
CO2 capture enhancement in MOFs via the confinement of molecules
Vanesa del C.
Cotlame-Salinas†
a,
Alfredo
López-Olvera†
b,
Alejandro
Islas-Jácome
 a,
Eduardo
González-Zamora
*a and
Ilich A.
Ibarra
*b
a,
Eduardo
González-Zamora
*a and
Ilich A.
Ibarra
*b
aDepartamento de Química, Universidad Autónoma Metropolitana-Iztapalapa, San Rafael Atlixco 186, Col. Vicentina, C.P. 09340, Iztapalapa, Ciudad de México, Mexico. E-mail: egz@xanum.uam.mx
bLaboratorio de Fisicoquímica y Reactividad de Superficies, Instituto de Investigaciones en Materiales, Universidad Nacional Autónoma de México, Circuito Exterior S/N, Ciudad Universitaria, Coyoacán, Ciudad de México C.P. 04510, Mexico. E-mail: argel@unam.mx
First published on 4th January 2021
Abstract
Metal–organic frameworks (MOFs) have become the most promising molecular sponges to capture gases contributing to the greenhouse effect, e.g. CO2, due to various desirable features such as tuneable pore shapes, sizes and functionalities, a great surface area, resistance to harsh conditions (wide ranges of pH, temperatures, humidity, etc.), ease of their preparation, and in most cases, a high degree of recovery. Thus, despite many MOFs adsorbing extensive amounts of CO2 in their pristine form, it has been demonstrated that their uptake capability can be considerably enhanced when they are post-synthetically modified by the confinement of molecules with different polarities, as a result of new physical–chemical interactions between the pre-confined species and the CO2 molecules within their cavities, for example, via the bottleneck effect, H-bonds and/or even π–π stacking interactions. Thus, this review covers selected studies, mainly from the last five years, highlighting the most significant advances on the CO2 enhanced uptake performance of selected MOFs with pre-adsorbed polar (water, alcohols, and amines) and non-polar (toluene and benzene) molecules, as well as some interesting findings from robust computational calculations behind understanding the nature of such host–guest interactions, with the latter one being a practical and useful tool in the research field.
1. Introduction
The impact of climate change on human health is one of the most important concerns in contemporary society. This issue not only causes direct damage to the world (severe storms, floods, prolonged heatwaves and droughts)1 but also can indirectly damage human health by deteriorating the environment and changing the balance of different ecosystems, which develops many diseases while hindering their prevention and treatment.2 Global warming is mainly caused by a group of gases better known as greenhouse gases, such as: methane (CH4); nitrous oxides (NOX), fluorinated gases and carbon dioxide (CO2).3 In fact, the increase of the levels of CO2 emissions in the atmosphere, due to the excessive consumption of fossil fuels (coal, natural gas and oil),4 has become a global environmental concern.5 According to the EPA's report,6 in 2018, the total CO2 emission was 6667 million metric tons (81% of all greenhouse gases), which comes mostly from anthropogenic sources. It is known that CO2 is removed from the atmosphere by different natural processes. Unfortunately, the large amounts of CO2 released into the atmosphere exceed natural remediation processes. Thus, the design and immediate implementation of new recovery systems is imperative.Over the years, numerous research studies and technologies have been implemented to capture CO2 from industry emissions and store it through chemical/physical strategies.7 One of the main techniques applied in CO2 sequestration is the use of alkanolamines in aqueous solution (by a chemisorption process), performing as a Lewis base.8 Nevertheless, such an absorption method has shown many drawbacks such as: deterioration of pipelines, by-product formation and high recovery costs.9 Therefore, it is essential to migrate towards more effective ways to decrease CO2 waste. Comparatively, physical adsorption with porous materials such as adsorbents is extensively used for CO2 capture due to environmental benefits and economic viability. Different porous solid adsorbent materials, such as zeolites, activated carbons and metal oxides, have been design for CO2 adsorption.10 Relevant features such as high selectivity and adsorption capacity, recyclability, good thermal and chemical stability, and fast kinetics, along with profitable costs, are necessary for a good CO2 adsorbent material.
Undoubtedly, zeolites are the most widely studied adsorbents for CO2 capture.11 These micro–mesoporous solids (pore size between 0–2 and 2–50 nm, respectively) are constructed from aluminosilicates with different polyvalent cations, in a tetrahedral array. Even though zeolites have been a great support in the development of the science of modern porous materials,12 some of these classical porous solids have shown critical constraints for CO2 adsorption (e.g., a high hydrophilic character and high dehydration temperature), meaning their exclusion for CO2 removal under humid conditions.13
In the last decade, several adsorbent materials have been developed, including ionic liquids (ILs), porous organic polymers (POPs), and metal–organic materials (MOMs), among others.14 Metal–organic frameworks (MOFs), also known as porous coordination polymers (PCPs), are a subclass of MOMs that have shown an exceptional adsorbent ability comparable to other porous materials. MOFs are a relatively new type of crystalline micro–mesoporous solid material constructed from strong interactions (coordination bonds) between metal clusters and organic ligands (classically carboxylic acids or azo compounds), generating robust arrays in 1, 2 or 3 dimensions.
Along with a diverse selection of inorganic metal nodes and organic ligands, and different synthetic methodologies, these materials have demonstrated tuneable physical/chemical properties, highlighting ultrahigh porosity, a large surface area and high thermal/chemical stability. These features clearly postulate MOFs as promising materials for CO2 capture.15 The combustion of fossils fuels, in many industrial processes, releases flue streams which include gases such as CO2, CO, CH4, N2 and water vapour (5 to 7 wt%).16 One of the main disadvantages of a large majority of MOF materials is their low stability toward H2O. Furthermore, H2O molecules can, in some cases, disrupt the coordination bonds between the organic ligands and metal nodes, followed by partial or total fragmentation of the MOF structure. For instance, MOFs containing Zn–COOH bonds, such as DMOF-1, DMOF-1-NH2 and UMCM-1, suffer from a complete loss of crystallinity under exposure to water.17 Thus, a fundamental prerequisite for MOF materials is water stability which is key to CO2 capture under humid conditions.18
In addition, the ideal adsorbent for CO2 capture should show two main characteristics: a high CO2 adsorption capacity at a low loading pressure between 0.05 to 0.15 bar;19 and a good balance with a reversible adsorption/desorption process with a moderate heat of adsorption of 30–60 kJ mol−1 (cycles).20 Therefore, to achieve these points in the field of MOF materials, it is necessary to use the modulation of their chemical–physical properties, by a new process or post-synthetic modification (PSM). According to several research groups,21 there are three foremost approaches to improve the CO2 capture in MOFs based on the increment of the strength of the interaction between CO2 and MOFs (primarily via their functional groups). These approaches are: (i) direct modification of organic linkers by insertion of polar functional groups (–NH2, –OH, –COOH, –CN, –SH, and –NO2).22 An improvement in the CO2 capture has been observed in different examples, such as amine functionalised CAU-1.23 (ii) Generation of metal uncoordinated sites, also known as open metal sites (OMSs).24 These OMSs are generated when a MOF is treated by a thermal or vacuum process to remove terminal labile linkers (usually synthetic solvents) binding to metal clusters. Thus, the quadrupole of CO2 molecules can interact with metal Lewis acid sites. (iii) The confinement of molecules within MOFs such as water, alcohols, and amines and even the use of corrosive gases (e.g., H2S) have demonstrated an improvement in the CO2 capture.21,25 This review is focused on the last approach and determinant topics as the nature of solvents and their polarity will be comprehensibly discussed in the following sections.
In this context, it is known that the affinity of MOFs for polar molecules can increase for MOFs based on inorganic cluster chains with μx-OH bridging groups.25,26 This architecture can form hydrogen bonds between such bridging (μx-OH) groups and confined molecules.27 In general, this functionalisation can enhance the CO2 adsorption properties of MOFs by confining small amounts of polar and non-polar molecules.
In the last decade, the main use of small alcohols (i.e., methanol and ethanol) as working fluids in MOFs for heat-pump chillers have increased considerably.27,28 Additionally, other uses of these polar molecules in MOFs have gained attention for the improvement of the CO2 adsorption performance (via the pre-confinement of alcohols).21 Following the strategy of confining small quantities of water and alcohols, new research has been carried out on the confinement of different polar molecules (amines) and non-polar molecules (hydrocarbons) for the enhancement of CO2 uptake.
Major breakthroughs by the confinement of small amounts of molecules have been achieved in the last 4 years. Therefore, this review aims to give a broad overview grouping these advances, taking water confinement as the starting point, and the use of other molecules (polar and non-polar), to explain the fundamental molecular interactions responsible for the enhancement of the CO2 capture in functionalised MOFs. Finally, with this review we try to encourage more research groups to explore and extend the knowledge on the effects of confining molecules within MOF materials.
2. Water pre-confinement
Despite the fact that the presence of water has a detrimental effect in gas adsorption processes of several MOFs due to structural collapse or a reduction in the amount of adsorption sites, some research groups have focused on the use of water as a fundamental tool in the enhancement of CO2 adsorption under closely related industrial conditions (vide supra). In this section, we provide a brief discussion about some studies that we consider as a benchmark in the development of pre-confinement of water to improve the CO2 adsorption properties of MOF materials. One of the first steps toward understanding the influence of pre-confinement of water inside of MOFs was published in 2009 by Snurr et al.28 As already recompiled in other reviews,25,26b this work shows how small amounts of water (4 wt% and 8 wt%) improve the CO2 capture on a MOF based on Cu(II) with OMS called HKUST-1. This early study demonstrated that the confinement of water inside MOFs could be a new and better alternative for capturing CO2 than using other classical materials (zeolites and activated carbons) that have shown several disadvantages in CO2 removal under humid conditions. A few years later, Llewellyn and co-workers29 reported an interesting systematic study, where they explored the effect of water on CO2 uptake and the variations in the adsorption enthalpy in two microporous and one mesoporous MOFs: HKUST-1, UiO-66 (Zr) and MIL-100 (Fe), respectively, under post-combustion conditions (0.2 bar of CO2 pressure and 298 K). This work can be considered as the cornerstone in the pre-confinement of water in MOF materials, due to the outstanding improvement of the CO2 adsorption under humid conditions showed for the mesoporous MOF (MIL-100(Fe)) (Table 1), and the intriguing CO2 adsorption mechanism that was suggested by the authors. Taking into account the significant advances made by Llewellyn and co-workers, our research group decided to delve into water confinement in microporous MOF materials. Thus, we developed several studies based on a set of microporous water-stable MOF materials decorated with hydroxo bridge groups (μx-OH) such as MFM-300 (In and Sc),30,31 NOTT-401,32 MIL-53(Al) (BDC and TDC),33,34 CAU-10 (ref. 35) and Mg-CUK-1,36 working in a minimal range of quantity of confined water from 0.5 to 4 wt% on average (Table 1). For example, it was demonstrated that although CAU-10 does not have a considerable surface area, compared to other representative MOFs,37 this porous solid can achieve a 1.3-fold CO2 enhancement (i.e., from 1.2 to 1.65 under dry and wet conditions respectively) only using 0.55 wt% water (Table 1). Besides, CAU-10 shows an excellent cyclability performance, being stable up to 5 cycles with minimal changes in its adsorption properties. Mg-CUK-1 (ref. 36) demonstrated an outstanding performance reaching a maximum for the CO2 capture of almost 1.9-fold with 0.9 wt% of preconfined water (Table 1). Overall, such enhancement showed for these materials is associated with the polar environment inside the pores which is provided by the hydroxo bridging groups (either μ3-OH or -μ2-OH), which form hydrogen bonds with water molecules at low-charge, and the water molecules fixed in the pores can accommodate very efficiently CO2 molecules by weak interactions. The possible adsorption mechanism of CO2 under humid conditions is supported by computational calculations. Thus, these findings are an interesting guideline to evaluate other molecules for a higher CO2 capture.| MOF material | Surface area BET [m2 g−1] | Press. [bar] | Temp [K] | CO2 adsorption under dry conditions [mmol g−1] | Amount of water pre-adsorbed | CO2 adsorption under wet conditions [mmol g−1] | Ref. |
|---|---|---|---|---|---|---|---|
| a Values calculated from the experimental dates (some have been converted to wt% from the originally reported units). K. C: kinetic conditions. | |||||||
| HKUST-1 | 1387 | 1 | 298 | 4.88a | 4 wt% | 8.63a | 38 |
| HKUST-1-pellet | 921 | 1 | 298 | 2.90a | 1.8 wt%a | 3.25a | 39 |
| Mg-MOF-74 | 1495 | 0.167 | 298 | 4.91a | 9% RH | 1.63a | 40 |
| 36% RH | 1.22a | ||||||
| 70% RH | 0.77a | ||||||
| Co-MOF-74 | 1080 | 0.167 | 298 | 2.78a | 70% RH | 2.37a | 39 |
| Co-MOF-74 | 1105 | 1 | 298 | 6.14 | 15.6 wt% | 4.46 | 41 |
| Zn-MOF-74 | 816 | 0.167 | 298 | 1.51a | 70% RH | 0.32a | 39 |
| Ni-MOF-74 | 1070 | 0.167 | 298 | 2.62a | 70% RH | 1.59a | 38 |
| NiMOF-74-pellet | 639 | 1 | 298 | 6.68a | 1.8 wt%a | 6.45a | 38 |
| MIL-100(Cr) | 1790 | 1 | 303 | 1.42 | 5.83 wta | 0.71 | 42 |
| MIL-100(Fe) | 2006 | 0.2 | 298 | 0.50 | 1 wt%a | 0.59 | 29 |
| 3 wt%a | 0.54 | ||||||
| 4.8 wt%a | 1.52 | ||||||
| 55 wt%a | 2.47 | ||||||
| UiO-66(Zr) | 1179 | 0.2 | 298 | 0.61 | 1 wt%a | 0.77 | 29 |
| 2 wt%a | 0.75 | ||||||
| 4 wt%a | 0.75 | ||||||
| 18 wt%a | 0.79 | ||||||
| MIL-101(Cr) | 3314 | 0.1 | 298 | 0.50 | 20% RH | 0.48 | 43 |
| [Cu(PF6)2(bpp)2]n | — | 10 | 298 | 1.31 | 17% RH | 1.34 | 44 |
| ZIF-68 | — | 0.1 | 298 | 0.029 | 50 wt%a | 0.029 | 45 |
| PCN-250(Fe3) | 1470 | 0.15 | 298 | 1.18 | 50% RH | 1.82 | 38 |
| 90% RH | 1.52 | ||||||
| PCN-250(Fe2Co) | 1653 | 0.15 | 298 | 1.32 | 50% RH | 2.23 | 37 |
| 90% RH | 2.27 | ||||||
| MFM-300(In) | 1060 | K. C | 298 | 1.23 | 30% RH | 2.5 | 30 |
| MFM-300(Sc) | 1356 | K. C | 303 | 0.93 | 1.7 wt%a | 1.8 | 31 |
| 2.2 wt%a | 2.31 | ||||||
| NOTT-401 | 1504 | K. C | 303 | 0.27 | 1.1% wta | 0.88 | 32 |
| 3.0 wt%a. | 0.43 | ||||||
| 4.2 wt%a | 0.09 | ||||||
| MIL-53(Al) | 1096 | K. C | 303 | 0.79 | 1.5 wt%a | 1.18 | 33 |
| MIL-53(Al)-TDC | 1443 | K. C | 303 | 1.13 | 0.60 wt% | 1.47 | 34 |
| CAU-10 | 615 | K. C | 303 | 1.22 | 0.55 wt% | 1.65 | 35 |
| Mg-CUK-1 | 600 | K. C | 303 | 1.04 | 0.2 wt% | 1.12 | 36 |
| 0.5 wt% | 1.20 | ||||||
| 0.8 wt% | 1.92 | ||||||
| 0.9 wt% | 1.94 | ||||||
| 1.4 wt% | 1.89 | ||||||
| 1.7 wt% | 1.82 | ||||||
| Fe(pz)[Pt(CN)4] | 431 | K. C | 298 | 2.11 | 11.7% | 0.6 | 46 |
Recently,38 in 2018, Chen and Li reported two water-stable microporous MOF materials: PCN-250 (Fe3) and PCN-250 (Fe2Co), which were evaluated using 50 and 90% RH at 298 K and dynamic adsorption conditions. These framework materials were constructed from Fe3(μ3-O)(CH3COO)6 or Fe2Co(μ3-O)(CH3COO)6 metal clusters, respectively, and the H4ABTC organic linker (H4ABTC = 3,3′,5,5′-azobenzenetetracarboxylic acid). Both materials showed substantial enhancement in the CO2 uptake under humid conditions (i.e., CO2 captures achieved at 50% RH were 1.82 and 2.23 mmol g−1while those at 90% RH were 1.52 and 2.27 mmol g−1 for PCN-250 (Fe3) and PCN-250 (Fe2Co), respectively). The amount of CO2 adsorbed for both materials under dry conditions was 1.18 and 1.23 mmol g−1, respectively. The authors attributed this improvement to the influence of the oxo bridged groups (μ3-O) within the pore environment for both frameworks in the presence of water, such functional group acts as a clamp-like mechanism. Finally, the authors mentioned that both microporous solids showed a remarkable cyclability performance (up to 10 cycles) without showing any change in their crystalline structure.
3. Alcohol and amine molecule confinement for CO2 capture
Following the previous approach, it was interesting to explore the effect on CO2 adsorption in MOFs when other polar solvents were confined in their channels. Thus, a series of studies have been reported since 2016, concerning the confinement of different alcohols (MeOH, EtOH, 1-PrOH and 2-PrOH) in MOF materials (MFM-300(In),47–50 HKUST-1,51 MIL-53(Al)52 and MIL-53(Al)-TDC).34The initial study involved the MOF material labeled MFM-300(In) (also known as InOF-1) and confined ethanol.47 MFM-300(In) is a microporous material constructed from binuclear [In2(μ2-OH)] building blocks linked by BPTC4− ligands (H4BPTC = biphenyl-3,3′,5,5′-tetracarboxylic acid), forming a 3D structure with channel dimensions of 7.5 Å (Fig. 1).
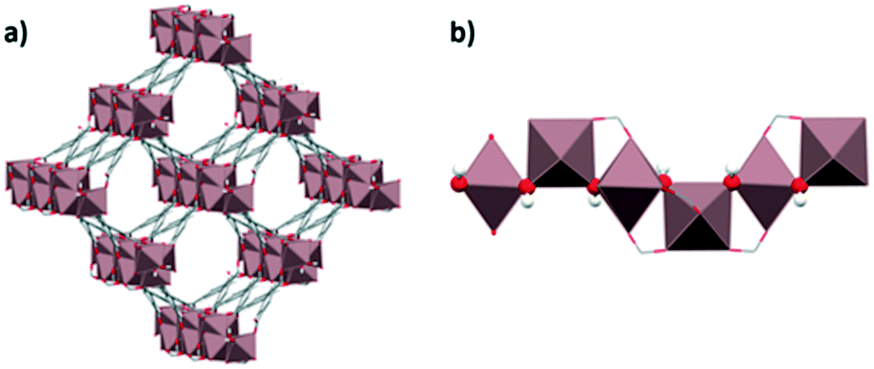 | ||
| Fig. 1 a) MFM-300(In) 3D structural view along the crystallographic axis c, showing the pore opening. b) Inorganic oxo-cluster chain with In(III) in octahedral coordination geometry, showing the position of the μ2-OH bridge groups. | ||
Kinetic and static CO2 adsorption studies of a pre-loaded (confined) MFM-300(In) sample with 2.63 wt% ethanol were carried out. The results showed an enhancement in the CO2 adsorption capabilities in both systems, reaching a maximum CO2 capture of 2.7-fold under kinetic conditions (3.21 mmol g−1) and 3.6-fold (3.11 mmol g−1) under static conditions, at 1 bar of CO2 pressure, compared to dry adsorption measurements (1.19 mmol g−1 and 0.86 mmol g−1, respectively). This behavior was attributed to the presence of hydrogen bonds between hydroxo bridging groups (μ-OH) and EtOH molecules that substantially diminished the dimensions of the channels and changed the shape of the pores, producing a partial obstruction. Consequently, when a flow of CO2 molecules crosses the channels, the obstructed pores reduce the self-diffusion coefficient of CO2 molecules. This effect was designated as the “bottleneck effect”. Fig. 2 shows the pore reduction of MFM-300(In) as a function of preconfined ethanol. An amount of 2.63 wt% ethanol represents approximately 1.35 molecules of ethanol per unit cell, and the distribution of the confined molecules creates “bottlenecks” separated in sections of 37 Å, forcing the CO2 molecules to stay longer around the bottleneck due to steric effects. Thus, CO2 molecules can be accommodated more efficiently inside the channels. Recently, in 2020, it was reported that such a bottleneck effect also occurs in Mg-CUK-1 (ref. 53) when water molecules were confined inside the pores and a CO2 uptake enhancement of 1.8-fold was achieved. This effect was successfully demonstrated by PXRD with Le Bail methodology refinements.
 | ||
| Fig. 2 Crystal structure of MFM-300(In) and the effect of the amount of ethanol confined in the pores via the formation of a hydrogen bond to the In2(μ2-OH) hydroxo groups on the pore diameter and shape. View along the crystallographic 001 direction with and without the channel surface (left) and along the 110-direction rotated additionally by 35° around 010 (right) to demonstrate the change from an almost cylindrical shape to a helicoidal shape (reproduced from ref. 47 with permission from The Royal Society of Chemistry). | ||
Later, some studies related to the confinement of alcohols with different kinetic diameters were presented. Thus, the confinement of methanol within MFM-300(In) was reported in 2017.48 The material loaded with 2 wt% MeOH showed an interesting CO2 uptake enhancement. Kinetic and static CO2 isotherms exhibited a 1.3-fold and 4.88-fold increase in the CO2 adsorption capacity, respectively (from 1.19 mmol g−1 to 1.56 mmol g−1 under kinetic conditions and from 0.26 mmol g−1 to 1.27 mmol g−1 under static conditions) (Fig. 3). This improvement was corroborated by computational Monte Carlo (MC) calculations, which illustrated the preferential adsorption sites for the MeOH molecules near the μ2-OH groups, tending to form lumps, and hence reducing the pore dimensions (Fig. 4). This explanation supported the bottleneck effect (vide supra), which led to the enhancement in the adsorption of CO2. More in-depth analysis of the hydrogen bonding between MeOH, CO2 molecules and μ-OH bridging groups and their fundamental role in increasing the CO2 uptake were detailed in a recent study.54 Along with density functional theory (DFT), periodic calculations, and experimental in situ DRIFTS, the authors postulated four feasible mechanisms of CO2 uptake in MFM-300(In) with MeOH pre-confined in its pores. According to the structure stability, the mechanisms are stated in the following order: (I) CO2 and MeOH molecules interact with different μ2-OH bridging groups of MFM-300(In), and they could not interact with each other; (II) CO2 interacts with MeOH; (III) CO2 is trapped by MeOH due to the weak CH⋯O interactions and van der Waals interactions with MFM-300(In) and (IV) a CO2 molecule replaces a MeOH molecule and forms a hydrogen bond with μ2-OH of InOF-1. This mechanism emphasises the importance of inorganic building units (μx-OH) which are part of other metal clusters.55
 | ||
| Fig. 3 Kinetic CO2 uptake experiments performed at 303 K with a CO2 flow of 60 mL min−1 for InOF-1 (blue curve) and MeOH@MFM-300(In) (orange curve) (a) static CO2 adsorption–desorption performed from 0 to 1 bar at 196 K on InOF-1 (black circles) and MeOH@MFM-300(In) (red circles) (b) (reproduced from ref. 48 with permission from The Royal Society of Chemistry). | ||
 | ||
| Fig. 4 van der Waals surface areas plotted for the DFT-optimized empty MFM-300(In) (right) and MeOH@MFM-300(In) (left) structures (reproduced from ref. 48 with permission from The Royal Society of Chemistry). | ||
Encouraged by the results, more recent studies explored the effect of confining 1-propanol49 and 2-propanol50 inside MFM-30O(In). Interestingly, dynamic and static experimental CO2 isotherms showed that the confinement of a small amount of 1-propanol (2 wt%) did not improve the CO2 adsorption capacity.49 Grand canonical Monte Carlo (GCMC) simulations were carried out in order to understand this behavior. It was found that 1-propanol mixed with CO2 tends to form clusters, instead of a localised distribution, causing blockage within the channels of MFM-300(In) and thus, a decrease of the CO2 uptake. A study of 2-propanol50 confinement in MFM-300(In) was recently reported in 2019. Dynamic experimental CO2 isotherms were collected with 2.05 wt% 2-propanol, and a 1.25-fold increase of the CO2 uptake (from 1.18 mmol g−1 to 1.48 mmol g−1) was observed. After 10 cycles of sorption–desorption, there was a slight reduction of the CO2 capacity uptake (0.4 wt%). To gain a deeper understanding of this CO2 capacity enhancement, quantum chemical models were calculated, demonstrating that this is a result of hydrogen bond interactions (2-PrOH with μ2-OH bridging groups, and 2-PrOH with CO2) and the reduction of the pore volume generated by the presence of confined 2-propanol, which causes the bottleneck effect previously discussed (Fig. 5).
 | ||
| Fig. 5 Solid (top) and transparent (bottom) isosurface of the electron density (ρ = 0.0003 a. u.), denoting the voids of the empty MFM-300(In) (left) and i-PrOH@MFM-300(In) (right) (reproduced from ref. 50 with permission from The Royal Society of Chemistry). | ||
Confinement of ethanol within HKUST-1 (ref. 51) was also investigated. This study is relevant because the structure is different from MFM-300(In). HKUST-1 is not a hydroxo-functionalized material and it shows a rigid structure and open metal sites. HKUST is formed by square Cu2 paddlewheel clusters linked by BTC ligands. This material has two pore size dimensions, a large one with around 10 and 11 Å and a second one with a diameter of 4 Å. For the CO2 dynamic isotherm, a sample with a small amount of EtOH (3 wt%) confined in HKUST-1 pores was used and a slight adsorption capacity enhancement was observed (from 2.93 mmol g−1 to 2.97 mmol g−1) (Fig. 6a). It is worth noting that after 10 cycles of sorption–desorption, the crystal structure was retained, and the CO2 capacity was maintained without the need for gas purging or thermal re-activation (Fig. 6b). A different behavior was observed when small quantities of water (3 wt%) were confined in the MOF, showing a decrease of CO2 capacity uptake (Fig. 6a).
 | ||
| Fig. 6 CO2 uptake experiments performed on HKUST-1 with small loadings of pre-adsorbed water and ethanol at 303 K (a). Inset: Zoom of CO2 adsorption emphasising the slightly enhanced CO2 capture with the use of ethanol. CO2 cycling measurements for the ethanol impregnated HKUS-1 sample at 303 K, showing the reversibility of the capture process (b) (reproduced from ref. 51 with permission from The Royal Society of Chemistry). | ||
The effect on the CO2 adsorption capacities when alcohol molecules (MeOH and 2-PrOH) were confined in MIL-53(Al) was also studied.52 MIL-53(Al) is a flexible material formed by μ2-OH bridging groups in trans orientation. As Fig. 7a depicts, dynamic isotherms revealed a CO2 adsorption enhancement when a small amount of MeOH and 2-propanol (2 wt%) was confined in MIL-53(Al) (1.07 mmol g−1 and 1.022 mmol g−1, respectively), in comparison to the fully activated material (0.79 mmol g−1) (Fig. 7a). The static isotherm also showed a CO2 capture enhancement when MeOH was confined (from 7.2 to 9.0 mmol g−1) (Fig. 7b). The sorption–desorption process of MIL-53(Al), with MeOH confined showed a small loss of the CO2 adsorption capacity (6.3% wt) after 10 cycles (Fig. 7c). Remarkably, this process only needed mild reactivation conditions. In addition, a structural model was optimised to visualise the hydrogen bonds expected between MeOH and the hydroxo functional groups of this MOF material. A minimal variation of the hydrogen bond length between CO2–MeOH (1.79 Å) and CO2–2-PrOH (1.85 Å) was found, and indeed this fact can explain the small difference in the CO2 adsorption obtained between both confined molecules (Fig. 8).
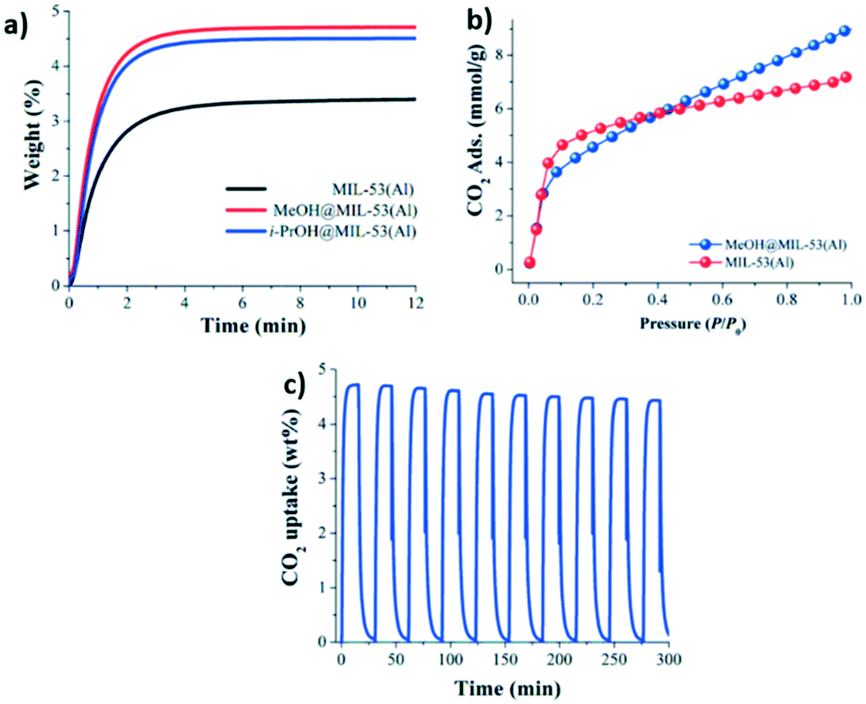 | ||
| Fig. 7 Kinetic CO2 uptake experiments at 30 °C with a CO2 flow of 60 mL min−1 in MIL-53(Al) (black curve); MeOH@MIL-53(Al) (red curve) and i-PrOH@MIL-53(Al) (blue curve) (a). Static CO2 adsorption from 0 to 1 bar at 196 K on MIL-53(Al) (red circles) and MeOH@MIL-53(Al) (blue circles) (b). Adsorption–desorption cycling for MeOH@MIL-53(Al) (c) (reproduced from ref. 52 with permission from The Royal Society of Chemistry). | ||
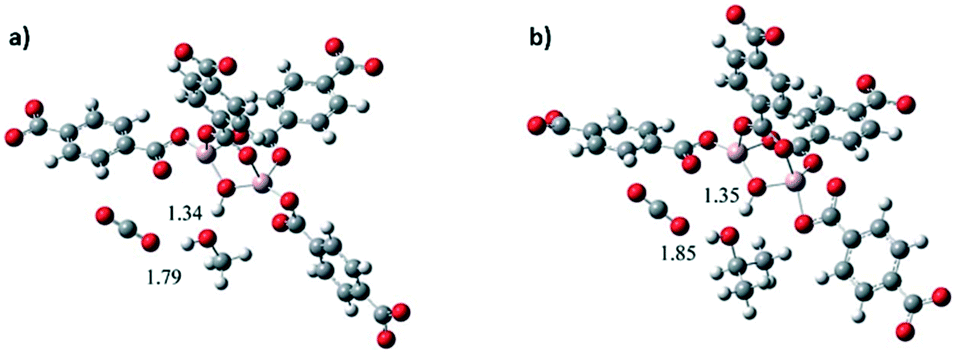 | ||
| Fig. 8 Optimised structures of MIL-53(Al): (a) proposed model interacting with MeOH and CO2 and (b) proposed model interacting with i-PrOH and CO2 (reproduced from ref. 52 with permission from The Royal Society of Chemistry). | ||
In 2018, a study on the CO2 adsorption properties (EtOH confined) of an Al(III)-based MOF material labeled MIL-53(Al)-TDC34 was performed. MIL-53(AL)-TDC is a microporous material, constructed with 2,5-thiophenedicarboxylate (TDC) and Al(III), which provides a rigid structure. Kinetic CO2 experimental isotherms were obtained with small amounts of EtOH confined. Interestingly, the CO2 capture was not enhanced. An optimised model was calculated to visualize relevant hydrogen bonds, revealing that the position of the interaction between EtOH and CO2 (EtOH⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO) was out of plane, in other words, these molecules were not well-positioned for a favorable interaction and, therefore, CO2 capture enhancement.
CO) was out of plane, in other words, these molecules were not well-positioned for a favorable interaction and, therefore, CO2 capture enhancement.
A new method was reported by Chen et al.56 in 2018. Using a slurry mixture of ZIF-8/mIm (methylimidazole)–glycol for the capture of CO2, via a hybrid absorption–adsorption method, they found that 5 w% mIm in the liquid phase increases the CO2 capture in the presence of water.
On the other hand, the use of aminated solvents has been shown to be an interesting alternative for CO2 capture. In this regard, a widely used technique is the introduction of alkyldiamine molecules inside MOF channels. Several studies have reported the increase of the CO2 adsorption capacity by open metal sites (OMSs) and the generation of amine adsorption sites.57–63 Herein, one amine group is coordinated to an open metal site, while the other one remains free to interact with CO2 molecules, taking advantage of the affinity of basic amines with CO2 molecules. However, this method requires the use of only MOFs with OMSs, which greatly hinders the recovery of CO2 adsorbed. Similar to the diamine use mentioned above, another CO2 adsorption approach has been investigated by observing the influence of alkylamine fixation in MOF materials with Brønsted acid–base pendent groups (such as UiO-66-NH2 and Cr-MIL-101-SO3H).64 In this study, a noticeable increase in the CO2 adsorption properties was observed and in the case of EDA-UiO-66 (EDA = ethylenediamine), a remarkable cyclability performance was shown. Nevertheless, in all adsorption experiments, the formation of carbamate as a by-product was detected.
Otherwise, amide molecules, e.g. N,N-dimethylformamide (DMF), N,N-diethylformamide (DEF), N,N-dimethylacetamide (DMA) and N-methyl-2-pyrrolidone (NMP), have shown a great influence on the physical and chemical properties of some MOFs.65 In this context, Garcia-Garibay and co-workers66 investigated the diffusion-controlled rotation of a triptycene-based MOF in a viscous environment induced by confined DMF. Accordingly, Lu and Usman67 demonstrated how the DMF loading increased the dielectric constant in three isostructural 1D-MOFs based on Sr(II).
Motivated by the preliminary investigations on the use of amides in MOF materials, our group performed, in 2017 (ref. 68), the pre-confinement of DMF within MFM-300(In) to study the CO2 adsorption properties. An enhancement in the CO2 uptake was observed when MFM-300(In) was loaded with a minimal quantity of DMF (4.2 wt%). Dynamic and static CO2 isotherms showed a 1.5-fold (1.83 mmol g−1) and 1.4-fold (7.5 mmol g−1) increase of the CO2 captures, respectively (Fig. 9a and b), in comparison to dry adsorption measurements (1.19 mmol g−1 and 5.5 mmol g−1, respectively). Adsorption–desorption measurements revealed the stability of CO2 adsorption even after 10 cycles under mild conditions (Fig. 9c). To provide evidence of host–guest hydrogen bonds, which are likely responsible for the increase of CO2 adsorption, the single crystal X-ray diffraction structure of MFM-300(In) with DMF confined was determined. It was corroborated that DMF molecules were situated in an ideal position in a “dent in the wall” between the ligands. The angle formed is 115°, close to the theoretical 120° from sp2 hybridization, and O–H⋯O is nearly linear (Fig. 10).
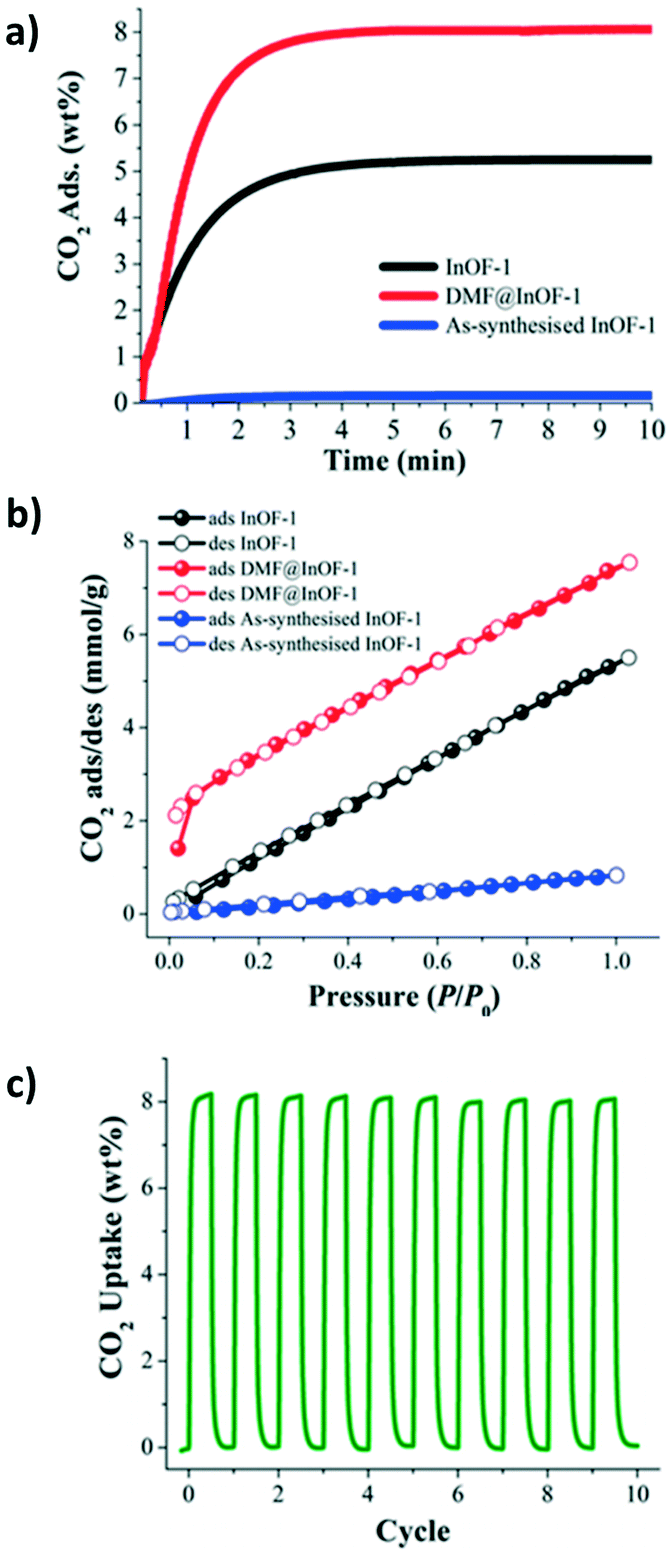 | ||
| Fig. 9 (a) Kinetic CO2 uptake experiments at 303 K on InOF-1, DMF@InOF-1, and as-synthesised InOF-1 (black, red, and blue lines respectively). (b) Static CO2 adsorption/desorption at 1 bar and 196 K on InOF-1, DMF@InOF-1, and as-synthesized InOF-1 (black, red, and blue circles respectively). (c) Adsorption/desorption cycling for DMF@InOF-1. Reprinted with permission from ref. 68 (copyright 2017, American Chemical Society). | ||
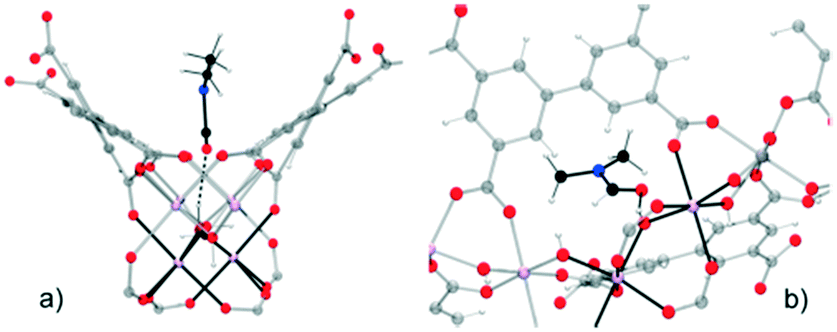 | ||
| Fig. 10 Mutual orientations of the DMF molecules and the MFM-300(In) framework, showing the hydrogen bonds between the DMF and In2(μ-OH) groups (a) in the direction of the channel and (b) showing details of the hydrogen bonds. Reprinted with permission from ref. 68 (copyright 2017, American Chemical Society). | ||
4. Confinement of aromatic and other molecules to enhance CO2 capture
Up to this point, we have only considered the influence of small polar molecules (water, alcohols and amines) on the CO2 uptake and how these pre-confined guest molecules interact simultaneously with the pore wall and the CO2 molecule, mainly by hydrogen bonding and other weak interactions leading to micro-pockets and bottleneck behaviours.29,47,68 It is worth mentioning that not only these types of pre-confinement molecules can enhance the CO2 capture performance, but also recent investigations have shown how non-polar molecules such as toluene,69 benzene70 and other slightly bigger molecules (ionic liquids and dyes),71 and even corrosive gases such as SO2 can improve CO2 capture72via reduction of the cross-sectional surface area and pore volume leading to a better packing of CO2 molecules into MOF channels.Following the same methodology, a small amount of toluene (1.5 wt%) was confined in MFM-300(In).69 Interestingly, a 1.38-fold increase of CO2 capture was obtained in contrast to the fully activated material under kinetic conditions (from 1.19 mmol g−1 to 1.65 mmol g−1) (Fig. 11a). When 6 cycles of adsorption–desorption were achieved, the CO2 adsorption capacity was not affected (Fig. 11b), and its crystallinity did not suffer from any change. The isosteric heat of adsorption (ΔHads = −46.81 kJ mol−1), experimentally evaluated, suggests the substantial affinity of MFM-300(In) toward toluene. Theoretical studies and DRIFTS experiments were carried out to investigate the predominant interactions in the adsorption process. DFT studies revealed π–π stacking interactions between the toluene molecule and the aromatic rings of the MFM-300(In) linkers. This was corroborated by electron density analysis, wherein weaker interactions were observed, such as C–H⋯O hydrogen bonds and C⋯C, and no typical H⋯H interactions were even found.73 The CO2 capture enhancement of toluene@MFM-300(In) was attributed to the bottleneck effect, and the interactions between toluene⋯CO2 and MFM-300(In)⋯CO2.47 The electron density isosurface showed a toluene molecule per unit cell confined in MFM-300(In), generating a reduction of 15% of the pore dimensions compared with the pristine material (Fig. 12). Moreover, an optimised model (Fig. 13) showed how the CO2 molecule interacts with the centroids of aromatic rings of toluene and MFM-300(In) linkers, contributing to the CO2 capture.
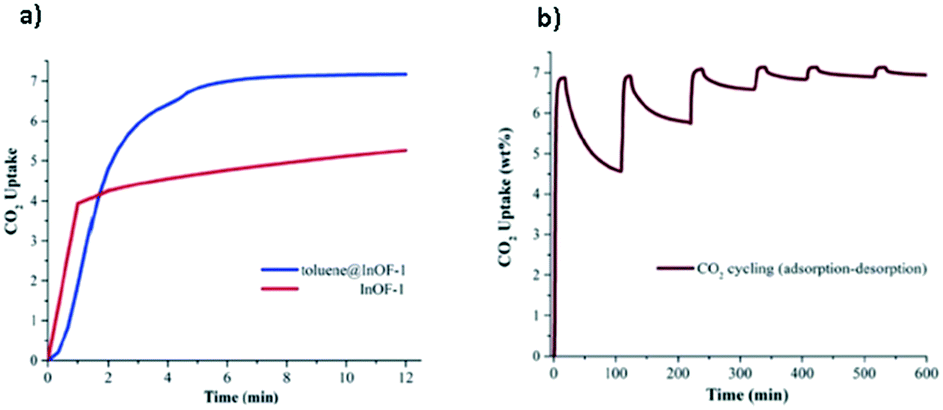 | ||
| Fig. 11 CO2 dynamic adsorption experiments performed at 298 K for InOF-1 (red curve) and toluene@InOF-1 (blue curve) (a). Adsorption–desorption CO2 cycling for toluene@InOF-1 (b) (reproduced from ref. 69 with permission from The Royal Society of Chemistry). | ||
 | ||
| Fig. 12 Electron density isosurfaces, denoting the void channels of the pristine (left) and toluene-functionalized MFM-300(In) (right) (reproduced from ref. 69 with permission from The Royal Society of Chemistry). | ||
 | ||
| Fig. 13 Close up (left) and pore channel (right) views of CO2 captured by toluene-functionalized InOF-1. Toluene, CO2, and the MFM-300 framework are depicted in blue, yellow and gray, respectively. Reproduced from ref. 69 with permission from The Royal Society of Chemistry. | ||
In addition, MFM-300(In) was pre-loaded with benzene to have a direct comparison. Thus, a small amount of benzene (1.5 wt%) was pre-confined and the CO2 adsorption was evaluated,65 resulting in a 1.6-fold increase under dynamic conditions (Fig. 14a). Fig. 14b shows eight CO2 adsorption–desorption cycles on benzene@MFM-300(In) with an outstanding CO2 reversibility performance. The isosteric heat of adsorption (ΔHads = −25.1 kJ mol−1) suggested a weaker interaction between benzene and the MOF material in comparison to that with toluene.69 According to computational studies, π⋯π interactions, weak C–H⋯O hydrogen bonds, and medium strength C–H⋯H–O hydrogen bonds were found by confining benzene in MFM-300(In), as previously stated by confining toluene.64 The optimized structures depicted in Fig. 15 show the most stable arrangement of CO2 molecules inside the channels of the MOF material. Therefore, the CO2 molecule can be found (i) interacting with the μ2-OH bridge group and with the centroid of the aromatic ring in the linker, and (ii) parallel to the aromatic ring of the linker.
 | ||
| Fig. 14 CO2 uptake experiments performed in InOF-1 (orange curve) and Bz@InOF-1 (blue curve) at 298 K (a). CO2 cycling measurements for Bz@InOF-1 at 298 K (b) (reproduced from ref. 70 with permission from The Royal Society of Chemistry). | ||
 | ||
| Fig. 15 Two possible interactions between a CO2 molecule and the MFM-300(In) material: μ2-OH⋯OCC interaction (left) and CO2⋯MFM-300(In) linker centroid (right). The CO2 molecule is depicted in green color. The aromatic ring mass centre is indicated with a black sphere (reproduced from ref. 70 with permission from The Royal Society of Chemistry). | ||
In this context, the confinement of ionic liquids (ILs) within MOFs to enhance the CO2 adsorption has also recently been investigated. In 2015, Yang and Ban71a used ZIF-8 nanocages, a material with limited CO2 capture performance, to introduce an imidazolium-based IL [bmim][Tf2N] by ionothermal synthesis (Fig. 16). The authors chose this IL because the Tf2N mono-anionic species has been shown to have a remarkable affinity for the CO2 molecule and its cationic counterpart (bmim) fits properly into the ZIF-8 microporous cavity. A reduction of the pore volume and surface area was observed. Despite these changes, the CO2 capture (particularly in the low-pressure range) was almost two-fold enhanced (from 0.28 mmol g−1 to 0.45 approximately at 0.3 bar and 298 K) with a molar ratio of 0.235 IL/ZIF-8. In addition, this improvement is due to two contributions: (i) the reduction of the pore dimensions of ZIF-8 and (ii) the ion-induced dipole interactions between the IL and the CO2 molecules, but more details are needed to confirm such a hypothesis. Although the confinement of ILs into ZIF-8 was not shown to be the best alternative to improve the CO2 capture compared with other pre-confined molecules (water, alcohols and amines), the use of larger molecules as ILs proves that the reduction of a cross section of pore volume or the formation of micropockets29 can be induced in different MOFs to modify their gas adsorption properties.
 | ||
| Fig. 16 Illustration of the cavity-occupying concept for tailoring the molecular sieving properties of ZIF-8 by incorporation of RTILs. The cut-off size shifts from the aperture size of the six-membered ring to the reduced effective cage size by confinement of [bmim][Tf2N] in a ZIF-8 cage. Reprinted with permission from ref. 71a (copyright 2015, Wiley-VCH Verlag GmbH & Co.). | ||
Similarly, Wu and co-workers71b confined dye molecules, such as cationic rhodamine B, anionic CR, and neutral melanin, which are aromatic molecules with different functional groups (–OH and –NH). The confinement of these dye molecules within a mesoporous MOF material, labeled CZJ-10, was performed in different quantities (0.28, 0.04, and 13.1 mol per unit of material, respectively). CZJ-10 is assembled from 4,4′,4′′-benzene-1,3,5-triyltricinnamic acid as a linker and Cu2(CO2) paddlewheels as the inorganic building unit. Unlike other materials with open metal sites that have been used in the small polar solvent pre-loading approach (e.g., water and alcohols within HKUST),38,51 the CZJ-10 pre-loaded with each dye molecule (RB@CZJ-10, CR@CZJ-10 and melanin@CZJ-10, respectively) showed a significant improvement of CO2 capture (Fig. 17). Melanin@CZJ-10 achieved slightly the best CO2 uptake compared to the other dye@CZJ-10 (1.46, 1.37 and 1.63 mmol g−1 for RB@CZJ-10, CR@CZJ-10 and melanin@CZJ-10, respectively), and an almost 2.5-fold increase compared to pristine CZJ-10 (0.67 mmol g−1) at 1 bar and 298 K. The authors attributed this enhancement to the addition of new binding sites inside the pores, resulting in a stronger affinity to CO2 molecules. To support this statement, the authors encapsulated triphenylmethane (TPM), a neutral aromatic molecule, into CZJ-10 (TPM@CZJ-10). The CO2 isotherm of TPM@CZJ-10 measured up to 1 bar and 298 K reached only 0.55 mmol g−1, resulting in a lower CO2 uptake compared to pristine CJZ-10.
 | ||
| Fig. 17 CO2 adsorption isotherms of CZJ-10, RB@CZJ-10, CR@ CZJ-10, melanin@CZJ-10, and TPM@CZJ-10, and N2 adsorption isotherm of CZJ-10 at 298 K. Reprinted with permission from ref. 71b (copyright 2017, American Chemical Society). | ||
On the other hand, the gas co-adsorption process in traditional porous materials has been suggested as an excellent alternative to improve the adsorption of toxic gases by either host–guest or guest–guest interactions.74 In this context, Son and Chihaia highlighted the fundamental role of computational research using a predictive tool in the improvement of CO2 capture.72 They evaluated the simultaneous adsorption of CO2 and SO2 in a Ni-based MOF (Ni(bdc)(ted)0.5) by DFT studies (Fig. 18). The authors observed that the material exhibited a higher affinity to SO2 over CO2. Additionally, the CO2 molecule can interact with the SO2 molecule by weak interaction between the oxygen atom of CO2 and the sulphur of SO2 (CO2⋯SO2) (Fig. 18b). This example suggests that the co-adsorption method can be one of the best alternatives to improve the CO2 gas adsorption in MOFs especially at the industrial level, since not only the SO2 molecule can be a good candidate, but also molecules with a relevant polar nature (e.g., NH3 and H2S which are present in some flue gas streams) can be used for this task.
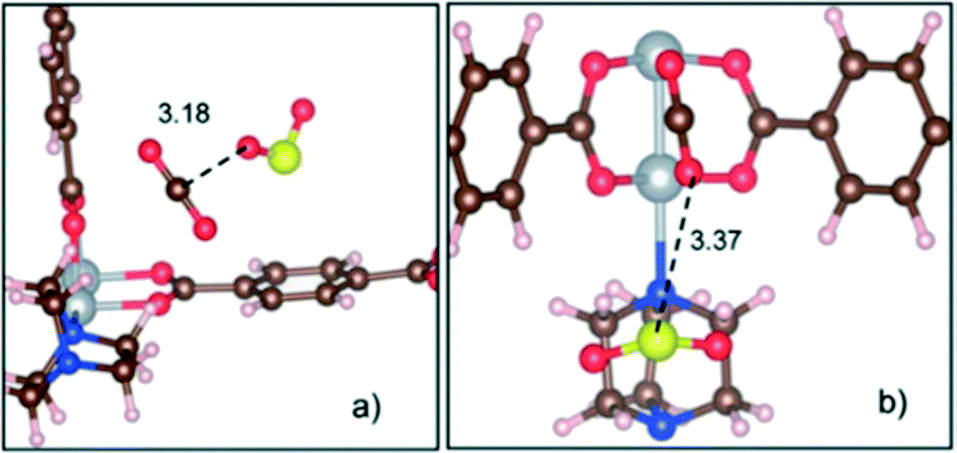 | ||
| Fig. 18 Two main configurations for co-adsorption of SO2 and CO2 molecules in the Ni(bdc)(ted)0.5 material. (a) Less stable configuration with an SO2⋯CO2 interaction type and a binding energy of −0.33 eV. (b) Most stable configuration with a CO2⋯SO2 interaction type and a binding energy of −0.65 eV and a length of 3.37 Å. C (brown), O (red), H (white), S (yellow), N (blue), and Ni (silver) (reproduced from ref. 72 with permission from The Royal Society of Chemistry). | ||
5. Conclusions and outlook
Concern behind designing better materials for environment harmful gases uptake, e.g. CO2, is growing. MOFs have been widely studied for this aim, due to the ease of modulating the chemical and physical environments of their pores, and therefore, the way they interact with CO2 molecules. In the recent years, a series of studies have reported the post-synthetic modification of MOFs by confining polar and non-polar molecules, achieving a significant improvement of CO2 capture. Herein, we had cited the progress for this new strategy.Water confinement has been studied in meso and microporous MOFs that contain hydroxo bridge groups (μx-OH) in their pores, which form hydrogen bonds with water molecules. These host–guest interactions are responsible for the so-called bottleneck effect, which is defined as a reduction of the pore volume, not allowing the free passage of CO2 flow, and as a result, CO2 molecules can be accommodated more effectively in the cavities.
These results encouraged the study of confining molecules with different polarities. A small quantity of molecules, such as EtOH, MeOH, 2-propanol and DMF (in the range of 2.0–4.2 wt%), were confined in hydroxo-functionalized MOFs ((MFM-300(In), MIL-53(Al) and MIL-53(Al)-TDC)) and a non-hydroxo-functionalized MOF (HKUST-1), showing an improvement of the CO2 capture. Hydrogen bonds between the polar molecules and the hydroxo bridge groups were demonstrated by computational studies. According to these results, the best enhancement was achieved with a 2.7-fold increase with the confinement of EtOH in MFM-300(In). Interestingly, an enhancement of CO2 capture was also found when non-polar molecules, such as toluene and benzene, were confined in the pores. Computational studies suggest that π⋯π stacking-type interactions, weak C–H⋯O hydrogen bonds, and medium strength C–H⋯H–O are responsible for the stronger interaction with CO2 molecules and the bottleneck effect.
Similarly, ILs were confined in the nanocages of ZIF-8 and the CO2 capture was also improved, despite a reduction of the pore volume being observed; the CO2 adsorption mechanism is not well-known. Furthermore, aromatic dye molecules confined in CZJ-10 also improved the CO2 capture because of the additional binding sites provided by the confined molecules.
Clearly, this new strategy of introducing molecules into MOF pores does not only significantly enhance the CO2 adsorption but also avoid the negative effect of water and other solvents in capturing CO2, enabling more real applications of MOFs in industry. Even though significant progress has been made in the last five years, some challenges lie ahead. For example, changing the chemical environment or the topology of the pores by functionalizing MOFs with other functional groups or introducing molecules with a different kinetic diameter or polarity. All these studies could provide a clear understanding of the CO2 adsorption mechanism.
In addition, the early post-modification approach has been shown to have some relevant advantageous/disadvantageous points. For example, the use of any pre-confined molecule (vide supra) represents a more suitable and cheaper strategy to improve the CO2 capture in MOFs than other pre- or post-modifications (i.e., the insertion of polar functional groups or the generation of OMSs), due to the easy adaptation of molecules into the cavities of MOFs and the low cost of organic/inorganic molecules (water, alcohols and amines). Furthermore, as far as we have investigated, the pre-confinement of alcohols can be considered the best option to improve the CO2 capture with an industrial profile, mainly due to three reasons: (i) relevant improvement of CO2 gas adsorption, especially at a low pressure range; (ii) low pre-confined amount of alcohol needed to achieve CO2 enhancement; and (iii) good recyclability, since this type of molecule has been shown to be present in at least ten CO2 adsorption–desorption cycles. However, a clear disadvantage of this methodology is the nature of the molecule to be used, e.g., the nature of water confinement requires the use of MOFs with a high structural stability to water (to avoid structural collapse) and a remarkable hydrophilic character to retain H2O into the cavities. Other concerns regarding this methodology are that the confined molecules should not exceed the minimum range to achieve such improvement (between 1 to 5 wt%, in most cases), and the toxicity of aromatic molecules such as benzene and toluene.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thanks PAPIIT UNAM (IN202820), México for financial support. We thank U. Winnberg (ITAM) and E. Martínez-Ahumada (IIM-UNAM) for scientific discussions and G. Ibarra-Winnberg for scientific encouragement.Notes and references
- K. Anderson and A. Bows, Philos. Trans. R. Soc., A, 2011, 369, 20–44 CrossRef CAS.
- N. Watts, M. Amann, N. Arnell, S. Ayeb-Karlsson and H. Montgomery, Lancet, 2019, 394, 1836–1879 CrossRef.
- B. R. Villanueva, M. B. Salvador and R. G. Huelgas, Rev. Clin. Esp., 2019, 219, 260–265 CrossRef.
- G. Astarita, Ind. Eng. Chem. Fundam., 1963, 2(4), 294–297 CrossRef.
- A. El-Sayed and M. Kamel, Environ. Sci. Pollut. Res., 2020, 1–17 CAS.
- U. S. Environmental Protection Agency, Inventory of U.S. Greenhouse Gas Emissions and Sinks, 2018, https://www.epa.gov/ghgemissions/inventory-us-greenhouse-gas-emissions-and-sinks Search PubMed.
- (a) B. Aghel, S. Sahraie, E. Heidaryan and K. Varmira, Process Saf. Environ. Prot., 2019, 131, 152–159 CrossRef CAS; (b) M. J. Kim, S. W. Choi, H. Kim, S. Mun and K. B. Lee, Chem. Eng. J., 2020, 397, 125404 CrossRef CAS; (c) S. T. Hosseini, H. Raissi and M. Pakdel, New J. Chem., 2020, 44, 7771–7779 RSC; (d) M. Younas, M. Sahail, L. K. Leong, M. J. K. Bashir and S. Sumathi, Int. J. Environ. Sci. Technol., 2016, 13, 1839–1860 CrossRef CAS.
- J. J. Chai, T. C. Johnstone, D. Voicu, P. Mehlman, F. Dielmann, E. Kumacheva and D. W. Stephan, Chem. Sci., 2017, 8, 3270–3275 RSC.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS.
- G. Singh, J. Lee, A. Karakoti, R. Bahadur and J. Yi, Chem. Soc. Rev., 2020, 49, 4360–4404 RSC.
- (a) G. T. Rochelle, Science, 2009, 325, 1652–1654 CrossRef CAS; (b) R. V. Siriwardane, M.-S. Shen, E. P. Fisher and J. Losch, Energy Fuels, 2005, 19(3), 1153–1159 CrossRef CAS; (c) S. Dinda, P. Murge and C. Paruchuri, Bull. Mater. Sci., 2019, 42, 1–9 CrossRef CAS.
- (a) S.-H. Hong, M.-S. Jang, S. J. Cho and W.-S. Ahn, Chem. Commun., 2014, 50, 4927–4930 RSC; (b) F. Su and C. Lu, Energy Environ. Sci., 2012, 5, 9021–9027 RSC.
- (a) C. M. Mfoumou, S. Mignard and T. Belin, Adsorpt. Sci. Technol., 2018, 36, 1246–1259 CrossRef CAS; (b) G. Li, P. Xiao, P. A. Webley, J. Zhang and R. Singh, Energy Procedia, 2009, 1, 1123–1130 CrossRef CAS.
- M. Ding, R. W. Flaig, H.-L. Jiang and O. M. Yaghi, Chem. Soc. Rev., 2019, 48, 2783–2828 RSC.
- (a) Z. Hu, Y. Wang, B. B. Shah and D. Zhao, Adv. Sustainable Syst., 2019, 3, 1800080 CrossRef; (b) J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC; (c) J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112(2), 869–932 CrossRef CAS.
- S. Keskin, T. M. Van Heest and D. S. Sholl, ChemSusChem, 2010, 3, 879–891 CrossRef CAS.
- P. M. Schoenecker, C. G. Carson, H. Jasuja, C. J. J. Flemming and K. S. Walton, Ind. Eng. Chem. Res., 2012, 51, 6513–6519 CrossRef CAS.
- Y. Xiong, Y. Gao, X. Guo, Y. Wang, X. Su and X. Sun, ACS Sustainable Chem. Eng., 2019, 7, 3120–3126 CrossRef CAS.
- D. Banerjee, B. J. Deibert and H. W. J. Li, Encycl. Inorg. Bioinorg. Chem., 2014, 1, 1–21 Search PubMed.
- Y. Belmabkhout, V. Guillerm and M. Eddaoudi, Chem. Eng. J., 2016, 296, 386–397 CrossRef CAS.
- C. G. Piscopo and S. Loebbecke, ChemPlusChem, 2020, 85, 538–547 CrossRef CAS.
- (a) Z. Zhang, Z.-Z. Yao, S. Xiang and B. Chen, Energy Environ. Sci., 2014, 7, 2868–2899 RSC; (b) J.-R. Li, Y. Ma, M. C. McCarthy, J. Sculley, J. Yu, H.-K. Jeong, P. B. Balbuena and H.-C. Zho, Coord. Chem. Rev., 2011, 255, 1791–1823 CrossRef CAS.
- X. Si, C. Jiao, F. Li, J. Zhang, S. Wang, S. Liu, Z. Li, L. Sun, F. Xu, Z. Gabelicad and C. Schick, Energy Environ. Sci., 2011, 4, 4522–4527 RSC.
- (a) M. P. Suh, H. J. Park, T. K. Prasad and D.-W. Lim, Chem. Rev., 2012, 112, 782–835 CrossRef CAS; (b) Y. Harada, Y. Hijikata, S. Kusaka, A. Hori, Y. Ma and R. Matsuda, Dalton Trans., 2019, 48, 2545–2548 RSC; (c) J. N. Hall and P. Bollini, React. Chem. Eng., 2019, 4, 207–222 RSC.
- E. González-Zamora and I. A. Ibarra, Mater. Chem. Front., 2017, 1, 1471–1484 RSC.
- (a) C. Wang, X. Liu, N. K. Demir, J. P. Chen and K. Li, Chem. Soc. Rev., 2016, 45, 5107–5134 RSC; (b) N. C. Burtch, H. Jasuja and K. S. Walton, Chem. Rev., 2014, 114, 10575–10612 CrossRef CAS.
- (a) W. Li, X. Xia, M. Cao and S. Li, J. Mater. Chem. A, 2019, 7, 7470–7479 RSC; (b) M. F. Lange, B. L. Velzen, C. P. Ottevanger, K. J. F. M. Verouden, L.-C. Lin, T. J. H. Vlugt, J. Gascon and F. Kaptein, Langmuir, 2015, 31, 12783–12796 CrossRef; (c) Y. I. Aristov, J. Chem. Eng. Jpn., 2007, 40, 1242–1251 CrossRef.
- A. O. Yazaydın, A. I. Benin, S. A. Faheem, P. Jakubczak, J. J. Low, R. R. Willis and R. Q. Snurr, Chem. Mater., 2009, 21, 1425–1430 CrossRef.
- E. Soubeyrand-Lenoir, C. Vagner, J. W. Yoon, P. Bazin, F. Ragon, Y. K. Hwang, C. Serre, J.-S. Chang and P. L. Llewellyn, J. Am. Chem. Soc., 2012, 134, 10174–10181 CrossRef CAS.
- R. A. Peralta, B. Alcántar-Vázquez, M. Sánchez-Serratos, E. González-Zamora and I. A. Ibarra, Inorg. Chem. Front., 2015, 2, 898–903 RSC.
- J. R. Álvarez, R. A. Peralta, J. Balmaseda, E. González-Zamora and I. A. Ibarra, Inorg. Chem. Front., 2015, 2, 1080–1084 RSC.
- E. Sánchez-González, J. R. Álvarez, R. A. Peralta, A. Campos-Reales-Pineda, A. Tejeda-Cruz, E. Lima, J. Balmaseda, E. González-Zamora and I. A. Ibarra, ACS Omega, 2016, 1, 305–310 CrossRef.
- M. Sánchez-Serratos, P. A. Bayliss, R. A. Peralta, E. González-Zamora, E. Lima and I. A. Ibarra, New J. Chem., 2016, 40, 68–72 RSC.
- G. A. González-Martínez, T. Jurado-Vázquez, D. Solís-Ibarra, B. Vargas, E. Sánchez-González, A. Martínez, R. Vargas, E. González-Zamora and I. A. Ibarra, Dalton Trans., 2018, 47, 9459–9465 RSC.
- V. B. López-Cervantes, E. Sánchez-González, T. Jurado-Vázquez, A. Tejada-Cruz, E. González-Zamora and I. A. Ibarra, Polyhedron, 2018, 155, 163–169 CrossRef.
- M. Sagastuy-Breña, P. G. M. Mileo, E. Sánchez-González, J. E. Reynods III, T. Jurado-Vázquez, J. Balmaseda, E. González-Zamora, S. Devautour-Vinot, S. M. Humphrey, G. Maurin and I. A. Ibarra, Dalton Trans., 2018, 47, 15827–15834 RSC.
- H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choj, E. Choi, A. Ö. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. M. Yagui, Science, 2010, 329, 424–428 CrossRef CAS.
- Y. Chen, Z. Quiao, J. Huang, H. Wu, J. Xiao, Q. Xia, H. Xi, J. Hu, J. Zhou and Z. Li, ACS Appl. Mater. Interfaces, 2018, 10, 38638–38647 CrossRef CAS.
- J. Liu, Y. Wang, A. I. Benin, P. Jakubczak, R. R. Willis and M. D. Le Van, Langmuir, 2010, 26, 14301–14307 CrossRef CAS.
- A. C. Kizzie, A. G. Wong-Foy and A. J. Matzger, Langmuir, 2011, 27, 6368–6373 CrossRef CAS.
- P. Vervoorts, A. Schneemann, I. Hante, J. Pirillo, Y. Hijikata, T. Toyao, K. Kon, K.-I. Shimizu, T. Nakamura, S.-I. Noro and R. A. Fisher, ACS Appl. Mater. Interfaces, 2020, 12(8), 9448–9456 CrossRef CAS.
- G. D. Pirngruberx, L. Hamon, S. Bourrelly, P. L. Llewelly, E. Lenoir, V. Guillerm, C. Serre and T. Devic, ChemSusChem, 2012, 5, 762–776 CrossRef.
- Q. Liu, L. Q. Ning, S. D. Zheng, M. N. Tao, Y. Shi and Y. He, Sci. Rep., 2013, 3, 1–6 Search PubMed.
- S.-I. Noro, R. Matsuda, Y. Hijikata, Y. Inubashi, S. Takeda, S. Kitagawa, Y. Takahashi, M. Yoshitake, K. Kubo and T. Nakamura, ChemPlusChem, 2015, 80, 1517–1524 CrossRef CAS.
- Y. Liu, J. Liu, Y. S. Lin and M. Chang, J. Phys. Chem. C, 2014, 118, 6744–6751 CrossRef CAS.
- D. Alvarado-Alvarado, J. H. González-Estefan, J. Gabriel-Flores, J. R. Álvarez, J. Aguilar-Pliego, A. Islas-Jácome, G. Chastanet, E. González-Zamora, H. A. Lara-García, B. Alcántar-Vázquez, M. Gonidec and I. A. Ibarra, Organometallics, 2020, 39(7), 949–955 CrossRef CAS.
- R. A. Peralta, A. Campos-Reales-Pineda, H. Pfeiffer, J. R. Álvarez, J. A. Zárate, J. Balmaseda, E. González-Zamora, A. Martínez, D. Martínez-Ortero, V. Jancik and I. A. Ibarra, Chem. Commun., 2016, 52, 10273–10276 RSC.
- E. Sánchez-González, P. G. M. Mileo, J. R. Álvarez, E. González-Zamora, G. Maurin and I. A. Ibarra, Dalton Trans., 2017, 46, 15208–15215 RSC.
- J. R. Álvarez, P. G. M. Mileo, E. Sánchez-González, J. A. Zárate, J. Rodríguez-Hernández, E. González-Zamora, G. Maurin and I. A. Ibarra, J. Phys. Chem. C, 2018, 122(10), 5566–5577 CrossRef.
- J. E. Sánchez-Bautista, B. Landeros-Rivera, T. Jurado-Vázquez, A. Martínez, E. González-Zamora, J. Balmaseda, R. Vargas and I. A. Ibarra, Dalton Trans., 2019, 48, 5176–5182 RSC.
- J. R. Álvarez, E. Sánchez-González, E. Pérez, E. Schneider-Revueltas, A. Martínez, A. Tejeda-Cruz, A. Islas-Jácome, E. González-Zamora and I. A. Ibarra, Dalton Trans., 2017, 46, 9192–9200 RSC.
- J. A. González-Martínez, J. A. Zárate, A. Martínez, E. Sánchez-González, J. R. Álvarez, E. Lima, E. González-Zamora and I. A. Ibarra, RSC Adv., 2017, 7, 24833–24840 RSC.
- E. Sánchez-González, J. G. Flores, J. C. Flores-Reyes, I. Morales-Salazar, R. E. Blanco-Carapia, M. A. Rincón-Guevara, A. Islas-Jácome, E. González-Zamora, J. Aguilar-Pliego and I. A. Ibarra, Materials, 2020, 13, 1840 CrossRef.
- H. A. Lara-García, B. Landeros-Rivera, E. González-Zamora, J. Aguilar-Pliego, A. Gómez-Cortés, A. Martínez, R. Vargas, G. Díaz and I. A. Ibarra, Dalton Trans., 2019, 48, 8611–8616 RSC.
- P. V. D. Voort, K. Leus, Y.-Y. Liu, M. Vandichel, V. V. Speybrock, M. Waroquier and S. Biswas, New J. Chem., 2014, 38, 1853–1867 RSC.
- C. Jia, H. Li, B. Liu, Z. Qiao, Z. Zhang, C. Sun, L. Yang and G. Chen, Ind. Eng. Chem. Res., 2018, 57(37), 12494–12501 CrossRef CAS.
- A. Demessence, D. M. D'Alessandro, M. Lin-Foo and J. R. Long, J. Am. Chem. Soc., 2009, 131(25), 8784–8786 CrossRef CAS.
- S. Choi, T. Watanabe, T. Bae, D. S. Sholi and C. W. Jones, J. Phys. Chem. Lett., 2012, 3, 1136–1141 CrossRef CAS.
- N. Wang, A. Mundstock, Y. Liu, A. Huang and J. Caro, Chem. Eng. Sci., 2015, 124, 27–36 CrossRef CAS.
- F. Martínez, R. Sanz, G. Orcajo, D. Briones and V. Yángüez, Chem. Eng. Sci., 2016, 142, 56–61 CrossRef.
- X. Su, L. Bromberg, V. Martis, F. Simeon, A. Huq and T. A. Hatton, ACS Appl. Mater. Interfaces, 2017, 9, 11299–11306 CrossRef CAS.
- N. Vrtovec, M. Mazaj, G. Buscarino, A. Terracina, S. Agnello, I. Arčon, J. Kovač and N. Z. Logar, Cryst. Growth Des., 2020, 20, 5455–5465 CrossRef CAS.
- V. Irani, A. Tavasoli, A. Maleki and M. Vahidi, Int. J. Hydrogen Energy, 2018, 43, 5610–5619 CrossRef CAS.
- (a) H. Molavi, F. A. Joukani and A. Shojaei, Ind. Eng. Chem. Res., 2018, 57, 7030–7039 CrossRef CAS; (b) H. Li, K. Wang, D. Feng, Y.-P. Chen, W. Verdeegal and H.-C. Zhou, ChemSusChem, 2016, 9, 1–10 CrossRef.
- (a) Z. Akimbekov, D. Wu, C. Brozek, M. Dinca and A. Navrotsky, Phys. Chem. Chem. Phys., 2016, 18, 1158–1162 RSC; (b) P. A. Konik, E. A. Berdonosova, I. M. Savvotin and S. N. Klyamkin, Microporous Mesoporous Mater., 2019, 277, 132–135 CrossRef CAS.
- X. Jiang, H.-B. Duan, S. I. Khan and M. A. Garcia-Garibay, ACS Cent. Sci., 2016, 2(9), 608–613 CrossRef CAS.
- M. Usman, P.-H. Feng, K.-R. Chiou, J.-W. Chen, L.-W. Lee, Y.-H. Liu and K.-L. Lu, ACS Appl. Electron. Mater., 2019, 1, 836–844 CrossRef CAS.
- E. Sánchez-González, E. González-Zamora, D. Martínez-Otero, V. Jancik and I. A. Ibarra, Inorg. Chem., 2017, 56(10), 5863–5872 CrossRef.
- L. P. Garrido-Olvera, J. E. Sánchez-Bautista, D. Alvarado-Alvarado, B. Laderos-Rivera, J. Raziel-Álvarez, R. Vargas, E. González-Zamora, J. Balmaseda, H. A. Lara-García, A. Martínez and I. A. Ibarra, RSC Adv., 2019, 9, 32864–32872 RSC.
- L. J. Barrios-Vargas, J. G. Ruíz-Montoya, B. Landeros-Rivera, J. Raziel-Álvarez, D. Alvarado-Alvarado, R. Vargas, A. Martínez, E. González-Zamora, L. M. Cáceres, J. C. Morales and I. A. Ibarra, Dalton Trans., 2020, 49, 2786–2793 RSC.
- (a) Y. Ban, Z. Li, Y. Li, Y. Peng, H. Jian, W. Jiao, A. Guo, P. Wang, Q. Yang, C. Zhong and W. Yang, Angew. Chem., Int. Ed., 2015, 54, 15483–15487 CrossRef CAS; (b) M. Zhao, S. Ou and C.-D. Wu, Cryst. Growth Des., 2017, 17, 2688–2693 CrossRef CAS.
- D. N. Son, T. T. T. Huong and V. Chihaia, RSC Adv., 2018, 8, 38648–38655 RSC.
- (a) C. F. Matta, J. Hernández-Trujillo, T. H. Tang and R. F. Bader, Chem. – Eur. J., 2003, 9, 1940–1951 CrossRef CAS; (b) J. D. Dunitz and A. Gavezzotti, Angew. Chem., Int. Ed., 2005, 44, 1766–1787 CrossRef CAS.
- K. V. Kumar and F. Rodríguez-Reinoso, Nanotechnology, 2012, 24, 035401 CrossRef.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |