
Metal-based catalysts for the non-oxidative dehydrogenation of light alkanes to light olefins
Sibao
Liu†
,
Bofeng
Zhang†
and
Guozhu
Liu
 *
*
Key Laboratory for Green Chemical Technology of Ministry of Education, School of Chemical Engineering and Technology, Tianjin University, Tianjin 300072, China. E-mail: gliu@tju.edu.cn
First published on 28th October 2020
Abstract
Catalytic non-oxidative dehydrogenation of light alkanes is one of the most promising routes for the production of value-added olefins because of high selectivity to the target alkenes and increased supply of natural and shale gas. In this review, we emphasize the recent advances in the development of metal-based catalysts for the direct dehydrogenation of light alkanes to olefins. Metal-based catalysts including Pt-, Pd-, Rh- and Ni-based catalysts, particularly Pt-based bimetallic catalysts, are summarized and exemplified. A detailed discussion of the structure–performance correlations of these catalysts is presented. We systematically discussed the geometric and electronic effects of the promoters on these metal-based catalysts. Moreover, we highlighted recent advanced strategies for improving the dehydrogenation performance and the stability of metal-based catalysts. In light of the existing contributions, a perspective was provided to evaluate some of the potential avenues into future research. We hope that this review will inspire the rational design of metal-based catalysts to realize efficient direct dehydrogenation of light alkanes for olefins production.
1. Introduction
Light olefins, such as ethylene, propylene and butene, are bulk commodities and platform building blocks for the production of value-adding polymers, oxygenates and important chemical intermediates.1–8 The demand for these useful olefins has increased rapidly.9–11 Currently, steam cracking of crude-oil-derived naphtha and fluid catalytic cracking of heavy oil are the main technologies for the production of light olefins.12 However, these two existing routes suffer from high-energy demands, enormous CO2 emissions and low selectivity towards light olefins. In addition, these routes right now cannot produce sufficient light olefins to satisfy the increasing market demand, which motivates an interest in seeking other means for producing light olefins. Catalytic non-oxidative dehydrogenation of light alkanes including ethane, propane and butane offers an alternative, practical and environmentally friendly approach for the production of light olefins,13 because cheap light alkanes separated from abundant shale gas or natural gas can be readily used as the feedstock in this process, and high olefin selectivity can be obtained.7The dehydrogenation of light alkanes involves the breaking of two carbon–hydrogen bonds with simultaneous release of a corresponding alkene together with a molecular hydrogen. This reaction is highly endothermic and typically operates at high temperature (400–750 °C), which makes it difficult to control the selectivity to the target olefins. Metal-based catalysts, Pt-based catalysts in particular, have been widely used for alkane dehydrogenation due to their excellent activity and high selectivity to olefins.5,7,14 The addition of a promoter to metal-based catalysts has been shown to enhance the catalytic performance. Tin (Sn) is the most frequently used modifier, which might be due to its positive effects on the selectivity towards the target alkene products and suppression of catalytic deactivation. The first industrial alkane dehydrogenation technology (the Pacol process) on the basis of an alumina-supported platinum catalyst was commercialized by UOP for producing linear alkenes for detergents in 1968.15 Later, an alkalized-Al2O3-supported Pt–Sn catalyst was commercialized by UOP and utilized in their Oleflex processes to produce light olefins.1,7 In addition, Pt–Sn bimetallic-based catalysts are also employed in two other industrial processes, the STAR process and the Linde–BASF process, for the direct dehydrogenation of light alkanes.1,7
The catalytic properties of metal-based catalysts strongly depend on their structures.14 In general, all the surface-accessible metal atoms can serve as the active sites for catalyzing the dehydrogenation of light alkanes, and thus the dehydrogenation is structure insensitive. In striking contrast, the accompanying side reactions, such as hydrogenolysis, isomerization and coke formation, are significantly affected by the structure of metal particles as well as the acid sites on the support. The deactivation of metal-based catalysts is primarily due to coke deposition and metal sintering. Therefore, careful catalyst design that ensures high metal dispersity, suppresses the surface sites responsible for hydrogenolysis, over-dehydrogenation and coking, and stabilizes the metal particles is the key to develop efficient catalysts for dehydrogenation.
Oxidative dehydrogenation of light alkanes is another promising route for the production of olefins. Several interesting reviews have already been published in this field.2–4,16,17 Compared with non-oxidative dehydrogenation, it suffers from deep oxidation of light alkanes and alkenes to COx, which results in loss of olefin selectivity and yield. In this regard, this review will focus on the non-oxidative dehydrogenation of light alkanes. We summarize the most recent and impactful progress in understanding the metal-catalyzed non-oxidative dehydrogenation of light alkanes, with a primary focus on Pt-, Pd-, Rh- and Ni-based catalysts. The structure–performance correlations of these catalysts are discussed. The roles of promoter addition are addressed. Some of the recent strategies, such as support engineering, advanced preparation methods and catalyst structural innovation to improve the dehydrogenation performance and the catalyst stability are also highlighted. It is noted that the dehydrogenation of cyclic alkanes and long-chain alkanes for the production of pure hydrogen and detergents, respectively, are also important topics in catalysis and the chemical industry. Several literature reviews dealing with these topics have been published.15,18–20 However, these topics are out of the scope of this review and will not be addressed here.
2. The reaction
The dehydrogenation of light alkanes (eqn (1)–(7)) is thermodynamically limited, strongly endothermic and prone to volume expansion. Consequently, a high reaction temperature (500–700 °C) and/or low alkane partial pressure are required to achieve economically relevant conversions of 30–50%. Under a certain condition, the dehydrogenation equilibrium conversions for the light alkanes increase with the carbon numbers and the degrees of branching (C2 < C3 < C4 < i-C4).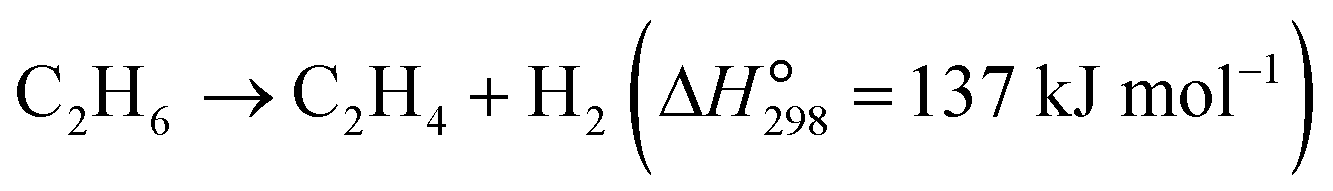 | (1) |
 | (2) |
 | (3) |
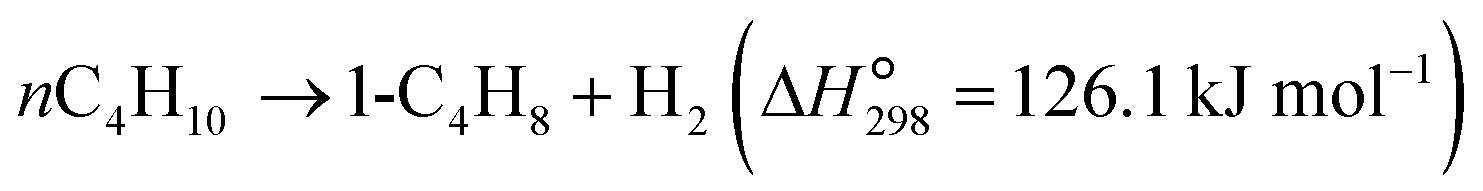 | (4) |
 | (5) |
 | (6) |
 | (7) |
Co-feeding hydrogen with light alkanes is frequently employed in the reaction systems. According to Le Chatelier's principle, the equilibrium conversion of light alkanes would decrease with co-feeding hydrogen. However, Bell et al. claimed that the addition of an optimal amount of hydrogen to the feed can enhance the formation of olefins by promoting the dehydrogenation of adsorbed alkyl species on the catalyst surface under kinetic conditions.21,22 In addition, DFT calculations revealed that co-feeding hydrogen decreases the propylene adsorption strength while it increases the energy barriers for further dehydrogenation of propylene.23 This could provide more free sites for the dehydrogenation and thus induce higher catalytic activity. In addition, co-feeding hydrogen can suppress coke formation through inhibiting the formation of coke precursors.
In general, the C–H bonds of light alkanes and olefins are more reactive than C–C bonds and a catalyst that possesses a higher activity of C–H scission over C–C cleavage is desirable. On the other hand, when the alkane molecules are effectively activated, the formed intermediates and the olefins are much more reactive than the alkanes and can easily undergo undesirable side reactions such as deep dehydrogenation, cracking, isomerization, and polymerization. These side reactions may result in low selectivity to alkenes and coke formation, which is a reason for catalyst deactivation. Fortunately, the active sites for the side reactions are not required for dehydrogenation. This makes it possible to potentially eliminate side reactions and exclusively enable C–H bond activation via rational catalyst design.
3. The catalysts
3.1 Pt-based catalysts
Pt is the most commonly utilized noble metal for the direct dehydrogenation of light alkanes, thanks to its facile C–H bond activation over C–C bond scission. While all surface-accessible Pt atoms can facilitate the dehydrogenation reaction in virtually the same manner, Pt–Pt ensembles with large enough domain sizes are active for side reactions (i.e. cracking, over-dehydrogenation, etc.). As a result, substantial effort has gone into the modification of the Pt surface geometric/electronic structures using metal/non-metal promoters, with bimetallic alloying being the predominate candidate developed in past decades. Table 1 lists the recently reported Pt-based catalysts for the non-oxidative dehydrogenation of propane.| Catalysts | Metal loading (%) | Temp (°C) | WHSV (h−1) | Feed composition | Conversion (%) | Selectivity (%) | Operation time (h) | Specific activitya (s−1) | Deactivation constantb (h−1) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
a Specific activity is defined as the moles of C3H6 formation per mole Pt atoms per second.
b
 , where ConFinal and ConInitial are the final and initial conversions of the PDH reaction, respectively; t is the duration of a PDH reaction in hours; and kd is the deactivation constant (h−1). The higher kd is, the faster the inactivation rate will be. , where ConFinal and ConInitial are the final and initial conversions of the PDH reaction, respectively; t is the duration of a PDH reaction in hours; and kd is the deactivation constant (h−1). The higher kd is, the faster the inactivation rate will be.
|
||||||||||
| PtSn/Al2O3-sheet | 0.36 | 590 | 9.4 | C3H8/H2/N2 = 1/1.25/4 | 48.7–44.6 | 99.1 | 24 | 1.58 | 0.007 | 48 |
| Pt–Sn/γ-Al2O3 | 0.5 | 590 | 3.0 | C3H8/H2 = 4/1 | 29.4–22.7 | 76 | 6 | 0.21 | 0.058 | 41 |
| 0.5Pt1.5Sn/Al2O3 | 0.5 | 600 | 3.2 | Pure C3H8 | 35.6 | 88.5 | 6 | 0.248 | — | 34 |
| 0.5Pt0.9Sn/Al2O3 | 0.9 | 590 | 5.2 | C3H8/H2/He = 1/1.25/4 | 48.5–42.5 | 97 | 20 | 0.609 | 0.012 | 49 |
| Pt–Sn/θ-Al2O3 | 0.5 | 550 | 11.8 | C3H8/H2/He = 1/1/8 | 41.1–37.6 | 97.7–98.3 | 72 | 1.14 | 0.002 | 36 |
| 0.3Pt–0.2Sn–0.5K/θ-Al2O3 | 0.3 | 600 | 4 | C3H8/H2 = 1/0.5 | 39.9–38.2 | 92.0–95.5 | 25 | 0.602 | 0.0029 | 52 |
| Pt–Sn/mesoporous Al2O3 | 0.5 | 590 | 3.0 | C3H8/H2 = 4/1 | 29.8–24.6 | 92 | 6 | 0.22 | 0.044 | 41 |
| Pt/Mg(Sn)(Al)O@Al2O3 | 0.5 | 600 | 14 | C3H8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2:Ar = 1:0.5:2 :H2:Ar = 1:0.5:2 |
48.3–43.0 | 86.4–98.1 | 47 | 1.46 | 0.0045 | 46 |
| PtSn/Mg(Al)O | 0.3 | 550 | 1.13 | Pure C3H8 | 27.9–27.7 | 95.6–96.5 | 5 | 0.12 | 0.0015 | 55 |
| Pt–Sn/MgAl2O4 | 0.39 | 580 | 2.2 | C3H8/H2/He = 1/1/8 | 45.0–38.9 | 99.5 | 90 | 0.34 | 0.0028 | 145 |
| Pt–Sn/CeO2 | 1 | 680 | 2.159 | C3H8/He = 2/10 | 39.5 | 84.5 | — | 0.0887 | — | 66 |
| K–PtSn@MFI | 0.4 | 600 | 29.5 | C3H8/He = 24/76 | 38.7–31.9 | >97 | 25 | 3.5 | 0.012 | 147 |
| K–PtSn@MFI | 0.42 | 600 | 1.7 | C3H8/N2 = 5/16 | 71–42 | 88–95 | 65 | 0.311 | 0.019 | 146 |
| Pt–Sn/ZSM-5 | 0.5 | 590 | 3.0 | C3H8/H2 = 4/1 | 33.1–26.3 | 47.7 | 6 | 0.24 | 0.054 | 41 |
| Pt/Sn-ZSM-5 | 0.3 | 600 | 5.2 | C3H8/N2 = 1/5.5 | 68–43 | 95.5–99.7 | 25 | 1.36 | 0.041 | 149 |
| Pt–Na/Sn-ZSM-5 | 0.5 | 590 | 3 | C3H8/H2 = 4/1 | 41.7–39.0 | 95.3–93.8 | 9 | 0.294 | 0.012 | 59 |
| 0.3Pt/0.5Sn–Si-Beta | 0.3 | 550 | 1 | C3H8/N2 = 5:95 |
27.5–25.2 | 99.1–99.9 | 24 | 0.13 | 0.007 | 62 |
| Pt/Sn2.00-Beta | 0.5 | 550 | 140 | C3H8/H2/He = 1/1/8 | 51.1–46.1 | 95.4–98.6 | 24 | 16.45 | 0.0083 | 58 |
| PtSnNa/Ce-ZSM-5 | 0.5 | 590 | 3 | C3H8/H2 = 4/1 | 43.5–40.7 | 84.5–96.9 | 6.92 | 0.291 | 0.017 | 63 |
| PtSnNa@SUZ-4 | 0.5 | 590 | 3 | C3H8/H2 = 1/3 | 24.7–20.2 | 90–94.7 | 9 | 0.164 | 0.029 | 65 |
| 0.5Pt2Sn–1Na/SUZ-4 | 0.5 | 590 | 3 | C3H8/H2 = 1/3 | 24.6–20.6 | 81.3–89.7 | 10 | 0.14 | 0.0229 | 64 |
| PtSn/TS-1 | 0.5 | 590 | 3 | C3H8/H2/N2 = 1/1/4 | 53.5–47.7 | 92.5 | 7 | 0.40 | 0.033 | 61 |
| Pt–Sn/SBA-15 | 0.5 | 590 | 3.0 | C3H8/H2 = 4/1 | 11.0–6.0 | 80 | 6 | 0.08 | 0.110 | 41 |
| Pt/0.5Sn-SBA-15 | 1 | 580 | 8.25 | C3H8/Ar = 7/3 | 43.8–38.2 | 97.2–97.3 | 6 | 0.42 | 0.0386 | 57 |
| PtZn4@S-1-H | 0.72 | 550 | 3.6 | C3H8/N2 = 1/3 | 47.7–40.4 | 93.2–99.2 | 216.7 | 0.25 | 0.001 | 74 |
| PtZn4@S-1-H | 0.72 | 550 | 108 | C3H8/N2 = 1/3 | 21.2–11.8 | 98.3–98.6 | 10.33 | 3.55 | 0.072 | 74 |
| 0.3Pt0.5Zn@S-1 | 0.3 | 550 | 6.5 | C3H8/N2 = 11/19 | 45–42 | 99.0–99.9 | 60 | 1.53 | 0.002 | 75 |
| Pt0Znδ+/SiO2 | 3 | 550 | 32 | C3H8/Ar = 1/4 | 35.3–26.6 | 97.6–96.3 | 30 | 0.45 | 0.014 | 77 |
| Pt0Znδ+/SiO2 | 3 | 550 | 75 | C3H8/Ar = 1/4 | 30.2–16.1 | 98.1–95.0 | 30 | 0.91 | 0.027 | 77 |
| PtNa/Zn-ZSM-5 | 0.5 | 590 | 3 | C3H8/H2 = 4/1 | 40.6–37.6 | 93–97 | 10 | 0.279 | 0.013 | 70 |
| PtGa–Pb/SiO2 | 3 | 600 | 30.7 | C3H8/H2/He = 3.9/5/40 | 30.0–28.4 | 99.6 | 96 | 0.38 | 0.001 | 88 |
| Pt/HT-10Ga | 3 | 600 | 8.8 | C3H8/He = 1/4 | 8–6 | 99 | 1.83 | 0.257 | 0.169 | 85 |
| Gaδ+Pt0/SiO2 | 1.55 | 550 | 98.3 | C3H8/Ar = 1:4 |
31.9–18.2 | 99 | 20 | 1.36 | 0.038 | 87 |
| Gaδ+Pt0/SiO2 | 1.55 | 550 | 43.3 | C3H8/Ar = 1:4 |
36.5–26.9 | 90.9 | 20 | 0.76 | 0.023 | 87 |
| Pt/Mg(Ga)(Al)O | 0.9 | 600 | 2.6 | C3H8/H2/He = 20/25/55 | 16–11.4 | 99.2 | 2 | 0.056 | 0.196 | 84 |
| PtGa/CeO2–Al2O3 | 1 | 600 | 10 | C3H8/H2/Ar = 26/26/48 | 41.1–32.2 | 99.6 | 14.17 | 0.504 | 0.026 | 82 |
| Pt/Mg(In)(Al)O | 0.7 | 600 | 2.6 | C3H8/H2/He = 20/25/55 | 20.4–16.3 | 98 | 2 | 0.183 | 0.14 | 90 |
| Pt3In/SiO2 | 0.3 | 600 | 3 | C3H8/H2/N2 = 14/14/72 | 17.5–16.5 | 96–97.5 | 2 | 0.207 | 0.073 | 93 |
| PtIn/Mg(Al)O-600 | 0.6 | 620 | 3.3 | C3H8/H2/Ar = 8/7/35 | 69–50 | 98 | 41.0 | 0.45 | 0.027 | 96 |
| 0.1Pt10Cu/Al2O3 | 0.1 | 550 | 4 | C3H8/H2/H2 = 8/8/34 | 24.1–21.6 | 88–90 | 11.67 | 0.95 | 0.012 | 99 |
| Pt–Cu/MgAl2O4 | 1 | 590 | 6.8 | C3H8/H2/He = 19/19/2 | 25.7–21.0 | 87.5 | 24 | 0.24 | 0.011 | 98 |
| PtFe@Pt/SBA-15 | 0.75 | 600 | 3.48 | C3H8/H2/N2 = 26/26/48 | 35–31 | 85–87 | 1.33 | 0.170 | 0.136 | 106 |
| Pt/0.5Mn-DMSN | 0.5 | 590 | 2.4 | C3H8/N2 = 1/2 | 56.1–40.1 | 95.6–99.3 | 100 | 0.31 | 0.0065 | 104 |
| Pt–Ag/MgAl2O4 | 1 | 590 | 6.8 | C3H8/H2/He = 19/19/2 | 30.6–16.7 | 95.1 | 24 | 0.28 | 0.034 | 98 |
| Pt–Au/MgAl2O4 | 1 | 590 | 6.8 | C3H8/H2/He = 19/19/2 | 33.7–16.6 | 60 | 24 | 0.31 | 0.040 | 98 |
| Pt/ND@G | 0.5 | 600 | 1.62 | C3H8/N2 = 1/19 | — | 88 | — | 0.064 | — | 31 |
| Pt/TiO2–Al2O3 | 1 | 600 | 10 | C3H8/H2/N2 = 26/26/48 | 43.7–25.9 | 77.5–89.5 | 9.67 | 0.45 | 0.0976 | 29 |
A typical catalyst for light alkane dehydrogenation is Pt/Al2O3.7 However, certain characteristics, such as electron-deficient Pt particles, weak interaction between Pt and Al2O3, and strong acidity of Al2O3 could lead to low selectivity to olefins, sintering and coking.24 Therefore, engineering of the supports is implemented in the development of high-performance Pt monometallic catalysts to improve the metal–support interaction and induce the electronic effects. Studies on support innovation have been focused on TiO2–Al2O3,29 zeolites,30 and nanocarbon materials,31–33 especially a nanocarbon (ND@G) composed of a nanodiamond core and a defective, ultrathin graphene nanoshell.31 Up to 90% alkene selectivity and enhanced catalyst stability have been reported on these catalysts with novel supports.29–33
3.1.2.1 Pt–Sn catalysts. Tin (Sn) is the most commonly used promoter of Pt-based bimetallic catalysts for light alkane dehydrogenation. All the known platinum-based catalysts that have been used on an industrial scale or a pilot scale contain Sn as a modifier.7 The addition of Sn greatly improves the selectivity towards target alkene products and slows down catalytic deactivation.7 The promotional role of Sn in Pt-based catalysts has been ascribed to both geometric and electronic effects, which will be discussed in the following sections. In addition, the presence of Sn atoms plays a positive effect on the regeneration of deactivated Pt–Sn/Al2O3 catalysts by providing nucleation sites for mobilized Pt atoms during oxidative regeneration.34 The catalyst structure, dispersion and the formation of an active phase significantly depend on the preparation methods,35,36 pretreatment conditions,37–40 support type41,42 and Sn content.28,43 The extent of alloy formation and the location of Sn in the catalysts have a great impact on the catalytic performances.28,39 Upon reduction, several alloy phases such as PtSn, Pt3Sn, Pt2Sn3, and PtSn4 alloys could be formed and the Pt3Sn alloy is widely recognized as the most active one for the dehydrogenation.44 In some cases, Sn could serve as a partition agent45 or a stabilizer for Pt clusters46 but does not alloy with Pt. A number of supports including Al2O3,47–49 alkalized Al2O3,50–52 rare-earth-oxide doped Al2O3,53 Mg(Al)O hydrotalcite,21,28,46,54,55 MgAl2O4 spinel,36 ZnAl2O4,7,56 SiO2,39,57 zeolites,58–65 CeO266 and carbon materials45 have been employed for light alkane dehydrogenation. Among them, alkalized θ-Al2O3, ZnAl2O4 and ZrO2 were used in the UOP, Uhde and BASF/Linde dehydrogenation processes, respectively.7 Novel supports, such as Al2O3 nanosheets,48 hydrotalcites,46 CeO2,66 siliceous zeolites58,62 and carbon materials, have been developed and engineered to improve the dispersion and provide specific sites for anchoring Pt or Pt–Sn clusters in recent years. The obtained Pt–Sn catalysts with engineered supports showed remarkable catalytic performances, particularly in terms of stability.
3.1.2.2 Pt–Zn catalysts. Zinc has also been considered to be an efficient promoter in Pt-based bimetallic catalysts for light alkane dehydrogenation to olefins. After reduction with hydrogen, a Pt–Zn alloy would be formed and brings about remarkable changes in the structural, geometric, and electronic properties of Pt catalysts. In addition, the unalloyed Zn species on the support, such as framework Zn in the zeolites or single site Zn on the silica support, play an essential role in the stabilization of the active Pt–Zn alloy. Consequently, the catalytic performances could be significantly improved.
In some of the pioneer work, workers at British Petroleum (BP) firstly disclose a series of catalysts composed of Pt on a zincosilicate molecular sieve support with MFI and MEL structures for the dehydrogenation of ethane, propane and isobutane.67 These catalysts possess a high dispersion of active sites, which might be Pt–Zn alloy, and give high activity, selectivity and coke resistance for light alkane dehydrogenation. Remarkably, the activity of the catalyst can be maintained even after 796 h time-on-stream. After that, a number of Pt–Zn alloys supported on various zeolites, including CIT-6,68 HZSM-5,69,70 Na-ZSM-5,70,71 Na-Beta,71 Na-mordenite,71 Na-MCM-22,72 Na–Y,71 KL71,73 and silicate-1 (ref. 74 and 75) zeolites, were reported. In some cases, alkali metal ions including K+ and Na+ were added to poison the Brønsted acid sites on zeolites in order to avoid cracking, oligomerization and coke formation.72 The addition of Zn can effectively increase the Pt dispersion.70 The selectivity to propene with silica–aluminum zeolites is not as high as that with silicious zeolites, apparently due to the adverse effects of the acidity on silica–aluminum zeolites.69–75 Remarkably, the Pt–Zn bimetallic particles encaged in silicate-1 zeolites exhibit extraordinary catalytic stability.74,75 Other supports, such as SiO2,76–78 Mg(Zn)AlOx hydrotalcites79 and titanate ETS-2,80 are also investigated in a number of studies. The Pt–Zn/SiO2 catalysts prepared by advanced preparation methods, such as atomic layer deposition (ALD)78 and surface organometallic chemistry (SOMC),77 showed excellent performance, particularly in terms of productivity.
3.1.2.3 Pt–Ga catalysts. Gallium is another frequently used promoter in Pt-based bimetallic catalysts for light alkane dehydrogenation to alkenes. Upon reduction at high temperatures (e.g., 500 °C), Pt–Ga alloys with variable compositions were formed. The introduction of Ga results in a remarkable enhancement of selectivity toward the desired products and catalytic stability, which is also attributed to the combination of geometric and electronic effects.
Scelza et al. used a series of Pt–Ga/Al2O3 catalysts for direct propane dehydrogenation.81 With the introduction of Ga, the selectivity to propene was enhanced and the catalyst deactivation and the carbon formation are suppressed. Given that the addition of Ga slightly modified the acidity of the Al2O3 support, they proposed that the geometric and electronic effects of the Pt–Ga alloy are the main reasons for the enhanced catalytic performance. Catalyst stability can be further improved by using CeO2–Al2O3 as a support.82 In this case, the incorporation of Ga3+ cations in the cubic fluorite structure of CeO2 increases both lattice oxygen storage capacity and surface oxygen mobility and thus eliminates coke deposition. Bell and his co-workers investigated Pt/Mg(Ga)(Al)O catalysts with various Ga/Pt molar ratios for ethane and propane dehydrogenation.22,83,84 They observed the formation of Pt–Ga alloyed nanoparticles at high reduction temperatures (>500 °C). The catalytic activity and selectivity towards olefins were dramatically enhanced with the addition of Ga. The selectivity could reach up to 100% at Ga/Pt = 5.4. The activity exhibited a volcano correlation to the Ga content with a maximum rate obtained at Ga/Pt = 1.4–5.4, presumably due to the dilution of the Pt ensemble by Ga. Further, Marin et al. found that Pt–Ga catalysts showed reversible alloy/segregation looping when exposed to a redox cycle alternating between 5%H2/He and 20%O2/N2 at 600 °C.85,86 This reversibility ensures catalyst stability during the reaction/regeneration process. Recently, a series of well-defined Pt–Ga/SiO2,87 PtGa–Pb/SiO2,88 and Ga37Pt/Al2O3 (ref. 89) catalysts have been carefully prepared. All the catalysts showed remarkable productivity and stability, which is primarily due to the high dispersion and specific structure.
3.1.2.4 Pt–In catalysts. Indium is a promising candidate to promote Pt-based catalysts for the dehydrogenation of light alkanes. Compared with Pt–Sn and Pt–Ga catalysts, Pt–In catalysts were found to be more homogeneous in composition, leading to a less sensitive performance to the preparation methods.90,91 Upon reduction, Pt and In can form seven intermetallic alloys, including Pt3In, Pt2In, Pt13In9, PtIn, Pt2In3, PtIn2 and Pt3In7, the relative shares of which depend in part on the global In content.92 DFT calculations indicate that Pt3In has a comparable activity to that of pure Pt and Pt3Sn, while it can achieve higher selectivity towards propene than that of pure Pt.93 Miller and his co-workers prepared a couple of silica-supported Pt–In intermetallic catalysts for ethane dehydrogenation.92 They proposed that the Pt3In phase is more active than the PtIn2 phase. Both phases showed higher intrinsic activity, selectivity to ethene (nearly 100%) and catalyst stability than those over monometallic Pt, which is due to the geometric and electronic effects of the Pt–In alloy.92
Bell et al. applied a series of Pt/Mg(In)(Al)O catalysts in light alkane dehydrogenation.90,94 They observed the formation of a Pt–In alloy with reduction in H2 at temperatures above 400 °C. Furthermore, the content of In in the Pt–In alloy particles increases as the reduction temperature and bulk In/Pt molar ratio increase. X-ray absorption near edge structure (XANES) showed an increasing electron transfer from In to Pt with increasing In content in the Pt–In alloy, which ensures enhancement of catalytic performances. When the In/Pt ratio is 0.48, the Pt/Mg(In)(Al)O catalyst exhibited the highest activity for ethane and propane dehydrogenation, with an alkene selectivity approaching 100%.90 The optimal ratio of In/Pt for n-butane dehydrogenation was between 0.33 and 0.88.94 Compared with Pt/Mg(Sn)(Al)O and Pt/Mg(Ga)(Al)O catalysts, Pt/Mg(In)(Al)O exhibits the best performance for light alkane dehydrogenation in terms of activity and catalyst stability.90 In addition, Guo et al. found that the Mg/Al molar ratio and calcination temperature significantly affect the structure and catalytic performance of Pt–In/Mg(Al)O catalysts.95–97 With the optimal Mg/Al molar ratio (4:1) and calcination temperature (600 °C), the catalysts showed the lowest fraction of strong acid, largest surface area and highest dispersion of metal particles, which result in the best catalytic performance. However, the exact structure of the Pt–In alloy was not determined.
3.1.2.5 Other Pt-based bimetallic catalysts. Besides Sn, Zn, Ga and In as promoters, Cu,98–102 Mn,103,104 Fe,105,106 Co,106,107 Ni,106,108 Ag,98,99 Ge,109,110 Cr,111 Bi,112 Sb,113 Ce,114 Y (ref. 114) and La (ref. 114) have been found to be good promoters for Pt-based catalysts in recent years. With the addition of these metals, the corresponding bimetallic alloys can be formed with various structures, such as random alloys, single-atom alloys (SAAs) and intermetallic compounds. Carefully controlling the surface and subsurface compositions can tune the catalytic performance. The geometric and electronic features of these alloys are responsible for the boosted catalytic performance.
Jung et al.102 and Gong et al.101 investigated Cu-modified Pt/Al2O3 for n-butane dehydrogenation and PDH, respectively. Copper modification enhanced alkene selectivity as well as catalyst stability. They attribute the improved performance to the electronic effects of the Pt–Cu alloy, which weakened the adsorption of propylene. Further, Pt–Cu/SiO2 (ref. 100) and Pt–Cu/Al2O3 (ref. 99) single-atom alloy catalysts, in which Pt atoms were isolated by Cu, could activate C–H efficiently and improve the selectivity to alkene and coke resistance in propane and n-butane dehydrogenation. Ren et al. studied the promotion role of group IB metals (Cu, Ag, Au) in Pt/MgAl2O4 catalysts for PDH.98 They found that the addition of Cu and Ag could significantly enhance the propane conversion and propylene selectivity while the introduction of Au does not, which can be explained by the dilution and electron donation of Cu and Ag to Pt. In addition, Pt–Cu/MgAl2O4 exhibited excellent regeneration stability, likely owing to the formation of thermally stable intermetallic compounds.
Gong's group investigated the promotion effect of 3d transition metals (Ni, Co, Fe) on Pt-based catalysts with controlled surface and subsurface compositions in PDH.106 They found that the well-defined Pt-skin catalysts with different 3d transition metal atoms in the subsurface showed notable enhancement of selectivity to propene in the order Pt < PtNi@Pt < PtCo@Pt < PtFe@Pt, which corresponds to the calculated trend of d-band center shifting. Miller's group reported that the addition of Mn to Pt catalysts with different subsurface compositions could alter the catalytic and adsorption properties of Pt-based catalysts. They found that the catalyst with a Pt3Mn intermetallic subsurface significantly improved C–H bond activation, leading to high selectivity to propene during PDH.103 In addition, this group also reported several Co,107 Fe,105 Cr,111 Bi,112 and Sb (ref. 113) modified Pt-based catalysts with various compositions and intermetallic structures that are promising for light alkane dehydrogenation.
3.2 Pd-based bimetallic catalysts
Pd-based bimetallic catalytic systems could be an alternative to Pt-based bimetallic catalysts for light alkane dehydrogenation. The supported monometallic Pd catalysts for the dehydrogenation of light alkanes have the same problems mentioned for Pt catalysts. Compared with Pt, Pd metal shows higher intrinsic activity towards C–C bond cracking, which results in poor selectivity to target olefins but high yield of methane. In addition, monometallic Pd catalysts rapidly deactivate due to coke formation. To overcome these shortcomings, recent studies focus on modifying Pd catalysts with the addition of promoters including Ga, Zn, In, Fe, Sn and Sb. The addition of these inert second metals can significantly enhance the selectivity as well as stability. Analogous to Pt-based bimetallic catalysts, the geometric and electronic effects of these promoters on Pd catalysts are responsible for the enhanced catalytic performances.Arteaga et al. tested a series of Ga-modified Pd/Al2O3 catalysts for the dehydrogenation of n-butane.120 They have shown that the selectively to butene and catalyst stability are improved significantly in the presence of Ga. Low molar ratios of Ga/Pd (<1.68) are critical to achieve these enhancements. However, the addition of Ga reduced the initial activity. In contrast, Wasserscheid et al. observed that the intrinsic activity can be remarkably increased (4-fold) at high molar atomic ratios of Ga/Pd (>10).121 Coke formation can be minimized. They suggested that the superior catalytic performance resulted from the unique liquid metallic nature on the support, where the atomically isolated Pd active sites were dynamically formed.
Miller and his co-workers demonstrated a series of supported Pd-based intermetallic alloy catalysts including Pd–Zn,122 Pd–In,123 Pd–Fe,124 Pd–Sn (ref. 125) and Pd–Sb125 for ethane and propane dehydrogenation. All of these catalysts showed higher activity for dehydrogenation, selectivity to olefins and stability than Pd monometallic catalysts. They suggested that the geometrically isolated Pd catalytic sites neighboring the inactive metallic promoters on these Pd-based intermetallic compounds are responsible for the high olefin selectivity, while the weaker adsorbate bond energies and lower reactant surface coverages due to the decrement in energy of the filled Pd 4d orbital lead to the increase in intrinsic activity and stability.122–125
Although Pd-based bimetallic catalytic systems showed some promising performance in the dehydrogenation of light alkanes, the activity is still lower than that over Pt-based bimetallic catalysts. In addition, the price of Pd is much higher than that of Pt,126 making it unappealing in large-scale applications.
3.3 Other bimetallic catalysts
Apart from Pt-based and Pd-based catalysts for the selective dehydrogenation, several recent studies report that effective catalysts composed of some other metals (e.g. Rh, Ni) as active sites were reported. The Ga promoter was often added to these catalyst systems to form either a single-atom diluted alloy or intermetallic compounds. These specific catalyst structures enable unusual metal sites active in C–H bond dissociation.Wasserscheid et al. developed supported catalytically active liquid metal solution (SCALMS) catalysts Ga–Rh/Al2O3 (Ga/Rh >80), which possess dynamic single Rh atoms at the surface during reaction, for propane dehydrogenation.127,128 Due to the unique structure during reaction, these Ga–Rh/Al2O3 SCALMS showed high catalytic activity, high propylene selectivity (∼92%) and moderate catalyst stability.
To lower the cost of the noble-metal-based catalysts, several Ni-based bimetallic catalysts have been developed. Ni-based catalysts possess appealing ability in the activation of alkene molecules. However, unmodified Ni-based catalysts exhibit a high intrinsic activity towards C–C hydrogenolysis, leading to methane, coke and hydrogen. This is due to the ensembles of Ni metal atoms on the catalyst surface. To improve olefin selectivity, several metal promoters such as Au,129 Sn (ref. 130) and Ga131,132 have been introduced to dilute the large nickel ensembles and thereby suppress the hydrogenolysis. When a Ni/SiO2 catalyst is incorporated with Au for propane dehydrogenation, the selectivity to propene remarkably increased from 0% to 46%, while methane selectivity dramatically decreased from 99.9% to 27%.129 The addition of Sn and Ga is more effective. Li et al. prepared a NiSn/SiO2 catalyst for isobutane dehydrogenation.130 Characterization indicated that the formation of a Ni2.67Sn2 alloy significantly breaks the large Ni ensembles. In addition, DFT calculations confirmed the electron transfer from Sn to Ni. The electron-rich Ni surface facilitates the desorption of iso-butene and thus inhibits undesired side reactions. Combined with geometric and electronic effects, 90.2% selectivity to iso-butene and high stability can be achieved on this catalyst. The Laursen group developed several non-noble Ni–Ga-based intermetallic compound catalysts for the dehydrogenation of ethane131 and propane.132 The catalysts have a core–shell structure, where the Ni3Ga intermetallic phase is surrounded by a shell with varied Ni and Ga compositions. The Ni atoms on the surface are requisite to drive the reaction while the Ga at the surface sustains the selectivity and catalyst stability. The optimal surface composition is critically important for achieving high performance. Conversion decreased and selectivity increased as Ga loading was increased from 3:1 to 1:2 Ni:Ga molar ratio (Fig. 1a and b). DFT calculations revealed that unselective dehydrogenation reactions, which lead to coke, C–C cracking and methane formation, are significantly inhibited with increasing amount of Ga. The catalyst with a 1:1 Ni:Ga ratio ((1:1 Ni:Ga)@/Ni3Ga/Al2O3) presented the best balance between activity and selectivity in both dehydrogenation of propane and ethane. In a propane dehydrogenation reaction prolonged to 82 h, this catalyst showed reasonably stable activity (∼13% to 9%) and a high steady-state selectivity towards olefins (propene and ethylene). The selectivity to propene decreased moderately from ∼94% to 81%, while the selectivity to ethylene increased slightly (Fig. 1c). The catalyst can be regenerated by in situ O2 and H2 treatment. Compared with commercial and in-house produced Pt–Sn catalysts, this catalyst showed comparable or higher selectivity and stability.
 | ||
| Fig. 1 (a and b) Catalytic performance of propane dehydrogenation over Ni3Ga/Al2O3 with different Ni:Ga actual loadings. (c) Catalytic activity, 82 h stability, and regenerability of (1:1 Ni:Ga)@/Ni3Ga/Al2O3. Reproduced from ref. 125 with permission from the American Chemical Society, copyright 2018. | ||
4. The functions of the promoters
To explain the modification of the catalytic properties by addition of the promoter in bimetallic catalysts, geometric effects and electronic effects have been discussed. It is very difficult to identify the contribution of each in the dehydrogenation reactions. Usually, the combination of both effects leads to significant changes in the catalytic properties.4.1 Geometric effect
The introduction of the promoters to the supported metal-based catalysts significantly influence the active metal structures through alloy formation and surface modification. The accompanying geometric effect leads to remarkable changes in the catalytic properties. Two kinds of explanations have been suggested to understand the geometric effect of the promoters. Both of these two geometric effects are able to suppress the hydrogenolysis and deep dehydrogenation reactions, thus reducing the production of shorter hydrocarbons and inhibiting coke formation.It is believed that only a small ensemble of active metal sites or even a single metal atom could catalyze the structure-insensitive dehydrogenation reactions. The structure-sensitive side reactions such as hydrogenolysis, deep dehydrogenation and coke formation can only occur on the large ensembles of the active metal. The formation of an alloy and/or the partial covering of the active metal particles by promoter species break the large ensembles of active metal and create small active metal surface ensembles or even isolated atoms. This ensemble effect favors the main alkane dehydrogenation and inhibits side reactions. Indeed, the deeper dehydrogenated intermediates require more active metal sites to bind due to the highly unsaturated nature of their C atoms. DFT studies also revealed that intermediates such as CHCHCH3 and CH3C binding on the PtGa2(111)82 and PtSn(111)133 without three-fold Pt hollow sites is much weaker than those on Pt(111) with three-fold Pt sites. Hence, the selectivity to the alkene is enhanced and coking is suppressed.
It is also suggested that side reactions are facile on the low-coordinated sites, such as edge sites and corner sites. These defect sites can be selectively covered by the addition of the promoters through alloy formation and surface modification, which can be confirmed by FT-IR spectroscopy with CO adsorption.25,39 The absence of low-coordinated defect metal sites contributes to the suppression of side reactions. In addition, DFT calculations by Honkala et al.134 revealed that the lack of highly active Pt step sites on Pt3Sn(211) weakens propene adsorption and suppresses further dehydrogenation of propene, thus improving catalyst selectivity and stability.
4.2 Electronic effect
Modification of the electronic properties of the active metal is the other role of the promoters. The electronic effect on the active metal is still under debate. Two different electronic effects have been proposed, which are electron donation from the promoter to the active metal and energy change of the Pt 5d orbitals or the filled Pd 4d orbitals.The electron transfer from the promoter to the 5d band of the Pt atoms is the most commonly accepted electronic promotion in Pt-based bimetallic catalysts. The electron transfer results in an increase in the electron density of Pt and can be identified by FT-IR spectroscopy with CO adsorption,39,48 XAS,90,112 XPS75 and DFT.133 In the case of FT-IR spectroscopy with CO adsorption, the absorption bands for CO linearly adsorbed onto Pt-based bimetallic catalysts, such as Pt3Sn,39 PtSn39 and PtZn (ref. 75) alloys, present at lower wavenumbers than that on monometallic Pt terraces. The redshifted CO vibration frequency demonstrates the electron donation from the promoter to Pt, which is due to a higher electron density available for back-donation to the 2π* orbitals in the CO bonding to Pt. Deng et al. analyzed the electronic property of Pt by calculating Pt d-band vacancy based on white line intensity in Pt LIII and LII XANES spectra. The lower the d-band vacancy, the higher the electron density.39 They revealed that the addition of Sn makes Pt electron-rich and the Pt3Sn alloy showed the highest electron density. The change in the electronic state of Pt modified with the promoters affects the adsorption fashion of intermediates and products. DFT calculations revealed that the presence of the promoters weakened the binding energies of alkene and coke precursors.133 The electron-enriched Pt surface thereby facilitates alkene formation and coke precursor desorption, concomitantly suppressing deep dehydrogenation, hydrogenolysis and coke formation. Consequently, the electron-enriched Pt surfaces improve the selectivity to target olefins and enhance the catalytic stability against coke deposits dramatically.
In contrast with electron donation, Miller and co-workers argued that the electronic effect of the promoter on the active metal is the energy change of the Pt 5d orbitals or the Pd 4d orbitals. They studied the electronic structure of Pt in Pt1Zn1,76 PtFe (ref. 105) and PtIn (Pt3In and PtIn2)92 intermetallic compounds by using resonant inelastic X-ray scattering, Pt LIII XANES and DFT calculations. They found that the energy of the filled Pt valence 5d orbitals decreased and the energy of the unfilled Pt 5d orbitals increased upon bonding with Zn, Fe or In. The lowered energy of the filled Pt 5d valence orbitals results in the decrease of Pt adsorbate bond energy, leading to faster alkene desorption and higher intrinsic reaction activity. Analogously, the addition of Zn,122 In,123 Fe (ref. 124) or Sb (ref. 125) decreases the energy of the filled Pd 4d valence orbitals in these Pd-based intermetallic compounds. This electronic promotion effect on these alloys is expected to weaken the Pd–C and Pd–H interactions and to enhance the catalytic activity but not the selectivity.
5. Reaction mechanism
The reverse Horiuti–Polanyi mechanism is the most commonly used mechanism to explain the dehydrogenation of alkanes over metal-based catalysts.135 The mechanism consists of four main steps (Scheme 1): (i) cleavage of the first C–H bond in a dissociative adsorption of the alkane, resulting in an adsorbed alkyl group; (ii) cleavage of the second C–H bond, resulting in a π- or di-σ-bonded olefin; (iii) desorption of the olefin molecule; and (iv) combinative desorption of the hydrogen molecule. The mechanism follows the Langmuir–Hinshelwood kinetics, where all the surface sites of the catalyst are considered identical. Either the dissociative adsorption of an alkane molecule136,137 or the second C–H cleavage step44,82,99 has been suggested as the rate-limiting step of the dehydrogenation reaction.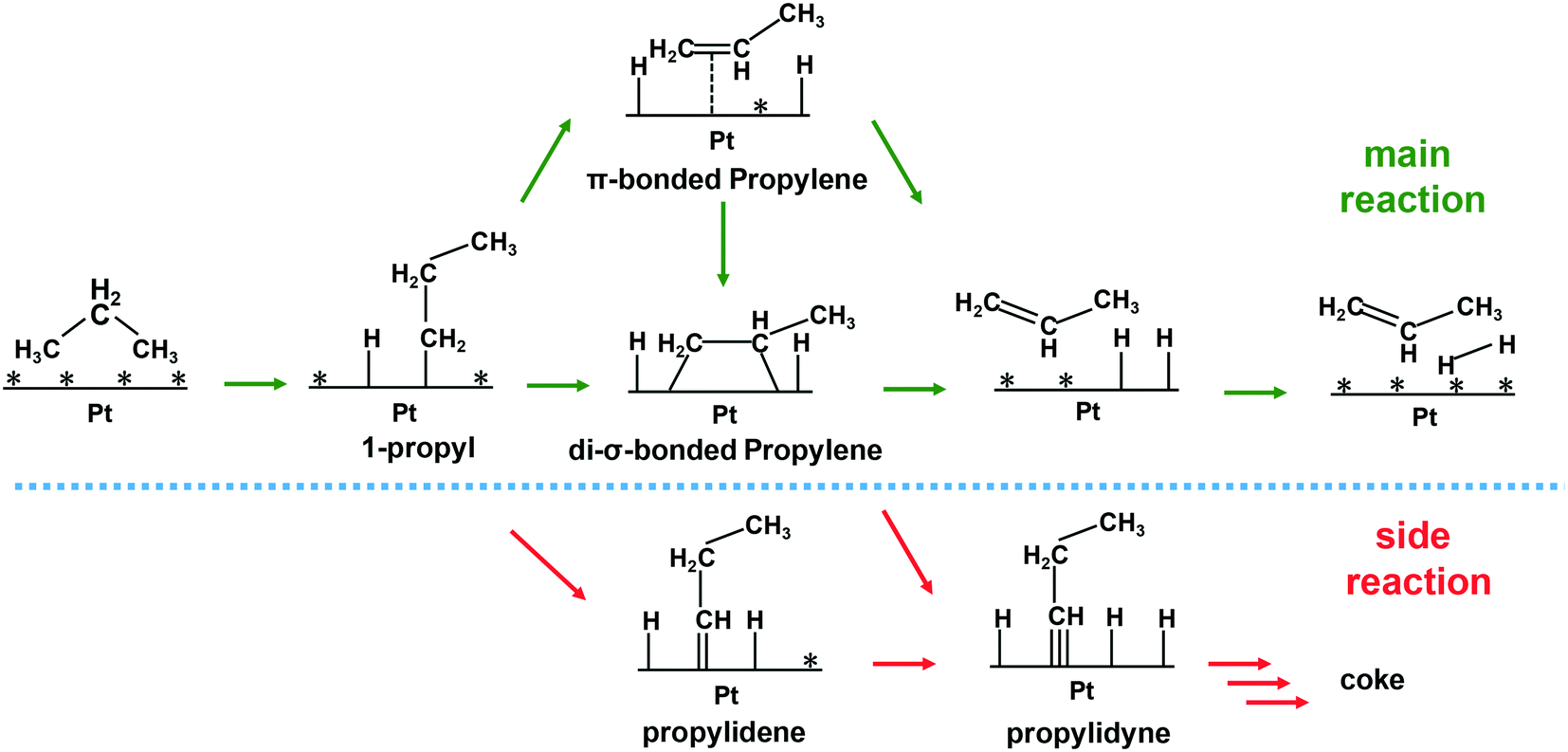 | ||
| Scheme 1 Proposed propane dehydrogenation mechanism. | ||
Several studies have been performed on Pt-based catalysts to gain a detailed understanding of the mechanism of the dehydrogenation and the effect of the promoter. Koel and co-workers studied alkene adsorption modes and desorption manner on Pt(111) and Pt–Sn surface alloys.138,139 They observed that ethylene binds in a di-σ ethylene binding geometry preferentially on Pt(111) and all the Pt–Sn alloy surface. Hook et al. also found that the di-σ ethylene binding fashion was preferred for both extended Pt and Pt–Sn surfaces by using DFT methods.133 As Sn content increased, the desorption temperature for the alkene decreased substantially, which facilitates alkene desorption. Gong and his co-workers revealed that the di-σ-bound propene adsorbed on the Pt(111) and Pt3Cu(111) surface while only π-bound propene existed on the Pt/Cu SAA surface based on DFT calculations.99 The binding strength of π-bound propene is weaker than that of the di-σ adsorbed ones, suggesting facile propylene desorption from Pt/Cu SAA. Furukawa et al. observed that PtGa–Pt1(111) with a single Pt atom on the surface binds propene in a π fashion as well.88 The alkene adsorption energy is below the barrier for dehydrogenation of alkene with the aid of promoters, making further dehydrogenation and cracking reactions less likely. In the cases of supported nanoparticles, the binding mode of alkene is totally different from the observation on extended metal surfaces. Dumesic et al. studied the adsorption geometries and energies of ethylene to silica-supported Pt and PtSn catalysts using microcalorimetry and IR spectroscopy at low temperatures.140 During the adsorption of ethylene, di-σ- and π-bound ethylene and ethylidyne are detected over both of the catalysts. The incorporation of Sn resulted in an increase in the fraction of π-bound ethylene and a decrease in the heat of adsorption. Similarly, Al2O3-supported Pt nanoparticles also resulted in three ethylene adsorption modes at 180 K.141 The di-σ-bound ethylene was converted to ethylidyne and hydrogen while all π-bound ethylene was found to desorb intact at room temperature. Anderson et al. observed similar adsorption and transformation modes of ethylene over the Pt4/SiO2 catalyst.142 However, they found that Pt4Sn3/SiO2 binds ethylene only in the π mode. Ha and Baxter et al. examined changes in ethylene binding/desorption over Ptn/Al2O3 and PtnBx/Al2O3 and found that the addition of boron suppressed di-σ-binding of ethylene and substantially lowered the desorption energy.117,118 Note that the di-σ-bonded alkene, adsorbed on the Pt surface strongly, is suspected to be an intermediate in further dehydrogenation while the π-bonded one is ready to desorb142 (Scheme 1). Moreover, ethylidyne is suggested to be an intermediate in the ethylene cracking to methane and coke during ethane dehydrogenation based on isotopic labeling experiments (Scheme 1).21,22 Although the adsorption mode of alkene over Pt-based catalysts is still under debate, one thing is apparent: that the addition of promoter always weakens the strength of alkene adsorption and favors desorption of alkene over further dehydrogenation, leading to high selectivity towards olefins and low carbon deposition. This is rationalized by a combination of geometric and electronic effects.142 On the other hand, DFT calculations showed that the addition of promoter increases the barrier for dissociative adsorption of light alkanes and lowers the rate of dehydrogenation of light alkanes.44,82,88,104 Therefore, the effect of the promoter on the overall reaction activity depends on the trade-off between the rates of dissociative adsorption of alkane and desorption of formed alkene.
6. Recent advanced strategies for the development of bimetallic catalysts
6.1 Novel support materials
Good support materials for light alkane dehydrogenation should have a high surface area, high thermal stability, and excellent mechanical strength. The properties of the supports affects the dispersion, stability and structure of bimetallic catalysts significantly. Thus, modulating the support is an alternative strategy to enhance catalytic performances. Besides alkalized θ-Al2O3, ZnAl2O4 and ZrO2 used in industrial and pilot scale, a number of novel supports including Al2O3 nanosheets, hydrotalcites, CeO2, zeolites and carbon materials have been developed and engineered, aiming to improve the dispersion and anchor the active sites.Alumina is a typical support for commercial Pt–Sn bimetallic catalysts. To improve metal dispersion and the metal–support interaction, Lu et al. synthesized γ-Al2O3 nanosheets rich in pentacoordinate Al3+ ions as a support for Pt–Sn bimetallic catalysts in propane direct dehydrogenation.48 The coordinatively unsaturated Al atoms act as anchoring sites to disperse and immobilize raft-like Pt–Sn clusters with mean diameters of 1.3 nm (Fig. 2A and B). The electronic density of Pt sites was increased with the addition of Sn and the electronic interaction between Pt and Sn on this unique nanosheet support was enhanced, leading to a lower desorption temperature of propene than that on conventional γ-Al2O3 supports (Fig. 2C). In addition, this unique nanosheet structure facilitates mass transfer and improves diffusion kinetics. Consequently, the catalyst displays a high productivity of propene with a selectivity of >99% and a low deactivation rate of 0.007 h−1 at 590 °C, which are much better than those over traditional Pt–Sn/γ-Al2O3 catalysts (Fig. 2D). To further improve the stability of Pt–Sn bimetallic catalysts, a Mg(Sn)(Al)O@Al2O3 support was used for Pt–Sn catalysts.46 The surface basicity of hydrotalcite is expected to suppress coke formation. The hydrotalcite support interacts strongly with Sn and stabilizes it in an oxidized state. The confined SnOx in the Mg(Al)O lattice can provide the anchoring sites for immobilizing highly dispersed Pt clusters by strong interactions and thus afford a catalyst with a lower deactivation rate of 0.0045 h−1 at 600 °C.
 | ||
| Fig. 2 (A) TEM image and (B) HAADF-STEM image of PtSn/Al2O3 sheet. (C) Propylene-TPD profiles of PtSn/Al2O3 ref. and PtSn/Al2O3 sheet. (D) Specific rate of C3H6 formation versus WHSV for PtSn/Al2O3 ref. and PtSn/Al2O3 sheet. Reproduced from ref. 41 with permission from John Wiley and Sons, copyright 2015. | ||
CeO2 as an alternative support for Pt–Sn catalysts was explored.66 The Pt–Sn/CeO2 catalysts showed a reversible alloy/segregation process in reaction/oxidation cycling. The CeO2 support can provide the trapping sites for atomically dispersing and re-dispersing Pt while Sn can act as a nucleation site for forming active Pt–Sn clusters during reaction. Additionally, the redox properties of CeO2 minimize coke formation by a co-feeding stream. In consequence, Pt–Sn/CeO2 showed remarkable stability and regenerability, alleviating the requirement of complex oxychlorination regeneration as practiced in industry.
Nanocarbon materials are appealing supports for metal catalysts, as they can provide abundant surface sites to host metal atoms. Liu et al. used a nanocarbon (ND@G) composed of a nanodiamond core and a defective, ultrathin graphene nanoshell as a support to prepare a Pt–Sn/ND@G catalyst for n-butane dehydrogenation.45 Due to the strong interaction with the support (Pt–C bonding) and the partition of monodispersed Sn species, the catalyst contains atomically dispersed Pt clusters with three Pt atoms on average (Fig. 3A). In contrast, without Sn, both Pt clusters and crystalline Pt nanoparticles coexisted on Pt/ND@G.31 In n-butane dehydrogenation, the bimetallic catalysts showed higher reactivity and selectivity to C4 olefins (>98%) than Pt/ND@G and Pt3Sn/Al2O3, which is due to the fully exposed Pt atom and easy desorption of butene (Fig. 2B). However, coke formation is still unavoidable in this dehydrogenation reaction, and the regeneration of carbon-supported catalysts is a critical issue in practical applications.
 | ||
| Fig. 3 (A) HAADF-STEM image of the Pt–Sn/ND@G catalyst; the atomic dispersion of Pt and Sn, scale bar = 0.5 nm. (B) Direct dehydrogenation of n-butane over different catalysts. Reproduced from ref. 45 with permission from the American Chemical Society, copyright 2019. | ||
Zeolites and mesoporous materials containing promoter atoms in the framework have been studied as supports in recent years. The promoter atoms in the framework provide anchoring sites to disperse and immobilize Pt. In addition, some of the promoter atoms in the framework might migrate and alloy with Pt during calcination and reduction processes. Yuan and coworkers used Sn–Si-Beta as a support to prepare a highly efficient Pt/Sn–Si-Beta catalyst for propane dehydrogenation (Scheme 2).62 The isolated Sn sites in the zeolite framework could interact strongly with Pt and stabilize ultra-small Pt nanoclusters. As a result, the catalyst gave a high propene formation rate and good stability. Jing et al. employed Sn-incorporated SBA-15 mesoporous materials as supports for Pt/Sn-SBA-15 in propane dehydrogenation.57 The catalyst activity and selectivity to propene were enhanced due to the promoted efficiency of Sn in the support. Recently, Zhao et al. synthesized a highly dispersed MnOx decorated dendritic mesoporous silica support by a simple in situ emulsion method.104 The support stabilizes the high dispersion of Pt clusters through the strong electrostatic interaction between the support and the Pt precursor. A Pt–Mn alloy was formed after reduction and gave excellent catalytic activity and stability owing to its balanced abilities of C–H activation and propene desorption.
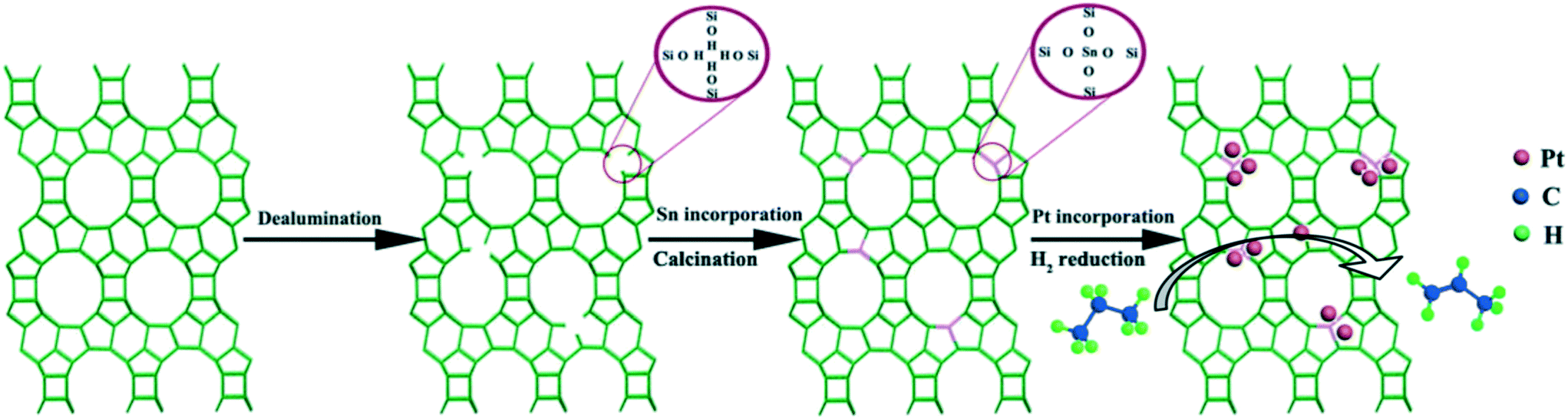 | ||
| Scheme 2 Illustration of the preparation of the Pt/Sn–Si-Beta catalyst. Reproduced from ref. 55 with permission from the Royal Society of Chemistry, copyright 2019. | ||
6.2 Novel preparation methods
The catalyst structure, dispersion and formation of the active phase significantly depend on the catalyst preparation and pretreatment methods. The impregnation method is the most commonly used approach for the preparation of bimetallic catalysts. However, with this method, it is very difficult to obtain homogeneously distributed nanoparticles with ultra-high dispersion and to result in satisfactory catalytic performances. To address this, several new strategies for the synthesis of supported metal catalysts have been developed.Basset and Zhu et al. prepared a Pt–Sn/MgAl2O4 catalyst with surface-enriched Sn by using K2PtCl4 and Bu3SH as precursors and LiB(C2H5)3H as a reduction agent.145 The catalyst exhibited high selectivity and long-term stability in propane dehydrogenation to propylene due to the dilution effect of the high concentration of inactive Sn on the surface of Pt nanoparticles. After that, they synthesized Pt–Sn/θ-Al2O3 catalysts with ultra-small bimetallic Pt–Sn clusters of around 0.75 nm by using Pt(COD)Me2 and HSnPh3 as precursors.36 The obtained catalysts showed high activity (TOF of 102 s−1), high selectivity (99%) and excellent stability in propane dehydrogenation. To further improve the activity, a completely dealuminated Beta zeolite was used as a support for Pt–Sn bimetallic catalysts.58 After SOMC and reduction with hydrogen, homogeneously dispersed Pt–Sn alloy clusters located at the Sn single site in the zeolite framework were formed. The catalyst exhibits slightly higher activity with a TOF of 114 s−1 than Pt–Sn/θ-Al2O3. The strong interaction between Pt–Sn bimetallic clusters and isolated Sn sites in the zeolite framework Sn conferred high catalytic stability and good regenerability.
Copéret's group prepared a uniform nanometric Pt–Ga alloy supported on silica containing Ga single sites for PDH by the SOMC method (Fig. 4a).87 The loading amount of Pt is up to 3 wt%, which is much higher than that of typical Pt-based bimetallic catalysts (∼0.5 wt%). XPS and in situ XAS revealed that the reduced catalysts possess GaxPt (0.5 < x < 0.9) alloy. The presence of Lewis acidic Ga single sites remaining at the surface could favor the nucleation and stabilization of nanometric particles, leading to highly dispersed nanometric alloyed particles with an average particle size of around 1.0 nm (Fig. 4b). Consequently, the catalyst exhibited a great propene productivity (1050 gC3H6 gcat−1 h−1) at high WHSV (98 h−1). To further improve the productivity, a Pt0–Znδ+/SiO2 catalyst with a similar structure to that of Pt0–Gaδ+/SiO2 has been developed by the same group recently.77 The catalyst gave a super high productivity (up to 703 gC3H6 gPt−1 h−1), which might be due to the smaller average Pt–Zn alloy particle size of around 0.8 nm. However, the stability of both catalysts is not good owing to coke formation as well as sintering. In addition, the catalytic activity cannot be fully recovered (ca. 75% of the initial activity) after oxidation/reduction regeneration, which might be ascribed to the structural change during the regeneration process. In contrast, the Pt–Sn/Beta can be fully restored after regeneration, which might be associated with robust Sn single sites in the zeolite framework.
 | ||
| Fig. 4 (a) Synthesis of the bimetallic Pt0–Gaδ+/SiO2 catalyst. (b) Representative TEM image and particle size distribution of Pt0–Gaδ+/SiO2 catalyst. Reproduced from ref. 80 with permission from the American Chemical Society, copyright 2018. | ||
An in situ ligand-stabilized method has been proven to be an effective method to encapsulate metal species in pure siliceous zeolite. Ethylenediamine is often used as a ligand to protect the metal species from premature colloidal precipitation during zeolite crystallization. Using this method, Corma and Liu et al. have synthesized K–PtSn@Silicate-1 catalysts for propane dehydrogenation.146 The loading amount of Pt is about 0.4 wt%. The introduction of K+ could compensate for the silanol groups and further stabilize the subnanometer Pt species. High-resolution high-angle annular dark-field scanning transmission electron microscopy (HR HAADF-STEM) and integrated differential phase contrast (iDPC) imaging techniques evidenced that subnanometric PtSn clusters encapsulated in sinusoidal channels. With the confinement of zeolites, the ultra-small size of PtSn clusters and the electronic interaction between Pt and Sn, the pretreated K–PtSn@Silicate-1 showed exceptional activity, selectivity and stability. Catalyst pretreatment with hydrogen at a long reduction time (>12 h), allowing Sn to incorporate with Pt, is essential to achieve high stability.147 To further improve the propylene yield, they prepared another K–PtSn@Silicate-1 with a high loading of Pt (∼1.4 wt%).148 They found that high-temperature calcination in air after hydrothermal synthesis is critical to disintegrate the Pt particles on the external surface into subnanometric Pt species and stabilize Pt in the sinusoidal channels, while high-temperature reduction treatment at 650 °C favors the migration of Sn species from the surface region to the internal region. The resultant high dense subnanometric bimetallic PtSn clusters in the sinusoidal 10 MR channels showed excellent catalytic performance for propane dehydrogenation, especially in terms of propene yield.
Similarly, Yu's group developed an ethylenediamine-protected direct hydrogen reduction method for encaging Pt–Zn bimetallic clusters into silicalite-1 zeolite (Fig. 5a).74 Cs-corrected STEM-HAADF images and EXAFS results of PtZn@Silicate-1 confirmed that subnanometric Pt–Zn clusters are encapsulated in the five/six-membered rings (MRs) of zeolites (Fig. 5b–g). They claimed that direct hydrogen reduction after hydrothermal synthesis is essential to obtain subnanometric Pt–Zn clusters. At almost the same time, Yuan et al. also reported a Pt–Zn@Silicate-1 catalyst prepared by a similar ethylenediamine-protected method. Unlike Yu's method, Yuan et al. applied calcination followed by hydrogen reduction.75 Ultra-small Pt–Zn clusters with an unknown particle size confined in silicate-1 could be obtained as well. In addition, the Zn species increase the electron density of Pt, as confirmed by the shift of the CO adsorption band, leading to facile desorption of propene on Pt–Zn clusters. Meanwhile, the small size of the Pt–Zn clusters could enhance the electron interaction between Pt and Zn. Consequently, combined with small Pt–Zn clusters caged in zeolites and facile desorption of propene, both of the catalysts showed superior performances in terms of activity, selectivity, stability and regenerability in propane dehydrogenation. Yu et al. indicated that the introduction of Cs+ ions located inside the zeolite channels is critical for maintaining the regeneration stability, as Cs+ ions could effectively suppress metal migrations to the outer surface of zeolite crystals and thereby inhibit the aggregation of the metal species. However, the addition of Cs+ was not needed in the catalyst made by Yuan et al. Remarkably, both subnanometric Pt–Sn and Pt–Zn bimetallic clusters (<1 nm) encapsulated in zeolites exhibit high activity at high weight hour space velocity (up to 108 h−1).74,147,148
 | ||
| Fig. 5 (a) Schematic representation of the synthesis of zeolite-encaged subnanometer Pt–Zn cluster catalysts. (b–d) HAADF-STEM images of PtZn4@S-1-H. (e) ABF-STEM image of PtZn4@S-1-H along the [010] projection with the model superimposed. (f and g) High-resolution HAADF-STEM images of PtZn4@S-1-H. Reproduced from ref. 67 with permission from John Wiley and Sons, copyright 2020. | ||
To prepare the catalysts more efficiently, Okubo and his co-workers have recently developed an ultrafast in situ encapsulation method to prepare PtSn@ZSM-5.149 They proposed this method based on ultrafast synthesis of zeolites developed previously. By passing a zeolite precursor solution after aging and seeding through a tubular reactor featuring fast heating, they are able to obtain crystallized zeolites in a few minutes. This ultrafast synthesis of zeolites could coordinate with the fast formation of metal nanoclusters to synthesize nanocluster-encapsulated zeolites. As a result, the ligand is not needed in this method. The synthesized PtSn@ZSM-5 catalysts showed excellent activity, selectivity and stability in the dehydrogenation of propane to propene.
6.3 Single-atom alloy catalysts for dehydrogenation
As discussed in the Geometric effect section, dehydrogenation reactions are structure–insensitive reactions, which means that even a single metal atom can drive the reaction. In contrast, undesired side reactions, such as hydrogenolysis and coke formation, are typically structure–sensitive reactions and require large ensembles of metal atoms. Based on these, single atom alloys (SAAs), composed of catalytically active noble metal atoms atomically dispersed in inert host metals, have been developed recently for light alkane dehydrogenation to eliminate the side reactions and improve the catalytic performance.Marcinkowski et al. studied both a model Pt–Cu SAA surface and real particles prepared by the galvanic replacement method for n-butane dehydrogenation.100 Gong et al. also prepared a Pt–Cu/Al2O3 SAA catalyst through a co-impregnation method for PDH (Fig. 6a–c).99 They both found that Pt–Cu SAAs can activate C–H bonds efficiently, improve the selectivity to alkene and retard coke formation. They proposed that the single Pt atoms diluted by Cu can drastically facilitate the desorption of propylene and suppress its further dehydrogenation to form coke. Consequently, the activity of the Pt–Cu SAA catalyst can be maintained in 120 h time-on-stream duration at 520 °C (Fig. 6d). However, the copper particles sintered quickly at temperatures higher than 600 °C, resulting in quick deactivation. To further improve the stability, a PtGa–Pb/SiO2 catalyst with a thermally stable intermetallic Pd–Ga alloy was prepared by Furukawa et al. via a simple impregnation method.88 Intermetallic PtGa displays three-fold Pt ensembles and single Pt atoms isolated by Ga on the surface. The former is selectively blocked and disabled by Pb deposition, which enables single-atom Pt in intermetallic PtGa to catalyze the dehydrogenation. The single-atom-like PtGa–Pb/SiO2 catalyst showed high selectivity (99.6%) towards propene and exceptional stability in a long-term operation at high temperature (600 °C, 96 h). Conversely, the PtGa/SiO2 without Pb modification showed lower selectivity and stability, suggesting the superior catalytic properties of such single-atom-like isolated Pt atoms in PtGa–Pb/SiO2. The performance is almost identical after oxidation/reduction regeneration, which is due to thermally stable intermetallic Pt–Ga. DFT calculations revealed that the isolated Pt atoms catalyze the dissociation of the first and the second C–H bonds, while they effectively inhibit the third one, which minimizes coke formation and thus drastically improves the selectivity and stability.
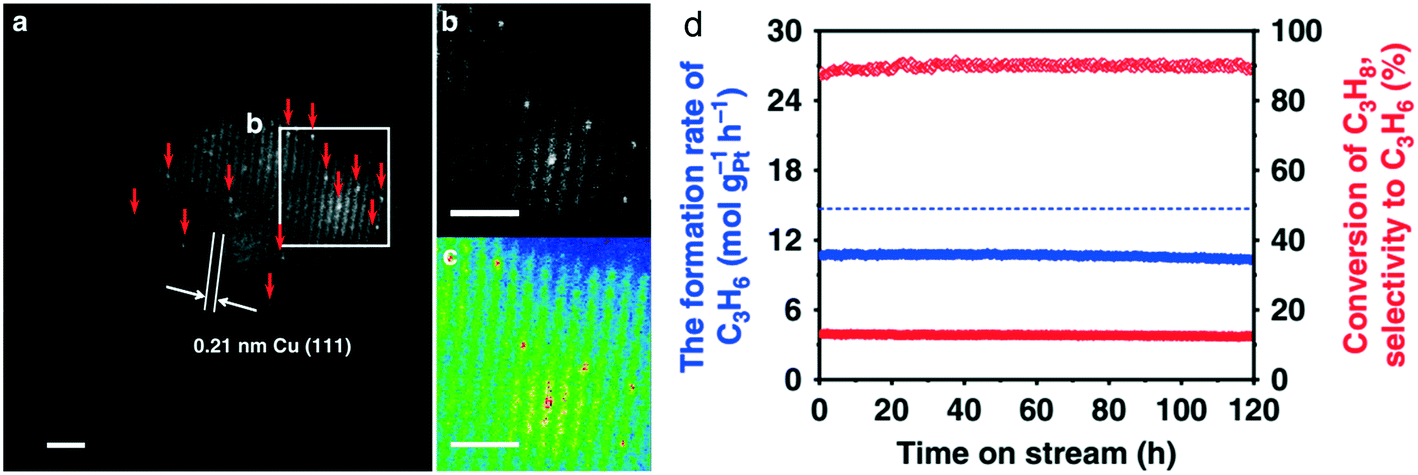 | ||
| Fig. 6 (a) HAADF-STEM images of a typical region of the reduced 0.1Pt10Cu/Al2O3 catalyst, showing Pt atoms individually dispersed on Cu(111). Pt atoms are highlighted by red arrows. (b) The enlarged image and (c) the colored intensity map from the selected region in (a). d) Long-term stability test for 0.1Pt10Cu/Al2O3 at 520 °C for 120 h. Reproduced from ref. 92 with permission from Nature, copyright 2018. | ||
Wasserscheid and co-workers first developed supported catalytically active liquid metal solution (SCALMS) catalysts Pt–Ga/Al2O3,89 Pd–Ga/glass,121 and Rh–Ga/Al2O3 (ref. 127 and 128) for propane and n-butane dehydrogenation. Unlike previous PtGa intermetallic compounds, these SCALMS are rich in Ga and possess lower melting points. They exhibit a liquid metal phase on the support and possess dynamic single Pt or Rh atoms at the surface under the dehydrogenation reaction. The porous support is important for dispersing the gallium droplets and preventing agglomeration. All these catalysts showed high activity, selectivity to alkenes and resistance to coke formation. The noble metal and Ga catalyze dehydrogenation synergistically. The isolated noble metal atoms activate alkane and the alkyl migrates to the Ga surface. Then, two hydrides recombined to H2 at the single noble metal site. In addition, Ga promotes the diffusion of the target alkene and thus results in high selectivity and catalytic stability.
7. Conclusion and perspectives
In this critical review, we presented the progress on metal-based catalysts for the non-oxidative dehydrogenation of light alkanes to corresponding olefins. These olefins can be used as chemical building blocks for the production of polymers, oxygenates and important chemical intermediates. We summarized and exemplified the metal catalysts including Pt-, Pd-, Rh- and Ni-based catalysts, especially in combination with the promoters (e.g., Sn, Zn, Ga, In, B), in the direct dehydrogenation of light alkanes. Among them, Pt-based catalysts are the most widely studied. The introduction of the promoters can remarkably improve their catalytic performances, especially in terms of selectivity to target olefins and catalytic stability. Next, we discussed the roles of the promoters, including geometric and electronic effects, in the modification of catalytic properties. The enhanced catalytic performances always rise from the combination of both effects. A brief reaction mechanism was also presented. Lastly, we highlighted the most recent advances in the development of novel high-performance dehydrogenation catalysts through three primary approaches: support engineering, advanced preparation methods and catalyst structure innovation.Although improved catalytic performances in the direct dehydrogenation of light alkanes have been obtained over metal-based catalysts in the past decades, significant efforts are still needed to develop more efficient novel catalysts and understand the mechanisms. Based on the current state of the art in the development of metal-based catalyst, especially Pt-based catalysts, it can be found that subnanometric metal catalysts and single-atom catalysts exhibit promising performance in the direct dehydrogenation of light alkanes. However, the loading amount of active metals is quite low (∼0.5 wt%), resulting in low catalytic productivity. In this regard, high-density subnanometric metal catalysts and single-atom catalysts are expected to improve the catalytic productivity. Although subnanometric metal catalysts (such as Pt–Zn/SiO2 (ref. 77) and Pt–Ga/SiO2 (ref. 87)) with high loading (3 wt%) have been prepared by SOMC methods and demonstrate significantly high propene productivity, the stability of these catalysts is still unsatisfactory at high reaction temperatures. Thus, developing novel economically viable synthesis methods, such as encapsulation of active sites in porous shells, to obtain stable subnanometric metal catalysts and single-atom catalysts with high metal loading is encouraged for practical applications. In addition, to lower the cost of expensive noble-metal dehydrogenation catalysts, it is urgent to develop many other efficient and durable catalysts using non-noble metals. In fact, a Ni–Ga-based intermetallic compound catalyst has been prepared by Laursen's group and showed comparable or higher selectivity and stability than commercial and in-house prepared Pt–Sn catalysts.131,132 Selecting the optimal combination of the promoters and non-noble metals and carefully controlling the catalyst structure could be the key criteria for the development of non-noble metal-based dehydrogenation catalysts. The reaction mechanisms and the roles of the promoters are still under debate. Therefore, future studies with the aid of advanced in situ and operando spectroscopy techniques and theoretical calculations are necessary to understand the catalytically active sites, the reaction mechanisms and the roles of the promoters. These will give insights into the rational design of highly efficient dehydrogenation catalysts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the National Natural Science Foundation of China (21522605 and 21776210) and the Tianjin Natural Science Foundation (18JCJQJC46800) is gratefully acknowledged.Notes and references
- J. C. Bricker, Top. Catal., 2012, 55, 1309–1314 CrossRef CAS
.
- C. A. Carrero, R. Schloegl, I. E. Wachs and R. Schomaecker, ACS Catal., 2014, 4, 3357–3380 CrossRef CAS
- F. Cavani, N. Ballarini and A. Cericola, Catal. Today, 2007, 127, 113–131 CrossRef CAS
- J. T. Grant, J. M. Venegas, W. P. McDermott and I. Hermans, Chem. Rev., 2018, 118, 2769–2815 CrossRef CAS
- Z.-P. Hu, D. Yang, Z. Wang and Z.-Y. Yuan, Chin. J. Catal., 2019, 40, 1233–1254 CrossRef CAS
- Z. Nawaz, Rev. Chem. Eng., 2015, 31, 413–436 CAS
- J. J. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Chem. Rev., 2014, 114, 10613–10653 CrossRef CAS
- G. Wang, X. Zhu and C. Li, Chem. Rec., 2020, 20, 604–616 CrossRef CAS
-
https://www.thebusinessresearchcompany.com/report/ethylene-global-market-report, 2020
-
https://www.thebusinessresearchcompany.com/report/propylene-global-market-report, 2020
-
https://www.globalinforesearch.com/Global-1-Butene_p482674.html, 2020
- A. Farshi, F. Shaiyegh, S. H. Burogerdi and A. Dehgan, Pet. Sci. Technol., 2011, 29, 875–885 CrossRef CAS
- E. McFarland, Science, 2012, 338, 340–342 CrossRef CAS
- S. Chen, C. Pei, G. Sun, Z.-J. Zhao and J. Gong, Acc. Mater. Res., 2020, 1(1), 30–40 CrossRef CAS
- M. M. Bhasin, J. H. McCain, B. V. Vora, T. Imai and P. R. Pujadó, Appl. Catal., A, 2001, 221, 397–419 CrossRef CAS
- M. A. Atanga, F. Rezaei, A. Jawad, M. Fitch and A. A. Rownaghi, Appl. Catal., B, 2018, 220, 429–445 CrossRef CAS
- E. Gomez, B. Yan, S. Kattel and J. G. Chen, Nat. Rev. Chem., 2019, 3, 638–649 CrossRef CAS
- E. Gianotti, M. Taillades-Jacquin, J. Rozière and D. J. Jones, ACS Catal., 2018, 8, 4660–4680 CrossRef CAS
- R. B. Biniwale, S. Rayalu, S. Devotta and M. Ichikawa, Int. J. Hydrogen Energy, 2008, 33, 360–365 CrossRef CAS
- O. O. James, S. Mandal, N. Alele, B. Chowdhury and S. Maity, Fuel Process. Technol., 2016, 149, 239–255 CrossRef CAS
- V. Galvita, G. Siddiqi, P. Sun and A. T. Bell, J. Catal., 2010, 271, 209–219 CrossRef CAS
- G. Siddiqi, P. Sun, V. Galvita and A. T. Bell, J. Catal., 2010, 274, 200–206 CrossRef CAS
- S. Saerens, M. K. Sabbe, V. V. Galvita, E. A. Redekop, M.-F. Reyniers and G. B. Marin, ACS Catal., 2017, 7, 7495–7508 CrossRef CAS
- H. Saito and Y. Sekine, RSC Adv., 2020, 10, 21427–21453 RSC
- A. Virnovskaia, S. Morandi, E. Rytter, G. Ghiotti and U. Olsbye, J. Phys. Chem. C, 2007, 111, 14732–14742 CrossRef CAS
- M. Santhosh Kumar, D. Chen, J. C. Walmsley and A. Holmen, Catal. Commun., 2008, 9, 747–750 CrossRef
- J. Zhu, M.-L. Yang, Y. Yu, Y.-A. Zhu, Z.-J. Sui, X.-G. Zhou, A. Holmen and D. Chen, ACS Catal., 2015, 5, 6310–6319 CrossRef CAS
- J. Wu, Z. Peng and A. T. Bell, J. Catal., 2014, 311, 161–168 CrossRef CAS
- F. Jiang, L. Zeng, S. Li, G. Liu, S. Wang and J. Gong, ACS Catal., 2014, 5, 438–447 CrossRef
- M. Santhosh Kumar, A. Holmen and D. Chen, Microporous Mesoporous Mater., 2009, 126, 152–158 CrossRef CAS
- J. Liu, Y. Yue, H. Liu, Z. Da, C. Liu, A. Ma, J. Rong, D. Su, X. Bao and H. Zheng, ACS Catal., 2017, 7, 3349–3355 CrossRef CAS
- A. Volynkin, M. Rønning and E. A. Blekkan, Top. Catal., 2015, 58, 854–865 CrossRef CAS
- J. Liu, J. Li, J. Rong, C. Liu, Z. Dai, J. Bao, Z. Da and H. Zheng, Appl. Surf. Sci., 2019, 464, 146–152 CrossRef CAS
- H. N. Pham, J. J. H. B. Sattler, B. M. Weckhuysen and A. K. Datye, ACS Catal., 2016, 6, 2257–2264 CrossRef CAS
- N. Prakash, M.-H. Lee, S. Yoon and K.-D. Jung, Catal. Today, 2017, 293–294, 33–41 CrossRef CAS
- Z. Xu, R. Xu, Y. Yue, P. Yuan, X. Bao, E. Abou-Hamad, J.-M. Basset and H. Zhu, J. Catal., 2019, 374, 391–400 CrossRef CAS
- L. Deng, H. Miura, T. Shishido, S. Hosokawa, K. Teramura and T. Tanaka, ChemCatChem, 2014, 6, 2680–2691 CrossRef CAS
- L. Deng, T. Shishido, K. Teramura and T. Tanaka, Catal. Today, 2014, 232, 33–39 CrossRef CAS
- L. Deng, H. Miura, T. Shishido, Z. Wang, S. Hosokawa, K. Teramura and T. Tanaka, J. Catal., 2018, 365, 277–291 CrossRef CAS
- L. Deng, H. Miura, T. Ohkubo, T. Shishido, Z. Wang, S. Hosokawa, K. Teramura and T. Tanaka, Catal. Sci. Technol., 2019, 9, 947–956 RSC
- Y. Zhang, Y. Zhou, J. Shi, S. Zhou, X. Sheng, Z. Zhang and S. Xiang, J. Mol. Catal. A: Chem., 2014, 381, 138–147 CrossRef CAS
- N. Kaylor and R. J. Davis, J. Catal., 2018, 367, 181–193 CrossRef CAS
- M.-H. Lee, B. M. Nagaraja, K. Y. Lee and K.-D. Jung, Catal. Today, 2014, 232, 53–62 CrossRef CAS
- M.-L. Yang, Y.-A. Zhu, X.-G. Zhou, Z.-J. Sui and D. Chen, ACS Catal., 2012, 2, 1247–1258 CrossRef CAS
- J. Zhang, Y. Deng, X. Cai, Y. Chen, M. Peng, Z. Jia, Z. Jiang, P. Ren, S. Yao, J. Xie, D. Xiao, X. Wen, N. Wang, H. Liu and D. Ma, ACS Catal., 2019, 9, 5998–6005 CrossRef CAS
- Y. Zhu, Z. An, H. Song, X. Xiang, W. Yan and J. He, ACS Catal., 2017, 7, 6973–6978 CrossRef CAS
- H. Liu, X. Dong, J. Xia, X. Li, Y. Sun and Q. Cai, Trans. Tianjin Univ., 2020, 26, 362–372 CrossRef CAS
- L. Shi, G. M. Deng, W. C. Li, S. Miao, Q. N. Wang, W. P. Zhang and A. H. Lu, Angew. Chem., Int. Ed., 2015, 54, 13994–13998 CrossRef CAS
- E. J. Jang, J. Lee, H. Y. Jeong and J. H. Kwak, Appl. Catal., A, 2019, 572, 1–8 CrossRef CAS
- B. M. Nagaraja, H. Jung, D. R. Yang and K.-D. Jung, Catal. Today, 2014, 232, 40–52 CrossRef CAS
- Y. Zhang, Y. Zhou, J. Shi, S. Zhou, Z. Zhang, S. Zhang and M. Guo, Fuel Process. Technol., 2013, 111, 94–104 CrossRef CAS
- Y. Shi, X. Li, X. Rong, B. Gu, H. Wei and C. Sun, RSC Adv., 2017, 7, 19841–19848 RSC
- B. K. Vu, M. B. Song, I. Y. Ahn, Y.-W. Suh, D. J. Suh, W.-I. L. Kim, H.-L. Koh, Y. G. Choi and E. W. Shin, Catal. Today, 2011, 164, 214–220 CrossRef CAS
- Q. Zhang, K. Zhang, S. Zhang, Q. Liu, L. Chen, X. Li, C. Wang and L. Ma, J. Catal., 2018, 368, 79–88 CrossRef CAS
- T. Srisakwattana, K. Suriye, P. Praserthdam and J. Panpranot, Catal. Today, 2019 DOI:10.1016/j.cattod.2019.06.058
- G. Aguilar-Ríos, M. Valenzuela, P. Salas, H. Armendáriz, P. Bosch, G. Del Toro, R. Silva, V. Bertín, S. Castillo, A. Ramírez-Solís and I. Schifter, Appl. Catal., A, 1995, 127, 65–75 CrossRef
- B. Li, Z. Xu, W. Chu, S. Luo and F. Jing, Chin. J. Catal., 2017, 38, 726–735 CrossRef CAS
- Z. Xu, Y. Yue, X. Bao, Z. Xie and H. Zhu, ACS Catal., 2019, 10, 818–828 CrossRef
- Y. Zhang, Y. Zhou, L. Huang, M. Xue and S. Zhang, Ind. Eng. Chem. Res., 2011, 50, 7896–7902 CrossRef CAS
- Z. Nawaz and F. Wei, J. Ind. Eng. Chem., 2010, 16, 774–784 CrossRef CAS
- J. Li, J. Li, Z. Zhao, X. Fan, J. Liu, Y. Wei, A. Duan, Z. Xie and Q. Liu, J. Catal., 2017, 352, 361–370 CrossRef CAS
- Y. Wang, Z.-P. Hu, W. Tian, L. Gao, Z. Wang and Z.-Y. Yuan, Catal. Sci. Technol., 2019, 9, 6993–7002 RSC
- Y. Zhang, M. Xue, Y. Zhou, H. Zhang, W. Wang, Q. Wang and X. Sheng, RSC Adv., 2016, 6, 29410–29422 RSC
- H. Zhou, J. Gong, B. Xu, S. Deng, Y. Ding, L. Yu and Y. Fan, Chin. J. Catal., 2017, 38, 529–536 CrossRef CAS
- H. Zhou, J. Gong, B. Xu, L. Yu and Y. Fan, Appl. Catal., A, 2016, 527, 30–35 CrossRef CAS
- H. Xiong, S. Lin, J. Goetze, P. Pletcher, H. Guo, L. Kovarik, K. Artyushkova, B. M. Weckhuysen and A. K. Datye, Angew. Chem., Int. Ed., 2017, 56, 8986–8991 CrossRef CAS
-
S. A. I. Barri and R. Tahir, US5208201, 1993
- P. Andy and M. E. Davis, Ind. Eng. Chem. Res., 2004, 43, 2922–2928 CrossRef CAS
- C. Chen, M. Sun, Z. Hu, J. Ren, S. Zhang and Z.-Y. Yuan, Catal. Sci. Technol., 2019, 9, 1979–1988 RSC
- Y. Zhang, Y. Zhou, L. Huang, S. Zhou, X. Sheng, Q. Wang and C. Zhang, Chem. Eng. Sci., 2015, 270, 352–361 CrossRef CAS
- P. L. De Cola, R. Gläser and J. Weitkamp, Appl. Catal., A, 2006, 306, 85–97 CrossRef CAS
- I. Kley and Y. Traa, Microporous Mesoporous Mater., 2012, 164, 145–147 CrossRef CAS
-
B. D. Alexander, J. George and A. Huff, US5453558, 1995
- Q. Sun, N. Wang, Q. Fan, L. Zeng, A. Mayoral, S. Miao, R. Yang, Z. Jiang, W. Zhou, J. Zhang, T. Zhang, J. Xu, P. Zhang, J. Cheng, D. C. Yang, R. Jia, L. Li, Q. Zhang, Y. Wang, O. Terasaki and J. Yu, Angew. Chem., Int. Ed., 2020, 59(44), 19450–19459 CrossRef CAS
- Y. Wang, Z.-P. Hu, X. Lv, L. Chen and Z.-Y. Yuan, J. Catal., 2020, 385, 61–69 CrossRef CAS
- V. J. Cybulskis, B. C. Bukowski, H.-T. Tseng, J. R. Gallagher, Z. Wu, E. Wegener, A. J. Kropf, B. Ravel, F. H. Ribeiro, J. Greeley and J. T. Miller, ACS Catal., 2017, 7, 4173–4181 CrossRef CAS
- L. Rochlitz, K. Searles, J. Alfke, D. Zemlyanov, O. V. Safonova and C. Copéret, Chem. Sci., 2020, 11, 1549–1555 RSC
- J. Camacho-Bunquin, M. S. Ferrandon, H. Sohn, A. J. Kropf, C. Yang, J. Wen, R. A. Hackler, C. Liu, G. Celik, C. L. Marshall, P. C. Stair and M. Delferro, ACS Catal., 2018, 8, 10058–10063 CrossRef CAS
- O. B. Belskaya, L. N. Stepanova, T. I. Gulyaeva, S. B. Erenburg, S. V. Trubina, K. Kvashnina, A. I. Nizovskii, A. V. Kalinkin, V. I. Zaikovskii, V. I. Bukhtiyarov and V. A. Likholobov, J. Catal., 2016, 341, 13–23 CrossRef CAS
- Z. Yu, J. A. Sawada, W. An and S. M. Kuznicki, AIChE J., 2015, 61, 4367–4376 CrossRef CAS
- E. L. Jablonski, A. A. Castro, O. A. Scelza and S. R. de Miguel, Appl. Catal., A, 1999, 183, 189–198 CrossRef CAS
- T. Wang, F. Jiang, G. Liu, L. Zeng, Z.-J. Zhao and J. Gong, AIChE J., 2016, 62, 4365–4376 CrossRef CAS
- M. Filez, E. A. Redekop, V. V. Galvita, H. Poelman, M. Meledina, S. Turner, G. Van Tendeloo, A. T. Bell and G. B. Marin, Phys. Chem. Chem. Phys., 2016, 18, 3234–3243 RSC
- P. Sun, G. Siddiqi, M. Chi and A. T. Bell, J. Catal., 2010, 274, 192–199 CrossRef CAS
- M. Filez, E. A. Redekop, H. Poelman, V. V. Galvita, R. K. Ramachandran, J. Dendooven, C. Detavernier and G. B. Marin, Chem. Mater., 2014, 26, 5936–5949 CrossRef CAS
- E. A. Redekop, V. V. Galvita, H. Poelman, V. Bliznuk, C. Detavernier and G. B. Marin, ACS Catal., 2014, 4, 1812–1824 CrossRef CAS
- K. Searles, K. W. Chan, J. A. Mendes Burak, D. Zemlyanov, O. Safonova and C. Copéret, J. Am. Chem. Soc., 2018, 140, 11674–11679 CrossRef CAS
- Y. Nakaya, J. Hirayama, S. Yamazoe, K. I. Shimizu and S. Furukawa, Nat. Commun., 2020, 11, 2838 CrossRef CAS
- T. Bauer, S. Maisel, D. Blaumeiser, J. Vecchietti, N. Taccardi, P. Wasserscheid, A. Bonivardi, A. Gorling and J. Libuda, ACS Catal., 2019, 9, 2842–2853 CrossRef CAS
- P. Sun, G. Siddiqi, W. C. Vining, M. Chi and A. T. Bell, J. Catal., 2011, 282, 165–174 CrossRef CAS
- M. C. Román-Martínez, J. A. Maciá-Agulló, I. M. J. Vilella, D. Cazorla-Amorós and H. Yamashita, J. Phys. Chem. C, 2007, 111, 4710–4716 CrossRef
- E. C. Wegener, Z. Wu, H.-T. Tseng, J. R. Gallagher, Y. Ren, R. E. Diaz, F. H. Ribeiro and J. T. Miller, Catal. Today, 2018, 299, 146–153 CrossRef CAS
- S. Zha, G. Sun, T. Wu, J. Zhao, Z. J. Zhao and J. Gong, Chem. Sci., 2018, 9, 3925–3931 RSC
- J. Wu, Z. Peng, P. Sun and A. T. Bell, Appl. Catal., A, 2014, 470, 208–214 CrossRef CAS
- K. Xia, W.-Z. Lang, P.-P. Li, L.-L. Long, X. Yan and Y.-J. Guo, Chem. Eng. Sci., 2016, 284, 1068–1079 CrossRef CAS
- L.-L. Shen, K. Xia, W.-Z. Lang, L.-F. Chu, X. Yan and Y.-J. Guo, Chem. Eng. Sci., 2017, 324, 336–346 CrossRef CAS
- K. Xia, W.-Z. Lang, P.-P. Li, X. Yan and Y.-J. Guo, RSC Adv., 2015, 5, 64689–64695 RSC
- G.-Q. Ren, G.-X. Pei, Y.-J. Ren, K.-P. Liu, Z.-Q. Chen, J.-Y. Yang, Y. Su, X.-Y. Liu, W.-Z. Li and T. Zhang, J. Catal., 2018, 366, 115–126 CrossRef CAS
- G. Sun, Z. J. Zhao, R. Mu, S. Zha, L. Li, S. Chen, K. Zang, J. Luo, Z. Li, S. C. Purdy, A. J. Kropf, J. T. Miller, L. Zeng and J. Gong, Nat. Commun., 2018, 9, 4454 CrossRef
- M. D. Marcinkowski, M. T. Darby, J. Liu, J. M. Wimble, F. R. Lucci, S. Lee, A. Michaelides, M. Flytzani-Stephanopoulos, M. Stamatakis and E. C. H. Sykes, Nat. Chem., 2018, 10, 325–332 CrossRef CAS
- Z. Han, S. Li, F. Jiang, T. Wang, X. Ma and J. Gong, Nanoscale, 2014, 6, 10000–10008 RSC
- S. Veldurthi, C.-H. Shin, O.-S. Joo and K.-D. Jung, Catal. Today, 2012, 185, 88–93 CrossRef CAS
- Z. Wu, B. C. Bukowski, Z. Li, C. Milligan, L. Zhou, T. Ma, Y. Wu, Y. Ren, F. H. Ribeiro, W. N. Delgass, J. Greeley, G. Zhang and J. T. Miller, J. Am. Chem. Soc., 2018, 140, 14870–14877 CrossRef
- X. Fan, D. Liu, X. Sun, X. Yu, D. Li, Y. Yang, H. Liu, J. Diao, Z. Xie, L. Kong, X. Xiao and Z. Zhao, J. Catal., 2020, 389, 450–460 CrossRef CAS
- E. C. Wegener, B. C. Bukowski, D. Yang, Z. Wu, A. J. Kropf, W. N. Delgass, J. Greeley, G. Zhang and J. T. Miller, ChemCatChem, 2020, 12, 1325–1333 CrossRef CAS
- W. Cai, R. Mu, S. Zha, G. Sun, S. Chen, Z.-J. Zhao, H. Li, H. Tian, Y. Tang, F. Tao, L. Zeng and J. Gong, Sci. Adv., 2018, 4, eaar5418 CrossRef CAS
- L. G. Cesar, C. Yang, Z. Lu, Y. Ren, G. Zhang and J. T. Miller, ACS Catal., 2019, 9, 5231–5244 CrossRef CAS
- X. Yang, G. Liu, Y. Li, L. Zhang, X. Wang and Y. Liu, Trans. Tianjin Univ., 2019, 25, 245–257 CrossRef CAS
- S. Rimaz, L. Chen, S. Kawi and A. Borgna, Appl. Catal., A, 2019, 588, 117266 CrossRef CAS
- E. Jimenez-Izal, J.-Y. Liu and A. N. Alexandrova, J. Catal., 2019, 374, 93–100 CrossRef CAS
- N. J. LiBretto, C. Yang, Y. Ren, G. Zhang and J. T. Miller, Chem. Mater., 2019, 31, 1597–1609 CrossRef CAS
- J. Zhu Chen, Z. Wu, X. Zhang, S. Choi, Y. Xiao, A. Varma, W. Liu, G. Zhang and J. T. Miller, Catal. Sci. Technol., 2019, 9, 1349–1356 RSC
- C. Ye, Z. Wu, W. Liu, Y. Ren, G. Zhang and J. T. Miller, Chem. Mater., 2018, 30, 4503–4507 CrossRef CAS
- R. Ryoo, J. Kim, C. Jo, S. W. Han, J. C. Kim, H. Park, J. Han, H. S. Shin and J. W. Shin, Nature, 2020, 585, 221–224 CrossRef CAS
- M. Aly, E. L. Fornero, A. R. Leon-Garzon, V. V. Galvita and M. Saeys, ACS Catal., 2020, 10, 5208–5216 CrossRef CAS
- C. Byron, S. Bai, G. Celik, M. S. Ferrandon, C. Liu, C. Ni, A. Mehdad, M. Delferro, R. F. Lobo and A. V. Teplyakov, ACS Catal., 2019, 10, 1500–1510 CrossRef
- E. T. Baxter, M.-A. Ha, A. C. Cass, H. Zhai, A. N. Alexandrova and S. L. Anderson, J. Phys. Chem. C, 2018, 122, 1631–1644 CrossRef CAS
- M. A. Ha, E. T. Baxter, A. C. Cass, S. L. Anderson and A. N. Alexandrova, J. Am. Chem. Soc., 2017, 139, 11568–11575 CrossRef CAS
- J. Dadras, E. Jimenez-Izal and A. N. Alexandrova, ACS Catal., 2015, 5, 5719–5727 CrossRef CAS
- L. Rodríguez, D. Romero, D. Rodríguez, J. Sánchez, F. Domínguez and G. Arteaga, Appl. Catal., A, 2010, 373, 66–70 CrossRef
- N. Taccardi, M. Grabau, J. Debuschewitz, M. Distaso, M. Brandl, R. Hock, F. Maier, C. Papp, J. Erhard, C. Neiss, W. Peukert, A. Gorling, H. P. Steinruck and P. Wasserscheid, Nat. Chem., 2017, 9, 862–867 CrossRef CAS
- J. R. Gallagher, D. J. Childers, H. Zhao, R. E. Winans, R. J. Meyer and J. T. Miller, Phys. Chem. Chem. Phys., 2015, 17, 28144–28153 RSC
- Z. Wu, E. C. Wegener, H.-T. Tseng, J. R. Gallagher, J. W. Harris, R. E. Diaz, Y. Ren, F. H. Ribeiro and J. T. Miller, Catal. Sci. Technol., 2016, 6, 6965–6976 RSC
- C. Yang, Z. Wu, G. Zhang, H. Sheng, J. Tian, Z. Duan, H. Sohn, A. J. Kropf, T. Wu, T. R. Krause and J. T. Miller, Catal. Today, 2019, 323, 123–128 CrossRef CAS
- G. Zhang, C. Ye, W. Liu, X. Zhang, D. Su, X. Yang, J. Z. Chen, Z. Wu and J. T. Miller, Nano Lett., 2019, 19, 4380–4383 CrossRef CAS
-
https://apps.catalysts.basf.com/apps/eibprices/mp/defaultmain.aspx
- M. Wolf, N. Raman, N. Taccardi, M. Haumann and P. Wasserscheid, ChemCatChem, 2020, 12, 1085–1094 CrossRef CAS
- N. Raman, S. Maisel, M. Grabau, N. Taccardi, J. Debuschewitz, M. Wolf, H. Wittkamper, T. Bauer, M. Wu, M. Haumann, C. Papp, A. Gorling, E. Spiecker, J. Libuda, H. P. Steinruck and P. Wasserscheid, ACS Catal., 2019, 9, 9499–9507 CrossRef CAS
- Z. Yan and D. W. Goodman, Catal. Lett., 2012, 142, 517–520 CrossRef CAS
- G. Wang, H. Wang, H. Zhang, Q. Zhu, C. Li and H. Shan, ChemCatChem, 2016, 8, 3137–3145 CrossRef CAS
- Y. He, Y. Song and S. Laursen, ACS Catal., 2019, 9, 10464–10468 CrossRef CAS
- Y. He, Y. Song, D. A. Cullen and S. Laursen, J. Am. Chem. Soc., 2018, 140, 14010–14014 CrossRef CAS
- A. Hook, J. D. Massa and F. E. Celik, J. Phys. Chem. C, 2016, 120, 27307–27318 CrossRef CAS
- L. Nykänen and K. Honkala, ACS Catal., 2013, 3, 3026–3030 CrossRef
- I. Horiuti and M. Polanyi, Trans. Faraday Soc., 1934, 30, 1164–1172 RSC
- R. D. Cortright, P. E. Levin and J. A. Dumesic, Ind. Eng. Chem. Res., 1998, 37, 1717–1723 CrossRef CAS
- J. M. Hill, R. D. Cortright and J. A. Dumesic, Appl. Catal., A, 1998, 168, 9–21 CrossRef CAS
- M. T. Paffett, S. C. Gebhard, R. G. Windham and B. E. Koel, Surf. Sci., 1989, 223, 449–464 CrossRef CAS
- Y.-L. Tsai, C. Xu and B. E. Koel, Surf. Sci., 1997, 385, 37–59 CrossRef CAS
- J. Shen, J. M. Hill, R. M. Watwe, B. E. Spiewak and J. A. Dumesic, J. Phys. Chem. B, 1999, 103, 3923–3934 CrossRef CAS
- S. B. Mohsin, M. Trenary and H. J. Robota, J. Phys. Chem., 1988, 92, 5229–5233 CrossRef CAS
- T. J. Gorey, B. Zandkarimi, G. Li, E. T. Baxter, A. N. Alexandrova and S. L. Anderson, ACS Catal., 2020, 10, 4543–4558 CrossRef CAS
- C. Coperet, Nat. Energy, 2019, 4, 1018–1024 CrossRef CAS
- M. K. Samantaray, V. D'Elia, E. Pump, L. Falivene, M. Harb, S. Ould Chikh, L. Cavallo and J.-M. Basset, Chem. Rev., 2020, 120, 734–813 CrossRef CAS
- H. Zhu, D. H. Anjum, Q. Wang, E. Abou-Hamad, L. Emsley, H. Dong, P. Laveille, L. Li, A. K. Samal and J.-M. Basset, J. Catal., 2014, 320, 52–62 CrossRef CAS
- L. Liu, M. Lopez-Haro, C. W. Lopes, C. Li, P. Concepcion, L. Simonelli, J. J. Calvino and A. Corma, Nat. Mater., 2019, 18, 866–873 CrossRef CAS
- L. Liu, M. Lopez-Haro, C. W. Lopes, S. Rojas-Buzo, P. Concepcion, R. Manzorro, L. Simonelli, A. Sattler, P. Serna, J. J. Calvino and A. Corma, Nat. Catal., 2020, 3, 628–638 CrossRef CAS
- L. Liu, M. Lopez-Haro, C. W. Lopes, D. M. Meira, P. Concepcion, J. J. Calvino and A. Corma, J. Catal., 2020, 391, 11–24 CrossRef CAS
- J. Zhu, R. Osuga, R. Ishikawa, N. Shibata, Y. Ikuhara, J. N. Kondo, M. Ogura, J. Yu, T. Wakihara, Z. Liu and T. Okubo, Angew. Chem., Int. Ed., 2020, 59(44), 19669–19674 CrossRef CAS
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |