
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of 7-amino-6-halogeno-3-phenylquinoxaline-2-carbonitrile 1,4-dioxides: a way forward for targeting hypoxia and drug resistance of cancer cells†
Galina I. Buravchenkoab,
Alexander M. Scherbakov c,
Lyubov G. Dezhenkovaa,
Lianet Monzoted and
Andrey E. Shchekotikhin*a
c,
Lyubov G. Dezhenkovaa,
Lianet Monzoted and
Andrey E. Shchekotikhin*a
aGause Institute of New Antibiotics, 11 B. Pirogovskaya Street, Moscow 119021, Russia. E-mail: shchekotikhin@mail.ru
bMendeleyev University of Chemical Technology, 9 Miusskaya Square, Moscow 125190, Russia
cBlokhin National Medical Research Center of Oncology, 24 Kashirskoye Sh., Moscow 115522, Russia
dDepartment of Parasitology, Pedro Kouri Tropical Medicine Institute, Havana, Cuba
First published on 3rd December 2021
Abstract
To establish a new approach for the synthesis of quinoxaline 1,4-dioxides as hypoxia-selective cytotoxic agents, an original multi-step preparation of derivatives possessing the diamine moiety at position 7 was evaluated. Herein, we present the synthesis of a series of novel 7-amino-6-halogeno-3-phenylquinoxaline-2-carbonitrile 1,4-dioxides 13a–h, 14a,b,g based on the regioselective Beirut reaction. Comparison of antitumor properties of derivatives possessing the diamine moiety at position 7 with structurally close congeners possessing the corresponding amino groups at position 6 revealed key differences in the cytotoxicity profiles and HIF-1α inhibition. All the synthesized 7-amino-6-halogeno derivatives 13a–h, 14a,b,g demonstrated significant cytotoxic activities against breast cancer cell lines (MCF7, MDA-MB-231) in normoxia and hypoxia with IC50 values ranging from 0.1 to 7.6 μM. Most of these novel derivatives can circumvent the multidrug resistance of tumor cells caused by P-glycoprotein over expression. The lead compounds 13a, 14a and 14b can suppress the expression of HIF-1α at low micromolar concentrations and induce apoptosis in breast cancer MCF7 cells. In addition, compound 14b effectively inhibits BCL2 and ERα expression in MCF7 cells. The current research opens a new direction for targeting hypoxia and drug resistance of cancer cells.
Introduction
The main challenge of rapidly proliferating solid tumors is hypoxia.1 This phenomenon promotes chemo- and radio-resistance of cancer cells in hypoxic niches. An increase in hypoxic regions in tumor tissue correlates with the worst outcome of the disease and low effectiveness of treatment.2,3 Moreover, hypoxia in cancers changes cellular metabolism, leading to a more aggressive phenotype, and increases the expression of genes associated with angiogenesis and invasion tending toward metastasis in human cancers. Thus, this general hallmark of tumor cells, hypoxia, is one of the main targets for the development of new selective cytotoxic drugs for hypoxic cells. One way of targeting tumor hypoxia is to design hypoxic prodrugs that are activated in hypoxic tissue and thus selectively kill hypoxic tumor cells.4 Cellular reductases can convert such prodrugs to agents with impressive antiproliferative potency against hypoxic tumor cells. Several classes of compounds display hypoxia selectivity in solid tumors, such as DNA alkylating agents, nitroaromatic compounds, and aromatic N-oxides.5 The most potent compound in this class is tirapazamine (TPZ) based on the benzotriazine 1,4-di-N-oxide scaffold (1, Fig. 1). The hypoxic prodrug, tirapazamine, has been extensively tested in clinical trials, but results have been disappointing because of its poor extravascular transport. However, the unique one-electron reduction activation mechanism and encouraging antitumor profile have stimulated intense research efforts in the design and synthesis of a variety of its analogues. Based on the structural similarity between TPZ and quinoxaline 1,4-dioxide, a series of quinoxaline-2-carbonitrile 1,4-dioxides, 2–4 (ref. 6–9), have been synthesized and evaluated. Selected derivatives have shown a high hypoxia selectivity and antiproliferative potency against a panel of wild-type and drug-resistant tumor cell lines.9 Additionally, in an in-depth study of the Beirut reaction between monosubstituted benzofuroxans and benzoylacetonitrile, we identified isomeric 6(7)-monosubstituted quinoxaline-2-carbonitrile 1,4-dioxide, 2, which demonstrated significant differences in biological activity. Thus, in search for new knowledge regarding the possibility of chemical modification of the quinoxaline scaffold and for further exploration of the structure–activity relationships for quinoxaline-2-carbonitrile 1,4-dioxides, a novel water-soluble derivative with residues of cyclic diamines was obtained.10 Lead compound 4b (for R = F, Fig. 1) suppressed HIF-1α expression and demonstrated a strong anti-estrogenic effect.10 The position of the pharmacophore group in the heterocyclic core of 3-phenylquinoxaline-2-carbonitrile 1,4-dioxides (e.g., compounds 3–4, Fig. 1) and its structure are of fundamental importance for antiproliferative potency and hypoxic selectivity of these derivatives.10,11 Thus, derivative 3 with the piperazine residue in position 7 was noticeably more potent against MCF7 and MDA-MB-231 under normoxic (10 times) and hypoxic conditions (20 times) than its regioisomer 4a (for R = H, Fig. 1). Similar differences in anticancer activity were observed for the series of isomeric derivatives, 2 (ref. 8).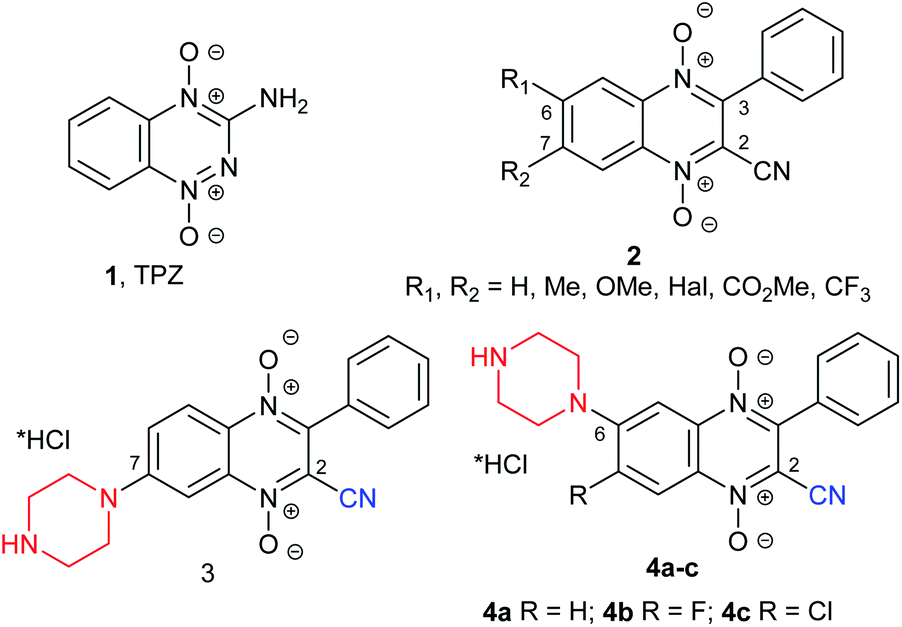 | ||
| Fig. 1 Structures of the previously obtained hypoxia-selective cytotoxic agents. | ||
Analysis of the antiproliferative properties of derivatives 2–4 (Fig. 1) showed that the position of the diamino residues in the quinoxaline ring may significantly effect interactions with targets and antiproliferative profiles. Thus, for in-depth analysis of the structural modifications of quinoxaline 1,4-dioxides, in this study, we aimed to estimate the role of the position of the cyclic diamino moiety in quinoxaline-2-carbonitrile 1,4-dioxides. For this purpose, a series of 6-halogeno-3-phenylquinoxaline-2-carbonitrile 1,4-dioxides bearing the basic fragment at position 7 in the quinoxaline ring were obtained and evaluated.
Results and discussion
Synthesis
Earlier, it was shown that halogen atoms at position 6 of quinoxaline-2-carbonitrile 1,4-dioxides (due to activation with the electron-withdrawal effect of the cyano group) were more active than in position 7 in the reaction of nucleophilic substitution. Thus, the treatment of 6,7-dihalogeno-3-phenylquinoxaline-2-carbonitrile 1,4-dioxides 2 (R1 = R2 = Hal) with amines exclusively leads to 6-amino derivatives 4b and 4c (ref. 10). Meanwhile, the revision of the Beirut reaction showed that condensation of benzofuroxans with electron-donating substituents (for example, methyl and methoxy groups) and benzoylacetonitrile leads only to the 7-isomers (derivatives 2, R1 = H, R2 = Me, OMe; Fig. 1).8 Therefore, for the preparation of the 7-aminoderivatives of quinoxalin-2-carbonitrile 1,4-dioxides, we developed an original scheme based on similar heterocyclization of 5-aminosubstituted benzofuroxans with benzoylacetonitrile via the Beirut reaction (Schemes 1, 2).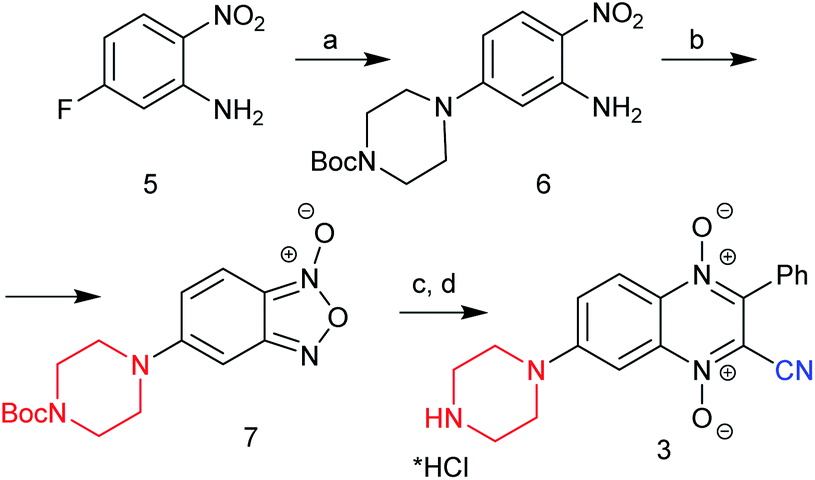 | ||
| Scheme 1 New route for the synthesis of 7-(piperazin-1-yl)-quinoxaline-2-carbonitrile 1,4-dioxide (3). Reagents and conditions: (a) 1-Boc-piperazine, DMF, TEA, 50 °C, 4 h, 86%; (b) NaOCl, KOH(aq), DMF, 0–5 °C, 10–15 min, 81%; (c) BzCH2CN, Et3N, CHCl3, r. t., 3 h, 71%; (d) HCl/MeOH, r.t., 68%. | ||
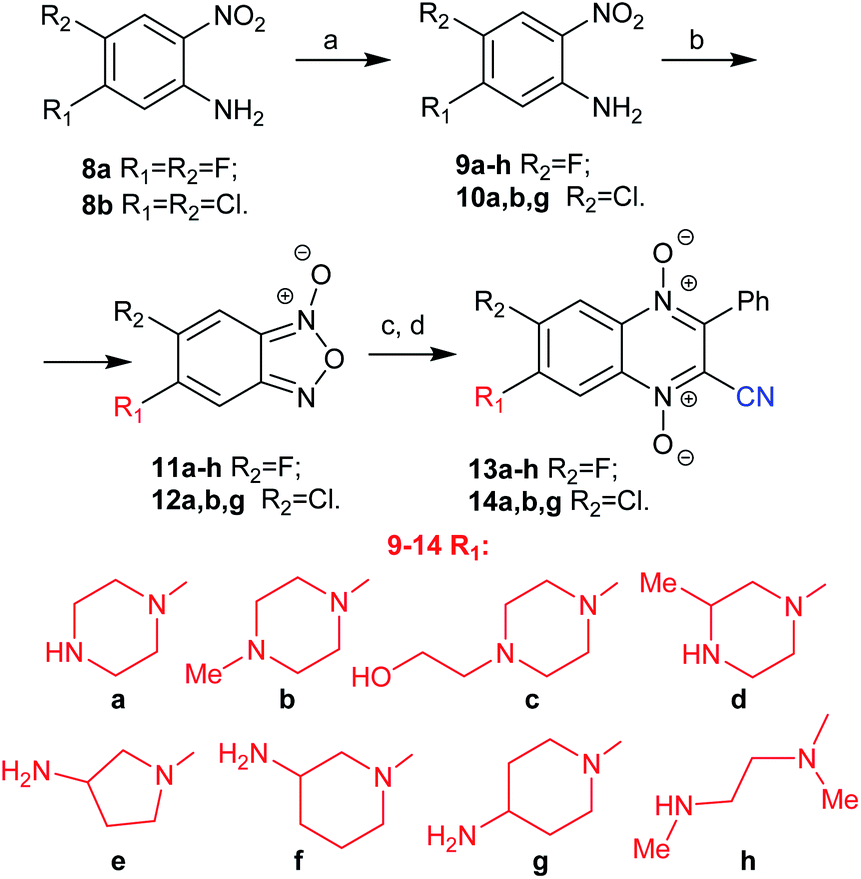 | ||
| Scheme 2 Synthesis of 7-amino-6-halogeno-3-phenyl-quinoxaline 1,4-dioxides 13a–h and 14a,b,g. Reagents and conditions: (a) N-Boc-protected diamine or diamine, DMF, TEA, r.t., 2–5 h, 61–90%; (b) NaOCl, KOH(aq), DMF, 0–5 °C, 10–15 min, 72–93%.; (c) benzoylacetonitrile, TEA, CHCl3, r.t., 2 h; (d) HCl/MeOH or MsOH, r.t., 23–82%. | ||
First, for confirmation of this approach for the synthesis of the target 7-aminoquinoxaline-2-carbonitrile 1,4-dioxides, we examined the efficacy of this scheme for the preparation of 7-piperazinyl derivative 3, which was obtained early from 7-fluoro derivative 2 (R1 = H, R2 = F; Fig. 1). Starting with 5-fluoro-2-nitroaniline (5) by nucleophilic substitution of an activated halogen atom at position 5 with Boc-piperazine, it converted into 2-nitro-5-(Boc-piperazinyl)aniline (6) in high yield (Scheme 1). The next step of this synthetic pathway was the preparation of amino-substituted benzofuroxans, which are key intermediates for the subsequent Beirut heterocyclization. Several methods can be successfully used for the synthesis of benzofuroxans.12–15 Among them, the most effective include thermolysis of 2-nitroarylazides and oxidative cyclization of 2-nitroanilines. However, the main limiting factors for the synthesis of benzofuroxans from 2-nitroarylazides are the two-stage method and the explosiveness of the arylazides used during isolation.15 Therefore, to obtain the key benzofuroxan, the one-stage method of oxidative cyclization of 2-nitroanilines was examined because it proceeds under mild conditions and does not require hazardous reagents. This method was found to be suitable for the preparation of aminobenzofuroxans. Thus, by treatment with sodium hypochlorite in the presence of a base, nitroaniline 6 can be cyclized into 5-Boc-aminobenzofuroxan 7 in high yield (Scheme 1). During the optimization of the conditions for the oxidation of 2-nitroaniline 6, solvents, such as MeOH, EtOH, THF and DMF, and bases, such as NaOH and KOH, were tested. The highest yield of the target benzofuroxan 7 was achieved using an aqueous solution of potassium hydroxide as a base in DMF.
Due to the high efficiency of the previously described method for the synthesis of quinoxaline-2-carbonitrile 1,4-dioxides by the Beirut reaction,8,9,16,17 a similar procedure was used for the cyclization of the benzofuroxan 7 to quinoxaline-2-carbonitrile 1,4-dioxide 3. Thus, the condensation of benzofuroxan 7 with benzoylacetonitrile in the presence of triethylamine afforded the corresponding Boc-derivative of 3 in a good yield (Scheme 1). The obtained Boc-intermediate of 3 was further purified by column chromatography and converted into the target quinoxaline-2-carbonitrile 1,4-dioxide 3 by treatment with a solution of HCl in methanol. Of note, the quinoxaline 1,4-dioxide 3 (Scheme 1), formed from benzofuroxan 7, according to the physicochemical and spectral properties, was identical to derivative 3 (Fig. 1) previously obtained from quinoxaline 1,4-dioxide 2 (R1 = H, R2 = F) (ESI, Fig. S105†).8,9 Thus, the identical result of the counter synthesis additionally confirmed the position of the amino group in the quinoxaline core and supported the previous conclusion that the presence of electron-donating groups in the benzofuroxans contribute to the exceptional formation of 7-substituted quinoxaline-2-carbonitrile 1,4-dioxides in the Beirut reaction.8
Because the developed method was suitable for the synthesis of 7-aminoquinoxaline-2-carbonitrile 1,4-dioxide 3 (Scheme 1), it was used for the synthesis of an extended series of analogues with variation of the diamine residue at position 7 and the halogen atom at position 6 of the heterocyclic core. Thus, by substitution of the halogen atoms at the 5 position of nitroanilines 8a and 8b with different diamines a–h, 5-aminonitroanilines 9a–h and 10a,b,g were obtained in good yields (Scheme 2). The mono Boc-protected diamines were used in the case of amines a and d–h. Additionally, to study the impact of the amino group on the biological properties of 3-phenylquinoxalin-2-carbonitrile 1,4-dioxide, linear diamine h was used. The procedure for the synthesis of benzofuroxan 7 (Scheme 1) was tested for the preparation of aminobenzofuroxans 11a–h and 12a,b,g. Thus, the oxidation of anilines 9a–h and 10a,b,g with sodium hypochlorite in the presence of potassium hydroxide proceeds smoothly even in the case of derivatives b and c with ternary amino groups on the side chain and gave the key aminobenzofuroxans 11a–h and 12a,b,g in good yield (Scheme 2).
The subsequent Beirut condensation of benzofuroxans 11a–h and 12a,b,g with benzoylacetonitrile in the presence of triethylamine in chloroform selectively yielded the derivatives of 7-amino-6-halogenoquinoxaline-2-carbonitrile 1,4-dioxides 13a–h and 14a,b,g without their regioisomers (Scheme 2).
In the case of fluorine derivatives, the Boc intermediates were transformed into hydrochlorides of the target 7-amino-6-fluoroquinoxaline-2-carbonitrile 1,4-dioxides 13a–h by treatment with HCl in methanol. Comparison of the solubility of derivative 4a with hydrochlorides of 4b–c, 13a and 14a showed that the introduction of a halogen atom into the benzene ring of quinoxaline 1,4-dioxides significantly decreased their solubility in water (ESI, Table S2†). Thus, the hydrochlorides of chlorine derivatives 14a,b,g were poorly soluble in water, while their methanesulfonates obtained by treatment of the Boc intermediates with methanesulfonic acid had noticeably higher solubility (ESI, Table S2†). Therefore, the studies of the biological and physicochemical properties of compounds 14a,b,g were carried out for their mesylates.
All final compounds of 13a–h and 14a,b,g were further purified by re-precipitation of their hydrochloride or mesylate salts. The purified compounds (purity >95%, HPLC) were used for analytical and biological studies. The structure of undescribed benzofuroxanes 7, 11a–h, and 14a,b,g were studied by 1H and 13C NMR analysis at 75 °C in DMSO-d6.18 When 1H and 13C NMR spectra of 11a were recorded at room temperature, the aromatic region demonstrated very broad and poorly interpreted signals (ESI, Fig. S23 and S24†). Thus, at high temperature (75 °C), the signals of the aromatic region in 1H and 13C NMR spectra were observed as precise narrow peaks, except for the signals regarding the quaternary carbon atoms of C-8 and C-9 (ESI, Fig. S25 and S26†).18 The structure of quinoxalin-2-carbonitrile 1,4-dioxide 3 (Fig. 1 and Scheme 1) was confirmed by HRMS ESI, HPLC, and NMR spectroscopies. Of note, in contrast to the earlier described 7(6)-monosubstituted quinoxaline-2-carbonitrile-1,4-dioxides 2 (R1 = H), 3, and 4a, 7-amino-6-halogenderivatives 13a, 14a, and their previously obtained regioisomers 4b–c showed differences in mobility on TLC and in retention times (tR) in HPLC. As in the case of 6(7)-aminoderivatives 3 and 4a, regioisomers 13a, 14a, 4b and 4c have identical 1H NMR spectra and different 13C NMR spectra, which made it possible to distinguish the isomers from each other.
Comparison of the chemical shifts of the signals in the 13C NMR spectra of 3-phenylquinoxaline-2-carbonitrile 1,4-dioxide8 with the corresponding signals of 7- and 6-amino derivatives 13a, 14a, 4b, and 4c showed that changes in the chemical shifts of the signals correspond with increments (IC) of the tertiary amino group in the 13C NMR spectra of arenes19,20 (ESI, Table S1†). Introduction of the amino group at position 7 (for 13a and 14a) led to a significant shift in signals in the para-position of the benzene ring (C-10) and pyrazine fragment (C-3). Thus, the analysis of the chemical shift increments in 13C NMR spectra confirmed the position of the amino group in the quinoxaline 1,4-dioxides obtained in the Beirut reaction.
Antiproliferative activity
Novel 7-amino-6-halogeno-3-penylquinoxaline-2-carbonitrile 1,4-dioxides 13a–h and 14a,b,g were tested against several mammalian cancer cell lines, including breast cancer cell lines MCF7 and MDA-MB-231, under normoxic (21% O2) and hypoxic (1% O2) conditions and human myeloid leukemia K562 and its isogenic drug-resistant counterpart K562/4 (selected for resistance to doxorubicin (Dox); Pgp-positive).For the antiproliferative potencies of derivatives 13a–h and 14a,b,g, reference compounds 4b, 4c, TPZ, Dox, and CisPt are summarized in Tables 1 and 2. All new derivatives 13a–h and 14a,b,g inhibited tumor cell proliferation at submicromolar or low micromolar concentrations. The majority of synthesized compounds demonstrated high antiproliferative activities against adenocarcinoma cells, which were close to that of the piperazine derivatives 4b and 4c (Table 1). Moreover, the cytotoxic potencies of the synthesized quinoxalines 13a–h and 14a,b,g were 4.2–34.6 times and 1.2–45 times (under normoxic and hypoxic conditions, respectively) higher than for the reference TPZ agent against estrogen-dependent MCF7 breast cancer cells. Of note, the triple-negative breast cancer cell line MDA-MB-231 was a more sensitive cell line for the obtained compounds 13a–h and 14a,b,g, as observed in the IC50 values, which were 5.2–49.4 times and 10–73 times, respectively, higher than those for TPZ. Additionally, as in the case of the previously obtained compounds (e.g., 3–4), the inhibitory potency of these derivatives increased under hypoxic conditions. The most hypoxia-selective was the 6-chloro-7-methylpiperazine derivative 14b, which showed 7.4- and 12.9-fold improvement compared with TPZ (HCR values of 5.4 and 1.8) with HCR values of 39.8 and 23.3, respectively. Of note, the structure of the diamine residue plays an important role in the biological activity of the derivatives of this scaffold. Thus, its analogues with different cyclic diamino groups in the side chain were less selective under hypoxic conditions but more potent for MCF7 and MDA-MB-231 with similar IC50 values against these tumor cell lines. In striking contrast, compound 13h with the linear N,N′-dimethylethylenediamino residue showed no activity against all tested cell lines. The relocation of the cyclic amino groups in the halogen-substituted 3-phenylquinoxaline-2-carbonitrile 1,4-dioxides 13a–g and 14a,b,g led to an increase in the cytotoxic activity of these derivatives compared to the previously obtained 6-amino regioisomers10 but significantly reduced their solubility. Thus, in contrast to the previously obtained monosubstituted quinoxaline-2-carbonitrile 1,4-dioxides 3 and 4a, fluoroderivatives 13a–g demonstrated 3–6 times higher IC50 values under normoxic conditions than the previously obtained regioisomers against the tested adenocarcinoma cell lines.10 Interestingly, replacement the fluorine atom at the 6 position with chlorine decreased the cytotoxicity of 14a,b,g under normoxic conditions but improved the activity and selectivity in hypoxia.
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | IC50b (μM) | |||||
| MCF7 | HCRa | MDA-MB-231 | HCRa | |||||
| Nc | Hd | Nc | Hd | |||||
| a HCR, hypoxic cytotoxicity ratio: IC50(N)/IC50(H).b IC50, μM (mean ± S.D. of 3 experiments).c N = Normoxia: 21% of oxygen.d H = Hypoxia: 1% of oxygen. | ||||||||
| 13a |  |
F | 0.78 ± 0.08 | 0.65 ± 0.07 | 1.2 | 1.41 ± 0.02 | 0.61 ± 0.07 | 2.3 |
| 13b | 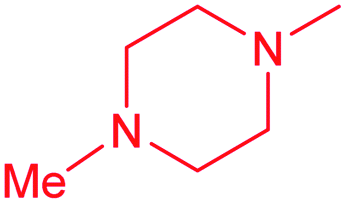 |
F | 1.1 ± 0.1 | 1.2 ± 0.1 | 0.9 | 1.72 ± 0.16 | 0.90 ± 0.08 | 1.9 |
| 13c | 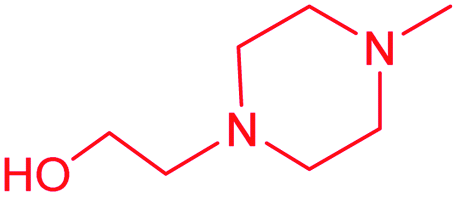 |
F | 1.6 ± 0.2 | 0.8 ± 0.07 | 2.0 | 3.8 ± 0.2 | 1.7 ± 0.1 | 2.2 |
| 13d | 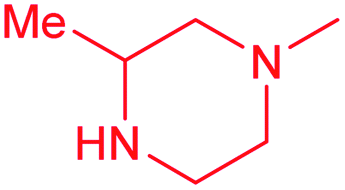 |
F | 0.93 ± 0.08 | 0.61 ± 0.07 | 1.5 | 0.89 ± 0.07 | 0.49 ± 0.06 | 1.8 |
| 13e | 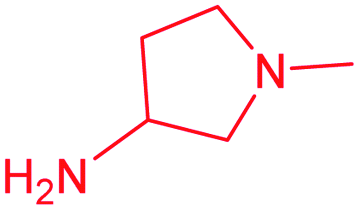 |
F | 1.9 ± 0.2 | 2.1 ± 0.2 | 0.9 | 3.1 ± 0.2 | 1.6 ± 0.2 | 1.9 |
| 13f | 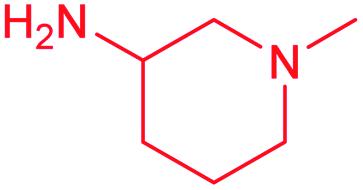 |
F | 1.6 ± 0.2 | 0.80 ± 0.08 | 2.0 | 2.0 ± 0.1 | 1.0 ± 0.1 | 2.0 |
| 13g | 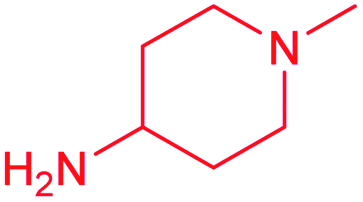 |
F | 0.74 ± 0.08 | 0.62 ± 0.08 | 1.2 | 1.2 ± 0.1 | 0.57 ± 0.06 | 2.1 |
| 13h |  |
F | 21.8 ± 2.0 | 22.8 ± 2.1 | 1.0 | 18.0 ± 2.0 | 19.0 ± 2.2 | 0.9 |
| 14a | 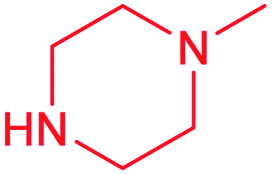 |
Cl | 0.92 ± 0.08 | 0.22 ± 0.03 | 4.2 | 1.0 ± 0.1 | 0.40 ± 0.05 | 2.5 |
| 14b | 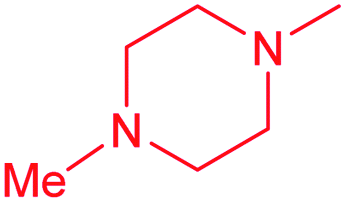 |
Cl | 5.8 ± 0.5 | 0.15 ± 0.1 | 39.8 | 7.0 ± 0.8 | 0.30 ± 0.04 | 23.3 |
| 14c | 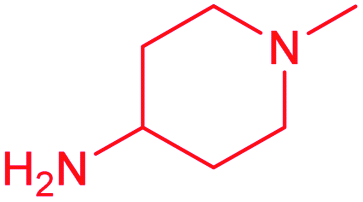 |
Cl | 5.0 ± 0.5 | 3.8 ± 1.1 | 1.3 | 7.6 ± 0.7 | 2.2 ± 0.2 | 3.5 |
| 4b | F | 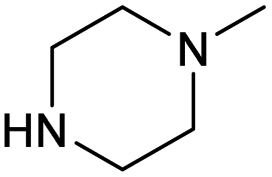 |
2.0 ± 0.1 | 0.42 ± 0.03 | 4.7 | 1.5 ± 0.1 | 0.35 ± 0.02 | 4.3 |
| 4c | Cl | 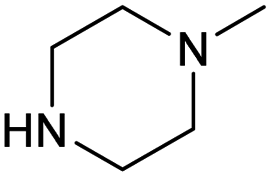 |
0.82 ± 0.05 | 0.23 ± 0.02 | 3.6 | 0.90 ± 0.05 | 0.29 ± 0.02 | 3.1 |
| TPZ | — | — | 24.2 ± 3.3 | 4.5 ± 1.1 | 5.4 | 39.5 ± 5.4 | 22.0 ± 2.1 | 1.8 |
| Dox | — | — | 0.31 ± 0.03 | 0.44 ± 0.03 | 0.7 | 0.32 ± 0.03 | 0.21 ± 0.01 | 1.5 |
| CisPt | — | — | 8.3 ± 0.8 | 11.4 ± 2.3 | 0.7 | 13.0 ± 1.2 | 16.2 ± 1.0 | 0.8 |
| Compound | IC50a (μM) | ||
|---|---|---|---|
| K562 | K562/4 | RIb | |
| a IC50, μM (mean ± S.D. of 3 experiments).b RI, resistance index: IC50(K562/4)/IC50(K562). | |||
| 13a | 1.0 ± 0.07 | 1.2 ± 0.07 | 1.2 |
| 13b | 0.6 ± 0.02 | 1.1 ± 0.07 | 1.8 |
| 13c | 2.5 ± 0.2 | 1.6 ± 0.09 | 0.6 |
| 13d | 1.8 ± 0.09 | 1.2 ± 0.06 | 0.7 |
| 13e | 1.0 ± 0.05 | 1.3 ± 0.05 | 1.3 |
| 13f | 1.0 ± 0.03 | 0.4 ± 0.03 | 0.4 |
| 13g | 1.1 ± 0.4 | 1.3 ± 0.08 | 1.2 |
| 13h | >50 | >50 | — |
| 14a | 3.0 ± 0.3 | 3.5 ± 0.5 | 1.1 |
| 14b | 3.0 ± 0.5 | 4.0 ± 0.6 | 1.3 |
| 14g | 5.5 ± 0.7 | 6.0 ± 0.8 | 1.1 |
| 4b | 4.2 ± 0.5 | 4.0 ± 0.6 | 0.9 |
| 4c | 2.0 ± 0.1 | 1.5 ± 0.2 | 0.8 |
| Dox | 0.3 ± 0.02 | 5.0 ± 0.7 | 11.7 |
Because decreased drug accumulation is one of the reasons for the MDR phenotype of tumor cells, the development of chemotherapeutic agents that can circumvent the P-glycoprotein (Pgp)-mediated efflux of numerous clinical drugs, including the “gold standard” doxorubicin (Dox), is an urgent goal in medicine. Importantly, most new derivatives are similarly potent for wild-type leukemia cells (K562) and sublines with an altered chemotherapeutic drug response (K562/4) with the expression of functional Pgp, which are critical for the cytotoxic properties of Dox.21–24 As shown in Table 2, 13a–g (except for derivative 13h) all effectively inhibited the growth of K562 cells as well as MDR-subline K562/4 at submicromolar or low micromolar concentrations. Overall, the introduction of a halogen atom at position 6 of the quinoxaline ring, as in the case of regioisomers 3 and 4b,c, led to an increase in cytotoxicity of derivatives 13a and 14a in comparison with their isomer 4a.10 Importantly, the majority of the obtained derivatives were more potent to Pgp expression of the K562/4 subline than for the paternal cell line of K562. Thus, derivatives 13c, 13d and 13f demonstrated a higher antiproliferative activity against Pgp-positive K562/4 cells than for the wild-type cell line K562, with a resistance index (RI) less than 1.
Interestingly, both the halogen atom and the amino group position had no significant effect on the ability to block the growth of leukemia cells, while the structure of the diamine in the heterocyclic nucleus was found to be of decisive importance for the overall level of activity of the compounds and, more importantly, showed a strong effect on overcoming the resistance of cells with the overexpression of transmembrane transporter Pgp. Compound 13f with the 3-aminopiperidine residue in the side chain notably stands out with its antiproliferative activity against Pgp-positive cells, whereas doxorubicin is 8.8-fold less active against the MDR subline. The resistance index (RI) for the 13f derivative is 30-fold higher compared to that of Dox (RI value of 11.7). Of note, the introduction of the linear diamine into the quinoxaline 1,4-dioxide moiety led to a dramatic decrease in activity against all tested tumor cell lines. Additionally, the 7-aminoderivatives of 6-halogenoquinoxaline-2-carbonitrile 1,4-dioxide showed higher apoptotic potency for both leukemia cell lines (K562; K562/4) than the previously obtained series of their regioisomeric analogues (3, 4).10 In striking contrast to the previously obtained regioisomers 3 and 4a (Fig. 1), compound 14a had similar IC50 values against all tested cell lines compared to its 6-substituted analogue 4b (Fig. 1 and Tables 1 and 2).10 Nevertheless, the location of the substituents also plays an important role in the cytotoxic properties.
Overall, comparison of the cytotoxicity of the obtained derivatives 13a–g with the previously obtained regioisomeric analogues10 showed that compounds 13a–g containing an amino group at position 7 of the quinoxaline core were 2–5 times more cytotoxic than their isomers.
HIF-1α targeting
Three compounds, 13a, 14a and 14b were selected for further in-depth study. The antiproliferative effects of the chosen compounds on MCF7 cells varied in IC50 values from 0.7 to 5.8 μM in normoxia and from 0.1 to 0.6 μM in hypoxia. To analyze the signaling pathways in the MCF7 line, cells were cultivated in hypoxia (1% O2), and then, protein expression was assessed by immunoblotting. Control MCF7 cells were incubated in normoxia (21% O2). As shown in Fig. 2, hypoxia induces a significant accumulation of HIF-1α in MCF7 cells.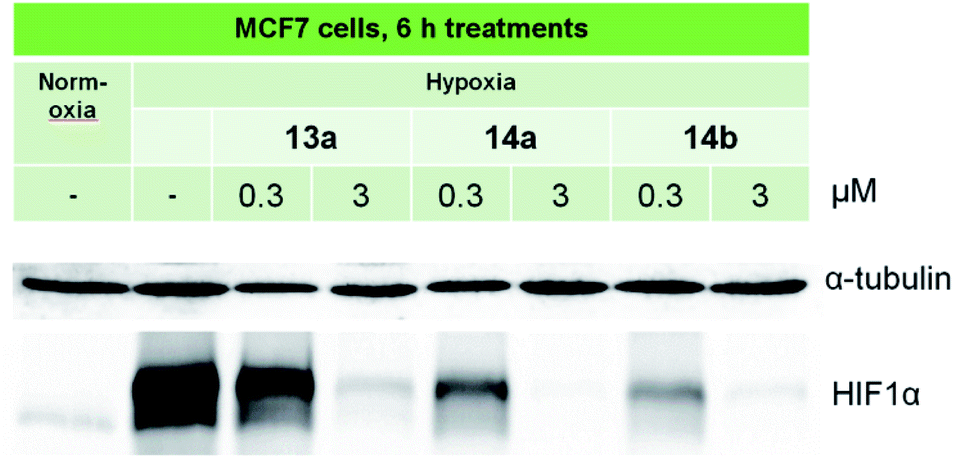 | ||
| Fig. 2 Immunoblotting of the HIF-1α protein in MCF7 cells treated with lead compounds. Antibodies against α-tubulin were used to control loading. | ||
At low concentration, 13a slightly decreased the hypoxia-induced expression of HIF-1α, and the effects of 14a and 14b were more pronounced. At high doses, all three selected compounds completely blocked HIF-1α expression, suggesting their potency as anti-HIF1 agents. More effective inhibition of HIF-1α was revealed for 14b, which was characterized by a high HCR (39.8, Table 1).
Research on HIF inhibition by quinoxaline derivative treatment is under way and is actively being developed by several research groups. Mona Diab-Assef and colleagues have shown that quinoxaline 1,4-dioxides are hypoxia-selective active compounds, and HIF-1α is considered one of their targets. The most cytotoxic 2-benzoyl-3-phenyl-6,7-dichloroquinoxaline 1,4-dioxide (DCQ) was potent at 1 μM with an HCR of 100 and significantly reduced the levels of HIF-1α transcript and protein.25 HIF-1α was inhibited in cell lysates of MCF7 and MDA-MB-231 after 6 h of exposure to 5 μM DCQ under normoxia or hypoxia in one study.26 Thus, DCQ is active at micromolar concentrations, while the new quinoxaline derivatives obtained in our study block the expression of HIF-1α at a concentration of 300 nM.
Hypoxia stimulates the active growth of blood vessels in tumors. The new vessels supply a tumor with oxygen and nutrients, supporting its growth. Blocking angiogenesis (vascular growth) is one of the promising approaches for the treatment of solid neoplasms. Quinoxaline 1,4-dioxides, due to their ability to inhibit HIF-1α, are considered potential agents targeting angiogenesis.27 DCQ showed cytotoxic effects against Lewis lung carcinoma and EMT-6 mammary adenocarcinoma cells under hypoxic conditions as described previously.27 Treatments with DCQ and 2-benzoyl-3-phenylquinoxaline 1,4-dioxide (BPQ) inhibited HIF-1α and VEGF secretion, which are key proangiogenic factors, with DCQ being more potent than BPQ. Another derivative, 3-(4-bromophenyl)-2-(ethylsulfonyl)-6-methylquinoxaline 1,4-dioxide (Q39), has shown great antiproliferative activity in extensive cell lines in normoxia and hypoxia.28 Weng Q. and colleagues showed that pretreatment of SMMC-7721 cells with Q39 at a micromolar concentration significantly inhibited hypoxia-induced HIF-1α protein accumulation in a dose-dependent manner.28 Suppression of HIF-1α accumulation by Q39 correlated with dephosphorylation of mTOR and 4E-BP1.
Assessment of apoptosis
Quinoxaline derivatives are active inducers of tumor cell death, as discussed in several papers.29–32 For instance, quinoxaline dioxide DCQ causes the death of various types of breast cancer. DCQ causes a significant accumulation of ROS in cells, which leads to their death. In the work of Khaled Ghattass and colleagues, it was revealed that 3–5 μM DCQ can induce the death of MCF7 and MDA-MB-231 cells.33 Apoptosis in cells is assessed by the presence of specific markers, and one of these markers is cleaved PARP.34 MCF7 cells were treated with compounds 13a, 14a and 14b for 3 days, and then, the level of full and cleaved PARP was determined in the samples. As shown in Fig. 3, all three selected compounds increased the accumulation of cleaved PARP. Compound 13a causes moderate accumulation of cleaved PARP, whereas 14a and 14b induce its significant accumulation. BCL2 belongs to a large family of proteins that are involved in the regulation of cell death. By blocking the mitochondrial pathway, BCL2 prevents apoptosis, and this protein is considered a target for future therapies.35,36 Quinoxaline drug candidates XK469 and CQS exhibit antiproliferative and proapoptotic effects against cancers, including through the mitochondria.37,38 In a recent work by Yukari Ono and colleagues,38 quinoxaline-1,3,4-oxadiazole hybrid derivatives were considered as potential inhibitors of the anti-apoptotic BCL2 protein. RT-PCR analysis demonstrated that novel derivatives inhibit BCL2 expression.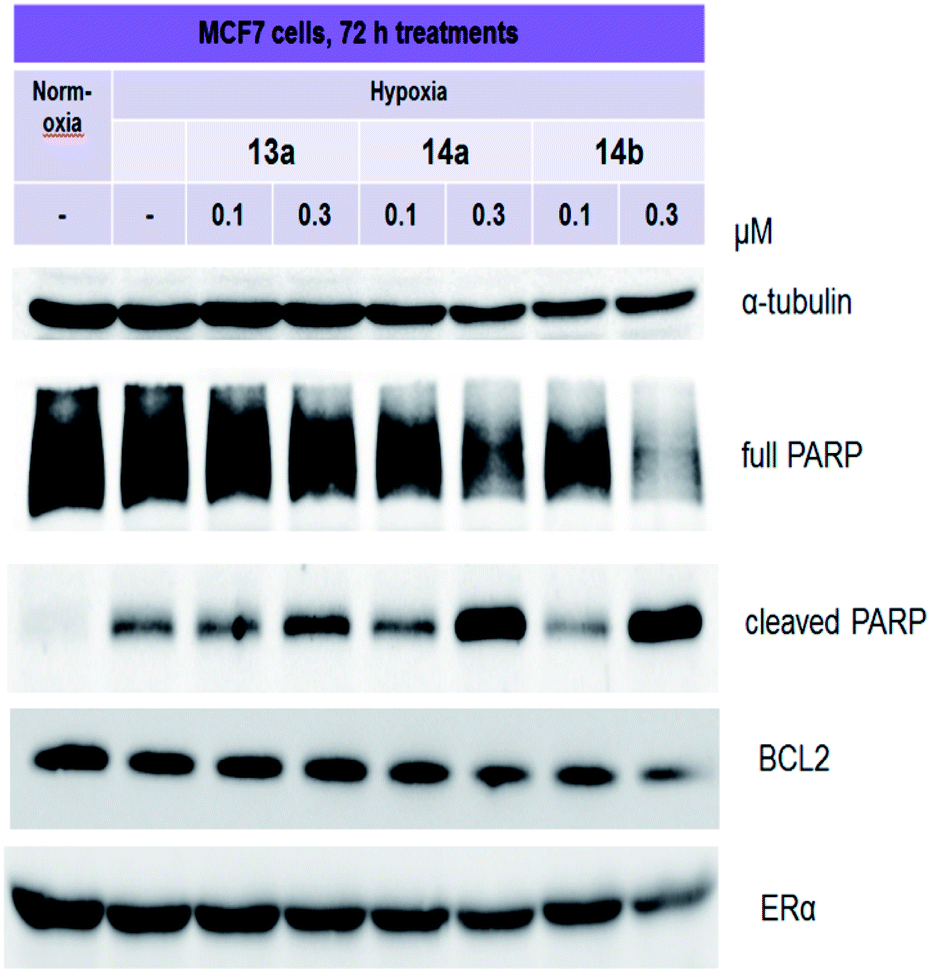 | ||
| Fig. 3 Immunoblotting of PARP, BCL2, and ERα in MCF7 cells treated with lead compounds 13a, 14a and 14b. Antibodies against α-tubulin were used to control loading. | ||
In our work, the expression of BCL2 was analyzed by immunoblotting. Derivative 14a caused a slight decrease in BCL2 expression, whereas the effects of compound 14b were more pronounced. Considering that ERα is the main driver of the growth of hormone-dependent cancers, we analyzed its expression in MCF7 cells. As shown in Fig. 3, at submicromolar concentrations, 14b decreased ERα expression.
Thus, the selected compounds applied at submicromolar concentrations have a complex effect on the signaling pathways in cancer cells: they block HIF1α and induce apoptosis. Moreover, the lead compound 14b inhibits BCL2 and ERα expression.
Conclusions
We developed a convenient method for preparing halogen-substituted quinoxaline-2-carbonitrile 1,4-dioxides bearing a diamino side chain at position 7 of the heterocyclic ring starting from amino-substituted 2-nitroanilines 9a–h and 10a,b,g. The high regioselectivity of the Beirut reaction was shown in the case of benzofuroxans with electron-donating substituents, which made it possible to obtain 7-amino-6-halogenoquinoxaline-2-carbonitrile 1,4-dioxides. The presence of an amino group in the quinoxaline core allowed us to improve the biological properties and the aqueous solubility of these compounds. All the obtained compounds (except 13h with the linear amine residue) were highly efficient against adenocarcinoma cell lines (MCF7, MDA-MB-231). The most hypoxia-selective 6-chlorosubstituted quinoxaline-2-carbonitrile 1,4-dioxide, 14b, bearing the N-methylpiperazine moiety, demonstrated superior HCR values for MCF7 and MDA-MB-231 cell lines at 39.8 and 23.3, respectively, and an IC50 < 0.5 μM under hypoxic conditions. This compound was also 7.4- and 12.9-fold more potent than the reference hypoxic cytotoxin, TPZ (HCR values of 5.4 and 1.8, respectively) (Table 1). Furthermore, compounds 13a–g demonstrated similar toxicity for the MDR cell line K562/4 and its K562 wild-type cells (RI values between 0.4 and 1.8; Table 2) as opposed to being otherwise resistant to Pgp-transported drugs, including doxorubicin (Dox) (RI = 11.7; Table 2). Of note, the introduction of a halogen atom independent of the position in the quinoxaline ring, as in the case of regioisomers 4a, 4b and 4c, leads to an increase in the cytotoxicity of derivatives 13a and 14a compared with that of compound 3.10 It was revealed that the structure of the diamine in the quinoxaline core is of critical importance for the biological activity of such compounds. Thus, the screening of cytotoxicity properties of the obtained compounds showed that derivative 13h with the linear diamine moiety (Tables 1 and 2) in the quinoxaline scaffold exhibited no activity against all tested cancer cells. The selected compounds 13a, 14a and 14b acted via HIF-1α inhibition and induced apoptosis under hypoxia. Moreover, ERα and BCL2 expression was inhibited in hormone-dependent breast cancer cells after treatment with lead compound 14b at submicromolar concentration. Therefore, these derivatives could be potential leaders for further structural modifications and biological studies, especially in vivo evaluations. Novel 7-amino-6-halogeno-3-phenylquinoxaline-2-carbonitrile 1,4-dioxides are of great interest for the development of new approaches for the targeting of tumors with large hypoxic regions.Experimental part
General methods
NMR spectra of all synthesized compounds were recorded on a Varian VXR-400 instrument at 400 MHz (1H NMR) and 100 MHz (13C NMR). Chemical shifts were measured in DMSO-d6 using TMS as an internal standard. NMR spectra of benzofuroxans 7, 11a–h, 10a,b,g were recorded at 80 °C. Analytical TLC was performed on silica gel F254 plates (Merck) and column chromatography on Silica Gel Merck 60. Melting points were determined on a Buchi SMP-20 apparatus and are uncorrected. High-resolution mass spectra were recorded by electron spray ionization on a Bruker Daltonics micro OTOF-QII instrument. UV spectra were recorded on a Hitachi-U2000 spectrophotometer. IR spectra were recorded on Nicolet iS10 Fourier transform IR spectrometer (Nicolet iS10 FT-IR, Madison, WI, USA). HPLC was performed using Shimadzu Class-VP V6·12SP1 system (GraseSmart RP-18, 6 × 250 mm). Eluents: A, H3PO4 (0.01 M); B, MeCN. All solutions were evaporated at reduced pressure on a Buchi-R200 rotary evaporator at a temperature below 50 °C. All solvents, chemicals, and reagents were obtained from Sigma-Aldrich (unless specified otherwise) and used without purification. The purity of compounds 13a–h and 14a,b,g was >95% as determined by HPLC analysis. All products were dried under vacuum at room temperature. The synthesis of compounds 9b, 10b based on the nucleophilic substitution of halogen atom in the nitroanilines 8a–b and subsequent oxidation of 9b, 10b into benzofuroxans 9b and 10b respectively, have been described previously.39–45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:1) and dried. Yield 0.73 g (2.1 mmol, 68%), red solid, mp 223–234 °C. HPLC (LW = 300 nm, gradient B 20/60% (30 min)) tR = 10.3 min, purity 95.2%. λmax (EtOH)/nm 222, 299, 361, 479. IR νmax, (film)/cm 3393, 2980, 2911, 2718, 2493, 1710, 1600, 1519, 1505, 1456, 1389, 1324, 1316, 1268, 1192, 1151, 1030, 996, 953. 1H NMR (400 MHz; DMSO-d6; Me4Si) δ 9.65 (2H, br s, NH2+), 8.32 (1H, d, J = 9.5 Hz, H-5), 7.88 (1H, d, J = 9.5 Hz, H-6), 7.70–7.44 (6H, m, C6H5, H-8), 3.82–3.75 (4H, m, CH2), 3.48–3.39 (4H, m, CH2); 13C NMR (100 MHz; DMSO-d6) δ 151.9 (7-C), 139.9 (3-C), 138.0 (10-C), 132.3 (9-C), 130.7 (4′-CH), 130.4 (2 × 2′CH), 128.4 (2 × 3′CH), 127.9 (1′-C), 123.6 (5-CH), 121.5 (6-CH), 120.1 (2-C), 111.4 (CN), 98.6 (8-CH), 43.8 (2 × CH2), 42.1 (2 × CH2). HRMS (ESI) calculated for C19H18N5O2+ (M + H)+ 348.1455, found. 348.1441.:3:1) and dried. The yellow solid was collected by filtration, successively washed with acetone, Et2O, n-hexane and dried. Yield 0.1 g (0.3 mmol, 66%), yellow solid, mp 218–220 °C. λmax (EtOH)/nm 220, 242, 291, 359. HPLC (LW = 290 nm, gradient B 15/70% (35 min)) tR = 15.7 min, purity 97.7%. 1H NMR (400 MHz; DMSO-d6; Me4Si) δ 11.48 (1H, br s, NH+), 8.28 (1H, d, J = 13.0 Hz; H-5), 7.82 (1H, d, J = 8.3 Hz, H-8), 7.75–7.70 (2H, m, C6H5), 7.64–7.59 (3H, m, C6H5), 3.91–3.81 (2H, m, CH2), 3.60–3.43 (4H, br m, CH2), 3.32–3.23 (2H, br m, CH2); 13C NMR (100 MHz; DMSO-d6) δ 158.3 (d, J = 260.1 Hz, 6-CF), 143.3 (d, J = 10.7 Hz, 7-C), 141.9 (3-C), 134.8 (d, J = 11.4 Hz, 10-C), 134.6 (9-C), 130.9 (4′-CH), 130.2 (2 × 2′CH), 128.5 (2 × 3′CH), 127.6 (1′-C), 120.1 (2-C), 111.1 (CN), 107.2 (d, J = 28.9 Hz, 5-CH), 106.9 (8-CH), 51.8 (2 × CH2), 46.5 (2 × CH2), 41.9 (CH3). HRMS (ESI) calculated for C20H19FN5O2+ (M + H)+ 380.1517, found. 380.1491.:3) and dried. The orange solid was collected by filtration, successively washed with acetone, Et2O, n-hexane and dried. Yield 0.98 g (2.4 mmol, 82%), yellow solid, mp 215–217 °C. λmax (EtOH)/nm 224, 296, 359. HPLC (LW = 3290 nm, gradient B 20/60% (33 min)) tR = 12.7 min, purity 98.5%. 1H NMR (400 MHz; DMSO-d6; Me4Si) δ 9.96 (1H, br s, NH+), 8.54 (1H, s, H-5), 7.95 (1H, s, H-8), 7.73 (2H, br m, C6H5), 7.62 (3H, br m, C6H5), 4.03–3.98 (4H, br m, CH2), 3.76 (2H, br d, J = 10.9 Hz, CH2), 3.63 (2H, br d, J = 10.9 Hz, CH2), 2.94 (3H, s, CH3), 2.40 (3H, s, CH3SO3−); 13C NMR (100 MHz; DMSO-d6) δ 151.1 (7-C), 142.5 (3-C), 136.5 (6-CCl), 135.3 (9-C), 135.0 (10-C), 131.2 (4′-CH), 130.3 (2 × 2′CH), 128.6 (2 × 3′CH), 127.5 (1′-C), 122.1 (8-CH), 120.7 (2-C), 111.1 (CN), 109.2 (5-CH), 52.4 (CH2), 47.7 (CH2), 42.4 (CH3), 39.8 (CH3SO3−). HRMS (ESI) calculated for C20H19ClN5O2+ (M + H)+ 396.1222, found 396.1259.
:3:1) and dried. Yield 0.73 g (2.1 mmol, 68%), red solid, mp 223–234 °C. HPLC (LW = 300 nm, gradient B 20/60% (30 min)) tR = 10.3 min, purity 95.2%. λmax (EtOH)/nm 222, 299, 361, 479. IR νmax, (film)/cm 3393, 2980, 2911, 2718, 2493, 1710, 1600, 1519, 1505, 1456, 1389, 1324, 1316, 1268, 1192, 1151, 1030, 996, 953. 1H NMR (400 MHz; DMSO-d6; Me4Si) δ 9.65 (2H, br s, NH2+), 8.32 (1H, d, J = 9.5 Hz, H-5), 7.88 (1H, d, J = 9.5 Hz, H-6), 7.70–7.44 (6H, m, C6H5, H-8), 3.82–3.75 (4H, m, CH2), 3.48–3.39 (4H, m, CH2); 13C NMR (100 MHz; DMSO-d6) δ 151.9 (7-C), 139.9 (3-C), 138.0 (10-C), 132.3 (9-C), 130.7 (4′-CH), 130.4 (2 × 2′CH), 128.4 (2 × 3′CH), 127.9 (1′-C), 123.6 (5-CH), 121.5 (6-CH), 120.1 (2-C), 111.4 (CN), 98.6 (8-CH), 43.8 (2 × CH2), 42.1 (2 × CH2). HRMS (ESI) calculated for C19H18N5O2+ (M + H)+ 348.1455, found. 348.1441.:3:1) and dried. The yellow solid was collected by filtration, successively washed with acetone, Et2O, n-hexane and dried. Yield 0.1 g (0.3 mmol, 66%), yellow solid, mp 218–220 °C. λmax (EtOH)/nm 220, 242, 291, 359. HPLC (LW = 290 nm, gradient B 15/70% (35 min)) tR = 15.7 min, purity 97.7%. 1H NMR (400 MHz; DMSO-d6; Me4Si) δ 11.48 (1H, br s, NH+), 8.28 (1H, d, J = 13.0 Hz; H-5), 7.82 (1H, d, J = 8.3 Hz, H-8), 7.75–7.70 (2H, m, C6H5), 7.64–7.59 (3H, m, C6H5), 3.91–3.81 (2H, m, CH2), 3.60–3.43 (4H, br m, CH2), 3.32–3.23 (2H, br m, CH2); 13C NMR (100 MHz; DMSO-d6) δ 158.3 (d, J = 260.1 Hz, 6-CF), 143.3 (d, J = 10.7 Hz, 7-C), 141.9 (3-C), 134.8 (d, J = 11.4 Hz, 10-C), 134.6 (9-C), 130.9 (4′-CH), 130.2 (2 × 2′CH), 128.5 (2 × 3′CH), 127.6 (1′-C), 120.1 (2-C), 111.1 (CN), 107.2 (d, J = 28.9 Hz, 5-CH), 106.9 (8-CH), 51.8 (2 × CH2), 46.5 (2 × CH2), 41.9 (CH3). HRMS (ESI) calculated for C20H19FN5O2+ (M + H)+ 380.1517, found. 380.1491.:3) and dried. The orange solid was collected by filtration, successively washed with acetone, Et2O, n-hexane and dried. Yield 0.98 g (2.4 mmol, 82%), yellow solid, mp 215–217 °C. λmax (EtOH)/nm 224, 296, 359. HPLC (LW = 3290 nm, gradient B 20/60% (33 min)) tR = 12.7 min, purity 98.5%. 1H NMR (400 MHz; DMSO-d6; Me4Si) δ 9.96 (1H, br s, NH+), 8.54 (1H, s, H-5), 7.95 (1H, s, H-8), 7.73 (2H, br m, C6H5), 7.62 (3H, br m, C6H5), 4.03–3.98 (4H, br m, CH2), 3.76 (2H, br d, J = 10.9 Hz, CH2), 3.63 (2H, br d, J = 10.9 Hz, CH2), 2.94 (3H, s, CH3), 2.40 (3H, s, CH3SO3−); 13C NMR (100 MHz; DMSO-d6) δ 151.1 (7-C), 142.5 (3-C), 136.5 (6-CCl), 135.3 (9-C), 135.0 (10-C), 131.2 (4′-CH), 130.3 (2 × 2′CH), 128.6 (2 × 3′CH), 127.5 (1′-C), 122.1 (8-CH), 120.7 (2-C), 111.1 (CN), 109.2 (5-CH), 52.4 (CH2), 47.7 (CH2), 42.4 (CH3), 39.8 (CH3SO3−). HRMS (ESI) calculated for C20H19ClN5O2+ (M + H)+ 396.1222, found 396.1259.Cell lines and antiproliferative assay
The MCF7 (ATCC HTB-22) and MDA-MB-231 (ATCC HTB-26) human breast cancer cells and K562 (ATCC CCL-243) chronic myelogenous leukaemia cells were obtained from the ATCC collection. The multidrug-resistant (MDR) subline K562/4 (kind gift of Dr Alexander Shtil, Blokhin National Medical Research Center of Oncology, Russia) was obtained by stepwise selection of K562 cells for survival under the continuous exposure to Dox. This subline expresses the MDR1 gene and functional P-glycoprotein and is characterized by a high resistance index for Dox.46 The MCF7 and MDA-MB-231 cells were cultured in standard 4.5 g L−1 glucose DMEM medium (Gibco) supplemented with 10% FCS (HyClone), 2 mM L-glutamine, 50 U mL−1 penicillin, 50 μg mL−1 streptomycin (PanEco), and 100 μg mL−1 sodium pyruvate (Santa Cruz) at 37 °C, 5% CO2, and 80–85% humidity in NuAir incubator. Suspension myelogenous leukaemia cells (K562, K562/4) were propagated in RPMI-1640 (PanEco) with 5% FCS (HyClone), 2 mM L-glutamine, 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin at 37 °C, 5% CO2, and 80–85% humidity in Binder incubator. Cells in the logarithmic phase of growth were used in the experiments. The growth inhibitory activity of compounds was assessed by the MTT test based on the metabolism of the MTT reagent (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) (Applichem) in living cells, with modifications as described previously in ref. 9.Briefly, the cells were seeded in 24-well plates (Corning) in 900 μL of the standard medium. Compounds at different concentrations in 100 μL of the appropriate medium were added, and the cells were grown for 72 h. For assessment of hypoxic cytotoxicity ratio, hypoxia (1% O2) conditions were simulated in Binder multigas incubator, as described in ref. 9. After incubation with compounds, the medium was removed, the MTT reagent that was dissolved in the medium was added to the final concentration of 0.2 mg mL−1 to each well, and the incubation was performed for 2 h. Then the cell supernatants were removed and purple formazan crystals were dissolved in DMSO (350 μL per well). Culture plates were gently shaken, and the absorbance was measured at 571 nm with a reference wavelength of 630 nm on a MultiScan reader (ThermoFisher). The viability of the cells was expressed as a percentage of control. Dose–response curves were analyzed by regression analysis using sigmoid curves (log(concentration) vs. normalized absorbance). In this study, the half-maximal inhibitory concentration (IC50) values were determined using GraphPad Prism (Version 7.0).
Immunoblotting
MCF7 cells were washed twice, and incubated for 10 min on ice in the total lysis buffer containing 50 mM Tris–HCl, pH 7.4, 1% SDS, 1% Igepal CA-630, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EDTA, 1 mM PMSF; 1 μg mL−1 each aprotinin, leupeptin, pepstatin; 1 mM Na-orthovanadate and 1 mM NaF. Samples were sonicated 5 times for 5 s each at 30% output, centrifuged for 5 min at 15 000g, and supernatants were then used as total cell extracts. Total protein content was determined by Bradford method.MCF7 cell lysates were separated in 10% SDS-PAGE under reducing conditions, transferred to a nitrocellulose membrane (GE HealthCare), and processed according to a standard protocol. HIF-1α, full and cleaved PARP, ERα and BCL2 antibodies were obtained from Cell Signaling Technology; the antibodies against α-tubulin (Cell Signaling Technology) were added to standardize loading. Goat anti-rabbit IgGs (Jackson ImmunoResearch) conjugated to horseradish peroxidase were used as secondary antibodies. Signals were detected using the ECL reagent as described in Mruk and Cheng's protocol47 and an ImageQuant LAS4000 system (GE HealthCare).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The study was partially financially supported by Russian Foundation for Basic Research, grant numbers 19-33-90186 (to G. I. B. for chemical synthesis), 18-29-09017 (to A. M. S. for ERα immunoblotting), 18-53-34005 (to G. I. B., A. M. S., L. G. D. and A. E. S. for chemical and biological study) and Ministry of Science, Technology and Environment of the Republic of Cuba (to L. M. for biological study). The authors thank Dr A. M. Korolev, N. M. Maliutina, Dr I. V. Ivanov and Dr Y. N. Lusikov (deceased) from Gause Institute of New Antibiotics for HRMS, HPLS analyses, and NMR spectra and D.V. Sorokin from Blokhin National Medical Research Center of Oncology for biological evaluation. The authors would like to thank Falcon Scientific Editing (https://falconediting.com) for proofreading of the text.Notes and references
- J. A. Bertout, S. A. Patel and M. C. Simon, Nat. Rev. Cancer, 2008, 8, 967–975, DOI:10.1038/nrc2540.
- D. M. Gilkes, G. L. Semenza and D. Wirtz, Nat. Rev. Cancer, 2014, 14, 430–439, DOI:10.1038/nrc3726.
- F. Spill, D. S. Reynolds, R. D. Kamm and M. H. Zaman, Curr. Opin. Biotechnol., 2016, 40, 41–48 CrossRef CAS PubMed.
- C. Wigerup, S. Påhlman and D. Bexell, Pharmacol. Ther., 2016, 164, 152–169, DOI:10.1016/j.pharmthera.2016.04.009.
- W. R. Wilson and M. P. Hay, Nat. Rev. Cancer, 2011, 11, 393–410 CrossRef CAS PubMed.
- C. R. Howg, W. R. Wilson and K. O. Hicks, Clin. Cancer Res., 2010, 16, 4946–4957 CrossRef PubMed.
- Q. J. Weng, D. D. Wang, P. Guo, L. Fang, Y. Z. Hu, Q. J. He and B. Yang, Eur. J. Pharmacol., 2008, 581, 262–269 CrossRef CAS PubMed.
- G. I. Buravchenko, A. M. Scherbakov, A. A. Korlukov, P. V. Dorovatovskii and A. E. Shchekotikhin, Cur. Org. Chem., 2020, 17, 29–39 CAS.
- A. M. Scherbakov, A. M. Borunov, G. I. Buravchenko, O. E. Andreeva, L. G. Dezhenkova and A. E. Shchekotikhin, Cancer Invest., 2018, 36, 199–209 CrossRef PubMed.
- G. I. Buravchenko, A. M. Scherbakov, L. G. Dezhenkova, E. E. Bykov, S. E. Solovieva, A. A. Korlukov, D. V. Sorokin, F. L. Monzote and A. E. Schekotikhin, Bioorg. Chem., 2020, 104, 104324 CrossRef CAS PubMed.
- Y. Hu, Q. Xia, S. Shangguan, X. Liu, Y. Hu and R. Sheng, Molecules, 2012, 17, 9683–9696 CrossRef CAS PubMed.
- J. H. Boyer and S. E. Ellzey, J. Org. Chem., 1961, 26, 4684–4685 CrossRef CAS.
- A. J. Boulton, A. C. Gripper, G. R. Katritzky and A. R. Katritzky, J. Chem. Soc., 1965, 1116, 5958–5962 RSC.
- E. Leyva, R. M. González-Balderas, D. A. Loera and R. Jiménez-Cataño, Tetrahedron Lett., 2012, 53, 2447–2461 CrossRef CAS.
- E. A. Chugunova, A. S. Gazizov, A. R. Burilov, L. M. Yusupova, M. A. Pudovik and O. G. Sinyashin, Russ. Chem. Bull., 2019, 68, 887–910 Search PubMed.
- L. M. Lima and D. N. Amaral, Rev. Virtual Quim., 2013, 5, 1075–1100 CAS.
- M. J. Haddadin and C. H. Issidorides, Heterocycles, 1993, 35, 1503–1525 CrossRef CAS.
- C. Jovené, M. Jacquet, J. Marrot, F. Bourdreux, M. E. Kletsky, O. N. Burov, A. M. Gonçalves and R. Goumont, Eur. J. Org. Chem., 2014, 29, 6451–6466 CrossRef.
- E. Pretsch, P. Bullmann and C. Affolter, Structure determination of organic compounds, fourth, revised and enlarged, Springer-Verlag Berlin Heideberg, New York, 2000 Search PubMed.
- P. E. Hansen, Org. Magn. Reson., 1979, 12, 109–142 CrossRef CAS.
- R. Krishna and L. D. Mayer, Eur. J. Pharm. Sci., 2000, 11, 265–283 CrossRef CAS PubMed.
- A. Arora and Y. Shukla, Cancer Lett., 2003, 189, 167–173 CrossRef CAS PubMed.
- A. A. Shtil, Curr. Drug Targets, 2001, 2, 57–77 CrossRef CAS PubMed.
- A. A. Shtil, J. Hematother. Stem Cell Res., 2002, 11, 437–439 CrossRef PubMed.
- M. Diab-Assef, M. J. Haddadin, P. Yared, C. Assaad and H. U. Gali-Muhtasib, Mol. Carcinog., 2002, 33, 198–205 CrossRef CAS PubMed.
- K. Ghattass, S. El-Sitt, K. Zibara, S. Rayes, M. J. Haddadin, M. El-Sabban and H. Gali-Muhtasib, Mol. Cancer, 2014, 13, 12 CrossRef PubMed.
- H. Gali-Muhtasib, M. Sidani, F. Geara, A. D. Mona, J. Al-Hmaira, M. J. Haddadin and G. Zaatari, Int. J. Oncol., 2004, 24, 1121–1131 CAS.
- Q. Weng, J. Zhang, J. Cao, Q. Xia, D. Wang, Y. Hu, R. Sheng, H. Wu, D. Zhu, H. Zhu, Q. He and B. Yang, Invest. New Drugs, 2011, 29, 1177–1187 CrossRef CAS PubMed.
- R. Zamudio-Vázquez, S. Ivanova, M. Moreno, M. I. Hernandez-Alvarez, E. Giralt, A. Bidon-Chanal, A. Zorzano, F. Albericio and J. Tulla-Puche, Chem. Sci., 2015, 6, 4537–4549 RSC.
- T. R. Mielcke, T. C. Muradás, E. C. Filippi-Chiela, M. Eduarda, A. Amaral, L. W. Kist, M. R. Bogo, A. Mascarello, P. D. Neuenfeldt, R. J. Nunes and M. M. Campos, Sci. Rep., 2017, 7, 15850 CrossRef PubMed.
- J. Qi, J. Huang, X. Zhou, W. Luo, J. Xie, L. Niu, Z. Yan, Y. Luo, Y. Men, Y. Chen, Y. Zhang and J. Wang, Chem. Biol. Drug Des., 2019, 93, 617–627 CrossRef CAS PubMed.
- M. A. Sibiya, L. Raphoko, D. Mangokoana, R. Makola, W. Nxumalo and T. M. Matsebatlela, Molecules, 2019, 24, 407–423 CrossRef PubMed.
- K. Ghattass, S. El-Sitt, K. Zibara, S. Rayes, M. J. Haddadin, M. El-Sabban and H. Gali-Muhtasib, Mol. Cancer, 2014, 13, 12–25 CrossRef PubMed.
- P. J. Duriez and G. M. Shah, Biochem. Cell Biol., 1997, 75, 337–349 CrossRef CAS PubMed.
- G. Radha and S. C. Raghavan, Acta Rev. Cancer, 2017, 1868, 309–314 CAS.
- A. Rossi, S. Orecchioni, P. Falvo, V. Tabanelli, E. Baiardi, C. Agostinelli, F. Melle, G. Motta, A. Calleri, S. Fiori, C. Corsini, B. Casadei, S. Mazzara, U. Vitolo, F. Bertolini, P. L. Zinzani, M. Alcalay, P. G. Pelicci, S. Pileri, C. Tarella and E. Derenzini, Leukemia, 2021 DOI:10.1038/s41375-021-01347-6.
- Z. Ding, J. Y. Zhou, W. Z. Wei, V. V. Baker and G. S. Wu, Oncogene, 2002, 21, 4530–4538 CrossRef CAS PubMed.
- Y. Ono, M. Ninomiya, D. Kaneko, A. D. Sonawane, T. Udagawa, K. Tanaka, A. Nishina and M. Koketsu, Bioorg. Chem., 2020, 104, 104245 CrossRef CAS PubMed.
- L. Wang, C. Li, Y. Zhang, C. Qiao and Y. Ye, J. Agric. Food Chem., 2013, 61, 8632–8640 CrossRef CAS PubMed.
- T. Fuente, M. Martín-Fontecha, J. Sallander, B. Benhamu, M. Campillo, R. A. Medina, L. P. Pellissier, S. Claeysen, A. Dumuis, L. Pardo and M. L. Lopez-Rodríguez, J. Med. Chem., 2010, 53, 1357–1369 CrossRef PubMed.
- V. Cecchetti, A. Fravolini, F. Schiaffella, O. Tabarrini and W. Zhou, J. Heterocycl. Chem., 1992, 29, 375–382 CrossRef CAS.
- O. Okamoto, K. Kobayashi, H. Kawamoto, S. Ito, A. Satoh, T. Kato, I. Yamamoto, S. Mizutani, M. Hashimoto, A. Shimizu, H. Sakoh, Y. Nagatomi, Y. Iwasawa, H. Takahashi, Y. Ishii, S. Ozaki and H. Ohta, Bioorg. Med. Chem. Lett., 2008, 18, 3278–3281 CrossRef CAS PubMed.
- R. G. Glushkov, T. I. Vozyakova, E. V. Adamskaya, S. A. Aleinikova, T. P. Radkevich, L. D. Shepilova, E. N. Padeiskaya and T. A. Gus'kova, Pharm. Chem. J., 1994, 28, 17–20 CrossRef.
- M. M. El-Abadelah, M. Z. Nazef, N. S. El-Abadla and H. Meier, Heterocycles, 1995, 41, 2203–2219, DOI:10.3987/COM-95-7128.
- S. K. Kotovskaya, S. A. Romanova, V. N. Charushin, M. I. Kodess and O. N. Chupakhin, J. Fluorine Chem., 2004, 125, 421–428 CrossRef CAS.
- A. E. Shchekotikhin, L. G. Dezhenkova, O. Yu. Susova, V. A. Glazunova, Y. N. Luzikov, Y. B. Sinkevich, V. N. Buyanov, A. A. Shtil and M. N. Preobrazhenskaya, Bioorg. Med. Chem., 2007, 15, 2651–2659 CrossRef CAS PubMed.
- D. D. Mruk and C. Y. Cheng, Spermatogenesis, 2011, 1, 121–122 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra07978f |
| This journal is © The Royal Society of Chemistry 2021 |