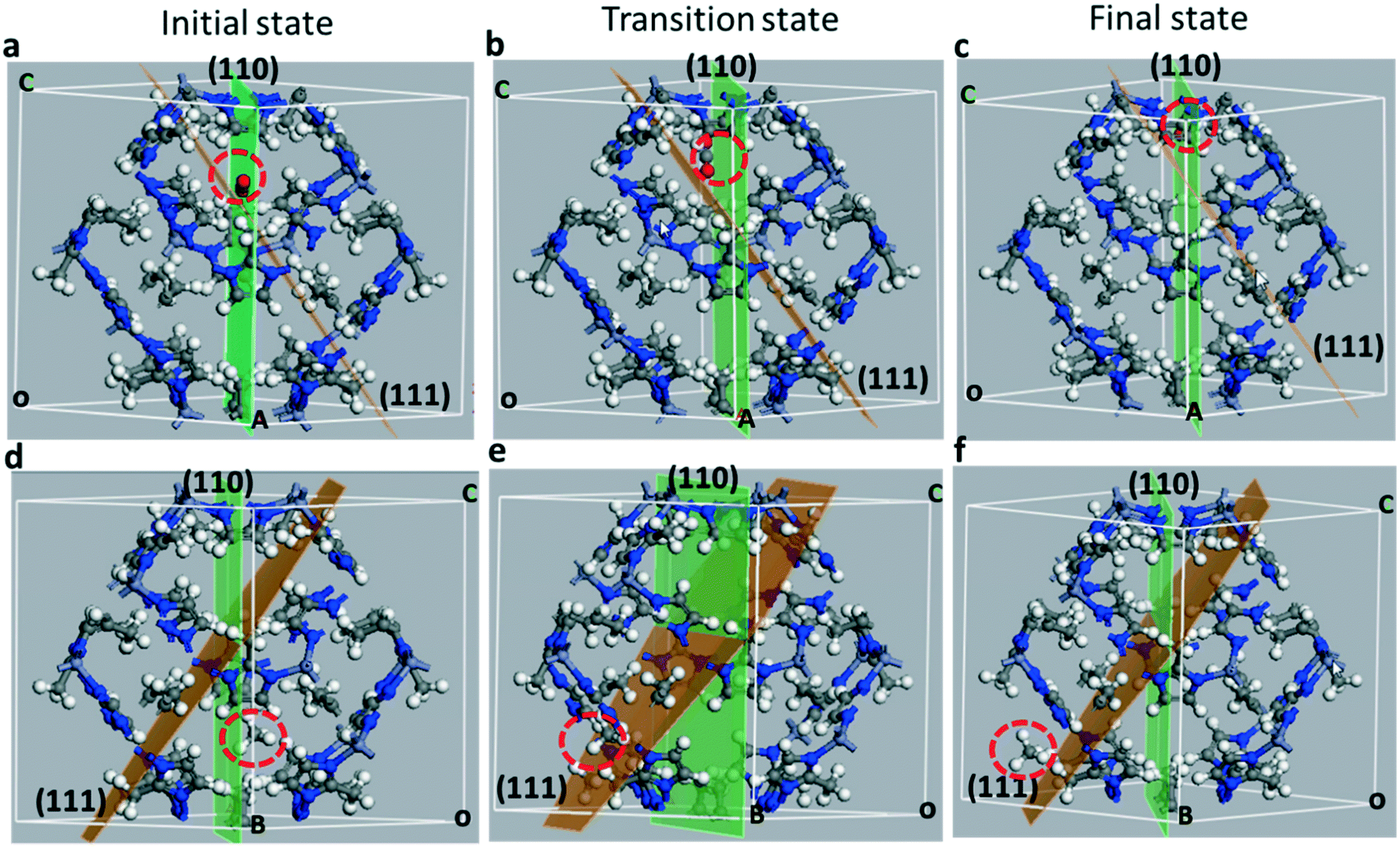 | ||
| Fig. 5 Initial (a and d), transition (b and e) and final (b and d) images for CO2 (a–c) and CH4 (d–f) in ZIF-8. The locations of diffusing molecules can be identified with reference to the (111) (orange) and (110) (green) Miller planes. The locations of the molecules in each figure is marked with a red dotted circle. | ||
To understand the diffusion of CO2 and CH4 from the initial state in one pore, and finally to the final state in the neighboring pore in ZIF-8, a number of steps between the initial and final states are mapped out under the NEB approach as shown in Fig. 5. These pathways are studied in two cases: (1) with the atomic positions of all atoms allowed to relax and (2) with the MOF atomic positions frozen and the CO2 or CH4 atomic positions relaxed. The lattice parameters are kept constant in both cases. In (1), the calculation yields the quasi-vibrational modes of molecules and frameworks together with a combined center of mass oscillation as all ions are relaxed. In (2), the calculations provide the vibrational modes of CO2 against the framework, which also reflects the attempt frequency of CO2 while diffusing through the pores. Fig. 6 shows the minimum energy pathways obtained for CO2 and CH4 for both cases (1) and (2).
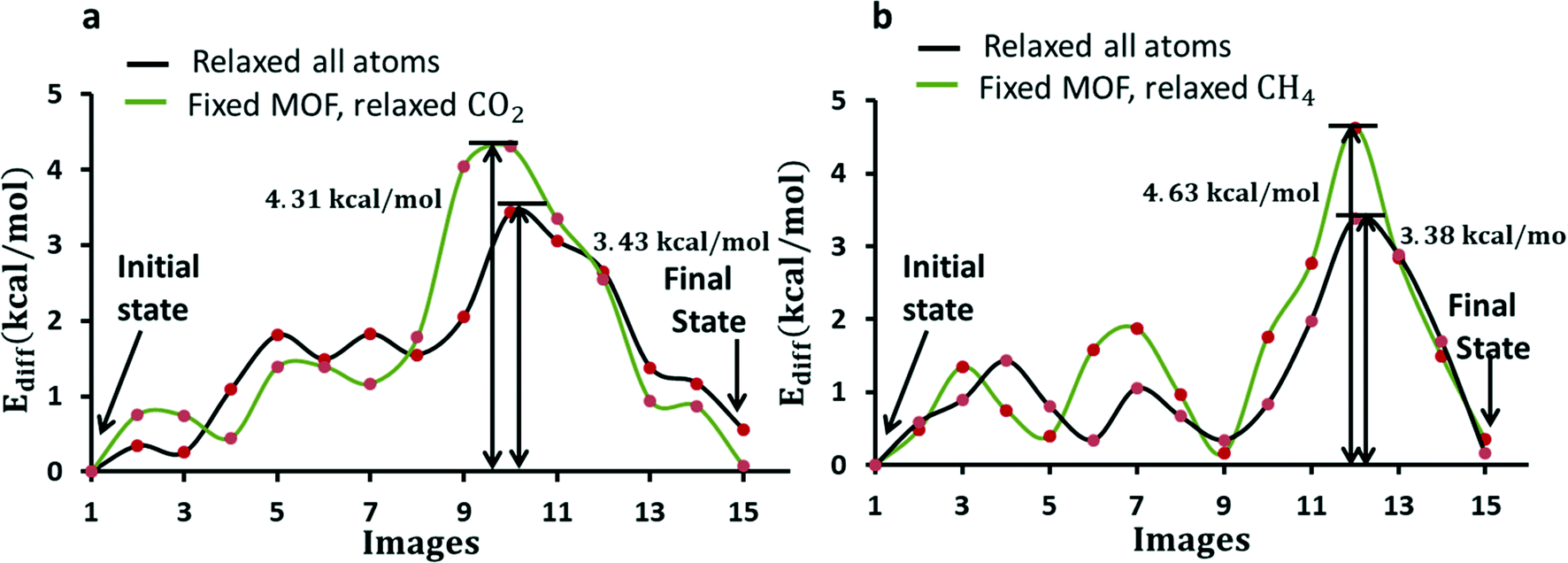 | ||
| Fig. 6 Diffusion barriers for CO2 (a) and CH4 (b) in ZIF-8 while all atoms are relaxed (black), and fixed ZIF-8 and relaxed CH4 (green) at constant lattice parameters. The initial and final states are images corresponding to the minimum of the energy path. The heights of the barriers are shown with vertical lines with respective values. | ||
The diffusion barrier for CO2 is found to be 3.43 kcal mol−1 in the flexible ZIF-8 (case (1)). The diffusion energy barrier is increased by 25% (∼1 kcal mol−1) in the fixed ZIF-8 (case (2)) as shown in Fig. 6a. Although other gas diffusion paths may exist, this CO2 diffusion energy barrier suggests that CO2 diffusion at 298 K in the flexible ZIF-8 is about 5.0 times larger than in the fixed ZIF-8. This CO2 diffusion difference in the flexible and fixed ZIF-8 structures obtained from the quantum chemistry calculations is similar to the results obtained from the molecular dynamics simulations by using a classical flexible force-field for ZIF-8. Similarly, the diffusion barrier for CH4 diffusion is found to be 3.38 kcal mol−1 in case (1) and is increased by 37% in case (2). Fixing framework atoms has a higher impact on the barrier in the case of CH4. This is expected as the kinetic diameter of the CH4 molecule is larger than that of the window. CH4 experiences a strong repulsion while passing through the window. The peak in the pathway in Fig. 6 for both CO2 and CH4 arises due to gas molecules interacting relatively strongly while they lie near the 6-MR window as shown in Fig. 5(b and e).
The diffusion coefficient can be calculated using the Einstein–Smoluchowski relation:69
 | (4) |
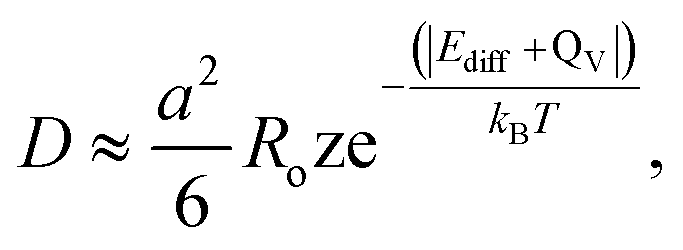 | (5) |
| Molecule | Methods | D diff (m2 s−1) | Sources |
|---|---|---|---|
| CO2 | Flexible framework | 8.77 × 10−10 | This work |
| Fixed framework | 1.7 × 10−10 | ||
| Infrared microscopy | 1.5 × 10−10 | Ref. 42 | |
| DFT optimized | 9.31 × 10−11 | Ref. 50 | |
| Flexible framework (MD) | 7.93 × 10−10 | Ref. 50 | |
| Flexible framework (MC/MD) | 6.2 × 10−9 | Ref. 16 | |
| Infrared microscopy | 1.56 × 10−10 | Ref. 70 | |
| CH4 | Flexible framework | 3.52 × 10−10 | This work |
| Fixed framework | 4.6 × 10−11 | ||
| Infrared microscopy | 1.0 × 10−10 | Ref. 42 | |
| Flexible framework (MD) | 4.47 × 10−12 | Ref. 50 | |
| Flexible framework (MC/MD) | 6.4 × 10−11 | Ref. 16 |
We extend the above calculation to capture the diffusion dynamics from one pore to the next nearest neighbor pore of the same size. The molecule diffuses in through one of the pores in a particular unit cell, migrates across the subsurface, and diffuses out through the neighbor pore of the same unit cell as shown by the initial and final states in Fig. 7 for CO2. The diffusion barrier is symmetrical with two lobes occurring across the two pores.
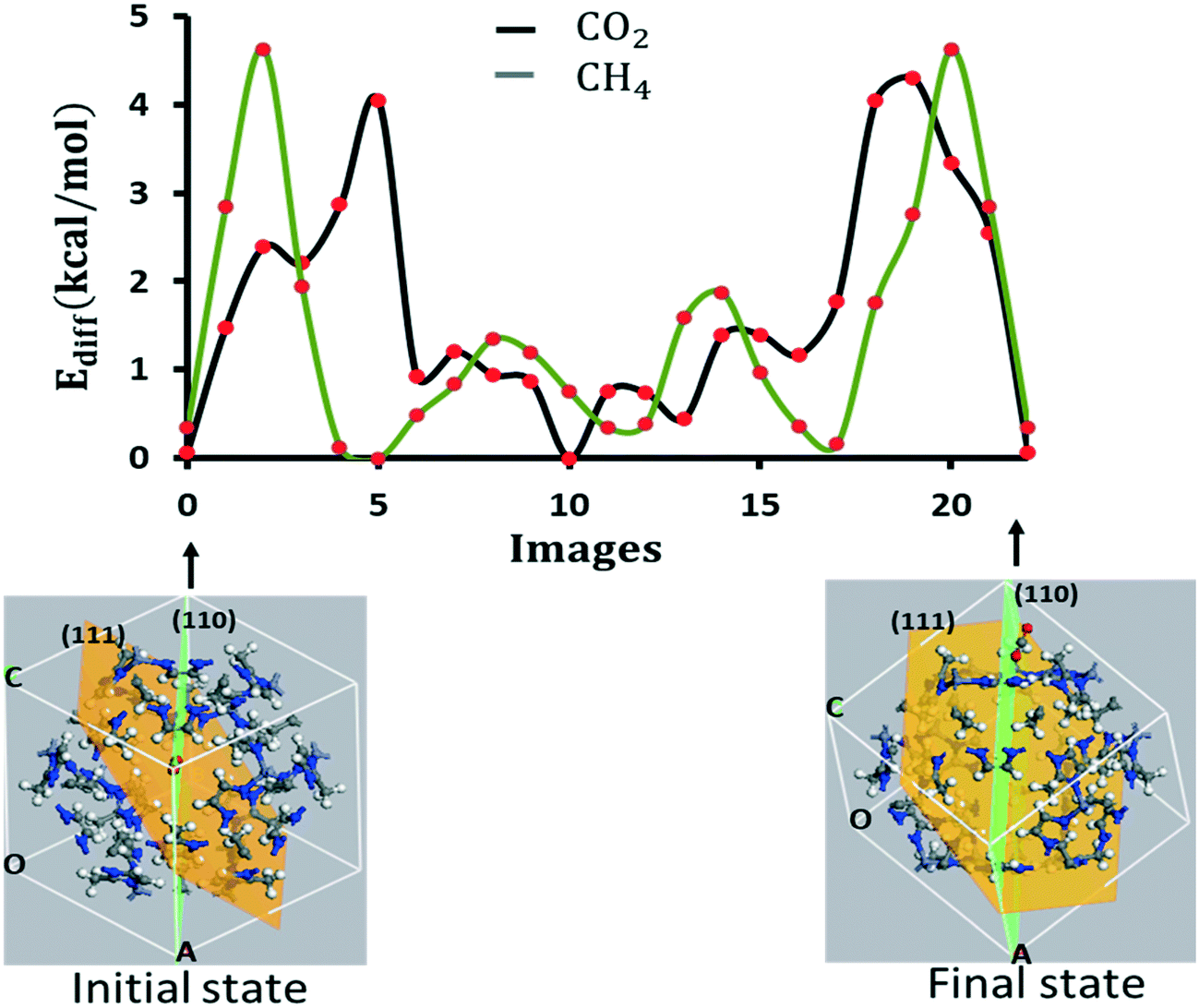 | ||
| Fig. 7 Diffusion barriers for diffusion of CO2 (black) and CH4 (green) from one pore to the nearest neighbor pore of the same size calculated by fixing the framework but relaxing the molecule. The initial and final sates for CO2 are shown with images. | ||
From the results presented here, we find that loading of individual gas molecules of CO2 until around 2% change in volume does not significantly affect its adsorption energy. At higher loading limit, after loading of about 17 molecules, the volume started changing dramatically with a simultaneous decrease in adsorption energy in the case of CH4 whereas a rapid decrease in adsorption energy is found to occur after loading of 23 molecules in the case of CO2. These results indicate that the maximum loading limit (as in experiment) is more sensitive to adsorption energy for CH4 loading than for CO2 where up to 2% of the framework's volume expansion change in adsorption energy is negligibly small. In the case of a large number of molecules diffusing in the framework, the crowding effect may alter the diffusion coefficient due to interactions among the gas molecules in addition to the molecule–framework interaction. Nevertheless, our results advance the understanding of diffusion mechanisms of gas molecules and can be potentially useful in designing membranes.
By default, DFT does not consider any temperature effect. There exist many closely spaced local energy minima within the ZIF-8 frameworks for the adsorbate molecule. The interaction between them is weak and is mostly of the van der Waal type. It could be challenging to locate global minima in such cases using the DFT method as it does not employ statistics for phase space sampling. Phase space sampling using MD simulation is helpful to locate a global minimum, where statistics is rigorously applied. However, the DFT method could be an alternative method to achieve a good estimate in the system like ZIF-8. This opens up an avenue for the possibility of using the DFT method for diffusion property calculations in other MOF materials for which force field parameters are not readily available and calculation of those parameters is a daunting task. The difference in many closely spaced energy sites is most likely within the DFT energy bar of 0.025 kcal mol−1. In addition, if we compare the computational costs and the significance of expected accuracy in the results, phase space sampling using MD by developing reasonable force field parameters in many instances would not improve the overall quality of the work appreciably. Importantly, ZIF-8 has a highly symmetrical conventional unit cell.
At 0 K, the Gibbs free energy G is equal to the enthalpy H of the system. As the temperature is increased, the entropy of the system changes and the change in entropy contributes to the Gibb's free energy ΔG = ΔH – TΔS. The error in adsorption and diffusion barriers due to a rise of temperature is substantially mitigated due to the cancellation of such error in total energy while talking a difference. In solid systems like fcc Al, the temperature dependence of enthalpy and entropy is shown to nearly cancel out each other under harmonic and quasi-harmonic approximations.71 Nevertheless, our results obtained for volume dependence of adsorption energy and diffusion coefficients provide good estimates and help in understanding of major diffusion channels in ZIF-8 materials.
4. Conclusions
The selective adsorption of CO2 in the presence of other gas species, such as CH4, is a subject of fundamental interest as CO2 is the main component of greenhouse gases. We performed a quantitative analysis of the adsorption and diffusion properties of CO2 and CH4 gas molecules in bulk ZIF-8 materials using density functional theory calculations. We identified the stability of the adsorption sites for given gas molecules. The geometry of the gas molecules does not alter noticeably, which indicates that the interaction of the gas molecules and framework is mainly of the van der Waals type. At the most stable site, we found that the adsorption energy per molecule for CO2 is 4.6 kcal mol−1 whereas for CH4 it is 3.7 kcal mol−1. Our calculated results closely corroborated with the experimental findings for loading with a given adsorption energy per mole when saturation in loading is achieved. We presented the loading of gas molecules both by relaxing and keeping fixed the atomic positions of the framework and molecules, and the volume of the unit cell. We found that the change in adsorption energy weakly depends on the volume change until about 17 gas molecules in the cage. Within about 2% of the volume change, there is still a negligibly small change in adsorption energy. The binding strength decreases as the change in volume increases with the number of adsorbed molecules. At higher loading, both the volume and adsorption energy were found to change rapidly. In addition to that, we presented the diffusion energy profiles of CO2 and CH4 in bulk ZIF-8 at low temperature. Our calculated results for diffusion coefficients obtained by relaxing the framework and gas molecules at 0 K were: DRdiff(CO2) = 8.77 × 10−10 m2 s−1 and DRdiff(CH4) = 3.52 × 10−10 m2 s−1 which were about five times higher for CO2 and two times higher for CH4 than the energy barriers observed in experiments. Both CO2 and CH4 diffusivities in the flexible ZIF-8 are 5–8 times larger than in the fixed ZIF-8. The presence of impurities or defects in the sample in the experiment could affect the measured diffusion barriers. It is to be noted that our calculated results should be taken as baselines in a pure ZIF-8 material at low temperatures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is supported by the National Energy Technology Laboratory's (NETL's) on-going research program in Carbon Capture. H. P. thanks the NETL Research and Innovation Center (R&IC) for providing computational resources administered by the Oak Ridge Institute for Science and Education (ORISE). The authors thank Dr. Dan Sorescu for fruitful discussions and suggestions, and professor Cerasela Zoica Dinu of West Virginia University for reading the manuscript and fruitful comments. This report was prepared as an account of work sponsored by an agency of the United States Government. Neither the United States Government nor any agency thereof, nor any of their employees, makes any warranty, express or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof. The views and opinions of the authors expressed herein do not necessarily state or reflect those of the United States Government or any agency hereof.References
- L. M. Robeson, J. Membr. Sci., 2008, 320, 390–400 CrossRef CAS.
- B. Y. Li, Y. Duan, D. Luebke and B. Morreale, Appl. Energy, 2013, 102, 1439–1447 CrossRef CAS.
- Y. Duan, J. Lekse, X. Wang, B. Li, B. Alcantar-Vazquez, H. Pfeiffer and J. W. Halley, Phys. Rev. Appl., 2015, 3, 044013 CrossRef.
- Y. Duan and K. Parlinski, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 104113 CrossRef.
- Y. Duan and D. C. Sorescu, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 014301 CrossRef.
- H. P. Hsieh, Inorgaic Membranes for Separation and Reaction, Elsevier, New York, 1996 Search PubMed.
- B. D. Freeman, Macromolecules, 1999, 32, 375–380 CrossRef CAS.
- G. Dong, H. Li and V. Chen, J. Mater. Chem. A, 2013, 1, 4610–4630 RSC.
- L. M. Robeson, W. F. Burgoyne, M. Langsam, A. C. Savoca and C. F. Tien, Polymer, 1994, 35, 4970–4978 CrossRef CAS.
- K. Aoki, K. Kusakabe and S. Morooka, Ind. Eng. Chem. Res., 2000, 39, 2245–2251 CrossRef CAS.
- R. Mahajan and W. J. Koros, Ind. Eng. Chem. Res., 2000, 39, 2692–2696 CrossRef CAS.
- S. Keskin and D. S. Sholl, Ind. Eng. Chem. Res., 2009, 48, 914–922 CrossRef CAS.
- G. P. Liu, V. Chernikova, Y. Liu, K. Zhang, Y. Belmabkhout, O. Shekhah, C. Zhang, S. L. Yi, M. Eddaoudi and W. J. Koros, Nat. Mater., 2018, 17, 283–289 CrossRef CAS.
- Y. H. Zhu, J. Ciston, B. Zheng, X. H. Miao, C. Czarnik, Y. C. Pan, R. Sougrat, Z. P. Lai, C. E. Hsiung, K. X. Yao, I. Pinnau, M. Pan and Y. Han, Nat. Mater., 2017, 16, 532–536 CrossRef CAS.
- S. R. Venna and M. A. Carreon, J. Am. Chem. Soc., 2010, 132, 76–78 CrossRef CAS.
- L. L. Zhang, G. Wu and J. W. Jiang, J. Phys. Chem. C, 2014, 118, 8788–8794 CrossRef CAS.
- R. Chen, J. Yao, Q. Gu, S. Smeets, C. Baerlocher, H. Gu, D. Zhu, W. Morris, O. M. Yaghi and H. Wang, Chem. Commun., 2013, 49, 9500–9502 RSC.
- J. Tang, R. R. Salunkhe, J. Liu, N. L. Torad, M. Imura, S. Furukawa and Y. Yamauchi, J. Am. Chem. Soc., 2015, 137, 1572–1580 CrossRef CAS.
- T. Rodenas, I. Luz, G. Prieto, B. Seoane, H. Miro, A. Corma, F. Kapteijn, F. Xamena and J. Gascon, Nat. Mater., 2015, 14, 48–55 CrossRef CAS.
- C. L. Hobday, C. H. Woodall, M. J. Lennox, M. Frost, K. Kamenev, T. Duren, C. A. Morrison and S. A. Moggach, Nat. Commun., 2018, 9, 1429 CrossRef.
- K. S. Park, Z. Ni, A. P. Cote, J. Y. Choi, R. D. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS.
- A. Phan, C. J. Doonan, F. J. Uribe-Romo, C. B. Knobler, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2010, 43, 58–67 CrossRef CAS.
- H. Yang, X. W. He, F. Wang, Y. Kang and J. Zhang, J. Mater. Chem., 2012, 22, 21849–21851 RSC.
- J. J. Low, A. I. Benin, P. Jakubczak, J. F. Abrahamian, S. A. Faheem and R. R. Willis, J. Am. Chem. Soc., 2009, 131, 15834–15842 CrossRef CAS.
- A. S. Huang, Q. Liu, N. Y. Wang, Y. Q. Zhu and J. Caro, J. Am. Chem. Soc., 2014, 136, 14686–14689 CrossRef CAS.
- D. Fairen-Jimenez, R. Galvelis, A. Torrisi, A. D. Gellan, M. T. Wharmby, P. A. Wright, C. Mellot-Draznieks and T. Duren, Dalton Trans., 2012, 41, 10752–10762 RSC.
- O. Karagiaridi, M. B. Lalonde, W. Bury, A. A. Sarjeant, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 18790–18796 CrossRef CAS.
- E. E. Sann, Y. Pan, Z. F. Gao, S. S. Zhan and F. Xia, Sep. Purif. Technol., 2018, 206, 186–191 CrossRef CAS.
- M. Hoop, C. F. Walde, R. Ricco, F. Mushtaq, A. Terzopoulou, X. Z. Chen, A. J. deMello, C. J. Doonan, P. Falcaro, B. J. Nelson, J. Puigmarti-Luis and S. Pane, Appl. Mater. Today, 2018, 11, 13–21 CrossRef.
- U. P. N. Tran, K. K. A. Le and N. T. S. Phan, ACS Catal., 2011, 1, 120–127 CrossRef CAS.
- G. Lu and J. T. Hupp, J. Am. Chem. Soc., 2010, 132, 7832–7833 CrossRef CAS.
- J. Devkota, K. J. Kim, P. R. Ohodnicki, J. T. Culp, D. W. Greve and J. W. Lekse, Nanoscale, 2018, 10, 8075–8087 RSC.
- B. L. Chen, Z. X. Yang, Y. Q. Zhu and Y. D. Xia, J. Mater. Chem. A, 2014, 2, 16811–16831 RSC.
- R. Semino, N. A. Ramsahye, A. Ghoufi and G. Maurin, ACS Appl. Mater. Interfaces, 2016, 8, 809–819 CrossRef CAS.
- A. Knebel, B. Geppert, K. Volgmann, D. I. Kolokolov, A. G. Stepanov, J. Twiefel, P. Heitjans, D. Volkmer and J. Caro, Science, 2017, 358, 347–351 CrossRef CAS.
- B. Zheng, Y. C. Pan, Z. P. Lai and K. W. Huang, Langmuir, 2013, 29, 8865–8872 CrossRef CAS.
- R. Chanajaree, T. Chokbunpiam, J. Karger, S. Hannongbua and S. Fritzsche, Microporous Mesoporous Mater., 2019, 274, 266–276 CrossRef CAS.
- C. Chmelik, J. van Baten and R. Krishna, J. Membr. Sci., 2012, 397, 87–91 CrossRef.
- C. Chmelik, D. Freude, H. Bux and J. Haase, Microporous Mesoporous Mater., 2012, 147, 135–141 CrossRef.
- C. Zhang, R. P. Lively, K. Zhang, J. R. Johnson, O. Karvan and W. J. Koros, J. Phys. Chem. Lett., 2012, 3, 2130–2134 CrossRef CAS.
- R. Poloni, K. Lee, R. F. Berger, B. Smit and J. B. Neaton, J. Phys. Chem. Lett., 2014, 5, 861–865 CrossRef CAS.
- E. Pantatosaki, F. G. Pazzona, G. Megariotis and G. K. Papadopoulos, J. Phys. Chem. B, 2010, 114, 2493–2503 CrossRef CAS.
- G. Garberoglio and S. Taioli, Microporous Mesoporous Mater., 2012, 163, 215–220 CrossRef CAS.
- R. J. Verploegh, A. Kulkarni, S. E. Boulfelfel, J. C. Haydak, D. Tang and D. S. Sholl, J. Phys. Chem. C, 2019, 123, 9153–9167 CrossRef CAS.
- D. S. Sholl and R. P. Lively, Nature, 2016, 532, 435–437 CrossRef.
- J. C. Liu, S. Keskin, D. S. Sholl and J. K. Johnson, J. Phys. Chem. C, 2011, 115, 12560–12566 CrossRef CAS.
- S. Keskin, J. Phys. Chem. C, 2011, 115, 800–807 CrossRef CAS.
- M. Fischer and R. G. Bell, CrystEngComm, 2014, 16, 1934–1949 RSC.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS.
- E. Haldoupis, T. Watanabe, S. Nair and D. S. Sholl, ChemPhysChem, 2012, 13, 3449–3452 CrossRef CAS.
- G. Chaplais, G. Fraux, J.-L. Paillaud, C. Marichal, H. Nouali, A. H. Fuchs, F.-X. Coudert and J. Patarin, J. Phys. Chem. C, 2018, 122, 26945–26955 CrossRef CAS.
- K. G. Ray, D. Olmsted, N. He, Y. Houndonougbo, B. B. Laird and M. Asta, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 085410 CrossRef.
- K. G. Ray, D. L. Olmsted, Y. Houndonougbo, B. B. Laird and M. Asta, J. Phys. Chem. C, 2013, 117, 14642–14651 CrossRef CAS.
- E. Haldoupis, S. Nair and D. S. Sholl, J. Am. Chem. Soc., 2010, 132, 7528–7539 CrossRef CAS.
- H. Demir, J. A. Greathouse, C. L. Staiger, J. J. Perry, M. D. Allendorf and D. S. Sholl, J. Mater. Chem. A, 2015, 3, 23539–23548 RSC.
- G. Henkelman and H. Jonsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jonsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- H. Jonsson, G. Mills and K. W. Jacobsen, in Classical and Quantum Dynamics in Condensed Phase Simulations, ed. B. J. Berne, G. Ciccotti and D. F. Coker, World Scientific, 1998, pp. 385–404 Search PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- H. P. Paudel and Y. Duan, J. Phys. Chem. C, 2018, 122, 28447–28459 CrossRef CAS.
- T. Jia, Z. Zeng, H. Paudel, D. J. Senor and Y. Duan, J. Nucl. Mater., 2019, 522, 1–10 CrossRef CAS.
- J. Perez-Pellitero, H. Amrouche, F. R. Siperstein, G. Pirngruber, C. Nieto-Draghi, G. Chaplais, A. Simon-Masseron, D. Bazer-Bachi, D. Peralta and N. Bats, Chem. – Eur. J., 2010, 16, 1560–1571 CrossRef CAS.
- S. Gadipelli, W. Travis, W. Zhou and Z. X. Guo, Energy Environ. Sci., 2014, 7, 2232–2238 RSC.
- M. Fernandez and A. S. Barnard, ACS Comb. Sci., 2016, 18, 243–252 CrossRef CAS.
- P. Shewmon, Diffusion in Solids, Springer International, Switzerland, 2016 Search PubMed.
- H. Bux, C. Chmelik, J. M. van Baten, R. Krishna and J. Caro, Adv. Mater., 2010, 22, 4741–4743 CrossRef CAS.
- M. Mantina, Y. Wang, R. Arroyave, L. Q. Chen, Z. K. Liu and C. Wolverton, Phys. Rev. Lett., 2008, 100, 215901 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0re00416b |
| This journal is © The Royal Society of Chemistry 2021 |